

Abstract
Black-rot disease caused by the phytopathogen Xanthomonas campestris pv. campestris (Xcc) continues to have considerable impacts on the productivity of cruciferous crops in Trinidad and Tobago and the wider Caribbean region. While the widespread occurrence of resistance of Xcc against bactericidal agrochemicals can contribute to the high disease burdens, the role of virulence and pathogenicity features of local strains on disease prevalence and severity has not been investigated yet. In the present study, a comparative genomic analysis was performed on 6 pathogenic Xcc and 4 co-isolated non-pathogenic Xanthomonas melonis (Xmel) strains from diseased crucifer plants grown in fields with heavy chemical use in Trinidad. Native isolates were grouped into two known and four newly assigned ribosomal sequence types (rST). Mobile genetic elements were identified which belonged to the IS3, IS5 family, Tn3 transposon, resolvases, and tra T4SS gene clusters. Additionally, exogenous plasmid derived sequences with origins from other bacterial species were characterised. Although several instances of genomic rearrangements were observed, native Xcc and Xmel isolates shared a significant level of structural homology with reference genomes, Xcc ATCC 33913 and Xmel CFBP4644, respectively. Complete T1SS hlyDB, T2SS, T4SS vir and T5SS xadA, yapH and estA gene clusters were identified in both species. Only Xmel strains contained a complete T6SS but no T3SS. Both species contained a complex repertoire of extracellular cell wall degrading enzymes. Native Xcc strains contained 37 T3SS and effector genes but a variable and unique profile of 8 avr, 4 xop and 1 hpa genes. Interestingly, Xmel strains contained several T3SS effectors with low similarity to references including avrXccA1 (~89%), hrpG (~73%), hrpX (~90%) and xopAZ (~87%). Furthermore, only Xmel genomes contained a CRISPR-Cas I-F array, but no lipopolysaccharide wxc gene cluster. Xmel strains were confirmed to be non-pathogenic by pathogenicity assays. The results of this study will be useful to guide future research into virulence mechanisms, agrochemical resistance, pathogenomics and the potential role of the co-isolated non-pathogenic Xanthomonas strains on Xcc infections.
Keywords: Xanthomonas campestris pv. campestris, Xanthomonas melonis, Genome plasticity, Integrated mobile elements, Type 3 secretion system, Bacterial secretion systems
Introduction
Xanthomonas is one of the most important and widespread genera of phytopathogenic bacteria which infects most commercial food crops (cabbage, citrus, tomato, peppers, and rice), trees, and ornamental plants (Ryan et al., 2011; Jacques et al., 2016; An et al., 2020). Being a successful, versatile, and globally distributed pathogen (Lamichhane et al., 2018) armed with a robust genomic arsenal, this genus requires constant attention to develop genomic atlases to identify features that influence complex host-pathogen interactions. Pathoadaptations afford multiple functions and capabilities, including host defence evasion, expression of a diverse repertoire of protein and DNA effector genes (transcription activator-like effectors (TALE’s)), degradative enzyme and biofilm production, and utilization of multiple secretion systems (Ryan et al., 2011; Vicente & Holub, 2013; Alvarez-Martinez et al., 2021). The ability to exchange and maintain mobile elements within the wider Xanthomonad family further increases its genomic fortitude with the ability to encode for copper, heavy metal, and agrochemical resistance factors (Ryan et al., 2011; Behlau et al., 2013). Of particular interest to the current study is native copper resistant Xanthomonas campestris pv. campestris (Xcc), the causal agent of black rot disease of crucifiers, and the co-isolated copper resistant Xanthomonas melonis, which has never previously been associated with crucifers. Originally part of the X. campestris group, Xmel is reported as a soft rot pathogen affecting melons but not much is known beyond this (https://www.cabi.org/isc/datasheet/56949, (Vauterin et al., 1995)). Both species have the potential to affect disease management via agrochemical resistance and potential dual-species plant-microbe interactions.
Of the ~3,736 Xanthomonas genomes available in GenBank, Xcc contributes only a small number (87) and is dominated by others including X. phaseoli (161), X. citri (216), X. arboricola (144), X. oryzae (457), and the tomato pathogens X. vesicatoria, X. perforans and X. euvesicatoria (239). Only 1 Xcc genome from the database originated from the Caribbean and it was from the strain BrA1 isolated from Trinidad (Lugo et al., 2013) while only 1 Xmel assembly exists in the GenBank as of September 2021. While Xanthomonas strains and closely related species display genomic collinearity, rearrangements and a diverse mobilome are common (Comas et al., 2006; Ryan et al., 2011). Mobilome factors, including IS elements, transposons, and plasmids, contribute to horizontal gene transfer (HGT), translocation, and acquisition of patho-capacities including LPS, Type 3 and 4 secretion systems (T3SS and T4SS respectively), and resistance elements (Vicente & Holub, 2013; An et al., 2020).
Virulence and pathogenicity in Xcc are influenced by the secretion systems, effectors, extracellular enzymes, and polysaccharides, which in turn affect the host range and characteristics of disease (Lu et al., 2008; White et al., 2009). Of particular importance to pathogenicity in Xcc are the Types 2 (T2SS), 3, and 5 secretion systems (T5SS) (Vicente & Holub, 2013). Although Type 1 (T1SS), Type 4 (T4SS), and Type 6 (T6SS) (partial) are also present, their strong links to virulence are yet to be established. The T3SS represents the most important pathogenic determinant, and its function includes evading host defence and translocating effectors into host cells (Büttner & Bonas, 2010). The T3SS pathogenicity island falls into the Hrp2 family T3SS (An et al., 2020), comprising of hrp (hypersensitivity response and pathogenicity), hrc (hrp-conserved), and hpa (hrp-associated) genes (Cornelis & Van Gijsegem, 2000; da Silva et al., 2002; Tampakaki et al., 2004). Associated secreted T3 effector diversity is highly correlated with host adaptation and is attributed to ~39 different Xop and Avr proteins (White et al., 2009). The T2SS secretes proteins from the periplasm to the extracellular milieu such as proteases, lipases, toxins, and plant cell wall degrading enzymes (PCWDEs). These enzymes, including endoglucanases, xylanases, cellulases, pectinases and cellulases, are thought to be essential in overcoming host defences, movement across physical barriers to other plant areas, and nutrient acquisition in limited states (Lu et al., 2008; Solé et al., 2015).
In Xcc, the T1, 4, and 5SS do not appear to play a role in virulence as they do in other species (He et al., 2007; Vicente & Holub, 2013; Alvarez-Martinez et al., 2021). However, the T1SS is linked to virulence in X. oryzae pv. oryzae and typically translocate effector proteins including bacteriocins, RTX-nonapeptide motif-containing proteins (possible toxins), and adhesins (Jarrod, Sondermann & O’Toolea, 2018). The T4SS translocates proteins and DNA-protein complexes into foreign cells as an antagonistic or conjugative response (Souza et al., 2011). Currently, this secretion system is split into the T4ASS (typical of Xanthomonas) and the T4BSS (typical of human pathogens) (Guglielmini et al., 2014). In conjunction with the T4ASS chromosomal genes, the T6SS in Xanthomonas appears to be involved in an antagonistic response towards other bacteria and amoeba (Bayer-Santos et al., 2018).
While not directly involved in virulence, the production of biofilm, EPS, and adhesions play a critical role in establishing colonies and adherence to host surfaces within a complex microbial community, which are essential for disease initiation (Cesbron et al., 2015). In Xcc, biofilms are essential for survival in-planta, on leaf surfaces, disease progression, binding of Ca2+ and other divalent ions, and inhibition of callose deposition (Aslam et al., 2008). Additionally, LPS (lipopolysaccharides) O-antigens, an integral membrane component necessary for survival, have been linked to virulence. LPS biosynthesis is controlled by >20 genes (Braun et al., 2005) with the entire biosynthetic cluster highly variable among species (Vorhölter, Niehaus & Pühler, 2001; Patil & Sonti, 2004; An et al., 2020).
The current study addresses the lack of representative Xcc genomes native to Trinidad and adds vital genome characteristics of these pathogenic strains to worldwide databases. From this study, genomes of pathogenic Xcc and non-pathogenic co-inhabitant Xanthomonas melonis (Xmel) have been sequenced and characterized. Although a previous study characterized a single Xcc strain, BrA1 (Behlau et al., 2017), analysis of multiple strains has not been attempted before. While not much is known on Xmel, the current study contributes to the knowledge on the ecological role of this species. The study presents detailed evidence of genome plasticity and virulence gene cluster diversity in native species. The data will be useful for future studies on specific genomic clusters related to pathogenicity, virulence, horizontal gene transfer and recombination events.
Materials and Methods
Bacterial strains used
Morphologically identified and PCR-validated Xcc isolates were selected from a larger collection made from 2015–2017 (unpublished) at the UWI-St. Augustine- Plant-Microbe laboratory. These isolates were obtained from infected crucifer plants from fields that were affected by black-rot disease despite the heavy use of copper-based fungicides. Bacteria were isolated from infected crucifer leaf tissue according to methods described by Lugo et al. (2013), and morphologically identified as Gram-negative, and displaying mucoid, convex, yellow colonies on Nutrient Agar. Copper sensitivity was determined using CuSO4 amended MGY agar (Behlau et al., 2012), according to the protocol outlined by Marin et al. (2019). Nine strains from three major crucifer cultivation areas in Trinidad with representative copper sensitivity profiles were chosen for whole-genome sequencing. The copper resistant native Xcc BrA1 characterised elsewhere (Behlau et al., 2017) was also included and re-sequenced in this study.
Genomic DNA preparation and Illumina sequencing
A modified CTAB protocol (Lugo et al., 2013) was used to extract total genomic DNA. DNA quality assessment, library preparation, and sequencing were carried out at Novogene Corporation (USA). High MW intact DNA (≥10 ng/µL) was assessed for quality and quantity by agarose gel electrophoresis and the use of a Qubit 2.0 fluorometer. The DNA samples were then fragmented using sonication, followed by end polishing, ligation to Illumina adaptors, and amplification with P5 and P7 index oligos. The AMPure XP system was used to purify amplified products and libraries were built using the NEBNext Ultra II DNA Library Prep Kit with a ~350 bp insert size. The size distribution and concentration of libraries were assessed using an Agilent 2100 Bioanalyzer and by qPCR. Illumina sequencing was performed on a Hiseq system (150 bp PE) to a depth of 1G. Adapters and ligation sequences were removed, and raw sequences were filtered to provide reads at Phred scores >30 and error ~0.03%.
Whole genome assembly, annotation, and species identification
Bioinformatic manipulations were carried out on the public server usegalaxy.eu (Afgan et al., 2018) unless otherwise stated. Read pair association and quality were assessed using FastQC 0.72 and trimmed with Cutadapt 3.4 to maintain a QC ≥30 and length ≥100 bp. Trimmed reads were assembled using Shovill 1.1.0 (Spades option with pilon enabled). Resultant multi-fasta contigs were annotated using Prokka 1.14.5 and RAST 2.0 (Overbeek et al., 2014). Annotated 16S rRNA gene sequences were then analyzed using the RDP Classifier (Wang et al., 2007). The Microbial genome atlas (Rodriguez-R et al., 2018) and GTDB-Tk 1.1.0 (Kbase) (Chaumeil et al., 2020) tools were used to determine species identity and to assess genome contamination.
The virulence capabilities of native Xanthomonas strains were assessed to further identify isolates. Cabbage (Tropicana variety) seedlings at the 2-week stage were sown in a 2:1 PRO-MIX and manure/potting mix combination in 16oz pots under open field conditions. Plants were fertilized at recommended levels as outlined by the Ministry of Agriculture, Trinidad and Tobago and irrigated daily. At 4 weeks old, uniform growth seedlings with at least 6 full leaves (past the 2-week stage) were inoculated under humid conditions using the leaf clipping method outlined by Lugo et al. (2013). Xcc and Xmel cultures were grown for 48 h in NB and standardized to an OD600 of 1 before use. Three plants were used per isolate and 3 leaves were clipped per plant on either side of the middle vein. Disease symptoms were assessed at 10 days post infection. Observed results and lesion measurements are contained in Table S11.
