

Abstract
Targeted delivery of dexamethasone to inflamed tissues using nanoparticles is much-needed to improve its efficacy while reducing side effects. To drastically improve dexamethasone loading and prevent burst release once injected intravenously, a lipophilic prodrug dexamethasone palmitate (DXP) was encapsulated into poly(DL-lactide-co-glycolide)-polyethylene glycol (PLGA-PEG) nanoparticles (NPs). DXP-loaded PLGA-PEG NPs (DXP-NPs) of about 150 nm with a drug loading as high as 7.5% exhibited low hemolytic profile and cytotoxicity. DXP-NPs were able to inhibit the LPS-induced release of inflammatory cytokines in macrophages. After an intravenous injection to mice, dexamethasone (DXM) pharmacokinetic profile was also significantly improved. The concentration of DXM in the plasma of healthy mice remained high up to 18 h, much longer than the commercial soluble drug dexamethasone phosphate (DSP). Biodistribution studies showed lower DXM concentrations in the liver, kidneys, and lungs when DXP-NPs were administered as compared with the soluble drug. Histology analysis revealed an improvement in the knee structure and reduction of cell infiltration in animals treated with the encapsulated DXP compared with the soluble DSP or non-treated animals. In summary, the encapsulation of a lipidic prodrug of dexamethasone into PLGA-PEG NPs appears as a promising strategy to improve the pharmacological profile and reduce joint inflammation in a murine model of rheumatoid arthritis.
Graphical abstract

Supplementary information
The online version contains supplementary material available at 10.1007/s13346-021-01112-3.
Keywords: PLGA, Dexamethasone palmitate, Pharmacokinetics, Rheumatoid arthritis
Introduction
Glucocorticoids (GCs) are potent anti-inflammatory and immunosuppressive drugs applied in the treatment of many inflammatory and autoimmune diseases [1], the latest application being the treatment of hyper inflammation in COVID-19 [2]. However, when administered intravenously, their unfavorable pharmacokinetics (PK), the high doses needed to reach a therapeutic effect, and the associated side effects have limited their prescription on a chronic basis [3]. Nevertheless, the therapeutic use of GC could greatly benefit from their delivery by NPs. Indeed, if NPs possess a prolonged blood circulation time, they could benefit from the extravazation through leaky vasculature and the subsequent inflammatory cell-mediated sequestration (ELVIS) effect to accumulate into inflamed tissues, release GCs, and therefore reduce the dose needed and all the associated side effects [4].
Dexamethasone (DXM) has already been encapsulated into polymeric NPs [5, 6]. Although the encapsulation efficiency was low and the non-encapsulated drug formed crystals, these formulations have already shown a controlled release of the drug. DXM has also been encapsulated into liposomes, however, the encapsulation efficiency and the drug loading were also very low [7, 8]. Nevertheless, these liposomal formulations have already proven a better therapeutic efficacy in rheumatoid arthritis (RA) animal models and a reduction of the side effects compared with the free drug.
As an alternative to DXM, we propose to use dexamethasone palmitate (DXP) a hydrophobic prodrug formed by DXM and a palmitate group linked by an ester bond, with a longer half-life in plasma than DXM [9]. The prodrug needs to be hydrolyzed into DXM by esterases, which are present in the site of inflammation, to release the active drug. DXP is commercialized as a phospholipid emulsion, named Limethason® [10]. The advantage of encapsulation is the avoidance of DXP crystal formation [9]. However, this formulation has shown a short to medium sustained release in healthy volunteers and patients with inflammatory osteoarthritis, with redistribution into the systemic circulation of 90% of the drug within 48 h after the intra-articular administration [9, 11]. Liposomes were also explored as nanocarriers for DXP, but they have shown many drawbacks such as short residence time in the inflamed joints [11, 12], low encapsulation efficiency, and poor stability [13].
The most recent approach in formulating DXP NPs was conducted in our group: it consists of turning DXP into NPs using distearoylphosphatidylethanolamine — polyethylene glycol as the sole stabilizing amphiphilic molecule. The NPs were highly stable and the PK much improved compared to the soluble form of DXM dexamethasone sodium phosphate (DSP) but not prolonged enough due to quick hydrolysis of the prodrug in blood [14, 15].
To further improve the encapsulation efficiency and the sustained release of the prodrug, in the present work, DXP was encapsulated into poly(DL-lactide-co-glycolide)-polyethylene glycol (PLGA-PEG) NPs. PLGA is one of the most widely used polymers for NP synthesis due to its good biocompatibility, biodegradability, and capability to induce sustained drug release [16]. Polylactide-co-glycolide (PLGA) and polyethylene glycol (PEG) are FDA-approved polymers and PLA-PEG nanoparticles have entered several clinical trials [17].
The formulation was characterized by dynamic light scattering (DLS) and transmission electron microscopy (TEM). The potential toxicological effect, the internalization of the NPs in macrophages, and the capacity of the formulation to inhibit the release of pro-inflammatory cytokines were studied in vitro in the murine macrophage cell line RAW 264.7. The biodistribution and the PK of the prodrug (DXP) and the active drug (DXM) were also characterized in healthy mice injected with DXP NPs, and compared with soluble DSP. Finally, the therapeutic efficacy was tested in collagen-induced arthritis (CIA), a murine model of RA [18].
Materials and methods
Materials
PLGA-PEG [poly(ethylene glycol)-poly(DL-lactide-co-glycolide) 50:50 Resomer PEG type RGP d 50,105 (MW: 50,000 g/mol) (diblock, 10% PEG with 5000 Dalton)] and the PLGA-Rhodamine (PLGA-Rho) polymers were purchased from Evonik Industries AG (Essen, Germany) and PolySciTech® Akina Inc. (Indiana, USA), respectively. Dexamethasone palmitate was provided by Interchim (France). Immunization grade chick type II collagen, Complete Freund’s Adjuvant 4 mg/mL, and Incomplete Freund’s Adjuvant were provided by AMSBio (Abingdon, UK). Paraformaldehyde (Antigenfix), EDTA-based decalcification solution (Microdec), and paraffin for paw fixation were provided by Microm Microtech (Brignais, France) as well as eosin-phloxine and hematoxylin for staining. Saffron in alcoholic solution was purchased from RAL Diagnostics (Martillac, France). Dichloromethane in analytical grade and acetonitrile in high-performance liquid chromatography (HPLC) grade were obtained from Carlo Erba Reagents (France). Water was purified using a Synergy system from Millipore (France). All the other reagents were from Sigma-Aldrich unless otherwise stated.
Cell culture
RAW 264.7 cells were purchased from the American Type Culture Collection (ATCC) and they were cultured in Dulbecco’s modified Eagle’s medium (DMEM), supplemented with 10% fetal bovine serum (FBS) and 10,000 unit/mL of penicillin and 10 mg/mL of streptomycin (Lonza, Switzerland). Cultures were maintained at 37 °C in a humidified atmosphere containing 5% CO2. The phosphate-buffered saline (PBS) was also from Lonza (Switzerland). After thawing, cells were used for experiments from passage 6 to 15.
Nanoparticle preparation
The unloaded NPs and DXP-NPs were prepared by the solvent emulsion evaporation technique as described before [19] with some modifications. Briefly, 100 mg of PLGA-PEG and different amounts of dexamethasone palmitate were dissolved into 4 mL of dichloromethane. This organic solution was pre-emulsified with 20 mL of a sodium cholate aqueous solution (1.5% w/v) by vortexing at 3200 rpm for 1 min (Mini Vortexer VWR, USA). The pre-emulsion was kept on ice and sonicated at 300 W for another minute using a Vibra cell sonicator (Bioblock Scientific, France). The organic phase was then evaporated at 20 °C in a water bath under gentle magnetic stirring (300 rpm). The final volume was completed to 20 mL with MilliQ water and the formulation was filtered by using 0.45-μm PVDF filters. For internalization experiments, NPs were prepared using 90/10 (w/w) PLGA-PEG and PLGA-Rhodamine and following the same procedure.
Removal of non-encapsulated dexamethasone palmitate
The non-encapsulated DXP was removed by adding an SDS aqueous solution (20% w/v) to yield a final concentration of 5% SDS. SDS role is to solubilize the hydrophobic non-encapsulated DXP. The sample was ultracentrifuged twice using an Optima LE-80 K centrifuge (Beckman Coulter Inc.) at 20,000 rpm (27,400 g) for 1 h to remove both sodium cholate and SDS. The final formulation was resuspended in 20 mL of water to yield a polymer concentration of 5 mg/mL for physicochemical characterization. For the in vitro and in vivo characterization, the formulation was further concentrated or diluted depending on the final concentration needed.
