

Abstract
Targeted inhibition of BRAF V600E achieves tumor control in a subset of advanced thyroid tumors. Nearly all tumors develop resistance, and some have been observed to subsequently undergo dedifferentiation. The molecular alterations associated with thyroid cancer dedifferentiation in the setting of BRAF inhibition are unknown. We analyzed targeted next-generation sequencing data from 639 advanced, recurrent and/or metastatic thyroid carcinomas, including 15 tumors that were treated with BRAF inhibitor drugs and had tissue sampled during or post treatment, 8 of which had matched pre-therapy samples. Pre- and post-therapy tissues from one additional patient were profiled with whole exome sequencing and RNA expression profiling. Mutations in genes comprising the SWI/SNF chromatin remodeling complex and the PI3K/AKT/mTOR, MAPK, and JAK/STAT pathways all increased in prevalence across more dedifferentiated thyroid cancer histologies. Of 7 thyroid cancers that dedifferentiated after BRAF inhibition, 6 had mutations in these pathways. These mutations were mostly absent from matched pre-treatment samples and were rarely detected in tumors that did not dedifferentiate. Additional analyses in one of the vemurafenib-treated tumors before and after anaplastic transformation revealed the emergence of an oncogenic PIK3CA mutation, activation of ERK signaling, dedifferentiation, and development of an immunosuppressive tumor microenvironment. These findings validate earlier preclinical data implicating these genetic pathways in resistance to BRAF inhibitors, and suggest that genetic alterations mediating acquired drug resistance may also promote thyroid tumor dedifferentiation.
Keywords: Thyroid cancer, Anaplastic transformation, BRAF inhibitors
Introduction
The vast majority of cancers arising from the thyroid gland are papillary thyroid carcinomas (PTC), a disease with generally indolent course and a high rate of cure after standard therapy. However, through a process of gradual microevolution, aggressive variants of PTC can develop and transition to dedifferentiated histologies with poorer prognoses, such as poorly differentiated thyroid carcinoma (PDTC) and anaplastic thyroid carcinoma (ATC) (1,2). After evolution to anaplastic histology, the disease generally becomes incurable, with 1-year overall survival rates of 35% in the era of concurrent chemoradiation therapy (3,4).
A deeper understanding of the genetic alterations underlying thyroid cancer has facilitated the development of targeted therapies for clinically aggressive tumors; for example, inhibition of oncogenic drivers such as the BRAF V600E oncoprotein. Tumor responses to these drugs are often short-lived and drug resistance is inevitable. Many tumors harbor genetic alterations that may confer primary resistance. Alternatively, other tumors develop acquired resistance, either via restoration of MAPK signaling via mutations in RAS (5) or other genes (6), or via pathway bypass by signaling through alternative pathways such as PI3K/AKT (7), JAK/STAT (8), EGFR (9) or HER2/HER3 (10).
A third mechanism of resistance to targeted therapy is the development of an alternative cell state, often involving tumor dedifferentiation as a result of selective pressures exerted by pathway blockade (11). Dedifferentiation has been demonstrated to occur after BRAF inhibition in melanoma (12,13) and thyroid tumors (9). Although rare, there have been a small number of reported cases of differentiated thyroid cancers transforming to ATC after BRAF inhibition (14). The mechanism underlying this phenomenon is unclear. While past studies have evaluated the molecular correlates of anaplastic evolution (15) and BRAF inhibitor resistance separately (16), anaplastic transformation in the setting of BRAF inhibitor therapy has yet to be specifically explored, to our knowledge.
To better understand possible mechanisms of this phenomenon, we performed genomic analyses in thyroid cancers across the spectrum of histologic differentiation, and in matched samples before and after exposure to BRAF inhibitors for thyroid cancers harboring BRAF V600E mutations, some of which underwent dedifferentiation.
MATERIALS AND METHODS
Study Oversight
This study was approved by the Memorial Sloan Kettering Cancer Center (MSKCC) Institutional Review Board. Patients provided written informed consent for collection and analysis of tumor and blood samples.
Specimen Collection
DNA was extracted from 639 formalin-fixed, paraffin-embedded advanced, recurrent and/or metastatic thyroid cancers treated at MSKCC between April 2015 and September 2020. Tumor DNA and patient-matched normal samples were profiled on the MSK-IMPACT (Integrated Mutational Profiling of Actionable Cancer Targets) targeted next-generation sequencing platform (details below).
Tumors from one additional patient (MSK-THY1), whose tumor underwent anaplastic transformation subsequent to vemurafenib therapy, were profiled with whole exome sequencing (WES) and RNA expression profiling. One specimen was collected 25 months prior to the first dose of vemurafenib, at which time the pathologic diagnosis was histologically confirmed to be PTC. Two additional specimens were collected 11 months after the start of vemurafenib therapy, at which time the tumor had transformed to ATC. Samples from MSK-THY1 were all immediately snap frozen in liquid nitrogen and stored at −80°C. For pathologic confirmation of diagnoses, specimens were formalin fixed, paraffin embedded and sectioned onto glass slides at 4-μm thickness. Slides were prepared with hematoxylin and eosin stains by standard procedures and reviewed by board-certified sub-specialty thyroid pathologists at our institution. Histologic diagnoses were made according to the World Health Organization’s classification of endocrine tumors (17). Briefly, PTCs are characterized by two cardinal morphologic features including the presence of papillae and alterations in nuclei (e.g., characteristic changes in size or shape, nuclear membrane, and/or chromosomal features). PDTCs are tumors of follicular cell origin that have solid, trabecular, or insular growth pattern, absence of typical nuclear features of PTC, and at least one of the following: convoluted nuclei, ≥ 3 mitoses per 10 high-power fields, and tumor necrosis. ATCs are undifferentiated at the histologic level, having lost the ability to form follicles, papillae or even solid nests of follicular monomorphic cells.
Of the 639 MSK-IMPACT profiled tumors, there were 15 (designated MSK-THY2 through MSK-THY16) that received treatment with the BRAF inhibitors vemurafenib or dabrafenib for non-ATC differentiated thyroid cancer and had tissue sampled during or after treatment available for sequencing. Of these 15 patients, 5 had tissue profiled on-treatment (median time from first BRAF inhibitor dose to tumor sampling: 9.9 months, range 0.1-66.5) and 10 had tissue profiled after treatment (median time from BRAF inhibitor end of treatment to tumor sampling: 20.8 months, range 0.2-44.4).
DNA Sequencing
DNA was extracted from tumor specimens of MSK-THY1 using the Blood and Tissue Kit (Qiagen) and assessed for quality and integrity with BioAnalyzer (Agilent Technologies). The SureSelectXT Library Preparation Kit (Agilent) was used to prepare WES libraries per manufacturer’s specifications. Extracted DNA was sheared using a LE220 Focused Ultrasonicator (Covaris). DNA fragments were end repaired, adenylated, ligated to Illumina sequencing adaptors, and amplified by PCR. The SureSelectXT v4 51 Mb capture probe set (Agilent) was used to build captured exome libraries, which were then amplified by PCR. The final libraries were quantified using the KAPA Library Quantification Kit (KAPA Biosystems), Qubit Fluorometer (Life Technologies), and 2100 BioAnalyzer (Agilent). Sequencing was performed using 2 × 125 bp cycles. Mutations were called using a previously described algorithm (18) and allele-specific copy-number analyses were performed using FACETS (19). All mutations were orthogonally validated with next-generation sequencing using a custom Ion AmpliSeq panel on the IonTorrent PGM sequencer at mean target coverage of 9546×. Intratumor genetic heterogeneity was analyzed based on cancer cell fractions in the validated mutations as determined using PyClone as previously described (20,21). To determine cancer cell fractions, we used aberrant cell fraction and major and minor allele copy number data from FACETS, and variant allele frequencies for validated somatic mutations. These data were inputed into PyClone, which estimates the cancer cell prevalence for each somatic mutation in each tumor sample, and subsequently uses Bayesian methodology to cluster cancer cell prevalence estimates into clonal populations. Intratumor heterogeneity was depicted as the number of clusters, or (sub)clonal populations.