Genome completeness and general statistics were obtained using BUSCO 5.0 and QUAST 5.0 (Mikheenko et al., 2018). Assembled genomes for all 9 native isolates were submitted to NCBI under BioProject PRJNA701249. Whole-genome SNP-Phylogenetic reconstruction with Xanthomonas reference genomes was carried out using CSIPhylogeny (Kaas et al., 2014) and FastTree 2.0 (GTR, SPR, 1000 bootstrap) (Price, Dehal & Arkin, 2010) via NGPhylogeny (Lemoine et al., 2019). Phylogenetic trees were edited in TreeGraph 2 (Stöver & Müller, 2010). Accurate sequence typing and grouping were further carried out using rMLST typing via PubMLST Species ID (Jolley et al., 2012).
Genome characterization and feature annotation
Genomic features of each strain were obtained from the RAST server. Pangenome analysis for core genes (95% similarity) and accessory genes (<50% strains) were determined using the Roary 3.13.0 pipeline (Page et al., 2015). Pan-genome functional categorization using COG (Cluster of orthologous groups) classification was described using EggNog mapper 2.0 (Huerta-Cepas et al., 2017) and the NCBI COG database (https://www.ncbi.nlm.nih.gov/research/cog/). Identification of potential secondary metabolite biosynthetic gene clusters (BGC’s) was done using the AntiSMASH 5.0 webserver (Blin et al., 2019). Genome alignments to determine regions of similarity, gain or loss of segments, and recombination events were carried out using progressiveMauve (Darling, Mau & Perna, 2010). Draft genomes were first ordered against the reference strains, X. campestris pv. campestris ATCC 33913 and X. melonis CFBP4644, using the Mauve Contig Mover (MCM). Secretion systems were identified based on cumulative annotations from Prokka and RAST, predictions using the EffectiveDB server (Eichinger et al., 2016), tblastn and blastn (>70% coverage and identity) analysis using manually curated databases of published sequences.
Genes for plant cell wall degrading enzymes (PCWDEs) were identified using the dbCAN2 web server (Zhang et al., 2018). Identification of mobile genetic elements was facilitated from annotated CDS and the ISFinder database (Siguier et al., 2006). The prediction of potential integrated and conjugated mobile elements was facilitated using ICEberg 2.0 (Liu et al., 2019). The location of mobile elements in each native strain was generated using blastn against ordered, concatenated contigs. Circular plots were then generated using this data and the CGView server (Grant & Stothard, 2008). Plasmid derived contigs were predicted using PlasmidVerify (Antipov et al., 2019), a Bayesian classifier trained on HMM’s associated plasmid sequences, and RFPlasmid (van der Graaf van Bloois, Wagenaar & Zomer, 2020), an implementation of a Random Forest model on multiple bacterial DNA species databases. Predicted plasmid derived contigs >1 kb were identified using both PLSDB (Galata et al., 2019), and blastn (%ID >80%) against a manually curated plasmid database (https://ftp.ncbi.nih.gov/refseq/release/plasmid/). The CRISPRCasTyper 1.4.1 server (Russel et al., 2020) was used to identify CRISPR arrays and Cas proteins, with identified spacers subjected to Blast analysis against the NCBI phage, plasmid, and IMGVR databases via the CRISPRTarget tool (Biswas et al., 2013). Heatmaps were generated using TBtools 1.082 (Chen et al., 2020) and synteny maps using Gene Graphics (Harrison, Crécy-Lagard & Zallot, 2018).
Results
Genome statistics of native Xanthomonas isolates from Trinidad
Nine Xanthomonas contig assemblies were generated from black-rot leaf lesion isolates with varying levels of copper tolerance/resistance using the Illumina Hiseq shotgun sequencing approach. To validate the identity in our collection, Xcc strain BrA1 (provided by Prof. Jeffery Jones, University of Florida, USA) was re-sequenced. General assembly metrics are presented in Table S1. Overall, 8.6–21 M reads with a Phred score of ~36 were obtained after quality filtering, yielding 107–257 de-novo assembled contigs (44–90 >1 kb). Two Xanthomonas species were identified with 98% ANI similarity to reference genomes using MIGA and GTDB-Tk (Table S1). Xcc strains included Ar1BCA1, Ar1PC2, BrA1, CaNP1C, Cf3C and Cf4B1, while CaNP1D, CaNP5B, CaNP6A and DMCX were classified as Xmel. All assembled genomes were 99.8% complete (BUSCO), showed typical GC% (65.1 to 66.4%) for each species, and were supported by 251–615X coverage (Table 1). The assemblies also had complete single 16S rRNA genes and less than 0.01% of reads were identified as contaminants, which were removed before downstream analysis. Pathogenicity against the tropicana cabbage variety was confirmed for all Xcc strains but Xmel isolates did not elicit any symptoms (Table S11).
Table 1. De-novo shotgun assembled genome statistics of Trinidadian strains of two Xanthomonas species isolated from infected crucifer leaves.
| Strain ∆ | Copper sensitivity† | Sequenced length (kb) | GC% | Coverage (X) | N50 | rMLST ST‡ | NCBI Accession | Total genes | Coding genes | RNA genes | tRNAs | ncRNAs | Total pseudogenes |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Species: Xanthomonas campestris pv. campestris (Xcc) | |||||||||||||
| Ar1PC21 | Tolerant | 5.23 | 65.01 | 367 | 166,424 | rST-171941 | JAFFQP000000000 | 4,621 | 4,310 | 150 | 53 | 94 | 161 |
| Ar1BCA11 | Tolerant | 4.98 | 65.2 | 274 | 130,896 | rST-132382 | JAFFQL000000000 | 4,374 | 4,100 | 139 | 53 | 84 | 135 |
| BrA12 | Resistant | 5.15 | 65.08 | 615 | 130,897 | rST-132382 | – | 4,410 | 4,283 | 153 | 59 | 94 | 138 |
| Cf3C3 | Resistant | 5.16 | 65.06 | 296 | 113,525 | rST-132382 | JAFFQN000000000 | 4,575 | 4,292 | 140 | 53 | 84 | 143 |
| Cf4B13 | Resistant | 5.16 | 65.07 | 317 | 129,122 | rST-132382 | JAFFQM000000000 | 4,563 | 4,278 | 140 | 53 | 84 | 145 |
| CaNP1C3 | Tolerant | 5.08 | 65.03 | 251 | 176,946 | rST-68128 | JAFFQO000000000 | 4,510 | 4,228 | 144 | 52 | 86 | 138 |
| Species: Xanthomonas melonis (Xmel) | |||||||||||||
| CaNP1D3 | Tolerant | 4.78 | 66.04 | 579 | 166,537 | rST- 171942 | JAFFQH000000000 | 4,120 | 3,938 | 99 | 52 | 44 | 83 |
| CaNP5B3 | Resistant | 4.84 | 65.99 | 276 | 156,120 | rST- 171943 | JAFFQJ000000000 | 4,182 | 3,980 | 102 | 56 | 43 | 100 |
| CaNP6A3 | Resistant | 4.84 | 65.99 | 335 | 172,216 | rST- 171942 | JAFFQI000000000 | 4,202 | 3,998 | 105 | 56 | 43 | 99 |
| DMCX4 | Tolerant * | 4.8 | 65.97 | 386 | 282,115 | rST- 171942 | JAFFQK000000000 | 4,195 | 3,990 | 103 | 56 | 44 | 102 |
Notes:
∆ - superscripts represent source crops: 1; Bok Choy, 2; Broccoli, 3; Cauliflower, 4; Cabbage.
Copper Tolerant isolates grew in the presence of up to 200 ppm CuSO4, while resistant strains grew in the presence of up to 360 ppm CuSO4.
rST of Ar1PC1 and all Xmel represent unique sequence types newly deposited in the PubMLST database, full allelic profiles are given in Table S2.
Xmel strain DMCX was only screened up to 200 ppm CuSO4.
Phylogenetic relationships among native isolates and Xanthomonas reference species were assessed using common whole-genome SNPs as seen in the cladogram in Fig. 1. Native Xcc isolates clustered with the type strain (ATCC 33913) and other RefSeq genomes (X. campestris cluster) while showing separation according to rMLST within this clade. A similar placement is seen with Xmel strains, where all native strains were grouped with the single reference assembly (CFBP4644). The rMLST typing of native isolates based on 53 ribosomal rps and rpl genes (Table S2) showed that the Xcc strains fell into three groups based on ribosomal allelic profiles (rST) (Table 1). Four of the 6 isolates (Ar1BCA1, BrA1, Cf3C, Cf4B1) together with one reference (Xcc 3811) were categorized as rST-132382, while one isolate (CaNP1C) and five references (including MAFF302021) had the rST-68128 profile. The remaining isolate Ar1PC2 did not match with any existing rST and has been added to a new group on the database. All native Xmel strains were classed as unique, and the newly identified rST were added to the database. Two rST’s were inferred from the four native Xmel isolates, where CaNP1D, CaNP6A and DMCX fell in the same group.
Figure 1. Phylogenetic reconstruction of whole genome SNPs of native Xanthomonas strains and reference genomes (cladogram).
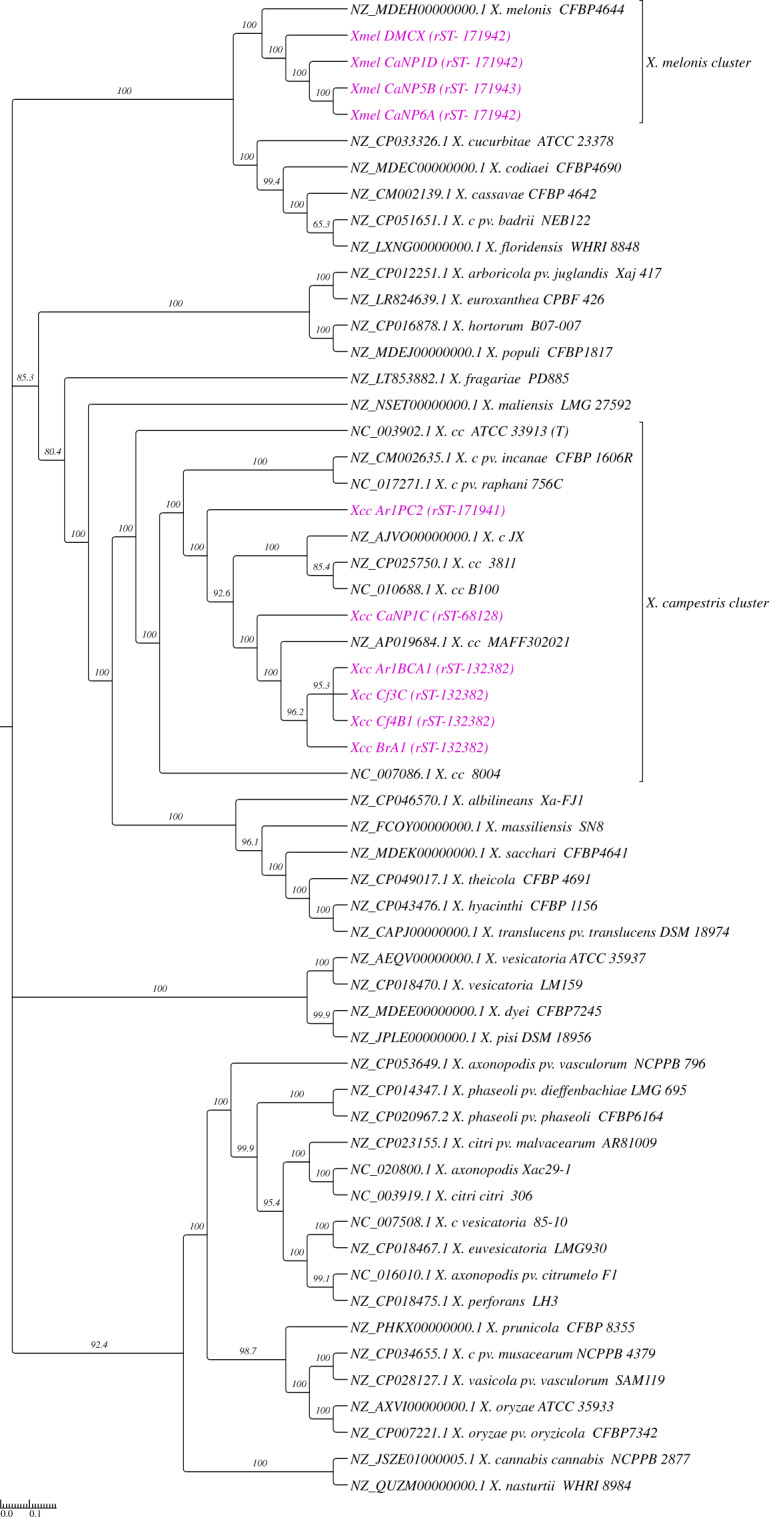
Native strains are represented in purple text with their respective rMLST in brackets. Called SNPs from 46 reference species and 10 native genomes were concatenated by CSIPhylogeny and ML Phylogenetic reconstruction carried out using FastTree with 1000 boostrap (SH-Like) replicates. Reference genomes consisted of RefSeq representative and/or next available assembly genomes from the full NCBI taxonomic listing of relevant Xanthomonas spp.
