

Abstract
The eukaryotic 43S pre-initiation complex (PIC) containing Met- in a ternary complex (TC) with eIF2-GTP scans the mRNA leader for an AUG codon in favorable “Kozak” context. AUG recognition triggers rearrangement of the PIC from an open conformation to a closed state with more tightly bound Met-. Yeast ribosomal protein uS5/Rps2 is located at the mRNA entry channel of the 40S subunit in the vicinity of mRNA nucleotides downstream from the AUG codon or rRNA residues that communicate with the decoding center, but its participation in start codon recognition was unknown. We found that nonlethal substitutions of conserved Rps2 residues in the entry channel reduce bulk translation initiation and increase discrimination against poor initiation codons. A subset of these substitutions suppress initiation at near-cognate UUG start codons in a yeast mutant with elevated UUG initiation, and also increase discrimination against AUG codons in suboptimal Kozak context, thus resembling previously described substitutions in uS3/Rps3 at the 40S entry channel or initiation factors eIF1 and eIF1A. In contrast, other Rps2 substitutions selectively discriminate against either near-cognate UUG codons, or poor Kozak context of an AUG or UUG start codon. These findings suggest that different Rps2 residues are involved in distinct mechanisms involved in discriminating against different features of poor initiation sites in vivo.
Keywords: initiation, accuracy, regulation, scanning, ribosome, uS5, Rps2, yeast
In eukaryotic protein synthesis, a preinitiation complex comprising the 40S ribosomal subunit, initiation factors, and initiator tRNAi scans the mRNA leader for an AUG codon in favorable context. The 40S protein uS5 (yeast Rps2) resides in the entry channel, but its role in start codon selection was unknown. Hinnebusch and Dong find that substituting uS5 residues near the entry channel increases discrimination against non-AUG codons and/or poor sequence context, both features of suboptimal start sites, implicating uS5/Rps2 in initiation accuracy.
Introduction
Accurate identification of the translation initiation codon is crucial for expressing the correct cellular proteins. This process typically operates in eukaryotic cells by a scanning mechanism, wherein the small (40S) ribosomal subunit recruits charged initiator tRNA (Met-) in a ternary complex (TC) with the GTP bound form of eukaryotic initiation 2 (eIF2-GTP) to form the 43S pre-initiation complex (PIC) in a manner stimulated by eIFs 1, 1A, 3, and 5. The 43S PIC then joins the 5’ end of the mRNA and scans the 5’-untranslated region (5’UTR), with Met- not fully engaged with the 40S “P” decoding site (the “POUT” state), for complementarity with the anticodon of Met-, to identify the AUG start codon. Nucleotides surrounding the AUG, particularly at the −3 and +4 positions (the Kozak context), influence the efficiency of AUG selection. eIF2 can hydrolyze GTP in the scanning complex, dependent on GTPase activating protein eIF5, but Pi release from GDP is blocked by eIF1, whose presence also impedes stable binding of Met- in the “PIN” state. Start-codon recognition triggers dissociation of eIF1 from the 40S subunit, allowing Pi release from eIF2-GDP·Pi, and rearrangement to the PIN state of the 48S PIC. Subsequent dissociation of eIF2-GDP and other eIFs from the 48S PIC enables eIF5B-catalyzed subunit joining and formation of an 80S initiation complex with Met- base-paired to AUG in the P site (reviewed in Hinnebusch 2014, 2017; Llacer et al. 2018). eIF1 plays a dual role in the scanning mechanism, promoting rapid TC loading in the POUT conformation while blocking rearrangement to PIN at both near-cognate start codons (e.g., UUG) and cognate (AUG) codons in poor Kozak context. Accordingly, eIF1 must dissociate from the 40S subunit for start codon recognition to be finalized. Consistent with this, structural analyses of partial PICs reveal that eIF1 physically clashes with Met- in the PIN state (Rabl et al. 2011; Lomakin and Steitz 2013), and is both deformed and displaced from its 40S location during the POUT to PIN transition (Hussain et al. 2014). Mutations that weaken eIF1 binding to the 40S subunit elevate initiation at both near-cognate codons (the Sui− phenotype) and AUGs in poor context in vivo, in a manner attributed to destabilizing the open/POUT conformation and favoring inappropriate rearrangement to the closed/PIN state at poor start codons during scanning (Martin-Marcos et al. 2011, 2013). Moreover, decreasing wild-type (WT) eIF1 abundance reduces initiation accuracy (the Sui− phenotype), whereas overexpressing eIF1 suppresses initiation at near cognates or AUGs in poor context (Valasek et al. 2004; Alone et al. 2008; Ivanov et al. 2010; Saini et al. 2010; Martin-Marcos et al. 2011). Eukaryotic cells exploit the mechanistic link between eIF1 abundance and initiation accuracy to autoregulate eIF1 expression, as the AUG codon of the eIF1 gene (SUI1 in yeast) occurs in poor context and the frequency of its recognition is inversely related to eIF1 abundance (Ivanov et al. 2010; Martin-Marcos et al. 2011).
The stability of the codon-anticodon duplex is an important determinant of initiation accuracy, as the rate of the POUT to PIN transition is accelerated, and the PIN state is stabilized, in presence of AUG vs non-AUG start codons (Kolitz et al. 2009). Favorable Kozak context might also contribute to PIN stability (Pisarev et al. 2006; Martin-Marcos et al. 2011), but the stimulatory effect of optimum context on initiation rate is not well understood. There is evidence that it requires the α-subunit of eIF2 in the mammalian system (Pisarev et al. 2006), and structural analyses of partial mammalian 43S (Hashem et al. 2013) and yeast 48S PICs (Hussain et al. 2014) place domain 1 (D1) of eIF2α near the AUG codon, with conserved arginine residues R55 and R57 of yeast eIF2α in proximity to the key −3 residue of the “Kozak” context. We found however that R55 and R57 substitutions reduce recognition of the poor-context AUG codon of SUI1 mRNA and also suppress enhanced initiation at UUG start codons in a Sui− mutant, conferring the Ssu− phenotype; and biochemical experiments confirmed that these substitutions destabilize TC binding at a UUG codon in the PIN state. Thus, in stabilizing the closed, PIN conformation of the PIC, R55/R57 interactions with mRNA enhance utilization of poor start codons rather than start codons with optimum Kozak context (Thakur et al. 2020). The conserved β-hairpin of the 40S ribosomal protein uS7 (Rps5 in yeast) also lies in proximity to the Kozak context, and the C-terminal helix of Rps5/uS7 interacts directly with D1 of eIF2α. The precise conformation of their interaction surface varies between two partial yeast 48S (py48S) complexes that appear to represent the open/POUT conformation conducive to scanning (py48S-open) and the closed/PIN state following AUG selection (py48S-closed) (Llacer et al. 2015). Genetic analysis showed that these different uS7/eIF2α-D1 contacts stabilize first the open conformation, and then the closed state of the PIC, to calibrate usage of near-cognate UUG start codons in vivo (Visweswaraiah and Hinnebusch 2017).
In addition to the role of uS7/Rps5 at the mRNA exit channel just described, the uS3/Rps3 protein found at the mRNA entry channel of the 40S subunit also participates in start codon selection. Substitutions of five conserved basic residues of uS3, which contact the mRNA as it enters the 40S subunit, reduce bulk initiation and suppress initiation at UUG start codons (Ssu− phenotype). Biochemical evidence indicates that two such residues, arginines R116 and R117, functionally cooperate with the a-subunit of the eIF3 complex to promote PIC:mRNA contacts at the 40S entry channel, which appear to stabilize the closed/PIN conformation to enable initiation at poor start codons (Dong et al. 2017).
The uS5/Rps2 protein is also located at the mRNA entry channel, where it directly contacts uS3/Rps3 and the mRNA (Supplementary Figure S1). In the recent py48S-5N structure, depicting a closed conformation of the PIC following selection of the AUG codon and dissociation of eIF1 (Llacer et al. 2018), 11 residues of Rps2/uS5 occur in proximity to mRNA nucleotides in the entry channel located 8–12 nt downstream of the AUG codon. Three of these residues, V80, I83, and F98, are located in strands β-1 or β-2 of the extended β-hairpin loop of Rps2, which forms part of the mRNA entry channel, and are within 4–5Å of the mRNA (Figure 1, A and B, red asterisks). Five other residues in this same hairpin are 6–8Å from the mRNA (D78, N82, Q87, Q94, and T96) (Figure 1B, gray asterisks); and three additional residues (V121, I125, and R126) in proximity to the same mRNA nucleotides reside at the N-terminus of the adjoining helix α4, found at the entry channel pore (Figure 1, A and B, red asterisks). Glu-120 (E120) in α4 is additionally within 4 Å of residue R116 of Rps3/uS3 at the entry channel pore (Supplementary Figure S1, green $), mentioned above for its role in stabilizing PIC:mRNA interactions that promote selection of near-cognate start codons (Dong et al. 2017). We hypothesized that Rps2 residues in proximity to mRNA play a similar role in 40S·mRNA interaction at the entry channel.
Figure 1.