Nanoparticle characterization
Particle size and polydispersity index were determined using a Malvern Zetasizer Nano ZS (Malvern Instrument, UK) based on quasi-elastic light scattering. Size measurements were performed in triplicate following a 1/100 (v/v) dilution of the NP suspension in Milli-Q water at 20 °C. Zeta potential was measured using the same instrument at 20 °C following a 1/50 (v/v) dilution in a 1 mM NaCl solution. Transmission electron microscopy was performed at Imagif (CNRS, Gif-sur-Yvette, France). A volume of 5 µL of the NP suspension was deposited for 1 min on formvar-coated copper grids. Negative staining was performed by the addition to the grid of a drop of uranyl acetate at 2% w/w for 30 s. Excess solution was removed and grids were left to dry before observation. The observations were carried out on a JEOL microscope at an acceleration voltage of 80 kV.
Characterization of DXP loading
NPs were dissolved in acetonitrile (1/100 v/v) and filtered with a 0.45-μm PVDF filter. The quantity of DXP in the NPs was determined by injecting 50 μL of the filtered solution in a Waters™ high-performance liquid chromatography (HPLC) system. The analysis was performed at 240 nm using a SymmetryShield™ column RP18 5 µm (250 × 4.6 mm) at 40 °C with a mobile phase composed of 85% acetonitrile and 15% water at 1.2 mL/min. The retention time for DXP was 23 min approximately and DXP solutions of known concentration were used for the calibration curve. An aliquot of the formulation before the SDS treatment was also measured to determine the total DXP amount.
The encapsulation efficiency was calculated as the percentage of encapsulated drug and the initial drug. The drug loading is the percentage, in mass, of the drug in the formulation (drug + polymer):
where DXPi is the initial amount of DXP, DXPe is the encapsulated DXP, and mNPs is the polymer mass in the NPs.
Cell viability
The influence of NPs and DXM on cell viability was studied in the RAW 264.7 cell line using an MTT assay that evaluates mitochondrial activity. Cells were seeded in 96-well plates at a density of 8 × 103 cells/well in 100 µL of culture medium, and incubated overnight. Afterward, DXP-NPs and unloaded NPs were added. The plates were incubated for 24 h and the yellow tetrazolium MTT (3-(4, 5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide) was added at a final concentration of 500 µg/mL and incubated for another hour. The MTT is reduced by metabolically active cells to form the purple formazan crystals. After the formation of the crystals, the medium was replaced by 100 µL of DMSO to dissolve them and the absorbance was measured at 570 nm. The absorbance of the cells treated with the NPs was divided by the absorbance of the control cells, after subtraction of the blank, and expressed as the percentage of viable cells.
Hemolysis
The potential hemolysis induced by DXP-NPs on mouse red blood cells was studied in vitro. Mouse whole blood was extracted from healthy mice (strain DBA-1) and washed three times in PBS at 1000 g, 10 min at 4 °C to obtain a suspension of 3% (w/v). TritonX-100 at 1% was used as a positive control. The erythrocyte suspension was seeded in a 96-well round-bottom plate (80 µL per well). Then, 80 µL of DXP-NPs or unloaded NPs at 800 µg/mL PLGA-PEG were added to test a final NP concentration of 400 µg/mL, or 80 µL of the TritonX-100 1% for the positive control (total lysis of the red blood cells). The plate was incubated at 37 °C for 4 h and centrifuged at 1000 g, 10 min at 4 °C. The supernatant was transferred to a 96-well flat-bottom plate and the absorbance at 570 nm, due to the release of the hemoglobin, was measured. The absorbance of the PBS samples was subtracted from the absorbance of the samples with NPs or with TritonX-100. The hemolysis was expressed as follows:
Nanoparticle uptake in RAW 264.7 cells
The uptake of DXP-NPs in RAW 264.7 cells was measured by flow cytometry using Rhodamine-labeled NPs. Cells were seeded in 12-well plates at 8 × 104 cells/well and incubated 24 h to reach 80% confluency. DXP-NPs were added at 100 µg/mL. PLGA-PEG and plates were incubated at different time intervals. After incubation, the medium was removed and cells were washed with PBS, detached with trypsin, and resuspended in PBS. The mean fluorescence intensity (MFI) of the treated cells was measured using a flow cytometer (Accuri C6, BD Biosciences, USA) and it was normalized to the non-treated cells to quantify the MFI fold increase.
Cytokine release
For the cytokine release experiment, RAW 264.7 cells were seeded in 24-well plates at a cell density of 4 × 104 cells/well in culture medium and were left in the incubator for 48 h. The medium was then replaced by fresh medium alone or fresh medium with LPS at 0.1 µg/mL, and the plates were incubated for another 3 h. Afterward, the DXP-NPs at 10 µg/mL of DXP (130 µg/mL PLGA-PEG) and unloaded NPs at the same concentration were added to some wells. Culture medium alone was used as negative control and LPS 0.1 µg/mL as a positive control. DSP at the same DXM concentration (8.2 µg/mL) was also tested to compare the encapsulated GC with the soluble drug. After 24 h of incubation with the treatments, the supernatants were collected and frozen at − 20 °C until the analysis was performed. The mouse inflammatory cytokines IL-12p70, TNFα, IFN-γ, MCP-1, IL-10, and IL-6 were quantified using the Cytometric Beads Array (CBA) mouse inflammation kit (BD Biosciences, USA). The supernatant was tested undiluted (50 µL) for all the cytokines except for TNFα, because for this cytokine the concentration was very high and out of the calibration curve. In this case, a dilution of 1/20 was used. In both cases, the experiment was performed following the manufacturer’s instructions.
The cytokine quantification (IL-4, TNFα, IL-17A, IL-10, IL-6, IL-1β, MCP-1) in serum samples from CIA mice was also performed by flow cytometry using custom-made bead assay (LEGENDplex from Biolegend, San Diego, USA). The samples were diluted 1:2 and the kit was performed as described by the manufacturer. Data were analyzed with the LEGENDplex software provided with the kit.
Pharmacokinetics and biodistribution
The PK studies were performed in 9-week-old male DBA-1 mice (Harlan, France). Seven animals per group were used and the equivalent of 12 mg/kg of DXM was injected as DXP NPs. The animals were sacrificed at different time points (10, 20, 40, 60, 120, 240, 360, 480, 1080 min) and plasma and organs (liver, spleen, kidneys, and lung) were collected for DXP and DXM quantification by HPLC. A group that received a DSP solution at the same DXM dose was used as a control.
The organs collected from mice were weighted, diluted with an equivalent volume of PBS, and homogenized with a tissue grinder coupled to a drill. Testosterone decanoate and dexamethasone acetate (DXA) at 4 µg/mL were used as internal standards for DXP and DXM, respectively.
An aliquot of 100 µL of plasma or 100 mg of the ground organs was used for the extraction, diluted with 100 µL of testosterone decanoate and DXA in ACN and mixed by vortexing. Then, 3 mL of CHCl3/CH4O 9:1 was added and mixed again by vortexing. Centrifugation was performed at 3500 rpm (= 1690 g) for 30 min (ST16R centrifuge, rotor TX-400, Thermo Scientific, France). The organic phase was collected in a glass vial and evaporated under an N2 atmosphere at 30 °C using a sample block heater. After evaporation, the sample was resuspended in 200 µL of acetonitrile and injected in the HPLC using the same conditions described for drug loading in the case of the DXP. For dexamethasone (DXM) measurement, the mobile phase was composed of 35% acetonitrile and 65% water, and pure dexamethasone was used for the calibration curve. The retention time was approximately 9 min for the drug at 30 °C. The retention times for the testosterone decanoate and DXA were 20 min and 30 min, respectively, in their corresponding mobile phase.
