

Abstract
Outbreaks of the Ebola virus (EBOV) continue to occur and while a vaccine and treatment are now available, there remains a dearth of options for those who become sick with EBOV disease. An understanding at the atomic and molecular level of the various steps in the EBOV replication cycle can provide molecular targets for disrupting the virus. An important step in the EBOV replication cycle is the transport of EBOV structural matrix VP40 protein molecules to the plasma membrane inner leaflet, which involves VP40 binding to the host cell’s Sec24c protein. Though some VP40 residues involved in the binding are known, the molecular details of VP40-Sec24c binding are not known. We use various molecular computational techniques to investigate the molecular details of how EBOV VP40 binds with the Sec24c complex of the ESCRT-I pathway. We employed different docking programs to identify the VP40 binding site on Sec24c and then performed molecular dynamics simulations to determine the atomic details and binding interactions of the complex. We also investigated how the inter-protein interactions of the complex are affected upon mutations of VP40 amino acids in the Sec24c binding region. Our results provide a molecular basis for understanding previous coimmunoprecipitation experimental studies. In addition, we found that VP40 can bind to a site on Sec24c that can also bind Sec23 and suggests that VP40 may use the COPII transport mechanism in a manner that may not need the Sec23 protein in order for VP40 to be transported to the plasma membrane.
1. Introduction
The Ebola virus (EBOV), a negative sense RNA virus, belongs to the Filoviridae family1,2. EBOV causes hemorrhagic fever with high fatality rates3 and despite recent approval of a vaccine against EBOV, only one monoclonal antibody has been approved to treat EBOV disease4–6. The 19-kb long EBOV genome encodes seven different proteins, including the peripheral protein VP40 that is involved in forming the viral filaments’ structural matrix7. There is also a glycoprotein (GP), as well as other proteins (VP24, VP30, VP35, nucleoprotein (NP), L protein, and an oligomer8 of VP40) that are responsible for the transcription and replication processes of the virus. VP40 associates with the plasma membrane (PM) of the host cell and alone can form virus-like particles (VLPs), indicating that VP40 is the most important protein for the structural assembly and budding of the virus9,10. The monomeric structure of VP40 consists of two domains: the C-terminal domain (CTD) that is involved in the lipid binding process, and the N-terminal domain (NTD) that is involved in the VP40 dimerization process11. Soon after an infected cell produces a VP40 monomer, it dimerizes and the VP40 dimer migrates and associates with PM lipids12–15 and forms oligomers16–18. At the PM, VP40 dimers oligomerize via CTD-CTD interactions into a matrix lattice and initiate the filamentous structure of a new VLP19.
Before interacting with the PM and forming the VLPs, the VP40 protein must be transported to the PM. As with other viral lifecycles, binding interactions between viral and host proteins play important roles20. One of the pathways utilized by viruses to transport proteins within the host cell is the coat protein complex II (COPII) pathway21,22. In general, COPII forms coat vesicles on the ER membrane. These coat vesicles transport cargo molecules such as VP40 from the site where they are synthesized on the endoplasmic reticulum (ER) to other locations in the cell such as the PM or the Golgi complex in human cells23. Various COPII proteins (Sec23, Sec24, Sec13, Sec31, Sar1, GTP) are required to form a coat vesicle. After insertion of Sar1 in the ER-membrane, it then interacts with GTP, which allows the recruitment of Sec23/Sec24 to form a Sar1-Sec23/Sec24 inner layer of the coat vesicle attached to the ER membrane. This inner layer of the coat vesicle binds with the cargo protein, and the transport vesicle is completed by the attachment of Sec13/Sec31 to form an outer layer. The coat vesicle initiates membrane curvature change in the ER membrane that then allows the budding of the coat vesicle and dissociation from the ER. The Sec24 protein plays a role in identifying the cargo proteins during coat vesicle formation. The Sec24 protein has four isoforms: Sec24a, Sec24b, Sec24c and Sec24d23,24. These isoforms of Sec24 are responsible for packaging and interacting with various proteins. For example, the entry of the Hepatitis virus, which is regulated by the co-receptor Claudin-1 has been shown to be dependent on transport of Sec24c21. Similarly, the Sec24c coat protein binds with other viral proteins, such as EBOV VP40 and transports them25.
Yamayoshi et al.26 demonstrated experimentally that EBOV VP40 is transported to the PM using the COPII transport system. VP40 interacts with the Sec24c protein in the coat vesicle. The important binding residues on VP40 are known to include residues 210 and 211 and residues 303 to 30826,27, however, the binding site on the Sec24c protein is still not identified and molecular details of VP40-Sec24c interactions are lacking. Therefore, the important protein-protein residue interactions for the VP40-Sec24c complex also remain elusive. As the interaction of Sec24c with VP40 occurs in the initial phase of the EBOV replication cycle, understanding the atomic level details of this interaction will be useful for determining targets for drug molecules on those binding regions of the complex. In this work, we investigated the molecular details of how EBOV VP40 binds with the human COPII protein Sec24c. We used various molecular computational techniques and employed different docking programs to identify the binding site on Sec24c using the known binding site information of VP40. We performed molecular dynamics simulations for the highest ranked Sec24c-VP40 dock complex to determine the atomic details and binding interactions of the complex. We also investigated how the inter-protein atomic interactions of the complex are affected upon mutations of VP40 amino acids in the binding region, which has been performed experimentally. For the L303A and I307A mutation, we observed a decrease in inter-protein interactions, as also observed experimentally. For the M305A mutation, we observed little change in the interactions. We describe the atomic level details that explain the changes in inter-protein interactions that we observe.
The sequence of molecular docking steps involving VP40, Sec24c and Sec23 that facilitates the transport of VP40 to the plasma membrane is not yet completely understood. To gain insight, we also performed docking of VP40 at the site on Sec24c that can also bind Sec23. The binding of VP40 in the trunk domain region of Sec24c that also binds Sec23 suggests that VP40 might use the COPII transport mechanism in a manner that may not need the Sec23 protein in order for VP40 to be transported to the plasma membrane.
2. Materials and methods
The human Sec24c (PDB ID: 3EH2)28 and EBOV VP40 (PDB ID: 4LDB)11 structures were obtained from the Protein Data Bank. The crystal structure of Sec24c is a trimer (Fig. 1a) and that of VP40 is a dimer (Fig. 1b). We used chain A of both crystal structures (Fig. 1c and Fig. 1d) for our study. The missing residues of the structures were added using the Modeller29 software package.
Figure 1.