DNA from 639 thyroid tumors treated at MSK was sequenced with MSK-IMPACT. The technical details for tumor collection, processing and sequencing by MSK-IMPACT have been previously described (22). MSK-IMPACT profiles matched tumor and normal (peripheral blood-derived) DNA samples for 341, 410 or 468 genes with known roles in oncogenesis and/or targeted therapy. Of 639 thyroid cancer cases profiled with MSK-IMPACT, there were 369 with PTC (mean target coverage: 575×), 164 with PDTC (627×), and 106 with ATC (724×). The prevalence of mutations with described roles in BRAF inhibitor resistance and/or ATC pathogenesis were compared between PTCs, PDTCs, and ATCs. Of the subset of 15 patients treated with BRAF inhibitors for non-ATC differentiated thyroid cancers, mean target coverage was 661×.
Transcriptomic Analyses
RNA was extracted from tumor specimens from patient MSK-THY1 using the RNeasy Plus Mini Kit (Qiagen), in vitro transcribed to cRNA using the Affymetrix IVT Kit, hybridized to Affymetrix U133 plus 2.0 microarrays, and data RMA normalized and analyzed in Partek Genomics Suite 6.1, as previously described (23).
Thyroid Differentiation Scores (TDS) were calculated using the normalized expression levels of 16 thyroid metabolism and function genes as previously described (24). TDS scores have been shown to accurately identify thyroid tumor dedifferentiation (25). Pathway analyses were performed using Gene Set Enrichment Analysis (GSEA) and Qiagen Ingenuity Pathway Analysis (IPA) with Benjamini-Hochberg false-discovery rate correction.
Tumor Immune Infiltration and Activity
Tumor microenvironment immune infiltration and immune activity were evaluated using several tools. Cell-type Identification by Estimating Relative Subsets of RNA Transcripts (CIBERSORT) estimates the prevalence of various cell types within complex tissues using a linear support vector regression to adaptively select genes from a reference gene expression signature (26). Single-Sample Gene Set Enrichment Analysis (ssGSEA) estimates infiltration levels of 24 immune cell types by calculating enrichment scores for sample and gene set pairs (27), also inferring aggregate immune infiltration scores (IIS) based on both innate and adaptive cell populations and T-cell infiltration scores (TIS) based on nine T-cell subtypes. Estimation of Stromal and Immune Cells in Malignant Tumor Tissues using Expression Data (ESTIMATE) is based on ssGSEA and infers the proportion of stromal and immune cells in tumor samples using a 141-gene signature derived from differential expression analysis of tumors with high and low immune cell infiltration (28). Tumor Immune Dysfunction and Exclusion (TIDE) models two primary mechanisms of immune evasion, T-cell dysfunction and exclusion, by applying genetic signatures of these two processes to batch-corrected normalized data (29).
Data Availability Statement
The sequencing variant calls and gene expression data used in this paper are provided in Supplementary Tables.
RESULTS
Genomic evolution during dedifferentiation to anaplastic carcinoma
The timeline of clinical events for patient MSK-THY1 is shown in Figure 1A–C. This patient was first diagnosed with classic PTC in 2001 at age 55 and treated with total thyroidectomy and adjuvant radioactive iodine (RAI) therapy. Between 2004 and 2010, she underwent additional surgical resections and RAI therapy for recurrences in regional lymph nodes in the neck, all of which revealed classic PTC on surgical pathology. One sample from the PTC nodal metastasis in 2010 was snap frozen for WES and RNA sequencing. In 2011, lung metastases were diagnosed, and she was treated with sorafenib and temsirolimus (30), with tumor experiencing partial response (31). Treatment was continued for 13 months until disease progression. In 2012, a BRAF V600E mutation was identified and vemurafenib therapy was initiated (32). Best response on vemurafenib was stable disease (−20% tumor regression from baseline) and treatment was discontinued after five months due to progression of disease. Subsequently, she was started on pazopanib, but was treated for less than one week before it was discontinued for toxicity. Two months after discontinuation of vemurafenib, she was first noted to have metastatic tumor in soft tissues, which quickly progressed to biopsy-proven ATC in multiple regions, including scalp, upper extremities, kidneys and brain. At this time, tissue from biopsies of 2 separate scalp metastases were snap-frozen for correlative studies.
Figure 1.
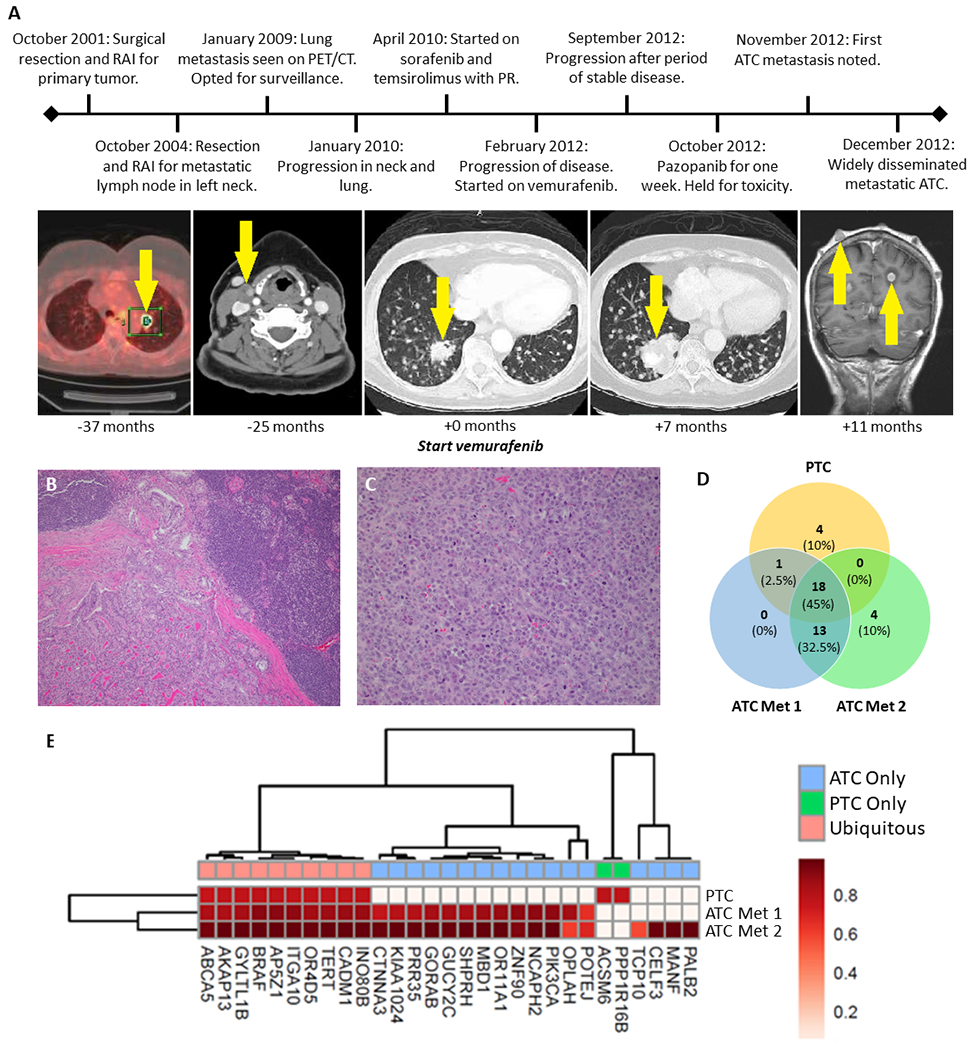
Clinical and genomic features of patient MSK-THY1 with differentiated thyroid cancer that underwent anaplastic transformation subsequent to treatment with vemurafenib. A, Clinical timeline with major events indicated. Serial radiographic images illustrate progression of papillary thyroid cancer to widely disseminated anaplastic malignancy. Time of imaging relative to vemurafenib start date is provided for each image and areas of disease are indicated by arrows. Panel 1: PET/CT, Panels 2-4: CT with contrast, Panel 5: T1-weighted MRI with contrast, RAI: radioactive iodine, PR: partial response, ATC: anaplastic thyroid cancer. B & C, Pre-treatment and post-transformation hematoxylin-and-eosin-stained slides show histologic transformation from papillary to anaplastic thyroid carcinoma. D, Number of shared and exclusive mutations in classic-type papillary thyroid cancer (PTC) compared to anaplastic metastases (ATC Met 1 & ATC Met 2). E, Landscape of single nucleotide variants of the PTC tumor and ATC metastases. Each mutation color coded by cancer cell fraction (CCF). Unsupervised hierarchical clustering identified subsets of related samples and mutations.
