

Abstract
Type 2 T helper (TH2) cells are protective against parasitic worm infections but also aggravate allergic inflammation. Although the role of dendritic cells (DCs) in TH2 cell differentiation is well established, the underlying mechanisms are largely unknown. Here, we show that DC induction of TH2 cells depends on membrane-associated RING-CH-1 (MARCH1) ubiquitin ligase. The pro-TH2 effect of MARCH1 relied on lymph node (LN)–resident DCs, which triggered T cell receptor (TCR) signaling and induced GATA-3 expression from naïve CD4+ T cells independent of tissue-driven migratory DCs. Mice with mutations in the ubiquitin acceptor sites of MHCII and CD86, the two substrates of MARCH1, failed to develop TH2 cells. These findings suggest that TH2 cell development depends on ubiquitin-mediated clearance of antigen-presenting and costimulatory molecules by LN-resident DCs and consequent control of TCR signaling.
INTRODUCTION
Dendritic cells (DCs) play an instrumental role in inducing antigen-specific T cell immunity (1). Encounter and capture of foreign antigens in the tissues activate DCs and induce migration to the draining lymph node (LN) (2). Migrated DCs present antigens to naïve T cells through the major histocompatibility complex (MHC) antigen-presenting molecules and provide the T cells with costimulatory signals that lower the threshold of activation (3). Activated T cells differentiate into specific types of effector cells depending on the nature of the antigens (4). Type 1 T helper (TH1) effector cells develop in response to intracellular pathogens, playing a critical role in activating macrophages to facilitate pathogen clearance and inducing robust cytotoxic CD8+ T cell responses (5, 6). DC production of interleukin-12 (IL-12) is important for differentiation of TH1 and cytotoxic CD8+ T cells (7, 8). TH17 cells develop in response to extracellular bacteria or fungi, and IL-1β, IL-6, IL-23, or transforming growth factor–β1 produced by DCs plays a key role in driving the development of these cells (9). TH2 cells develop in response to extracellular parasites and environmental substances including common allergens (10). Unlike the defined role for DC-derived cytokines in driving TH1 or TH17 cell differentiation, specific cytokines produced by DCs that drive TH2 cell differentiation are not known (11). DC expression of the transcription factors interferon regulatory factor 4 (IRF4) or Krüppel-like factor 4 (KLF4) has been implicated in TH2 cell development, but these factors also profoundly alter DC development (12–14). DC surface molecules such as OX40 ligand and CD40 have been also implicated for TH2 cell development (15, 16), but many of these molecules serve as general costimulatory molecules rather than specifically promoting TH2 cell development (17, 18). Weak T cell receptor (TCR) signal strength has been proposed to foster TH2 cell differentiation (4). Priming of naïve antigen-specific T cells with a low dose of antigens promoted IL-4 secretion and GATA-3 expression from the T cells in vitro (19, 20). Adoptive transfer of DCs loaded with a low dose of antigens preferentially gave rise to TH2 over TH1 cells in vivo (21). However, whether TCR signal strength is modulated during TH2 cell development under physiological conditions and, if so, how such modulation is achieved have not been defined.
DCs control the surface expression of the antigen-presenting molecule MHCII and the costimulatory molecule CD86 through ubiquitination by membrane-associated RING-CH-1 (MARCH1) E3 ligase (22, 23). MARCH1 enables transfer of ubiquitin moieties from a ubiquitin-conjugating enzyme to a cytoplasmic lysine amino acid on its substrates (24). MARCH1 targets MHCII only after the MHCII is loaded with antigenic peptide and transported to the plasma membrane, thus mediating surface turnover of peptide-MHCII complexes (25). Despite being a well-characterized molecular pathway and mechanism, the functional role of MHCII ubiquitination is not clearly understood (26). We have reported that deficiency of MARCH1 or MHCII ubiquitination reduces the competitiveness of germinal center B cells during immune responses, implicating MHCII turnover in B cell function of producing high-affinity antibodies (27). Mice deficient in MARCH1 or the ubiquitin-accepting lysine of MHCII in DCs exhibited a decrease in the number and repertoire of thymic regulatory T cells, implicating MARCH1 in DC function of selecting natural regulatory T cells (28, 29). However, the role of MARCH1 in driving antigen-specific T cell immunity has not been explored extensively.
Here, we investigated the role of MARCH1 in the development of T cell immunity using mouse models of microbial and parasitic infections or immune-stimulatory diseases. This study reveals that MARCH1 plays an essential role in the development of TH2 cells by enabling LN-resident DCs to transmit TCR signaling optimal for TH2 cell differentiation involving turnover of MHCII and CD86.
RESULTS
MARCH1 is required for the development of TH2 cell immunity but not TH1, TH17, or cytotoxic CD8+ T cell immunity
We explored the role of MARCH1 in the development of type 2 T cell immunity by using a mouse model of allergic asthma driven by house dust mite (HDM) allergen (Fig. 1A). MARCH1-deficient mice failed to induce IL-4– or IL-13–producing TH2 cells in the lungs (Fig. 1B and fig. S1A). These mice also failed to elicit eosinophilia in the lungs (Fig. 1C and fig. S1B). Total and HDM-specific immunoglobulin E (IgE) antibody responses were also impaired (Fig. 1D). However, airway infiltration of neutrophils, DCs, or monocyte-derived macrophages occurred to a similar extent to wild-type (WT) mice (Fig. 1, E to G), indicating that immune activation was not entirely impaired in these mice. These mice were resistant to developing airway hyperresponsiveness (AHR) or severe airway goblet cell metaplasia (Fig. 1, H and I), denoting that MARCH1 contributes to the pathology associated with allergic asthma. To determine whether the observed defect is restricted to HDM or reflects a general type 2 immune development defect, we treated mice with two additional type 2 immune stimuli, the fungal cellular extract from Aspergillus fumigatus or the parasitic worm Nippostrongylus brasiliensis. MARCH1-deficient mice elicited poor TH2 cells, eosinophilia, or IgE antibodies against both stimuli (fig. S2, A to F). MARCH1 thus plays an essential role in the development of type 2 immunity independent of specific antigens.
Fig. 1. MARCH1 is required for the development of TH2 cell immunity and experimental asthma.
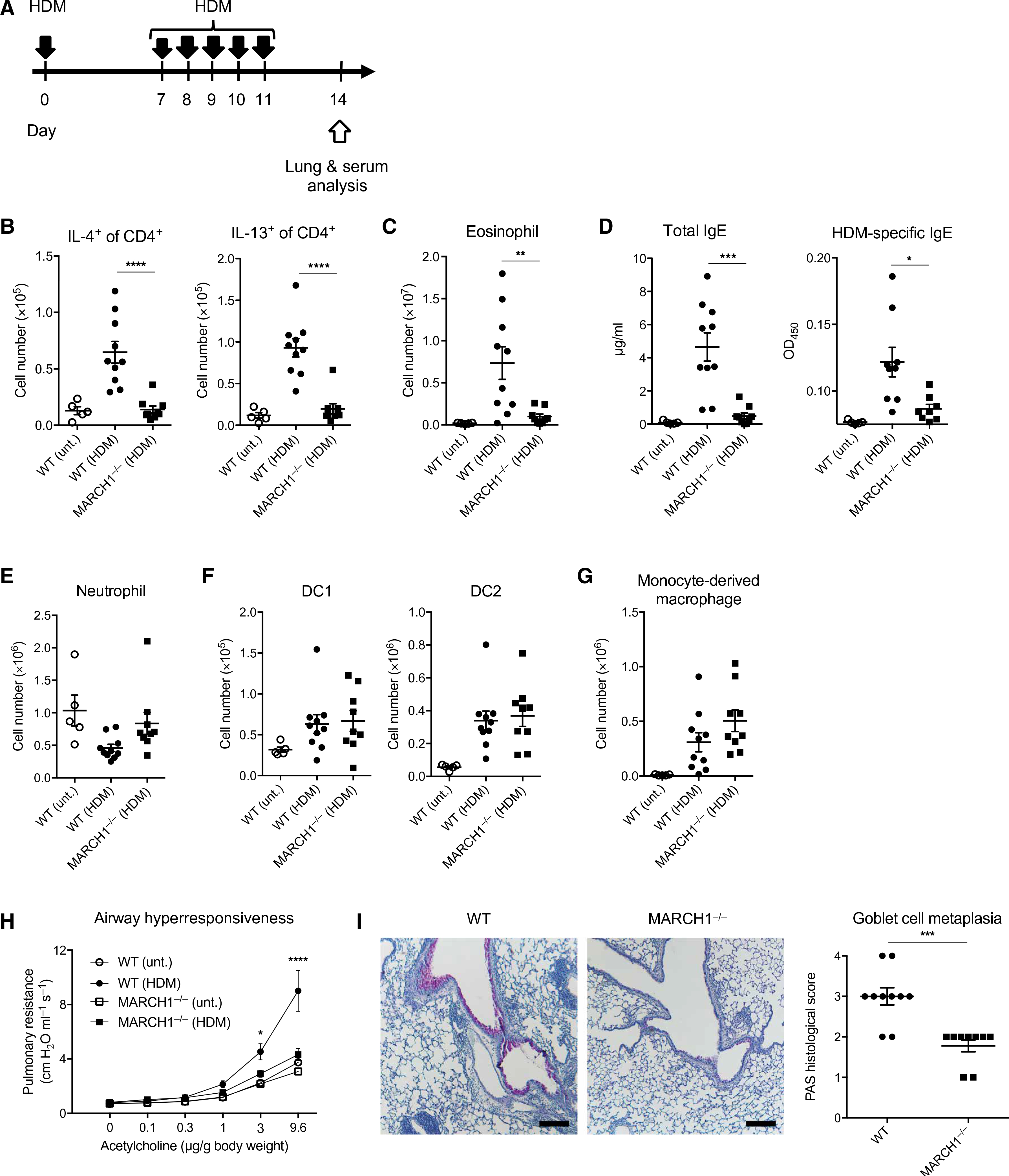
(A) Experimental outline of HDM extract challenge. Solid black arrows indicate oropharyngeal aspiration of 10 μg of HDM. (B) Total numbers of IL-4– or IL-13–competent CD4+ T cells in lungs of WT or MARCH1−/− mice untreated (unt.) or treated with HDM. (C) Total numbers of eosinophils in lungs. (D) Serological IgE titers and optical density (OD) values for HDM-specific IgE antibodies as determined by ELISA. (E to G) Total numbers of neutrophils (E), DC1 or DC2 (F), and monocyte-derived macrophages (G) in lungs. Data in (B) to (G) are pooled from two independent experiments with each experiment having four to five mice of each genotype administered with HDM and two to three untreated control mice. (H) Pulmonary resistance to acetylcholine. (I) Representative images of PAS-stained lung sections (magnification, ×5; scale bars, 200 μm) and scoring of the stain. Data in (H) and (I) are obtained from one experiment with 7 to 10 mice of each genotype for every condition. Data are shown as means ± SEM. Statistical significance was determined by one-way ANOVA with Tukey’s multiple comparisons test (B to D), two-way ANOVA with Tukey’s multiple comparisons test (H), or unpaired Student’s t test (I). *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
We next examined the role of MARCH1 in the development of type 1 immunity using a mouse model of viral infection induced by the influenza virus (X31). MARCH1-deficient mice elicited interferon-γ (IFN-γ)–producing TH1 cells at similar levels to WT mice (fig. S3A). MARCH1-deficient mice also elicited robust clonal expansion of viral nucleoprotein (NP)–specific CD8+ T cells and production of IFN-γ and granzyme B by these cells (fig. S3B). Influenza-reactive IgG antibodies were produced similarly to WT mice (fig. S3C), and the kinetics of viral clearance were also similar (fig. S3D). Thus, MARCH1 is not required for the development of type 1 immunity and host protection against the influenza virus. We further assessed the role of MARCH1 in the development of experimental autoimmune encephalomyelitis (EAE), a mouse model of multiple sclerosis driven by TH1 and TH17 cells. MARCH1-deficient mice expanded myelin-specific CD4+ T cells as robustly as WT mice (fig. S3E), and these T cells committed to IFN-γ, IL-17A, or granulocyte-macrophage colony-stimulating factor (GM-CSF) production (fig. S3F). MARCH1-deficient mice developed this neuroinflammatory disease as severely as WT mice as determined by daily assessments of the clinical score of EAE (fig. S3G). Together, MARCH1 is required for the development of TH2 cell immunity but not TH1, TH17, or cytotoxic CD8+ T cell immunity.
The role of MARCH1 in promoting TH2 cell immunity depends on its expression by DCs
To understand the cellular mechanism by which MARCH1 selectively supports TH2 cell immune development, we aimed to identify the specific cell type(s) that uses MARCH1 for this event. Among the antigen-presenting cells that express MARCH1, DCs are best implicated in their role in type 2 immunity by their abilities of priming TH2 cells (30). To examine the role of DC-expressed MARCH1, we generated mice lacking MARCH1 in cells expressing CD11c (MARCH1fl/fl CD11cCre). We found that MARCH1fl/fl CD11cCre mice recapitulated the absence of TH2 cells, eosinophils, IgE, and HDM-specific IgE antibodies observed in MARCH1-deficient mice (Fig. 2, A to C), supporting a role for DC-expressed MARCH1 in type 2 immune development. Because CD11c is not only expressed in DCs but also in some macrophages (31, 32), we generated mice in which MARCH1 is deleted in cells expressing the more selective marker for DCs, Zbtb46 (33). Unexpectedly, MARCH1fl/fl Zbtb46Cre mice displayed incomplete MARCH1 deletion with about 30% of DCs persistently expressing MARCH1 (fig. S4A). This incomplete deletion was also shown by the surface expression of MHCII, a substrate of MARCH1, which increased only in a fraction of DCs (fig. S4, B and C). Despite this partial deletion, we examined whether MARCH1fl/fl Zbtb46Cre mice would still exhibit appreciable defects in developing type 2 immune responses to HDM. We performed correlation analyses between MHCII surface levels in pulmonary DCs, an indicator of MARCH1 deletion efficiency, and the extent of HDM-induced inflammatory and IgE responses. We found that the numbers of TH2 cells, eosinophils, and IgE titers were each inversely correlated with the surface expression of MHCII on pulmonary DCs (Fig. 2, D to F), corroborating a crucial role for DC-expressed MARCH1 in the development of type 2 immunity.
Fig. 2. The role of MARCH1 in promoting TH2 cell immunity depends on its expression by DCs.

