

Abstract
Radicals in biology, once thought to all be bad actors, are now known to play a central role in many enzymatic reactions. Of the known radical-based enzymes, ribonucleotide reductases (RNRs) are pre-eminent as they are essential in the biology of all organisms by providing the building blocks and controlling the fidelity of DNA replication and repair. Intense examination of RNRs has led to the development of new tools and a guiding framework for the study of radicals in biology, pointing the way to future frontiers in radical enzymology.
Graphical Abstarct
Introduction
Radicals in biology are often vilified in the popular press as agents of aging and even death. For such portrayals, radicals are often categorized without distinction, and if mentioned, they are described as reactive oxygen or nitrogen species (ROS, RNS) such as O2•−, HO•, NO•.1 The origins of the small molecule oxygen radicals are traced to reactions with reduced metals2 during the transition from an anaerobic to aerobic world >800 million years ago.3 Whereas O2 is essential to respiratory organisms, when left uncontrolled, its reductive metabolites can result in DNA damage, mutations, and cell death (Figure 1A). NO• is produced intracellularly from arginine by NO synthases. It can react with O2 or O2•− to produce RNS such as N2O3 and ONO2− (peroxynitrite), respectively which can damage DNA (Figure 1A).4 O2•− and NO• are Janus-faced molecules. The former places an essential role in metal cluster assembly in enzymes and the latter plays an important role as a signaling agent.5
Figure 1. The dichotomy of radicals in biology.

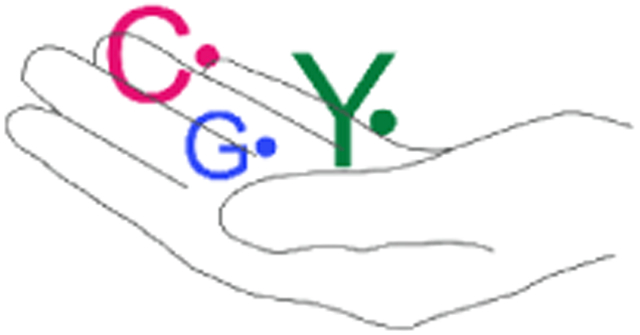
(A) Reactive oxygen radicals such as O2•−/HO• are often generated as the inadvertent consequence of our external or intracellular environments. While O2 is stable, in the presence of Fe(II) and Cu(I), ROS are produced. NO• is produced intracellularly by NO synthase. Its reaction with O2 or O2•− generate RNS. Both ROS and RNS if left to their own demise, can modify DNA, proteins, and lipids and contribute to the aging process and diseased states. (B) In contrast, Nature harnesses the reactivity of radicals to mediate challenging, essential reactions in biology with exquisite specificity. Enzymes involved in primary and secondary metabolism often require cofactors such as flavins (FAD), thiamin pyrophosphate (TPP), pyridoxal phosphate (PLP) that can be involved in controlled radical-based reactions. Stable and transient amino acid radicals of tyrosine (Y•), glycine (G•), cysteine (C•) and 5′-deoxyadenosine (5′-dA•) play essential roles in ribonucleotide reductases (RNRs). These enzymes make the deoxynucleotides essential for DNA replication and are central in the fidelity of the damaged DNA repair. The RNRs and their associated amino acid radicals are the focus of this Perspective.
Radicals are actually found throughout biology. They play a central role in cofactor biosynthesis used to expand the repertoire of enzyme catalyzed reactions6 and are essential for many metabolic transformations in primary and secondary metabolism and in cell signaling. In addition, stable and transient amino acid radicals of tyrosine, cysteine, glycine and tryptophan within proteins (Figure 1B) play an important role in metabolism.7 The significance of radical based reactions was initially underappreciated because they are highly reactive species with “fleeting” lifetimes, making them difficult to observe on timescales accessible with common biochemical and biophysical methods of earlier times. The first inkling of “radicals” in proteins remained unidentified due to their exchange coupling to metals.8 However, in 1978, the first stable protein radical,9 a tyrosyl radical (Y•) in the E. coli ribonucleotide reductase (RNR) was structurally identified. Tyrosyl radicals of varying stability were subsequently established to be essential in electron transport from the reaction center to the O2 evolving complex of Photosystem II10 and in the electron/proton chemistry of the reduction of O2 to water by cytochrome c oxidase of the respiratory chain.11 The explosion of information from genome sequencing and analysis has revealed >500,000 enzymes and proteins known to derive function from radicals,6,12 giving birth to the field of radical enzymology. This Perspective provides an overview of how Nature has harnessed the reactivity of radicals to carry out difficult and essential reactions with exquisite specificity, beginning with RNR as the exemplar, and then moving to new frontiers in radical chemistry and biology.
RNRs Central Role in DNA Replication and Repair
RNRs are essential for the conversion of four nucleoside di- and tri-phosphates (ND(T)Ps) to deoxynucleotides (dND(T)Ps) in all three domains of life and in all organisms. They play essential roles in the regulation of the fidelity of DNA replication, repair and cell survival.13,14 As with all things biological, homeostasis is paramount to controlling radical reactivity and is central to an organism’s biological function and survival.
A general mechanistic model for NDP(NTP) reduction evolved from early studies on the E. coli and L. leichmannii RNRs (class Ia and II, Figures 2)7,15 in conjunction with their x-ray structures that revealed the location of the three important cysteines shown in Figure 2.16-18 These studies led to the classification of RNRs based on the metallocofactor involved in generating a cysteine radical (C•) (red C in Figure 2).19 This cysteine (C408 in L. leichmannii in class II, C439 in E. coli in class Ia and C290 in T4 phage in class III) in all RNRs is located at the tip of a finger loop inserted into a 10 stranded α/β barrel of their conserved α structures. This C• is central to initiating ND(T)P reduction by removal of the 3′-H atom (red H in NDP, Figure 2).7,15 The prominent role of RNRs in a broad range of radical-based reactions with avoidance of their self-destruction suggests that lessons learned from studies of these enzymes could provide a roadmap to Nature’s designs for controlled radical reactivity in all enzymes and offers new opportunities for drug design targeting the control of radicals.
Figure 2. Thyl radical initiation in RNRs.

RNRs catalyze de novo deoxynucleoside (di or triphosphates) biosynthesis in all organisms initiated by a cysteine thiyl radical, red C•, in the protein α. It is generated by distinct metallocofactors that define the three classes of RNR (I, II and III). In class I, this process requires a second subunit β (which is part of a homodimer β2) with the metallocofactor initiating C• formation over 32 Å. This process involves a reversible RT pathway with multiple PCET steps involving 4Ys (122, 356, 731, 730), a C (439) and possibly a tryptophan (W48). Residue numbers are for E. coli Ia RNR. The wavy lines indicate that the amino acid radicals are part of the protein. The class II RNRs use adenosylcoblamin (AdoCbl, with a DMB axial ligand(dimethylbenzimidazole). The class III RNRs also require a second protein, the activating enzyme, that uses S-adenosylmethionine and an [4Fe4S] + cluster to generate its glycyl radical (G•). The G• then generates the C• in α. In general, the reducing equivalents to make the deoxynucleotides are supplied by oxidation of two additional cysteines in α to a disulfide. They are recycled by reducing equivalents supplied by redoxin systems such as thioredoxin (Trx), thioredoxin reductase (TrxR) and NADPH. In a subclass of class III RNRs, formate is oxidized to CO2 and supplies the reducing equivalents (gray).