Conserved uS5/Rps2 residues in proximity to the mRNA or rRNA at the 40S mRNA entry channel. (A) Cartoon depiction of the segment of Rps2 at the entry channel and its interactions with mRNA or rRNA constructed in PyMol from a partial yeast 48S complex in closed conformation (PDB: 6FYY), showing selected rRNA residues in h28 or the h28–h44 linker (wheat or pale yellow), mRNA nucleotides from +6 to +17 relative to the AUG (+1) (magenta), Rps3-R116 (green), and selected Rps2 residues (orange, cyan, or black). Side-chains are shown for Rps2 residues subjected to mutagenesis and colored black if only lethal substitutions were identified, light cyan if nonlethal substitutions were identified that increase discrimination against near-cognate UUG codons, and dark cyan (T90, Q94, and T96) for nonlethal substitutions that exclusively increase discrimination against start codons in poor Kozak context. (B) Multiple sequence alignment of Rps2 from the indicated species, with residues colored according to the chemical properties of the side chains, with boundaries and numbering of α-helices and β-strands indicated above the residue numbering for the S. cerevisiae sequence. Residues are annotated above the sequence as follows: red asterisks, 4–5Å from mRNA; gray asterisks, 6–8Å from mRNA; #, 4–5Å from rRNA; green $, 4 Å from Rps3-R116; gray $, 6 Å from Rps3-R116; black squares, recessive lethal; black ovals, dominant lethal; gray squares, Slg−; cyan ovals, strong Ssu−; cyan triangles, weak Ssu−.
Other Rps2 residues of interest, Q89, T90, and R91, occur in the loop of the β1-β2 hairpin and contact rRNA residues belonging to the first three base pairs in helix 28 (U1145:A1633, G1146:C1632, A1147:U1630 S.cerevisiae numbering) or A1635 in the linker connecting helix 28 (h28) to helix 44 (h44) (Figure 1A). The h28 forms the “neck” connecting the head and body domains of the 40S subunit, and we showed previously that substitutions in certain h28 rRNA residues perturb binding of TC and AUG recognition during scanning (Dong et al. 2008). Thus, we also considered that substituting these Rps2 residues could alter the fidelity of start codon recognition. Here, we provide genetic evidence that these latter residues, and other Rps2 residues in the vicinity of mRNA in the 40S entry channel mentioned above, all promote the closed conformation of the PIC in a manner required for efficient recognition of poor initiation sites in vivo. Interestingly, certain of these residues appear to discriminate specifically against near-cognate UUG start codons, while others primarily disfavor start codons in poor Kozak context, suggesting distinct molecular mechanisms for these two aspects of initiation fidelity.
Materials and methods
Plasmid constructions
Plasmids used in this work are listed in Supplementary Table S2. Plasmid pDH12-1 was constructed as follows. The RPS2 gene, including 528 bp upstream of the ATG start codon and 220 bp downstream of the stop codon, was amplified by PCR using the two primers 5′-GAA TGC GGC CGC GAA GAA GGT TTA-3′ and 5′-AAC TGC AGC GTT TGA TAG GCAAAA G-3′ and genomic DNA from yeast WT strain BY4741 as template. The ∼1.5-kb amplicon was digested with NotI and PstI, and the resulting fragment was inserted between the NotI and PstI sites of plasmid pRS315 to produce pDH12-1. The DNA sequences of the entire open reading frame (ORF), as well as the 5′-noncoding region and 200 bp downstream of the stop codon, were verified. pDH12-50 was constructed by inserting the NotI-PstI fragment containing RPS2 isolated from pDH12-1 between the NotI and PstI sites of pRS316. It was shown that both pDH12-1 and pDH12-50 fully rescue growth of PGAL-RPS2 strain HD2729 on glucose medium. Plasmids pDH12-72, pDH12-78, pDH13-80, pDH12-73, pDH12-75, pDH12-76, pDH12-79, pDH12-80, pDH13-81, pDH13-82, pDH12-81, pDH13-83, pDH12-96, pDH12-97, pDH13-85, pDH13-100, pDH13-86, pDH13-88, pDH13-87, pDH12-89, pDH13-89, pDH13-90, pDH13-92, pDH13-91, pDH12-90, pDH12-92, pDH13-93, pDH13-94, pDH13-95, pDH13-96, pDH12-93, pDH13-97, and pDH12-94 were derived from pDH12-1 by site-directed mutagenesis using the QuikChange XL Kit (Stratagene) and the corresponding primers in Supplementary Table S3, and the mutations were verified by sequencing the entire ORF and both the 5′ noncoding region and region downstream of the stop codon.
Yeast strain constructions
Yeast strains used in this work are listed in Supplementary Table S1. Strain HD2729 was derived from H2995 by replacing the promoter of chromosomal RPS2 with the GAL1 promoter by one-step gene replacement, as follows. Primers 5′-GCA CGC AGC TTC CCA GGA CGC CTA GCT ATT TTT TCA TCT ATT CCC CTC TGT AGT AAC GTA AGA GTT TTC GAA TTC GAG CTC GTT TAA AC-3′ and 5′-CTG TTT GGA CGG CCT CTG TTA CGG CCA CCG AAA CCA CCT CTC TTT TGT TGT TGA GCT TCT GGA GCA GAC ATT TTG AGA TCC GGG TTT T′ were used to amplify by PCR the appropriate DNA fragment from plasmid pFA6a-kanMX6-PGAL1 (p3218), which was used to transform strain H2995 to kanamycin resistance. The presence of PGAL1-RPS2 in the resulting strain (HD2738) was established by demonstrating that the lethality on glucose medium can be complemented by low-copy (lc) RPS2 LEU2 plasmid pDH12-1; and also confirmed by PCR analysis of chromosomal DNA with the appropriate primers.
Strains HD3365, HD3367, HD3391, HD3392, HD3410, HD3385, HD3386, HD3387, HD3369, HD3388, HD3393, HD3394, HD3412, HD3429, HD3378, HD3379, HD3396, HD3519, HD3397, HD3400, HD3873, HD3874, HD3389, HD3390, HD3401, HD3402, HD3404, HD3403, HD3871, HD3382, HD3405, HD3406, HD3407, HD3408, HD3376, HG3398, and HD3377 were derived from HD2729 by transformation with a LEU2 plasmid containing the appropriate RPS2 allele, as indicated in Supplementary Table S2. Strains HD3414, HD3415, HD3409, HD3426, HD3424, HD3411, HD3428, HD3412, HD3429, HD3419, HD3420, HD3430, HD3431, HD3453, HD3421, HD3422, HD3455, HD3457, HD3461, HD3459, HD3416, HD3423, HD3516, HD3517, and HD3467 were derived from the foregoing strains by transformation with single-copy (sc) TRP1 SUI5 plasmid p4281, or empty vector YCplac22, as indicated in Supplementary Table S1.
To produce strain HD2976, pDH12-50 (lc URA3 RPS2+) was introduced into the RPS2/rps2Δ diploid strain YSC1021-674246 purchased from Research Genetics. The Ura+ transformants were sporulated and subjected to tetrad analysis. HD2976 was identified as a KanR Ura+ ascospore clone incapable of growth on medium containing 5-fluoroorotic acid (5-FOA) unless transformed with RPS2+ LEU2 plasmid pDH12-1 but not with empty LEU2 vector pRS315. The presence of rps2Δ::kanMX in HD2976 was verified by PCR analysis of chromosomal DNA. The strains HD3867, HD3868, HD33869, HD3870, HD3873, and HD3874 were derived from HD2976 by plasmid shuffling to replace pDH12-50 with the appropriate lc LEU2 plasmid containing the appropriate RPS2 allele, as indicated in Supplementary Table S2.
Biochemical methods
Assays of β-galactosidase activity in whole-cell extracts (WCEs) were performed as described previously (Moehle and Hinnebusch 1991). For Western analysis, WCEs were prepared by trichloroacetic acid extraction as described (Reid and Schatz 1982) and immunoblot analysis was conducted as described previously (Martin-Marcos et al. 2011) with antibodies against eIF1 (Valasek et al. 2004) and Hcr1 (Valášek et al. 2001). The signal intensities were quantified using a LI-COR Odyssey infrared scanner.
For polysome analysis, the rps2Δ::KanMX deletion strains harboring plasmid-borne WT RPS2+ (HD3867), or rps2 alleles HD3869 (Q89K), HD3870 (K119A), HD3871(V121D), HD3872 (I125K), HD3873 (Q94D), and HD3874 (T96K) were cultured in SC-Leu medium at 30°C to A600 ≈1. Cycloheximide was added to 50 μg/mL 5 min before harvesting, and WCE was prepared in breaking buffer (20 mM Tris⋅HCl, pH 7.5, 50 mM KCl, 10 mM MgCl2, 1 mM DTT, 5 mM NaF, 1 mM phenylmethylsulfonyl fluoride, 1 cOmplete EDTA-free Protease Inhibitor Tablet (Roche) per 50 mL buffer). Fifteen A260 units of WCE were separated by velocity sedimentation on a 4.5%–45% sucrose gradient by centrifugation at 39,000 rpm for 3 h in an SW41Ti rotor (Beckman). Gradient fractions were scanned at 254 nm to visualize ribosomal species. To analyze total 40S/60S profiles, the same strains were cultured in yeast extract peptone dextrose (YEPD) at 30°C to A600≈1, but cycloheximide was omitted. WCEs were prepared in the absence of Mg+2, and 15 A260 units of WCE were resolved by velocity sedimentation through 5–30% (wt/vol) sucrose gradients at 40,000 rpm for 4 h in an SW41Ti rotor (Beckman).
Results
RPS2 mutations restore discrimination against near-cognate UUG codons in a hypoaccurate eIF5 Sui− mutant in vivo
To examine the role of uS5/Rps2 residues in start codon selection in vivo, we constructed 39 RPS2 mutations that introduced single substitutions of residues found in proximity to mRNA or rRNA residues visualized in the py48S-5N structure (Llacer et al. 2018). These residues are located between Q77 and F98 in the β1-β2 hairpin, between K119 and R126 in the linker between β3 and α4, or at the N-terminus of α4 (Figure 1A), and all are highly conserved among diverse eukaryotes (Figure 1B). Residues were substituted with Ala to shorten the side-chain, or with acidic or basic residues to alter side-chain charge (Table 1, column 1). The mutations were generated in an RPS2 allele under its own promoter on a low-copy plasmid and examined in a yeast strain containing wild-type (WT) chromosomal RPS2 under a galactose-inducible promoter (PGAL1). Mutant phenotypes were scored following a switch from galactose to glucose, where chromosomal PGAL1-RPS2+ expression is repressed.