Progressive Mauve genome alignment, pangenome analysis, and Biosynthetic gene cluster (BGC) prediction
The annotated genome statistics (Table 1) show that not only were the Xcc genomes larger, but apart from tRNAs, they also contained more CDS than native Xmel isolates. To maximize local collinear block (LCB) size and arrangement, multi-fasta assemblies were ordered against the most curated reference genomes. Collinear LCB’s among strains within species, as well as distinct patterns of rearrangements via inversions and translocations within contigs, were identified (Figs. 2 and 3).
Figure 2. Progressive Mauve alignment of Xcc genomes of native isolates ordered against the reference Xcc ATCC 33913.

Figure 3. Progressive Mauve alignment of Xmel genomes of native isolates ordered against the reference Xmel CFBP4644.

The Xcc and Xmel genomes differed to such an extent that combined alignments for both species were not possible. The genomes of the native Xcc isolates had a higher level of conserved structural organization and fewer rearrangements compared to Xmel. This was supported by the congruent arrangement and mostly complete similarity plots of each Xcc LCB. Furthermore, these differences in similarity plots and gaps between LCB’s (incomplete/white regions) indicate that there are more cases of unique genomic regions within native strains than the reference. Xmel genomes had a larger number of smaller LCBs with a more complex arrangement.
The pangenome of each species contained 5,019 and 4,491 genes respectively. Approximately 83% of genes made up the core genome for each species (90–100% occurrence in strains within species), with ~900 accessory genes having <50% occurrence across strains. CDS were found in ~94% and ~62% of core and accessory genes, respectively, with 10% and 27% remaining unclassified. Figure 4 depicts COG categories that provide functional descriptions of the core genome, with major functions including energy production, amino acid, lipid, and cell wall biosynthesis and metabolism, inorganic ion transport and metabolism and signal transduction. Except for secondary metabolite biosynthesis and metabolism, as well as intracellular vesicular transport, the Xcc genomes had more genes in all categories. High copy numbers of genes in both species were functionally classified as outer membrane Fe transporters, MDR/outer-membrane efflux and transporters (TolC, AcrA), transcriptional regulators (LysR, AcrR, OmpR, LacI/PurR, NarL/FixJ), NAD(P)-dependent dehydrogenases, MFS family permeases, signal transduction histidine kinases, chemotaxis proteins (MCP, CheY), GGDEF domain secondary messengers, cell wall biosynthesis, SAM-dependent methyltransferases, glycosyltransferases, and detoxification enzymes (See Table S3 for full details).
Figure 4. COG functional characterization of native Xanthomonas pangenome elements.

Accessory genes of both species were more functionally variable than core genes, with Xcc strains dominating in most categories. Mobile elements made up a large portion of the pangenome but were more prevalent in the accessory genome. COG classification of accessory genes revealed that they were involved in extracellular stress response, transcription, DNA repair and recombination, and cell motility. In Xcc strains, a greater number of accessory genes were involved in intracellular trafficking and secretion, and secondary metabolite transport. Other Xcc gene-specific functions included Par based chromosome segregation and plasmid partitioning, NRPS enzymes, Fe transport receptors, and T4SS proteins involved in mobile pilus biogenesis. In Xmel, the accessory genes were mostly involved in putative lipase activity, biofilm formation, adhesion, Fe transport, and type II secretion system (T4SS, T2/4SS ATPases, GspE) (Table S3). Most functional categories and members in the accessory genome were also found in the core genome indicating possible functional redundancy.
Table 2 summarizes the BGC and predicted products based on syntenic similarity to reference Xanthomonas genome clusters in the MiBIG database as output from the AntiSMASH webserver. The number of BGC’s in Xmel genomes (4–14) was higher than in Xcc genomes (3–8). All Xanthomonas genomes contained the hallmark BGC’s relating to xanthoferrin and xanthomonadin production. Differences in other BGC’s include the production of pseudopyronine seen only in Xcc BrA1, while production of rhizomide A, B, and C was predicted in three Xmel isolates and Xcc Ar1PC2. Multiple NRPS clusters were found in all strains. While some were homologous to RefSeq NRPS genes from the same or other Xanthomonas species, others appear to be linked to uncharacterised compounds. Interestingly, putative class II lasso peptide clusters were predicted only in Xmel genomes.
Table 2. Predicted Biosynthetic gene clusters (BGC’s) and secondary metabolites in Xcc and Xmel genomes.
| Strain | # BGC | BGC type | Predicted metabolites* |
|---|---|---|---|
| Species: Xcc | |||
| Cf4B1 | 3 | siderophore, NRPS, aryl polyene | Xanthoferrin, Xanthomonadin I, putative NRPS |
| Cf3C | 3 | siderophore, NRPS, aryl polyene | Xanthoferrin, Xanthomonadin I, putative NRPS |
| CaNP1C | 3 | siderophore, NRPS, aryl polyene | Xanthoferrin, Xanthomonadin I, putative NRPS |
| Ar1BCA1 | 3 | siderophore, NRPS, aryl polyene | Xanthoferrin, Xanthomonadin I, putative NRPS |
| Ar1PC2 | 8 | siderophore, NRPS, aryl polyene | Xanthoferrin, Xanthomonadin I, putative NRPS, Kedarcidin (4%), cyanopeptin (50%), rhizomide A/rhizomide B/rhizomide C (multiple) |
| BrA1 | 3 | siderophore, NRPS, aryl polyene | Xanthoferrin, pseudopyronine A/pseudopyronine B (synteny to Xanthomonadin clusters of reference Xcc), putative NRPS |
| Species: Xmel | |||
| DMCX | 4 | siderophore, lassopeptide, NRPS, aryl polyene | Xanthoferrin, Xanthomonadin I, putative lassopeptide |
| CaNP6A | 12 | siderophore, lassopeptide, aryl polyene, NRPS, T1PKS (NRPS) | Xanthoferrin, Xanthomonadin I, putative lassopeptide, xenoamicin A/xenoamicin B (multiple, 25%), xenotetrapeptide, cichopeptin (50%), rhizomide A/rhizomide B/rhizomide C (multiple) |
| CaNP5B | 14 | siderophore, lassopeptide, aryl polyene, NRPS, T1PKS (NRPS) | Xanthoferrin, Xanthomonadin I, putative lassopeptide, xenoamicin A/xenoamicin B (multiple, 25%), xenotetrapeptide, rhizomide A/rhizomide B/rhizomide C (multiple), lokisin (14%), bananamide 1/bananamide 2/bananamide 3 (37%) |
| CaNP1D | 11 | siderophore, lassopeptide, aryl polyene, NRPS, T1PKS (NRPS) | Xanthoferrin, Xanthomonadin I, putative lassopeptide, xenoamicin A/xenoamicin B (25%), xenotetrapeptide, rhizomide A/rhizomide B/rhizomide C, teixobactin (20%), tolaasin A (66%) |
Note:
Predicted metabolites are based on homology to characterized BGC in the MIBiG database with linked chemical isolation data. Percentages indicate the % similarity to those gene clusters in the database and were 90–100% similar to reference clusters unless otherwise indicated.
Mobile elements and CRISPR-Cas systems
Mobile elements in bacterial genomes represent units of genetic diversity and potentially exogenous gene sequences. Figure 5 shows the distribution of mobile elements detected from RAST annotations and database queries including insertion sequences (IS), transposases and Tn related proteins, plasmid and phage related proteins, recombinases, integrases, and plasmid-derived contigs. The Xcc isolates had overall more genetic elements and more on plasmid-derived contigs than Xmel genomes. Integrases (including phage-derived proteins), a plasmid stabilization protein, and site-specific tyrosine recombinases (XerC, D) were common in both species and most prevalent on chromosomal contigs. The only Xmel species-specific elements were a putative tniA transposase and an un-identified element with a transposase and integrase Pfam domain. Other transposon elements, Tn4652 and Tn4651 tnpT were more prevalent in Xcc plasmid contigs compared to Xmel.
Figure 5. Distribution of annotated mobile elements (non-IS) in Xcc and Xmel chromosomes and plasmid derived contigs (A), and total element numbers in strains from both species (B).

(A) Vertical colour legends refer to gene counts for each respective mobile element occurring on either chromosomal or predicted/mapped plasmid contigs (coloured bar to the left of (A)). (B) Cumulative gene counts for each mobile element in (A), per species.
Identification of plasmid-derived contigs was accepted if contig length was >2 kb and contained >2 plasmid-associated Pfam HMMs with >80% identity to reference plasmid sequences. Sequence % ID and length (kb) of partial or whole contigs mapped to RefSeq plasmid sequences are provided in Tables S4A and S4B. While contigs mapped only to plasmids characterised in Xanthomonas spp., only two homologous plasmids could be reliably predicted in native strains of both species. Native Xcc (BrA1, Cf3C, Cf4B1) contained fragments of NZ_CP018463.1 X. euvesicatoria pLMG930.2 (77–82 kb, 96% ID) whereas Xmel (CaNP5B and CaNP6A) contained 41 kb from NZ_CP018472.1 X. perforans pLH3.1 (99.9% ID).
Seventy-four IS elements (12-plasmid borne, 62-chromosome borne) from eight families (IS407, IS5, IS1031, IS51, IS10, IS3, IS427, IS1595) were found at least once in the genome of native Xanthomonas isolates (Table S5). The IS element families IS1031, IS407, IS5, and IS51 were found in greater abundance in all strains (Fig. 6A). Only chromosomal-based elements were commonly found in isolates of both species, but these same elements were also found on plasmid-derived contigs in Xcc (Fig. 6A). Overall, IS elements outnumbered other mobile elements, both of which were greater in Xcc than Xmel (Fig. 6B).
Figure 6. IS element families and groups present in Xcc and Xmel chromosome and plasmid derived contigs (A), and total IS and other mobile elements per strain (B).

(A) Vertical colour legends refer to gene counts for each respective mobile element occurring on either chromosomal or predicted/mapped plasmid contigs (coloured bar to left of (A)). (B) Cumulative IS elements vs other mobile elements in each native strain from both species.
A greater occurrence of the IS1031 and IS407 family elements was seen in Xcc strains, while IS5 and IS51 family elements were found in greater abundance in Xmel strains. These elements in Xcc strains were homologous to those characterized from X. campestris, X. axonopodis, X. oryzae, and X. fuscans (Table S5). Xmel associated chromosomal IS elements were homologous to those from Azospirillum sp., Azotobacter sp., Burkholderia sp., Janthinobacterium sp., P. aeruginosa; alcaligenes, stutzeri, syringae, Ralstonia solanacearum, S. maltophilia and, X. citri. Figure 7 depicts the location of all mobile elements in each Xcc (A) and Xmel (B) genome. The presence of transposon, IS elements, and integrases in clusters in Xcc genomes suggests a more complex organisation of integrative mobile elements than in Xmel isolates. The latter species’ genome, on the other hand, revealed more instances of IS-rich loci in more than one location. Integrative mobile and conjugative elements were predicted in isolates of both species (using IceBerg2), except strain DMCX, and displayed a lower GC% than other genomic regions (Table S5).
Figure 7. Location and organization of mobile element loci in native Xcc (A) and Xmel (B) genomes.