Radical Transport in RNR
L. leichmannii RNR,17 a monomeric α, possesses the simplest pathway for C• generation (Figure 2) and was a keystone to generalizing radical initiation, transport and substrate activation for all other RNR classes. Stopped flow visible and rapid freeze quench EPR spectroscopic studies on the enzyme revealed formation of a C• that was exchange coupled to cob(II)alamin and was generated in a kinetically competent fashion.20
Class I RNRs, in contrast to the class II enzymes, require two subunits, α and β (Figures 2 and 3A), for activity. Prior to a cryo-EM structure on a trapped, active α2β2 complex,21 the field was guided by a symmetrical docking model based on the shape complementarity between the individual α2 and β2 structures.16 The oxidant to generate the C439•, is a diferric-Y122• cofactor located in β that resides 32 A from C439 (Figure 2). The generation of C439• by Y122• is reversible and radical transport (RT) between them occurs on every turnover of the enzyme. The pathway involves the five and possibly six amino acids (if W48 is involved) shown in Figure 2, that form transiently oxidized radical intermediates.16,22 Obtaining evidence in support of this model for RT and the subsequent chemistry of NDP reduction has been challenging as the rate-limiting step(s) in all RNRs is(are) conformationally gated, masking the chemistry of the long-range radical transfer pathway.
Figure 3. Subunit, active, and inactive structures of class I RNRs.

(A) The α (cyan/blue) and β (red/orange) subunits in the E. coli class Ia RNRs are each homodimers. α has three nucleotide binding sites: the catalytic site (C-site), the specificity site (S-site) and the activity site (A site or cone domain, green) in α2, and the radical initiation metallocofactor in β2. The C- and N-termini tails are disordered, indicated by colored dashed lines. (B) The only RNR structure of an active α2β2 complex is an E. coli double mutant that has been trapped in the presence of substrate and effector and solved by cryo-EM. C Structures of dATP inhibited states α4β4 in E. coli and α6 in human Ia RNRs have been solved crystallographically and by cryo-EM.
Quaternary RNR Structures
Diverse quaternary structures of RNRs regulate active and inactive states, mediated by dNTPs, ATP, and likely other small molecules. An allosteric specificity site (S-site, Figure 3A) is essential for determining which dNTP or ATP effector controls which NDP is reduced.14,16,23 A second allosteric site in α, the activity site (A-site or cone domain in green, Figure 3A), requires further examination.24 ATP was initially proposed to be a universal activator and dATP a universal inhibitor. It is now clear, however, that quaternary structural changes of α, and α and β together, have evolved to generate “new” inhibited structures that can be controlled not only by dATP and combinations of ATP/dNTPs,25 but by dAMP and other metabolites that remain to be discovered.26 The cone domain architecture is mobile and is also present in many bacterial transcription factors involved in regulation of deoxynucleotide metabolism, although the small molecule regulators remain unknown.24 Recently, studies with the clinically used nucleoside anticancer therapeutic (clofarabine, ClF)27,28 and prospective antibacterials (2-hydroxypyridone derivatives)29 have been proposed to work by trapping inhibited RNR states (Figure 3C); evidence suggests that an α6 hexameric state30 is trapped in the former case and that an α4β4 state31 in the latter case. These two inhibited RNR states have also been shown to occur inside cells.29,32
Breakthroughs Enabling Radical Investigations in RNR
Rate limiting conformational steps often mask radical chemistry. The amino acid radical propagates in biology by transferring a H•, arising from the coupling of a proton to an electron. The high-energy barriers encountered by transferring an electron independent of a proton dictate that the coupled process must occur to maintain compatibility with physiological conditions. Because the proton resting mass is ~2,000 times larger than the electron, the wavelength of the proton is ~40 times shorter than that of an electron at a fixed energy. Consequently, proton transfer is fundamentally limited to short distances whereas the lighter electron may transfer over very long distances.33 Thus, small changes in proton distance induced by conformational changes can have significant impact on RT and overall enzymatic and cellular function. The RT pathway in RNR is exemplar of how small changes in the proton transfer distance can disrupt RT and enzyme function. As such, the study of radicals in the class I enzymes require conformational gating to be unmasked. Another complexity in class I RNRs is the mechanism of their cofactor assembly in β. Diferric-Y• cofactor formation in class Ia (Figure 2) requires in vitro, Fe2+, O2 and a reductant, and in vivo likely proteins involved in a biosynthetic pathway, both of which are incompletely understood.34 Accordingly, new methods (Figure 4) have been essential to unmasking radical chemistry in RNR. These methods are general and will be useful to study other radical-based systems.
Figure 4. Methods to investigate radicals in RNRs:

(A) site-specific incorporation of UAA; (B) mechanism-based inhibitors; (C) cyro-EM; (D) radical photogeneration with a covalently attached photooxidant (PO, a [ReI] complex bound to C355 in β; and, (E) high field and multifrequency (9.4, 34, 94, 263 GHz) paramagnetic resonance methods (figure adapted from multifrequency studies of Bennati and coworkers42,43,45).
Biochemical Methods.
Site-directed incorporation of unnatural amino acids (UAA) using orthogonal tRNA synthetase technology35 has played a central role in studying the E. coli class Ia RNRs. tRNA synthetases (RS)/tRNAs were evolved to incorporate UAA radical traps such as NH2Y, DOPA and FnYs (n=2,3) , which provide altered pKas and redox potentials (Figure 4A).22,36,37 Placement and trapping of FnYs• in the RT pathway due to their unique EPR and vis spectroscopic handles have been especially central to understanding RT.
Cryo-EM.
The cryo-EM structure of an active E. coli RNR21 with both subunits, α and β present (Figure 3B), has displaced the Uhlin and Ekund docking model.16 Disorder of the C-terminal tails of both subunits in all class I RNRs has presented a major obstacle to understanding RT and reactivity. In α, the tail is essential for re-reduction of the active site disulfide (Figure 2) that provides the reducing equivalents for dNDP produced during each turnover. For β, the terminal ~35 amino acids of the tail are disordered with the last 15 paramount to α2 interaction,38,39 which is weak and dynamic. Studies with several trapped radicals in the RT pathway resulted in enhanced α/β affinity40 and in conjunction with the kinetics of their radical transfer, suggested a time course for freezing each sample for cryo-EM analysis. Indeed, an active α2β2 complex with a β-Y356• was successfully trapped during back radical transfer with a double mutant (F3Y122•/E52Q-β2) with α2, GDP and TTP.41 In this trapped state, one dGDP was formed along one arm of the α′/β′ complex. Switching radical transport to the second arm of the α/β pair caused alignment of the radical transport pathway. This structure (Figure 3B) provided the first visualization of the subunit interface and the entire C-terminal tail of β, which moves into α enclosing the active site when the correct substrate and effector pair are bound (orange, Figure 4C). Within this sequestered environment, the radical chemistry involved in dNDP formation occurs rapidly and with exquisite specificity.