WES of the pre-treatment PTC nodal metastasis and post-transformation ATC distant metastases, followed by orthogonal next-generation deep sequencing (9,546× mean target coverage) for validation of mutations, demonstrated aspects of genomic evolution during the course of anaplastic transformation (Figures 1D–E, Supplemental Tables 1–2). The BRAF V600E mutation was identified in all samples, as was a TERT promoter mutation (at position 1,295,228). In addition, 17 new mutations emerged in the ATC metastases, including genes involved in PI3K/AKT/mTOR signaling (PIK3CA H1047R, KIAA1024 P412Q), cell-cell adhesion (CTNNA3 M568I), cell division (GORAB P119L, NCAPH2 N533S), epigenetic regulation (MBD1 I550N), transcriptional regulation (ZNF90 A321V), RNA splicing and editing (CELF3 T392N), cell survival (MANF M1I), and DNA repair (PALB2 R753Q).
PIK3CA mutations have been previously described as a mechanism of resistance to BRAF inhibition in thyroid carcinoma (33) and melanoma (34). In both ATC metastases, the PIK3CA H1047R mutation was observed to have high cancer cell fraction (0.90 and 0.97). Orthogonal next-generation sequencing of the index PTC tumor did not identify PIK3CA mutations in the PTC tumor, despite high-depth coverage of this gene target (>15,000×). These data suggest that the PIK3CA mutation was likely to have been present, but spatially heterogeneous, in the original PTC tumor and that subclonal populations containing the PIK3CA mutation later seeded the ATC metastases. It is possible that selective pressure exerted by BRAF inhibitor therapy may have contributed to the outgrowth of cellular subpopulations that were initially spatially constrained, such as PIK3CA-mutated cells.
To examine this more broadly, we performed analyses of intra-tumor genetic heterogeneity (Supplemental Figure 1), which demonstrated limited genetic heterogeneity in the PTC sample (2 clonal populations), which increased during the course of anaplastic evolution (ATC Metastasis 1 had 5 clonal populations; ATC Metastasis 2 had 4 clonal populations). In concert with the increase in mutations in the ATC tumors, many more copy number alterations were observed in the ATC metastases (mean ploidy 2.88 and 1.75, total copy number altered segments 311 and 108), compared to the PTC primary tumor, which exhibited fewer copy number alterations (mean ploidy 1.91, total copy number altered segments 63; Supplemental Figure 2), indicative of increased chromosomal instability.
Transcriptomic changes during dedifferentiation to anaplastic carcinoma
Gene expression data confirmed that cell cycle and DNA replication pathways were enriched in ATC metastases compared to the pre-treatment PTC, consistent with increased cellular proliferation (Supplemental Figure 3, Supplemental Table 3). Transcriptomic data were used to determine the degree of thyroid cellular differentiation. The TDS (Thyroid Differentiation Score), consisting of 16 thyroid metabolism and function genes (24), was significantly lower (difference in z-scores: p < 0.001) in the ATC metastases compared to the pre-treatment PTC sample (Figure 2A), consistent with the histologic observation of dedifferentiation. Nearly all 16 genes were markedly downregulated in the ATC metastases, with the exception of 2 genes that were low at baseline and did not change: SLC5A5 (encoding the sodium-iodide symporter) and THRB (thyroid hormone receptor beta) (Figure 2A). While iodine uptake and retention are governed by multiple genes within the TDS, the low baseline expression of SLC5A5 was observed in all samples, consistent with its downregulation by BRAF V600E signaling (24). Of note, DUOX1, DUOX2, and TPO—all of which play key roles in thyroid hormone production—were amongst the genes markedly downregulated in the ATC metastases.
Figure 2.
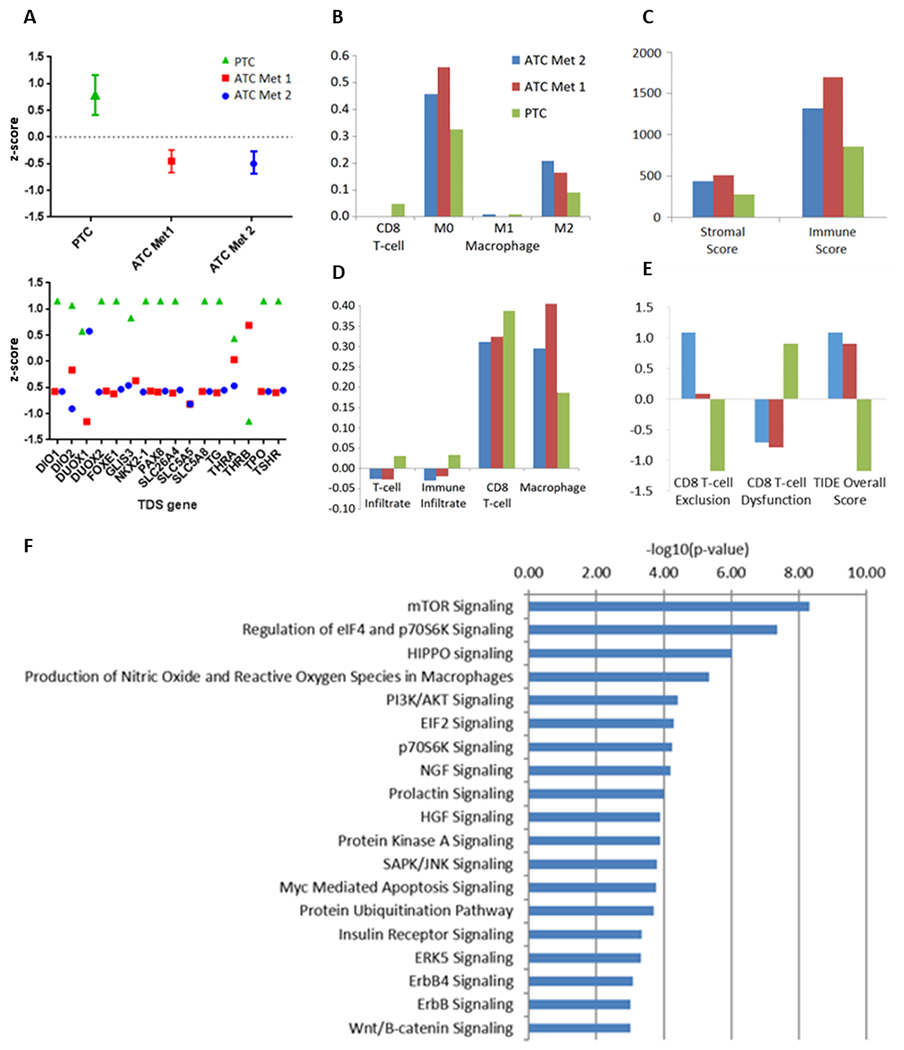
RNA expression profiling for MSK-THY1. A, Thyroid differentiation scores (TDS), overall and stratified by gene, for the papillary thyroid carcinoma (PTC) sample and anaplastic metastases (ATC Met 1 and ATC Met 2). Immune deconvolution by CIBERSORT (B), ESTIMATE (C), ssGSEA (D), and T cell dysfunction and exclusion (TIDE) (E) for the PTC and ATC samples. F, Most enriched molecular pathways among alterations in ATC metastases compared to the PTC sample
Confirming the functional relevance of the PIK3CA mutations in the ATC samples, Ingenuity Pathway Analysis of transcriptomic data indicated that the ATC metastases had undergone coordinate enrichment in signaling through the PI3K/AKT (FDR-corrected p = 3.8 x 10−5) and mTOR (FDR-corrected p = 4.9 x 10−9) pathways, together with the gene set regulating downstream targets eIF4E binding protein and p70 ribosomal protein S6 kinase (p70S6K; FDR-corrected p = 4.5 x 10−8) (Figure 2F). Hippo signaling (p = 9.5 x 10−7) was also one of the most significantly enriched pathways. Both PI3K and Hippo signaling play key roles in the dedifferentiation, and aggressive behavior, of thyroid cancers (25,35).