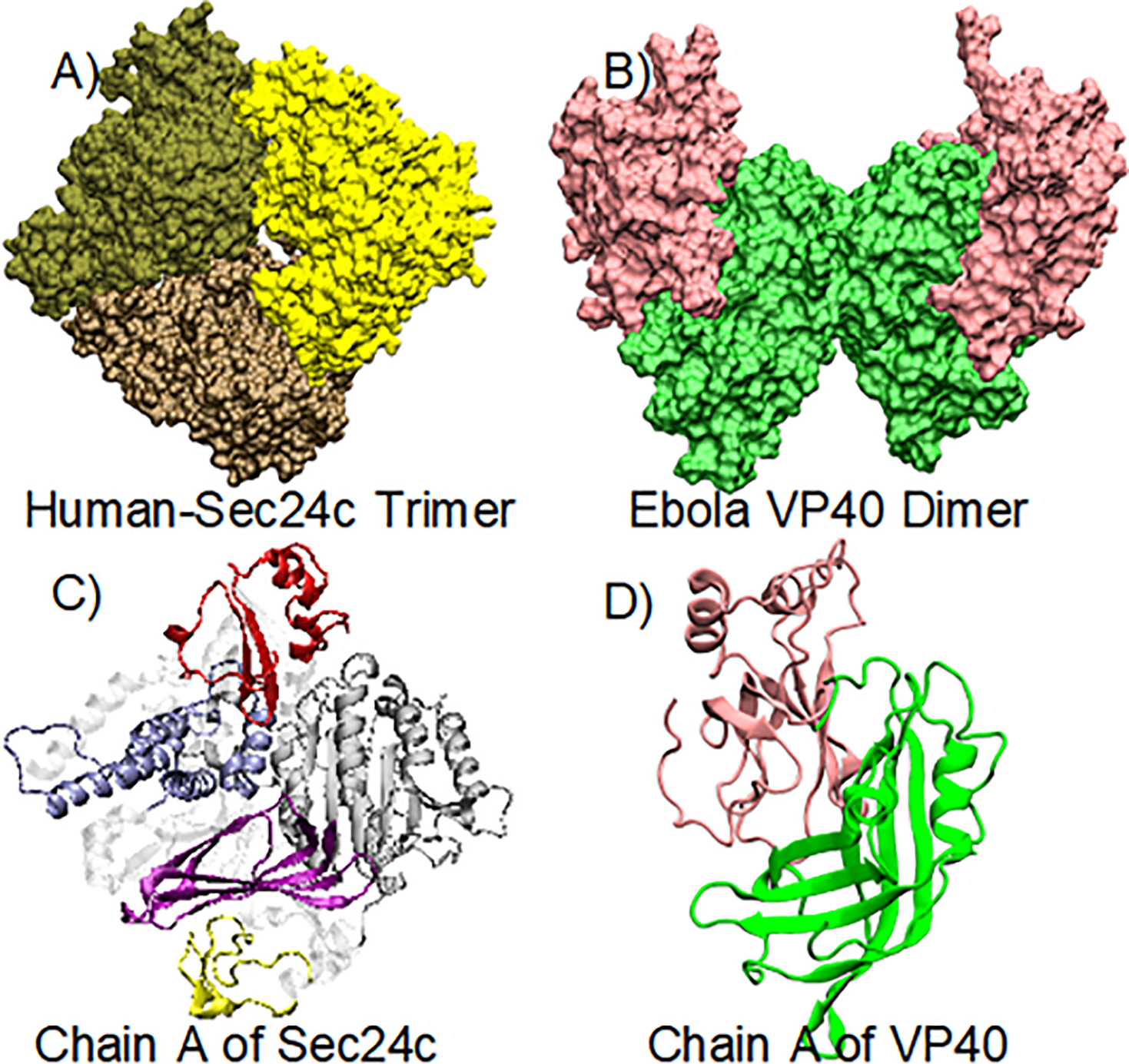
(A) Crystal structure of the human Sec24c trimer (PDB: 3EH2); the three chains are colored differently. (B) Crystal structure of the Ebola virus VP40 dimer: NTDs (green) and CTDs (pink). (C) Monomer of Sec24c: zinc finger (yellow), trunk domain (gray), beta-sandwich (purple), helical-domain (blue), gelsolin-like domain (red). (D) Monomeric structure of the VP40 protein.
2.1. Protein-protein docking
The protein structures used for docking contained residues 329−1094 for Sec24c chain A and 44−326 for VP40 chain A. The first 328 residues are missing in the crystal structure of Sec24c because they are disordered as well as hypervariable. In order to determine the reliability of our docking sites, we used three different docking software programs. Protein-protein docking was performed with the use of ZDOCK, CLUSPRO, and Patch Dock with the Fire Dock refinement30–33. Top ranked docking complexes were obtained from each docking program and analyzed for the interactions between protein-protein complexes, and the best complex was selected for molecular dynamics simulations that is consistent with experimental information26.
2.2. Molecular dynamics (MD) simulation:
Since the Sec24c binding region of VP40 is in the CTD, we considered the CTD region of VP40 for our molecular dynamics (MD) simulations. This smaller system enables us to sample for longer time scales while performing MD simulations. We also performed MD simulations on three separate VP40 point-mutations that were experimentally investigated previously. The three different mutations are in the CTD domain of VP40: L303A, M305A, and I307A. Therefore, there are four different systems; the wild type and the three separate mutations.
The CHARMM-GUI web-server34 was used to set up input files for the all-atom simulations. The system was solvated using the TIP3 water model in a cubic box and was neutralized with counter ions. The final solvated and ionized system of the Sec24c-VP40 complex contained 72,000 atoms. All-atom MD simulations was performed with NAMD 2.1235 using the CHARMM36 force field36. Each system was minimized for 10,000 steps and equilibrated for 100 ps with backbone and sidechain restraints. The particle mesh Ewald37 method was used for the long-range electrostatic interactions, and the SHAKE algorithm38 was employed for constraining the covalent bonds. The Nose−́Hoover Langevin 39 method with a piston period of 50 fs and a decay of 25 fs was used to control the pressure. Similarly, Langevin temperature coupling with a friction coefficient of 1 ps−1 was used to control the temperature. A 2 fs time step was used to propagate the simulations. For each system, a 500 ns MD simulation was performed. Image rendering, visualization and data analysis were performed with the use of the Visual MD (VMD)40 software package.
3. Results and Discussion
The crystal structure of Sec24c (PDB ID: 3EH2)28, as shown in Fig. 1a, has three identical chains colored differently. We performed domain prediction analysis with chain A of Sec24c (Fig. 1c) using InterPro41. Since the first 328 residues are missing in the crystal structure of Sec24c, for our computations we renumbered the residues so that residue 329 is now residue 1 and residue 1094 is now residue 767. The domain prediction identified five different domains in chain A of Sec24c: zinc finger (residues 94 to 132, yellow), trunk domain (171 to 417, gray), beta-sandwich (420 to 503, purple), helical domain (516 to 615, blue) and gelsolin domain (634 to 706, red). The crystal structure of the EBOV dimeric VP40 (PDB ID: 4LDB) is shown in Fig. 1b and its monomeric structure is shown in Fig. 1d. The monomer has two domains: N-terminal domain (residues 44 to 196, green) and C-terminal domain (197 to 326, pink)11.
3.1. Sec24c and VP40 docking
Before performing docking of VP40 with Sec24c, we explored the different binding regions in the Sec24 protein (PDB ID:1M2V42) with various other proteins23,24,42–45. Figure 2 shows the different binding sites in Sec24 (blue) and the corresponding region in Sec24c (red). Of the five different binding sites, sites A and B are the most studied. The A–site is responsible for binding the Sed5 protein, whereas the B-site binds Sys1, Bet1 and Yor143. Another studied site, which is also known as the IxM binding site, contains the LIL motif and is shown to be important for binding of the Hepatitis virus co-receptor protein Claudin-121. Recently, Pagent et. al.43 found a new site in Sec24 which is referred to as the D-site and is responsible for binding Erv14. Apart from binding several other proteins, Sec24 possess a trunk domain which is responsible for binding with the Sec23 protein39, an essential interaction for COPII transport.
Figure 2.
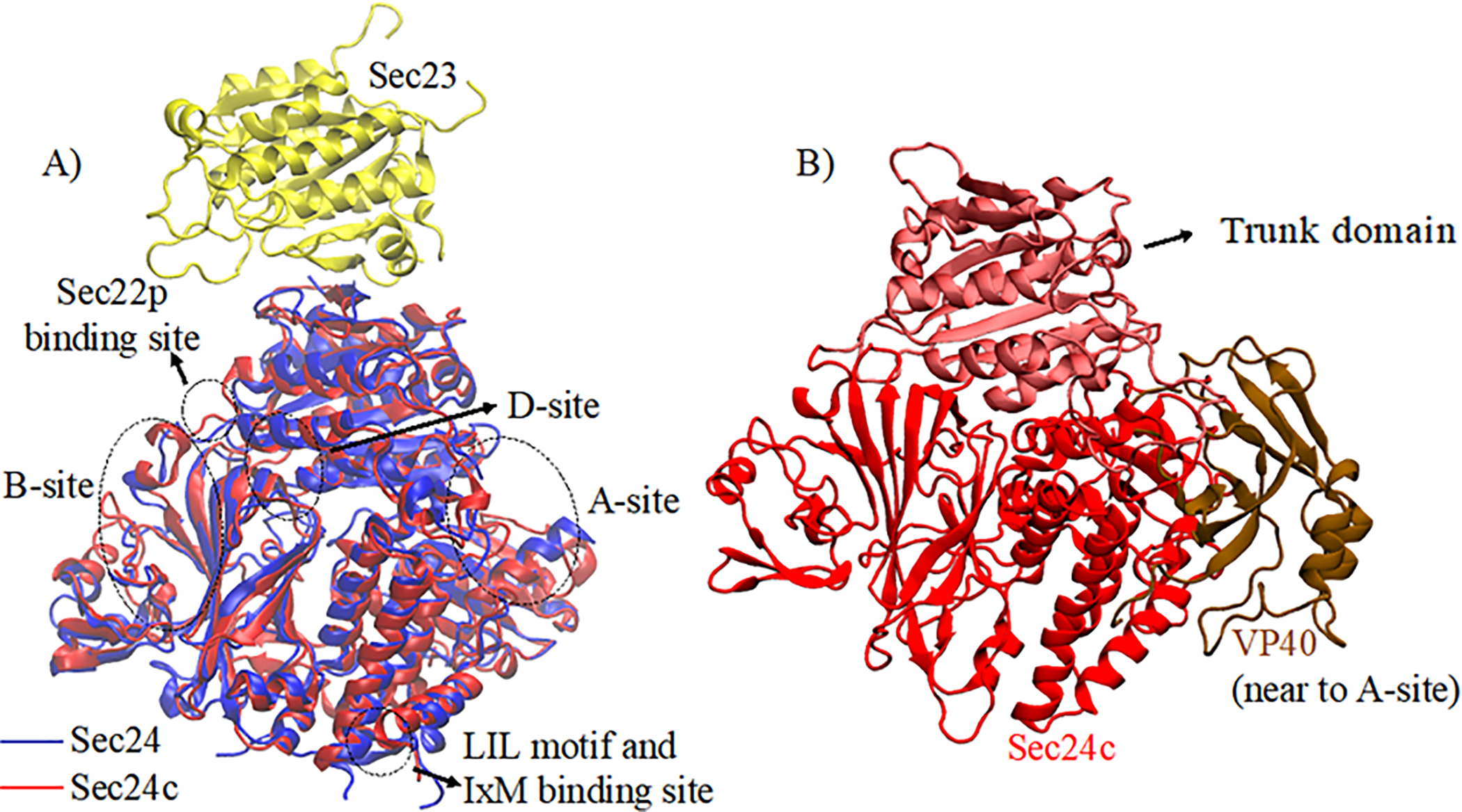
A) Sequence alignment of Sec24 (blue) and Sec24c (red) highlighting different binding sites. Sec23 (yellow) binding to Sec24 in the trunk domain region is also shown. (B) Docking complex of Sec24c (red) with VP40-CTD (brown).