Multi-Frequency and High-Field (HF) EPR.
Multifrequency HF-EPR methods (Figure 4E) have been used in conjunction with specifically located FnYs and NH2Ys within the RT pathway.42-45 They have been central to elucidating the mechanism of RT and revealing the flexibility of RT pathway residues (e.g., Y731-α) and the importance of structured water at the α/β interface. Trapping of FnY•s within the RT pathway and monitoring their equilibration with other pathway Ys have been possible due to HF-EPR and the hyperfine interactions associated with the 19F magnetic nuclei.22,46 These studies are consistent with the proposed uphill thermodynamic landscape for the journey of the radical from Y122• to C439• in the forward direction and the rapid downhill regeneration of Y122• in reverse direction.47 ENDOR has allowed examination of the interaction of magnetic nuclei (1H, 2H, 19F) with trapped radicals in the pathway giving distances and orientations.42,45 PELDOR methods have provided the first picture of an active, asymmetric α2β2 complex and the importance of stacked Y731/Y730 pairs within α2 of the complex.48 These studies and rapid chemical quench studies to monitor product formation led to a model where dNDP formation in one α/β pair happens prior to dNDP formation in the second pair. The mechanism of conformational switching from α′/β′ to α/β (Figure 3B) remains a mystery.
Photo-RNRs.
Photo-RNRs uncouple conformational gating from radical generation and transport. The photo-RNRs are effectively a “RNR photosystem” that allows radicals to be generated promptly and kinetics to be observed with precision (ns to ms timescales). A photooxidant (PO) attached site-specifically to the C-terminal tail of β2 enables the light-mediated generation of Y356• at the α/β interface (Figure 4D).49 The success of the method in retrospect is due to the asymmetry in the active RNR structure, allowing for the PO to be accommodated without significantly interfering with subunit association (Figure 4C and 4D). Rapid kinetics experiments provided the first measurements of the “fast chemistry” of RT and of 3′C-H bond cleavage of NDP reduction with the normal substrate and mechanism-based inhibitor within the C-site of α.50 Variants of photo-RNR studies using FnY356 amino acids as a function of pH, have also allowed direct interrogation of RT across the subunit interface and the importance of water channels in managing proton transfer within the subunit interface.51
Proton-Coupled Electron Transfer (PCET).
RT may be deconvolved into PCET events embodying the proton and the electron.33 Prior to the discovery of radicals in biology, PCET had been treated at a thermodynamic level, as it was known that protons affect redox potentials of cofactors.52 Mitchell first recognized the importance of the energetics of electron-proton coupling with his proposal of the chemiosmotic principle.53 From a kinetics perspective, PCET had been inferred from isotope effects on the overall rates of reaction, but had not been isolated until supramolecular donor-acceptor complexes, which allowed for the movement of an electron coupled to a proton to be temporally resolved.54,55 On this experimental foundation, the first theory of PCET emerged33 in which the electron transfer was treated on one coordinate and the proton transfer was treated on an orthogonal coordinate – giving rise to the “square scheme” for PCET.
Within this framework of PCET, a model for RT in the RNR complex was proposed to involve different PCET mechanisms for RT in the α and β subunits.22,56 In β, oxidation and reduction of the Y122 and Y356 are coupled to orthogonal proton hops (to water in the interface for Y356 and water bound to the diiron cofactor for Y122), coupled to a long-distance electron transfer through β. Conversely, PCET in α is unidirectional in which a proton shift among amino acids and possibly structured waters accompany the transfer of the electron between the Y731 interface residue and active site C439. PCET has also provided a guiding framework for deciphering the active site chemistry where redox events are tightly coupled to proton transfers to glutamates and cysteines within the active site.48
Frontiers in Radical Biology Emerging from RNR
The remainder of this Perspective describes four targets of opportunity in basic and translational science associated with the radical chemistry of RNR and other enzyme systems, and a final thought on the enigmatic role that RNR may play in the origin of life.
Metallocofactor Assembly in Cells and in vitro.
An estimated one third of all proteins require metallocofactors of varying complexity for activity.34 These cofactors often utilize biosynthetic machinery for their assembly and repair if they become damaged. Identifying the protein networks and obtaining homogeneous metallocofactors are essential for understanding their biology in the cellular context and the chemistry of their catalytic transformations.
Class I RNRs have a diverse array of metallocofactors that must be assembled correctly to produce active enzyme. The diferric-Y• cofactor of the E. coli class Ia in an α2β2 complex was once thought to be a paradigm for all class I RNRs. However, recent discoveries of class Ib-Ie RNRs with very different cofactors for radical generation (Table 1),18,57- 62 despite sharing the same β framework, challenges this premise. For class I RNRs, new method(s) to obtain a homogenous metallocluster in β2 is(are) essential for obtaining structural insight about active α2β2 and its dynamics or other active states, the role of the metallocluster in long range RT, and to target metallocofactor assembly with therapeutics.
Table 1.
Class I cofactors for radical generation.
The importance of homogenous metallocluster formation in cells and in vitro extend far beyond RNR. Non-heme iron mononuclear, binuclear and FeS species are ubiquitous and recent studies in S. cerevisiae, show they are linked to each other through regulatory mechanisms, which are actively being investigated.63 Development of new methods to provide greater insight about metallocofactor assembly strategies will set a foundation for a quantitative biochemistry for a large expanse of iron-based and other metalloenzymes.
Dynamics of Regulatory Proteins.
The structural basis for conformational gating of active and inactive states of regulatory enzymes in biology remain difficult to define due to the challenges associated with the lifetime of the states and their interconversions, and the sensitivity of the methods for the detection of mixtures of their often closely related structures. Cryo-EM offers, a unique opportunity to trap conformationally-gated active and inactive states under physiological conditions. While still early days, an understanding of regulation by conformation will be forthcoming by combining trapped cryo-EM structures with new biochemical, spectroscopic, and computational methods.
RNR is paradigmatic in its ability to use conformation to control the reactivity of radicals. The biochemistry enabling the E. coli Ia RNR cryo-EM structure sets the stage for trapping other distinct conformations of α2β2 during both forward and reverse RT. The tools to study RNR chemistry, mechanism-based inhibitors and UAA trapping of RT residues, may provide ways to trap RNRs in distinct states.48 Obtaining a number of moderately homogenous trapped states analyzed by cryo-EM in conjunction with method development for deconvolution of structurally similar states within one sample, may lead to a model for the conformational dynamics of the E. coli RNR during turnover.