(A) Total numbers of IL-4– or IL-13–competent CD4+ T cells in lungs of MARCH1fl/fl or MARCH1fl/fl CD11cCre mice untreated or treated with HDM. (B) Total numbers of eosinophils in lungs. (C) Serological IgE titers and OD values for HDM-specific IgE antibodies as determined by ELISA. Data in (A) to (C) are pooled from two independent experiments with three to six mice administered with HDM and two to three untreated control mice. Data are shown as means ± SEM. Statistical significance was determined by one-way ANOVA with Tukey’s multiple comparisons test. **P < 0.01; ***P < 0.001; ****P < 0.0001. (D to F) Total numbers of IL-4– or IL-13–competent CD4+ T cells (D), eosinophils (E), and serological IgE titers and HDM-specific IgE antibodies (F) in MARCH1fl/fl or MARCH1fl/fl Zbtb46Cre mice treated with HDM. Data are shown in relation to the surface expression of MHCII [geometric mean fluorescence intensity (geoMFI)] in lung DC1 (gated as shown in fig. S1). Data in (D) to (F) are pooled from two independent experiments with each experiment having four to seven mice administered with HDM. Data in (D) to (F) are shown with statistical r2 values, P values, and trend lines determined from linear regression analysis.
MARCH1 promotes TH2 cell development through LN-resident DCs
We sought to identify the specific role that MARCH1 plays in DC activation of type 2 immunity. We examined whether MARCH1 would be necessary for DC instruction of TH2 cell polarization in the LN. We found that MARCH1fl/fl CD11cCre mice had significantly fewer IL-4+CD4+ T cells in the mediastinal LN (medLN) compared with control mice at day 7 after HDM administration (Fig. 3, A and B), suggesting DC MARCH1 involvement in TH2 cell polarization. We corroborated this finding by using mixed bone marrow (BM) chimeric mice reconstituted with MARCH1−/−:Zbtb46DTR BM, in which, after diphtheria toxin (DT) treatment, all remaining DCs lack MARCH1 (Fig. 3, C and D). These mice failed to prime as many IL-4+CD4+ T cells in the medLN as control mice (Fig. 3E). MARCH1 in macrophages was not required as MARCH1fl/fl MafBCre mice, in which MARCH1 was specifically ablated in macrophages mounted normal IL-4+CD4+ T cell development (fig. S5A). MARCH1 in plasmacytoid DCs (pDCs) was also dispensable because mixed BM chimeric mice reconstituted with MARCH1−/−:CLEC4CDTR BM and treated with DT, which would confine MARCH1 deficieny to pDCs, were fully competent at inducing IL-4+CD4+ T cells (fig. S5B). MARCH1 in B cells was also dispensable because mixed BM chimeric mice reconstituted with MARCH1−/−:μMT BM, in which all developing B cells lack MARCH1, were also fully competent at inducing IL-4+CD4+ T cells (fig. S5C).
Fig. 3. MARCH1 promotes TH2 cell development through LN-resident DCs.
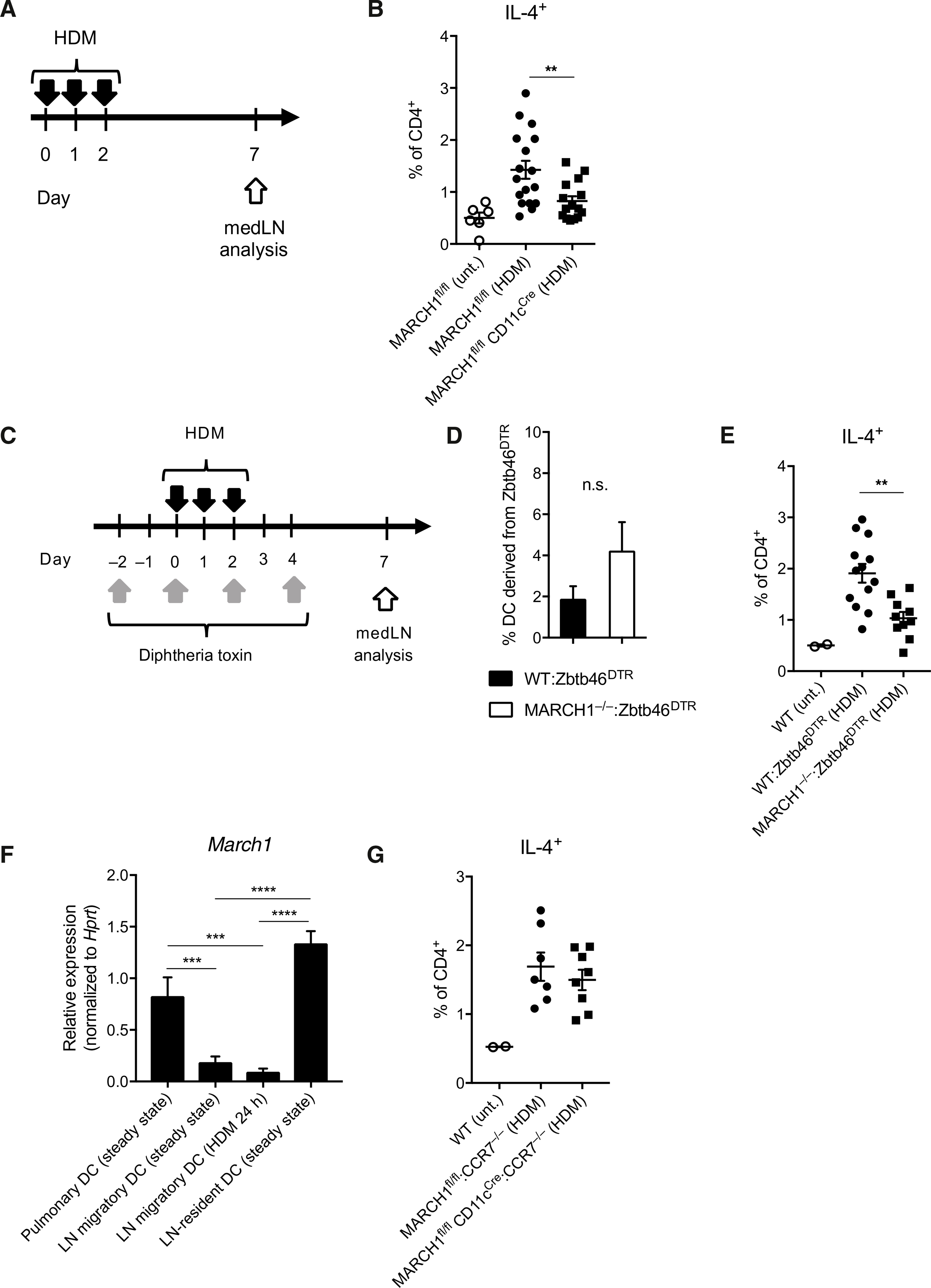
(A) Experimental outline of HDM administration. Solid black arrows indicate oropharyngeal aspiration of 10 μg of HDM. (B) Percentage of IL-4–competent CD4+ T cells in medLNs of MARCH1fl/fl and MARCH1fl/fl CD11cCre mice. Untreated mice were used as negative controls. Data are pooled from three independent experiments with each experiment having five to six mice per treated group. (C) Experimental outline of DT and HDM treatment for WT:Zbtb46DTR or MARCH1−/−:Zbtb46DTR mixed BM chimeric mice. (D) Percentage of DCs in the medLN derived from the Zbtb46DTR BM after DT treatment of WT:Zbtb46DTR or MARCH1−/−:Zbtb46DTR mixed BM chimeric mice. Not significant (n.s.), P > 0.1. (E) Percentage of IL-4–competent CD4+ T cells in medLNs of WT:Zbtb46DTR or MARCH1−/−:Zbtb46DTR mixed BM chimeric mice after DT and HDM treatment. Untreated mice were used as negative controls. Data are from one experiment with 10 to 12 mice per treated group. (F) March1 mRNA levels in FACS-purified pulmonary or medLN DCs normalized to the levels of Hprt housekeeping gene. Each bar represents at least three measurements. (G) Percentage of IL-4–competent CD4+ T cells in medLNs of MARCH1fl/fl:CCR7−/− or MARCH1fl/fl CD11cCre:CCR7−/− mixed BM chimeric mice. Untreated mice were used as negative controls. Data are from one experiment with seven to eight mice per treated group. Data are shown as means ± SEM. Statistical significance was determined by one-way ANOVA with Tukey’s multiple comparisons test (B and E to G) or unpaired Student’s t test. **P < 0.01; ***P < 0.001; ****P < 0.0001.
Pulmonary DCs that capture allergens and migrate to medLN have been implicated in driving TH2 cell polarization (34). We investigated whether MARCH1 is involved in homeostasis of pulmonary DCs, allergen-capturing activity of the DCs, or their migration to medLN. The number of DCs in the lungs or the efficiency of these DCs to capture HDM was not impaired by MARCH1 deficiency (fig. S6, A to C). The frequency of HDM-bearing DCs in the medLN of MARCH1-deficient mice was not significantly different from that of WT mice (fig. S6, D and E). We then investigated whether MARCH1 is involved in transcriptional reprograming of migratory DCs, which has been suggested to condition the DCs to drive TH2 cell development (12). Single-cell transcriptome analysis of HDM-bearing medLN DCs revealed that these DCs consist of two subsets marked by differential expression of genes highly expressed by DC1 but not DC2, such as IRF8, and vice versa, genes highly expressed in DC2 but not DC1, such as IRF4 (fig. S6, F and G) (35). Subsequent analysis indicated that the gene AY036118 was the only differentially expressed gene between WT and MARCH1-deficient DCs by more than twofold (fig. S6H). Together, pulmonary DC homeostasis, capture of allergens, migration to medLN, or transcriptional reprograming during migration does not require MARCH1.
DCs rapidly down-regulate the expression of MARCH1 after maturation (36). Because DC migration to LN is accompanied by maturation (2), we examined whether MARCH1 is perhaps down-regulated during pulmonary DCs’ migration to the medLN. Pulmonary DCs and medLN migratory DCs were isolated from mice at the steady state (fig. S7, A and B). HDM-bearing medLN migratory DCs were isolated from mice 24 hours after instillation with HDM (fig. S7C). medLN-resident DCs were also isolated for comparison (fig. S7D). The isolated DCs were analyzed for the expression of MARCH1 by quantitative reverse transcription polymerase chain reaction (PCR). We detected appreciable amount of MARCH1 in pulmonary DCs and LN-resident DCs but a very minor amount in migratory DCs both in the steady state and after HDM administration (Fig. 3F), suggesting that pulmonary DCs down-regulate MARCH1 during travel from the airway to the medLN. This finding raised a possibility that the effect of MARCH1 in promoting TH2 cell development may be exerted independent of migratory DCs. To test this, we generated chimeric mice reconstituted with MARCH1fl/fl CD11cCre:CCR7−/− BM, in which all migratory DCs are MARCH1-deficient. These mice were fully competent at eliciting IL-4+CD4+ T cells to HDM (Fig. 3G), indicating that the TH2-promoting effect of MARCH1 does not depend on migratory DCs; then, it must depend on nonmigratory LN-resident DCs.
MARCH1 expression in LN-resident DCs promotes up-regulation of GATA-3 and down-regulation of T-bet in developing TH2 cells
To explore the role of LN-resident DCs in TH2 cell priming, we blocked migration of DCs by treating mice with pertussis toxin (PTx) (37, 38) and subsequently monitored the activation and polarization of allergen-specific CD4+ T cells. PTx inhibited accumulation of HDM-bearing DCs in the medLN up to 48 hours (Fig. 4, A and B), validating its efficacy as a DC migration blocker. To monitor the activation of antigen-specific T cells, mice were transferred with CD4+ T cells isolated from ovalbumin (OVA)–specific TCR transgenic OT-II mice and subsequently challenged with HDM mixed with OVA. The activation of OT-II T cells in the medLN was examined by determining the expression of Nur77, CD44, and CD69, the early or later T cell activation markers. PTx treatment did not impair the extent of expression of these markers nor altered the kinetics of expression (Fig. 4C, left three panels), indicating that migratory DCs are not necessary to induce activation of antigen-specific naïve CD4+ T cells. Moreover, GATA-3 and T-bet, the transcription factors responsible for guiding TH2 or TH1 cell polarization, respectively, were also induced in mice treated with PTx at comparable levels to the mice not treated with PTx (Fig. 4C, right two panels). This finding suggests that migratory DCs are not necessary to initiate TH cell polarization.
Fig. 4. MARCH1 expression in LN-resident DCs promotes up-regulation of GATA-3 and down-regulation of T-bet in developing TH2 cells.