To determine the VP40 docking site on Sec24c and understand the interactions between EBOV VP40 and Sec24c on the atomic level, we performed molecular docking using several docking programs, including ZDOCK, ClusPro, and Patch Dock with the Fire Dock refinement. It has been proposed that residues 196–326 in VP40 may be important for docking to Sec24c. However, mutagenesis studies showed that the region spanning residues 303 to 308 are essential for coimmunoprecipitation of Sec24c24. With this VP40 binding information, we performed docking of Sec24c to a VP40 CTD domain.
We performed blind docking of chain A of Sec24c with the VP40-CTD. Each docking program ranked Sec24c-VP40 complexes according to their docking score. Top ranked docking complexes from each of the docking programs were visually inspected, and the orientation and interactions between Sec24c and VP40 were analyzed. We visually inspected the orientation of region 303 to 308 in VP40 with respect to Sec24c for all complexes. Based on the visualization and interaction of VP40 residues with Sec24c, the second complex from Patch Dock was selected for further analysis. As shown in Fig. 2b, in this docked complex the VP40 binding site was observed to be near the A-site (Fig. 2a) of Sec24c. The interactions between Sec24c and VP40 in the docked complex selected was further optimized using molecular dynamics simulations.
3.2. Molecular interactions determined from MD computational simulations
To investigate the structural stability of the VP40-Sec24c complex, we performed a 500 ns all-atom, explicit solvent molecular dynamics (MD) simulation for the selected complex. Figure 3A shows the structure used for the MD simulation, which includes the CTD domain of VP40 and the trunk domain of Sec24c.
Figure 3.
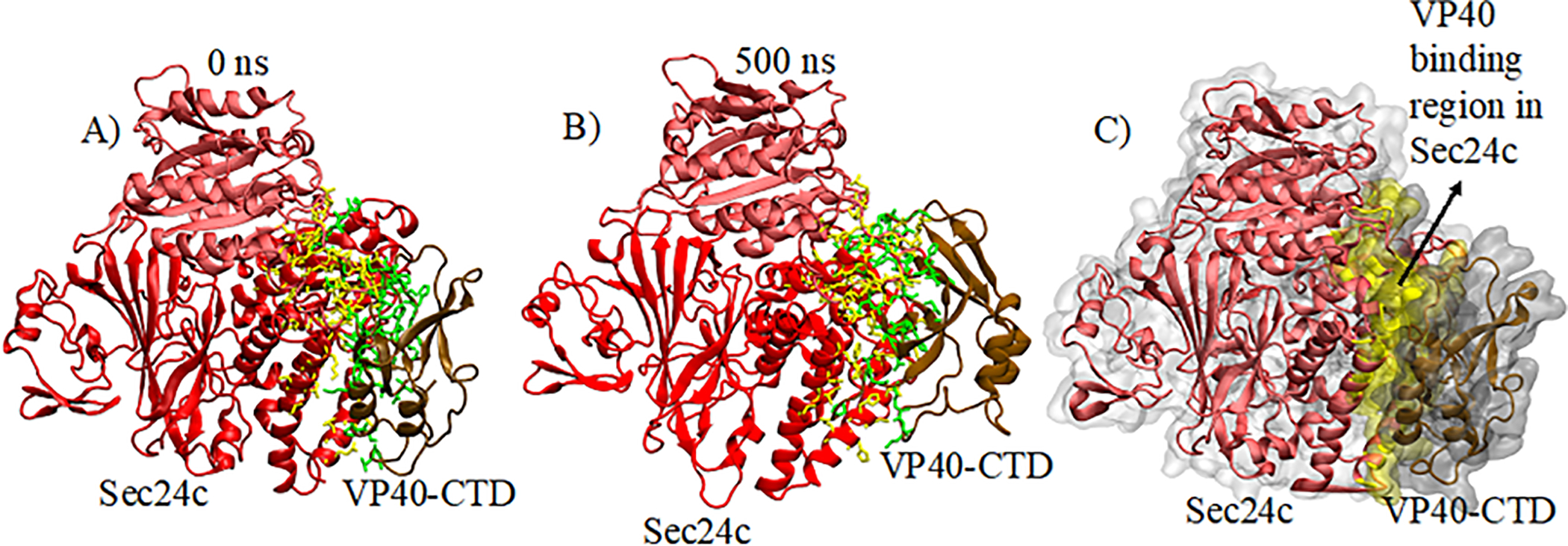
For (A) and (B), residues involved in the inter-protein interactions of the complex are highlighted: Sec24c (yellow) and VP40 (green). (A) Docked Sec24c-VP40 complex at 0 ns. (B) Sec24c-VP40 complex after 500 ns of MD simulation. (C) Surface representation of (B).
Figures 3A and 3B show the structure of the Sec24c-VP40 complex at the initial and final frames of the 500 ns MD simulation, respectively. Inter-protein interactions within 3.5Å of the protein at the binding region are highlighted: yellow represents the residues in Sec24c and green represents the residues for the VP40 protein. The complex remained intact for the entire 500 ns MD simulation, indicating that the complex maintains inter-protein interactions throughout the simulation time.
The binding regions in Sec24c that we identified are comprised of residues 331 to 343, 535 to 546, 580 to 603, and 729 to 767. Most of these segments are close to the Sec24c A-site. Consistent with Yamayoshi et al.26, we found that the binding region in VP40 consists mainly of residues 288 to 326. VP40 residues 196 to 224 also interact with Sec24c, which also supports the idea that the proline rich region 205 to 219 is important for binding with Sec24c as discussed by Reynard et al.27 Figure 3C shows the VP40 binding region in Sec24c in surface representation (yellow) at 500 ns. Expanded views of the interaction region with additional details are given in Fig. 5 and Fig. 6.
Figure 5.