A non-compartmental analysis was used to study the pharmacokinetic behavior of DXP and DXM in the plasma of animals injected with DXP-NPs or DSP. The pharmacokinetic parameters were calculated using Excel’s PK solver program. The calculated parameters from the concentration–time curve were area under plasma concentration–time curve (AUC) using the linear trapezoid method, area under the first moment curve (AUMC), the terminal elimination rate constant (Ke) which was calculated from the slope of the logarithm of the plasma concentration versus time profile, the elimination half-life (t1/2), mean residence time (MRT) as AUMC/AUC both extrapolated to infinity, clearance (Cl) as dose/AUC, and volume of distribution in steady state (Vd) as MRT × Cl.
Therapeutic efficacy of the DXP nanoparticles in mice with CIA
For the therapeutic efficacy studies, thirty 9-week-old DBA-1 mice were immunized with collagen type II emulsified with complete Freund’s adjuvant to induce arthritis. The collagen was dissolved at 4 mg/mL in 10 mM acetic acid at 4 °C and under constant stirring the day previous to the immunization. The animals were shaved at the base of the tail using isoflurane to anesthetize them.
The CFA was placed in a 5-mL syringe and the collagen suspension was added dropwise at a ratio of 1:1 and emulsified using an ultraturrax (IKA) with an 8-G rod coupled. All the material used was pre-cooled to avoid denaturation of the protein. The mixture was emulsified for 30 s and chilled in ice for another 30 s performing several cycles until a homogeneous white emulsion was formed. Then, the emulsion was transferred into a 1-mL Hamilton syringe for intradermal injection of 50 µL at the back of each mouse, near the tail. The animals were anesthetized with isoflurane before the injection and forceps were used to lift the skin.
A booster injection was performed after 21 days to induce a high prevalence of CIA using the same concentration and the same procedure but avoiding injecting in the inflamed tissue from the previous immunization. The previous day to the booster injection and every 2 or 3 days after, the arthritic score and paw volume were monitored for all mice. The paw volume was measured using a plethysmometer which calculates the volume based on the displacement of the liquid after dipping the paw in a liquid-filled cell.
The mice were divided into three groups and were treated with DXP-NPs, DSP, or PBS as a negative control, respectively. The DXM dose administered to the animals treated with DXP NPs or DSP was 1 mg/kg and the treatments were administered intravenously three times every 2 days starting at day 32 after the first immunization. During the treatment period, the arthritis score and paw volume were monitored daily. The mice were bled before and after the treatments at days 31 and 37, respectively, to collect the serum for cytokine quantification. The serum samples were frozen at − 20 °C until the analysis was performed. All mice were sacrificed at day 38; the hind paws were collected for histology studies and fixed in paraformaldehyde 4% 24 h after removing the skin and the muscles.
Histological characterization of the hind paws
The hind paws in paraformaldehyde 4% were rinsed twice with Milli-Q water and the bones were softened by incubation with a calcium chelator (Microdec) for 8 h. After decalcification, the paws were washed again and dehydrated with ethanol at increasing concentrations (70–100%). Finally, samples were immersed in xylene and fixed in paraffin blocks. The blocks were cut into 7-µm slices by using a microtome (Leica RM2255) and placed on crystal slides. Some consecutive slides were selected for histology characterization. The samples were rehydrated by immersion sequentially in xylene, ethanol solutions of decreasing concentrations (100–70%), and distilled water for 5 min in each solvent. The slides selected for histological studies were immersed in hematoxylin for 3 min to stain the cell nucleus. Afterward, they were rinsed with abundant water from the tap before their immersion in eosin-phloxine for 8 min to stain the interstitial tissues. The slides were rinsed in distilled water for 20 s and 95% ethanol for 2 min before their immersion in a saffron solution for 7 min to stain the bone and cartilage. The samples were dehydrated again by performing three immersions in 100% ethanol followed by two immersions in xylene for 5 min each and fixed with DPX mounting medium. A coverslip was placed over the slides and they were allowed to dry under the hood.
Statistical analysis
All measurements were repeated three times. The values were expressed as the mean ± standard deviation. Multiple comparisons by two-way ANOVA have been used to assess the statistically significant differences between the means (p < 0.05) when appropriate.
Results and discussion
Nanoparticle characterization and dexamethasone palmitate loading
The unloaded NPs and DXP-NPs were prepared by the solvent emulsion evaporation technique, varying the amount of initial DXP in the formulation. After removal of free DXP, nanoparticles were characterized for size, polydispersity index (PDI), and zeta potential. All NPs had an average size of approximately 150 nm, with a PDI around 0.1, indicative of a highly homogenous formulation, and a negative zeta potential (− 25 to − 28 mV) (Fig. 1A). The homogeneity of the DXP-NPs was confirmed by transmission electron microscopy (TEM; Fig. 1B) and their shape was spherical. The size of about 150 nm, and the low PdI, makes the NPs adequate for an intravenous treatment and a passive delivery in the inflamed tissues through extravasation via leaky vasculature followed by inflammatory cell sequestration (ELVIS) [20]. The high negative charge allows a more stable colloidal suspension, avoiding the coalescence and aggregation of the NPs due to the electrostatic repulsion between them. Indeed, the stability of DXP-NPs stored at 4 °C was monitored for more than 1 month by measuring the size and the PdI of both unloaded and DXP-NPs (10 mg initial DXP) (Supporting information Figure S1). Both formulations were stable for up to 30 days without the appearance of any drug crystals that could be observed under the optical microscope. Encapsulation efficiency and drug loading were measured by HPLC. The drug loading followed a linear behavior (Fig. 1C), and the encapsulation efficiency went up to 85% for low DXP amount and down to 75% for high DXP amount. The DXP was encapsulated into PLGA-PEG NPs with high encapsulation efficiency and good drug loading. The non-encapsulated drug could easily be removed using 5% SDS and removing the supernatant after ultracentrifugation. The encapsulation efficiency of the DXP into PLGA-PEG NPs and the removal of the non-encapsulated drug were much better than for DXM [5] or dexamethasone acetate [6], with, for the latest, an efficiency of about 3% and the formation of crystals in the NP suspension by the non-encapsulated drug. Besides, in the DXM and dexamethasone acetate formulations, there was a rapid saturation when the initial amount of GC was increased while, in the case of the DXP, the encapsulation was linear up to the highest concentration tested (20 mg/mL). As a compromise between good encapsulation efficiency and high drug loading, the remaining experiments were conducted with DXP-NPs formulated with an initial mass of 10 mg DXP; these NPs named DXP-NPs hereby exhibit a drug loading of 7.5% w/w.
Fig. 1.

A Size, PdI, and Zeta-potential of the NPs. B Transmission electron microscopy images of the DXP-NPs 10 mg. The scale bar represents 200 nm. C Dexamethasone palmitate loading (DL) and encapsulation efficacy (EE) into PLGA-PEG nanoparticles as a function of initial DXP amount
Cell viability and hemolysis
The potential toxicity of the unloaded PLGA-PEG and DXP-NPs was tested in the RAW 264.7 cells using the tetrazolium MTT assay (Fig. 2A). The highest DXP concentration tested was approximately 75 µg/mL for the DXP-NPs at 1 mg/mL. There was a reduction of the viability at the highest polymer concentrations (0.25–1 mg/mL) of about 30%. Nevertheless, the viability was always higher than 80% for both formulations (Fig. 2A). DXP does not reduce the cell viability further than PLGA-PEG NPs at high concentrations; hence, the encapsulated DXP was not toxic for the macrophage cells at any concentration tested.
Fig. 2.