Despite therapy with BRAF inhibition, the post-treatment ATC tumors demonstrated enrichment of the 52-gene thyroid-specific ERK signaling signature (24) which was derived from BRAF V600E-mutated thyroid cells and evaluated in thyroid cancers (Supplemental Figure 3). ERK signaling activity was enriched in the ATC metastases (FDR-corrected q = 0.005), in line with prior findings in mouse models that ERK signaling can be paradoxically upregulated by BRAF inhibition in the setting of upstream activating mutations, such as PIK3CA (33,36,37). Taken together, these data reveal upregulated signaling through several pathways in concert with BRAF inhibition and the expansion of PIK3CA-mutated cells during the evolution of this thyroid cancer.
Immune microenvironmental evolution during dedifferentiation to anaplastic carcinoma
Gene expression data were then used for deconvolution of infiltrating immune populations (Figures 2B–2E). Overall, there was a mixed picture of change in immune infiltration during the evolution from PTC to ATC. The ATC tumors had an increased presence of infiltrating M0 and M2 macrophages, myeloid-derived suppressor cells, as well as fibroblasts, all of which are consistent with an immune-suppressive environment. M1 macrophage prevalence was similar in both pre-vemurafenib and post-anaplastic transformation specimens, and CD8+ T-cells became less prevalent in ATC metastases. Despite a higher degree of CD8+ T-cell exclusion, T cell dysfunction scores were also lower in ATC metastases compared to pre-vemurafenib PTC. Overall, infiltration by immunosuppressive cell types may have contributed to immune evasion during the evolution of this thyroid cancer.
Mutations associated with dedifferentiated thyroid cancers
Given the potential mechanistic role of PIK3CA mutations in resistance to BRAF inhibitors, further analyses in an expanded cohort were conducted to investigate the association between PIK3CA mutations and thyroid cancer dedifferentiation. We examined PIK3CA mutations in 496 patients with primary PTC from The Cancer Genome Atlas cohort (24) and compared these primary tumors to 639 advanced, recurrent and/or metastatic thyroid cancers (including PTC, PDTC, and ATC) undergoing targeted next-generation sequencing on the MSK-IMPACT platform.
In the primary PTCs in the TCGA cohort, PIK3CA mutations were uncommon (2/496; 0.4%, Supplemental Figure 4, Supplemental Table 4). PIK3CA mutations were more than 10 times as common in advanced/recurrent/metastatic PTCs in the MSK-IMPACT cohort (19/369, 5.1%; OR = 13.4, p < 0.001) and in PDTCs (9/164, 5.5%; OR = 14.3, p < 0.001); and over 40 times as common in ATCs (17/106, 16.0%; OR = 47.2, p < 0.001).
The prevalence of other previously described alterations linked to BRAF inhibitor primary or acquired resistance or ATC pathogenesis (25,35,36,38–42) were also explored in these cohorts (Supplemental Figure 4, Supplemental Table 4). Mutations in ARID2, MTOR, NF2, NRAS, and PBRM1 were all observed to increase in prevalence in more dedifferentiated categories of thyroid cancers, consistent with prior findings (25,38).
Mutations in BRAF inhibitor-treated thyroid cancers
Of the 639 advanced, recurrent, and/or metastatic thyroid tumors, 77 were cases of PTC or PDTC that were treated with BRAF inhibitor targeted therapy (vemurafenib or dabrafenib). Of these, 15 had tissue sampled during or after treatment available for sequencing on the MSK-IMPACT platform (MSK-THY2 through MSK-THY16). Of these 15 patients, 8 also had matched pre-therapy tissue available for sequencing.
Subsequent to BRAF inhibitor therapy, 6 of 15 patients had tumors that underwent transformation to more dedifferentiated histology, which was confirmed with biopsy (Table 1, Supplemental Figure 5), and 9 patients had no clinical or histological evidence of dedifferentiation. It is important to clarify that this proportion (6/15) does not necessarily represent the rate of transformation among all treated patients. Additional biopsies after BRAF inhibitor therapy are not routine and are often prompted by observations of disease progression. As such, patients for whom such biopsies are pursued may be enriched for dedifferentiated tumors. Also, 1 of the patients (MSK-THY3) also received systemic therapies other than BRAF inhibitors prior to post-therapy sequencing. Therefore, genomic changes in these patients may be in part attributable to these other agents.
Table 1.
Clinical and genomic features of patients whose tumors dedifferentiated after BRAF inhibitor targeted therapy. Genes listed represented mutated genes unless otherwise noted. PTC: Papillary Thyroid Cancer; PDTC: Poorly Differentiated Thyroid Cancer; ATC: Anaplastic Thyroid Cancer
| ID | Age1 | Gender | Timing2 | Intervening systemic therapies3 | Radiation3 | Initial Pathology | Final Pathology | Potential Mechanisms of Resistance |
|---|---|---|---|---|---|---|---|---|
| MSK-THY1 | 66 | F | After / +4 months | Pazopanib | No | PTC | ATC | PIK3CA |
| MSK-THY2 | 70 | F | During / +5 months | None | No | PTC | PDTC | NF2, MET amplification |
| MSK-THY3 | 68 | M | After / +31 months | Lenvatinib | Yes | PTC | ATC | NF2 |
| MSK-THY4 | 68 | M | After / +34 months | None | No | PTC | ATC | None |
| MSK-THY5 | 68 | F | During / +40 months | None | No | PTC | PDTC | NRAS |
| MSK-THY6 | 52 | M | After / +7 months | None | No | PDTC | ATC | ARID2, JAK1, PBRM1 |
| MSK-THY7 | 66 | M | After / +3 months | None | Yes | PDTC | ATC | RASA1, MTOR |
Age at first dose of BRAF inhibitor
When sequencing was performed relative to course of BRAF inhibition; duration from BRAF inhibitor start date reported for those sequenced during BRAF inhibition, and duration from BRAF inhibitor stop date for those sequenced after BRAF inhibition.
Therapies administered between BRAF inhibition and subsequent treatment sequencing
The mutational landscapes (Figure 3, Supplementary Figure 6A, Supplemental Table 5) of these dedifferentiated tumors were reviewed for alterations that have been linked to resistance to BRAF inhibition and/or ATC pathogenesis. These tumors underwent high coverage next-generation sequencing (mean target coverage 659×) which offered high sensitivity for the detection of subclonal variants with low allelic fraction. As expected in a cohort of thyroid tumors with aggressive clinical behavior and treated with BRAF inhibitor drugs, all tumors had mutations in BRAF as well as the TERT promoter (43–47). Genetic alterations of potential relevance identified in this cohort include mutations in genes involved in the SWI/SNF chromatin remodeling complex (ARID2, PBRM1), PI3K/AKT/mTOR pathway (MTOR, PIK3CA), MAPK/ERK pathway (MET amplification, NF2, NRAS, RASA1), and JAK/STAT pathway (JAK1).