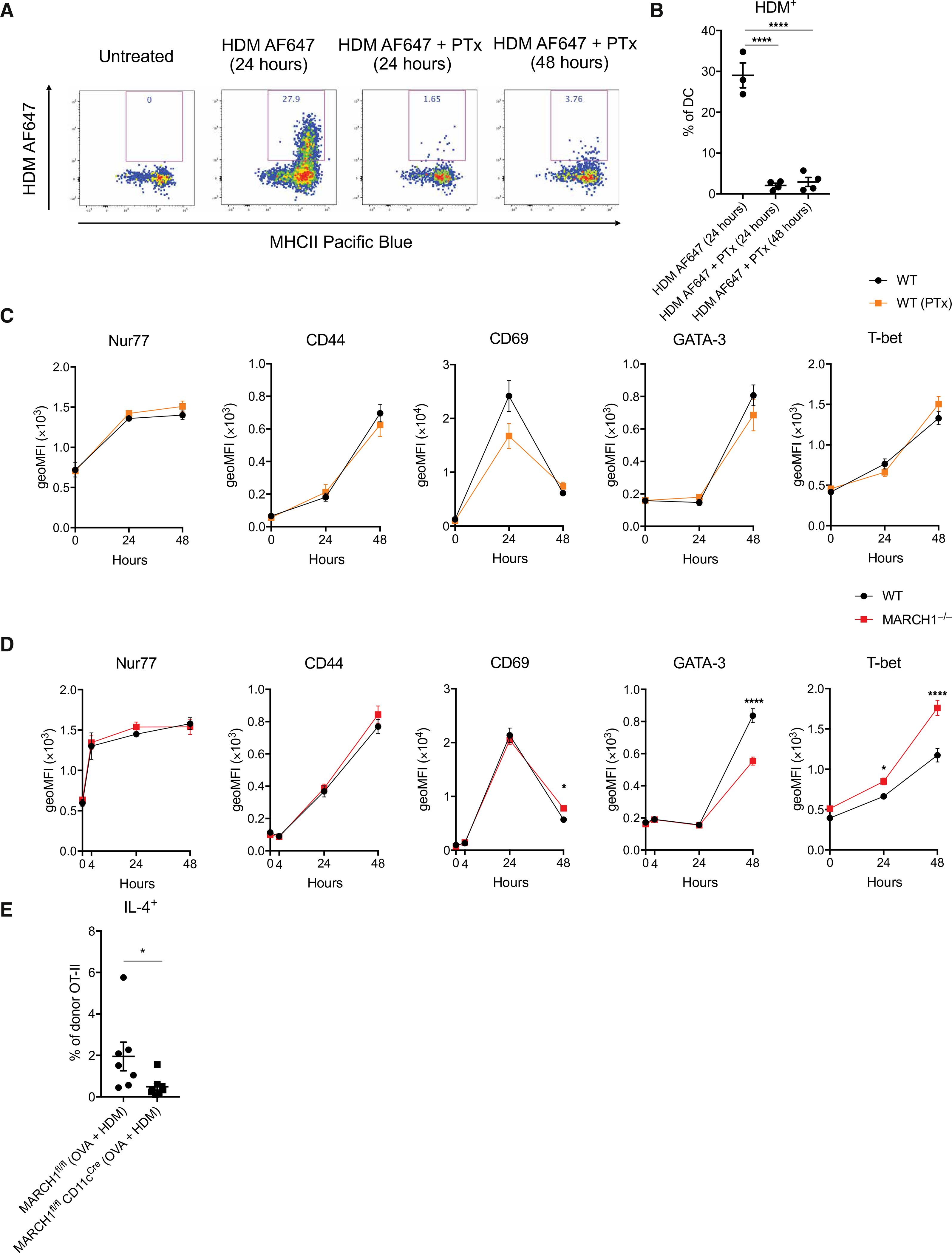
(A and B) WT mice were oropharyngeally administered with HDM conjugated to Alexa Fluor 647 (HDM AF647) alone or together with PTx. medLNs were isolated 24 or 48 hours later, and the frequency of HDM+ DCs was determined by flow cytometry. A WT mouse remained untreated for gating purposes. Representative flow cytometry plots (A) and percentages of medLN DCs bearing HDM (B) are shown. Data are from one experiment with three to four mice per group. (C) Expression kinetics of CD69, CD44, Nur77, GATA-3, or T-bet in adoptively transferred OT-II cells. Mice that had received OT-II CD4+ T cells were oropharyngeally administered with the mixture of HDM and OVA alone or together with PTx. medLNs were isolated, and the expression of designated molecules in OT-II CD4+ T cells was determined by geoMFI. Data are from two independent experiments with each experiment having two to five mice at each time point per group. (D) Expression kinetics of CD69, CD44, Nur77, GATA-3, or T-bet in adoptively transferred OT-II cells. WT or MARCH1−/− mice that had received OT-II CD4+ T cells were oropharyngeally administered with the mixture of HDM and OVA. medLNs were isolated, and the expression of designated molecules in OT-II CD4+ T cells was determined by geoMFI. Data are from three independent experiments with each experiment having three to five mice at each time point per group. (E) Percentages of IL-4–competent OT-II T cells in the medLNs. MARCH1fl/fl or MARCH1fl/fl CD11cCre mice that had received OT-II CD4+ T cells were oropharyngeally administered with the mixture of HDM and OVA. medLNs were isolated on day 7, and the frequency of IL-4–competent OT-II T cells was determined. Data are from one experiment with seven to eight recipient mice per group. Data are shown as means ± SEM. Statistical significance was determined by one-way ANOVA with Tukey’s multiple comparisons test (B), two-way ANOVA with Fisher’s LSD test (C and D), or unpaired Student’s t test (E). *P < 0.05; ****P < 0.0001.
Having found that early activation and polarization of TH cells do not depend on migratory DCs, we tested whether these events depend on MARCH1. MARCH1 deficiency resulted in a decrease in the expression of GATA-3 and an increase in the expression of T-bet. This alteration was accompanied by sustained expression of CD69, whereas no difference was observed in the expression of Nur77 or CD44 (Fig. 4D and fig. S8, A and B). A follow-up analysis on day 7 confirmed poor differentiation of these T cells to TH2 cell fate (Fig. 4E). These findings suggest that LN-resident DCs are sufficient to activate and initiate polarization of TH2 cells during allergic airway sensitization and that MARCH1 in the LN-resident DCs plays a key role by promoting GATA-3 and repressing T-bet in developing TH2 cells.
Ubiquitin-dependent turnover of MHCII and CD86 promotes up-regulation of GATA-3 and down-regulation of T-bet in developing TH2 cells
The sustained expression of CD69 in OT-II T cells in MARCH1-deficient mice suggested that these T cells might have received persistent TCR signaling compared with those in WT mice. Because MARCH1 mediates ubiquitin-dependent turnover of MHCII and CD86 from DC surface, deficiency of MARCH1 would result in excessive accumulation of MHCII and CD86 on the DCs, which, in turn, would result in persistent TCR signaling in antigen-specific CD4+ T cells. LN-resident DCs of MARCH1-deficient mice displayed MHCII and CD86 molecules 7- to 10-fold higher than those of WT mice (Fig. 5A and fig. S9). To determine whether this accumulation reflects delayed MHCII turnover, we used DCs cultured from BM of mice using Fms-related tyrosine kinase 3 ligand (FLT3L) (BMDCs), the in vitro counterpart of lymphoid organ–resident DCs (39), and compared the rate of MHCII degradation in DCs derived from WT and MARCH1-deficient BM. For this assay, we made retrovirus encoding MHCII β chain fused with a photoconvertible tag (I-Ab-mKikGR) (27) and transduced the BMDCs with this virus. Violet light exposure irreversibly photoconverted I-Ab-mKikGR on both WT and MARCH1-deficient DCs. At 16 hours after photoconversion, WT DCs had degraded most of the photoconverted I-Ab-mKikGR molecules, whereas MARCH1-deficient DCs retained substantial amounts of the photoconverted fusion protein (fig. S10, A and B). This finding confirms that MARCH1 deficiency significantly delays MHCII turnover in DCs.
Fig. 5. Ubiquitin-dependent turnover of MHCII and CD86 promotes up-regulation of GATA-3 and down-regulation of T-bet in developing TH2 cells.
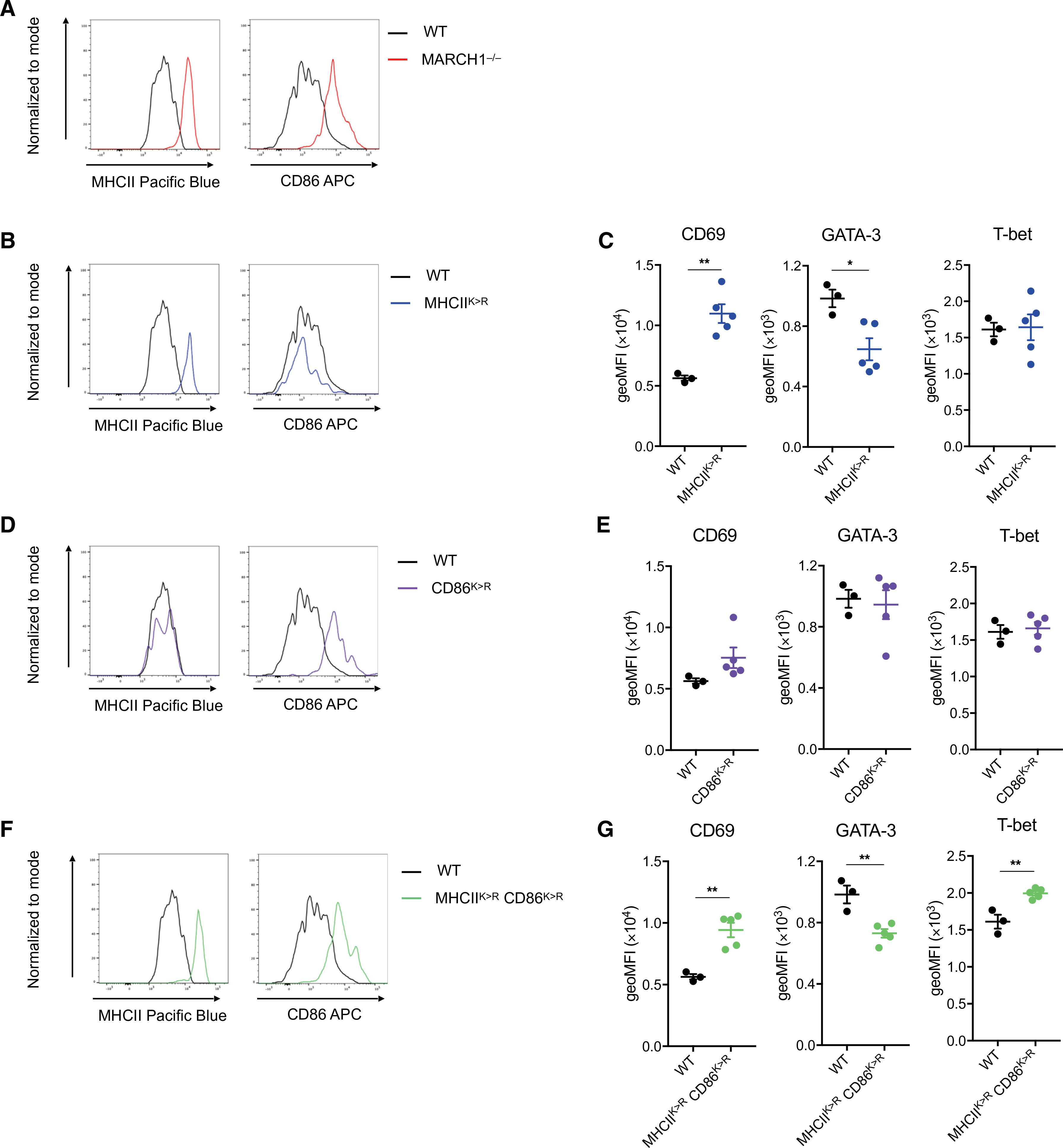
(A) Representative histograms showing MHCII and CD86 abundance on the surface of medLN-resident DCs in a MARCH1−/− mouse in comparison to a WT mouse. (B) Representative histograms showing MHCII and CD86 abundance on the surface of medLN-resident DCs in a MHCIIK>R mouse in comparison to a WT mouse. (C) Expression level of CD69, GATA-3, or T-bet in adoptively transferred OT-II cells in medLN of WT or MHCIIK>R mice at 48 hours after challenge with the mixture of HDM and OVA. (D) Representative histograms showing MHCII and CD86 abundance on the surface of medLN-resident DCs in a CD86K>R mouse in comparison to a WT mouse. (E) Expression level of CD69, GATA-3, or T-bet in adoptively transferred OT-II cells in medLN of WT or CD86K>R mice at 48 hours after challenge with the mixture of HDM and OVA. (F) Representative histograms showing MHCII and CD86 abundance on the surface of medLN-resident DCs in a MHCIIK>RCD86K>R mouse in comparison to a WT mouse. (G) Expression level of CD69, GATA-3, or T-bet in adoptively transferred OT-II cells in medLN of WT or MHCIIK>RCD86K>R mice at 48 hours after challenge with the mixture of HDM and OVA. Data in (A), (B), (D), and (F) are obtained from one experiment with one mouse per genotype. Data in (C), (E), and (G) are obtained from one experiment with three to five mice per group. Data are shown as means ± SEM. Statistical significance was determined by unpaired Student’s t test. *P < 0.05; **P < 0.01.
Having confirmed delayed MHCII turnover in MARCH1-deficient DCs, we tested whether this delay is the cause of sustained CD69, decreased GATA-3, and increased T-bet expression in OT-II T cells in the mice. To test this, we used MHCIIK>R knock-in mice, which have the ubiquitin-accepting lysine (K) of MHCII replaced by arginine (R). These mice exhibited markedly elevated surface MHCII on LN-resident DCs similar to that observed in MARCH1-deficient mice, whereas the level of CD86 was not altered (Fig. 5B and fig. S9). OT-II cells transferred to MHCIIK>R mice sustained CD69 and expressed less GATA-3 than those transferred to WT mice, although T-bet level did not differ (Fig. 5C). Next, we tested whether delayed turnover of CD86 may also alter the expression or regulation of CD69, GATA-3, or T-bet in T cells. CD86K>R mice, which have all ubiquitin-accepting lysines (K) of CD86 replaced by arginines (R), exhibited an increase in the level of CD86 in the LN-resident DCs similar to that observed in MARCH1-deficient mice, whereas that of MHCII was not altered (Fig. 5D and fig. S9). OT-II cells transferred to CD86K>R mice did not exhibit any significant difference in the expression of CD69, GATA-3, or T-bet from those transferred to WT mice (Fig. 5E). Last, we generated MHCIIK>RCD86K>R mice, in which MHCII and CD86 both fail to be ubiquitinated and thus both highly accumulate on the surface of LN-resident DCs (Fig. 5F and fig. S9). OT-II cells transferred to MHCIIK>RCD86K>R mice fully recapitulated the retention of CD69, reduced GATA-3, and enhanced T-bet (Fig. 5G) observed in MARCH1-deficient mice (Fig. 4D). This finding suggests that ubiquitin-mediated turnover of MHCII and CD86 contributes to regulating activation and polarization of developing TH2 cells in a cooperative manner.
Ubiquitination of MHCII and CD86 promotes TH2 cell immune development in a cooperative manner
We further tested the individual or cooperative effect of MHCII or CD86 ubiquitination in TH2 cell immune development and inflammation after HDM exposure. MHCIIK>R mice elicited IL-4+CD4+ T cells in the medLN normally (Fig. 6A) but exhibited a partial deficit in IL-4– or IL-13–producing TH2 cells in the lung (Fig. 6B). CD86K>R mice were competent at both of these responses (Fig. 6, C and D). MHCIIK>RCD86K>R mice exhibited significant deficits both in TH2 cell priming in the medLN and in TH2 cell inflammation in the lungs (Fig. 6, E and F). These findings demonstrate a cooperative role of MHCII and CD86 ubiquitination in driving TH2 cell immune development against respiratory allergens.