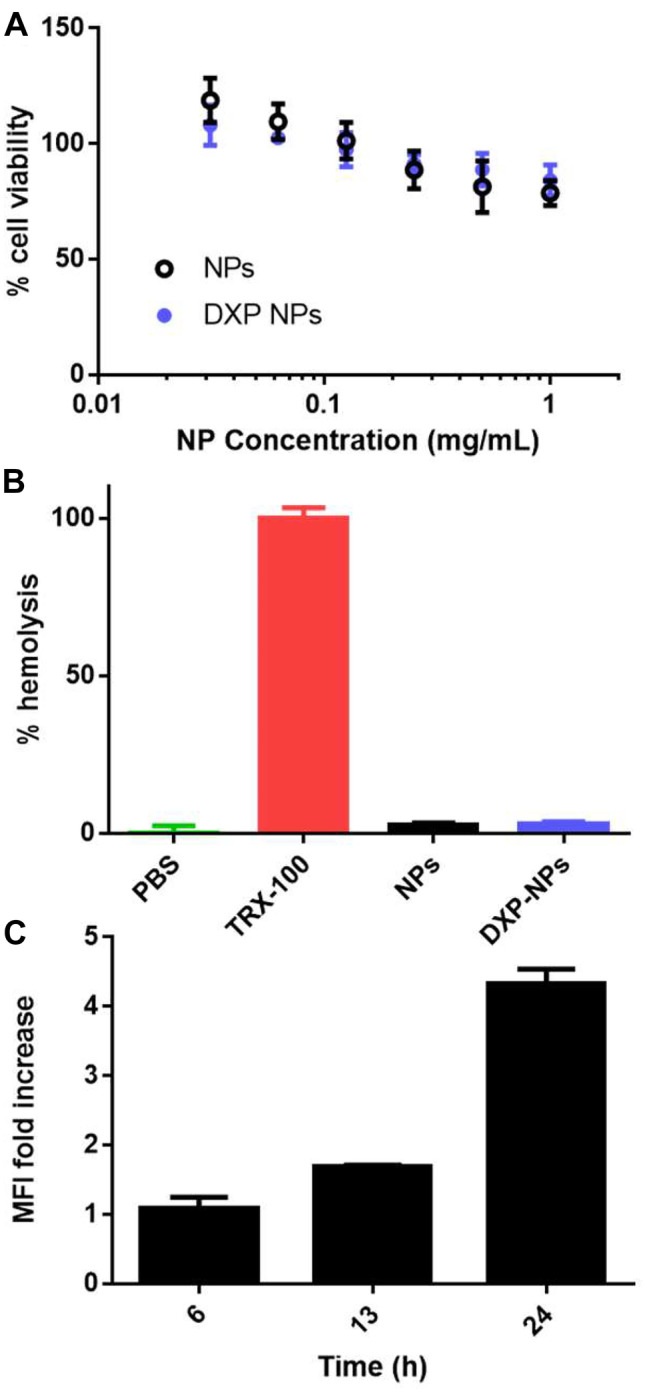
A Cell viability of the RAW 264.7 cell line incubated with the unloaded NPs or DXP-NPs at different concentrations. B Hemolysis induced by unloaded and DXP-NPs at a polymer concentration of 400 µg/mL, and approximately 30 µg/mL of DXP for the loaded NPs. TritonX-100 (TRX-100) was used as a positive control. C Internalization of the DXP-NPs at 100 µg/mL in the mouse macrophage cell line RAW 264.7 at different time points presented as mean fluorescence increase compared to untreated cells
The potential hemolysis of the formulation was also tested in mouse erythrocytes incubated with NPs at 400 µg/mL PLGA-PEG. This concentration was slightly higher than the concentration used in the CIA model studies to confirm the safe use of the formulation in animals. Both formulations, unloaded and DXP-NPs, did not show any relevant toxicity for the erythrocytes at the concentration tested, and the percentage of hemolysis remained lower than 5% (Fig. 2B). Unloaded and DXP-NPs did not show toxicity to RAW 264.7 macrophages (Fig. 2) at physiologically relevant concentrations. They also demonstrated to have a safe profile for the parenteral administration because they did not induce hemolysis in mouse erythrocytes. Hence, the DXP formulation seems to be safe for intravenous administration.
Internalization of the nanoparticles in mouse macrophages
The internalization of DXP-NPs in RAW 264.7 macrophages was evaluated using flow cytometry. The internalization of the NPs was slow and the first clear increase in the fluorescence was only detected after 6 h of NP incubation with cells (Fig. 2C). The fluorescence increased almost twice after 13 h of incubation and it was high after 24 h, with an MFI about 4.5 times higher than the initial fluorescence (Fig. 2C). DXP-NPs were highly internalized by the macrophages, which are the main immune cells recruited in the inflamed tissues and the main ones responsible for the cytokine release [21]. However, the internalization in the macrophages was slow due to the presence of the PEG at the NP surface [22–25]. Despite this slow internalization, PEG is necessary to benefit from long circulation in the bloodstream and accumulation in inflamed tissues thanks to the ELVIS effect [20]. It could be an advantage to allow a more specific targeting to the inflamed cells by avoiding a rapid internalization by the cells of the mononuclear phagocyte system (MPS) [25–27].
Inhibition of the inflammatory cytokine release by DXP-NPs
To characterize the capacity of the encapsulated DXP to inhibit the release of inflammatory cytokines, the RAW 264.7 cells were activated with LPS at 0.1 µg/mL for 3 h. After this time, the NPs were added. As a control, the unloaded NPs were also tested. NPs were also added to non-activated cells to check if they could induce the release of the cytokines and be inflammatory to the macrophages. As a positive control, cells treated only with LPS 0.1 µg/mL were used and non-treated cells were used as a negative control. Finally, the equivalent DXM concentration was also tested by using the soluble DSP to compare the encapsulated prodrug with the free drug as DXP is very poorly water-soluble.
A multiplex flow cytometer kit was used to study six cytokines simultaneously, but only four of them were induced by the LPS in the RAW 264.7 cells (TNFα, MCP-1, IL-10, and IL-6). IL-6 was expressed at very low concentrations and it was difficult to detect the basal concentration. Only in the positive control and in the samples with LPS which were not able to inhibit the cytokine release, the concentration was above the detection limit. DXP-NPs were able to inhibit the cytokines in a significant manner. At a dose of 10 µg/mL of DXP, the inhibition was significant concerning the positive control for all the cytokines tested (Fig. 3) and similar to the inhibition induced by an equivalent DSP concentration. The unloaded NPs were only able to slightly inhibit IL-10 and IL-6 to a much lower extent than the DXP-NPs. This very surprising result might arise from nonspecific interactions between cytokines and NPs. Nevertheless, the increased anti-inflammatory effect of the DXP-NPs, very similar to the free drug, could only be attributed to the encapsulated DXP and not to the NPs themselves.
Fig. 3.

Inhibition of the cytokine release in RAW 264.7 cells treated with the DXP nanoparticles. The cells were stimulated with 0.1 µg/mL LPS for 3 h before adding the nanoparticles at 10 µg/mL of DXP. C + positive control, LPS 0.1 µg/mL. C − negative control, culture medium. DSP dexamethasone sodium phosphate
TNFα and MCP-1 are key cytokines in the RA disease [28, 29]. Anti-TNFα antibodies have been the first successful biological response modifiers used in the therapy of RA because these biological molecules can induce remission or very low disease activity [30]. However, their potential to increase the risk of serious infections and malignancies, mainly when used in high concentrations, were also described in patients receiving anti-TNF therapies [31, 32]. MCP-1 is one of the chemokines involved in inflammation and angiogenesis in RA [33]. This chemokine is secreted by synovial stromal cells and, together with IL-8, attracts peripheral monocytes into the inflamed synovium [34].
IL-6 is a pleiotropic cytokine involved in B and T-cell proliferation and antibody production [35] and is another relevant pro-inflammatory cytokine in RA [36]. Production of IL-6 and its receptor, IL-6R, by effector cells cause and prolong inflammation [37]. Hence, anti-IL-6 agents are another type of biological response modifiers that are being explored as an alternative for patients with RA refractory to anti-TNFα agents [37, 38]. IL-10 and family-related cytokines are also overexpressed in patients with RA and they have a regulatory role in the inflammation [39]. Both the unloaded and DXP-NPs did not induce the release of the inflammatory cytokines in non-treated cells (Supporting information Figure S2) and thus confirming their safe profile, together with the absence of toxicity and hemolysis.
DXP-NPs were highly internalized by the macrophages (Fig. 2C), the main cells at the inflammation tissues in RA, and they were able to inhibit the release of all the cytokines studied (TNF, MCP-1, IL-10, and IL-6) and expressed by these cells in the presence of the endotoxin LPS (Fig. 3). This proves the efficiency of the encapsulated drug because the inhibition of the inflammatory cytokines is one of the main anti-inflammatory pharmacological activities of the GC [40]. Besides, GCs induce the expression of anti-inflammatory proteins such as Annexin I (also called lipocortine-1) and MAPK phosphatase 1 through the binding in the DNA to the glucocorticoid-responsive elements (GRE) [41]. Other anti-inflammatory and immunosuppressive mechanisms of the GC are the inhibition of several transcription factors, such as NF-kB and AP1, and the interaction with other second messengers, such as the PI3K-Akt-eNOS that induces the release of nitric oxide [41]. Because some of those mechanisms are also involved in physiologic signaling, prolonged use or high doses of GC have significant side effects [41]. Thus, GC formulations with more targeted delivery, together with an improvement of the PK and the use of lower doses, would be beneficial for the treatment of inflammatory diseases.