In the class Ia RNRs the active state(s) is(are) in dynamic equilibrium with inactive state(s), providing an important mechanism to manage the ratios and amounts of dNTP pools. The recent discovery of dATP-α4β4 inhibited state in the E.coli RNR31 and its perturbation by 2-hydroxypyridone inhibitors29 will provide insight about the dynamics of nucleotide-state interconversions and mechanisms of small molecule perturbations. In the class Ia human RNRs, despite the α,β structural homology with the subunits of E. coli, no high-resolution structures of the metallocofactor centers of β2s or any active α/β complexes, have been reported. The dATP-inactivated state is an α6 ring structure with no β (Figure 3C)30 and α is also hexameric in the presence of ATP and other dNTPS.25 The dynamics of α6 and its interaction with an undetermined number of β2s to generate an active state(states) also remain undefined.30,64 The central role of RNR in DNA replication and repair biology provides an imperative for an understanding of the structures of the human RNR subunits, their active and inactive states and the dynamics of quaternary structural interconversions.
Radical Chemistry Beyond Tyrosine.
Tyrosyl radical has been preeminent in studies of radical-based enzymes due to the ease of their detection by visible and EPR spectroscopies. However, RNRs have also highlighted the importance of C• and glycyl radicals (G•). The glycyl radical enzymes65 a small subset of the rSAM superfamily (SF) of enzymes,6,12,66 often catalyze C• formation, and are of interest in the human microbiome and metabolic engineering.67 Sulfur radicals have been proposed in a number of enzymatic transformations, but have eluded detection as they are not easily observed due to their weak absorption features and unusual electronic structure and EPR parameters.68 Their importance has firmly been established within RNRs by trapping them with bond formation to adjacent residues allowing for their detection when subsequent transformations are blocked.69 In the absence of such protein protective mechanisms, advanced methods will be needed to uncover them in biology (Figure 4A, 4D, 4E), including among others, site-specific incorporation of selenocysteine using UAA engineering.70 Development of these methods will provide ways to further probe the importance of amino acid C• and other radical-mediated transformations, many, of which will involve, unprecedented chemistries, as with RNRs. Genomics and bioinformatics have revealed that the rSAM SF of enzymes has >500,000 members.66,71 They employ the iron-sulfur cluster to bind SAM to reductively cleave it to generate 5-deoxyadenosyl radical via an organometallic intermediate,71 which then abstracts H-atoms to give rise to a large swath of carbon centered radicals to drive diverse chemistry and enzymology. Though the function of most members of this SF remain to be discovered, those characterized to date are harbingers of an exciting frontier in radical chemistry.
Radicals as Targets of Advanced Therapeutics.
RNRs are essential in the viability of all organisms as they are key responders to states arising from DNA mutations. RNRs control the amounts and ratios of dNTP pools in collaboration with editing domains of DNA polymerases, the mismatch repair pathway, and damage response pathways that deal with replication stress.72,73 Distinct features between the human, bacterial, and viral RNRs also suggest that these enzymes provide unique targets for selective therapeutic intervention.48 RNR function involves controlled radical chemistry, disruption of which could lead to a number of therapeutic targets (Figure 5) including: (A) interruption of the assembly of the essential metallocofactor or its repair, (B) inhibition of radical chemistry in the active site, and (C) promotion of inactive states that disrupt the RT pathway. For all drug design investigations, high throughput assay development inside the cell and in vitro will be essential.74 Promising paths for advanced therapeutics are:
Figure 5. Therapeutics that inhibit RNRs by distinct mechanisms.

Chlofarabine (ClF) as the diphosphate (ClFDP) and ClFTP reversibly bind to the C-site and A-site of α respectively resulting in formation of “stable” inhibited α6 states. Gemcitabine (F2C) as F2CDP binds to the C-site of α and requires the presence of β to form an active RNR and become a mechanism-based, irreversible inhibitor. Both ClF and F2C are used clinically. Hydroxyurea (HU) and triapine (TP) target β and interfere with the assembly and/or stability of its diferric-tyrosyl radical cofactor. HU is used clinically and triapine is in ongoing clinical trials. The peptide-X analog targets uniquely the herpes simplex virus (HSV) RNR, by specifically binding to its α, interfering with α/β subunit interactions and thus active HSV RNR formation.
(A). Selective disruption of cluster assembly.
The human β2, a tumor promoter often overexpressed in chemoresistant tumors, is a therapeutic target. Drugs that interfere with the assembly of the iron-Y• cofactor provide a unique opportunity for therapeutic intervention.75,76 However, extensively investigated compounds such as triapine (TP)77 and hydroxyurea (HU)78 (Figure 5) suffer from lack of specificity. Thus, cofactor targeting alone will be challenging, given the connection of this process to the machinery that controls Fe homeostasis and oxidative stress.
(B). Small molecules that reversibly bind to the C-site in α.
Napthylsalicylacyl hydrazine reversibly binds the C-site of human α279 and has been a starting point to screen for and design more potent non-nucleoside inhibitors. However, a singular focus on α2 may not be sufficient as E. coli Ia cryo-EM structure has recently shown that the disordered C-terminus of β closes transiently over the C-site in α to form a tightly enclosed cavity.21 If a similar strategy is used by the β tail of human RNR, then consideration of the β tail in conjunction with the α active site reversible binders represent a new way to target RNRs. The β tail (C-terminal, ~last 15 amino acids) in itself presents an opportunity to inhibit subunit interaction, as peptide-X in Figure 5 has been shown to be a potent therapeutic of Herpes simplex virus RNR.80,81
(C). Small molecules that promote inhibited quaternary structures.
Mechanism-based inhibitors such as gemcitabine (F2C), which targets both the human and E. coli Ia RNRs, are used clinically.82 One equivalent F2CDP (Figure 5) per α2β2 is sufficient for inactivation and the enzymes are trapped in distinct states in the presence and in the absence of reductants.83 Whereas mechanism-based inhibitors have been the historical path of RNR drug design, the design of drugs that promote inhibited quaternary structures provide a new opportunity for the development of selective therapeutics, as bacterial and human RNRs have different inactive states (Figure 3C). The ClF therapeutic in its triphosphate (ClFTP) and diphosphate (ClFDP) states (Figure 5) can trap human RNR in stabilized, inhibited α6 state(s) in vitro27,28 and in cells.32 ClFDP binds in the C-site and ClFTP binds to the A-site, cone domain (Figure 5).27 Since E. coli RNR does not form an α6 state, it is not inhibited by ClF nucleotides. Recently, multidrug resistant N. gonorrhoeae strains have been shown to be responsive to several 2-hydroxypyridone analogs. Their targets appear to be the α4β4 inactive state in vitro and they show in vivo efficacy in a mouse model.29 Recent reports of unusual inhibited quaternary structures of class Ia, Ib and Id RNRs26,84-86 suggest that trapping other RNRs may be possible for obtaining structures for cryo-EM analysis and for guided inhibitor (re)design.