Fig. 6. Ubiquitination of MHCII and CD86 promotes TH2 cell immune development in a cooperative fashion.
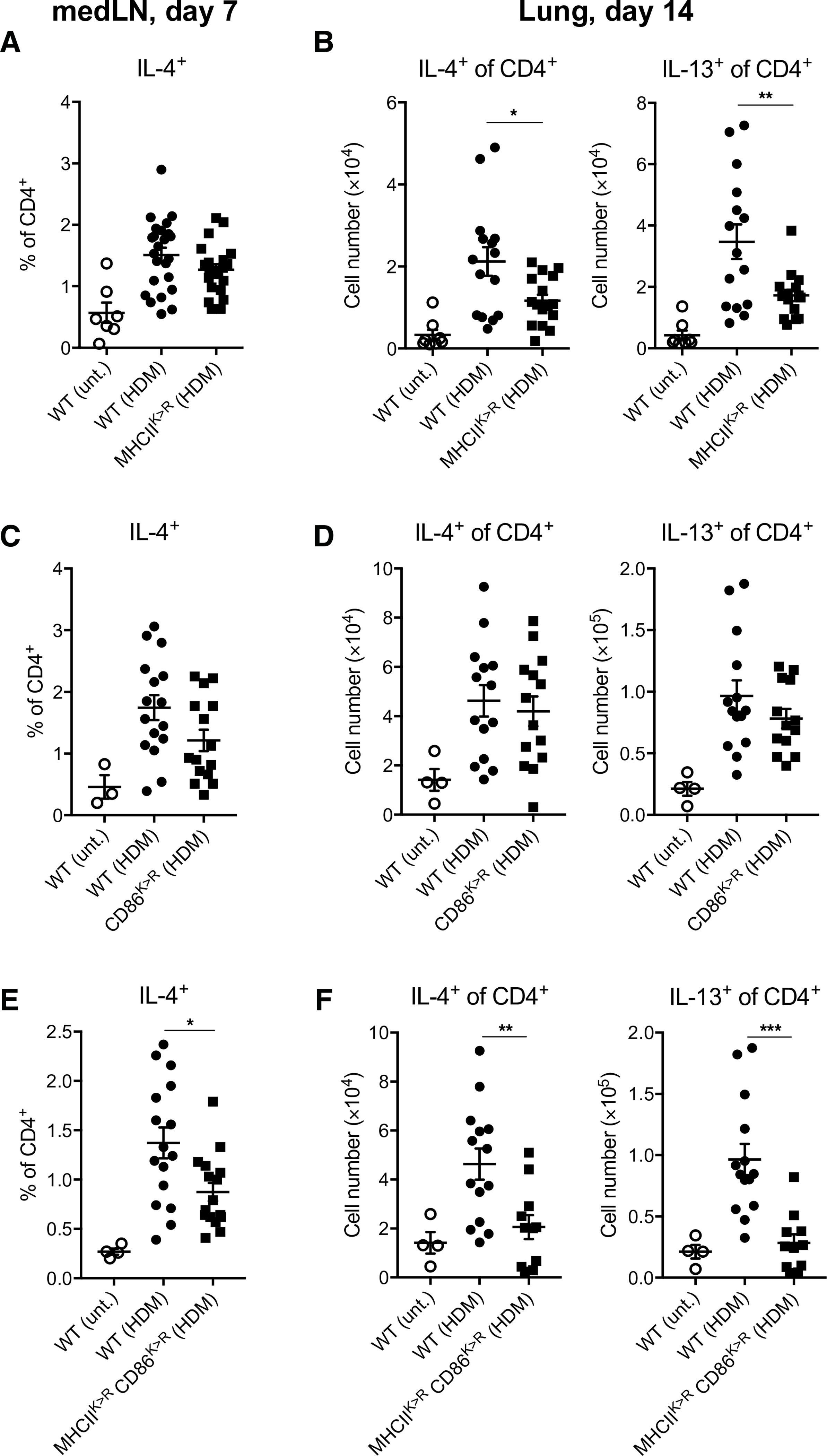
(A) Percentages of IL-4–competent CD4+ T cells in medLNs of WT or MHCIIK>R mice at day 7 after HDM challenge (experimental outline shown in Fig. 3A). Untreated mice were used as negative controls. Data are pooled from four independent experiments with each experiment having five to six mice per treated group. (B) Total numbers of IL-4– or IL-13–competent CD4+ T cells in lungs of WT or MHCIIK>R mice at day 14 after repetitive HDM challenge (experimental outline shown in Fig. 1A). Untreated mice were used as negative controls. Data are pooled from three independent experiments with each experiment having five to six mice per treated group. (C) Percentages of IL-4–competent CD4+ T cells in medLNs of WT or CD86K>R mice after HDM challenge. Untreated mice were used as negative controls. Data are pooled from two independent experiments with each experiment having seven to eight mice per treated group. (D) Total numbers of IL-4– or IL-13–competent CD4+ T cells in lungs of WT or CD86K>R mice at day 14 after repetitive HDM challenge. Untreated mice were used as negative controls. Data are pooled from two independent experiments with each experiment having six to eight mice per treated group. (E) Percentages of IL-4–competent CD4+ T cells in medLNs of WT or MHCIIK>RCD86K>R mice at day 7 after HDM challenge. Untreated mice were used as negative controls. Data are pooled from two independent experiments with each experiment having eight mice per treated group. (F) Total numbers of IL-4– or IL-13–competent CD4+ T cells in lungs of WT or MHCIIK>RCD86K>R mice at day 14 after repetitive HDM challenge. Untreated mice were used as negative controls. Data are pooled from two independent experiments with each experiment having four to seven mice per treated group. Data are shown as means ± SEM. Statistical significance was determined by one-way ANOVA with Tukey’s multiple comparisons test. *P < 0.05; **P < 0.01; ***P < 0.001.
Combined GWAS and eQTL analysis implicates low expression of MARCH1 associates with a low risk of asthma in humans
We tested for the association between genetic variants in MARCH1 and physician-diagnosed asthma status using asthma case and healthy control composed of 1353 Mexican American, 2118 Puerto Rican, and 1379 African American youths aged 8 to 21 (table S1). Association analyses were performed in each population separately and then combined in a trans-ethnic meta-analysis. The top 10 genetic associations with asthma are shown in table S2. Sensitivity analyses showed that downsampling to sample size without missingness in smoking status (~80% of the original sample size), rather than smoking status itself, had an impact on the meta-analysis results (data file S1), consistent with the relatively young study participants and hence low percentage of smokers (<10% overall). Three of the top 10 genetic variants (rs7666847, rs4691973, and rs2036903) were replicated at nominal significance level (P < 0.05) using published asthma genome-wide association studies (GWASs) from the Hartford–Puerto Rico study (table S3) (40) or Pan-UK Biobank (41). Flip-flop associations (nominally significant associations with different direction of effect) (42) were also observed from Pan-UK Biobank asthma GWAS (data file S2), indicating potential heterogeneous effect size due to different study design and/or older populations (between 40 and 69 of age) who were likely to experience different asthma subtypes. Three of the top 10 genetic variants, rs6815724, rs6815345, and rs2036903, were expression quantitative trait loci (eQTLs) in more than 29 tissues in the Genotype-Tissue Expression Project. Their alternative alleles were both associated with lower asthma risk and lower MARCH1 gene expression (fig. S11 and data file S3). Together, these findings support that asthma risk in humans is associated with genetic variants of MARCH1.
DISCUSSION
In the present study, we investigated the role of the MARCH1 ubiquitin ligase in the development of antigen-specific T cell immunity in vivo. We found that MARCH1 is not necessary for the development of TH1, TH17, or cytotoxic CD8+ T cell immunity but is required for TH2 cell immune development. This TH2-promoting effect depended on the expression of this molecule in LN-resident DCs. LN-resident DCs but not migratory DCs expressed MARCH1, and this molecule mediated active turnover of MHCII and CD86, enabling the DCs to provide allergen-specific T cells with modulated TCR signaling, which, in turn, resulted in a decrease in the expression of T-bet and an increase in the expression of GATA-3, leading to TH2 cell differentiation. These findings not only reveal a specific and essential role of MARCH1 in the development of TH2 cell immunity but also uncover the role of LN-resident DC in TH2 cell development together with an underlying mechanism.
MARCH1-deficient BMDCs have been shown to be incompetent at inducing production of IFN-γ and IL-17 from naïve CD4+ T cells (43) and poor at activating antigen-specific CD8+ T cells in vitro (44). More recently, splenic DCs derived from MHCIIK>R mice have been shown to poorly stimulate naïve CD4+ T cells in vitro and fail to expand adoptively transferred TCR transgenic T cells in vivo (45). Using mice immunized with OVA with alum and later challenged with OVA intranasally, Kishta et al. (46) identified that certain features of allergic asthma such as antigen-specific IgE antibody, IL-13–competent CD4+ T cells, and AHR developed poorly in MARCH1-deficient mice compared with WT mice, consistent with our findings. However, the biological relevance of these findings has not been firmly established. We used various mouse models of infection and immune-stimulatory diseases and showed that MARCH1-deficient mice were competent at eliciting IFN-γ–producing TH1 cells, IL-17–producing TH17 cells, or cytotoxic CD8+ T cells against virus or autoimmune self-antigens. However, these mice were resistant to developing TH2 cell immune responses to environmental allergens or parasitic worms. Moreover, MARCH1-deficient mice cleared influenza virus as effectively as WT mice and developed EAE as severely as WT mice but were resistant to developing allergic airway inflammation and experimental asthma. Consistent with the asthma-resistant property of MARCH1-deficient mice, our data suggest that the alleles of two genetic variants in human MARCH1 gene (rs6815724 and rs6815345), which were associated with lower MARCH1 gene expression, were also associated with lower asthma risk in the trans-ethnic meta-analysis. These findings reveal a selective role of MARCH1 in inducing type 2 T cell immunity. Although the mechanisms underlying this selective role remain to be defined, MARCH1 being down-regulated after DC maturation could be an attributing factor. DCs stop expressing MARCH1 upon Toll-like receptor (TLR) or other DC maturation–inducing signals (36, 47). Because TH1, TH17, and cytotoxic CD8+ T cell responses often develop after TLR-mediated activation and maturation of DCs (48, 49), MARCH1 would disappear from DCs during these immune responses, rendering its role irrelevant for those responses. In contrast, type 2 immune stimuli often lack DC maturation stimuli and even have an activity of suppressing it. For example, papain, a well-known TH2 immune stimulus, poorly induces DC maturation (50). The soluble egg antigen driven by Schistosoma mansoni parasite Omega-1 suppresses TLR signaling and inhibits DC maturation (51, 52). Because allergens are a complex mixture of many different antigens yet often contain endotoxin and other TLR-stimulating agents, there might be some mechanisms used directly by allergens or activated in the host to suppress TLR signaling in DCs.
The T cell priming function of DCs has been largely attributed to migratory DCs that capture antigens in the peripheral tissue, migrate to draining LN, and present the antigens to antigen-specific CD4+ T cells (2). However, other studies have shown that LN-resident DCs can directly capture antigens from lymphatics, present the antigens to antigen-specific CD4+ T cells, and induce adaptive T cell immunity (37, 53). Most of these studies were done after skin immunization, and no prior studies exist regarding the role of LN-resident DCs in immune response to respiratory antigens. We showed that allergens administered intratracheally can reach the medLN and be presented to allergen-specific CD4+ T cells as early as 4 hours after the administration. The T cells expressed TH cell–polarizing transcription factors such as GATA-3 and T-bet even when immigration of lung DCs to the LN was inhibited. Moreover, the expression of MARCH1 in DCs was crucial for TH2 cell development, but its expression in migratory DCs was not, suggesting that LN-resident DCs play a significant role in early instruction of TH2 cell development involving a MARCH1-dependent mechanism. Prevailing models for TH2 cell development rely on migration of allergen-bearing tissue DCs to the draining LN, and recent models have also suggested the role of various cell types in tissue in promoting DC migration in association with type 2 immune development (50, 54). Although, undoubtedly, many TH2 immune stimuli enhance DC migration, the specific role of these migratory DCs in TH2 cell development has not been clearly defined (55). Identifying the specific role played by LN-resident versus migratory DCs represents a future avenue of investigation for a superior understanding of the mechanism of TH2 cell development.
We found that the role of MARCH1 in supporting TH2 cell immune development depended on its activity of ubiquitinating MHCII and CD86 in DCs. MARCH1-deficient LN-resident DCs exhibited marked accumulation of MHCII and CD86 on the surface, and T cells activated by these DCs showed sustained expression of the TCR signaling reporter CD69, increased expression of T-bet, and repression of GATA-3. Because MARCH1-deficient DCs process and present antigens by the same kinetics as MARCH1-sufficient DCs (28), T cells that see either DCs would elicit the same strength of early TCR signaling, which seems to be reflected by the identical level of increase in the expression of Nur77 by these T cells. However, as time passes, the amounts of antigens staying on the surface of MARCH1-deficient and MARCH1-sufficient DCs would differ; more amounts of antigens would be present on MARCH1-deficient DCs than on MARCH1-sufficient DCs because of a defect in endocytosis and degradation of antigen-MHCII complexes in MARCH1-deficient DCs (36). This prolonged antigen presentation is likely to result in previously activated T cells receiving TCR signaling repeatedly. This persistent signaling appears to be reflected by the sustained expression of CD69 and also by the increased expression of T-bet and the decreased expression of GATA-3. Although the previous studies have suggested that stronger TCR signaling preferentially induces the expression of T-bet to GATA-3 in activated T cells (56, 57), our finding suggests that sustained TCR signaling may also exert the similar effect.
To confirm that the accumulation of MHCII and/or CD86 in DCs drove alteration in TCR signaling and TH cell polarization, we used mice in which ubiquitin-accepting sites of MHCII, CD86, or both were specifically mutated. T cells activated in mice where MHCII ubiquitination was ablated sustained the expression of CD69 and reduced expression of GATA-3, although the expression of T-bet was not altered. In contrast, T cells activated in mice where CD86 ubiquitination was ablated did not show any alteration in the expression of CD69, GATA-3, or T-bet, suggesting that the lack of CD86 ubiquitination alone is not sufficient to alter TH cell differentiation. However, in mice where MHCII and CD86 ubiquitination were both ablated, activated T cells expressed CD69 in a sustained manner, down-regulated the expression of GATA-3, and up-regulated that of T-bet. This finding suggests that MHCII ubiquitination plays a dominant role in modulating TCR signaling and controlling the expression of TH2-polarizing transcription factor, whereas CD86 ubiquitination plays a secondary role by contributing to the repression of TH1 polarization.