Pharmacokinetics and biodistribution
DXP-NPs and free DSP were injected in mice for PK and biodistribution studies at an equivalent dose of 12 mg/kg of DXM. Both DXP and DXM were quantified by HPLC. For DXP-NPs, DXP and DXM concentrations in plasma remained high for almost 18 h, much longer than the commercial injectable form of dexamethasone sodium phosphate, which only reaches a DXM concentration of 56 µM (Cmax) after injection (Fig. 4).
Fig. 4.
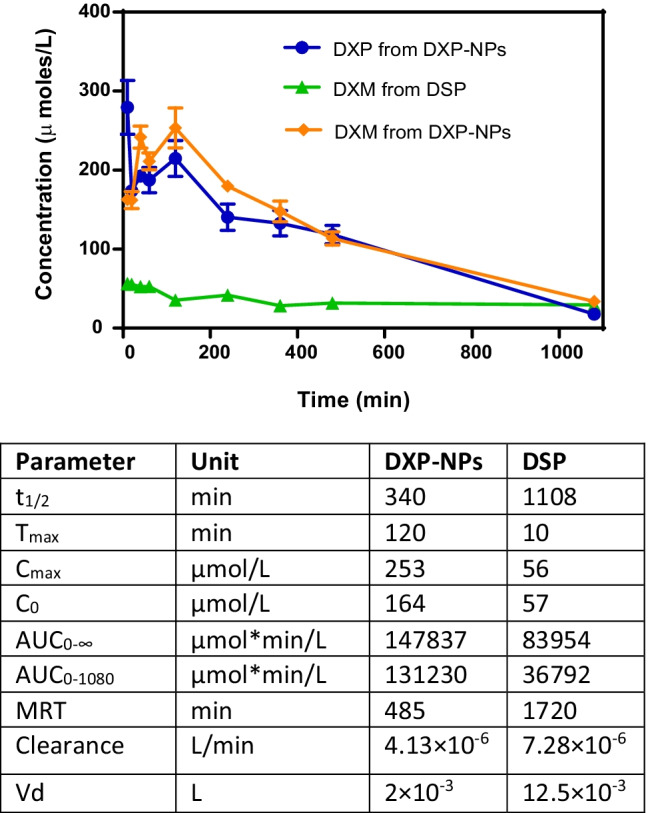
(top) Pharmacokinetics of the DXP nanoparticles in the plasma of healthy mice. The prodrug (DXP) and the active drug (DXM) were measured in the animals injected with the nanoparticles, and compared with the concentration of DXM in animals injected with the commercial soluble dexamethasone phosphate (DSP). (bottom) Pharmacokinetics parameters of DXM in plasma for DXP-NPs and DSP, calculated with a non-compartmental analysis. Cmax maximum plasma concentration of drug, Tmax time for drug concentration to reach Cmax, C0 initial concentration, AUC area under the plasma concentration vs time curve, t1/2 half-life, MRT mean residence time, Cl clearance of drug, and Vd volume of distribution
DXP-NPs showed a burst release effect and the initial DXM concentration (Co) in plasma was 164 µM. The DXP concentration was also high after injection. Two hours after injection, the concentration of both DXP and DXM started to decrease. However, it remained always higher than the DXM concentration from DSP up to 18 h post-injection, which is indicative of a sustained release of the drug from the NPs. Eighteen hours after injection, the concentration of both compounds became similar to the DXM concentration in animals injected with the commercial DSP.
In general, the concentration of DXP and DXM was similar during all the pharmacokinetics in animals injected with the DXP-NPs which is indicative of an equilibrium between the inactive and the active form of the drug. A similar pharmacokinetic profile was also observed with a lipophilic nanoformulation of the drug consisting of the formation of DXP NPs stabilized by distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(poly(ethylene glycol))-2000] (DSPE-PEG2000) [14]. However, the DXP concentration decreased faster and the DXM concentration was higher in the lipophilic NPs, probably due to a higher exposure to esterases leading to faster degradation of the prodrug into the active DXM form [14]. On the contrary, here the prodrug is more embedded within the PLGA matrix that slows its release and subsequent degradation by esterases.
A non-compartmental analysis was used to compare the pharmacokinetic behavior of DXM in the plasma of animals injected with DXP-NPs or DSP. The PK parameters were summarized in Table 1. For DXP-NPs the maximum concentration (Cmax) was about 4 times higher than for DSP and it was reached after 2 h (Tmax), while for DSP the Cmax was reached 10 min after the administration of the drug and, after 2 h, the concentration started to decrease. The clearance (Cl) was much faster for the soluble drug (0.0073 mL/min) than for the encapsulated DXP (0.0041 mL/min) and the volume of distribution of the drug (Vd) was about 6 times higher for DSP. The Vd is the apparent volume in which a drug is distributed. Hence, the encapsulated drug is not distributed from blood to other organs and/or eliminated as fast as the soluble drug which confirms the capacity of NPs to stay longer in the bloodstream. Although the prodrug loading is much lower for DXP-NPs than for the lipophilic nanoformulation (7.5 vs 50% w/w), the use of PLGA-PEG for the encapsulation of the prodrug avoid the initial burst release observed in the lipophilic NPs, in which the Cmax was reached 10 min after administration, like in the soluble drug [14]. Besides, the polymer encapsulation reduced the Vd about 3 times in comparison with the lipophilic nanoformulation and the conversion of the prodrug into the active drug, indicating a more protected and sustained release of the GC. The area under the curve for the measured kinetics (AUC0–1080) was about 3.6 times higher for the encapsulated drug than the soluble drug, and the AUC extrapolated to the infinite (AUC0–∞) was almost 1.8 times than for the soluble drug. Similar values were also obtained for lipophilic nanoformulation, indicative of a higher exposure of the drug in animals injected with the encapsulated prodrug in comparison with the DSP formulation. Contrary, the half-life (t1/2) in the plasma and the MRT were superior for the soluble drug, but this could be due to the plateau observed for DSP, as described before [14].
Table 1.
Scoring system for subjective evaluation of disease severity in CIA mice
| Score | Observation |
|---|---|
| 0 | No sign of inflammation |
| 1 | Paw swelling and/or swelling or redness confined to 1 digit |
| 2 | Swelling or redness confined to 2 digits |
| 3 | Swelling or redness in 3 digits and/or the entire paw |
The biodistribution of the GC in the liver, spleen, kidneys, and lung was also characterized in animals injected with DXP-NPs or DSP (Fig. 5). In both groups, DXM accumulated mainly in the liver. This accumulation was higher for the soluble drug (DSP), reaching almost 30% of the injected dose, while for the encapsulated drug was always inferior to 20%. The opposite was observed for the other organs studied. The encapsulated drug reached higher concentrations, mainly in the spleen, with an accumulation of almost 6% compared to less than 0.5% in the commercial drug. A higher accumulation of DPX and DXM in the spleen, in comparison with the soluble drug, was also observed for the lipophilic nanoformulation but not in the kidney or lungs [14]. There were some differences in the kinetics of the drug in the liver and spleen between NPs and DSP. While for DSP the concentration decreased all the time, for DXP-NPs the concentration increased up to 8 h and then decreased until the end of the pharmacokinetics, according to a slower clearance from the plasma (Fig. 4). Yet, for the lung and kidneys, the concentration of the drug decreased for both formulations. The preferential accumulation in the liver and spleen is a common feature for most particles in the nanometer range due to their rapid internalization by resident macrophages of the MPS and the fenestrated epithelia of these organs [42]. This accumulation depends on particle size, among other properties, i.e., particles in the range of 50–100 nm and 200–500 nm are mainly accumulated in the liver and spleen, respectively, while particles in the micrometer range accumulate preferentially in the lungs [43]. For that reason, monodisperse nanostructures with a size of 100–200 nm are preferred for intravenous administration, such as in the case of DXP-NPs.
Fig. 5.