Figure 3.
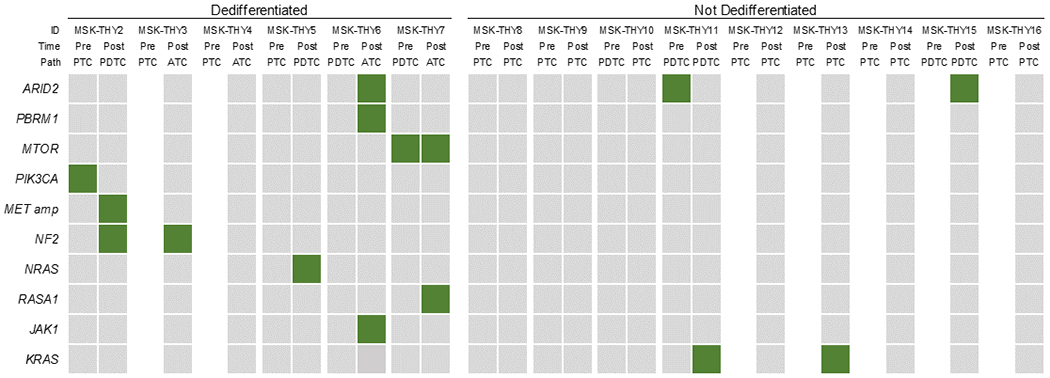
Frequency of genetic alterations linked to BRAF inhibitor resistance and/or anaplastic thyroid cancer pathogenesis for tumors that dedifferentiated after vemurafenib or dabrafenib (MSK-THY2-7) compared to those that did not dedifferentiate (MSK-THY8-16). Filled in squares represent mutated genes, with the exception of MET amp, indicating amplification. Pre: before BRAF inhibition, Post: during or after BRAF inhibition, PTC: Papillary Thyroid Cancer, PDTC: Poorly Differentiated Thyroid Cancer, ATC: Anaplastic Thyroid Cancer
The data from these 15 patients are summarized in Figure 3. Including patient MSK-THY1 (in Table 1), a total of 6 of 7 (86%) tumors that dedifferentiated after BRAF inhibition were observed to harbor mutations in one or more of these genes. In the dedifferentiated tumors with matched sequencing (MSK-THY1-2 & 5-7), 8 of the 9 (89%) mutations appeared to be acquired in on-therapy or post-therapy tissues, with the exception of 1 pre-treatment tumor that also had a mutation in MTOR. Notably, 1 tumor had a PIK3CA mutation before BRAF inhibition that was not detected in subsequent sequencing. In contrast, among tumors that did not undergo dedifferentiation (Figure 3, Supplementary Figure 6B), fewer tumors (3 of 9, 33%) had mutations in any of these genes, including one sample with a mutation in ARID2 and two samples with mutations in KRAS (9). The rate of mutations in these genes was enriched, although not to a statistically significant degree (RR=2.57, 95%CI 0.97-6.80, p=.06), in those tumors that de-differentiated. These data indicate that among tumors exposed to BRAF inhibitor therapy, mutations associated with resistance to BRAF inhibition were more frequently observed in those that evolved to more dedifferentiated states.
DISCUSSION
Papillary thyroid cancers are genetically defined by alterations that converge on the mitogen-activated protein kinase (MAPK) pathway (24), most commonly (60%) mutations in BRAF, followed by mutations in RAS genes (15%), and less commonly, rearrangements affecting other kinase genes such as RET and NTRK. Most aggressive variants of PTC have mutations in both BRAF and the TERT promoter (43–47), and this combination is commonly associated with evolution of differentiated thyroid carcinoma to more aggressive, poorly differentiated histology. The accumulation of additional genetic alterations such as TP53 mutation and alterations in the PI3K/AKT/MTOR pathway, the SWI/SNF complex and other epigenetic regulators, are associated with the evolution to ATC (25,39). At the same time, these genetic alterations are also associated with thyroid cancer resistance to BRAF inhibition (25,35,36,38–42). Here, by examining tumors that were treated with BRAF inhibitor drugs, we describe the evolution of genetic alterations, gene expression landscape, and immune microenvironment that occurs among tumors that undergo transformation to dedifferentiated or ATC histology.
We found that these genetic alterations were associated with both resistance to therapy and dedifferentiation after BRAF inhibitor therapy. Our findings in clinical samples validate a large body of preclinical research implicating several genetic programs and pathways. Thyroid tumors that dedifferentiated commonly had, or acquired, mutations in genes comprising the SWI/SNF chromatin remodeling complex (25,39) and PI3K/AKT/mTOR (33), MAPK/ERK (35,36,40,41), and JAK/STAT signaling pathways (42). In contrast, tumors that were not observed to undergo dedifferentiation did not commonly harbor these mutations. Mutations in these pathways, uncommon in PTC, were also observed to increase in prevalence in a stepwise fashion across the spectrum of more clinically aggressive and histologically dedifferentiated thyroid carcinomas.
It is unknown whether the mechanisms of drug resistance and microevolution to more dedifferentiated histologies are related, and whether BRAF inhibition promotes these processes. One mechanism of resistance to targeted therapy is the development of an alternative cell state, such as tumor dedifferentiation, as a result of selective pressures exerted by pathway blockade (11). After BRAF inhibition, dedifferentiation has been demonstrated to occur in melanoma (12,13). The findings in this study lend themselves to a hypothesis that inhibition of BRAF may, in some cases, drive the acquisition of aberrant oncogenic signaling that can both mediate drug resistance and accelerate the outgrowth of more aggressive subclonal cellular populations.
The transformation of differentiated thyroid cancer in the context of BRAF inhibition is a rare event, which to our knowledge, has only been formally reported in 2 prior cases (14). The 7 cases in this study were drawn from a cohort of 639 genomically profiled advanced thyroid cancers and represent a reasonably sized cohort of a rare phenotype.
This phenotype was most comprehensively examined in patient MSK-THY1. In this patient’s tumor, after treatment with BRAF inhibition, a PIK3CA H1047R-mutated cellular population was observed to undergo clonal expansion, driving PI3K/AKT/mTOR signaling in the tumor. At the same time, the tumor concomitantly underwent marked dedifferentiation, as evidenced by the downregulation of thyroid metabolism and function genes, and morphologic transformation to anaplastic thyroid carcinoma. Confirming the importance of this pathway, we found mutations affecting the PI3K/AKT/mTOR pathway to be present in tumors transforming subsequent to BRAF inhibition, with 2 of 7 (29%) dedifferentiating tumors in this series harboring alterations in PIK3CA or MTOR, compared to 0 of 9 (0%) in the non-dedifferentiating group. Across the spectrum of progressively less differentiated thyroid carcinoma histologies, we found that mutations in PIK3CA increased markedly from a low prevalence in localized PTC (0.4%) to higher rates in advanced/recurrent/metastatic PTC (5.1%), PDTC (5.5%) and ATC (16.0%).
These clinical and genomic data validate an earlier preclinical finding that PIK3CA contributes both to therapeutic resistance and the evolution of ATC. Roelli and colleagues used a transgenic mouse model of thyroid cancer with conditional tamoxifen-inducible Cre-mediated Braf V600E and Pik3ca H1047R mutations under a thyroid-specific promoter, and found these tumors to be resistant to treatment with PLX-4720, a BRAF inhibitor (33). After BRAF inhibition, these tumors were observed to develop paradoxically increased ERK pathway activation. This is attributable to release of feedback inhibition of ERK after BRAF V600E inhibition, which can then facilitate enhanced signaling of an upstream signal, such as activated PI3 kinase, through the wild-type BRAF allele and CRAF (48). Interestingly, these Braf/Pik3ca co-mutated thyroid tumors treated with PLX-4720 were observed to develop foci of ATC, which were also observed, but to a lesser degree, in the Braf/Pik3ca co-mutated tumors that did not receive BRAF inhibitor therapy.