Table 1.
Summary of phenotypes of all RPS2 alleles
| 1 | 2 | 3 | 4 | 5 | 6 | 7 |
|---|---|---|---|---|---|---|
| RPS2 allele | Cell Growth on SC at 30°Ca | His+/ Sui− b | Suppression of SUI5 His+ (Ssu−) | Suppression of SUI5 Slg− at 37°C | HIS4-lacZ UUG:AUG in SUI5 cells | Suppression of SUI5 UUG:AUG (Ssu−) |
| WT | ++++ | None | None | None | 0.17 ± 0.001 | None |
| Q77D | Dom. Lethal | NA | NA | NA | NA | NA |
| Q77K | ++++ | None | None | None | ND | ND |
| D78K | ++++ | None | None | ND | ND | ND |
| D78A | ++++ | None | Moderate | Strong | 0.025 ± 0.002 | Strong |
| E79K | ++++ | None | None | ND | ND | ND |
| V80D | Dom. Lethal | NA | NA | NA | NA | NA |
| V80K | Lethal | NA | NA | NA | NA | NA |
| N82D | ++++ | None | Weak | None | ND | ND |
| N82K | Lethal | NA | NA | NA | NA | NA |
| I83D | ++++ | None | None | None | NA | NA |
| I83K | Lethal | NA | NA | NA | NA | NA |
| P85K | ++++ | None | None | ND | ND | ND |
| P85D | ++++ | None | Weak | None | 0.144 ± 0.011 | None |
| Q87D | Dom. Lethal | NA | NA | NA | NA | NA |
| Q87K | ++++ | None | Weak | Weak | 0.346 ± 0.026 | None |
| Q89K | ++++ | None | Strong | Strong | 0.021 ± 0.002 | Strong |
| Q89D | Dom. Lethal | NA | NA | NA | NA | NA |
| T90D | ++ | None | ? | Moderate | 0.351 ± 0.048 | None |
| T90R | ++++ | None | Weak | None | 0.654 ± 0.102 | None |
| R91D | ++++ | None | Weak | None | ND | ND |
| A92D | ++++ | None | Weak | None | ND | ND |
| Q94D | +++ | None | Weak | Strong | 0.258 ± 0.037 | Nonec |
| Q94K | ++++ | None | ? | None | ND | ND |
| T96D | Dom. Lethal | NA | NA | NA | NA | NA |
| T96K | ++++ | None | Weak | Moderate | 0.278 ± 0.046 | Nonec |
| F98D | Lethal | NA | NA | NA | NA | NA |
| F98K | Lethal | NA | NA | NA | NA | NA |
| K119D | ++++ | None | Moderate | none | 0.202 ± 0.036 | None |
| K119A | ++++ | None | Moderate | Strong | 0.020 ± 0.002 | Strong |
| E120K | ++ | None | ? | Moderate | 0.664 ± 0.081 | None |
| E120A | ++++ | None | Moderate | Moderate | 0.141 ± 0.013 | None |
| V121D | ++ | None | ? | Moderate | 0.044 ± 0.0014 | Strong |
| V121K | + | None | ? | ? | ND | ND |
| G123K | ++++ | None | None | None | 0.096 ± 0.0006 | Moderate |
| G123D | ++++ | None | None | None | 0.092 ± 0.0006 | Moderate |
| I125K | + | None | ? | Moderate | 0.022 ± 0.0005 | Strong |
| I125D | Lethal | NA | NA | NA | NA | NA |
| R126A | Dom. Lethal | NA | NA | NA | NA | NA |
| R126D | ++++ | None | ND | ND | ND | ND |
Growth rate of viable mutants relative to the WT strain was summarized qualitatively with different numbers of plus signs.
As judged by plate tests in strains lacking SUI5.
Suppression was observed on assaying the ACG-UUG HIS4-lacZ reporter (Figure 4C).
?, suppression could not be evaluated because the mutation conferred a marked slow-growth phenotype in the presence of SUI5 on medium containing histidine at 30∘C; NA, not applicable; ND, not determined.
Six of the resulting Rps2 substitutions were recessive lethal, as the yeast strains harboring the corresponding RPS2 alleles could not support growth following the shift from galactose to glucose, including V80K, N82K, I83K, F98D, F98K, and I125D (Supplementary Figure S2; summarized in Table 1, column 2; marked with black squares in Figure 1B). Six other Rps2 substitutions confer dominant-lethal phenotypes, as the transformants harboring such alleles could not be obtained on galactose-containing medium where PGAL1-RPS2+ is expressed (Table 1, column 2; black ovals in Figure 1B). Except for R126A, the other five dominant-lethal substitutions (Q77D, V80D, Q87D, Q89D, and T96D) replace noncharged residues with negatively charged Asp, which might introduce deleterious electrostatic repulsion with nearby mRNA or rRNA nucleotides. This interpretation is consistent with the finding that substituting these residues with basic lysine, instead of acidic aspartic acid, is either viable (Q77K, Q87K, Q89K, and T96K) or confers recessive rather than dominant lethality (V80K) (Table 1, column 2). Recessive lethality presumably indicates a less severe defect than dominant lethality, as cells can survive with 40S subunits containing WT Rps2 produced during growth on galactose despite the presence of the mutant 40S subunits harboring a recessive-lethal Rps2 variant. Exceptions to this pattern of lethality include the F98K and F98D substitutions, both of which are recessive lethal; and the N82K/N82D and I83K/I83D pairs of substitutions where Lys, but not Asp substitutions, are recessive lethal. It is possible that Lys substitutions at these latter three positions in Rps2 introduce an electrostatic attraction with nearby mRNA or rRNA nucleotides that hinders an aspect of scanning or start codon recognition through an excessively stable PIC:mRNA association, or by perturbing rRNA structure and function. The I125K substitution and five other nonlethal substitutions, T90D, Q94D, E120K, V121D, and V121K all confer slow-growth phenotypes on glucose medium (Supplementary Figure S2; Table 1, column 1; Figure 1B, gray squares). As described below, three of these substitutions (I125K, Q94D, and V121D) and five others that do not impair growth (D78A, Q89K, T90R, T96K, and K119A) alter some aspect of the accuracy of start codon selection and thus appear to perturb the function of Rps2 during scanning.
To assess changes in initiation accuracy, the nonlethal RPS2 mutations were tested for the ability to suppress the histidine auxotrophy of the his4-301 allele, containing ACG in place of the AUG start codon, by increasing initiation at the third, in-frame UUG codon and thereby restoring expression of the histidine biosynthetic enzyme His4. Suppression of the histidine auxotrophy conferred by his4-301 by this mechanism indicates the Sui− phenotype (suppressor of initiation codon mutation) (Donahue and Cigan 1988). None of the RPS2 mutations allowed detectable growth on glucose medium lacking histidine (Table 1, column 3), suggesting that they do not demonstrably increase utilization of UUG start codons. Accordingly, we tested them for the Ssu− (Supressor of Sui−) phenotype, signifying suppression of the increased UUG initiation on his4-301 mRNA and attendant His+ phenotype conferred by a known Sui− mutation, in this case the dominant SUI5 allele encoding the eIF5-G31R variant (Huang et al. 1997). The His+ phenotype conferred by plasmid-borne SUI5 was evident in the strain containing WT RPS2, allowing growth on medium lacking histidine [Figure 2, A–E, panels (iii), cf. rows 1 and 2 in each panel]. This His+ phenotype was diminished to varying degrees by 19 of the Rps2 substitutions [Figure 2, A–E, (iii), for each panel cf. rows 3 and below with row 1; summarized in Table 1, column 4]. This finding was complicated however for seven of the 19 substitutions by the fact that they confer a slow-growth phenotype in the presence of SUI5 on medium containing histidine [Figure 2, A–E, (i)]. Excluding these ambiguous cases, it appears that the Sui− phenotype of SUI5 is essentially eliminated by Q89K; markedly diminished by D78A, Q94D, T96K, K119A/K119D, and E120A; and reduced weakly by N82D, Q87K, T90R, R91D, A92D, and P85D, indicating a range of Ssu− phenotypes (Table 1, column 4, entries listed as “Strong,” “Moderate,” “Weak,” respectively).
Figure 2.
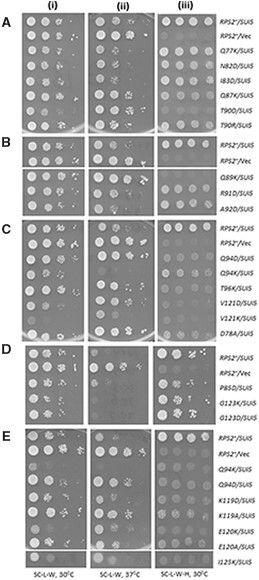
Certain RPS2 alleles suppress both the His+ and Slg− phenotypes conferred by eIF5 Sui− mutation SUI5 in his4-301 cells. (A–E) Serial dilutions of derivatives of PGAL-RPS2 his4-301 strain HD2729 harboring sc SUI5 plasmid p4281 or empty TRP1 vector YCplac22 (Vec), and the indicated plasmid-borne RPS2 allele or empty vector (Vec), were spotted on synthetic complete media lacking leucine and tryptophan with glucose as carbon source (SC-L-W; +His plates) and incubated at 30°C (i) or 37°C (ii), or on SC-L-W-H supplemented with 0.0015 mM histidine (0.5% of the standard supplement) and incubated at 30°C.