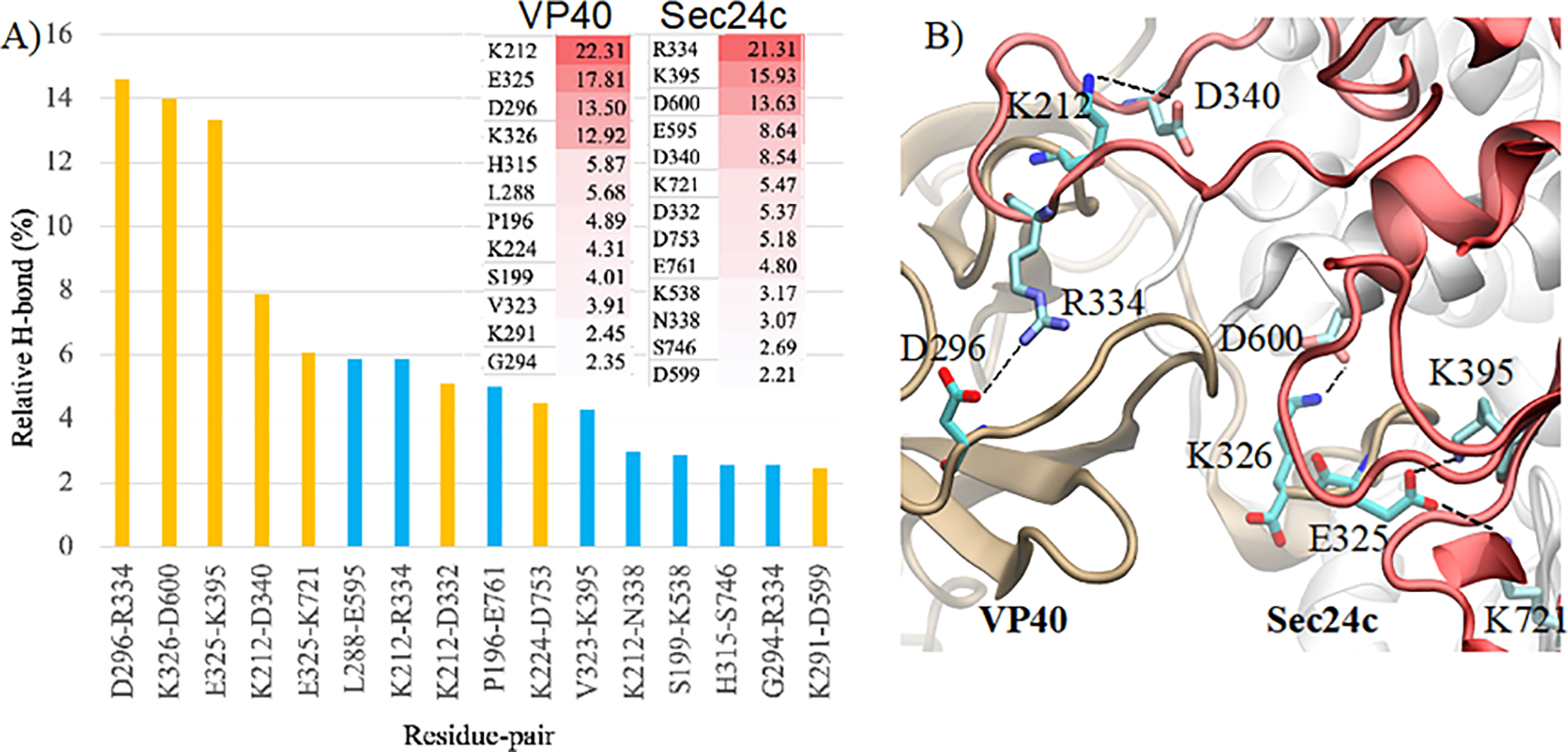
(A) Percentage of hydrogen-bond occupancy for different inter-protein amino acid pairs where charged interactions are colored in yellow. Inset: Hydrogen-bond occupancy of specific residues on VP40 (left column) and Sec24c (right column). (B) Residues involved in important hydrogen-bonding and salt-bridge interactions for the Sec24c-VP40-Wt complex. VP40 is on the left and Sec24c on the right.
Figure 6.
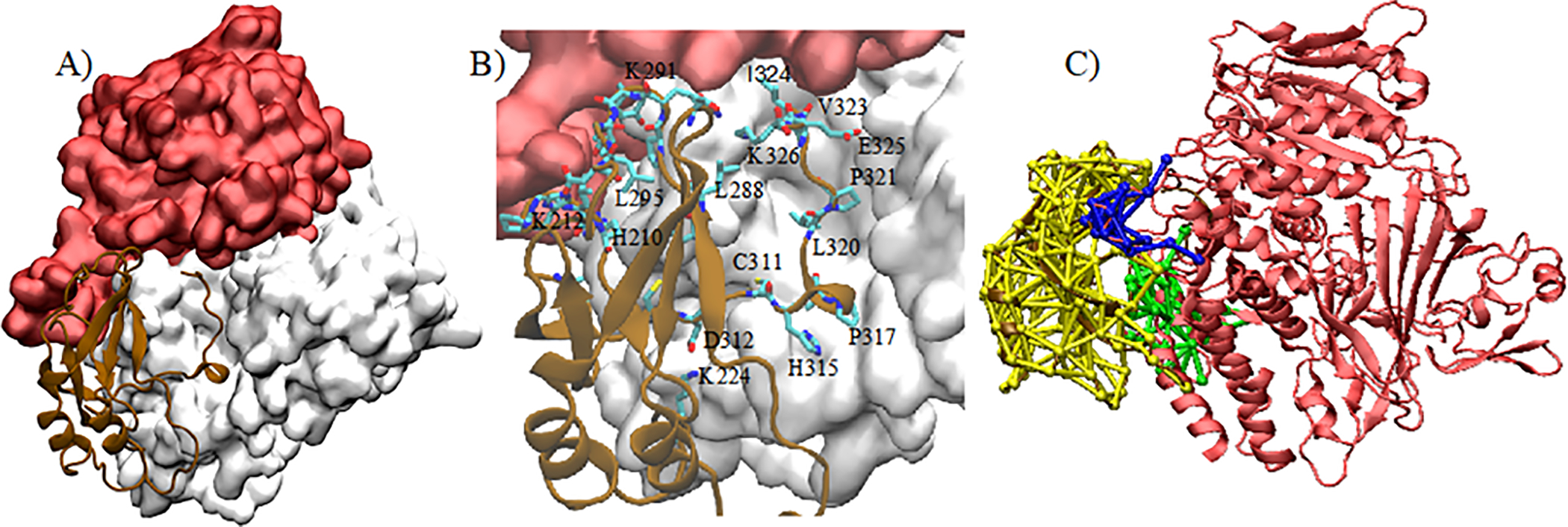
(A): VP40-CTD (brown) in the binding pocket of Sec24c (white). The trunk domain of Sec24c is displayed in red. (B) Residues in VP40 interacting in the binding pocket of Sec24c. (c) DNA communities between Sec24c-VP40-WT, where each community is colored differently (green, blue, yellow).
To quantify the binding between Sec24c and VP40, we calculated the root mean square deviation (RMSD) of the Cα atoms of both proteins. Figure 4A shows the RMSD as a function of time. The significant amount of fluctuations before 250 ns suggest that the complex is undergoing structural rearrangements, which is also reflected in the large increase in RMSD. After 250 ns, the smoothness in the RMSD indicates the stability of the Sec24c-VP40 complex. The system is now stabilized by different types of interactions, such as electrostatic, hydrophobic, and hydrogen bonding interactions. To gain further insight into the binding affinity of the complex, we plot in Fig. 4B as a function of time the total interaction energy (black line) between Sec24c and VP40, as well as two types of interaction energies: electrostatic (cyan) and Van der Waals (grey). Similar to the RMSD of Fig. 4A, the large decreases in the total energy and electrostatic energy plots before 250 ns again signify that the complex is rearranging and equilibrating. After 250 ns, the system has settled into a stable configuration.
Figure 4.