Crispr-Cas arrays of the Type I-F subtype were only found in Xmel strains (Table 3). The cas1, cas2-3, and csy clusters (csy1-4) were detected preceding a variable length CRISPR region. Direct repeats (DR) of these arrays were 96% identical across the entire region and strains. CaNP5B contained a separate DR entirely that was also part of an orphan array in DMCX. The number of spacers and diversity varied among strains, but the majority overwhelmingly targeted plasmids from X. oryzae, X. hortorum, X. perforans, X. campestris, X. citri; Xylella and Xanthomonas phages, and IMG/VR uncultured phylloplane viruses (IMG/VR).
Table 3. DR sequences of the CRISPR-Cas 1F arrays in native Xmel strains.
| Strain | DR sequence | # Repeats |
|---|---|---|
| CaNP1D | GTTCACTGCCGCGTAGGCAGCTCAGAAA | 71 |
| CaNP5B | TTTCTGAGCTGCCTACGCGGCAGTGAAC | 124 |
| CaNP6A | GTTCACTGCCGCGTAGGCAGCTCAGAAA | 128 |
| DMCX | GTTCACTGCCGCGTAGGCAGCTCAGAAA and orphan: TTTCTGAGCTGCCTACGCGGCAGTGAAC | 69 and orphan: 62 |
T3SS and effector profile of native Xcc and Xmel strains
Native Xcc and Xmel genomes along with complete Xcc references (Table S7) were screened for the presence of 51 T3SS and effector genes (xanthomonas.org; White et al., 2009). Only Xcc isolates had the T3SS and effector genes (Table S8) including avrBS2, PhpE/xopAL1, XccA1/Xca, avrXccA2; hpa4/xopF1, A, B, G/xopA, hpaP; hrcC, J, N, Q, R, S, T, U, hrcV; hrpB1, B2, B5, D, E, F, G, W, hrpX; and xopAM, AY, AZ, D, K, L/LR, N, Q, xopZ1. All native Xcc had one xopX gene with 94–100% similary to Xcc 8004, but Ar1PC2 and references B100, 3811, CFBP 1869, and CN03, also contained another with 62% similarity to Xcc 8004 downstream.
Figure 8 depicts the collinear organization of the conserved ~23 kb hrp T3SS gene cluster of native Xcc strains. The genes in three strains (Ar1BCA1, Ar1PC2, and Cf4B1) were organized in one direction as shown in Fig. 8, whereas those in the remaining isolates (BrA1, CaNP1C, and Cf3C) were oriented in the opposite direction. Interestingly, in all native strains, a Tn element was found downstream of hpa2. Aside from these T3 genes, Fig. 9 illustrates the varied presence and copy number of some avr, xop and, hpaF genes. When using relaxed protein queries, only 4 homologs of Xcc T3 secretion, one avr, and one effector were found in Xmel genomes (Table S9). These genes were <90% similar to Xcc 8004 homologues avrXccA1 (~89%), hrpG (~73%), hrpX (~90%) and xopAZ (~87%). No other T3 secretion and effector proteins were identified in Xmel isolates using homology-based or Pfam hmm-based searches against reference genes. EffectiveDB predictions revealed the presence of potential T3 effectors but only weak support for T3SS function in Xmel genomes. The T3SS from Xcc were predicted to be functional and have a greater number of potential effectors, as shown in Table S10.
Figure 8. hrp gene cluster map of native Xcc strains.

Figure 9. Differential occurrence of selected T3 effectors in native Xcc isolates and GenBank reference genomes.

The different colours in the vertical legend indicate copy number of homologs.
Multiple alignments of Avr, HrpG, HrpX and some Xop proteins were analysed to determine potential variation in effectors shown in Fig. 9 with documented links to virulence (Table 4). Unless otherwise specified, protein sequences were compared to those from reference genome Xcc 8004.
Table 4. Amino acid variation of select T3SS and effector proteins present across native Xcc and Xmel strains.
| Protein | Comment | Function | References |
|---|---|---|---|
| AvrAC/XopAC | All similar (2 substitutions), T358A (except ATCC 33913) and, R70S (ATCC 33913 only) | Mutants infect Chinese cabbage and resistant Arabidopsis Col-0 ecotype | (Xu et al., 2008; Oh, Lee & Heu, 2011) |
| AvrBS1 | Xcc Ar1BCA1 truncated (321 vs 445). Reference and, Ar1PC2 and CaNP1C share 1 substitution | AvrBs2 -full virulence variable copy number. AvrBS3 - TALE. Korean Xcc cabbage strains (AvrBS1 and 2), Chinese cabbage strains (AvrBs1 and 3), radish strains (AvrBs2). | |
| AvrBS2 | All strains except Xcc ATCC 33913 show an R712K substitution | ||
| AvrBS3 | Homology in ~ last 500 aa only and to CN03. Xcc strains Cf3C, Cf4B1, BrA1 similar. Both from Ar1PC2 are different from other native proteins. | ||
| AvrXccB/XopJ5 | All similar (2 substitutions- G96V and V197A) | Yop-J like targets host plasma membrane immune suppression (Arabidopsis) not required for full virulence | (Liu et al., 2017) |
| AvrXccC/AvrXccAH/xopAH | Native strains contained an additional (identical) 109 aa. Identical except for one substitution (CN03 and Ar1PC2, S12G) | inhibits host immune flg22-callose deposition promotes growth of Xcc in Arabidopsis. | |
| AvrXccE1 | All proteins identical | HR response in mustard cultivars affected by Chinese strains. Putative transglutaminase | (He et al., 2007; Vicente & Holub, 2013) |
| AvrXccE2/xopE3 | All proteins identical | HR response in Chinese cabbage. Putative transglutaminase | |
| HrpG | native Xcc identical, native Xmel proteins shorter and not homologous to Xcc | Omp-R type regulator of hrp gene expression. | (Büttner & Bonas, 2010; Feng et al., 2009) |
| HrpX | Native Xcc and Xmel proteins longer than references. Xcc homologous but Xmel proteins show multiple substitutions. | AraC-type transcriptional regulator, upregulated by hrpG. | |
| XopP | proteins from native strains and Xcc 17 appear truncated at the N terminal. Otherwise, identical except for the following substitutions: C85R, D146A, S442R, M566V, I730V | Target ubiquitin ligase in rice and inhibits host enzyme function | (Adlung et al., 2016) |
The T1-6SS, extracellular enzymes, and LPS biosynthesis cluster
The prevalence of T1-6SS and extracellular enzymes found in native Xcc and Xmel genomes are depicted in Fig. 10. Only the T1SS hlyDB gene cluster was found in native genomes. The highly conserved xps and xcs T2SS gene clusters composed of genes C, D, E, F, G, H, I, J, K, L, M, and N were found on separate contigs in all strains of Xcc and Xmel. T4ASS-virB1-11, virD4 genes were also found associated with isolates, but Xcc Ar1PC2 contained the extra virD2 with all but 2 strains containing the virJ gene. Interestingly, 3 Xcc strains possessed T4BSS icm-J/T like genes. Furthermore, all genomes contained the associated and complete T4 pilus gene cluster (pilA-Z) except for Xcc Cf3C which lacked pilI. The pilX gene was also found only in the Xcc isolates. Three T5SS-related genes, xadA, yapH and estA were also detected in both species but only 3 Xmel isolates contained the fhaBC gene cluster. Additionally, while all strains had the T6SS gene tssA, the entire gene cluster tssB-M, and associated hcp, ppkA, and pppA genes were only found in Xmel genomes. The presence of a functional T6SS is supported by EffectiveDB predictions, but only weakly in Xcc strains (data not shown).
Figure 10. Presence of T1-6SS genetic elements and extracellular enzymes in Trinidadian Xcc and Xmel isolates’ genomes.

Blue indicates presence while red indicates absence, “H_” denotes a hypothetical protein.
Native isolates of Xcc (97) Xmel (80) contained putative extracellular carbon targeting enzymes, mainly in the Glycosyl hydrolase (GH) CAZy module with others classified as polysaccharide lyases (PL’s), carbohydrate esterases (CE’s), and those with carbohydrate-binding motifs (CBM’s). PCWDEs targeting plant cell wall components with known SignalP sequences were selected from these results and are depicted in Fig. 10. The enzymes xylanases, glucanases, pectinases, protease, esterases, lipases, and rhamnogalturonases were found in both species, with a higher prevalence in Xcc strains. The Xmel strains lacked the extracellular cellulase repertoire found in Xcc.
The schematic organization of the LPS biosynthesis gene cluster in Xcc B100 with highly conserved flanking genes is shown in Fig. 11A. The LPS cluster is made up of 15 genes that were organized into 3 regions. All Xcc strains had a full gene cluster (Fig. 11B) of ~17.6 kb on a single contig, except BrA1. The WxcB protein sequences in Xcc strains Ar1BCA1, BrA1, Cf3C, and Cf4B1 were up to 60% identical to that of Xcc B100. In Xmel strains, an incomplete LPS biosynthesis cluster was discovered (Fig. 11C), containing 4/15 genes from region 1. In addition, IS401 transposase and other mobile elements were found in this region of DMCX. The LPS biosynthesis clusters in both species’ genomes were flanked by the highly conserved xanAB, rmlDCAB, and metB genes which shared collinearity with Xcc B100.
Figure 11. Schematic representation of the LPS biosynthesis gene cluster present in Xcc (B) and Xmel (C) genomes ordered against the Xcc B100 reference sequence (A).

Only the wxc gene cluster from reference Xcc B100 are showed in (A), flanking genes displayed in (B) are present in this strain.
Discussion
Genomic features of native Xcc and Xmel isolates display a high degree of structural conservation
All isolates were originally obtained from black rot infected crucifer leaf tissue and putatively identified as Xanthomonas spp. This study aimed to characterise pathogenicity and virulence genes, and other genetic factors that allow for survival and adaptation to host and associated environmental ecological niches of native Xanthomonas isolates. Multiple analysis tools were used to confirm the species identity of assembled genomes of native Xanthomonas isolates. This resulted in the identification of 5 Xanthomonas campestris pv. campestris (Xcc) and 4 Xanthomonas melonis (Xmel) isolates. Co-infection of Xcc with other species is not known to occur for black rot disease, and except for cases of secondary infection with soft rot pathogens, the phenomenon is not widely reported in the literature (Vicente & Holub, 2013). While Xcc is characterised as the causal agent of black rot in crucifers (Staub & Williams, 1972) other Xanthomonas campestris pathovars have also been associated with these crops (Vicente et al., 2001; Vicente, Everett & Roberts, 2006). However, there are no reports of Xmel causing disease or being associated with crucifers. Co-infection with different species of Xanthomonas has been described in other host systems where variations in virulence of pathoadapted strains were observed (Cesbron et al., 2015; Bansal, Kumar & Patil, 2020). However, the overall impact of non-pathogenic Xanthomonas as reservoirs of genetic diversity and resources for other species is not well understood but is an important factor to consider in plant-pathogen interactions (Lee, Yang & Huang, 2020; Fernandes et al., 2021). In fact, non-pathogenic species are commonly isolated from many plant sources, but these are overwhelmingly identified as X. aboricola, although at least one study has found non-pathogenic Xcc on crucifer planting material (Lee, Yang & Huang, 2020). This interesting association of Xcc and Xmel in infected crucifer tissues highlights the need to study both pathogenic and potentially non-pathogenic/opportunistic pathogens isolated from the same environment. Analysis of Xmel will help in understanding the ecological role of this species, explore its function as co-inhabitants in the pathogenesis of Xcc, and further add to baseline data on native Xanthomonas isolates.