Biodistribution of DXP and DXM after injection of DXP-NPs in healthy mice, or the soluble dexamethasone phosphate (DSP) as a control. Results are expressed as the percentage of the injected dose
A higher accumulation of the drug in the lungs of animals injected with DXP-NPs compared to animals injected with DSP could be indicative of some coalescence or crystallization process in the formulation after injection. However, the concentration of DXP and DXM was always below 2% of the injected dose in the lungs, where the larger particles could accumulate, and it decreased fast in a time-dependent manner. The same trend was observed for kidneys, and in the liver and spleen after reaching the maximum concentration. Therefore, all the organs studied were able to clear the GC and avoid long-term accumulation. Besides, the GC was mainly accumulated as the prodrug (DXP) in animals injected with the DXP-NPs, probably due to a lack of esterases in the healthy tissues that catalyze the degradation of the PLGA NPs and metabolize the prodrug into the active drug. This could be an advantage compared to the soluble or encapsulated DXM form because the prodrug is not active and it would avoid the undesirable effects associated with the use of GC in those organs.
Therapeutic efficacy
The therapeutic efficacy of DXP-NPs was tested in vivo in the CIA mouse model and compared to soluble DSP and non-treated animals (PBS group). The treatments or PBS were administered three times every other day starting at day 32 after the first immunization and the animals were sacrificed at day 38. For the DXP-NPs and DSP group, the equivalent DXM dose injected was 1 mg/kg each time. Figure 6 shows how the animals treated with DXP-NPs were able to reduce both the arthritic score of the disease from 8 to 1 and the paw volume from 0.1935 mL to basal levels (0.1385 mL) compared with PBS-treated animals (p < 0.0001). Moreover, the animals treated with DSP also reduced significantly both parameters compared to the untreated animals (p < 0.05 and p < 0.001, respectively), but the therapeutic efficacy of the DXP-NPs was higher than that of DSP. Although the difference in the reduction of the paw volume was not statistically significant, the decrease of the arthritis score was more relevant for the encapsulated drug compared to the soluble DSP (p < 0.01). Hence, the therapeutic efficacy of the encapsulated GC is higher than that of the soluble drug at the dose of 1 mg/kg DXM, in agreement with the results obtained for the lipophilic nanoformulation [15]. Moreover, the reduction of the arthritic score is more significant for DXP-NPs in comparison with the lipophilic nanoformulation at the same dose (p < 0.01 vs p < 0.05), in agreement with the more sustained GC release and slower clearance from the blood. The results confirmed the efficiency of PLGA-PEG NPs to deliver a therapeutic drug in the inflamed tissues in rheumatoid arthritis, likely due to the leaky vasculature and the angiogenesis that occurs in the inflamed joints and the associated ELVIS effect [20]. Indeed, the capacity of DXP-NPs to undergo extravazation and accumulation in the inflamed joints was demonstrated for lipophilic nanoformulation, with a size of 130 nm, by using fluorescent labeling [14].
Fig. 6.

Arthritis score A and paw volume B for DSP and DXP-NPs treated animals and non-treated animals after the induction of the disease and during the treatment period. The treated animals received three intravenous injections of 1 mg/kg DXM every 2 days (arrows and dotted lines) and the non-treated animals received a similar volume of PBS. The rheumatoid arthritis was induced by immunization of the mice with collagen at days 0 and 21. The treatments were administered on days 32, 34, and 36. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
Cytokine release in CIA mouse sera
The cytokine release in CIA-mouse serum was quantified using a custom-made flow cytometry bead assay. Apart from TNFα and MCP-1 (CCL2) that were highly expressed in the inflammation model in vitro and inhibited by DSP and DXP-NPs, IL-6, IL-4, IL-10, IL-17A, and IL-1β were also tested because they are relevant cytokines in RA disease [44, 45]. The serum samples were taken before the administration of the treatments, day 31, and the day after receiving the third and last dose, day 37, to check if the encapsulated DXP was able to inhibit the release of these cytokines and to compare with the soluble drug. Besides a clear inhibition of the IL-6 by the GC, which was only significant for DXP-NPs but not for DSP, the treated animals preserved or decreased to a little extent the concentration of all the other cytokines tested compared with the non-treated animals (Fig. 7). Interestingly, in the case of IL-1β, the concentration was significantly higher at day 37 for all the three groups compared to day 31 (Supplementary figure S3) and MCP-1 that was not significantly modified by the treatments (Fig. 7) contrary to what was observed for the RAW 264.7 cells treated with LPS.
Fig. 7.

Changes on cytokine concentration in CIA-mouse serum of animals treated with DSP, DXP-NPs, or PBS before (day 31) and after the last treatment (day 37)
TNFα and IL-10 were inhibited in animals treated with encapsulated DXP but not DSP compared to the untreated group (PBS) at day 37 and IL-4 was inhibited by both DSP and DXP-NPs. Moreover, IL-17A increased significantly from day 31 to day 37 for PBS and DSP-treated animals but not for animals treated with DXP-NPs.
The higher therapeutic efficacy of the encapsulated prodrug into DXP-NPs is also reflected by a more efficient inhibition of inflammatory cytokines compared to the soluble drug. Yet, DXP-NPs were not efficient in the inhibition of all the cytokines measured and in all the animals treated, at least at the selected dose. Nevertheless, a kinetic study from the onset of the disease to the end of the treatments would reflect more accurately the changes in the evolution of the cytokine expression for the three different groups (PBS, DSP, and DXP-NPs) because cytokine expression in the CIA model is time-dependent [46]. In situ studies in the inflamed joints would also reflect better the changes in cytokine expression induced by the GC treatment.
Histology of the knee in CIA mice
After the sacrifice of the animals, the hind paws were collected for histology characterization. Changes in the knee structure caused by the inflammation and the immune process associated with RA can be observed in non-treated animals (Fig. 8). The main changes, besides bone and cartilage erosion and reduction of the synovial space, are the infiltration of immune cells in the bone marrow (green arrows) and the fat pad (black arrows).
Fig. 8.

Histology of the knee of CIA mice treated with PBS, DSP, or DXP-NPs. The treatments were administered intravenously three times every other day and the DXM dose injected was 1 mg/kg each time for DSP and DXP-NPs. Cell infiltration in the bone marrow and fat pad (green and black arrows, respectively). F femur, BM bone marrow, M meniscus, T tibia
The soluble DSP was not able to reverse the histological changes in the knee of the CIA mice (Fig. 8). Side effects induced by the GC are a drawback in their use for the treatment of RA [3]. Among those side effects, bone erosion would be the greatest disadvantage in their clinical use for RA treatment.
On the contrary, the encapsulated DXP has shown a better histological outcome with a reduction of cell infiltration and bone distortion in the responding animals (Fig. 8). However, the histology of the knee was not completely recovered and the cartilage and bone erosion was not entirely avoided. This bone and cartilage erosion, together with the synovial space reduction, could appear early after the onset of the disease. Hence, the histological damage could have occurred before the administration of the treatment. However, the presence of chondrocytes (yellow rectangle) in DXP-NPs administered to animals could be indicative of the activation of a regeneration process in the animals to recover the cartilage.
In summary, the use of a prodrug (DXP) encapsulated into PEGylated NPs, for an increased circulation time, improved the therapeutic efficacy compared to soluble DSP (Fig. 5). Moreover, the presence of specific enzymes in the inflamed tissue that allows the activation of the prodrug, such as the case of the esterases, and the sustained release (Fig. 4) were relevant to the therapeutic effect observed in CIA animals and the improvement of the histological damage induced by the disease (Fig. 8). This formulation appears as a relevant strategy for the development of future therapeutic approaches in the treatment of RA using novel and more disease-specific drugs.