This study did not distinguish the contribution of MARCH1 expressed by DC1 or DC2 subsets to TH2 cell development. Preliminary results using mixed BM chimeric mice reconstituted with MARCH1−/−:XCR1DTR BM support a contribution of MARCH1 expressed by DC1s. Whether MARCH1 expressed by DC2s also contributes to TH2 cell development was not examined because the tools to address this question are not readily available. Given the facts that MARCH1 is expressed by both DC1 and DC2 lineages and that we observed that the defect in TH2 cell development in MARCH1−/−:XCR1DTR chimeras was not as severe as what was seen in MARCH1−/−:Zbtb46DTR chimeras, MARCH1 expressed by DC2 subset most likely also contributes to TH2 cell development, although this needs confirmation.
In conclusion, we reveal an unappreciated role of LN-resident DCs in TH2 cell immune development and demonstrate that MARCH1-dependent ubiquitination of MHCII and CD86 grants these DCs an ability to promote TH2 cell differentiation through modulated TCR signaling. On the basis of these findings, we propose that LN-resident DCs and MARCH1 could serve as novel therapeutic targets for the prevention or treatment of type 2 immune-stimulatory diseases.
MATERIALS AND METHODS
Study design
The study aimed to identify the role of MARCH1 in DC function in a T cell immune response. We used mouse models of allergic airway inflammation, pulmonary viral infection, systemic worm parasitic infection, and neuroinflammation to investigate the role of MARCH1 in TH2 cell immunity. We focused on the underlying molecular and cellular mechanisms in development of TH2 cells in the LN to the HDM allergen using genetically engineered mouse models and other experimental manipulations of mice in vivo. Sample sizes for each experiment are indicated in the figure legends.
Mice
WT C57BL/6J and B6.SJL-PtprcaPepcb/BoyJ (CD45.1+) mice were purchased from the Jackson Laboratory. MARCH1−/−, 2D2, MARCH1fl/fl, CD11cCre, Zbtb46Cre, OT-II, Zbtb46DTR, MafBCre, CLEC4CDTR, μMt, Zbtb46GFP, CCR7−/−, MHCIIK>R, and CD86K>R mice were previously described (28, 29, 33, 43, 58–67). MARCH1fl/fl Zbtb46Cre and MARCH1fl/fl MafBCre mice were generated by breeding MARCH1fl/fl mice with Zbtb46Cre or MafBCre mice, respectively. Our laboratory routinely genotypes the mice for adventitious germline deletion by checking the presence of intact loxp/loxp alleles in ear explants by PCR. We also examine the surface level of MHCII on blood B cells by flow cytometry because B cells are not supposed to be targeted by CD11cCre or MafBCre, but they express MARCH1 and thus increase MHCII surface level when MARCH1 is targeted. The mice that show surface MHCII expression on B cells increased compared with WT mice were excluded from experiments. CD45.1+ mice were mated to OT-II mice to generate CD45.1+ OT-II mice. MHCIIK>R CD86K>R mice were generated by breeding MHCIIK>R mice and CD86K>R mice. Mice were housed in a specific pathogen–free facility in the Laboratory Animal Research Center at the University of California, San Francisco (UCSF). Mice of both sexes were used at 5 to 12 weeks of age, except for EAE, AHR, and periodic acid–Schiff (PAS) staining experiments, in which only female mice were used. Sample sizes were guided by previous studies. Mouse genotypes were not blinded from the investigator, except for EAE, AHR, and PAS stain scoring. All experiments conformed to ethical principles and guidelines approved by the Institutional Animal Care and Use Committee at UCSF.
HDM-induced allergic asthma protocol
On days 0 and 7 to 11, naïve mice were anesthetized with isoflurane and given 10 μg of HDM extract (Dermatophagoides pteronyssinus, Stallergenes Greer, catalog no. NC0419530) by oropharyngeal aspiration. On day 14, mice were euthanized, and lungs and sera were collected for flow cytometry and enzyme-linked immunosorbent assays (ELISAs).
Aspergillus extract–induced allergic inflammation protocol
On days 0 and 7 to 10, naïve mice were anesthetized with isoflurane and given 10 μg of Aspergillus cellular extract (A. fumigatus, Stallergenes Greer, catalog no. NC1677927) by oropharyngeal aspiration. On day 13, mice were euthanized, and lungs and sera were collected for flow cytometry and ELISA.
N. brasiliensis infection
On day 0, naïve mice were infected subcutaneously with 500 N. brasiliensis (N.b.) stage 3 larvae (L3). On day 13, mice were euthanized, and lungs and sera were collected for flow cytometry and ELISA.
Influenza virus infection
On day 0, naïve mice were sedated with ketamine/xylazine and infected intranasally with 105 TCID50 (median tissue culture infectious dose) of the mouse attenuated H3N2 X31 influenza virus. On day 4, some mice were euthanized, and lungs were collected for qPCR. On day 10, the remaining mice were euthanized, and lungs and sera were collected for flow cytometry, ELISA, and qPCR.
Experimental autoimmune encephalomyelitis
On day 0, naïve mice were injected subcutaneously with 100 μg of myelin oligodendrocyte glycoprotein (MOG) peptide (35–55) (AnaSpec, catalog no. AS-60130–5) emulsified in complete Freund’s adjuvant (CFA) consisting of 100 μg of heat-killed Mycobacterium tuberculosis (HKMT) (InvivoGen, catalog no. tlrl-hkmt-1)–supplemented incomplete Freund’s adjuvant (Sigma-Aldrich, catalog no. F5506). On days 0 and 2, mice were also injected intraperitoneally with 200 ng of PTx (EMD Millipore, catalog no. 516560). Mice were monitored for clinical signs of disease beginning on day 5 until day 20 using the following scores: grade 0, no disease; grade 1, loss of tail tone; grade 2, abnormal gait and mild hindlimb paralysis; grade 3, hindlimb paralysis; grade 4, front limb paralysis; grade 5, moribund.
Preparation of lung and LN cells for flow cytometry
Lungs were digested with collagenase D (Roche, catalog no. 11088882001) and deoxyribonuclease I (Roche, catalog no. 10104159001) for 30 min at 37°C and dissociated in C tubes with a gentleMACS dissociator (Miltenyi Biotec). The digest was stopped with cold fluorescence-activated cell sorting (FACS) buffer [0.5% bovine serum albumin (BSA) in phosphate-buffered saline (PBS)] and centrifuged at 500g for 5 min at 4°C. Cell pellets were resuspended in red blood cell lysis buffer for 5 min at room temperature (RT) and washed with FACS buffer. Cell suspensions were filtered with 70-μm nylon mesh strainers. Cells were centrifuged and resuspended in 1 ml of FACS buffer and counted with a Z2 particle counter (Beckman Coulter). LNs were dispersed into cell suspensions with double-frosted microscope slides in RPMI and filtered in 5-ml cell strainer tubes. Cells were centrifuged at 500g for 5 min at 4°C, and cells were resuspended in FACS buffer. Lung and LN cells were prepared for surface or intracellular cytokine staining.
Surface staining of lung cells
Fc receptors of lung cells were blocked with anti-mouse CD16/32 (TruStain fcX, clone 93, catalog no. 101320, RRID: AB_1574975) antibody for 10 min on ice. For allergic inflammation or worm infection experiments, lung cells were stained with anti-mouse Pacific Blue I-A/I-E MHCII (M5/114.15.2, catalog no. 107620, RRID: AB_493527), anti-mouse BV605 CD4 (GK1.5, catalog no. 100451, RRID: AB_2564591 or RM4–5, catalog no. 116027, RRID: AB_2800581), anti-mouse BV650 Ly6G (1A8, catalog no. 127641, RRID: AB_2565881), anti-mouse fluorescein isothiocyanate (FITC) CD103 (2E7, catalog no. 121420, RRID: AB_10714791), anti-mouse phycoerythrin (PE) CD86 (GL-1, BD Pharmingen, catalog no. 553692), anti-mouse PerCP/Cy5.5 CD11b (M1/70, catalog no. 101228, RRID: AB_893232), anti-mouse PE/Cy7 B220 (RA3–6B2, catalog no. 103222, RRID: AB_313005), anti-mouse allophycocyanin (APC) CD64 (X54–5/7.1, catalog no. 139306, RRID: AB_11219391), anti-mouse Alexa Fluor 700 CD11c (N418, catalog no. 117320, RRID: AB_528736), and anti-mouse APC/Cy7 Siglec-F (E50–2440; BD Pharmingen, catalog no. 565527) and with propidium iodide (BioLegend, catalog no. 421301 or Invitrogen, catalog no. P3566) as a live/dead stain for 30 min on ice. For influenza virus infection experiments, lung cells were stained with an APC tetramer with the immunodominant NP-ASNENMETM epitope (MBL Life Science, catalog no. TS-M508–2) for 30 min on ice and subsequently stained with anti-mouse Pacific Blue I-A/I-E MHCII, anti-mouse BV605 CD4, anti-mouse BV650 Ly6G, anti-mouse FITC CD8a (53–6.7, catalog no. 100706, RRID: AB_312745), anti-mouse PE CD86, anti-mouse PerCP/Cy5.5 CD11b, anti-mouse PE/Cy7 CD44 (IM7, catalog no. 103030, RRID: AB_830787), anti-mouse Alexa Fluor 700 CD11c, and anti-mouse APC/Cy7 Siglec-F and with propidium iodide as a live/dead stain for 30 min on ice. Lung cells were washed and resuspended in FACS buffer for flow cytometry analysis on an LSRII machine. Antibodies were purchased from BioLegend unless stated otherwise.
Intracellular cytokine staining
Lung or LN cells were stimulated ex vivo with stimulation cocktail of phorbol 12-myristate 13-acetate (10 ng/ml; Sigma-Aldrich, catalog no. P1585) and ionomycin (1 μg/ml; Sigma-Aldrich, catalog no. I3909) in the presence of protein transport blocker brefeldin A (10 μg/ml; Sigma-Aldrich, catalog no. B5936) for 3 hours in a 37°C CO2 incubator. After ex vivo stimulation, lung or LN cells were blocked with anti-mouse CD16/32 antibody for 10 min on ice and then stained with anti-mouse BV605 CD4 and anti-mouse Alexa Fluor 700 CD8α (53–6.7, catalog no. 100730, RRID: AB_493703) and with eFluor780 Fixable Viability Dye (eBioscience, catalog no. 65–0865-14) as a live/dead stain for 30 min on ice. Lung or LN cells were fixed with IC fixation buffer (Invitrogen, catalog no. FB001) and permeabilized with permeabilization buffer (Invitrogen, catalog no. 00–8333-56). For allergic inflammation or worm infection experiments, lung cells were stained intracellularly with anti-mouse eFluor450 or FITC IFN-γ (XMG1.2, eBioscience, catalog no. 48–7311-82, RRID: AB_1834367 or catalog no. 11–7311-82, RRID: AB_465412), anti-mouse FITC IL-5 (TRFK5, Leinco Technologies), anti-mouse PE IL-13 (eBio13A, eBioscience, catalog no. 12–7133-82, RRID: AB_763559), anti-mouse PE/Cy7 IL-17A (TC11–18H10.1, catalog no. 506922, RRID: AB_2125010), and anti-mouse APC IL-4 (11B11, catalog no. 504106, RRID: AB_315320) for 30 min on ice. medLN cells were stained intracellularly with anti-mouse eFluor450 IFN-γ, anti-mouse PE/Cy7 IL-13 (eBio13A, eBioscience, catalog no. 25–7133-82, RRID: AB_2573530), anti-mouse PerCP/Cy5.5 IL-17A (TC11–18H10.1, catalog no. 506920, RRID: AB_961384), and anti-mouse APC IL-4 for 30 min on ice. For influenza virus infection experiments, lung cells were also surface stained with an APC tetramer with the immunodominant NP-ASNENMETM epitope for 30 min on ice and stained intracellularly with anti-mouse eFluor450 IFN-γ and anti-human/mouse PE granzyme B (GB12, Thermo Fisher Scientific, catalog no. MHGB04, RRID: AB_10372671). Lung or LN cells were washed and resuspended in FACS buffer for flow cytometry analysis on an LSRII machine. Antibodies were purchased from BioLegend unless stated otherwise.
Enzyme-linked immunosorbent assay
Nunc or Immulon 96-well plates were coated overnight at 4°C with anti-mouse IgE (R35–72, BD Pharmingen, catalog no. 553413) antibody. Plates were washed with PBST (0.05% Tween in PBS) and blocked with 1% BSA for 1 hour. Serum samples were incubated at 1:25 dilution along with a mouse IgE standard (C48–2, BD Pharmingen, catalog no. 557080) to determine total IgE or at 1:4 dilution to determine HDM-specific IgE at RT. Plates were washed with PBST and incubated with biotin-conjugated anti-mouse IgE (R35–118, BD Pharmingen, catalog no. 553419) for total IgE or biotin-conjugated HDM (prepared in-house with the EZ-Link Sulfo-NHS-Biotinylation Kit, Thermo Fisher Scientific, catalog no. 21425), for HDM-specific IgE. Plates were washed with PBST and incubated with horseradish peroxidase (HRP) streptavidin (BioLegend, catalog no. 405210) for 30 min at RT. Plates were washed with PBST and developed with trimethylboron substrate (eBioscience, catalog no. 00–4201-56). The reaction was stopped with 2 N H2SO4, and plates were read at 450 nm with a plate reader. For influenza virus–specific ELISA, plates were coated overnight with heat-inactivated X31 virus, serum samples were incubated at 1:1000, and HRP-conjugated anti-mouse IgG light chain (Jackson ImmunoResearch, catalog no. 115–035-174, RRID: AB_2338512) was used for detection.