Multiple metrics strongly supported the identification of native Xanthomonas strains, including ANI and full 16S rRNA gene % ID greater than the species identification threshold (>98%) (Konstantinidis & Tiedje, 2005; Edgar, 2018), and whole-genome SNP phylogenetic reconstruction with reference sequences and rMLST typing (Jolley et al., 2012). The genomes of the Xcc and Xmel isolates had sizes and GC % that were typical for each species. Native Xcc isolates formed three distinct genotypes, with one new rST. The Xmel isolates were grouped into 3 distinct genotypes which only included native isolates, all of which were newly recorded rST’s. Despite belonging to different species with unique genotypes, a core genome with shared COG functions, characteristic of the genus was observed in the Xcc and Xmel isolates. Accessory genes were linked to processes involved in DNA repair and to mobile elements. Such factors point to exogenous DNA and HGT events in native strains which can account for the intraspecific structural and genome size differences. Overall structural genome collinearity was maintained between reference and native Xcc strains, though rearrangements and unique organisation of genomic loci within native strains were observed. Genomic rearrangements are common within Xcc and have been linked to host adaptation within geographic regions (Jacques et al., 2016; An et al., 2020), highlighting the enormous potential of the genus to exist and thrive in different ecological niches. Xmel genomes contained collinear segments and a more chaotic series of arrangements further compounded by the lack of a complete reference genome for this species. However, the small size of LCB’s in both native and the reference assembly, points to greater levels of variation in Xmel compared to Xcc.
Native Xcc and Xmel mobilomes
In both native species, eight IS families were discovered, with Xcc IS elements outnumbering those in Xmel. The IS family complement in Xanthomonas is highly variable displaying species-specific family repertoires with a noticeable reduction in non-pathogenic strains (Parkhill et al., 2001; Cesbron et al., 2015). IS3 and IS5 family elements are commonly found in abundance in Xcc (Thieme et al., 2005; Cesbron et al., 2015), which were also most abundant in native Xcc and Xmel strains. The specific elements in Xcc were first identified in X. campestris, X. oryzae, and other Xanthomonas spp. While those in Xmel were identified from other diverse bacterial genera including Xanthomonas. This repertoire of elements indicates greater instances of HGT between more diverse bacterial species and Xmel strains, where that in Xcc appears to be constrained to closely related species. Mobile genetic elements such as transposable elements (TE’s), phage integrases and extrachromosomal sequences (plasmids) play a substantial role in genome evolution (Dziewit et al., 2012). These elements facilitate autonomous genome restructuring through inversions and enhanced homologous recombination, even with exogenous plasmid sequences (Cesbron et al., 2015). In Xanthomonas, TE’s are linked to the acquisition and modification of resistance and virulence genes (Mahillion & Chandler, 1998; Monteiro-vitorello et al., 2005; An et al., 2020). Of note is the occurrence of IS and other mobile elements flanking the T3SS in Xcc, a phenomenon that adds to the genomic diversity potential of these elements in this genus (Cesbron et al., 2015; Merda et al., 2017; Bansal, Kumar & Patil, 2020). There is a clear capacity for genomic rearrangements and plasticity in both native species. The repertoire and copy number of IS elements have been linked to a more streamlined host-adapted lifestyle in many other bacterial species (Siguier, Gourbeyre & Chandler, 2014). Thus, the greater diversity of these features implies a less host-adapted lifestyle of Xmel.
Tn3 elements, which are commonly associated with the acquisition of antibiotic and heavy metal resistance genes (Nicolas et al., 2015), were most abundant in Xcc genomes while Xmel appears to have unique Tn elements. Integrative conjugative and mobile regions were present in all native strains and displayed characteristic structure, organisation, and lower GC%. These regions in Xcc contained heavy metal transporters, but mostly hypothetical proteins with unknown genomic importance. Exogenous sequences of plasmid origin were predicted with strong support (~98% ID to references) in three (each) Xcc and Xmel genomes from X. euvesicatoria and X. perforans respectively. High homology of these regions to these reference plasmids gives evidence of HGT events. This in combination with the characterised mobilome highlights the complex nature of lateral gene transfer in Xanthomonas. Bacterial genomic phage content and plasmid presence can be influenced by CRISPR-Cas systems. In Xanthomonas, two subtypes (1-C and 1-F) have been found which are also present in other members of Xanthomonadaceae but not in Xcc. Some species also contain complex arrays which tend to target plasmids from other species and phages (Martins et al., 2019). In native strains, only the 1-F subtype CRISPR-Cas system was found in Xmel, with complex arrays targeting diverse plasmid sequences from other species. Its absence has been linked to the presence of multiple diverse plasmids in other Xanthomonas species (Martins et al., 2019) and possibly accounts for the greater number of predicted plasmid contigs in Xcc as well as, the reduced phage content in Xmel isolates.
Pathogenicity island conservation and variable T3 effector repertoire in Xcc
The T3SS pathogenicity island was only detected in native Xcc and was syntenic to reference Xcc islands of average length (~23 kb), with no internal rearrangements. The Xcc effector repertoire comprised 14 common and 12 genes of variable prevalence. These consisted of the 9 core effectors established for this genus (Ryan et al., 2011). Notably, only native strains contained a xopR gene and 3 were missing the xopP gene, both of which are involved in the suppression of PTI (Akimoto-Tomiyama et al., 2012; Üstün & Börnke, 2014). Furthermore, the additional 8 effectors found in all native and reference isolates can be considered an extended set of core effectors for Xcc specifically as noted by Ryan et al. (2011).
While the function of all effectors is not known, those found in native strains are involved in host specificity, HR responses in certain crucifers and PTI evasion. Interestingly, a homologue of xopAG/avrGf1 was present in native strains but has only been characterised in X. citri to cause HR response in grapefruit (Gochez et al., 2015). Furthermore, hrpG and X transcriptional regulators of the T3SS were found in all Xcc. While the operon structure was conserved, sequence differences in native hrpX proteins (extra terminal amino acids) imply some adaptation to local host environments. Non-pathogenic Xanthomonas are routinely found to contain a reduced T3 effector repertoire, no pathogenicity island, but also contain the hrp transcriptional regulators (Merda et al., 2017; Lee, Yang & Huang, 2020).
A severely reduced effector repertoire was found in native Xmel strains along with hrpG and X genes. The T3SS pathogenicity island in Xmel was shown in previous work to have been lost at a later evolutionary event, despite the genus showing 3 ancestral acquisitions of this cluster (Merda et al., 2017). HGT events aided by mobile elements are mostly implicated for the gain and spread of these clusters, where they can explain relatively recent acquisition events and possibly account for in-planta co-inhabitation of non-pathogenic species (Lee, Yang & Huang, 2020). It has been proposed that Xcc obtained its T3SS clusters relatively recently with recombination with other species in the genus and that of certain Pantoea sp. strains were obtained from plasmids. Furthermore, the diversity of T3 systems in phytopathogenic bacteria have not been fully described and some without classified systems may simply have a different pathogenicity cluster type (Merda et al., 2017). Despite evidence for later acquisition in other members of the genus, native Xmel strains indicate that this still has not occurred and that no other T3SS type is present, adding further evidence to its non-pathogenic co-inhabitant status. A lack of T3SS and reduced effectors does not imply a lack of environmental fitness, as native Xmel strains possess other factors enabling effective in-planta colonization. Moreover, strains such as X. albilineans effectively colonizes xylem vessels despite its reduced T3SS and effectors (Merda et al., 2017).
Transfer and effective usage of pathogenicity islands have been demonstrated between pathogenic and non-pathogenic strains (Lee, Yang & Huang, 2020), highlighting the need to consider these commensal species as potential factors in the broad Xanthomonas gene pool. Additionally, multiple sources have shown that extracellular enzyme production can be regulated by the hrp regulon (Cesbron et al., 2015; Bansal, Kumar & Patil, 2020). While this supports the T2SS and excreted proteins as important pathogenicity factors, it also highlights the opportunistic lifestyle potential of Xmel and its ability to colonise actively infected/dead tissues. The entire xps and xcs T2SS gene clusters were found in all Xcc and Xmel genomes. Previous studies have found variable evidence of a link to AA substitutions in XpsD and T2SS substrate specificity in some Xanthomonas species (Lu et al., 2008), and in Xcc this was highly homologous but in Xmel there were many substitutions. Genes encoding PCWDEs (xylanases, pectinases, and glucanases) secreted by this system were found in both species, with Xcc strains containing more enzymes in each category as well as cellulases. Cellobiosidases, active in xylem targeting pathogens, were also found in both species. PCWDE differences are thought to reflect different metabolic niches during virulence lifestyles and to change with different host cell wall components (Ryan et al., 2011). For Xmel, these enzymes may be involved in nutrient acquisition in saprophytic/opportunistic lifestyles.
Comparative genomics revealing differences in T3SS and effector repertoire among other genetic differences are necessary but cannot replace functional studies. A virulence screen of native isolates revealed that Xmel strains did not produce symptoms and inoculated plants were similar to uninfected controls. Xcc strains were able to cause infection and some variations in symptomologies were observed. Thus, Xmel strains characterised in this study can be considered non-pathogenic within the context of the screening method and current genomic characterisation. Functional infection studies to elucidate its potential agricultural impact and its effect on co-infection, would be the subject of future research by our group. However, these types are severely underreported in relation to pathogenic strains and the current study demonstrates their genetic potential in agrochemical resistant populations. Additional focus to understand the ecological role of Xmel and other potential commensal species and their contribution to Xcc genetic diversity in infectious lifestyles is required.
Competitive capabilities of native Xanthomonas strains based on bacterial secretion system components
While collinearity of the X. citri pv. citri hylDB T1SS homologs (Fonseca et al., 2019) were maintained in native strains, no hemolysin-like toxin was found downstream. T1SS are primarily responsible for bacteriocins, adhesins, enzymes, and toxin secretion. This system is thought to play a role in the early stages of infection and might allow native isolates an advantage in competing phylloplane communities. NRPS BGC entF homologs (E. coli) predicted to produce enterobactin were more prevalent in Xmel but are common in Xanthomonas (Royer et al., 2013). Unidentified toxins inferred from non-homologous NRPS clusters may have phytotoxic and antimicrobial properties (Royer et al., 2013). Furthermore, the potential of some native strains as useful bioprotectant agents (Li et al., 2020) and the associated characterisation of novel NRPS clusters requires attention.
The variable T5SS in Xanthomonas mediates adhesion production. The yapH and xadAB genes are generally more prevalent in pathogenic Xcc (Bansal, Kumar & Patil, 2020) but were found in both native species, with Xmel additionally containing fhaBC genes. The yapH gene promotes attachment and reduced in-planta movement while xadA allows cells to establish within xylem structures (Alvarez-Martinez et al., 2021). The presence of the T4 pilus biogenesis cluster (T4P) is linked to plant colonization and possibly virulence as a preceding success factor (Fernandes et al., 2021). Most of this gene cluster was found in native isolates, but key genes such as pilI were not present in one Xcc, and pilX was only found in Xmel. While the potential for attachment to external and intracellular surfaces, and colonization potential is inferred from the T5SS and T4P, differences in gene profiles suggest disparities in these capabilities.
Interestingly, only Xmel contained the complete T6SS. This system delivers antagonistic effectors into competing bacteria (Bayer-Santos et al., 2018; Alvarez-Martinez et al., 2021). Contrastingly, previous research has observed a greater occurrence of T6SS in other pathogenic Xanthomonas (Bansal, Kumar & Patil, 2020; Alvarez-Martinez et al., 2021). Complex plant-microbe interactions, preferential survival in host tissues and other factors may compensate for native XCC lacking T6SS in competitive bacterial communities. The T4SS provides ecological advantages to bacteria by performing two major functions namely, conjugative transfer of DNA-protein complexes and, delivery of toxic proteinaceous effectors to competing bacteria. Except for DMCX, conjugative T4SS genes were identified in putative integrative mobile elements in all strains for both species A fully functional but variable T4SS relating to conjugative transfer is assumed in all native strains. Furthermore, homologs of the T4BSS Dot/Icm genes in some native Xcc strains have been associated with plasmids from X. vesicatoria (Jalan et al., 2011) and self-transmissible plasmids in other species (Voth, Broederdorf & Graham, 2012).