Because of the overlap in molecular alterations that contribute to resistance to BRAF inhibitors and occur during thyroid tumor dedifferentiation (16), mutation in PIK3CA is only one of multiple potential mechanisms for this observed phenotype. In addition to PIK3CA mutations, a number of other resistance mechanisms have been described in prior preclinical literature. For example, mutations affecting the SWI/SNF chromatin remodeling complex (e.g., ARID2, PBRM1) are suspected to promote stem-cell like properties, negating the effects of BRAF inhibitors in re-establishing regular thyroid iodine metabolism and function (25,39). Mutations of NF2 (35), RASA1 (40), and NRAS (41), and amplification of MET (36) have been shown to reactivate MAPK/ERK signaling. JAK1 deregulation through inhibition of RNF125 has been described as a mechanism of resistance to BRAF inhibitors in melanoma (42). The results of this study, albeit derived from a small cohort of a rare phenotype, confirm these earlier findings and illustrate the suite of genetic alterations that can converge on these three pathways.
These findings have potential implications for the management of thyroid malignancies, particularly related to current areas of active investigation, including combination targeted therapies and immunotherapy. Given the near inevitable progression of disease with single-agent targeted therapy (49), consistent with the diversity of molecular alterations that may lead to resistance and/or dedifferentiation observed in our cohort, recent studies have focused on addressing complimentary pathways concurrently. A number of the resistance mechanisms described above are potentially targetable, including: MTOR with everolimus, PI3K with alpelisib and buparlisib, MET with crizotinib, and JAK1/2 with momelotinib. In addition, based on gene expression data, cell-cycle targeted therapies such as palbociclib, targeting CDK4 and CDK6, may have utility, possibly when combined with PI3K inhibition (50). Consistent with computational models (49) and experience with dual therapy in other cancer types (51,52), combination therapy may provide superior disease control in thyroid cancer. In BRAF V600E-mutated ATC, the combination of dabrafenib with trametinib is now commonly used, achieving high rates of partial response, albeit with few complete responses (53,54). However, the benefit of combination therapy is not clear in PTC—a randomized phase II study in radioiodine-refractory PTC did not show clinical benefit to the addition of trametinib compared to dabrafenib alone (55). Importantly, it is not known whether such approaches would mitigate the selective pressure that may drive dedifferentiation or anaplastic transformation. For example, in the study of Braf and Pik3ca co-mutated mouse tumors, treatment with dual inhibition of Braf and Pik3ca with PLX-4072 and GDC-0941 was more effective at achieving tumor size reduction, but not in preventing the development of ATC foci (33). These data suggest a treatment strategy of combination targeted therapy in the neoadjuvant setting to achieve tumor volume reduction, followed by complete surgical resection, an approach that is being investigated in localized but unresectable, aggressive thyroid cancers.
Combination therapy with BRAF inhibition and immunotherapy is currently under investigation (56); in addition to data with BRAF inhibitors, the widely used multi-kinase inhibitor lenvatinib has also been shown to increase CD8+ T-cell infiltration (57). In the serial analyses of the tumor immune microenvironment in patient MSK-THY1, we observed evidence of immune escape as well as infiltration by immunosuppressive cellular populations. These findings may reflect mechanisms of immune escape during dedifferentiation of this thyroid tumor but may also in part represent effects of BRAF inhibition on the tumor immune microenvironment. While BRAF inhibition is generally associated with a shift toward stronger anti-tumor immunity, such as enhanced T cell infiltration and function (58), it also drives an increase in infiltration by macrophages, which are able to mediate resistance to BRAF inhibition via macrophage-derived secreted factors TNF-α and VEGF (59). Ongoing clinical investigation will help determine whether enhanced immune surveillance can potentially constrain the transformation of PTCs to more dedifferentiated forms.
There are several important caveats to these findings; because of these limitations, we note that our data do not prove a causal link between BRAF inhibition and thyroid tumor dedifferentiation. Although our data do provide clinical validation of the prior mouse data indicating the role of PIK3CA mutations in both mediating resistance to BRAF inhibition as well as driving anaplastic transformation of PTC, conclusive data for other pathways is currently incomplete. While it is quite plausible that BRAF inhibition promoted the upregulation of these compensatory signaling pathways, it is also possible that these mutations and signaling pathways were already active in the primary tumor and were merely unmasked by undergoing clonal expansion during treatment with BRAF inhibitors. With these data, we cannot conclusively distinguish between dedifferentiation that was an inevitable part of the natural history of these tumors, as opposed to a phenomenon that was driven by BRAF inhibition. For example, the interval between the development of dedifferentiation and the last dose of BRAF inhibitor therapy was close for most, but not all patients—2 patients had an interval of over 2 years. In some cases, other factors may have in part contributed to the observed genomic changes, such as other systemic therapies in two patients.
Nevertheless, these data suggest that those genetic pathways that mediate resistance to BRAF inhibition may also be linked to transformation to ATC. The genetic alterations in the above pathways, which were observed in nearly all dedifferentiating tumors, were not commonly observed in the heavily-treated BRAF-mutated thyroid tumors that did not undergo dedifferentiation after BRAF inhibitor therapy. An additional important caveat is that this is a selected cohort, and these data should not be extrapolated to estimate the frequency of dedifferentiation after BRAF inhibition, since the decision to re-biopsy after therapy was generally performed after tumor progression, and not universally performed. Lastly, insights into tumor immune microenvironment were made by deconvolution of immune cells with transcriptomic data rather than immunohistochemical markers; however, these methods have been shown to be highly correlated in prior studies (26–29).
In conclusion, we have analyzed genomic data from thyroid cancers that progressed to more advanced histologies subsequent to BRAF inhibitor targeted therapy, and found that this event is associated with a diverse array of genetic alterations with known roles in either BRAF inhibitor resistance or ATC pathogenesis. These data indicate that alterations in SWI/SNF, PI3K/AKT/MTOR, MAPK/ERK and JAK/STAT may underlie both the processes of drug resistance and dedifferentiation to ATC. The diversity of potential mechanisms that may lead to resistance and/or dedifferentiation may explain the elusiveness of disease control with single-agent targeted therapy for advanced thyroid malignancies, and emphasizes the need to investigate combination therapies. Further research will be needed to determine the degree to which these genomic changes can be attributed to BRAF inhibitor therapies and resulting selective pressure. To this end, studies that incorporate long term clinical outcomes and genomic analyses in thyroid cancer patients with BRAF mutations will be informative.
Supplementary Material
Implications:
The possibility that thyroid cancer dedifferentiation may be attributed to selective pressure applied by BRAF inhibitor targeted therapy should be investigated further.
Acknowledgements
We are grateful to our patients and their families for their bravery and support of cancer research. We thank members of the Molecular Diagnostics Service in the Department of Pathology, the Marie-Josée and Henry R. Kravis Center for Molecular Oncology, and members of the Morris, Chan and Fagin Labs for helpful discussions.
This work was supported by Fundación Alfonso Martín Escudero (to CV, no grant number), the National Institutes of Health (K08 DE024774 and R01 DE027738), the Sebastian Nativo Fund, the Jayme and Peter Flowers Fund (to LGTM), the Pershing Square Sohn Cancer Research Foundation, the PaineWebber Chair, Stand Up To Cancer, the STARR Cancer Consortium, the National Institutes of Health (R01 CA205426 and R35 CA232097) (to TAC), and the National Institutes of Health Cancer Center Support Grant (P30 CA008748) (to MSKCC).