Evolution of a DNA World from a RNA World
The transformation of a RNA world to a DNA world is the subject of lively scientific debate.87 RNRs are currently the only way in biology to make deoxynucleotides in all organisms, and as depicted in Figure 2, C• chemistry is central to promoting the conversion of RNA to DNA building blocks. This conserved C• chemistry and the SAM bound-FeS-metallocofactor central to the large rSAM SF of “ancient” enzymes, suggest that long overlooked sulfur chemistry deserves further study.
Prebiotic chemistry studies have long indicated the building blocks for RNA and proteins can be made abiotically. Newer studies suggest that ara- and deoxy-pyrimidine nucleotide building blocks can be made using thiouridines, light, and H2S reductant.87 RNR has begun to reemerge in the continued discussions about the conversion of an RNA to a DNA/protein world. Using the current understanding of the RNR mechanism involving thiols, radicals, and a putative prebiotic environment (H2S, S•), computational methods have led to the proposition of several feasible mechanisms for nucleoside/ deoxynucleoside interconversion.88
A second issue concerns the evolution of the unprecedented long-range RT pathway (Figure 2) observed in the class I RNRs and brings this Perspective full circle to the control of radical reactions for challenging transformations and the avoidance of oxidative stress. The β2 subunit of class I RNRs are members of the ferritin SF of enzymes, with all members sharing a four-helix bundle architecture and binding sites for two redox active metals. 89 This SF may have evolved from an anoxic ocean 1.8-0.8 billion years ago during the transition to an aerobic world,3 to perhaps help ameliorate self-destruction by the “bad radicals” produced by the Fe(II) and Cu(I)-mediated reductive metabolites of O2. Ferritin currently sequesters iron and is an important contributor to protecting organisms from the “bad” radicals. So how did this four-helix bundle scaffold become the current structural basis for β2s that generate an active dimetallocofactor using O2, or O2•− or H2O2 to initiate a reversible oxidation over the 32 Å that involves the amino acid radical transport chain shown in Figure 2? Both cluster assembly and long-range RT in all class I RNRs are essential to making DNA building blocks, essential for fidelity of DNA replication and repair, while avoiding self-destruction. While the answer to this issue remains elusive, RNRs have provided us with new ways to think about the importance and the central role of controlled radical chemistry in biology and chemistry.
Concluding Remarks
A large number of enzymes are now known to or postulated to derive their function from radical chemistry. These enzymes perform diverse metabolic roles while maintaining specificity and limiting the production of reactive oxygen and other destructive species. Penetrating studies on the radical chemistry and biology of RNRs have provided the community with new tools and a guiding framework for future studies aimed at understanding the chemistry and biology of radicals, and the treatment of their associated disease states.
ACKNOWLEDGEMENTS
Studies on RNRs have been the collective efforts of ingenuity and commitment of our many graduate students and postdoctoral fellows. We are especially grateful for our collaborations with Marina Bennati and Catherine Drennan.
Funding Sources
This work was supported by the National Institutes of Health Grants GM047274 (DGN) and GM029595 (JS).
Footnotes
The authors declare no competing financial interests.
REFERENCES
- (1).Winterbourn CC Reconciling the Chemistry and Biology of Reactive Oxygen Species. Nat. Chem. Biol 2008, 4, 278–286. [DOI] [PubMed] [Google Scholar]
- (2).Frey PA; Reed GH The Ubiquity of Iron. ACS Chem. Biol 2012, 7, 1477–1481. [DOI] [PubMed] [Google Scholar]
- (3).Anbar AD Elements and Evolution. Science 2008, 322, 1481–1484. [DOI] [PubMed] [Google Scholar]
- (4).Burney S; Caulfield JL; Niles JR; Wishnok JS; Tannebaum SR The Chemistry of DNA Damage from Nitric Oxide and Peroxynitrite. Mut. Res. – Found. Mol. Mech. Mut 1999, 424, 23–49. [DOI] [PubMed] [Google Scholar]
- (5).Marletta MA Revisiting Nitric Oxide Signaling: Where Was It, and Where Is It Going? Biochemistry 2021, 10.1021/acs.biochem.1c00276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Holliday GL; Ajuva E; Meng EC; Brown SD; Cahoun S; Pieper U; Sali A; Booker SJ; Babbitt PC Atlas Radical of the Radical SAM Superfamily: Divergent Evolution of Functions Using a “Plug and Play” Domain. Meth. Enzymol 2018, 606, 1–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Stubbe J; van der Donk WA Protein Radicals in Enzyme Catalysis. Chem. Rev 1998, 98, 705–762. [DOI] [PubMed] [Google Scholar]
- (8).Orme-Johnson WH; Beinert H; Blakley RL Cobamides and Ribonucleotide Reduction. J. Biol. Chem 1974, 249, P2338–2343. [PubMed] [Google Scholar]
- (9).Sjöberg BM; Reichard P; Gräslund A; Ehrenberg A The Tyrosine Free Radical in Ribonucleotide Reductase from Escherichia coli. J. Biol. Chem 1978, 253, 6863–6865. [PubMed] [Google Scholar]
- (10).Barry BA; Babcock GT Tyrosyl Radicals are Involved in the Photosynthetic Oxygen Evolving System. Proc. Natl. Acad. Sci. U.S.A 1987, 84, 7099–7013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Sharma V; Wikström M The Role of the K-Channel and the Active Site Tyrosine is Essential in Proton Pumping in the Cytochrome c Oxidase. Biochem. Biophys. Acta Bioenerg 2016, 1857, 1111–1115. [DOI] [PubMed] [Google Scholar]
- (12).Zallot R; Cheng N; Gerlt JA The EFI Web Resource for Genomic Enzymology Tools: Leveraging Protein, Genome, and Metagenome Data Bases to Discover Novel Enzymes and Metabolic Pathways. Biochemistry 2019, 58, 4169–4182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Hofer A; Crona M; Logan DT; Sjöberg BM DNA Building Blocks: Keeping Control of Manufacture. Crit. Rev. Biochem. Mol. Biol 2012, 47, 50–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Nordlund P; Reichard P Ribonucleotide Reductases. Annu. Rev. Biochem 2006, 75, 681–706. [DOI] [PubMed] [Google Scholar]
- (15).Licht S; Stubbe J Mechanistic Investigations of Ribonucleotide Reductases. Compr. Nat. Prod. Chem 1999, 5, 163–203. [Google Scholar]