SUI5 also confers a slow-growth phenotype (Slg−) in histidine-replete (+His) medium at the elevated temperature of 37°C (Martin-Marcos et al. 2011) [Figure 2, A–E, (ii), cf, rows 1 and 2]; and this phenotype was also diminished by six of the substitutions shown above to confer strong or medium Ssu− phenotypes: D78A, Q89K, Q94D, T96K, K119A, and E120A [Figure 2, A–E, (ii), cf. rows 3 and below with row 1; summarized in Table 1, column 5]. Suppression of the Slg− at 37°C was also observed for T90D, V121D, and E120K, whose Ssu− phenotypes were regarded above as being ambiguous [Figure 2, A–C and E, (ii); Table 1]. The restoration of growth at 37°C is particularly noteworthy for T90D, V121D, and E120K considering that they reduce growth in the presence of SUI5 on His+ medium at 30°C [Figure 2, A, C, and E, cf. results in (i) and (ii)]. Together, these results suggest that the potent Ssu− substitutions D78A, Q89K, Q94D, T96K, K119A, K119D, and E120A are promising candidates for Rps2 variants that diminish the effect of SUI5 in allowing inappropriate selection of UUG start codons. Based on their suppression of the SUI5 Slg− phenotype at 37°C, the ambiguous Ssu− substitutions T90D, R91D, E120K, and V121D might also confer a similar increase in initiation accuracy.
The effect of SUI5 on the fidelity of start codon selection can be quantified by an increase in the expression of a HIS4-lacZ reporter containing a UUG start codon relative to a matched AUG reporter (Figure 3A). As expected, plasmid-borne SUI5 increased the UUG:AUG HIS4-lacZ expression ratio by ∼7- to 8-fold compared to that observed when the same RPS2+ strain carries an empty vector (Figure 3B, columns 1 and 2). Importantly, three of the substitutions with strong His−/Ssu− phenotypes described above, D78A, Q89K, and K119A, conferred substantial reductions in the UUG:AUG expression ratio (Figure 3, B–D; summarized in Table 1, columns 6 and 7). Marked reductions in the UUG/AUG ratio were also conferred by the ambiguous Ssu− substitutions V121D and I125K (Figure 3D; Table 1), of which V121D was also shown above to be a strong suppressor of the Slg− at 37°C of SUI5. These reductions in UUG:AUG ratio were primarily attributable to a greater reduction in expression of the UUG vs AUG reporter for all of these substitutions (Supplementary Figure S3, A–F). Thus, these findings generally support the conclusion that these five Rps2 Ssu− variants, D78A, Q89K, K119A, V121D, and I125K, suppress the hypoaccuracy phenotype of the SUI5 mutation by specifically reducing initiation at the near-cognate UUG triplet.
Figure 3.
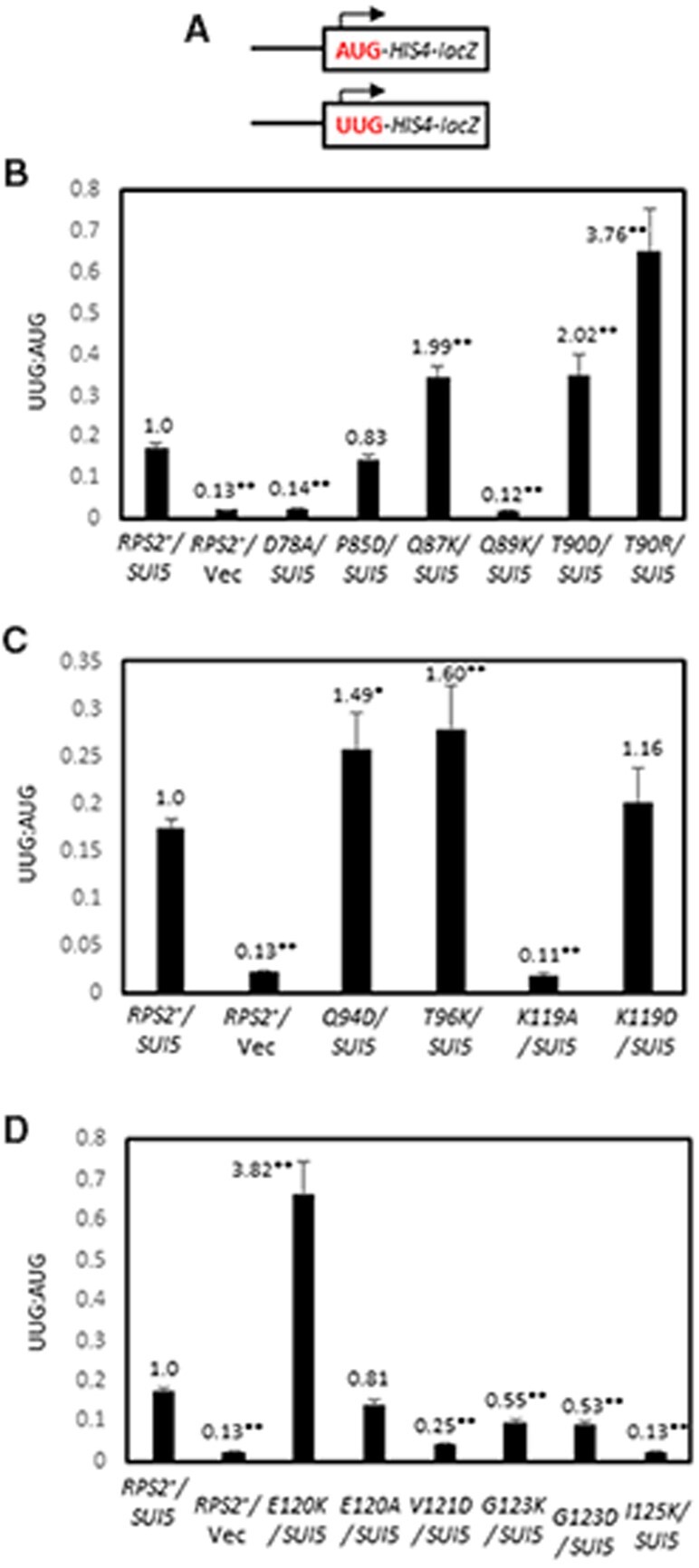
Certain RPS2 alleles suppress the elevated UUG:AUG initiation ratio of HIS4-lacZ reporters conferred by SUI5. (A–D) Derivatives of PGAL-RPS2 his4-301 strain HD2729 with the indicated RPS2 alleles and either SUI5 plasmid p4281 or empty vector, and also harboring one or the other HIS4-lacZ reporters shown schematically in (A) encoding AUG or UUG start codons at the position of the native HIS4 AUG codon (on plasmids p367 and p391, respectively), were cultured in SD+His medium and β-galactosidase specific activities were measured in whole cell extracts (WCEs), in units of nanomoles of o-nitrophenyl-β-D-galactopyranoside (ONPG) cleaved per min per mg of total protein, of six independent transformants. Mean ratios (with S.E.M.s shown as error bars) of expression of the UUG vs AUG reporter were plotted; and the ratios normalized to that obtained for the WT RPS2 SUI5 strain are given above each histogram bar. Asterisks indicate statistically significant differences between each mutant and the WT RPS2 SUI5 strain determined by a two-tailed, unpaired Student’s t-test (*P < 0.05; **P < 0.01).
Surprisingly, the strong Ssu− substitutions Q87K, Q94D, and T96K conferred a modest increase of ≤2-fold, rather than a decrease, in the UUG/AUG ratio in SUI5 cells (Figure 3, B and C). A similar or even larger increase in UUG: AUG ratio of ∼2- to ∼4-fold was also observed for the T90D, T90R, and E120K substitutions (Figure 3, B and D), of which T90D and E120K were shown above to also suppress the Slg− at 37°C but were ambiguous for their His−/ Ssu− phenotypes in SUI5 cells. These increases in UUG:AUG ratio were attributable to small increases in expression of the UUG reporter of <2-fold, coupled with <2-fold decreases for the AUG reporter for each of these anamolous substitutions, except for T90R that increased UUG reporter expression by nearly 3-fold. This discrepancy between suppression of the His+ and/or Slg− (37°C) phenotypes of SUI5 and the inability to reduce the elevated UUG/AUG HIS4-lacZ expression ratio conferred by SUI5 indicates a more complex suppression mechanism for this second group of Ssu− substitutions.
We reasoned that the aforementioned discrepancy might be related to the fact that the standard HIS4-lacZ reporter contains a UUG at both the first and third HIS4 codons, whereas the chromosomal his4-301 allele whose His− phenotype is suppressed by SUI5 contains a UUG only at the third codon and ACG at the first codon. As SUI5 appears to increase initiation only at UUG codons, it is expected to stimulate translation of his4-301 mRNA primarily by increasing recognition of the UUG triplet at the third codon rather than the ACG at the first codon (Huang et al. 1997). This occurs despite the fact that the ACG at codon 1 is in much better Kozak context, (−3AUA−1, G+4), with a context score of 0.701, vs the UUG at codon 3, (−3GUU−1, C+4), with a much lower context score of 0.200 (0.086 is the worst possible context score for yeast) (Miyasaka 1999; Zur and Tuller 2013). Accordingly, we examined the effects of SUI5 and Rps2 Ssu− substitutions on UUG initiation employing a different HIS4-lacZ reporter described previously that mimics his4-301 in containing ACG and UUG at the first and third codons, respectively (Dong et al. 2014).