Author disclosures
ALH received funding from Eisai, BMS, Kura Oncology, AstraZeneca, Genentech, Roche, Celldex, Pfizer, Lilly, Bayer, consulting fees from BMS, Merck, Novartis, AstraZeneca, Regeneron, Sanofi Aventis, Sun Pharmaceuticals, Eisai, Genentech/Roche, Genzyme, Ayala Pharmaceuticals, and travel reimbursement from Ignyta and Kura Oncology. TAC acknowledges grant funding from Bristol-Myers Squibb, AstraZeneca, Illumina, Pfizer, An2H, and Eisai, has served as an advisor for Bristol-Myers Squibb, Illumina, Eisai, and An2H, holds equity in An2H, and is a co-founder of Gritstone Oncology and holds equity. TAC and LGTM are inventors on a patent held by Memorial Sloan Kettering related to the use of TMB in cancer immunotherapy. LGTM reports laboratory research funding from AstraZeneca. All remaining authors have declared no conflicts of interest.
References
- 1.Shah JP, Patel SG, Singh B. Head and neck surgery and oncology. 5th ed. Elsevier Health Sciences; 2012. [Google Scholar]
- 2.Fagin JA, Wells SA Jr. Biologic and clinical perspectives on thyroid cancer. N Engl J Med 2016;375(11):1054–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fan D, Ma J, Bell AC, Groen AH, Olsen KS, Lok BH, et al. Outcomes of multimodal therapy in a large series of patients with anaplastic thyroid cancer. Cancer 2020;126(2):444–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maniakas A, Dadu R, Busaidy NL, Wang JR, Ferrarotto R, Lu C, et al. Evaluation of overall survival in patients with anaplastic thyroid carcinoma, 2000-2019. JAMA Oncol 2020;6(9):1397–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cabanillas ME, Dadu R, Iyer P, Wanland KB, Busaidy NL, Ying A, et al. Acquired secondary RAS mutation in BRAFV600E-mutated thyroid cancer patients treated with BRAF inhibitors. Thyroid 2020;30(9):1288–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baitei EY, Zou M, Al-Mohanna F, Collison K, Alzahrani AS, Farid NR, et al. Aberrant BRAF splicing as an alternative mechanism for oncogenic B-Raf activation in thyroid carcinoma. J Pathol 2009;217(5):707–15. [DOI] [PubMed] [Google Scholar]
- 7.Byeon HK, Na HJ, Yang YJ, Kwon HJ, Chang JW, Ban MJ, et al. c-Met-mediated reactivation of PI3K/AKT signaling contributes to insensitivity of BRAF (V600E) mutant thyroid cancer to BRAF inhibition. Mol Carcinog 2016;55(11):1678–87. [DOI] [PubMed] [Google Scholar]
- 8.Sos ML, Levin RS, Gordan JD, Oses-Prieto JA, Webber JT, Salt M, et al. Oncogene mimicry as a mechanism of primary resistance to BRAF inhibitors. Cell Rep 2014;8(4):1037–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Danysh BP, Rieger EY, Sinha DK, Evers CV, Cote GJ, Cabanillas ME, et al. Long-term vemurafenib treatment drives inhibitor resistance through a spontaneous KRAS G12D mutation in a BRAF V600E papillary thyroid carcinoma model. Oncotarget 2016;7(21):30907–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Montero-Conde C, Ruiz-Llorente S, Dominguez JM, Knauf JA, Viale A, Sherman EJ, et al. Relief of feedback inhibition of HER3 transcription by RAF and MEK inhibitors attenuates their antitumor effects in BRAF-mutant thyroid carcinomas. Cancer Discov 2013;3(5):520–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Konieczkowski DJ, Johannessen CM, Garraway LA. A convergence-based framework for cancer drug resistance. Cancer Cell 2018;33(5):801–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tsoi J, Robert L, Paraiso K, Galvan C, Sheu KM, Lay J, et al. Multi-stage differentiation defines melanoma subtypes with differential vulnerability to drug-induced iron-dependent oxidative stress. Cancer Cell 2018;33(5):890–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Konieczkowski DJ, Johannessen CM, Abudayyeh O, Kim JW, Cooper ZA, Piris A, et al. A melanoma cell state distinction influences sensitivity to MAPK pathway inhibitors. Cancer Discov 2014;4(7):816–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Patham B, Bible K, Waguespack S, Cabanillas M. Anaplastic transformation (ATC-t) of papillary thyroid cancer (PTC) after treatment with BRAF inhibitors (BRAFi). Endocr Rev 2015;36:SAT-011. [Google Scholar]
- 15.Sera N, Ashizawa K, Ando T, Ide A, Abe Y, Usa T, et al. Anaplastic changes associated with p53 gene mutation in differentiated thyroid carcinoma after insufficient radioactive iodine (131I) therapy. Thyroid 2000;10(11):975–9. [DOI] [PubMed] [Google Scholar]
- 16.Crispo F, Notarangelo T, Pietrafesa M, Lettini G, Storto G, Sgambato A, et al. Braf inhibitors in thyroid cancer: clinical impact, mechanisms of resistance and future perspectives. Cancers 2019;11(9):1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lloyd R, Osamura R, Klöppel G, Rosai J. WHO Classification of Tumours of Endocrine Organs. World Health Organization; 2017. [Google Scholar]
- 18.Dalin MG, Katabi N, Persson M, Lee K-W, Makarov V, Desrichard A, et al. Multi-dimensional genomic analysis of myoepithelial carcinoma identifies prevalent oncogenic gene fusions. Nat Commun 2017;8(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shen R, Seshan VE. FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res 2016;44(16):e131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roth A, Khattra J, Yap D, Wan A, Laks E, Biele J, et al. PyClone: statistical inference of clonal population structure in cancer. Nat Methods 2014;11(4):396–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morris LG, Riaz N, Desrichard A, Şenbabaoğlu Y, Hakimi AA, Makarov V, et al. Pan-cancer analysis of intratumor heterogeneity as a prognostic determinant of survival. Oncotarget 2016;7(9):10051–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): a hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J Mol Diagn 2015;17(3):251–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fang F, Turcan S, Rimner A, Kaufman A, Giri D, Morris LG, et al. Breast cancer methylomes establish an epigenomic foundation for metastasis. Sci Transl Med 2011;3(75):75ra25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Agrawal N, Akbani R, Aksoy BA, Ally A, Arachchi H, Asa SL, et al. Integrated genomic characterization of papillary thyroid carcinoma. Cell 2014;159(3):676–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Landa I, Ibrahimpasic T, Boucai L, Sinha R, Knauf JA, Shah RH, et al. Genomic and transcriptomic hallmarks of poorly differentiated and anaplastic thyroid cancers. J Clin Invest 2016;126(3):1052–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods 2015;12(5):453–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Şenbabaoğlu Y, Gejman RS, Winer AG, Liu M, Van Allen EM, de Velasco G, et al. Tumor immune microenvironment characterization in clear cell renal cell carcinoma identifies prognostic and immunotherapeutically relevant messenger RNA signatures. Genome Biol 2016;17(1):1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yoshihara K, Shahmoradgoli M, Martínez E, Vegesna R, Kim H, Torres-Garcia W, et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun 2013;4(1):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiang P, Gu S, Pan D, Fu J, Sahu A, Hu X, et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat Med 2018;24(10):1550–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sherman EJ, Ho AL, Fury MG, Baxi SS, Haque S, Korte SH, et al. A phase II study of temsirolimus/sorafenib in patients with radioactive iodine (RAI)-refractory thyroid carcinoma. J Clin Oncol; 2012. [Google Scholar]
- 31.Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 2009;45(2):228–47. [DOI] [PubMed] [Google Scholar]