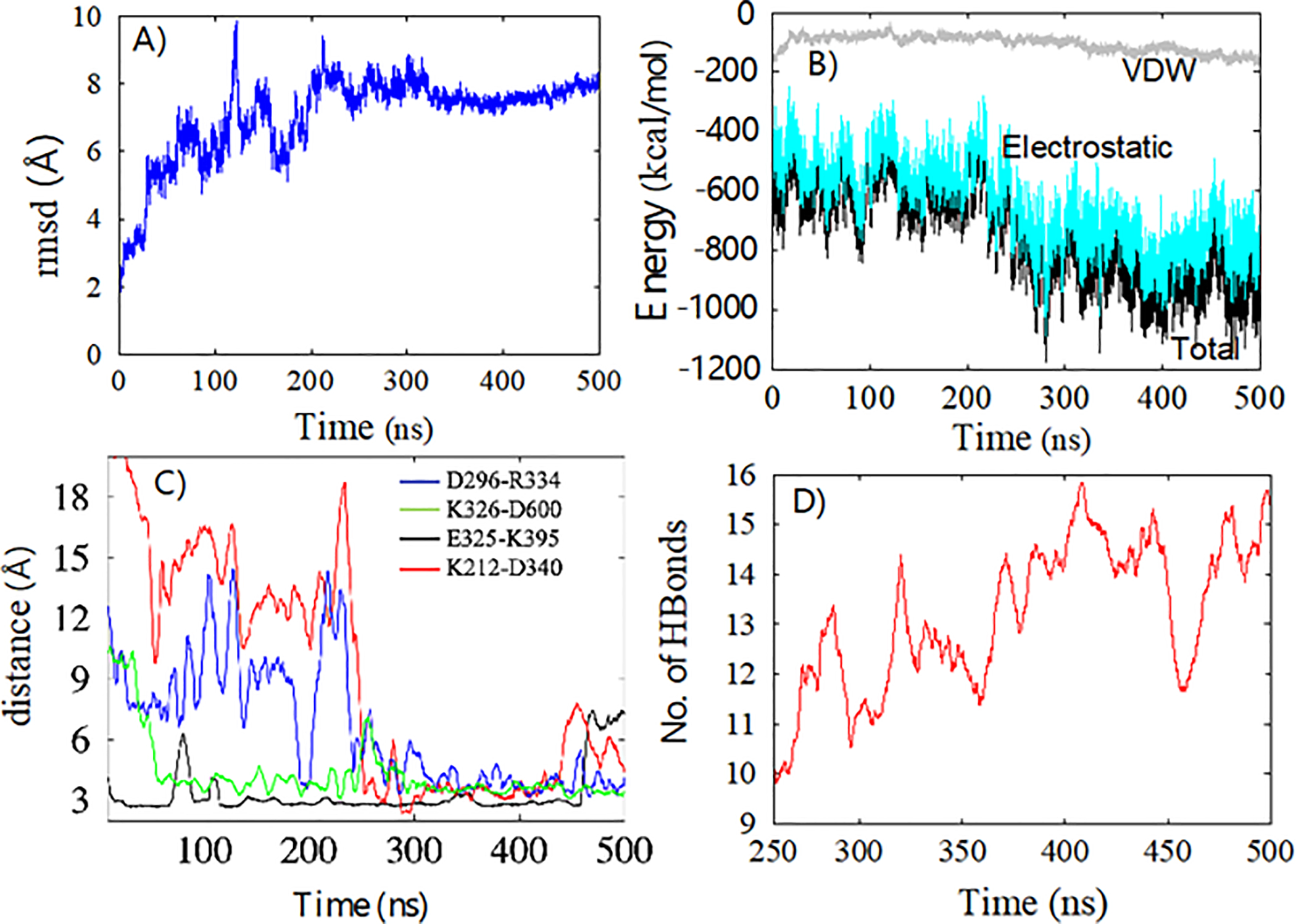
(A) Root mean square deviation of Cα atoms of Sec24c and VP40 with respect to time. (B) Interaction energy between Sec24c and VP40 as a function of time: Total Energy (black), Electrostatic (cyan), van der Waals (VDW) (grey). (C) The distance between the N and O atoms in the residues involved in four important salt-bridges. (D) Number of hydrogen bonds between Sec24c and VP40 continues to increase after the system has stabilized.
After quantifying that the interaction between Sec24c and VP40 complex was stable after 250 ns of MD simulation, we began more detailed analysis which involved characterizing the interactions between the proteins in the complex. As shown in Fig. 4B, electrostatic interactions are the primary reason for the stabilization of the Sec24c and VP40 system. The electrostatic interactions are dominated by salt-bridges between amino acids. More than eight salt-bridges were formed between Sec24c and VP40. The most stable salt-bridges were: D296-R334, K326-D600, E325-K395 and K212-D340. Figure 4C plots the distance between the nitrogen and oxygen atoms on the residues for four important salt-bridges as a function of time. Figure 4C shows that the D296-R334 separation and the E325-K395 separation each become much closer after 240 ns, and therefore both of these salt-bridges become stronger and help to stabilize the complex. We also observed a significant number of hydrogen bonds between Sec24c and VP40 (Fig 4D). Figure 4D shows that after the system has stabilized around 250 ns, the number of hydrogen bonds continues to increase. This suggests that the complex was stabilized mainly due to salt-bridge interactions and that hydrogen bonds play a role in fine-tuning the docking. A more detailed analysis for hydrogen bonds is given below.
3.3. H-bond analysis for the Sec24c-VP40 wild-type complex
Figure 4 A–C shows that during the 500 ns MD simulation, the Sec24c and VP40 complex has stabilized its large-scale structural arrangement by 250 ns. However, Fig. 4D shows that the hydrogen bond network continues to change after 250 ns, implying that small scale structural changes continue. We performed a detailed analysis of the inter-protein hydrogen bonds for the last 250 ns of the MD simulation. For the hydrogen bond analysis, a distance cut-off of 3.5 Å and an angle cut-off of 30o was used. Additionally, only heavy atoms were considered for the calculation. Figure 5A represents the percentage of time (percent occupancy) during the final 250 ns that a specific H-bond existed between the amino acids of Sec24c and VP40, including both sidechain and backbone H-bonds. The important interacting residue-pairs between VP40 and Sec24c include: D296-R334, K326-D600, E325-K395, K212-D340, E325-K271, L288-E595, K212-R334, K212-D332 and P196-E761. The inset of Fig. 5A shows the hydrogen-bond occupancy percentage of individual amino acids on VP40 and Sec24c. We found VP40 residue K212 is especially important for hydrogen binding between VP40 and Sec24c binding. Interestingly, K212 has also been found experimentally to be important in the formation of VLPs46.
4. Binding region Dynamic Network Analysis:
The binding region for VP40 in Sec24c lies in a small pocket in Sec24c, as shown in Fig. 6. In Fig 6A, the binding region in Sec24c is highlighted in white and the trunk domain of Sec24c is shown in red. The CTD region of VP40 that binds to Sec24c is displayed in brown. Figure 6B is an expanded version of the binding region and shows the VP40 residues that are within 3.5Å of Sec24c. These VP40 residues include I324, K326, L288, L320, K224, K212, P317, E325, V323, C311, D312, H210. As described above, the VP40 docking in the Sec24c pocket is stabilized by various electrostatic interactions.
To further understand the connections and contacts between Sec24c and VP40, we performed a dynamic network analysis (DNA) using the Network plugin in VMD44. Network analysis is a tool which highlights communities of amino acids that display correlated motion. Analyzing the details of the communities provides information about amino acids on each protein that are interacting and the strength of their connections. The number of nodes (circles) in a community is proportional to the number of amino acids composing that community, and line thickness denotes the weight of the connection between the amino acids. For this analysis, we considered the last 100 ns of the MD simulation, thereby allowing the protein complex to be fully stabilized. Different communities were generated using Carma45 and Catdcd software. We focused on the communities which were responsible for connecting the two proteins (Sec24c and VP40). We observe three communities, colored green, blue and yellow (Fig. 6C) with a total of 20 inter-protein connections. Important connections were made by VP40 K212 with eight amino acids on Sec24c (N338, D333, K335, L336, I337, R334, D340, T339). Other important amino acids on Sec24c are A749 (with VP40 D312, T313), G748 (with VP40 T313), M733 and L734 (with VP40 P317), H737 (with VP40 A318), and P592 and A594 (with VP40 R214). Additionally, strong network connections that stabilize the complex are made between VP40 residue M305 with Sec24c residues V555, V572, and between VP40 residue I307 with Sec24c residue V572. We also performed MD simulations with mutations of VP40 residues M305 and I307 and compare out computational results with experimental results, below.