Owing to the presence of only one UUG start codon, expression of the ACG-UUG HIS4-lacZ reporter is 5- to 10-fold lower than the corresponding UUG-UUG reporter, falling below the detection limit of the assay in WT cells; and its expression is ∼2-fold lower than the UUG-UUG reporter in SUI5 cells. The Ssu− substitutions Q89K and K119A reduced expression of the ACG-UUG reporter in SUI5 cells by ∼2.5-fold for K119A and below the detection limit for Q89K (Figure 4B), consistent with the marked reductions in expression they each conferred for the UUG-UUG HIS4-lacZ reporter (Figure 3B) and their ability to suppress the His+ phenotype of his4-301 (Table 1, columns 3 and 4). Importantly, the Ssu− substitutions Q94D and T96K reduced the UUG:AUG ratio for the ACG-UUG reporter by ∼2.5-fold (Figure 4C), again consistent with their ability to suppress the His+ phenotype of SUI5 (Table 1, columns 3 and 4). Thus, these last two substitutions can be added to the group of five described above that appear to diminish the hypoaccuracy phenotype of SUI5 by decreasing initiation at the near-cognate UUG start codon. The ability of Q94D and T96K to decrease utilization of the UUG triplet at codon 3 (in poor context) in the ACG-UUG reporter, but not the UUG triplet at codon 1 (with much better context) in the standard UUG-UUG reporter, thus appears to reflect increased discrimination against the poor context of the UUG triplet at codon 3.
Figure 4.
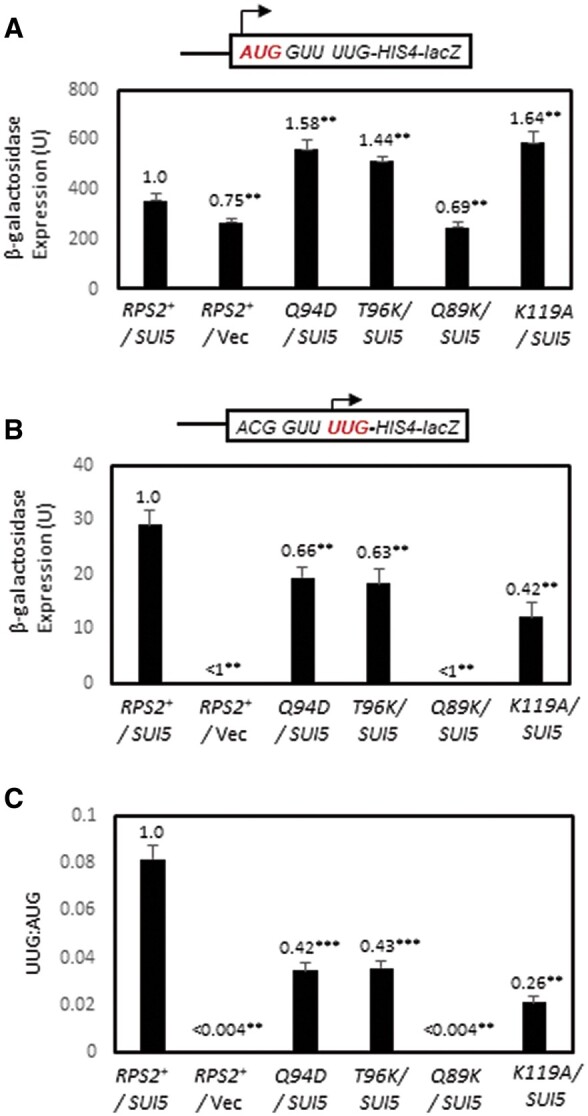
Certain RPS2 alleles suppress initiation at the third, poor-context UUG codon of native HIS4 in SUI5 cells. (A–C) Derivatives of PGAL-RPS2 his4-301 strain HD2729 with the indicated RPS2 alleles and either SUI5 plasmid p4281 or empty vector, and also harboring the HIS4-lacZ reporter shown schematically in panels A and B encoding AUG at the first codon (A, on plasmid p367) or UUG at the third codon with ACG replacing AUG at the first codon (B, on plasmid pDH9-42), were cultured in SD+His medium and β-galactosidase specific activities were measured in WCEs. Mean activities with S.E.M.s (shown as error bars) were determined from six independent transformants. In (C), mean ratios (with S.E.M.s) of expression of the UUG vs AUG reporter were calculated from the results in (A,B). For (A–C), values normalized to that obtained for the WT RPS2 SUI5 strain are given above each histogram bar. Asterisks indicate statistically significant differences between each mutant and the WT RPS2 SUI5 strain determined by a two-tailed, unpaired Student’s t-test (**P < 0.05; ***P < 0.01).
RPS2 Ssu− substitutions increase discrimination against the eIF1 AUG codon in suboptimal context
In addition to reducing initiation at near-cognate UUG codons in Sui− mutants, other previously identified Ssu− substitutions in eIF1 and eIF1A were shown to increase discrimination against the AUG start codon of the SUI1 gene, encoding eIF1, which occurs in very poor Kozak context (Figure 5A, context score of 0.11). This feature of SUI1 initiation underlies negative autoregulation of eIF1 synthesis, dampening the ability to overexpress eIF1 in WT cells, as the excess eIF1 suppresses initiation at the SUI1 start codon (Martin-Marcos et al. 2011). To determine whether Rps2 Ssu− substitutions discriminate against poor Kozak context, we examined a subset of these variants belonging to different phenotypic groups by Western blot analyses of eIF1. Relative to the Hcr1 protein analyzed as a loading control, we observed significantly reduced eIF1 expression conferred by Q94D and T96K (Figure 5A–B; data quantified in Figure 5C, columns 4 and 6), which are the two strong Ssu− substitutions described above that selectively reduced the UUG/AUG HIS4-lacZ expression ratio for the ACG-UUG reporter. This finding supports the idea that Q94D and T96K specifically increase discrimination against start codons (either UUG or AUG) present in poor Kozak context. The V121D and I125K substitutions also reduced eIF1 expression (Figure 5, B and C, columns 8 and 9), which together with their ability to suppress initiation of the UUG-UUG reporter suggests that they increase discrimination against near-cognate UUG start codons in good context and AUG codons in poor context, in the manner observed previously for Ssu− substitutions in eIF1A or eIF1 (Martin-Marcos et al. 2011). Interestingly, the strong Ssu− substitutions D78A, Q89K, and K119A, which reduced the UUG/AUG expression ratio for the UUG-UUG reporter, and also the ACG-UUG reporter for Q89K and K119A, did not affect eIF1 expression (Figure 5, B and C, columns 2 and 7), suggesting that they specifically increase discrimination against a near-cognate UUG codon regardless of its context, but not an AUG codon in poor Kozak context.
Figure 5.
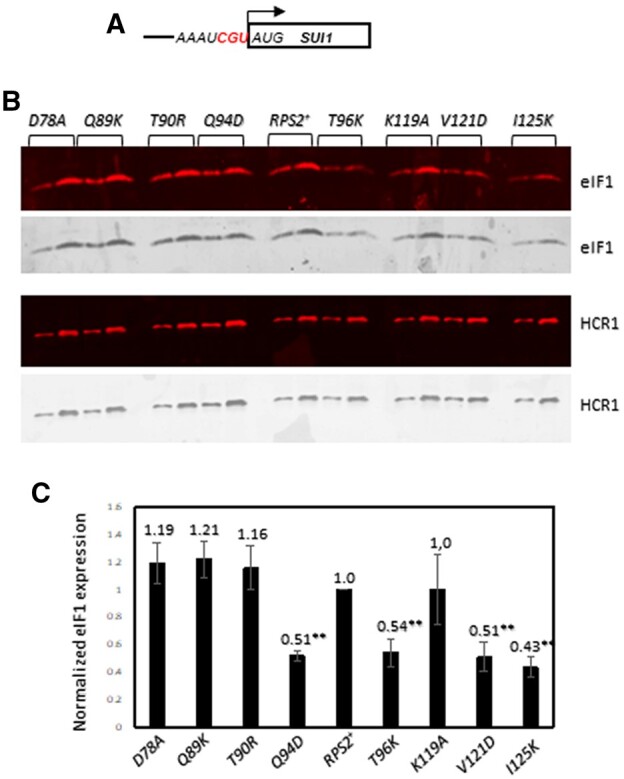
Certain RPS2 alleles reduce eIF1 expression from the native chromosomal SUI1 gene. (A) Schematic of chromosomal SUI1. (B) Derivatives of PGAL-RPS2 his4-301 strain HD2729 with the indicated RPS2 alleles were cultured in SD+His+Trp+Ura medium to an OD600 of ∼1.0 and WCEs were subjected to Western analysis using antibodies against eIF1 or Hcr1 (as loading control). Two amounts of each extract differing by a factor of two were loaded in successive lanes. (C) eIF1 expression, normalized to that of Hcr1, was obtained for each strain by quantifying the Western signals in (B), and mean values (±S.E.M.) were calculated from three biological replicates. Asterisks indicate significant differences between mutant and WT as judged by the Student’s t-test (P < 0.005).
In addition to reducing eIF1 expression, substitutions T90R, Q94D and T96K decreased expression of a SUI1-lacZ fusion containing the native, poor context of the SUI1 AUG (Figure 6A), but not the SUI1-opt-lacZ reporter in which the native context was replaced with optimum context by introducing A nucleotides at the −1 to −3 positions upstream of the AUG (Figure 6B). Accordingly, these three Rps2 substitutions, as well as I125K, significantly increased the SUI1-opt-lacZ/SUI1-lacZ expression ratio (Figure 6C, columns 4, 5, 6, 9 vs 1). These findings support the idea that these four substitutions increase discrimination against the SUI1 AUG in native, poor context. The other substitution that reduced eIF1 expression, V121D, reduced expression of both the native SUI1-lacZ and SUI1-opt-lacZ reporters similarly, making unclear its underlying mechanism for reducing eIF1 expression.