- 32.Brose MS, Cabanillas ME, Cohen EE, Wirth LJ, Riehl T, Yue H, et al. Vemurafenib in patients with BRAFV600E-positive metastatic or unresectable papillary thyroid cancer refractory to radioactive iodine: a non-randomised, multicentre, open-label, phase 2 trial. Lancet Oncol 2016;17(9):1272–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roelli MA, Ruffieux-Daidié D, Stooss A, ElMokh O, Phillips WA, Dettmer MS, et al. PIK3CAH1047R-induced paradoxical ERK activation results in resistance to BRAFV600E specific inhibitors in BRAFV600E PIK3CAH1047R double mutant thyroid tumors. Oncotarget 2017;8(61):103207–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Irvine M, Stewart A, Pedersen B, Boyd S, Kefford R, Rizos H. Oncogenic PI3K/AKT promotes the step-wise evolution of combination BRAF/MEK inhibitor resistance in melanoma. Oncogenesis 2018;7(9):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garcia-Rendueles ME, Ricarte-Filho JC, Untch BR, Landa I, Knauf JA, Voza F, et al. NF2 loss promotes oncogenic RAS-induced thyroid cancers via YAP-dependent transactivation of RAS proteins and sensitizes them to MEK inhibition. Cancer Discov 2015;5(11):1178–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Knauf JA, Luckett KA, Chen K-Y, Voza F, Socci ND, Ghossein R, et al. Hgf/Met activation mediates resistance to BRAF inhibition in murine anaplastic thyroid cancers. J Clin Invest 2018;128(9):4086–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McFadden DG, Vernon A, Santiago PM, Martinez-McFaline R, Bhutkar A, Crowley DM, et al. p53 constrains progression to anaplastic thyroid carcinoma in a Braf-mutant mouse model of papillary thyroid cancer. Proc Natl Acad Sci U S A 2014;111(16):E1600–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pozdeyev N, Gay LM, Sokol ES, Hartmaier R, Deaver KE, Davis S, et al. Genetic analysis of 779 advanced differentiated and anaplastic thyroid cancers. Clin Cancer Res 2018;24(13):3059–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saqcena M, Leandro-Garcia LJ, Maag JL, Tchekmedyian V, Krishnamoorthy GP, Tamarapu PP, et al. SWI/SNF complex mutations promote thyroid tumor progression and insensitivity to redifferentiation therapies. Cancer Discov 2020;11(5):1158–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jouenne F, De Moura CR, Lorillon G, Meignin V, Dumaz N, Lebbe C, et al. RASA1 loss in a BRAF-mutated Langerhans cell sarcoma: a mechanism of resistance to BRAF inhibitor. Ann Oncol 2019;30(7):1170–2. [DOI] [PubMed] [Google Scholar]
- 41.Kaplan FM, Kugel CH, Dadpey N, Shao Y, Abel EV, Aplin AE. SHOC2 and CRAF mediate ERK1/2 reactivation in mutant NRAS-mediated resistance to RAF inhibitor. J Biol Chem 2012;287(50):41797–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim H, Frederick DT, Levesque MP, Cooper ZA, Feng Y, Krepler C, et al. Downregulation of the ubiquitin ligase RNF125 underlies resistance of melanoma cells to BRAF inhibitors via JAK1 deregulation. Cell Rep 2015;11(9):1458–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu X, Qu S, Liu R, Sheng C, Shi X, Zhu G, et al. TERT promoter mutations and their association with BRAF V600E mutation and aggressive clinicopathological characteristics of thyroid cancer. J Clin Endocrinol Metab 2014;99(6):E1130–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xing M, Liu R, Liu X, Murugan AK, Zhu G, Zeiger MA, et al. BRAF V600E and TERT promoter mutations cooperatively identify the most aggressive papillary thyroid cancer with highest recurrence. J Clin Oncol 2014;32(25):2718–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu R, Bishop J, Zhu G, Zhang T, Ladenson PW, Xing M. Mortality risk stratification by combining BRAF V600E and TERT promoter mutations in papillary thyroid cancer: genetic duet of BRAF and TERT promoter mutations in thyroid cancer mortality. JAMA Oncol 2017;3(2):202–8. [DOI] [PubMed] [Google Scholar]
- 46.Landa I, Ganly I, Chan TA, Mitsutake N, Matsuse M, Ibrahimpasic T, et al. Frequent somatic TERT promoter mutations in thyroid cancer: higher prevalence in advanced forms of the disease. J Clin Endocrinol Metab 2013;98(9):E1562–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Song YS, Lim JA, Choi H, Won JK, Moon JH, Cho SW, et al. Prognostic effects of TERT promoter mutations are enhanced by coexistence with BRAF or RAS mutations and strengthen the risk prediction by the ATA or TNM staging system in differentiated thyroid cancer patients. Cancer 2016;122(9):1370–9. [DOI] [PubMed] [Google Scholar]
- 48.Hatzivassiliou G, Song K, Yen I, Brandhuber BJ, Anderson DJ, Alvarado R, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature 2010;464(7287):431–5. [DOI] [PubMed] [Google Scholar]
- 49.Bozic I, Reiter JG, Allen B, Antal T, Chatterjee K, Shah P, et al. Evolutionary dynamics of cancer in response to targeted combination therapy. Elife 2013;2:e00747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wong K, Di Cristofano F, Ranieri M, De Martino D, Di Cristofano A. PI3K/mTOR inhibition potentiates and extends palbociclib activity in anaplastic thyroid cancer. Endocr Relat Cancer 2019;26(4):425–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Larkin J, Ascierto PA, Dréno B, Atkinson V, Liszkay G, Maio M, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med 2014;371(20):1867–76. [DOI] [PubMed] [Google Scholar]
- 52.Long GV, Stroyakovskiy D, Gogas H, Levchenko E, De Braud F, Larkin J, et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: a multicentre, double-blind, phase 3 randomised controlled trial. Lancet 2015;386(9992):444–51. [DOI] [PubMed] [Google Scholar]
- 53.Subbiah V, Kreitman RJ, Wainberg ZA, Cho JY, Schellens JH, Soria JC, et al. Dabrafenib and trametinib treatment in patients with locally advanced or metastatic BRAF V600–mutant anaplastic thyroid cancer. J Clin Oncol 2018;36(1):7–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Keam B, Kreitman R, Wainberg Z, Cabanillas M, Cho D, Italiano A, et al. Updated efficacy and safety data of dabrafenib (D) and trametinib (T) in patients (pts) with BRAF V600E–mutated anaplastic thyroid cancer (ATC). Ann Oncol 2018;29:VIII645–6. [Google Scholar]
- 55.Shah MH, Wei L, Wirth LJ, Daniels GA, De Souza JA, Timmers CD, et al. Results of randomized phase II trial of dabrafenib versus dabrafenib plus trametinib in BRAF-mutated papillary thyroid carcinoma. J Clin Oncol 2017;35(15):6022. [Google Scholar]
- 56.Pembrolizumab, dabrafenib, and trametinib before surgery for the treatment of BRAF-mutated anaplastic thyroid cancer. ClinicalTrials.gov identifier: NCT04675710. Updated January 31, 2020. Accessed January 31, 2021. https://www.clinicaltrials.gov/ct2/show/NCT04675710?term=pembrolizumab+dabrafenib&cond=Thyroid+Cancer&draw=2&rank=1
- 57.Gunda V, Gigliotti B, Ashry T, Ndishabandi D, McCarthy M, Zhou Z, et al. Anti-PD-1/PD-L1 therapy augments lenvatinib’s efficacy by favorably altering the immune microenvironment of murine anaplastic thyroid cancer. Int J Cancer 2019;144(9):2266–78. [DOI] [PubMed] [Google Scholar]
- 58.Callahan MK, Masters G, Pratilas CA, Ariyan C, Katz J, Kitano S, et al. Paradoxical activation of T cells via augmented ERK signaling mediated by a RAF inhibitor. Cancer Immunol Res 2014;2(1):70–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Smith MP, Sanchez-Laorden B, O’Brien K, Brunton H, Ferguson J, Young H, et al. The immune microenvironment confers resistance to MAPK pathway inhibitors through macrophage-derived TNFα. Cancer Discov 2014;4(10):1214–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The sequencing variant calls and gene expression data used in this paper are provided in Supplementary Tables.