5. Comparison of Sec24c-VP40-WT complex with VP40 point mutations
Experimental studies by Yamayoshi et al.26, examined the effect of mutations on several residues in the binding region (303–308) of the VP40 protein. Out of several mutations carried out, they found L303A and I307A decreased the interaction with Sec24c, with L303A showing the bigger decrease, while M305A increased the propensity of binding. However, the atomic-level details of the changes of the inter-protein interactions of the mutated-complexes compared to VP40-WT are unknown. We therefore investigated the effect of mutations (L303A, I307A and M305A) on the Sec24c-VP40 binding using molecular dynamics simulations. We set up three additional simulations using VP40 mutants: Sec24c-VP40-L303A (L303A), Sec24c-VP40-I307A (I307A) and Sec24c-VP40-M305A (M305A), and performed 500 ns MD computational simulations on each complex for comparison with Sec24c-VP40-WT. Figures 7A–D represent the Sec24c-VP40-complex at the end of 500ns for all four complexes. Figure 7D shows that L303A has large structural differences in the binding region compared with the wild type. Structural differences in the binding region compared to the wild type are also displayed by I307A in Fig. 7C, whereas Fig. 7B shows that M305A looks most similar to the wild type. To further quantify the differences in the binding for all four complexes, we plotted inter-protein hydrogen bonds and interaction energy between Sec24c and VP40 for the last 250 ns of the MD simulation in Figs. 7E and 7F. As shown in Figs. 7E and 7F, the Sec24c-VP40-WT complex has the largest number of hydrogen bonds and strongest interaction energy of the four complexes. Consistent with the experimental results, we find that L303A has the largest decrease in the number of hydrogen bonds and in interaction binding energy. We also find that I307A has noticeable decreases in the number of hydrogen bonds and in interaction binding energy compared to the wild type. In contrast, we find the M305A complex to have similar characteristics to the wild type. Thus, the computational studies are very much in line with the previously published coimmunoprecipitation analysis (ref. 26).
Figure 7.
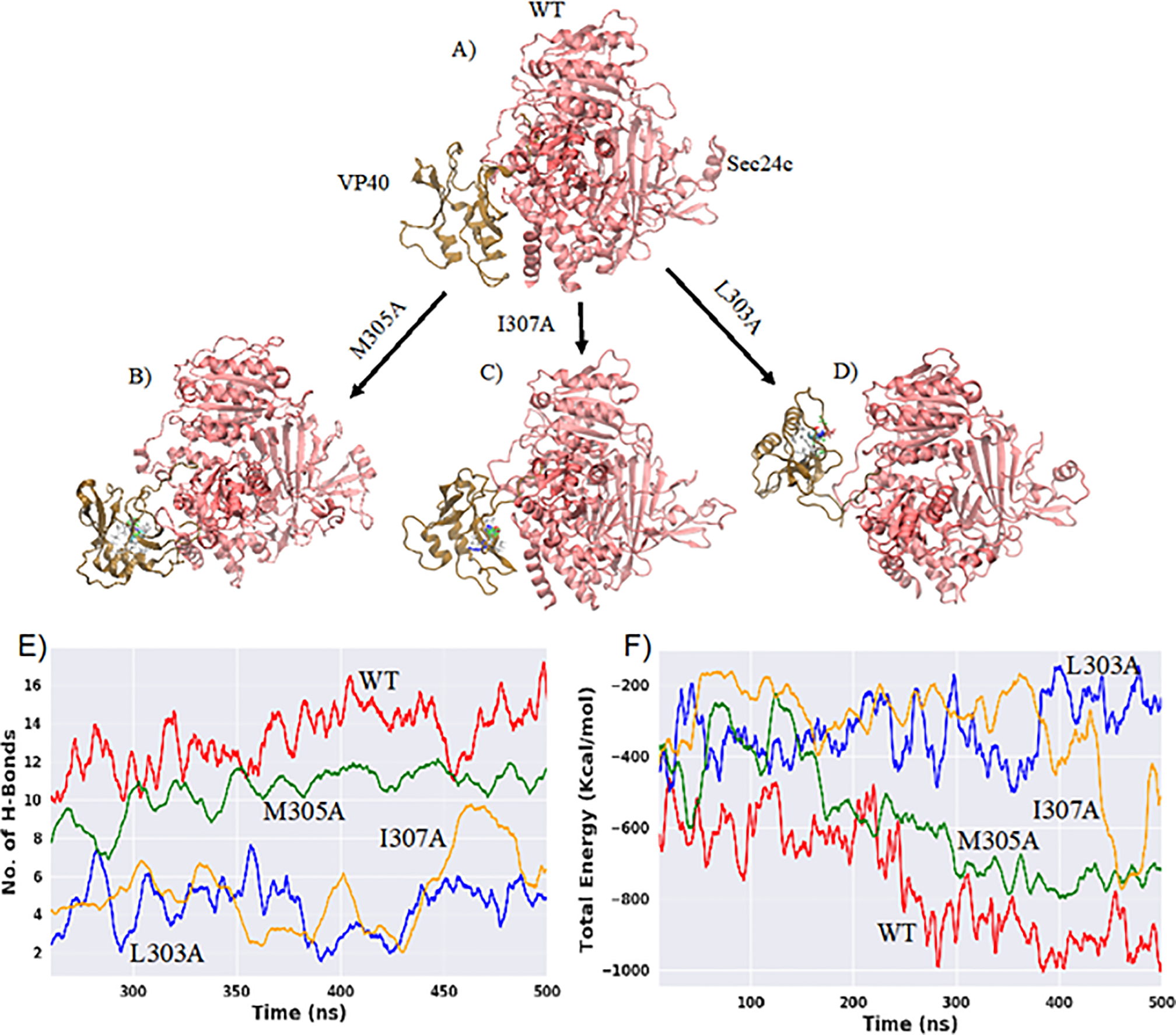
Sec24c and VP40 complex: A) WT B) M305A C) I307A D) L303A where Sec24c is represented in red color and VP40 in brown. The mutated residue on VP40 (Figs. B-D) is colored differently. E) Number of inter-protein hydrogen bonds as a function of time for all four complexes. F) Total interaction energy between Sec24c and VP40 with respect to simulation time.
6. Additional Docking complex
The sequence of molecular docking steps involving VP40, Sec24c and Sec23 that facilitates the transport of VP40 to the plasma membrane is not yet completely understood. To gain insight, we also performed docking of VP40 at the site on Sec24c that can also bind Sec23. This site is on the trunk domain of Sec24c (Fig. 2). Multiple docking complexes were generated using various docking servers like Patch dock, Z-dock and Cluspro. We observed that the VP40 region of amino acids 303–308 binds strongly in the trunk domain region (residues 171–417) of Sec24c. Specifically, the beta-sheet region 303 to 308 in VP40 interacts with the Sec24c trunk domain’s beta-sheet region (241 to 247) and helical region (276 to 286) as shown in Figs. 8A and 8B. The Sec24c region (241 to 247) is also found to be the binding region of Sec2343. In order to optimize the interactions and to study the stability of the system, we performed simulations of this additional docked complex (Sec24c-VP40-AD) for 500 ns. Figures 8A and 8B show the Sec24c-VP40-AD complex at 0 ns and 500 ns, respectively and the regions of inter-protein interactions are highlighted. For theVP40 protein, the binding region of residues 303 to 308 determined experimentally and from the computational molecular docking simulations remained stable during the MD simulation. In addition to VP40 residues 303–308, VP40 residues 233–243 also interact with Sec24c. When the alpha-helix on VP40 (residues 233–240) aligns with the alpha-helix on Sec24c (residues 276 to 286), several favorable, stabilizing inter-protein interactions are able to form involving residues within these alpha-helices as well as residues close to the alpha-helices. The important interacting residue-pairs between VP40 and Sec24c include T232-Q242, I307-V245, T232-M243, T242-D284, S233-Q242, D302-K227, A229-Q242, T304-S247, Q238-E280, S233-P241, K212-D251. Figure 8C displays a view that includes some of these interprotein bonds.
Figure 8.