Figure 6.
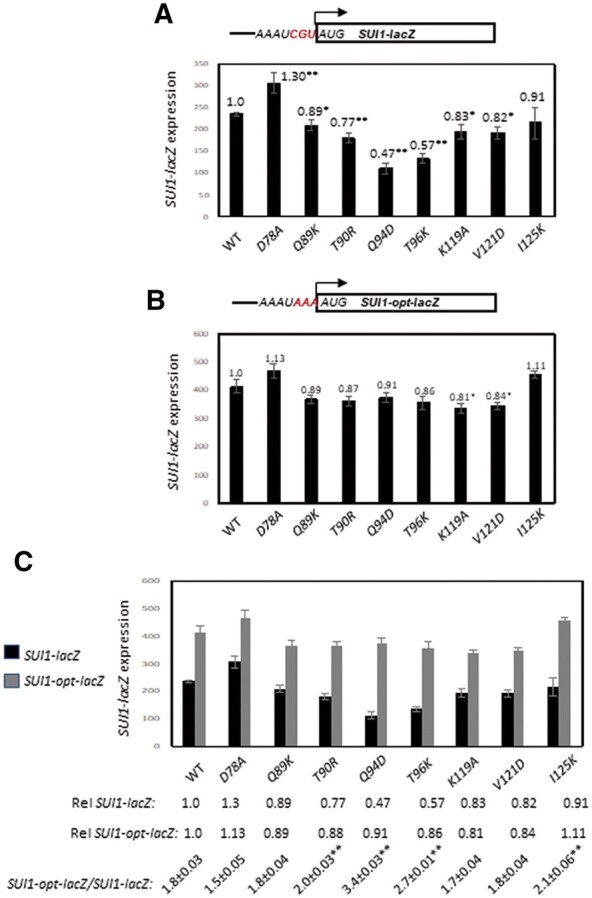
Certain RPS2 alleles increase discrimination against the poor-context AUG start codon of a SUI1-lacZ reporter. (A, B) Derivatives of PGAL-RPS2 his4-301 strain HD2729 with the indicated RPS2 alleles and also harboring (A) the SUI1-lacZ reporter with the AUG codon in native, poor context (pPMB24) or (B) the SUI1-opt-lacZ reporter with the AUG in optimum context (pPMB25) were cultured and assayed for β-galactosidase expression as described in Figure 3, except using SD+His+Trp medium. Mean expression levels and S.E.M.s from 6 transformants are plotted. (C) The data from (A, B) are replotted with the relative (Rel) mean expression levels normalized to that of the WT strain listed below the histogram, along with the expression ratios for the SUI1-lacZ vs SUI1-opt-lacZ reporters. Asterisks indicate significant differences between the ratios for mutant and WT as judged by a two-tailed, unpaired Student’s t-test (*, P < 0.05; **, P < 0.01).
As summarized in Table 2, based on the ability to suppress initiation at UUG codons in different contexts and the eIF1 AUG codon in native or optimal context, the Rps2 substitutions can be divided into three phenotypic classes. I125K and V121D, assigned to Class I, resemble previously described Ssu− substitutions in eIF1 and eIF1A (Martin-Marcos et al. 2011) that increase discrimination against both a near-cognate UUG codon and an AUG codon in poor context (Table 2, columns 1–7). D78A, Q89K, and K119A, assigned to class II, increase discrimination against near-cognate UUG codons regardless of their context, but not against a poor-context AUG codon. T90R, Q94D and T96K, assigned to class III, appear to increase discrimination against poor context regardless of the start codon, but not against a near-cognate UUG codon in optimum context.
Table 2.
Three phenotypic classes of RPS2 mutations that alter the accuracy of start codon recognition
| Near-cognate start |
Poor context AUG |
||||||
|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
| RPS2 allele | Phenotype Class | Suppression of SUI5 His+ (Ssu−) | Suppression of SUI5 UUG/AUG | Suppression of SUI5 UUG/AUG | Reduced eIF1 expression | Increased SUI1opt-lacZ/SUI1-lacZ | Increased el.-uORF1-GCN4-lacZ |
| (Two UUG reporter) (Ssu−) | (One UUG reporter) (Ssu−) | ||||||
| D78A | II | Moderate | Strong | ND | No | No | No |
| Q89K | II | Strong | Strong | Very Strong | No | No | No |
| T90R | III | Weak | None | ND | No | Weak | Moderate |
| Q94D | III | Weak | None | Moderate | Yes | Strong | Strong |
| T96K | III | Weak | None | Moderate | Yes | Moderate | Strong |
| K119A | II | Moderate | Strong | Strong | No | No | No |
| V121D | I | ? | Strong | ND | Yes | No | Moderate |
| I125K | I | ? | Strong | ND | Yes | Weak | Strong |
?, suppression could not be evaluated because these mutations impaired growth in the presence of SUI5 on medium containing histidine.
To provide further evidence that the class III substitutions Q94D and T96K increase discrimination against an AUG in poor context, we examined whether they decrease initiation at the AUG codon of an upstream ORF in suboptimal context and thereby increase expression of the downstream main ORF on the same mRNA by enhancing “leaky scanning” through the uORF AUG. To this end, we assayed expression of a GCN4-lacZ reporter containing a single, modified version of upstream ORF1 elongated to overlap the GCN4 main ORF (el.uORF1) and containing a suboptimal U nucleotide at position −3. In WT cells, replacing only the native, optimal A with U at the −3 position of el.uORF1 increases leaky scanning of uAUG-1 to produce a 10-fold increase in GCN4-lacZ translation, from 5.7 U to 57 U (cf. column 1 in Figure 7, A and B, noting difference in y-axis scales; summarized in Figure 7C-i, WT, rows 1 and 2). Interestingly, the four Rps2 substitutions found above to decrease eIF1 expression, Q94D, T96K, V121D, and I125K, plus the T90R substitution that increased the SUI1-opt-lacZ/SUI1-lacZ expression ratio, all increased expression of both el.-uORF1 GCN4-lacZ constructs (Figure 7, A and B, columns 4–6 and 8-9 vs column 1; summarized in Figure 7C-i, columns 1-6, rows 1 and 2), indicating decreased initiation at the el.-uORF1 AUGs in both optimum and weak contexts for these mutants. Consistently, the three Rps2 substitutions that did not affect eIF1 expression, D78A, Q89K, and K119A (Figure 5, A and B), did not increase expression of the el.-uORF1 GCN4-lacZ constructs (Figure 7, A and B, columns 2-3 and 7 vs 1).
Figure 7.

Certain RPS2 alleles increase leaky scanning of the upstream AUG codon of an elongated version of GCN4 uORF1, which overlaps the main ORF, in a manner modulated by the context of the uAUG. (A, B) Derivatives of PGAL-RPS2 his4-301 strain HD2729 with the indicated RPS2 alleles and also harboring an el.uORF1 GCN4-lacZ reporter with (A) optimum context (pC3502) or (B) weak context (pC3503) of the uAUG were cultured and assayed for β-galactosidase activities as in Figure 6. Mean expression levels and S.E.M.s from 6 transformants are plotted. Asterisks indicate significant differences between the ratios for mutant and WT as judged by a two-tailed, unpaired Student’s t-test (*, P < 0.05; **, P < 0.01). (Ci) Mean expression values from panels A and B are replotted on lines 1 and 2, respectively, for the WT RPS2 and selected mutant strains (columns 1–6). Line 3 contains expression data for a control GCN4-lacZ reporter with a point mutation in the uAUG (uORF-less construct, pC3505) assayed in the same strains as described in (A,B). (ii,iii) The percentages of scanning ribosomes that translate el.uORF1 (iii) or leaky-scan uAUG-1 and translate GCN4-lacZ instead (ii) were determined from the data in (i) by calculating the percentage of GCN4-lacZ expression observed for each el.uORF-containing reporter relative to the uORF-less construct, yielding the percentages in (ii), and subtracting the values in (ii) from 100 to obtain the percentages in (iii).
Comparing expression of the two GCN4-lacZ reporters containing el.-uORF1 to one lacking el.-uORF1 allows us to calculate the percentages of scanning ribosomes that either translate el.uORF1 or leaky-scan past uAUG-1 and translate GCN4-lacZ instead. Thus, in WT cells, 98.5% and 85% of scanning ribosomes recognize uAUG-1 in optimum or weak context, respectively, (Figure 7C-iii, WT, rows 7 and 8) and, accordingly, ∼1.5% and 15% bypass uAUG-1 and initiate at the GCN4 AUG instead (Figure 7C-ii, WT, rows 4 and 5). The fraction of ribosomes that translate el.-uORF1 in weak context decreases from 85% in WT to between 57% and 78% in the five different RPS2 mutants (Figure 7C-iii, row 8, mutants vs WT). The fraction of ribosomes that translate el.-uORF1 in optimum context decreases by a much smaller percentage from 98.5% in WT to between 97.3% and 98.1% for the same group of mutants. These findings suggest that these Rps2 substitutions preferentially discriminate against weak vs optimal AUG start codons.