- (16).Uhlin U; Eklund H Structure of Ribonucleotide Reductase Protein R1. Nature 1994, 370, 533–539. [DOI] [PubMed] [Google Scholar]
- (17).Sintchak MD; Arjara G; Kellogg BA; Stubbe J; Drennan DL The Crystal Structure of Class II Ribonucleotide Reductase Reveals How an Allosterically Regulated Monomer Mimics a Dimer. Nat. Struct. Biol 2002, 9, 293–300. [DOI] [PubMed] [Google Scholar]
- (18).Nordlund P; Sjöberg BM; Eklund H Three-Dimensional Structure of the Free Radical Protein of Ribonucleotide Reductase. Nature 1990, 345, 593–598. [DOI] [PubMed] [Google Scholar]
- (19).Stubbe J Ribonucleotide Reductases in the Twenty First Century. Proc. Natl. Acad. Sci. U.S.A 1998, 95, 2723–2724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Licht S; Gerfen GJ; Stubbe J Thiyl Radicals in Ribonucleotide Reductases. Science 1996, 271, 477–481. [DOI] [PubMed] [Google Scholar]
- (21).Kang G; Taguchi AT; Stubbe J; Drennan CL Structure of a Trapped Radical Transfer Pathway within a Ribonucleotide Reductase Holocomplex. Science 2020, 368, 424–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Minnihan EC; Nocera DG; Stubbe J Reversible, Long-Range Radical Transfer in E. coli Class Ia Ribonucleotide Reductase. Acc. Chem. Res 2013, 46, 2524–2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Zimanyi CM; Chen PY; Kang G; Funk MA; Drennan CL Molecular Basis for Allosteric Specificity Regulation in Class Ia Ribonucleotide Reductase from Escherichia coli. eLife 2016, 5, e07141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Jonna V; Crona M; Rofougaran R; Linden D; Joahansson S; Branstrom K; Sjoberg BM; Hofer A Diversity in Overall Activity Regulation of Ribonucleotide Reductase. J. Biol. Chem 2015, 290, 17339–17348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Rofougaran R; Crona M; Vodnala M; Sjöberg BM; Hofer A Oligomerization Status Directs Overall Activity Regulation of the Eschericia coli Class Ia Ribonucleotide Reductase. J. Biol. Chem 2008, 283, 35310–35318. [DOI] [PubMed] [Google Scholar]
- (26).Thomas WC; Brooks FP; Burnim AA; Bacik JP; Stubbe J; Kaelber JT; Chen JZ; Ando N Convergent Allostery in Ribonucleotide Reductase. Nat. Commun 2019, 10, 2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Aye Y, Stubbe J. Clofarabine 5′-Di- and -Triphosphates Inhibit Human Ribonucleotide Reductase by Altering the Quaternary Structure of its Large Subunit. Proc. Natl. Acad. Sci. U.S.A 2011, 108, 9815–9820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Wisitpitthaya S; Zhao Y; Long MJC; Li M; Fletcher EA; Blessing WA; Weiss RS; Aye Y Cladribine and Fludarabine Nucleotides Induce Distinct Hexamers Defining a Common Mode of Reversible RNR Inhibition. ACS Chem. Biol 2016, 11, 2021–2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Narasimhan J; Letinsk L; Jung S; Gerasyuto A; Wang J; Arnold M; Chen G; Hedrick J; Dumble M; Ravichandran K; Levitz TS; Chang C; Drennan CL; Stubbe J; Karp G; Branstrom A Ribonucleotide Reductase, A Novel Target for Gonorrhea. BioRxiv 2021, posted 8 February 2021, doi: 10.1101/2020.11.19.38957, (accessed 30 July 2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Fairman JW; Wijerathna SR; Ahmad MF; Xu H; Nakano R; Jha S; Prendergast J; Welin RM; Flodin S; Roos A; Nordlund P; Li Z; Walz T; Dealwis CG Structural Basis for Allosteric Regulation of Human Ribonucleotide Reductase by Nucleotide-Induced Oligomerization. Nat. Struct. Mol. Biol 2011, 18, 316–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Ando N; Brignole EJ; Zimanyi CM; Funk MA; Yokoyama K; Asturias FJ; Stubbe J; Drennan CL Structural Interconversions Modulate Activity of Escherichia coli Ribonucleotide Reductase. Proc. Natl. Acad. Sci. U.S.A 2011, 108, 21046–21051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Aye Y; Brignole EJ; Long MJ; Chittuluru J; Drennan CL; Asturias FJ; Stubbe J Clofarabine Targets the Large Subunit (α) of Human Ribonucleotide Reductase in Live Cells by Assembly into Persistent Hexamers. Chem. Biol 2012, 19, 799–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Cukier RI; Nocera DG Proton-Coupled Electron Transfer. Annu. Rev. Phys. Chem 1998, 49, 337–369. [DOI] [PubMed] [Google Scholar]
- (34).Cotruvo JA; Stubbe J Class I Ribonucleotide Reductases: Metallocofactor Assembly and Repair in vitro and in vivo. Annu. Rev. Biochem 2011, 80, 733–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Young DD; Schultz PG Playing with the Molecules of Life. ACS Chem. Biol 2018, 13, 854–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Minnihan EC; Young DD; Schultz PG; Stubbe J Incorporation of Fluorotyrosines into Ribonucleotide Reductase Using an Evolved, Polyspecific Aminoacyl-tRNA Synthetase. J. Am. Chem. Soc 2011, 133, 15797–16318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Oyala PH; Ravichandran KR; Funk MA; Stucky PA; Stich TA; Drennan CL; Britt RD; Stubbe J Biophysical Characterization of Fluorotyrosine Probes Site-Specifically Incorporated into Enzymes: E. coli Ribonucleotide Reductase as an Example. J. Am. Chem. Soc 2016, 138, 7951–7964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Climent I; Sjöberg BM; Huang CY Carboxyl-Terminal Peptides as Probes for Escherichia coli Ribonucleotide Reductase Subunit Interaction: Kinetic Analysis of Inhibition Studies. Biochemistry 1991, 30, 5164–5171. [DOI] [PubMed] [Google Scholar]
- (39).Climent I; Sjöberg BM; Huang CY Site-Directed Mutagenesis and Deletion of the Carboxyl Terminus of Escherichia coli Ribonucleotide Reductase Protein R2–Effects on Catalytic Activity and Subunit Interaction. Biochemistry 1992, 31, 4801–4807. [DOI] [PubMed] [Google Scholar]