Conclusions
DXP has been encapsulated into PLGA-PEG NPs with high encapsulation efficiency. This formulation was stable for about 1 month and showed a spherical shape, a homogeneous size, and a negative zeta potential similar to the unloaded PLGA-PEG NPs. DXP-NPs were highly internalized by a macrophage cell line and were able to inhibit the release of inflammatory cytokines in vitro. Moreover, the unloaded and DXP-loaded NPs were not toxic for those cells and not able to induce hemolysis. The formulation also exhibited a better pharmacokinetic profile than the soluble drug with a sustained drug release. The concentration in plasma after injection in healthy animals remained high up to 18 h, much longer than the commercial soluble drug, and the DXP-loaded NPs accumulated in the organs mainly as the prodrug. The therapeutic efficacy of DXP-NPs was also increased significantly compared with the soluble drug. The arthritis score and the paw volume after the injection of three doses of the treatment were much lower than those obtained with soluble DSP. Moreover, some inflammatory cytokines were also reduced significantly after 37 days compared with non-treated and DSP-treated animals. Besides, the histological damage and cell infiltration were also reduced better by DXP-NPs. In summary, DXP-NPs have proven to be a good formulation for a more rational delivery of DXM that could be used in inflammatory diseases such as RA.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
The authors would like to thank Stéphanie Denis for the excellent technical support in cell culture experiments. RSV acknowledges a postdoctoral contract from the regional government of Galicia (Xunta de Galicia, ref. ED481D-2017/017). The present work has benefited from the facilities and expertise of the Electron Microscopy facilities of Imagerie-Gif (http://www.i2bc.paris-saclay.fr/spip.php?article282) with the precious help of C. Boulogne. This core facility is a member of the Infrastructures en Biologie Santé et Agronomie (IBiSA), and is supported by the French National Research Agency under Investments for the Future programs “France-BioImaging,” and the Labex “Saclay Plant Science” (ANR-10-INSB-04-01 and ANR-11-IDEX-0003-02, respectively).
Author contribution
Conceptualization: Elias Fattal; methodology: Rosana Simón-Vázquez, Mathilde Lorscheider, Ainhoa Rodríguez, Patricia Calleja, Ludivine Mousnier, Encarnación de Miguel Villegas; formal analysis and investigation: Rosana Simón-Vázquez, Nicolas Tsapis, Elias Fattal; writing — original draft preparation: Rosana Simón-Vázquez; writing — review and editing: Rosana Simón-Vázquez, Nicolas Tsapis, Elias Fattal, África González-Fernández; funding acquisition: Elias Fattal; supervision: Nicolas Tsapis, Elias Fattal, África González-Fernández.
Availability of data and materials
Raw data will not be made available due to its confidential nature.
Declarations
Ethics approval and consent to participate
In vivo experimental procedures were approved by the Ethical Committee No. 026 and by the French Ministry of Education and Research (accepted protocol no. 2842-2015110914248481_v5).
Consent for publication
Not applicable.
Conflict of interest
The authors declare no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Chan ED, Chan MM, Chan MM, Marik PE. Use of glucocorticoids in the critical care setting: science and clinical evidence. Pharmacol Ther [Internet]. 2020;206:107428. Available from: http://www.sciencedirect.com/science/article/pii/S0163725819301809 [DOI] [PubMed]
- 2.Johnson RM, Vinetz JM. Dexamethasone in the management of COVID-19. BMJ. 2020. [DOI] [PubMed]
- 3.Townsend HB, Saag KG. Glucocorticoid use in rheumatoid arthritis: benefits, mechanisms, and risks. Clin Exp Rheumatol. 2004;22:S77–82. [PubMed] [Google Scholar]
- 4.Yu Z, Reynaud F, Lorscheider M, Tsapis N, Fattal E. Nanomedicines for the delivery of glucocorticoids and nucleic acids as potential alternatives in the treatment of rheumatoid arthritis. WIREs Nanomedicine and Nanobiotechnology [Internet]. John Wiley & Sons, Ltd; 2020;12:e1630. Available from: 10.1002/wnan.1630 [DOI] [PubMed]
- 5.Gómez-Gaete C, Tsapis N, Besnard M, Bochot A, Fattal E, Gomez-Gaete C, et al. Encapsulation of dexamethasone into biodegradable polymeric nanoparticles. Int J Pharm. 2007;331:153–159. doi: 10.1016/j.ijpharm.2006.11.028. [DOI] [PubMed] [Google Scholar]
- 6.Gomez-Gaete C, Fattal E, Silva L, Besnard M, Tsapis N, Gómez-Gaete C, et al. Dexamethasone acetate encapsulation into Trojan particles. J Control Release [Internet]. 2008;128:41–9. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18374442 [DOI] [PubMed]
- 7.Rauchhaus U, Schwaiger FW, Panzner S. Separating therapeutic efficacy from glucocorticoid side-effects in rodent arthritis using novel, liposomal delivery of dexamethasone phosphate: long-term suppression of arthritis facilitates interval treatment. Arthritis Res Ther [Internet]. 2009;11:1–9. Available from: 10.1186/ar2889 [DOI] [PMC free article] [PubMed]
- 8.Koning GA, Schiffelers RM, Wauben MHM, Kok RJ, Mastrobattista E, Molema G, et al. Targeting of angiogenic endothelial cells at sites of inflammation by dexamethasone phosphate–containing RGD peptide liposomes inhibits experimental arthritis. Arthritis Rheum. 2006;54:1198–1208. doi: 10.1002/art.21719. [DOI] [PubMed] [Google Scholar]
- 9.Bias P, Labrenz R, Rose P. Sustained-release dexamethasone palmitate — pharmacokinetics and efficacy in patients with activated inflammatory osteoarthritis of the knee. Clin Drug Investig. 2001;21:429–436. doi: 10.2165/00044011-200121060-00006. [DOI] [Google Scholar]
- 10.Yokoyama K, Watanabe M. Limethason as a lipid microsphere preparation: an overview. Adv. Drug Deliv. Rev. Elsevier Science B.V. 1996;195–201.
- 11.Edwards SHR. Intra-articular drug delivery: the challenge to extend drug residence time within the joint. Vet J [Internet]. 2011;190:15–21. Available from: http://www.sciencedirect.com/science/article/pii/S1090023310003102 [DOI] [PubMed]
- 12.De Silva M, Page Thomas DP, Hazleman BL, Wraight P. Liposomes in arthritis: a new approach. Lancet [Internet]. 1979;313:1320–2. Available from: https://www.scopus.com/inward/record.uri?eid=2-s2.0-0018747845&partnerID=40&md5=82463d631da1455f9003f20c367e22c9 [DOI] [PubMed]
- 13.Larsen C, Østergaard J, Larsen SW, Jensen H, Jacobsen S, Lindegaard C, et al. Intra‐articular depot formulation principles: role in the management of postoperative pain and arthritic disorders. J Pharm Sci [Internet]. 2008;97:4622–54. Available from: http://www.sciencedirect.com/science/article/pii/S0022354916327794 [DOI] [PubMed]
- 14.Lorscheider M, Tsapis N, Simón-Vázquez R, Guiblin N, Ghermani N, Reynaud F, et al. Nanoscale lipophilic prodrugs of dexamethasone with enhanced pharmacokinetics. Mol Pharm. 2019;16:2999–3010. doi: 10.1021/acs.molpharmaceut.9b00237. [DOI] [PubMed] [Google Scholar]
- 15.Lorscheider M, Tsapis N, ur-Rehman M, Gaudin F, Stolfa I, Abreu S, et al. Dexamethasone palmitate nanoparticles: an efficient treatment for rheumatoid arthritis. J Control Release [Internet]. Elsevier; 2019;296:179–89. Available from: https://www.sciencedirect.com/science/article/pii/S0168365919300331?dgcid=raven_sd_aip_email [DOI] [PubMed]
- 16.Kumari A, Yadav SK, Yadav SC. Biodegradable polymeric nanoparticles based drug delivery systems. Colloids Surf B Biointerfaces [Internet]. 2010;75:1–18. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19782542 [DOI] [PubMed]
- 17.Ashford MB, England RM, Akhtar N. Highway to success—developing advanced polymer therapeutics. Adv Ther. 2021;4:1–27. [Google Scholar]
- 18.Harris EDJ. Rheumatoid arthritis. N Engl J Med [Internet]. 1990;322:1277–89. Available from: 10.1056/NEJM199005033221805