We examined this last conclusion more rigorously using a thermodynamic model in which translation of el.-uORF1 is dictated by an equilibrium between the open and closed states of the 48S PIC at uAUG-1 (Thakur et al. 2020). The equilibrium constant for this reaction (Kclosed/open) is calculated as the ratio of the proportion of ribosomes that translate el.-uORF1 (taken from Figure 7C-iii, rows 7 and 8) to that which bypass uAUG-1 and translate GCN4-lacZ (from Figure 7C-ii, rows 4 and 5). The resulting constants for optimum and weak contexts for uAUG-1, Kopt and Kweak, were used to calculate the differences in free energy between the closed and open states (Δ) at uAUG-1 for optimal or weak contexts. Performing this calculation for WT and each mutant strain yields the change in Δ values between mutant and WT (ΔΔ) for each uAUG-1 context. The results, presented in greater detail in the Supplementary Discussion, indicated that the free energy of the closed complex is increased by each of the Rps2 substitutions to a greater extent for weak context vs optimum context, with the greatest increase for Q94D. This analysis supports the conclusion that the subset of Rps2 substitutions that decrease eIF1 expression (Q94D, T96K, V121D, and I125K), plus T90R that selectively reduces expression of the SUI1-lacZ reporter in native poor context, all increase leaky scanning of the AUG codon of el.-uORF1 by increasing discrimination against poor context.
Several RPS2 Ssu− mutations reduce bulk translation initiation without affecting 40S subunit abundance
We wished to determine the effects of RPS2 mutations on bulk translation initiation and 40S subunit levels. To this end, we generated strains in which the chromosomal RPS2 allele was deleted and the WT or mutant allele of interest was expressed from a plasmid, including the class I rps2 alleles V121D and I125K, class II alleles Q89K and K119A, and class III alleles Q94D and T96K. All six RPS2 mutations conferred significant reductions in the ratio of polysomes to 80S monosomes (P/M), measured in cycloheximide-stabilized extracts resolved by sedimentation through sucrose gradients (Figure 8, A–G), suggesting a reduced rate of bulk translation initiation relative to elongation in the mutants. The class I mutants V121D and I125K also displayed an obvious reduction in the ratio of free 40S to free 60S subunits (Figure 8, B and C), suggesting impaired biogenesis or stability of 40S subunits. This last inference was supported by measuring levels of total 40S and 60S subunits in extracts prepared without cycloheximide and magnesium, in which polysomes and 80S monosomes dissociate into free subunits, as only V121D and I125K conferred a significant reduction in the ratio of total 40S to 60S subunits (Supplementary Figure S4). Consistent with these findings, among the six mutations, only V121D and I125K conferred a marked Slg− phenotype (Table 1, column 2). These findings suggest that the decreased rate of initiation and selective increased discrimination against either UUG codons by Q89K and K119A or poor-context AUG codons by Q94D and T96K result from altered 40S function rather than abnormalities in expression of Rps2, 40S biogenesis, or stability of mature 40S subunits. The marked impairment of polysome assembly observed in the V121D and I125K mutants might well involve their reduced 40S abundance; however, because scanning occurs only after assembly and attachment of 43S PICs to the mRNA, their defects in start-codon recognition during scanning is most likely unrelated to the reduced concentration of 43S PICs expected in these two mutants. Among the four Rps2 substitutions we examined that do not appear to affect 40S biogenesis (Supplementary Figure S4, D–G), only Q94D confers slow growth. However, because Q94D impairs polysome assembly to nearly the same degree as Q89K, K119A, and T96K (Figure 8, D–G); the Slg− phenotype of Q94D might involve defects in elongation rate or fidelity in addition to the initiation defects we uncovered here.
Figure 8.
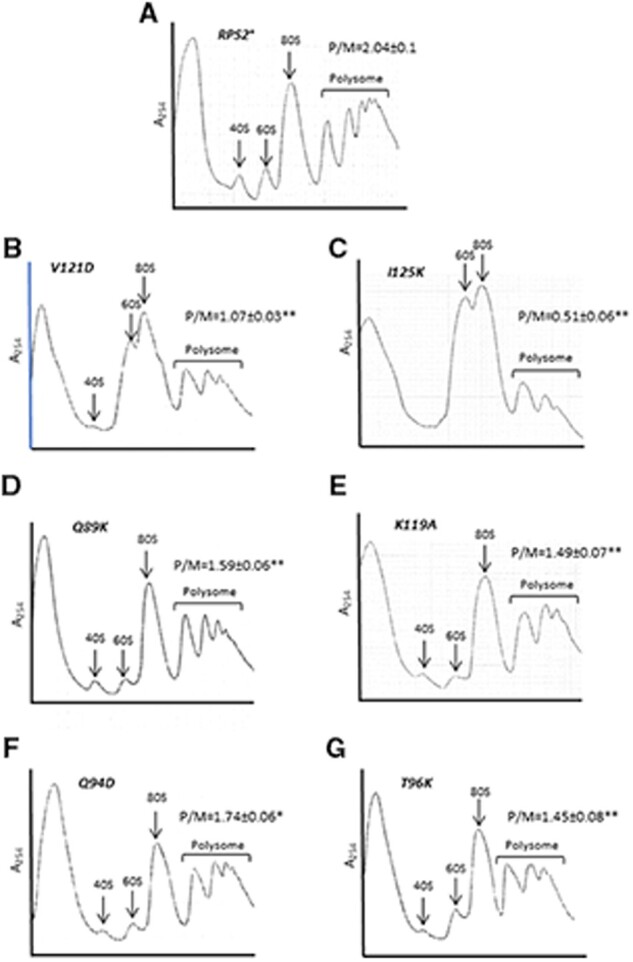
RPS2 mutations that alter start codon selection reduce bulk translation initiation. (A–G) Derivatives of rps2Δ strain HD2976 containing the indicated RPS2 alleles were cultured in SC-Leu at 30°C to A600 of ∼1.0, and cycloheximide was added (50 µg/mL) prior to harvesting. WCEs were separated by sucrose density gradient centrifugation and scanned at 254 nm. Mean (± S.E.M.) polysome/monosome (P/M) ratios from three biological replicates are indicated. Asterisks indicate significant differences between mutant and WT as judged by a two-tailed, unpaired Student’s t-test (*, P < 0.05; **, P < 0.0).
Discussion
In this study, we obtained genetic evidence implicating uS5/Rps2 residues located near the 40S mRNA entry channel in promoting bulk translation initiation and maintaining wild-type accuracy of start codon recognition in vivo. In a recent cryo-EM structure depicting the closed conformation of the PIC following AUG recognition (Llacer et al. 2018), six Rps2 residues were visualized in proximity (<5 Å) to the mRNA at the entry channel pore on the solvent-exposed surface of the 40S subunit, likely contacting mRNA nucleotides +8 to +13 (U36-U41) downstream of the AUG start codon: V80, I83, F98, V121, I125, R126. We identified lethal substitutions in five of these six residues, and the nonlethal substitutions V121D and I125K confer phenotypes indicating enhanced discrimination against both a UUG codon in good context and AUG start codons in poor context. The latter combination of phenotypes, designated here as class I (Table 2), was observed previously for Ssu− substitutions in eIF1 and eIF1A (Martin-Marcos et al. 2011). Substitutions in five other Rps2 residues in the vicinity of mRNA, but not close enough to make direct contacts in that PIC structure (D78, N82, Q87, Q94, and T96), also conferred either lethal phenotypes or increased discrimination against poor start codons. Interestingly, the nonlethal substitutions of this second group selectively reduced initiation either at a UUG codon in good context (D78A, class II phenotype), or at both UUG and AUG codons in poor context but not at a UUG codon in good context (Q94D and T96K, class III phenotype; Table 2). Thus, these latter substitutions appear to separate effects on near-cognate triplets vs start codon context. This separation of fidelity defects was also observed for nonlethal substitutions Q89K and T90R in the β1-β2 hairpin loop that contact rRNA residues in h28 or between h28 and h44 near the decoding center of the 40S subunit, with Q89K selectively increasing discrimination against UUG (class II) and T90R only affecting discrimination against poor-context (class III). The class II substitution K119A might indirectly alter the conformation or position of the β1-β2 loop based on its proximity to the loop (Figure 1A).
Previously, we showed that substitutions of two conserved Arg residues in uS3/Rps3 that contact mRNA at the entry channel, R116/R117, diminish initiation at both a UUG start codon and an AUG codon in poor context, resembling the class I Rps2 substitutions V121D and I125K described here. Additional evidence indicated that the substitutions of Rps3 R116/R117 destabilize the closed conformation of the PIC by weakening 40S interactions with the mRNA at the 40S entry channel (Dong et al. 2017). We proposed that Rps3 R116/R117 interactions with mRNA help to clamp the mRNA into the entry channel, which becomes constricted in the transition to the closed conformation of the PIC on start codon recognition, in which the P site is fully formed and encloses the Met- in the PIN state (Llacer et al. 2015). Thus, eliminating these mRNA interactions by substitutions of Rps3 R116/R117 would destabilize the closed conformation and indirectly destabilize Met- binding in the P site. Combining this destabilizing effect with the less stable codon: anticodon duplex formed at UUG codons could then account for the increased discrimination against UUG codons (Ssu− phenotype) conferred by these substitutions in vivo. Considering that Rps2 residues V121 and I125 contact mRNA at nearly the same location in the entry channel as does Rps3 R116 (Figure 1A), it seems plausible that Rps2 substitutions V121D/I125K reduce selection of near-cognate UUG codons by disrupting the same function proposed for Rps3 R116/R117 in clamping the mRNA in the entry channel (Dong et al. 2017) (Supplementary Figure S5A).