- (40).Minnihan EC; Ando N; Brignole EJ; Olshansky L; Chittuluru J; Asturias FJ; Drennan CL; Nocera DG; Stubbe J Generation of a Stable, Aminotyrosyl Radical-Induced α2β2 Complex of E. coli Class Ia Ribonucleotide Reductase. Proc. Natl. Acad. Sci. U.S.A 2013, 110, 3835–3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Lin Q; Parker MJ; Taguchi AT; Ravichandran K; Kim A; Kang G; Shao J; Drennan CL; Stubbe J Glutamate 52-β at the α/β Subunit Interface of Escherichia coli Class Ia Ribonucleotide Reductase Is Essential for Conformational Gating of Radical Transfer. J. Biol. Chem 2017, 292, 9229–9239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Nick TU; Lee W; Koßmann S; Neese F; Stubbe J; Bennati M Hydrogen Bond Network between Amino Acid Radical Intermediates on the Proton-Coupled Electron Transfer Pathway of E. coli α2 Ribonucleotide Reductase. J. Am. Chem. Soc 2015, 137, 289–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Seyedsayamdost MR; Chan CT; Mugnaini V; Stubbe J; Bennati M PELDOR Spectroscopy with DOPA-β2 and NH2Y-α2s: Distance Measurements Between Residues Involved in the Radical Propagation Pathway of E. coli Ribonucleotide Reductase. J. Am. Chem. Soc 2007, 129, 15748–15749. [DOI] [PubMed] [Google Scholar]
- (44).Greene BL; Taguchi AT; Stubbe J; Nocera DG Conformationally Dynamic Radical Transfer within Ribonucleotide Reductase. J. Am. Chem. Soc 2017, 139, 16657–16665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Hecker F; Stubbe J; Bennati M Detection of Water Molecules on the Radical Transfer Pathway of Ribonucleotide Reductase by 17O Electron-Nuclear Double Resonance Spectroscopy. J. Am. Chem. Soc 2021, 143, 7237–7241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Yokoyama K; Smith AA; Corzilius B; Griffin RG; Stubbe J Equilibration of Tyrosyl Radicals (Y356•, Y731•, Y730•) in the Radical Propagation Pathway of the Escherichia coli Class Ia Ribonucleotide Reductase. J. Am. Chem. Soc 2011, 133, 18420–18432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Ravichandran KR; Taguchi AT; Wei Y; Nocera DG; Stubbe J A >200 meV Uphill Thermodynamic Landscape for Radical Transport in E. coli Ribonucleotide Reductase Determined Using Fluorotyrosine-Substituted Enzymes. J. Am. Chem. Soc 2016, 138, 13706–13716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Greene BL; Kang G; Cui C; Bennati M; Nocera DG; Drennan CL; Stubbe J Ribonucleotide Reductases: Structure, Chemistry, and Metabolism Suggest New Therapeutic Targets, Annu. Rev. Biochem 2020, 89, 45–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Pizano AA; Lutterman DA; Holder PG; Teets TS; Stubbe J; Nocera DG Photo-Ribonucleotide Reductase β2 by Selective Cysteine Labeling with a Radical Phototrigger. Proc. Natl. Acad. Sci. U.S.A 2011, 109, 39–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Olshansky L; Pizano AA; Wei Y; Stubbe J; Nocera DG Kinetics of Hydrogen Atom Abstraction from Substrate by an Active Site Thiyl Radical in Ribonucleotide Reductase. J. Am. Chem. Soc 2014, 136, 16210–16216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Cui C; Greene BL; Kang G; Drennan CL; Stubbe J; Nocera DG Gated Proton Release during Radical Transfer at the Subunit Interface of Ribonucleotide Reductase. J. Am. Chem. Soc 2021, 143, 176–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Pourbaix M Atlas of Electrochemical Equilibria in Aqueous Solutions, 2nd ed., National Association of Corrosion Engineers, New York, NY, 1974. [Google Scholar]
- (53).Mitchell P Coupling of Phosphorylation to Electron and Hydrogen Transfer by a Chemiosmotic Type of Mechanism. Nature 1961, 191, 144–148. [DOI] [PubMed] [Google Scholar]
- (54).Chang CJ; Brown JDK; Chang MCY; Baker EA; Nocera DG Electron Transfer in Hydrogen-Bonded Donor-Acceptor Supramolecules, In Electron Transfer in Chemistry, Balzani V, Ed., Wiley-VCH: Weinheim, Germany, 2001, Vol. 3, Ch. 2.4, pp 409–461. [Google Scholar]
- (55).Reece SY; Nocera DG Proton-Coupled Electron Transfer in Biology: Results from Synergistic Studies in Natural and Model Systems. Annu. Rev. Biochem 2009, 78, 673–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Stubbe J; Nocera DG; Yee CS; Chang MCY Radical Initiation in the Class I Ribonucleotide Reductase: Long-Range Proton-Coupled Electron Transfer? Chem. Rev 2003, 103, 2167–2201. [DOI] [PubMed] [Google Scholar]
- (57).Boal AK; Cotruvo JA, Stubbe J; Rosenzweig AC Structural Basis for Activation of Class Ib Ribonucleotide Reductase. Science 2010, 329, 1526–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Jiang W; Yun D; Saleh L; Barr EW; Xing G; Hoffart LM; Maslak MA; Krebs C; Bollinger JM A Stable Manganese(IV)/Iron(III) Cofactor Initiates Substrate Radical Production in Chlamydia trachomatis Ribonucleotide Reductase. Science, 2007, 316, 1188–1191. [DOI] [PubMed] [Google Scholar]
- (59).Rose HR; Ghosh MK; Maggiolo AO; Pollock CJ; Blaesi EJ; Hajj V; Wei Y; Rajakovich LJ; Chang W; Han Y; Hajj M; Krebs C; Silakov A; Pandelia ME; Bollinger JM; Boal AK Structural Basis for Superoxide Activation of Flavobacterium johnsoniae Class I Ribonucleotide Reductase and for Radical Initiation by Its Dimanganese Cofactor. Biochemistry 2018, 57, 2679–2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Rozman-Grinberg I; Lundin D; Hasan M; Crona M; Jonna VR; Loderer C; Sahlin M; Markova N; Borovok I; Berggren G; Hofer A; Logan DT; Sjöberg BM Novel ATP-Cone-Driven Allosteric Regulation of Ribonucleotide Reductase via the Radical-Generating Subunit. eLife 2018, 7, e31529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Srinivas V; Lebrette H; Lundin D; Kutin Y; Sahlin M; Lerche M; Eirich J; Branca RMM; Cox N Sjöberg BM Högbom M Metal-Free Ribonucleotide Reduction Powered by a DOPA Radical in Mycoplasma Pathogens. Nature 2018, 563, 416–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Blaesi EJ; Palowitch GM; Hu K; Kim AJ; Rose HR; Alapati R; Lougee MG; Kim HJ; Taguchi AT; Tan KO; Laremore TN; Griffin RG; Krebs C; Matthews ML; Silakov A; Bollinger JM; Allen BD; Boal AK Metal-Free Class Ie Ribonucleotide Reductase from Pathogens Initiates Catalysis with a Tyrosine-Derived Dihydroxyphenyl-alanine Radical. Proc. Natl. Acad. Sci. U.S.A 2018, 115, 10022–10027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Li H; Stumpfig M; Zhang C; An X; Stubbe J; Lill R; Huang M The Diferric Tyrosyl Cluster of Ribonucleotide Reductase and Cytosoloic Iron Sulfur Clusters Have Distinct and Similar Biogenesis Requirements. J. Biol. Chem 2017, 253, 6863–6865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Brignole EJ; Tsai KL; Chittuluru J; Li H; Aye Y; Penczek PA; Stubbe J; Drennan CL; Francisco A 3.3-Å Resolution Cryo-EM Structure of Human Ribonucleotide Reductase with Substrate and Allosteric