- 19.Gomez-Gaete C, Tsapis N, Besnard M, Bochot A, Fattal E. Encapsulation of dexamethasone into biodegradable polymeric nanoparticles. Int J Pharm. 2007;331:153–159. doi: 10.1016/j.ijpharm.2006.11.028. [DOI] [PubMed] [Google Scholar]
- 20.Wang Q, Sun X. Recent advances in nanomedicines for the treatment of rheumatoid arthritis. Biomater Sci [Internet]. 2017/06/21. RSC Adv. 2017;5:1407–20. Available from: http://xlink.rsc.org/?DOI=C7BM00254H [DOI] [PubMed]
- 21.McInnes IB, Schett G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat Rev Immunol [Internet]. 2007; [cited 2017 Jan 29]7:429–42. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17525752 [DOI] [PubMed]
- 22.Yang Q, Jones SW, Parker CL, Zamboni WC, Bear JE, Lai SK. Evading immune cell uptake and clearance requires PEG grafting at densities substantially exceeding the minimum for brush conformation. J. Am. Chem. Soc. 2014;11:1250–8. doi: 10.1021/mp400703d. [DOI] [PubMed] [Google Scholar]
- 23.Tian X, Ma P, Sui C-G, Meng F-D, Li Y, Fu L-Y, et al. Suppression of tumor necrosis factor receptor-associated protein 1 expression induces inhibition of cell proliferation and tumor growth in human esophageal cancer cells. FEBS J. 2014;281:2805–2819. doi: 10.1111/febs.12822. [DOI] [PubMed] [Google Scholar]
- 24.Sanchez L, Yi Y, Yu Y. Effect of partial PEGylation on particle uptake by macrophages. Nanoscale [Internet] RCS Adv. 2017;9:288–97. doi: 10.1039/C6NR07353K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Perry JL, Reuter KG, Kai MP, Herlihy KP, Jones SW, Luft JC, et al. PEGylated PRINT nanoparticles: the impact of PEG density on protein binding, macrophage association, biodistribution, and pharmacokinetics. Nano Lett [Internet]. J Am Chem Soc. 2012;12:5304–10. 10.1021/nl302638g [DOI] [PMC free article] [PubMed]
- 26.Oh N, Park J-H. Endocytosis and exocytosis of nanoparticles in mammalian cells. Int J Nanomedicine [Internet]. Dove Medical Press; 2014;9:51–63. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4024976/ [DOI] [PMC free article] [PubMed]
- 27.Owens DE, Peppas NA. Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int J Pharm [Internet]. Elsevier; 2006 [cited 2019 Oct 30];307:93–102. Available from: https://www.sciencedirect.com/science/article/pii/S037851730500668X?via%3Dihub [DOI] [PubMed]
- 28.Feldmann M, Maini RN. TNF defined as a therapeutic target for rheumatoid arthritis and other autoimmune diseases. Nat Med [Internet]. Nature Publishing Group; 2003 [cited 2017 Jan 29];9:1245–50. Available from: http://www.nature.com/doifinder/10.1038/nm939 [DOI] [PubMed]
- 29.Teichmann A, Jacobi U, Ossadnik M, Richter H, Koch S, Sterry W, et al. Differential stripping: determination of the amount of topically applied substances penetrated into the hair follicles. J Invest Dermatol. 2005;125:264–269. doi: 10.1111/j.0022-202X.2005.23779.x. [DOI] [PubMed] [Google Scholar]
- 30.Geiler J, Buch MF, McDermott M. Anti-TNF treatment in rheumatoid arthritis. Curr Pharm Des [Internet]. 2011;17:3141–51. Available from: http://www.ingentaconnect.com/content/ben/cpd/2011/00000017/00000029/art00005 [DOI] [PubMed]
- 31.Bongartz T, Sutton AJ, Sweeting MJ, Buchan I, Matteson EL, Montori V. Anti-TNF antibody therapy in rheumatoid arthritis and the risk of serious infections and malignancies: systematic review and meta-analysis of rare harmful effects in randomized controlled trials. JAMA [Internet]. 2006;295:2275–85. Available from: 10.1001/jama.295.19.2275 [DOI] [PubMed]
- 32.Kroesen S, Widmer AF, Tyndall A, Hasler P. Serious bacterial infections in patients with rheumatoid arthritis under anti‐TNF‐α therapy. Rheumatology [Internet]. 2003;42:617–21. Available from: http://rheumatology.oxfordjournals.org/content/42/5/617.abstract [DOI] [PubMed]
- 33.Szekanecz Z, Kim J, Koch AE. Chemokines and chemokine receptors in rheumatoid arthritis. Semin Immunol [Internet]. 2003;15:15–21. Available from: http://www.sciencedirect.com/science/article/pii/S1044532302001240 [DOI] [PubMed]
- 34.Hayashida K, Nanki T, Girschick H, Yavuz S, Ochi T, Lipsky PE. Synovial stromal cells from rheumatoid arthritis patients attract monocytes by producing MCP-1 and IL-8. Arthritis Res. 2001;3:118–126. doi: 10.1186/ar149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kishimoto T. Interleukin-6: discovery of a pleiotropic cytokine. Arthritis Res Ther. 2006;8:S2. doi: 10.1186/ar1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yoshizaki K, Nishimoto N, Mihara M, Kishimoto T. Therapy of rheumatoid arthritis by blocking IL-6 signal transduction with a humanized anti-IL-6 receptor antibody. Springer Semin Immunopathol. 1998;20:247–259. doi: 10.1007/BF00832010. [DOI] [PubMed] [Google Scholar]
- 37.Emery P, Keystone E, Tony HP, Cantagrel A, Van Vollenhoven R, Sanchez A, et al. IL-6 receptor inhibition with tocilizumab improves treatment outcomes in patients with rheumatoid arthritis refractory to anti-tumour necrosis factor biologicals: results from a 24-week multicentre randomised placebo-controlled trial. Ann Rheum Dis. 2008;67:1516–1523. doi: 10.1136/ard.2008.092932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hashimoto J, Garnero P, van der Heijde D, Miyasaka N, Yamamoto K, Kawai S, et al. Humanized anti-interleukin-6-receptor antibody (tocilizumab) monotherapy is more effective in slowing radiographic progression in patients with rheumatoid arthritis at high baseline risk for structural damage evaluated with levels of biomarkers, radiograp. Mod Rheumatol [Internet]. 2011;21:10–5. Available from: 10.1007/s10165-010-0325-3 [DOI] [PMC free article] [PubMed]
- 39.Alanärä T, Karstila K, Moilanen T, Silvennoinen O, Isomäki P. Expression of IL-10 family cytokines in rheumatoid arthritis: elevated levels of IL-19 in the joints. Scand J Rheumatol [Internet]. Taylor & Francis; 2010;39:118–26. Available from: 10.3109/03009740903170823 [DOI] [PubMed]
- 40.Brattsand R, Linden M. Cytokine modulation by glucocorticoids: mechanisms and actions in cellular studies. Aliment Pharmacol Ther [Internet]. Blackwell Science Ltd; 1996;10:81–90. Available from: 10.1046/j.1365-2036.1996.22164025.x [DOI] [PubMed]
- 41.Rhen T, Cidlowski JA. Antiinflammatory action of glucocorticoids — new mechanisms for old drugs. N Engl J Med [Internet]. 2005;353:1711–23. Available from: 10.1056/NEJMra050541 [DOI] [PubMed]
- 42.Blanco E, Shen H, Ferrari M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat Biotechnol [Internet]. 2015;33:941–51. Available from: https://pubmed.ncbi.nlm.nih.gov/26348965 [DOI] [PMC free article] [PubMed]
- 43.Koster VS, Kuks PFM, Lange R, Talsma H. Particle size in parenteral fat emulsions, what are the true limitations? Int J Pharm [Internet]. 1996;134:235–8. Available from: http://www.sciencedirect.com/science/article/pii/0378517395044094
- 44.Hwang S-Y, Kim J-Y, Kim K-W, Park M-K, Moon Y, Kim W-U, et al. IL-17 induces production of IL-6 and IL-8 in rheumatoid arthritis synovial fibroblasts via NF-κB- and PI3-kinase/Akt-dependent pathways. Arthritis Res Ther [Internet]. London: BioMed Central; 2004;6:R120–8. Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC400429/ [DOI] [PMC free article] [PubMed]
- 45.Buchs N, di Giovine FS, Silvestri T, Vannier E, Duff GW, Miossec P. IL-1B and IL-1Ra gene polymorphisms and disease severity in rheumatoid arthritis: interaction with their plasma levels. Genes Immun [Internet]. 2001;2:222–8. Available from: http://www.ncbi.nlm.nih.gov/pubmed/11477478 [DOI] [PubMed]
- 46.Williams RO, Inglis JJ, Simelyte E, Criado G, Sumariwalla PF. Analysing the effect of novel therapies on cytokine expression in experimental arthritis. Int J Exp Pathol [Internet]. Blackwell Science Inc; 2005;86:267–78. Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2517440/ [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw data will not be made available due to its confidential nature.