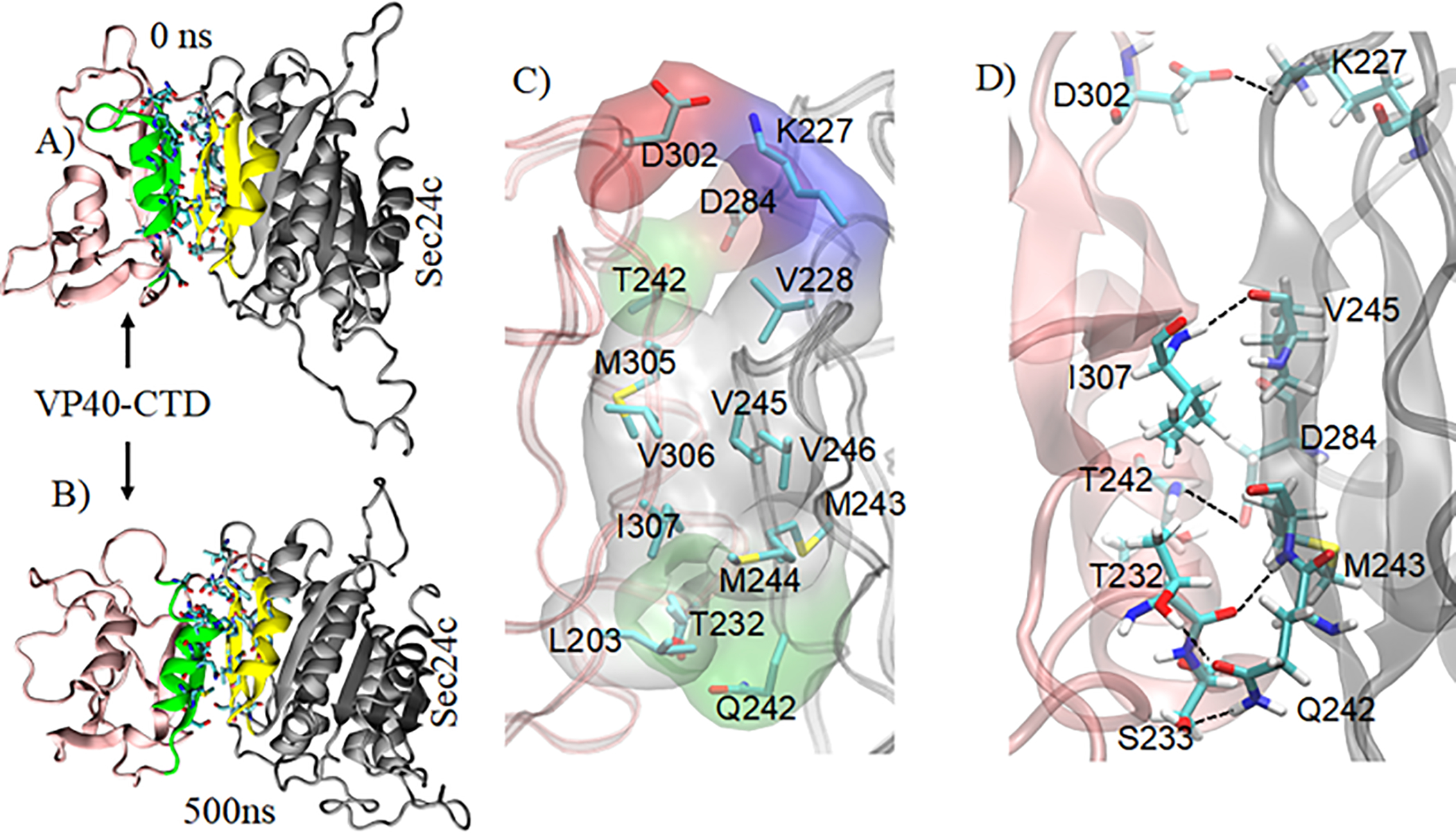
Sec24c-VP40-AD complex with highlighting of the VP40 binding region (green) and the Sec24c binding region (yellow). MD structure of the complex at: A) 0 ns, B) 500 ns. C) Important residues at the protein-protein interface. Hydrophobic residues (gray) form a strong hydrophobic core that contributes to the stability of the complex. The polar, negative, and positive residues at the interface are shown in green, red, and blue surface representation, respectively. D) View showing some interprotein residue-residue interactions between VP40 and Sec24c.
Among the amino acids involved in inter-protein interactions in the complex, 63% of the Sec24c residues are in the trunk domain’s beta-sheet region 241 to 247. For the VP40 CTD, 50% of the contributing residues lie within the 303–308 region and the other 50% are in an alpha-helical region (233 to 240). This result is consistent with the experimental study26 that showed that VP40 residues 303–308 are important for the binding to Sec24c. Additionally, the interfacial region of the complex contains a hydrophobic core involving VP40 residues M305, V306, I307 and L203, and Sec24c residues V228, M243, M244, V245, V246. These hydrophobic residues in the interface are shown in surface representation (gray) in Fig. 8D. The binding of VP40 in the trunk domain region of Sec24c that also binds Sec23 suggests that VP40 might use the COPII transport mechanism in a manner that may not need the Sec23 protein in order for VP40 to be transported to the plasma membrane. However, how VP40 actually utilizes the COPII mechanisms and whether it binds Sec24c before Sec23 and Sec24c interact still remains unknown.
7. Conclusion
The EBOV life-cycle requires transport of VP40 proteins to the plasma membrane and the COPII transport system plays a key role in this process. Sec24c is one of the proteins of the COPII transport system. Sec24c binds with VP40 and assists in the transport process to the intracellular leaflet of the PM where VP40 interacts with lipid molecules and oligomerizes to form the structural matrix filaments of the virus’ cylindrical lattice11,47. Experimentally, it has been shown that Sec24c binds with the VP40 C-terminal domain (CTD), particularly in the region spanning VP40 amino acids 303 to 308. In this study, we investigated which regions of Sec24c are most likely to be involved in the binding process using various computational techniques. The binding regions in Sec24c that we identified are comprised of residues 331 to 343, 535 to 546, 580 to 603, and 729 to 767. There is also another possible VP40 docking site in the trunk domain of Sec24c that can also bind Sec23. The site on the trunk domain of Sec24c involves a beta-sheet region composed of amino acids 241 to 247 and an alpha-helical region consisting of amino acids 276 to 286. Interestingly, in addition to the known VP40 binding region including the amino acids 303–308, we also observed binding interactions involving VP40 residues 232–243, which are part of an alpha-helical secondary structure. Our MD computations showed that after the initial docking of VP40 at this alternative site on Sec24c, the secondary structures (beta-sheet and alpha-helix) on both proteins at the interface undergo structural rearrangements which allow the Sec24c-VP40 complex to optimize the interactions and make the complex stable via inter-protein hydrogen bonds, hydrophobic interactions, and salt-bridges.
We also examined the effects of three VP40 mutations on the inter-protein interactions of the complex. We investigated the Sec24c-VP40-L303, Sec24c-VP40-M305A, and the Sec24c-VP40-I307A complexes. Consistent with experimental results on binding interactions, we find that L303A has the largest decrease in the number of inter-protein hydrogen bonds and in interaction binding energy. We also find that I307A has noticeable decreases in the number of hydrogen bonds and in interaction binding energy compared to the wild type. In contrast, we find the M305A complex to have similar characteristics to the wild type.
The sequence of molecular docking steps involving VP40, Sec24c and Sec23 that facilitates the transport of VP40 to the plasma membrane is not yet completely understood. The atomic-level findings provide insight into the mechanism of how Sec24c binds with VP40, a necessary step for the transport of VP40 to the cell’s PM. These molecular details about a critical step in the EBOV life-cycle provides information that can be helpful in developing drug molecules that target EBOV interactions with host proteins. Further, these studies should help guide molecular studies geared at the mechanism by which VP40 hijacks the COPII transport pathway. The binding of VP40 in the trunk domain region of Sec24c that also binds Sec23 suggests that VP40 might use the COPII transport mechanism in a manner that may not need the Sec23 protein in order for VP40 to be transported to the plasma membrane. More rigorous in vitro biophysical as well as cellular studies will be needed to determine how VP40 actually utilizes the COPII transport mechanisms and whether VP40 binds with Sec24c before Sec23 and Sec24c interact.
Acknowledgements
These studies were supported by the NIH (AI081077) to R.V.S.