AHR and PAS staining
Mice were sedated with ketamine/xylazine, attached to a rodent ventilator and pulmonary mechanics analyzer (FlexiVent; SCIREQ Inc.), and ventilated at a tidal volume of 9 ml/kg, a frequency of 150 breaths/min, and 2 cm H2O positive end-expiratory pressure. Mice were paralyzed with pancuronium (0.1 mg/kg, ip). A 27-gauge needle was placed in the tail vein, and measurements of airway mechanics were made continuously using the forced oscillation technique. Mice were given increasing doses of acetylcholine (0.1, 0.3, 1, 3, and 9.6 μg/g of body weight) administered through the tail vein to generate a concentration response. HDM-induced AHR was measured. Lungs were then isolated, and paraffin-embedded lung sections were stained with PAS reagent and scored on the basis of the percentage of PAS-positive cells among airway epithelial cells using the following scores: grade 0, none; grade 1, <25% of airway epithelial cells; grade 2, 25 to 50%; grade 3, 51 to 75%; and grade 4, >75%. Mice and samples were deidentified for blinding to the researcher performing these experiments.
qPCR for assessing viral burden
Lung lobes from influenza-infected mice were snap-frozen with liquid nitrogen, pulverized into powder, and dissolved in TRIzol LS reagent (Invitrogen, catalog no. 10296010). The RNA was purified and reverse-transcribed on the basis of equal amounts of RNA per sample into complementary DNA (cDNA) with oligo(dT) primers using the SuperScriptIV First-Strand Synthesis System (Invitrogen, catalog no. 18091050). The cDNA was used as template for qPCR reactions in triplicates using SYBR Green reagents (Bio-Rad Laboratories, catalog no. 1725120) with primers to the influenza gene PA encoding the polymerase acidic (PA) protein (PA sense: cggtccaaattcctgctgat; PA antisense: cattgggttccttccataca) and normalized to qPCR reactions with primers to endogenous Hprt. qPCR was performed on a Bio-Rad CFX Connect System, and relative viral titers were normalized to the sample from the least virally burdened mouse.
qPCR for assessing Cre-mediated MARCH1 deletion
Splenic DCs from MARCH1fl/fl, MARCH1fl/fl Zbtb46Cre, and MARCH1fl/fl CD11cCre mice were enriched using CD11c MicroBeads (Miltenyi Biotec, catalog no. 130–125-835) and sorted with a FACSAria from single-cell suspensions stained with anti-mouse Alexa Fluor 488 I-Ab MHCII (AF6–120.1, BD Pharmingen, catalog no. 553551), anti-mouse Alexa Fluor 647 CD11c (N418, BioLegend, catalog no. 117312, RRID: AB_389328), and anti-mouse PE/Cy7 B220. Genomic DNA from the sorted splenic DCs was purified using the PureLink Genomic DNA Mini Kit (Invitrogen, catalog no. K182001). The genomic DNA was used as template for qPCR reactions in triplicates using SYBR Green reagents with primers spanning the 3′ loxp site (MARCH1 sense: agctgtaagaactgacctcaa; MARCH1 antisense: ggaggatgcttgtcctgtaaa) and normalized to qPCR reactions to control noncoding DNA site. qPCR was performed on a Bio-Rad CFX Connect System, and MARCH1 expression was normalized to the MARCH1fl/fl samples.
qPCR for assessing MARCH1 expression
Pulmonary, LN migratory, or LN-resident DCs from naïve or Alexa Fluor 647–labeled HDM-treated WT or Zbtb46GFP mice (24 hours after oropharyngeal aspiration) were enriched from cell suspensions by depleting B220-, TCR-, and Ly6G-expressing cells through labeling with biotin-conjugated antibodies (RA3–6B2, BioLegend, catalog no. 103204, RRID: AB_312989; H57–597; catalog no. 13–5961-82, RRID: AB_466819; 1A8, catalog no. 127604, RRID: AB_1186108) and magnetic negative selection using EasySep RapidSpheres (STEMCELL Technologies, catalog no. 19860) and sorted with a FACSAria from the single-cell suspension stained with anti-mouse Pacific Blue I-A/I-E MHCII, anti-mouse PE/Cy7 CD11c (N418, BioLegend, catalog no. 117318, RRID: AB_493568), and anti-mouse PE Siglec-F (E50–2440, BD Pharmingen, catalog no. 562068, RRID: AB_394341) directly into TRIzol LS reagent. The RNA was purified and reverse-transcribed on the basis of equal amounts of RNA per sample into cDNA with oligo(dT) primers using the SuperScriptIV First-Strand Synthesis System. The cDNA was used as template for qPCR reactions in triplicates using SYBR Green reagents with primers to March1 isoform 2 (March1 sense: gaccagcagccacatttgctgtaa; March1 antisense: gctgcttgcctggagaaacaagtta) and normalized to qPCR reactions with primers to Hprt. qPCR was performed on a Bio-Rad CFX Connect System, and relative expression of March1 was calculated using the 2−ΔΔCq method.
HDM capture assay
HDM extract was labeled with the Alexa Fluor 488 Protein Labeling Kit (Invitrogen, catalog no. A10235) or with Alexa Fluor 647 NHS Ester (Invitrogen, catalog no. A37573), and 10 μg of Alexa Fluor 488–labeled or Alexa Fluor 647–labeled HDM was administered to mice by oropharyngeal aspiration. Mice were euthanized 24 or 48 hours after, and their lungs or medLNs were prepared for flow cytometric analysis. Control mice were untreated or given unlabeled HDM as negative controls.
2D2 T cell polarization
T cells were isolated using the EasySep Mouse Naïve CD4+ T cell Isolation Kit (STEMCELL Technologies, catalog no. 19765) from single-cell suspensions of spleens derived from CD45.1+ 2D2 donor mice. To determine IFN-γ+, IL-17A+, or GM-CSF+ OT-II cells, mice were transferred with CD45.1+ OT-II cells via retro-orbital injection. One day later, recipient mice were injected subcutaneously with 100 μg of MOG peptide (35–55) emulsified in 100 μg of HKMT-supplemented CFA. On days 0 and 2, mice were also injected intraperitoneally with 200 ng of PTx. At day 6 after challenge, recipient mice were euthanized, and draining inguinal LNs were collected and stimulated ex vivo for intracellular cytokine staining (as mentioned previously). Briefly, the LN cells were surface stained with anti-mouse BV605 CD4, anti-mouse PE Vα3.2 (RR3–16, BioLegend, catalog no. 135406, RRID: AB_1937255), and anti-mouse Alexa Fluor 700 CD45.1 (A20, BioLegend, catalog no. 110724, RRID: AB_493733) and stained intracellularly with anti-mouse eFluor450 IFN-γ, anti-mouse PE/Cy7 IL-17A, and anti-mouse APC GM-CSF (MP122E9, BioLegend, catalog no. 505414, RRID: AB_2721461), and 2D2 polarization was determined by flow cytometry.
OT-II T cell polarization
T cells were isolated using the EasySep Mouse CD4+ Naïve CD4+ T cell Isolation Kit from single-cell suspensions of spleens derived from CD45.1+ OT-II donor mice. To determine IL-4+ OT-II cells, mice were transferred with CD45.1+ OT-II cells via retro-orbital injection. One day later, recipient mice were challenged with 50 μg of OVA (Worthington, catalog no. LS003056) mixed with 10 μg of HDM by oropharyngeal aspiration. On day 7 after challenge, recipient mice were euthanized, and medLNs were collected and stimulated ex vivo for intracellular cytokine staining (as mentioned previously). Briefly, the medLN cells were surface stained with anti-mouse BV605 CD4, anti-mouse PerCP/Cy5.5 Vβ5 (MR9–4, BioLegend, catalog no. 139510, RRID: AB_2566807), and anti-mouse Alexa Fluor 700 CD45.1 and stained intracellularly with anti-mouse APC IL-4, and OT-II polarization was determined by flow cytometry. To determine TCR activation and polarizing transcription factors, at 4, 24, or 48 hours after challenge, recipient mice were euthanized, and medLNs were collected and surface stained with anti-mouse eFluor450 CD44 (IM7, eBioscience, catalog no. 48–0441-82, RRID: AB_1272246), anti-mouse BV605 CD4, anti-mouse FITC CD69 (H1.2F3, BioLegend, catalog no. 104506, RRID: AB_313109), anti-mouse PerCP/Cy5.5 Vβ5, and anti-mouse Alexa Fluor 700 CD45.1 and then prepared with the Foxp3 Transcription Factor Staining Buffer Kit (eBioscience, catalog no. 00–5523-00) for intracellular staining with anti-mouse PE GATA-3 (TWAJ, eBioscience, catalog no. 12–9966-42, RRID: AB_1963600), anti-mouse PE/Cy7 Nur77 (12.14, eBioscience, catalog no. 25–5965-82, RRID: AB_2811785), and anti-mouse eFluor660 T-bet (4B10, eBioscience, catalog no. 50–5825-82, RRID: AB_10596655), and OT-II activation and polarization were determined by flow cytometry.
BM chimeras and DT treatment
Mice were lethally irradiated (2 × 5.5 grays), and ~2 × 106 to 6 × 106 BM donor cells were injected through retro-orbital injection. Chimeras were reconstituted for at least 6 weeks before experimenting and were assessed for effective chimerism. Mice without effective chimerism were excluded from experiments. If chimeric mice received BM from Zbtb46DTR or CLEC4CDTR mice, then the mice were treated with DT (20 ng/g; Sigma-Aldrich, catalog no. D0564) on days −2, 0, 2, and 4, and medLNs were collected on day 7.
PTx-induced DC migration blockade
Mice were administered with 1 μg of PTx mixed with 50 μg of OVA and 10 μg of Alexa Fluor 647–labeled or unlabeled HDM through oropharyngeal aspiration.
MHCII turnover assay
BMDCs were generated from the BM of WT CD45.1+ or MARCH1−/− CD45.2+ mice and cultured in 1:1 ratio with FLT3L (100 ng/ml; PeproTech, catalog no. 250–31L) in 24-well plates. At day 2 or 3 of culture, the cells were spin-infected with a retrovirus with MSCV-I-Thy1.1 plasmid encoding an I-Ab-mKikGR fusion protein. At day 9 or 10 of culture, BMDCs were exposed to 415-nm violet light source set to a maximum intensity from a high–numerical aperture polymer optical fiber (1.5 mm core diameter) light guide and fiber collimator (Silver LED 415; Prizmatix). The cells were stained with anti-mouse Pacific Blue I-A/I-E MHCII, anti-mouse PerCP/Cy5.5 Thy1.1 (OX-7, BioLegend, catalog no. 202516, RRID: AB_961437), and anti-mouse Alexa Fluor 647 CD45.2 (104, BioLegend, catalog no. 109818, RRID: AB_492870), and the cells were analyzed by flow cytometry using an LSRII machine. In the retrovirally transduced BMDCs, mKikGRgreen fluorescence was detected using a blue laser, whereas mKikGRred fluorescence was detected using yellow-green laser, and MHCII turnover was calculated as the percentage of mKikGRred fluorescence left at 16 hours after photoconversion compared with mKikGRred fluorescence at 0 hours after photoconversion.
Study cohorts for genetic association with asthma
We examined 1353 Mexican American, 2118 Puerto Rican, and 1379 African American youths between 8 and 21 years of age with physician-diagnosed asthma from the Study of Genes-Environments and Admixture in Latino Americans (GALA II) and the Study of African Americans, Asthma, Genes and Environments (SAGE). A full description about the study design and recruitment has been previously described elsewhere (68–70). Briefly, participants were eligible for GALA II (SAGE) if they were 8 to 21 years of age and self-identified as African American (Latino) and had four African American (Latino) grandparents. Study exclusion criteria included the following: (i) any smoking within 1 year of the recruitment date, (ii) 10 or more pack-years of smoking, (iii) pregnancy in the third trimester, and (iv) history of lung diseases other than asthma (for cases) or chronic illness (for cases and controls). Local institutional review boards approved the studies (10–00889 for GALA II and IRB# 10–02877 for SAGE). All subjects and their legal guardians provided written informed consent.
Genetic association with asthma
Whole-genome sequencing was performed in batches using whole-blood samples from GALA II and SAGE participants under the Trans-Omics for Precision Medicine (TOPMed) program and the Centers for Common Disease Genomics of the Genome Sequencing Program as previously described (71–73) (see Supplementary Methods for further details).
Single-cell RNA sequencing
HDM+ WT DCs or HDM+ MARCH1−/− DCs were enriched from the medLNs of CD45.1/.1 Zbtb46GFP:CD45.2/.2 Zbtb46GFP MARCH1−/− mixed BM chimeric mice (24 hours after oropharyngeal aspiration), and the DCs were sorted using a FACSAria, as mentioned previously. Immediately after sorting, cells were run on the 10X Chromium (10X Genomics) and then through library preparation by the Institute for Human Genetics at UCSF following the recommended protocol for the Chromium Single Cell 3′ Reagent Kit (v3 Chemistry). Libraries were run on the NovaSeq 6000 for Illumina sequencing. Postprocessing and quality control were performed by the Genomics Core Facility at the Institute for Human Genetics at UCSF using the 10X Cell Ranger package (Cell Ranger version 3.0.2, 10X Genomics). Primary assessment with this software for WT DC sample reported 2441 cell barcodes with 7651 median unique molecular identifiers (transcripts) per cell and 2004 median genes per cell sequenced to 61.4% sequencing saturation with 48,559 mean reads per cell. Primary assessment with this software for MARCH1−/− DC sample reported 681 cell barcodes with 6309 median unique transcripts per cell and 1774 median genes per cell sequenced to 73.1% sequencing saturation with 202,410 mean reads per cell.
RNA sequence data analysis
Using Seurat R package v3 (Satija Lab), we filtered cells to only keep those that had less than 7% mitochondrial genes and cells with numbers of features greater than 1800 and numbers of counts greater than 7500. For clustering, we determined the principal components (principal components analysis), then constructed a shared nearest neighbor graph, identified clusters with a resolution of 0.4, and lastly visualized the cells using uniform manifold approximate and projection (UMAP), per the typical Seurat workflow. To identify cluster-specific markers after the creation of UMAP plots, we used normalized RNA counts of all clusters, scaled the data, and performed differential gene expression (DE) testing by applying the Wilcoxon rank sum test using Seurat’s FindMarkers function. To compare cell clusters of WT with MARCH1−/−, we once again used normalized/scaled RNA counts and performed DE testing with FindMarkers.
Quantification and statistical analysis
Flow cytometry data were analyzed by FlowJo v5 or v10 software (TreeStar). Graphs were drawn using Prism v7 software (GraphPad). Data are presented as means ± SEM. One-way analysis of variance (ANOVA) with Tukey’s multiple comparisons tests, two-way ANOVA with Tukey’s multiple comparisons tests, two-way ANOVA with Fisher’s least significant difference (LSD) tests, or unpaired twotailed Student’s t tests were performed for most statistical analysis using Prism. Pearson’s correlation coefficients and linear regression were also calculated for some statistical analysis using Prism. P values less than 0.05 were considered statistically significant.