Regulators Bound. eLife 2018, 7, e31502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Backman LRF; Funk MA; Dawson CD; Drennan CL New Tricks for the Glycyl Radical Family. Crit. Rev. Biochem 2017, 52, 674–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Sophia HJ; Chen O; Hetzler BG; Reyes-Spindola JF; Miller NE Radical SAM, A Novel Protein Superfamily Linking Radical Activations: Functional Characterization Using New Analysis and Information Visualization Methods. Nucl. Acid Res 2001, 29, 1097–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Fu B; Balskus EP Discovery of C–C Bond-Forming and Bond-Breaking Radical Enzymes: Enabling Transformations for Metabolic Engineering. Curr. Opin. Biotech 2020, 5, 94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).van Gastel M; Lubitz W; Lassmann G; Neese F Electronic Structure of the Cysteine Thiyl Radicals: A DFT and Correlated ab initio Study. J. Am. Chem. Soc 2004, 126, 2237–2246. [DOI] [PubMed] [Google Scholar]
- (69).Wei Y; Mathies G; Yokoyama K; Chen J; Griffin RG; Stubbe J A Chemically Competent Thiosulfuranyl Radical on the Class II Ribonucleotide Reductase. J. Am. Chem. Soc 2014, 136, 9001–9013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Green BL; Stubbe J; Nocera DG Selecocysteine Substitution in Class I Ribonucleotide Reductases. Biochemistry 2019, 58, 5074–5084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Broderick JB; Hoffman BM; Broderick WE Mechanism of Radical Initiation in the Radical S-Adenosylmethionine Superfamily. Acc. Chem. Res 2018, 51, 2611–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Aye Y; Li M; Long MJC; Weiss RS Ribonucleotide Reductase and Cancer: Biological Mechanisms and Targeted Therapies. Oncogene 2014, 34, 2011–2021. [DOI] [PubMed] [Google Scholar]
- (73).Ewald B; Sampath D; Plunkett W Nucleoside Analogs: Molecular Mechanisms Signaling Cell Death. Oncogene 2008, 27, 6522–6537. [DOI] [PubMed] [Google Scholar]
- (74).Ravichandran KR; Olshansky L; Nocera DG; Stubbe J Subunit Interaction Dynamics of Class Ia Ribonucleotide Reductases: In Search of a Robust Assay. Biochemistry 2020, 59, 1442–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Aye Y; Long MJ; Stubbe J Mechanistic Studies of Semicarbazone Triapine Targeting Human Ribonucleotide Reductase in vitro and in Mammalian Cells: Tyrosyl Radical Quenching Not Involving Reactive Oxygen Species. J. Biol. Chem 2012, 287, 35768–35778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Chen MC; Zhou B; Zhang K; Yuan YC; Un F; Hu S; Chou CM; Chen CH; Wu J; Wang Y; Liu X; Smith DL; Li H; Liu Z; Warden CD; Su L; Malkas LH; Chung YM; Hu MCT; Yen Y The Novel Ribonucleotide Reductase Inhibitor COH29 Inhibits DNA Repair in vitro. Mol. Pharmacol 2015, 87, 996–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Popović-Bijelić A; Kowol CR; Lind ME; Luo J; Himo F; Enyedy EA; Arion VB; Gräslund A Ribonucleotide Reductase Inhibition by Metal Complexes of Triapine (3-Aminopyridine-2-Carboxaldehyde Thiosemicarbaside): A Combined Experimental and Theoretical Study. J. Inorg. Biochem 2011, 105, 1422–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Davies BW; Kohanski MA; Simmons LA; Winkler JA; Collins JJ; Walker GC Hydroxyurea Induces Hydroxyl Radical-Mediated Cell Death in Escherichia coli. Mol. Cell 2009, 36, 845–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Yang M; Agrawal P; Pink J; Harris ME Dealwis C; Visvoncthan R Structure-Guided Synthesis and Mechanistic Studies on Napthyl Salicyl Hydrazine Scaffold as a Non-Nucleoside Competitive Inhibitor of Human Ribonucleotide Reductase. J. Med. Chem 2018, 61, 666–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Dutia MM; Frame MC; Subak-Sharppe JH; Clavic WN; Marsden HS Specific Inhibition of Herpes Virus Ribonucleotide Reductase by Synthetic Peptides. Nature 1986, 321, 439–441. [DOI] [PubMed] [Google Scholar]
- (81).Moss N; Beaulieu P; Duceppe S; Ferland JM; Garneu M; Gunhier J; Ghiro E; Gouled S; Guse I; Jaramillo J; Llinas-Brunet M; Malenfont E; Plante R; Poirer M; Soucy F; Wenic D; Yoakim C; Dezie R Peptideomimetic Inhibitors of Herpes Simplex Virus Ribonucleotide Reductase with Improved in vivo Antiviral Activity. J. Med. Chem 1996, 39, 4173–4180. [DOI] [PubMed] [Google Scholar]
- (82).Hertel LW; Boder GB; Kroin JS; Rinzel SM; Poore GA; Todd GC; Grindey GB Evaluation of the Antitumor Activity of Gemcitabine (2′,2′-Difluoro-2′-Deoxycytidine). Cancer Res. 1990, 50, 4417–4422. [PubMed] [Google Scholar]
- (83).Wang J; Lohman GJS; Stubbe J Enhanced Subunit Interactions with Gemcitabine-5′-Diphosphate Inhibit Ribonucleotide Reductases. Proc. Natl. Acad. Sci. U.S.A 2007. 104, 14324–14329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Rose HR; Maggiolo AO; McBride MJ; Palowitch GM; Pandelia ME; Davis KM; Yennawar NH; Boal AK Structures of Class Id Ribonucleotide Reductase Catalytic Subunits Reveal Minimal for Deoxynucleotide Synthesis. Biochemistry 2019, 58, 1845–1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Johansson R; Jona VR; Kuma R; Nayeri N; Lundin D; Sjöberg BM; Logan DT Structural Mechanism of Allosteric Activity Regulation in a Ribonucleotide Reductasewith a Double ATP Cone. Structure 2016, 24, 906–917. [DOI] [PubMed] [Google Scholar]
- (86).Martínez-Carranza M; Venkateswara RJ; Lundin D; Sahlin M; Carlson LA; Jemal N; Högbom M; Sjöberg BM; Stenmark P; Hofer A A Ribonucleotide Reductase from Clostridium botulinum Reveals Distinct Evolutionary Pathways to Regulation via the Overall Activity Site. J. Biol. Chem 2020, 295, 15576–15587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Xu J; Green NJ; Givard C; Krishnamurthy R; Sutherland JD Prebiotic Phosphorylation of 2-Thiuridine Provides Either Nucleotides or DNA Building Blocks via Photoreduction. Nat. Chem 2019, 11, 457–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Dragicevic I; Baric D; Kovacevic B; Golding B; Smith DM Non-enzymatic Ribonucleotide Reduction in the Prebiotic Context. Chem. Eur. J 2015, 21, 6131–6143. [DOI] [PubMed] [Google Scholar]
- (89).Lundin D; Berggren G; Logan DT Sjöberg BM The Origin and Evolution of Ribonucleotide Reductase. Life 2015, 5, 604–636. [DOI] [PMC free article] [PubMed] [Google Scholar]