Our thermodynamic analysis of leaky scanning of the AUG codon of an extended version of GCN4 uORF1 (el.-uORF1) in either optimal or weak context suggested that these two Rps2 substitutions (V121D/I125K) destabilize the closed conformation of the PIC with both optimal and weak contexts but have a relatively greater effect on the weak context AUG (Supplementary Discussion and Supplementary Table S4). As noted in the Introduction, the molecular basis for the stimulatory effect of optimum Kozak context remains obscure. Substituting the arginine residues in the loop of eIF2α that directly contacts the context nucleotides of the mRNA of the closed conformation of the PIC (Llacer et al. 2015) increased discrimination against poor context rather than eliminating the stimulatory effect of optimum context, and also increased discrimination against near-cognate UUG. It was suggested that by eliminating electrostatic attraction with the mRNA phosphodiester backbone the eIF2α arginine substitutions destabilize the closed conformation of the PIC in a manner that exacerbates the instability conferred by either poor context or a noncognate UUG codon (Thakur et al. 2020). We proposed previously that, by removing mRNA contacts in the entry channel and reducing the clamping of mRNA into the PIC, the Rps3 R116/R117 substitutions might reduce selection of poor-context AUG codons by decreasing the dwell-time of the AUG triplet in the P site during scanning, which is known to be an important factor in determining the impact of poor context in mammals (Kozak 1990). This latter mechanism could also apply to leaky scanning phenotypes of the Rps2 substitutions V121D/I125K.
Greater destabilization of the closed PIC conformation for weak vs optimal context of the el.-uORF1 AUG codon also seems to characterize the class III Rps2 substitutions Q94D and T96K (Supplementary Table S4), which selectively enhance discrimination against poor context. These nearly adjacent residues in the β-2 strand of the β1-β2 hairpin, and the third class III substitution T90R located within the loop of the β1-β2 hairpin (Figure 1A), are all positioned somewhat closer to the start codon and 40S decoding center compared to the mRNA contacts made by class I substitutions V121D and I125K (Figure 1A); however, they are still distant from the -3 mRNA nucleotide that dominates the effects of Kozak context (Supplementary Figure S5B). It is surprising that the class II substitutions Q89K, D78A, and K119A, which have no detectable effect on poor context while strongly increasing discrimination against UUG codons, are also located within or nearby the β1-β2 hairpin (Figure 1A). Given that both the class II and class III substitutions could affect either the conformation or orientation of the β1-β2 hairpin, it seems possible that they differentially perturb interactions of the hairpin loop with rRNA helices h28 or h44, which communicate with the decoding center (Supplementary Figure S5B), in a manner that selectively alters recognition of either the context nucleotides in mRNA or the codon-anticodon helix in the P site (Supplementary Figure S5B). Additional high-resolution PIC structures for mRNAs that differ in Kozak context sequence, and molecular dynamics simulations to predict differences in free energy associated with optimum vs weak context in addition to UUG vs AUG start codons (Kameda et al. 2021), should be instructive in deciphering the mechanisms involved for the different Rps2 substitutions.
Data availability
Strains and plasmids are available upon request. The authors affirm that all data necessary for confirming the conclusions of the article are present within the article, figures, and tables.
Supplementary material is available at GENETICS online.
Supplementary Material
Acknowledgments
The authors are grateful to Ms. Fan Zhang for conducting GCN4-lacZ reporter assays.
Funding
This work was supported in part by the Intramural Research Program of the National Institutes of Health.
Conflicts of interest
The authors declare that there is no conflict of interest.
Literature cited
- Alone PV, Cao C, Dever TE. 2008. Translation initiation factor 2gamma mutant alters start codon selection independent of Met-tRNA binding. Mol Cell Biol. 28:6877–6888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donahue TF, Cigan AM. 1988. Genetic selection for mutations that reduce or abolish ribosomal recognition of the HIS4 translational initiator region. Mol Cell Biol. 8:2955–2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J, Aitken CE, Thakur A, Shin BS, Lorsch JR, et al. 2017. Rps3/uS3 promotes mRNA binding at the 40S ribosome entry channel and stabilizes preinitiation complexes at start codons. Proc Natl Acad Sci USA. 114: E2126–E2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J, Munoz A, Kolitz SE, Saini AK, Chiu WL, et al. 2014. Conserved residues in yeast initiator tRNA calibrate initiation accuracy by regulating preinitiation complex stability at the start codon. Genes Dev. 28:502–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J, Nanda JS, Rahman H, Pruitt MR, Shin BS, et al. 2008. Genetic identification of yeast 18S rRNA residues required for efficient recruitment of initiator tRNA(Met) and AUG selection. Genes Dev. 22:2242–2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashem Y, Des Georges A, Dhote V, Langlois R, Liao HY, et al. 2013. Structure of the mammalian ribosomal 43S preinitiation complex bound to the scanning factor DHX29. Cell. 153:1108–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinnebusch AG. 2014. The scanning mechanism of eukaryotic translation initiation. Annu Rev Biochem. 83:779–812. [DOI] [PubMed] [Google Scholar]
- Hinnebusch AG. 2017. Structural insights into the mechanism of scanning and start codon recognition in eukaryotic translation initiation. Trends Biochem Sci. 42:589–611. doi: 10.1016/j.tibs.2017.03.004. [DOI] [PubMed] [Google Scholar]
- Huang H, Yoon H, Hannig EM, Donahue TF. 1997. GTP hydrolysis controls stringent selection of the AUG start codon during translation initiation in Saccharomyces cerevisiae. Genes Dev. 11:2396–2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain T, Llácer JL, Fernández IS, Munoz A, Martin-Marcos P, et al. 2014. Structural changes enable start codon recognition by the eukaryotic translation initiation complex. Cell. 159:597–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov IP, Loughran G, Sachs MS, Atkins JF. 2010. Initiation context modulates autoregulation of eukaryotic translation initiation factor 1 (eIF1). Proc Natl Acad Sci USA. 107:18056–18060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kameda T, Asano K, Togashi Y. 2021. Free energy landscape of RNA binding dynamics in start codon recognition by eukaryotic ribosomal pre-initiation complex. PLoS Comput Biol. 17:e1009068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolitz SE, Takacs JE, Lorsch JR. 2009. Kinetic and thermodynamic analysis of the role of start codon/anticodon base pairing during eukaryotic translation initiation. RNA. 15:138–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak M. 1990. Downstream secondary structure facilitates recognition of initiator codons by eukaryotic ribosomes. Proc Natl Acad Sci USA. 87:8301–8305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llacer JL, Hussain T, Marler L, Aitken CE, Thakur A, et al. 2015. Conformational differences between open and closed states of the eukaryotic translation initiation complex. Mol Cell. 59:399–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llacer JL, Hussain T, Saini AK, Nanda JS, Kaur S, et al. 2018. Translational initiation factor eIF5 replaces eIF1 on the 40S ribosomal subunit to promote start-codon recognition. Elife. 7:e39273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomakin IB, Steitz TA. 2013. The initiation of mammalian protein synthesis and mRNA scanning mechanism. Nature. 500:307–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Marcos P, Cheung YN, Hinnebusch AG. 2011. Functional elements in initiation factors 1, 1A, and 2β discriminate against poor AUG context and non-AUG start codons. Mol Cell Biol. 31:4814–4831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Marcos P, Nanda J, Luna RE, Wagner G, Lorsch JR, et al. 2013. β-hairpin loop of eIF1 mediates 40S ribosome binding to regulate initiator tRNAMet recruitment and accuracy of AUG selection in vivo. J Biol Chem. 288:27546–27562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyasaka H. 1999. The positive relationship between codon usage bias and translation initiation AUG context in Saccharomyces cerevisiae. Yeast. 15:633–637. [DOI] [PubMed] [Google Scholar]
- Moehle CM, Hinnebusch AG. 1991. Association of RAP1 binding sites with stringent control of ribosomal protein gene transcription in Saccharomyces cerevisiae. Mol Cell Biol. 11:2723–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisarev AV, Kolupaeva VG, Pisareva VP, Merrick WC, Hellen CU, et al. 2006. Specific functional interactions of nucleotides at key −3 and +4 positions flanking the initiation codon with components of the mammalian 48S translation initiation complex. Genes Dev. 20:624–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabl J, Leibundgut M, Ataide SF, Haag A, Ban N. 2011. Crystal structure of the eukaryotic 40S ribosomal subunit in complex with initiation factor 1. Science. 331:730–736. [DOI] [PubMed] [Google Scholar]
- Reid GA, Schatz G. 1982. Import of proteins into mitochondria. J Biol Chem. 257:13062–13067. [PubMed] [Google Scholar]
- Saini AK, Nanda JS, Lorsch JR, Hinnebusch AG. 2010. Regulatory elements in eIF1A control the fidelity of start codon selection by modulating tRNA(i)(Met) binding to the ribosome. Genes Dev. 24:97–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakur A, Gaikwad S, Vijjamarri AK, Hinnebusch AG. 2020. eIF2alpha interactions with mRNA control accurate start codon selection by the translation preinitiation complex. Nucleic Acids Res. 48:10280–10296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valasek L, Nielsen KH, Zhang F, Fekete CA, Hinnebusch AG. 2004. Interactions of eukaryotic translation initiation factor 3 (eIF3) subunit NIP1/c with eIF1 and eIF5 promote preinitiation complex assembly and regulate start codon selection. Mol Cell Biol. 24:9437–9455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valášek L, Phan L, Schoenfeld LW, Valásková V, Hinnebusch AG. 2001. Related eIF3 subunits TIF32 and HCR1 interact with an RNA recoginition motif in PRT1 required for eIF3 integrity and ribosome binding. EMBO J. 20:891–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visweswaraiah J, Hinnebusch AG. 2017. Interface between 40S exit channel protein uS7/Rps5 and eIF2alpha modulates start codon recognition in vivo. Elife. 6:e22572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zur H, Tuller T. 2013. New universal rules of eukaryotic translation initiation fidelity. PLoS Comput Biol. 9:e1003136. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Strains and plasmids are available upon request. The authors affirm that all data necessary for confirming the conclusions of the article are present within the article, figures, and tables.
Supplementary material is available at GENETICS online.