Supplementary Material
Acknowledgments:
We wish to acknowledge the following GALA II and SAGE study collaborators: S. Thyne, UCSF; H. J. Farber, Texas Children’s Hospital; D. Serebrisky, Jacobi Medical Center; R. Kumar, Lurie Children’s Hospital of Chicago; E. Brigino-Buenaventura, Kaiser Permanente; M. A. LeNoir, Bay Area Pediatrics; K. Meade, UCSF Benioff Children’s Hospital, Oakland; W. Rodríguez-Cintrón, VA Hospital, Puerto Rico; P. C. Ávila, Northwestern University; J. R. Rodríguez-Santana, Centro de Neumología Pediátrica; L. N. Borrell, City University of New York; A. Davis, UCSF Benioff Children’s Hospital, Oakland; and S. Sen, University of Tennessee. We acknowledge the families and patients for the participation and thank the numerous health care providers and community clinics for the support and participation in GALA II and SAGE. In particular, we thank the recruiters who obtained the data: D. Alva, G. Ayala-Rodríguez, L. Caine, E. Castellanos, J. Colón, D. DeJesus, B. López, B. López, L. Martos, V. Medina, J. Olivo, M. Peralta, E. Pomares, J. Quraishi, J. Rodríguez, S. Saeedi, D. Soto, and A. Taveras. We thank D. A. Nickerson from the University of Washington Northwest Genomics Center and S. Germer, M. C. Zody, and L. W. and C. Reeves from the New York Genome Center for whole-genome sequencing. We thank G. Abecasis and H. M. Kang from the TOPMed Informatics Research Center for whole-genome sequencing data quality control and processing. We thank the Institute of Human Genetics (UCSF, San Francisco, CA) including C. Chu, C. Julian, and E. Wan for single-cell RNA sequencing. We thank I. Mellman (Genentech, South San Francisco, CA) for MHCIIK>R mice. We thank A. K. Proebstel (UCSF, San Francisco, CA) for technical assistance with EAE experiments. We thank M. Fassett, K. M. Ansel, D. Shepard, and A. Ma (UCSF, San Francisco, CA) for helpful discussion. We dedicate this work to the memory of X. Huang (UCSF, San Francisco, CA), who passed away during the study. We acknowledge the studies and participants who provided biological samples and data for TOPMed.
Funding:
This work was supported by Sandler Asthma Basic Research Center (to J.-S.S.), NIH (R01GM105800 and R35GM131702 to J-S.S.; R01HL117004, X01HL134589, and R01MD010443 to E.G.B.; R01AI026918 to R.M.L.), DoD (W81XWH1810110 to J.-S.S.), the American Association of Immunologists (Careers in Immunology Fellowship to J.-S.S. and C.A.C.), the UCSF Biomedical Sciences (BMS) graduate program (to C.A.C.), and the IMSD program funded by NIH (to C.A.C.). The GALA II Study, SAGE, and E.G.B. were supported by the Sandler Family Foundation; the American Asthma Foundation; the RWJF Amos Medical Faculty Development Program; the Harry Wm. and Diana V. Hind Distinguished Professor in Pharmaceutical Sciences II; the National Heart, Lung, and Blood Institute (NHLBI) (R01HL117004 and X01HL134589); and the National Institute on Minority Health and Health Disparities (NIMHD) (R01MD010443). Whole-genome sequencing for the TOPMed program was supported by the National Heart, Lung, and Blood Institute (NHLBI). Whole-genome sequencing for “NHLBI TOPMed: Gene-Environment, Admixture and Latino Asthmatics Study” (phs000920) and “NHLBI TOPMed: Study of African Americans, Asthma, Genes and Environments” (phs000921) was performed at the New York Genome Center (3R01HL117004–02S3) and the University of Washington Northwest Genomics Center (HHSN268201600032I). Centralized read mapping and genotype calling, along with variant quality metrics and filtering, were provided by the TOPMed Informatics Research Center (3R01HL-117626–02S1; contract HHSN268201800002I). Phenotype harmonization, data management, sample-identity QC, and general study coordination were provided by the TOPMed Data Coordinating Center (3R01HL-120393–02S1; contract HHSN268201800001I). Whole-genome sequencing of part of GALA II was performed by the New York Genome Center under The Centers for Common Disease Genomics of the Genome Sequencing Program (GSP) grant (UM1 HG008901). The GSP Coordinating Center (U24 HG008956) contributed to cross-program scientific initiatives and provided logistical and general study coordination. GSP is funded by the National Human Genome Research Institute; the National Heart, Lung, and Blood Institute; and the National Eye Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
Competing interests: R.M.L. is a member of the Scientific Advisory Board at Genentech. The authors declare that they have no competing interests.
Data and materials availability: The GEO accession number for single-cell RNA sequencing data is GSE183861. All other data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. MHCII (K>R) mice are available from J.-S.S. under a material transfer agreement with Genentech.
REFERENCES AND NOTES
- 1.Banchereau J, Steinman RM, Dendritic cells and the control of immunity. Nature 392, 245–252 (1998). [DOI] [PubMed] [Google Scholar]
- 2.Alvarez D, Vollmann EH, von Andrian UH, Mechanisms and consequences of dendritic cell migration. Immunity 29, 325–342 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lanzavecchia A, Sallusto F, The instructive role of dendritic cells on T cell responses: Lineages, plasticity and kinetics. Curr. Opin. Immunol. 13, 291–298 (2001). [DOI] [PubMed] [Google Scholar]
- 4.Zhu J, Yamane H, Paul WE, Differentiation of effector CD4 T cell populations (*). Annu. Rev. Immunol. 28, 445–489 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tubo NJ, Jenkins MK, CD4+ T cells: Guardians of the phagosome. Clin. Microbiol. Rev. 27, 200–213 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nakanishi Y, Lu B, Gerard C, Iwasaki A, CD8+ T lymphocyte mobilization to virus-infected tissue requires CD4+ T-cell help. Nature 462, 510–513 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu CH, Fan YT, Dias A, Esper L, Corn RA, Bafica A, Machado FS, Aliberti J, Cutting edge: Dendritic cells are essential for in vivo IL-12 production and development of resistance against Toxoplasma gondii infection in mice. J. Immunol. 177, 31–35 (2006). [DOI] [PubMed] [Google Scholar]
- 8.Henry CJ, Ornelles DA, Mitchell LM, Brzoza-Lewis KL, Hiltbold EM, IL-12 produced by dendritic cells augments CD8+ T cell activation through the production of the chemokines CCL1 and CCL17. J. Immunol. 181, 8576–8584 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hu W, Troutman TD, Edukulla R, Pasare C, Priming microenvironments dictate cytokine requirements for T helper 17 cell lineage commitment. Immunity 35, 1010–1022 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Palm NW, Rosenstein RK, Medzhitov R, Allergic host defences. Nature 484, 465–472 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Durai V, Murphy KM, Functions of murine dendritic cells. Immunity 45, 719–736 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gao Y, Nish SA, Jiang R, Hou L, Licona-Limon P, Weinstein JS, Zhao H, Medzhitov R, Control of T helper 2 responses by transcription factor IRF4-dependent dendritic cells. Immunity 39, 722–732 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Williams JW, Tjota MY, Clay BS, Vander Lugt B, Bandukwala HS, Hrusch CL, Decker DC, Blaine KM, Fixsen BR, Singh H, Sciammas R, Sperling AI, Transcription factor IRF4 drives dendritic cells to promote Th2 differentiation. Nat. Commun. 4, 2990 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tussiwand R, Everts B, Grajales-Reyes GE, Kretzer NM, Iwata A, Bagaitkar J, Wu X, Wong R, Anderson DA, Murphy TL, Pearce EJ, Murphy KM, Klf4 expression in conventional dendritic cells is required for T helper 2 cell responses. Immunity 42, 916–928 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoshino A, Tanaka Y, Akiba H, Asakura Y, Mita Y, Sakurai T, Takaoka A, Nakaike S, Ishii N, Sugamura K, Yagita H, Okumura K, Critical role for OX40 ligand in the development of pathogenic Th2 cells in a murine model of asthma. Eur. J. Immunol. 33, 861–869 (2003). [DOI] [PubMed] [Google Scholar]
- 16.MacDonald AS, Patton EA, La Flamme AC, Araujo MI, Huxtable CR, Bauman B, Pearce EJ, Impaired Th2 development and increased mortality during Schistosoma mansoni infection in the absence of CD40/CD154 interaction. J. Immunol. 168, 4643–4649 (2002). [DOI] [PubMed] [Google Scholar]
- 17.Jenkins SJ, Perona-Wright G, Worsley AGF, Ishii N, MacDonald AS, Dendritic cell expression of OX40 ligand acts as a costimulatory, not polarizing, signal for optimal Th2 priming and memory induction in vivo. J. Immunol. 179, 3515–3523 (2007). [DOI] [PubMed] [Google Scholar]
- 18.Grewal IS, Xu J, Flavell RA, Impairment of antigen-specific T-cell priming in mice lacking CD40 ligand. Nature 378, 617–620 (1995). [DOI] [PubMed] [Google Scholar]
- 19.Constant S, Pfeiffer C, Woodard A, Pasqualini T, Bottomly K, Extent of T cell receptor ligation can determine the functional differentiation of naive CD4+ T cells. J. Exp. Med. 182, 1591–1596 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yamane H, Zhu J, Paul WE, Independent roles for IL-2 and GATA-3 in stimulating naive CD4+ T cells to generate a Th2-inducing cytokine environment. J. Exp. Med. 202, 793–804 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Panhuys N, Klauschen F, Germain RN, T-cell-receptor-dependent signal intensity dominantly controls CD4+ T cell polarization in vivo. Immunity 41, 63–74 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shin JS, Ebersold M, Pypaert M, Delamarre L, Hartley A, Mellman I, Surface expression of MHC class II in dendritic cells is controlled by regulated ubiquitination. Nature 444, 115–118 (2006). [DOI] [PubMed] [Google Scholar]
- 23.Baravalle G, Park H, McSweeney M, Ohmura-Hoshino M, Matsuki Y, Ishido S, Shin JS, Ubiquitination of CD86 is a key mechanism in regulating antigen presentation by dendritic cells. J. Immunol. 187, 2966–2973 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Samji T, Hong S, Means RE, The membrane associated RING-CH proteins: A family of E3 ligases with diverse roles through the cell. Int. Sch. Res. Notices 2014, 637295 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van Niel G, Wubbolts R, Ten Broeke T, Buschow SI, Ossendorp FA, Melief CJ, Raposo G, van Balkom BW, Stoorvogel W, Dendritic cells regulate exposure of MHC class II at their plasma membrane by oligoubiquitination. Immunity 25, 885–894 (2006). [DOI] [PubMed] [Google Scholar]
- 26.Oh J, Shin JS, Molecular mechanism and cellular function of MHCII ubiquitination. Immunol. Rev. 266, 134–144 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bannard O, McGowan SJ, Ersching J, Ishido S, Victora GD, Shin JS, Cyster JG, Ubiquitin-mediated fluctuations in MHC class II facilitate efficient germinal center B cell responses. J. Exp. Med. 213, 993–1009 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oh J, Wu N, Baravalle G, Cohn B, Ma J, Lo B, Mellman I, Ishido S, Anderson M, Shin JS, MARCH1-mediated MHCII ubiquitination promotes dendritic cell selection of natural regulatory T cells. J. Exp. Med. 210, 1069–1077 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oh J, Perry JSA, Pua H, Irgens-Moller N, Ishido S, Hsieh CS, Shin JS, MARCH1 protects the lipid raft and tetraspanin web from MHCII proteotoxicity in dendritic cells. J. Cell Biol. 217, 1395–1410 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pulendran B, Artis D, New paradigms in type 2 immunity. Science 337, 431–435 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.von Garnier C, Filgueira L, Wikstrom M, Smith M, Thomas JA, Strickland DH, Holt PG, Stumbles PA, Anatomical location determines the distribution and function of dendritic cells and other APCs in the respiratory tract. J. Immunol. 175, 1609–1618 (2005). [DOI] [PubMed] [Google Scholar]
- 32.van Rijt LS, Jung S, Kleinjan A, Vos N, Willart M, Duez C, Hoogsteden HC, Lambrecht BN, In vivo depletion of lung CD11c+ dendritic cells during allergen challenge abrogates the characteristic features of asthma. J. Exp. Med. 201, 981–991 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Loschko J, Schreiber HA, Rieke GJ, Esterhazy D, Meredith MM, Pedicord VA, Yao KH, Caballero S, Pamer EG, Mucida D, Nussenzweig MC, Absence of MHC class II on cDCs results in microbial-dependent intestinal inflammation. J. Exp. Med. 213, 517–534 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lambrecht BN, Hammad H, Biology of lung dendritic cells at the origin of asthma. Immunity 31, 412–424 (2009). [DOI] [PubMed] [Google Scholar]
- 35.Guilliams M, Ginhoux F, Jakubzick C, Naik SH, Onai N, Schraml BU, Segura E, Tussiwand R, Yona S, Dendritic cells, monocytes and macrophages: A unified nomenclature based on ontogeny. Nat. Rev. Immunol. 14, 571–578 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.De Gassart A, Camosseto V, Thibodeau J, Ceppi M, Catalan N, Pierre P, Gatti E, MHC class II stabilization at the surface of human dendritic cells is the result of maturation-dependent MARCH I down-regulation. Proc. Natl. Acad. Sci. U.S.A. 105, 3491–3496 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Itano AA, McSorley SJ, Reinhardt RL, Ehst BD, Ingulli E, Rudensky AY, Jenkins MK, Distinct dendritic cell populations sequentially present antigen to CD4 T cells and stimulate different aspects of cell-mediated immunity. Immunity 19, 47–57 (2003). [DOI] [PubMed] [Google Scholar]
- 38.Jakubzick C, Helft J, Kaplan TJ, Randolph GJ, Optimization of methods to study pulmonary dendritic cell migration reveals distinct capacities of DC subsets to acquire soluble versus particulate antigen. J. Immunol. Methods 337, 121–131 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Naik SH, Proietto AI, Wilson NS, Dakic A, Schnorrer P, Fuchsberger M, Lahoud MH, O’Keeffe M, Shao Q.-x., Chen W.-f., Villadangos JA, Shortman K, Wu L, Cutting edge: Generation of splenic CD8+ and CD8− dendritic cell equivalents in Fms-like tyrosine kinase 3 ligand bone marrow cultures. J. Immunol. 174, 6592–6597 (2005). [DOI] [PubMed] [Google Scholar]
- 40.Yan Q, Brehm J, Pino-Yanes M, Forno E, Lin J, Oh SS, Acosta-Perez E, Laurie CC, Cloutier MM, Raby BA, Stilp AM, Sofer T, Hu D, Huntsman S, Eng CS, Conomos MP, Rastogi D, Rice K, Canino G, Chen W, Barr RG, Burchard EG, Celedon JC, A meta-analysis of genome-wide association studies of asthma in Puerto Ricans. Eur. Respir. J. 49, 1601505 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pan-UKB team (2020); https://pan.ukbb.broadinstitute.org.