EPS and LPS biosynthetic gene clusters
Biofilm and its components promote Xanthomonas colonization of plant surfaces and tissues, as well as survival under adverse conditions and disease initiation (An et al., 2020). This extra polymeric substance is primarily composed of xanthan and is highly dynamic in response to signalling from bacterial cells induced by the environment. In Xcc, xanthan is linked to reduced Ca2+ related defence response in plants e.g., callose deposition (Aslam et al., 2008) and antagonizing plant triggered immunity by O-antigen detection of cell wall LPS (Girija et al., 2017). The gum operon, which regulates xanthan biosynthesis, was found in all Xcc and Xmel genomes, in addition to the xanthomonadin gene cluster. This corresponds to growth characteristics in the lab that are typical of this species (data not shown). LPS are important structural constituents of the bacterial outer membrane that influence resistance to antimicrobials and stresses. The biosynthesis of LPS is controlled by several gene clusters, all of which were conserved in the Xcc and Xmel genomes except for the wxc cluster. The latter is involved in O-antigen synthesis and varies between Xanthomonas species but shares a complex role in plant disease (Ryan et al., 2011; An et al., 2020). However, wxc gene mutants showed impaired T3SS function and disease development. The reduced wxc gene cluster in Xmel suggests an impaired ability to directly combat host defences in correlation to the lack of a T3SS. Interestingly, the wxcB gene in some native Xcc strains were not highly homologous to reference genes. This gene has been linked to O-antigen variability and structure which was also linked to reduced virulence in mutants (Park, Jung & Han, 2014). This in addition to the previously discussed variation in other virulence-related systems, points to disparities in virulent phenotypes which needs to be investigated in further functional studies.
Conclusion
This study represents the first comprehensive genome characterization of Xanthomonas campestris pv. campestris and co-inhabitant, Xanthomonas melonis. Both were isolated from crucifer leaves with black rot symptoms collected from copper agrochemical-impacted fields. While Xmel strains were obtained from the infected tissues, the results presented here reveal a lack of key genes known to be associated with pathogenicity and evasion of host defences. Regardless, its association with infections caused by Xcc is significant in the context of genomic reservoirs contributing to diversity within the genus and potential synergies between the two species. Characterization of hallmark features of the Xanthomonas genus such as secretion system variability, genome structural features, and the mobilome repertoire were presented in detail. A unique profile of T3SS effectors in native Xcc and Xmel suggests unique host adaptation traits which might influence host range or disease progression. This comprehensive genome study of native strains will contribute to a better understanding of the pathogenomics of Xcc and associated co-inhabitant species in Trinidad. The data presented here could be useful for future studies on key genomic features, selecting specific targets for virulence analysis and resistance breeding. The study also expands our understanding of combined genome characterization, virulence and resistance gene profiling for xanthomonads in the Caribbean region.
Supplemental Information
Acknowledgments
We thank Professor Jeffrey Jones (University of Florida, Gainesville) for providing the Xcc BrA1 strain and Mr Omar Ali (University of the West Indies, St. Augustine) for critical reading of the manuscript.
Abbreviations
- Xcc
Xanthomonas campestris pv. campestris
- Xmel
Xanthomonas melonis
- T1-6SS
Type 1–Type 6 secretion system
- PCWDE
Plant cell-wall degrading enzymes
- BGC
Biosynthetic gene cluster
- NRPS
Non-ribosomal peptide synthetase
Funding Statement
This work was supported by the Campus Research and Publication Fund (CRP.5.APR17.45) of The University of the West Indies, St. Augustine, Trinidad and the ACP-EU Research Project Grant (#17069) awarded to J.J and AR. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Contributor Information
Stephen D. B. Jr. Ramnarine, Email: stephen.ramnarine@my.uwi.edu.
Adesh Ramsubhag, Email: Adesh.Ramsubhag@sta.uwi.edu.
Additional Information and Declarations
Competing Interests
The authors declare that they have no completing interests.
Author Contributions
Stephen D. B. Jr. Ramnarine conceived and designed the experiments, performed the experiments, analyzed the data, prepared figures and/or tables, authored, and approved the final draft.
Jayaraj Jayaraman conceived and designed the experiments, acquired research funds, authored or reviewed drafts of the paper, and approved the final draft.
Adesh Ramsubhag conceived and designed the experiments, acquired research funds, authored or reviewed drafts of the paper, and approved the final draft.
Field Study Permissions
The following information was supplied relating to field study approvals (i.e., approving body and any reference numbers):
Diseased plant material from which Xanthomonas bacteria were isolated was sourced from farmers’ fields.
DNA Deposition
The following information was supplied regarding the deposition of DNA sequences:
The genomes of the native isolates from this study are available at GenBank for Ar1PC2, Ar1BCA1, Cf3C, Cf4B1, CaNP1C, CaNP1D, CaNP5B, CaNP6A, DMCX (PRJNA701249): JAFFQP000000000, JAFFQL000000000, JAFFQN000000000, JAFFQM000000000, JAFFQO000000000, JAFFQH000000000, JAFFQJ000000000, JAFFQI000000000, JAFFQK000000000.
Data Availability
The following information was supplied regarding data availability:
The raw data are available in the Supplemental Files.
References
- Adlung et al. (2016).Adlung N, Prochaska H, Thieme S, Banik A, Blüher D, John P, Nagel O, Schulze S, Gantner J, Delker C, Stuttmann J, Bonas U. Non-host resistance induced by the xanthomonas effector XopQ is widespread within the genus nicotiana and functionally depends on EDS1. Frontiers in Plant Science. 2016:7. doi: 10.3389/fpls.2016.01796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afgan et al. (2018).Afgan E, Baker D, Batut B, Van Den Beek M, Bouvier D, Ech M, Chilton J, Clements D, Coraor N, Grüning BA, Guerler A, Hillman-Jackson J, Hiltemann S, Jalili V, Rasche H, Soranzo N, Goecks J, Taylor J, Nekrutenko A, Blankenberg D. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Research. 2018;46(W1):W537–W544. doi: 10.1093/nar/gky379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akimoto-Tomiyama et al. (2012).Akimoto-Tomiyama C, Furutani A, Tsuge S, Washington EJ, Nishizawa Y, Minami E, Ochiai H. XopR, a type III effector secreted by Xanthomonas oryzae pv. oryzae, suppresses microbe-associated molecular pattern-triggered immunity in Arabidopsis thaliana. Molecular Plant-Microbe Interactions. 2012;25(4):505–514. doi: 10.1094/MPMI-06-11-0167. [DOI] [PubMed] [Google Scholar]
- Alvarez-Martinez et al. (2021).Alvarez-Martinez CE, Sgro GG, Araujo GG, Paiva MRN, Matsuyama BY, Guzzo CR, Andrade MO, Farah CS. Secrete or perish: The role of secretion systems in Xanthomonas biology. Computational and Structural Biotechnology Journal. 2021;19(Pt. 6):279–302. doi: 10.1016/j.csbj.2020.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An et al. (2020).An SQ, Potnis N, Dow M, Vorhölter FJ, He YQ, Becker A, Teper D, Li Y, Wang N, Bleris L, Tang JL. Mechanistic insights into host adaptation, virulence and epidemiology of the phytopathogen Xanthomonas. FEMS Microbiology Reviews. 2020;44(1):1–32. doi: 10.1093/femsre/fuz024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antipov et al. (2019).Antipov D, Raiko M, Lapidus A, Pevzner PA. Plasmid detection and assembly in genomic and metagenomic data sets. Genome Research. 2019;29(6):961–968. doi: 10.1101/gr.241299.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aslam et al. (2008).Aslam SN, Newman MA, Erbs G, Morrissey KL, Chinchilla D, Boller T, Jensen TT, De Castro C, Ierano T, Molinaro A, Jackson RW, Knight MR, Cooper RM. Bacterial polysaccharides suppress induced innate immunity by calcium chelation. Current Biology. 2008;18(14):1078–1083. doi: 10.1016/j.cub.2008.06.061. [DOI] [PubMed] [Google Scholar]
- Bansal, Kumar & Patil (2020).Bansal K, Kumar S, Patil PB. Phylogenomic insights into diversity and evolution of nonpathogenic Xanthomonas strains associated with citrus. mSphere. 2020;5(2):1–13. doi: 10.1128/msphere.00087-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer-Santos et al. (2018).Bayer-Santos E, Lima Lídia dos P, Ceseti Lde M, Ratagami CY, de Santana ES, da Silva AM, Farah CS, Alvarez-Martinez CE. Xanthomonas citri T6SS mediates resistance to Dictyostelium predation and is regulated by an ECF σ factor and cognate Ser/Thr kinase. Environmental Microbiology. 2018;20(4):1562–1575. doi: 10.1111/1462-2920.14085. [DOI] [PubMed] [Google Scholar]
- Behlau et al. (2012).Behlau F, Canteros BI, Jones JB, Graham JH. Copper resistance genes from different xanthomonads and citrus epiphytic bacteria confer resistance to Xanthomonas citri subsp. citri. European Journal of Plant Pathology. 2012;133(4):949–963. doi: 10.1007/s10658-012-9966-8. [DOI] [Google Scholar]
- Behlau et al. (2017).Behlau F, Gochez AM, Lugo AJ, Elibox W, Minsavage GV, Potnis N, White FF, Ebrahim M, Jones JB, Ramsubhag A. Characterization of a unique copper resistance gene cluster in Xanthomonas campestris pv. campestris isolated in Trinidad, West Indies. European Journal of Plant Pathology. 2017;147(3):671–681. doi: 10.1007/s10658-016-1035-2. [DOI] [Google Scholar]
- Behlau et al. (2013).Behlau F, Hong JC, Jones JB, Graham JH. Evidence for acquisition of copper resistance genes from different sources in citrus-associated xanthomonads. Phytopathology. 2013;103(5):409–418. doi: 10.1094/PHYTO-06-12-0134-R. [DOI] [PubMed] [Google Scholar]
- Biswas et al. (2013).Biswas A, Gagnon JN, Brouns SJJ, Fineran PC, Brown CM. CRISPRTarget. RNA Biology. 2013;10(5):817–827. doi: 10.4161/rna.24046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blin et al. (2019).Blin K, Shaw S, Steinke K, Villebro R, Ziemert N, Lee SY, Medema MH, Weber T. AntiSMASH 5.0: updates to the secondary metabolite genome mining pipeline. Nucleic Acids Research. 2019;47(W1):W81–W87. doi: 10.1093/nar/gkz310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun et al. (2005).Braun SG, Meyer A, Holst O, Pühler A, Niehaus K. Characterization of the Xanthomonas campestris pv. campestris lipopolysaccharide substructures essential for elicitation of an oxidative burst in tobacco cells. Molecular Plant-Microbe Interactions. 2005;18(7):674–681. doi: 10.1094/MPMI-18-0674. [DOI] [PubMed] [Google Scholar]
- Büttner & Bonas (2010).Büttner D, Bonas U. Regulation and secretion of Xanthomonas virulence factors. FEMS Microbiology Reviews. 2010;34(2):107–133. doi: 10.1111/j.1574-6976.2009.00192.x. [DOI] [PubMed] [Google Scholar]