References
- 1.Feldmann HJNEJoM. Ebola—a growing threat? 2014;371(15):1375–1378. [DOI] [PubMed] [Google Scholar]
- 2.Peters CJ, Peters JW. An Introduction to Ebola: The Virus and the Disease. The Journal of Infectious Diseases. 1999;179(Supplement_1):ix–xvi. [DOI] [PubMed] [Google Scholar]
- 3.Kadanali A, Karagoz GJNcoI. An overview of Ebola virus disease. 2015;2(1):81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tomori O, Kolawole MO. Ebola virus disease: current vaccine solutions. Current Opinion in Immunology. 2021;71:27–33. [DOI] [PubMed] [Google Scholar]
- 5.Gulland A First Ebola treatment is approved by WHO. In: British Medical Journal Publishing Group; 2014. [DOI] [PubMed] [Google Scholar]
- 6.Gaudinski MR, Coates EE, Novik L, et al. Safety, tolerability, pharmacokinetics, and immunogenicity of the therapeutic monoclonal antibody mAb114 targeting Ebola virus glycoprotein (VRC 608): an open-label phase 1 study. The Lancet. 2019;393(10174):889–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mühlberger E, Weik M, Volchkov VE, Klenk H-D, Becker SJJov. Comparison of the transcription and replication strategies of Marburg virus and Ebola virus by using artificial replication systems. 1999;73(3):2333–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dror I, Shazman S, Mukherjee S, Zhang Y, Glaser F, Mandel-Gutfreund Y. Predicting nucleic acid binding interfaces from structural models of proteins. Proteins: Structure, Function, and Bioinformatics. 2012;80(2):482–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Timmins J, Scianimanico S, Schoehn G, Weissenhorn WJV. Vesicular release of Ebola virus matrix protein VP40. 2001;283(1):1–6. [DOI] [PubMed] [Google Scholar]
- 10.Jasenosky LD, Neumann G, Lukashevich I, Kawaoka YJJov. Ebola virus VP40-induced particle formation and association with the lipid bilayer. 2001;75(11):5205–5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bornholdt ZA, Noda T, Abelson DM, et al. Structural rearrangement of ebola virus VP40 begets multiple functions in the virus life cycle. 2013;154(4):763–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gerstman BS, Chapagain PPJBeBA-B. Membrane association and localization dynamics of the Ebola virus matrix protein VP40. 2017;1859(10):2012–2020. [DOI] [PubMed] [Google Scholar]
- 13.Stahelin RV. Could the Ebola virus matrix protein VP40 be a drug target? In: Taylor & Francis; 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Panchal RG, Ruthel G, Kenny TA, et al. In vivo oligomerization and raft localization of Ebola virus protein VP40 during vesicular budding. 2003;100(26):15936–15941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Noda T, Sagara H, Suzuki E, Takada A, Kida H, Kawaoka YJJov . Ebola virus VP40 drives the formation of virus-like filamentous particles along with GP. 2002;76(10):4855–4865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pavadai E, Bhattarai N, Baral P, Stahelin RV, Chapagain PP, Gerstman BS. Conformational Flexibility of the Protein-Protein Interfaces of the Ebola Virus VP40 Structural Matrix Filament. J Phys Chem B. 2019;123(43):9045–9053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gc JB, Johnson KA, Husby ML, et al. Interdomain salt-bridges in the Ebola virus protein VP40 and their role in domain association and plasma membrane localization. Protein Sci. 2016;25(9):1648–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Radzimanowski J, Effantin G, Weissenhorn W. Conformational plasticity of the Ebola virus matrix protein. Protein Science. 2014;23(11):1519–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wan W, Clarke M, Norris MJ, et al. Ebola and Marburg virus matrix layers are locally ordered assemblies of VP40 dimers. Elife. 2020;9:e59225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.May ER, Armen RS, Mannan AM, Brooks CL III. The flexible C-terminal arm of the Lassa arenavirus Z-protein mediates interactions with multiple binding partners. Proteins: Structure, Function, and Bioinformatics. 2010;78(10):2251–2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yin P, Li Y, Zhang LJJov. Sec24C-dependent transport of claudin-1 regulates hepatitis C virus entry. 2017;91(18):e00629–00617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li J, Fuchs S, Zhang J, Wellford S, Schuldiner M, Wang XJJCS. An unrecognized function for COPII components in recruiting the viral replication protein BMV 1a to the perinuclear ER. 2016;129(19):3597–3608. [DOI] [PubMed] [Google Scholar]
- 23.Bickford LC, Mossessova E, Goldberg JJCoisb. A structural view of the COPII vesicle coat. 2004;14(2):147–153. [DOI] [PubMed] [Google Scholar]
- 24.Mossessova E, Bickford LC, Goldberg J. SNARE selectivity of the COPII coat. Cell. 2003;114(4):483–495. [DOI] [PubMed] [Google Scholar]
- 25.Zanetti G, Prinz S, Daum S, et al. The structure of the COPII transport-vesicle coat assembled on membranes. 2013;2:e00951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamayoshi S, Noda T, Ebihara H, et al. Ebola virus matrix protein VP40 uses the COPII transport system for its intracellular transport. 2008;3(3):168–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reynard O, Nemirov K, Page A, et al. Conserved proline-rich region of Ebola virus matrix protein VP40 is essential for plasma membrane targeting and virus-like particle release. 2011;204(suppl_3):S884–S891. [DOI] [PubMed] [Google Scholar]
- 28.Mancias JD, Goldberg J. Structural basis of cargo membrane protein discrimination by the human COPII coat machinery. EMBO J. 2008;27(21):2918–2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eswar N, Webb B, Marti-Renom MA, et al. Comparative protein structure modeling using Modeller. 2006;15(1):5.6. 1–5.6. 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kozakov D, Hall DR, Xia B, et al. The ClusPro web server for protein-protein docking. Nat Protoc. 2017;12(2):255–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pierce BG, Wiehe K, Hwang H, Kim B-H, Vreven T, Weng ZJB. ZDOCK server: interactive docking prediction of protein–protein complexes and symmetric multimers. 2014;30(12):1771–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schneidman-Duhovny D, Inbar Y, Nussinov R, Wolfson HJJNar. PatchDock and SymmDock: servers for rigid and symmetric docking. 2005;33(suppl_2):W363–W367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Duhovny D, Nussinov R, Wolfson HJ. Efficient unbound docking of rigid molecules. Paper presented at: International workshop on algorithms in bioinformatics2002. [Google Scholar]
- 34.Lee J, Cheng X, Swails JM, et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J Chem Theory Comput. 2016;12(1):405–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Phillips JC, Braun R, Wang W, et al. Scalable molecular dynamics with NAMD. 2005;26(16):1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang J, MacKerell AD Jr. CHARMM36 all-atom additive protein force field: validation based on comparison to NMR data. J Comput Chem. 2013;34(25):2135–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LGJTJocp. A smooth particle mesh Ewald method. 1995;103(19):8577–8593. [Google Scholar]
- 38.Andersen HC. Rattle: A “velocity” version of the shake algorithm for molecular dynamics calculations. Journal of Computational Physics. 1983;52(1):24–34. [Google Scholar]
- 39.Feller SE, Zhang Y, Pastor RW, Brooks BR. Constant pressure molecular dynamics simulation: The Langevin piston method. The Journal of Chemical Physics. 1995;103(11):4613–4621. [Google Scholar]
- 40.Humphrey W, Dalke A, Schulten KJJomg. VMD: visual molecular dynamics. 1996;14(1):33–38. [DOI] [PubMed] [Google Scholar]
- 41.Mitchell AL, Attwood TK, Babbitt PC, et al. InterPro in 2019: improving coverage, classification and access to protein sequence annotations. 2018;47(D1):D351–D360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bi X, Corpina RA, Goldberg J. Structure of the Sec23/24-Sar1 pre-budding complex of the COPII vesicle coat. Nature. 2002;419(6904):271–277. [DOI] [PubMed] [Google Scholar]
- 43.Pagant S, Wu A, Edwards S, Diehl F, Miller EA. Sec24 is a coincidence detector that simultaneously binds two signals to drive ER export. Curr Biol. 2015;25(4):403–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jiang J, Patarroyo C, Garcia Cabanillas D, Zheng H, Laliberte JF. The Vesicle-Forming 6K2 Protein of Turnip Mosaic Virus Interacts with the COPII Coatomer Sec24a for Viral Systemic Infection. J Virol. 2015;89(13):6695–6710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miller EA, Beilharz TH, Malkus PN, et al. Multiple cargo binding sites on the COPII subunit Sec24p ensure capture of diverse membrane proteins into transport vesicles. Cell. 2003;114(4):497–509. [DOI] [PubMed] [Google Scholar]
- 46.McCarthy SE, Johnson RF, Zhang YA, Sunyer JO, Harty RN. Role for amino acids 212KLR214 of Ebola virus VP40 in assembly and budding. J Virol. 2007;81(20):11452–11460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pavadai E, Gerstman BS, Chapagain PP. A cylindrical assembly model and dynamics of the Ebola virus VP40 structural matrix. Sci Rep. 2018;8(1):9776. [DOI] [PMC free article] [PubMed] [Google Scholar]