- 42.Lin PI, Vance JM, Pericak-Vance MA, Martin ER, No gene is an island: The flip-flop phenomenon. Am. J. Hum. Genet. 80, 531–538 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ishikawa R, Kajikawa M, Ishido S, Loss of MHC II ubiquitination inhibits the activation and differentiation of CD4 T cells. Int. Immunol. 26, 283–289 (2014). [DOI] [PubMed] [Google Scholar]
- 44.Wilson KR, Liu H, Healey G, Vuong V, Ishido S, Herold MJ, Villadangos JA, Mintern JD, MARCH1-mediated ubiquitination of MHC II impacts the MHC I antigen presentation pathway. PLOS ONE 13, e0200540 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim HJ, Bandola-Simon J, Ishido S, Wong NW, Koparde VN, Cam M, Roche PA, Ubiquitination of MHC class II by March-I regulates dendritic cell fitness. J. Immunol. 206, 494–504 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kishta OA, Sabourin A, Simon L, McGovern T, Raymond M, Galbas T, Majdoubi A, Ishido S, Martin JG, Thibodeau J, March1 E3 ubiquitin ligase modulates features of allergic asthma in an ovalbumin-induced mouse model of lung inflammation. J. Immunol. Res. 2018, 3823910 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Young LJ, Wilson NS, Schnorrer P, Proietto A, ten Broeke T, Matsuki Y, Mount AM, Belz GT, O’Keeffe M, Ohmura-Hoshino M, Ishido S, Stoorvogel W, Heath WR, Shortman K, Villadangos JA, Differential MHC class II synthesis and ubiquitination confers distinct antigen-presenting properties on conventional and plasmacytoid dendritic cells. Nat. Immunol. 9, 1244–1252 (2008). [DOI] [PubMed] [Google Scholar]
- 48.Iwasaki A, Medzhitov R, Regulation of adaptive immunity by the innate immune system. Science 327, 291–295 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dalod M, Chelbi R, Malissen B, Lawrence T, Dendritic cell maturation: Functional specialization through signaling specificity and transcriptional programming. EMBO J. 33, 1104–1116 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Perner C, Flayer CH, Zhu X, Aderhold PA, Dewan ZNA, Voisin T, Camire RB, Chow OA, Chiu IM, Sokol CL, Substance P release by sensory neurons triggers dendritic cell migration and initiates the type-2 immune response to allergens. Immunity 53, 1063–1077.e7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kane CM, Cervi L, Sun J, McKee AS, Masek KS, Shapira S, Hunter CA, Pearce EJ, Helminth antigens modulate TLR-initiated dendritic cell activation. J. Immunol. 173, 7454–7461 (2004). [DOI] [PubMed] [Google Scholar]
- 52.Everts B, Perona-Wright G, Smits HH, Hokke CH, van der Ham AJ, Fitzsimmons CM, Doenhoff MJ, van der Bosch J, Mohrs K, Haas H, Mohrs M, Yazdanbakhsh M, Schramm G, Omega-1, a glycoprotein secreted by Schistosoma mansoni eggs, drives Th2 responses. J. Exp. Med. 206, 1673–1680 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gerner MY, Torabi-Parizi P, Germain RN, Strategically localized dendritic cells promote rapid T cell responses to lymph-borne particulate antigens. Immunity 42, 172–185 (2015). [DOI] [PubMed] [Google Scholar]
- 54.Halim TYF, Steer CA, Matha L, Gold MJ, Martinez-Gonzalez I, McNagny KM, McKenzie ANJ, Takei F, Group 2 innate lymphoid cells are critical for the initiation of adaptive T helper 2 cell-mediated allergic lung inflammation. Immunity 40, 425–435 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Miller HL, Andhey PS, Swiecki MK, Rosa BA, Zaitsev K, Villani AC, Mitreva M, Artyomov MN, Gilfillan S, Cella M, Colonna M, Altered ratio of dendritic cell subsets in skin-draining lymph nodes promotes Th2-driven contact hypersensitivity. Proc. Natl. Acad. Sci. U.S.A. 118, e2021364118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Walker JA, McKenzie ANJ, TH2 cell development and function. Nat. Rev. Immunol. 18, 121–133 (2018). [DOI] [PubMed] [Google Scholar]
- 57.Bhattacharyya ND, Feng CG, Regulation of T helper cell fate by TCR signal strength. Front. Immunol. 11, 624 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Matsuki Y, Ohmura-Hoshino M, Goto E, Aoki M, Mito-Yoshida M, Uematsu M, Hasegawa T, Koseki H, Ohara O, Nakayama M, Toyooka K, Matsuoka K, Hotta H, Yamamoto A, Ishido S, Novel regulation of MHC class II function in B cells. EMBO J. 26, 846–854 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bettelli E, Pagany M, Weiner HL, Linington C, Sobel RA, Kuchroo VK, Myelin oligodendrocyte glycoprotein-specific T cell receptor transgenic mice develop spontaneous autoimmune optic neuritis. J. Exp. Med. 197, 1073–1081 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Caton ML, Smith-Raska MR, Reizis B, Notch-RBP-J signaling controls the homeostasis of CD8− dendritic cells in the spleen. J. Exp. Med. 204, 1653–1664 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Barnden MJ, Allison J, Heath WR, Carbone FR, Defective TCR expression in transgenic mice constructed using cDNA-based alpha- and beta-chain genes under the control of heterologous regulatory elements. Immunol. Cell Biol. 76, 34–40 (1998). [DOI] [PubMed] [Google Scholar]
- 62.Meredith MM, Liu K, Darrasse-Jeze G, Kamphorst AO, Schreiber HA, Guermonprez P, Idoyaga J, Cheong C, Yao KH, Niec RE, Nussenzweig MC, Expression of the zinc finger transcription factor zDC (Zbtb46, Btbd4) defines the classical dendritic cell lineage. J. Exp. Med. 209, 1153–1165 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wu X, Briseno CG, Durai V, Albring JC, Haldar M, Bagadia P, Kim KW, Randolph GJ, Murphy TL, Murphy KM, Mafb lineage tracing to distinguish macrophages from other immune lineages reveals dual identity of Langerhans cells. J. Exp. Med. 213, 2553–2565 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Swiecki M, Gilfillan S, Vermi W, Wang Y, Colonna M, Plasmacytoid dendritic cell ablation impacts early interferon responses and antiviral NK and CD8+ T cell accrual. Immunity 33, 955–966 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kitamura D, Roes J, Kuhn R, Rajewsky K, A B cell-deficient mouse by targeted disruption of the membrane exon of the immunoglobulin mu chain gene. Nature 350, 423–426 (1991). [DOI] [PubMed] [Google Scholar]
- 66.Satpathy AT, Kc W, Albring JC, Edelson BT, Kretzer NM, Bhattacharya D, Murphy TL, Murphy KM, Zbtb46 expression distinguishes classical dendritic cells and their committed progenitors from other immune lineages. J. Exp. Med. 209, 1135–1152 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Höpken UE, Droese J, Li J-P, Joergensen J, Breitfeld D, Zerwes HG, Lipp M, The chemokine receptor CCR7 controls lymph node-dependent cytotoxic T cell priming in alloimmune responses. Eur. J. Immunol. 34, 461–470 (2004). [DOI] [PubMed] [Google Scholar]
- 68.Nishimura KK, Galanter JM, Roth LA, Oh SS, Thakur N, Nguyen EA, Thyne S, Farber HJ, Serebrisky D, Kumar R, Brigino-Buenaventura E, Davis A, LeNoir MA, Meade K, Rodriguez-Cintron W, Avila PC, Borrell LN, Bibbins-Domingo K, Rodriguez-Santana JR, Sen S, Lurmann F, Balmes JR, Burchard EG, Early-life air pollution and asthma risk in minority children. The GALA II and SAGE II studies. Am. J. Respir. Crit. Care Med. 188, 309–318 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Thakur N, Oh SS, Nguyen EA, Martin M, Roth LA, Galanter J, Gignoux CR, Eng C, Davis A, Meade K, LeNoir MA, Avila PC, Farber HJ, Serebrisky D, Brigino-Buenaventura E, Rodriguez-Cintron W, Kumar R, Williams LK, Bibbins-Domingo K, Thyne S, Sen S, Rodriguez-Santana JR, Borrell LN, Burchard EG, Socioeconomic status and childhood asthma in urban minority youths. The GALA II and SAGE II studies. Am. J. Respir. Crit. Care Med. 188, 1202–1209 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Oh SS, Tcheurekdjian H, Roth LA, Nguyen EA, Sen S, Galanter JM, Davis A, Farber HJ, Gilliland FD, Kumar R, Avila PC, Brigino-Buenaventura E, Chapela R, Ford JG, LeNoir MA, Lurmann F, Meade K, Serebrisky D, Thyne S, Rodriguez-Cintron W, Rodriguez-Santana JR, Williams LK, Borrell LN, Burchard EG, Effect of secondhand smoke on asthma control among black and Latino children. J. Allergy Clin. Immunol. 129, 1478–1483.e7 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Taliun D, Harris DN, Kessler MD, Carlson J, Szpiech ZA, Torres R, Taliun SAG, Corvelo A, Gogarten SM, Kang HM, Pitsillides AN, LeFaive J, Lee SB, Tian X, Browning BL, Das S, Emde AK, Clarke WE, Loesch DP, Shetty AC, Blackwell TW, Smith AV, Wong Q, Liu X, Conomos MP, Bobo DM, Aguet F, Albert C, Alonso A, Ardlie KG, Arking DE, Aslibekyan S, Auer PL, Barnard J, Barr RG, Barwick L, Becker LC, Beer RL, Benjamin EJ, Bielak LF, Blangero J, Boehnke M, Bowden DW, Brody JA, Burchard EG, Cade BE, Casella JF, Chalazan B, Chasman DI, Chen Y-DI, Cho MH, Choi SH, Chung MK, Clish CB, Correa A, Curran JE, Custer B, Darbar D, Daya M, de Andrade M, DeMeo DL, Dutcher SK, Ellinor PT, Emery LS, Eng C, Fatkin D, Fingerlin T, Forer L, Fornage M, Franceschini N, Fuchsberger C, Fullerton SM, Germer S, Gladwin MT, Gottlieb DJ, Guo X, Hall ME, He J, Heard-Costa NL, Heckbert SR, Irvin MR, Johnsen JM, Johnson AD, Kaplan R, Kardia SLR, Kelly T, Kelly S, Kenny EE, Kiel DP, Klemmer R, Konkle BA, Kooperberg C, Köttgen A, Lange LA, Lasky-Su J, Levy D, Lin X, Lin K-H, Liu C, Loos RJF, Garman L, Gerszten R, Lubitz SA, Lunetta KL, Mak ACY, Manichaikul A, Manning AK, Mathias RA, McManus DD, McGarvey ST, Meigs JB, Meyers DA, Mikulla JL, Minear MA, Mitchell BD, Mohanty S, Montasser ME, Montgomery C, Morrison AC, Murabito JM, Natale A, Natarajan P, Nelson SC, North KE, O’Connell JR, Palmer ND, Pankratz N, Peloso GM, Peyser PA, Pleiness J, Post WS, Psaty BM, Rao DC, Redline S, Reiner AP, Roden D, Rotter JI, Ruczinski I, Sarnowski C, Schoenherr S, Schwartz DA, Seo JS, Seshadri S, Sheehan VA, Sheu WH, Shoemaker MB, Smith NL, Smith JA, Sotoodehnia N, Stilp AM, Tang W, Taylor KD, Telen M, Thornton TA, Tracy RP, Van Den Berg DJ, Vasan RS, Viaud-Martinez KA, Vrieze S, Weeks DE, Weir BS, Weiss ST, Weng LC, Willer CJ, Zhang Y, Zhao X, Arnett DK, Ashley-Koch AE, Barnes KC, Boerwinkle E, Gabriel S, Gibbs R, Rice KM, Rich SS, Silverman EK, Qasba P, Gan W; NHLBI Trans-Omics for Precision Medicine (TOPMed) Consortium, Papanicolaou GJ, Nickerson DA, Browning SR, Zody MC, Zollner S, Wilson JG, Cupples LA, Laurie CC, Jaquish CE, Hernandez RD, O’Connor TD, Abecasis GR, Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature 590, 290–299 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mak ACY, Sajuthi S, Joo J, Xiao S, Sleiman PM, White MJ, Lee EY, Saef B, Hu D, Gui H, Keys KL, Lurmann F, Jain D, Abecasis G, Kang HM, Nickerson DA, Germer S, Zody MC, Winterkorn L, Reeves C, Huntsman S, Eng C, Salazar S, Oh SS, Gilliland FD, Chen Z, Kumar R, Martinez FD, Wu AC, Ziv E, Hakonarson H, Himes BE, Williams LK, Seibold MA, Burchard EG, Lung function in African American children with asthma is associated with novel regulatory variants of the KIT ligand KITLG/SCF and gene-by-air-pollution interaction. Genetics 215, 869–886 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lee EY, Mak ACY, Hu D, Sajuthi S, White MJ, Keys KL, Eckalbar W, Bonser L, Huntsman S, Urbanek C, Eng C, Jain D, Abecasis G, Kang HM, Germer S, Zody MC, Nickerson DA, Erle D, Ziv E, Rodriguez-Santana J, Seibold MA, Burchard EG, Whole-genome sequencing identifies novel functional loci associated with lung function in Puerto Rican youth. Am. J. Respir. Crit. Care Med. 202, 962–972 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Willer CJ, Li Y, Abecasis GR, METAL: Fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.GTEx Consortium, The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318–1330 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.