- Cesbron et al. (2015).Cesbron S, Briand M, Essakhi S, Gironde S, Boureau T, Manceau C, Fischer-Le Saux M, Jacques MA. Comparative genomics of pathogenic and nonpathogenic strains of Xanthomonas arboricola unveil molecular and evolutionary events linked to pathoadaptation. Frontiers in Plant Science. 2015;6(431):1. doi: 10.3389/fpls.2015.01126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaumeil et al. (2020).Chaumeil PA, Mussig AJ, Hugenholtz P, Parks DH. GTDB-Tk: a toolkit to classify genomes with the genome taxonomy database. Bioinformatics. 2020;36:1925–1927. doi: 10.1093/bioinformatics/btz848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen et al. (2020).Chen C, Chen H, Zhang Y, Thomas HR, Frank MH, He Y, Xia R. TBtools: an integrative toolkit developed for Interactive analyses of big biological data. Molecular Plant. 2020;13(8):1194–1202. doi: 10.1016/j.molp.2020.06.009. [DOI] [PubMed] [Google Scholar]
- Comas et al. (2006).Comas I, Moya A, Azad RK, Lawrence JG, Gonzalez-Candelas F. The evolutionary origin of Xanthomonadales genomes and the nature of the horizontal gene transfer process. Molecular Biology and Evolution. 2006;23(11):2049–2057. doi: 10.1093/molbev/msl075. [DOI] [PubMed] [Google Scholar]
- Cornelis & Van Gijsegem (2000).Cornelis GR, Van Gijsegem F. Assembly and function of Type III secretory systems. Annual Review Microbiology. 2000;54:735–774. doi: 10.1146/annurev.micro.54.1.735. [DOI] [PubMed] [Google Scholar]
- da Silva et al. (2002).da Silva ACR, Ferro JA, Reinach FC, Farah CS, Furlan LR, Quaggio RB, Monteiro-Vitorello CB, Van Sluys MA, Almeida NF, Alves LM, do Amaral AM, Bertolini MC, Camargo LE, Camarotte G, Cannavan F, Cardozo J, Chambergo F, Ciapina LP, Cicarelli RM, Coutinho LL, Cursino-Santos JR, El-Dorry H, Faria JB, Ferreira AJ, Ferreira RC, Ferro MI, Formighieri EF, Franco MC, Greggio CC, Gruber A, Katsuyama AM, Kishi LT, Leite RP, Lemos EG, Lemos MV, Locali EC, Machado MA, Madeira AM, Martinez-Rossi NM, Martins EC, Meidanis J, Menck CF, Miyaki CY, Moon DH, Moreira LM, Novo MT, Okura VK, Oliveira MC, Oliveira VR, Pereira HA, Rossi A, Sena JA, Silva C, de Souza RF, Spinola LA, Takita MA, Tamura RE, Teixeira EC, Tezza RI, Trindade dos Santos M, Truffi D, Tsai SM, White FF, Setubal JC, Kitajima JP. Comparison of the genomes of two Xanthomonas pathogens with differing host specificities. Nature. 2002;417:459–463. doi: 10.1038/417459a. [DOI] [PubMed] [Google Scholar]
- Darling, Mau & Perna (2010).Darling AE, Mau B, Perna NT. Progressivemauve: multiple genome alignment with gene gain, loss and rearrangement. PLOS ONE. 2010;5(6):e11147. doi: 10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dziewit et al. (2012).Dziewit L, Baj J, Szuplewska M, Maj A, Tabin M, Czyzkowska A, Skrzypczyk G, Adamczuk M, Sitarek T, Stawinski P, Tudek A, Wanasz K, Wardal E, Piechucka E, Bartosik D. Insights into the transposable mobilome of Paracoccus spp. PLOS ONE. 2012;7(2):e32277. doi: 10.1371/journal.pone.0032277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar (2018).Edgar RC. Updating the 97% identity threshold for 16S ribosomal RNA OTUs. Bioinformatics. 2018;34(14):2371–2375. doi: 10.1093/bioinformatics/bty113. [DOI] [PubMed] [Google Scholar]
- Eichinger et al. (2016).Eichinger V, Nussbaumer T, Platzer A, Jehl MA, Arnold R, Rattei T. EffectiveDB: updates and novel features for a better annotation of bacterial secreted proteins and Type III, IV, VI secretion systems. Nucleic Acids Research. 2016;44(D1):D669–D674. doi: 10.1093/nar/gkv1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng et al. (2009).Feng JX, Song ZZ, Duan CJ, Zhao S, Wu YQ, Wang C, Dow JM, Tang JL. The xrvA gene of xanthomonas oryzae pv. oryzae, encoding an H-NS-like protein, regulates virulence in rice. Microbiology. 2009;155(9):303344. doi: 10.1099/mic.0.028910-0. [DOI] [PubMed] [Google Scholar]
- Fernandes et al. (2021).Fernandes C, Martins L, Teixeira M, Blom J, Pothier JF, Fonseca NA, Tavares F. Comparative genomics of Xanthomonas euroxanthea and Xanthomonas arboricola pv. juglandis strains isolated from a single walnut host tree. Microorganisms. 2021;9(3):1–15. doi: 10.3390/microorganisms9030624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonseca et al. (2019).Fonseca NP, Patané JSL, Varani AM, Felestrino É.B, Caneschi WL, Sanchez AB, Cordeiro IF, CGdC Lemes, RdAB Assis, Garcia CCM, Belasque J, Martins J, Facincani AP, Ferreira RM, Jaciani FJ, NFd Almeida, Ferro JA, Moreira LM, Setubal JC. Analyses of seven new genomes of xanthomonas citri pv. aurantifolii strains, causative agents of citrus canker B and C, show a reduced repertoire of pathogenicity-related genes. Frontiers in Microbiology. 2019;10:115. doi: 10.3389/fmicb.2019.02361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galata et al. (2019).Galata V, Fehlmann T, Backes C, Keller A. PLSDB: a resource of complete bacterial plasmids. Nucleic Acids Research. 2019;47(D1):D195–D202. doi: 10.1093/nar/gky1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girija et al. (2017).Girija AM, Kinathi BK, Madhavi MB, Ramesh P, Vungarala S, Patel HK, Sonti RV. Rice leaf transcriptional profiling suggests a functional interplay between Xanthomonas oryzae pv. oryzae lipopolysaccharide and extracellular polysaccharide in modulation of defense responses during infection. Molecular Plant-Microbe Interactions. 2017;30(1):16–27. doi: 10.1094/MPMI-08-16-0157-R. [DOI] [PubMed] [Google Scholar]
- Gochez et al. (2015).Gochez AM, Minsavage GV, Potnis N, Canteros BI, Stall RE, Jones JB. A functional XopAG homologue in Xanthomonas fuscans pv. aurantifolii strain C limits host range. Plant Pathology. 2015;64(5):1207–1214. doi: 10.1111/ppa.12361. [DOI] [Google Scholar]
- Grant & Stothard (2008).Grant JR, Stothard P. The CGView Server: a comparative genomics tool for circular genomes. Nucleic Acids Research. 2008;36(Web Server):181–184. doi: 10.1093/nar/gkn179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guglielmini et al. (2014).Guglielmini J, Néron B, Abby SS, Garcillán-Barcia MP, La Cruz DF, Rocha Key components of the eight classes of type IV secretion systems involved in bacterial conjugation or protein secretion. Nucleic Acids Research. 2014;42(9):5715–5727. doi: 10.1093/nar/gku194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison, Crécy-Lagard & Zallot (2018).Harrison KJ, Crécy-Lagard VDe, Zallot R. Gene Graphics: a genomic neighborhood data visualization web application. Bioinformatics. 2018;34(8):1406–1408. doi: 10.1093/bioinformatics/btx793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He et al. (2007).He Y-Q, Zhang L, Jiang B-L, Zhang Z-C, Xu R-Q, Tang D-J, Qin J, Jiang W, Zhang X, Liao J, Cao J-R, Zhang S-S, Wei M-L, Liang X-X, Lu G-T, Feng J-X, Chen B, Cheng J, Tang J-L. Comparative and functional genomics reveals genetic diversity and determinants of host specificity among reference strains and a large collection of Chinese isolates of the phytopathogen Xanthomonas campestris pv. campestris. Genome Biology. 2007;8(10):R218. doi: 10.1186/gb-2007-8-10-r218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huerta-Cepas et al. (2017).Huerta-Cepas J, Forslund K, Coelho LP, Szklarczyk D, Jensen LJ, Von Mering C, Bork P. Fast genome-wide functional annotation through orthology assignment by eggNOG-mapper. Molecular Biology and Evolution. 2017;34(8):2115–2122. doi: 10.1093/molbev/msx148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacques et al. (2016).Jacques MA, Arlat M, Boulanger A, Boureau T, Carrère S, Cesbron S, Chen NWG, Cociancich S, Darrasse A, Denancé N, Fischer-Le Saux M, Gagnevin L, Koebnik R, Lauber E, Noël LD, Pieretti I, Portier P, Pruvost O, Rieux A, Robène I, Royer M, Szurek B, Verdier V, Vernière C. Using ecology, physiology, and genomics to understand host specificity in Xanthomonas. Annual Review of Phytopathology. 2016;54(1):163–187. doi: 10.1146/annurev-phyto-080615-100147. [DOI] [PubMed] [Google Scholar]
- Jalan et al. (2011).Jalan N, Aritua V, Kumar D, Yu F, Jones JB, Graham JH, Setubal JC, Wang N. Comparative genomic analysis of Xanthomonas axonopodis pv. Citrumelo F1, which causes citrus bacterial spot disease, and related strains provides insights into virulence and host specificity. Journal of Bacteriology. 2011;193(22):6342–6357. doi: 10.1128/JB.05777-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarrod, Sondermann & O’Toolea (2018).Jarrod TS, Sondermann H, O’Toolea GA. Type 1 does the two-step: Type 1 secretion substrates with a functional periplasmic intermediate. Journal of Bacteriology. 2018;200(18):1–14. doi: 10.1128/JB.00168-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolley et al. (2012).Jolley KA, Bliss CM, Bennett JS, Bratcher HB, Brehony C, Colles FM, Wimalarathna H, Harrison OB, Sheppard SK, Cody AJ, Maiden MCJ. Ribosomal multilocus sequence typing: Universal characterization of bacteria from domain to strain. Microbiology. 2012;158(4):1005–1015. doi: 10.1099/mic.0.055459-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaas et al. (2014).Kaas RS, Leekitcharoenphon P, Aarestrup FM, Lund O. Solving the problem of comparing whole bacterial genomes across different sequencing platforms. PLOS ONE. 2014;9(8):1–8. doi: 10.1371/journal.pone.0104984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstantinidis & Tiedje (2005).Konstantinidis KT, Tiedje JM. Genomic insights that advance the species definition for prokaryotes. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(7):2567–2572. doi: 10.1073/pnas.0409727102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamichhane et al. (2018).Lamichhane JR, Osdaghi E, Behlau F, Köhl J, Jones JB, Aubertot J. Thirteen decades of antimicrobial copper compounds applied in agriculture: a review. Agronomy for Sustainable Development. 2018;38:28. doi: 10.1007/s13593-018-0503-9. [DOI] [Google Scholar]
- Lee, Yang & Huang (2020).Lee YA, Yang PY, Huang SC. Characterization, phylogeny, and genome analyses of nonpathogenic Xanthomonas campestris strains isolated from brassica seeds. Phytopathology. 2020;110(5):981–988. doi: 10.1094/PHYTO-08-19-0319-R. [DOI] [PubMed] [Google Scholar]
- Lemoine et al. (2019).Lemoine F, Correia D, Lefort V, Doppelt-Azeroual O, Mareuil F, Cohen-Boulakia S, Gascuel O. NGPhylogeny.fr: new generation phylogenetic services for non-specialists. Nucleic Acids Research. 2019;47(W1):W260–W265. doi: 10.1093/nar/gkz303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li et al. (2020).Li T, Mann R, Sawbridge T, Kaur J, Auer D, Spangenberg G. Novel Xanthomonas species from the perennial ryegrass seed microbiome – assessing the bioprotection activity of non-pathogenic relatives of pathogens. Frontiers in Microbiology. 2020;11:1–13. doi: 10.3389/fmicb.2020.01991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu et al. (2017).Liu L, Wang Y, Cui F, Fang A, Wang S, Wang J, Wei C, Li S, Sun W. The type III tffector AvrXccB in xanthomonas campestris pv. campestris targets putative methyltransferases and suppresses innate immunity in arabidopsis. Molecular Plant Pathology. 2017;18(6):76882. doi: 10.1111/mpp.12435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu et al. (2019).Liu M, Li X, Xie Y, Bi D, Sun J, Li J, Tai C, Deng Z, Ou HY. ICEberg 2.0: an updated database of bacterial integrative and conjugative elements. Nucleic Acids Research. 2019;47(D1):D660–D665. doi: 10.1093/nar/gky1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu et al. (2008).Lu H, Patil P, Van Sluys MA, White FF, Ryan RP, Dow JM, Rabinowicz P, Salzberg SL, Leach JE, Sonti R, Brendel V, Bogdanove AJ. Acquisition and evolution of plant pathogenesis-associated gene clusters and candidate determinants of tissue-specificity in Xanthomonas. PLOS ONE. 2008;3(11):e3828. doi: 10.1371/journal.pone.0003828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugo et al. (2013).Lugo AJ, Elibox W, Jones JB, Ramsubhag A. Copper resistance in Xanthomonas campestris pv. campestris affecting crucifers in Trinidad. European Journal of Plant Pathology. 2013;136(1):61–70. doi: 10.1007/s10658-012-0138-7. [DOI] [Google Scholar]
- Mahillion & Chandler (1998).Mahillion J, Chandler M. Insertion sequences. Microbiology and Molecular Biology Reviews. 1998;62:725–774. doi: 10.1016/B978-0-12-374984-0.00799-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin et al. (2019).Marin TGS, Galvanin AL, Lanza FE, Behlau F. Description of copper tolerant Xanthomonas citri subsp. citri and genotypic comparison with sensitive and resistant strains. Plant Pathology. 2019;68(6):1088–1098. doi: 10.1111/ppa.13026. [DOI] [Google Scholar]
- Martins et al. (2019).Martins PMM, Xavier Ada S, Takita MA, Alfemas-Zerbini P, de Souza AA. CRISPR-Cas systems in the plant pathogen Xanthomonas spp. and their impact on genome plasticity. bioRxiv. 2019 doi: 10.1385/1-59259-763-7:071. [DOI] [Google Scholar]
- Merda et al. (2017).Merda D, Briand M, Bosis E, Rousseau C, Portier P, Barret M, Jacques MA, Fischer-Le Saux M. Ancestral acquisitions, gene flow and multiple evolutionary trajectories of the type three secretion system and effectors in Xanthomonas plant pathogens. Molecular Ecology. 2017;26(21):5939–5952. doi: 10.1111/mec.14343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikheenko et al. (2018).Mikheenko A, Prjibelski A, Saveliev V, Antipov D, Gurevich A. Versatile genome assembly evaluation with QUAST-LG. Bioinformatics. 2018;34(13):i142–i150. doi: 10.1093/bioinformatics/bty266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteiro-vitorello et al. (2005).Monteiro-vitorello CB, Oliveira MCDE, Zerillo MM, Varani AM, Civerolo E. Xylella and Xanthomonas Mobil’omics 9. OMICS: A Journal of Integrative Biology. 2005;9:146–159. doi: 10.1089/omi.2005.9.146. [DOI] [PubMed] [Google Scholar]
- Nicolas et al. (2015).Nicolas E, Lambin M, Dandoy D, Galloy C, Nguyen N, Oger CA, Hallet B. The Tn3-family of replicative transposons. Mobile DNA III. 2015;49(Pt. 8):693–726. doi: 10.1128/9781555819217. [DOI] [PubMed] [Google Scholar]
- Oh, Lee & Heu (2011).Oh CS, Lee S, Heu S. Genetic diversity of AvrBs-like genes in three different xanthomonas species isolated in Korea. Plant Pathology Journal. 2011;27(1):2632. doi: 10.5423/PPJ.2011.27.1.026. [DOI] [Google Scholar]
- Overbeek et al. (2014).Overbeek R, Olson R, Pusch GD, Olsen GJ, Davis JJ, Disz T, Edwards RA, Gerdes S, Parrello B, Shukla M, Vonstein V, Wattam AR, Xia F, Stevens R. The SEED and the rapid annotation of microbial genomes using subsystems technology (RAST) Nucleic Acids Research. 2014;42(D1):206–214. doi: 10.1093/nar/gkt1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page et al. (2015).Page AJ, Cummins CA, Hunt M, Wong VK, Reuter S, Holden MTG, Fookes M, Falush D, Keane JA, Parkhill J. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics. 2015;31(22):3691–3693. doi: 10.1093/bioinformatics/btv421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, Jung & Han (2014).Park HJ, Jung HW, Han SW. Functional and proteomic analyses reveal that wxcB is involved in virulence, motility, detergent tolerance, and biofilm formation in Xanthomonas campestris pv. vesicatoria. Biochemical and Biophysical Research Communications. 2014;452(3):389–394. doi: 10.1016/j.bbrc.2014.08.076. [DOI] [PubMed] [Google Scholar]
- Parkhill et al. (2001).Parkhill J, Wren BW, Thomson NR, Titball RW, Holden MTG, Prentice MB, Sebaihia M, James KD, Churcher C. Genome sequence of Yersinia pestis, the causative agent of plague. Nature. 2001;413(6855):523–527. doi: 10.1038/35097083. [DOI] [PubMed] [Google Scholar]
- Patil & Sonti (2004).Patil PB, Sonti RV. Variation suggestive of horizontal gene transfer at a lipopolysaccharide (lps) biosynthetic locus in Xanthomonas oryzae pv. oryzae, the bacterial leaf blight pathogen of rice. BMC Microbiology. 2004;4(1):1–14. doi: 10.1186/1471-2180-4-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price, Dehal & Arkin (2010).Price MN, Dehal PS, Arkin AP. FastTree 2: approximately maximum-likelihood trees for large alignments. PLOS ONE. 2010;5(3):e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-R et al. (2018).Rodriguez-R LM, Gunturu S, Harvey WT, Rosselló-Mora R, Tiedje JM, Cole JR, Konstantinidis KT. The Microbial Genomes Atlas (MiGA) webserver: taxonomic and gene diversity analysis of Archaea and Bacteria at the whole genome level. Nucleic Acids Research. 2018;46(W1):W282–W288. doi: 10.1093/nar/gky467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royer et al. (2013).Royer M, Koebnik R, Marguerettaz M, Barbe V, Robin GP, Brin C, Carrere S, Gomez C, Hügelland M, Völler GH, Noëll J, Pieretti I, Rausch S, Verdier V, Poussier S, Rott P, Süssmuth RD, Cociancich S. Genome mining reveals the genus Xanthomonas to be a promising reservoir for new bioactive non-ribosomally synthesized peptides. BMC Genomics. 2013;14(1):1–19. doi: 10.1186/1471-2164-14-658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russel et al. (2020).Russel J, Pinilla-Redondo R, Mayo-Muñoz D, Shah SA, Sørensen SJ. CRISPRCasTyper: automated identification, annotation, and classification of CRISPR-Cas Loci. The CRISPR Journal. 2020;3(6):462–469. doi: 10.1089/crispr.2020.0059. [DOI] [PubMed] [Google Scholar]
- Ryan et al. (2011).Ryan RP, Vorhölter F-J, Potnis N, Jones JB, Van Sluys M-A, Bogdanove AJ, Dow JM. Pathogenomics of Xanthomonas: understanding bacterium-plant interactions. Nature Reviews Microbiology. 2011;9(5):344–355. doi: 10.1038/nrmicro2558. [DOI] [PubMed] [Google Scholar]
- Siguier, Gourbeyre & Chandler (2014).Siguier P, Gourbeyre E, Chandler M. Bacterial insertion sequences: their genomic impact and diversity. FEMS Microbiology Reviews. 2014;38(5):865–891. doi: 10.1111/1574-6976.12067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siguier et al. (2006).Siguier P, Perochon J, Lestrade L, Mahillon J, Chandler M. ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Research. 2006;34(90001):32–36. doi: 10.1093/nar/gkj014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solé et al. (2015).Solé M, Scheibner F, Hoffmeister AK, Hartmann N, Hause G, Rother A, Jordan M, Lautier M, Arlat M, Büttner D. Xanthomonas campestris pv. vesicatoria secretes proteases and xylanases via the Xps type II secretion system and outer membrane vesicles. Journal of Bacteriology. 2015;197(17):2879–2893. doi: 10.1128/JB.00322-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souza et al. (2011).Souza DP, Andrade MO, Alvarez-Martinez CE, Arantes GM, Farah CS, Salinas RK. A component of the Xanthomonadaceae type IV secretion system combines a VirB7 motif with a N0 domain found in outer membrane transport proteins. PLOS Pathogens. 2011;7(5):e1002031. doi: 10.1371/journal.ppat.1002031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staub & Williams (1972).Staub T, Williams PH. Factor influencing black rot lesion development in resistant and susceptible cabbage. Phytopathology. 1972;62(7):722–728. doi: 10.1094/Phyto-62-722. [DOI] [Google Scholar]
- Stöver & Müller (2010).Stöver BC, Müller KF. TreeGraph 2: combining and visualizing evidence from different phylogenetic analyses. BMC Bioinformatics. 2010;11(1):275. doi: 10.1186/1471-2105-11-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tampakaki et al. (2004).Tampakaki AP, Fadouloglou VE, Gazi AD, Panopoulos NJ, Kokkinidis M. Conserved features of type III secretion. Cellular Microbiology. 2004;6(9):805–816. doi: 10.1111/j.1462-5822.2004.00432.x. [DOI] [PubMed] [Google Scholar]
- Thieme et al. (2005).Thieme F, Koebnik R, Bekel T, Berger C, Boch J, Bu D, Caldana C, Gaigalat L, Goesmann A, Kay S, Kirchner O, Lanz C. Insights into genome plasticity and pathogenicity of the plant pathogenic bacterium. Society. 2005;187:7254–7266. doi: 10.1128/JB.187.21.7254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Üstün & Börnke (2014).Üstün S, Börnke F. Interactions of Xanthomonas type-III effector proteins with the plant ubiquitin and ubiquitin-like pathways. Frontiers in Plant Science. 2014;5(e1003952):1–6. doi: 10.3389/fpls.2014.00736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Graaf van Bloois, Wagenaar & Zomer (2020).van der Graaf van Bloois L, Wagenaar JA, Zomer AL. RFPlasmid: predicting plasmid sequences from short read assembly data using machine learning. bioRxiv. 2020 doi: 10.1101/2020.07.31.230631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vauterin et al. (1995).Vauterin L, Hoste B, Kersters K, Swings J. Reclassification of Xanthomonas. International Journal of Systematic Bacteriology. 1995;45(3):472–489. doi: 10.1099/00207713-45-3-472. [DOI] [Google Scholar]
- Vicente et al. (2001).Vicente JG, Conway J, Roberts SJ, Taylor JD. Identification and origin of Xanthomonas campestris pv. campestris races and related pathovars. Phytopathology. 2001;91(5):492–499. doi: 10.1094/PHYTO.2001.91.5.492. [DOI] [PubMed] [Google Scholar]
- Vicente, Everett & Roberts (2006).Vicente JG, Everett B, Roberts SJ. Identification of isolates that cause a leaf spot disease of brassicas as Xanthomonas campestris pv. raphani and pathogenic and genetic comparison with related pathovars. Phytopathology. 2006;96(7):735–745. doi: 10.1094/PHYTO-96-0735. [DOI] [PubMed] [Google Scholar]
- Vicente & Holub (2013).Vicente JG, Holub EB. Xanthomonas campestris pv. campestris (cause of black rot of crucifers) in the genomic era is still a worldwide threat to brassica crops. Molecular Plant Pathology. 2013;14(1):2–18. doi: 10.1111/j.1364-3703.2012.00833.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorhölter, Niehaus & Pühler (2001).Vorhölter FJ, Niehaus K, Pühler A. Lipopolysaccharide biosynthesis in Xanthomonas campestris pv. campestris: a cluster of 15 genes is involved in the biosynthesis of the LPS O-antigen and the LPS core. Molecular Genetics and Genomics. 2001;266(1):79–95. doi: 10.1007/s004380100521. [DOI] [PubMed] [Google Scholar]
- Voth, Broederdorf & Graham (2012).Voth DE, Broederdorf LJ, Graham JG. Bacterial TYpe IV secretion systems: verstaile virulence machines. Future Microbiology. 2012;7:241–257. doi: 10.2217/fmb.11.150.Bacterial. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang et al. (2007).Wang Q, Garrity GM, Tiedje JM, Cole JR. Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Applied and Environmental Microbiology. 2007;73(16):5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White et al. (2009).White FF, Potnis N, Jones JB, Koebnik R. The type III effectors of Xanthomonas. Molecular Plant Pathology. 2009;10(6):749–766. doi: 10.1111/j.1364-3703.2009.00590.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu et al. (2008).Xu RQ, Blanvillain S, Feng JX, Jiang BL, Li XZ, Wei HY, Kroj T, Lauber E, Roby D, Chen B, He YQ, Lu GT, Tang DJ, Vasse J, Arlat M. AvrACXcc8004, a type III efector with a leucine-rich repeat domain from xanthomonas campestris pathovar campestris confers avirulence in vascular tissues of arabidopsis thaliana ecotype col-0. Journal of Bacteriology. 2008;190(1):34355. doi: 10.1128/JB.00978-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang et al. (2018).Zhang H, Yohe T, Huang L, Entwistle S, Wu P, Yang Z, Busk PK, Xu Y, Yin Y. DbCAN2: A meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Research. 2018;46(W1):W95–W101. doi: 10.1093/nar/gky418. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The following information was supplied regarding data availability:
The raw data are available in the Supplemental Files.