

Abstract
This single-center study aims to determine the time, diagnostic procedure, and cost saving potential of early exome sequencing in a cohort of 111 individuals with genetically confirmed neurodevelopmental disorders. We retrospectively collected data regarding diagnostic time points and procedures from the individuals’ medical histories and developed criteria for classifying diagnostic procedures in terms of requirement, followed by a cost allocation. All genetic variants were re-evaluated according to ACMG recommendations and considering the individuals’ phenotype. Individuals who developed first symptoms of their underlying genetic disorder when Next Generation Sequencing (NGS) diagnostics were already available received a diagnosis significantly faster than individuals with first symptoms before this cutoff. The largest amount of potentially dispensable diagnostics was found in genetic, metabolic, and cranial magnetic resonance imaging examinations. Out of 407 performed genetic examinations, 296 (72.7%) were classified as potentially dispensable. The same applied to 36 (27.9%) of 129 cranial magnetic resonance imaging and 111 (31.8%) of 349 metabolic examinations. Dispensable genetic examinations accounted 302,947.07€ (90.2%) of the total 335,837.49€ in potentially savable costs in this cohort. The remaining 32,890.42€ (9.8%) are related to non-required metabolic and cranial magnetic resonance imaging diagnostics. On average, the total potentially savable costs in our study amount to €3,025.56 per individual. Cost savings by first tier exome sequencing lie primarily in genetic, metabolic, and cMRI testing in this German cohort, underscoring the utility of performing exome sequencing at the beginning of the diagnostic pathway and the potential for saving diagnostic costs and time.
Subject terms: Epilepsy, Neurodevelopmental disorders, Genetic testing, Genetics research, Paediatrics
Introduction
Neurodevelopmental disorders (NDD) and epilepsy are frequent causes for medical presentations/diagnostics [1, 2]. NDD affect ~3% of children and ~1/150 develop epilepsy in the first ten life years [3, 4]. Genetically they are heterogeneous including hundreds of disorders [5, 6].
Establishing a diagnosis involves coordinated interplay between pediatricians, neurologists, geneticists, and other physicians in centers for rare diseases. This process is time consuming, requires many diagnostic procedures and burdens affected families [7–10]. The associated costs and risks are considerable, underscoring the scope of this “diagnostic odyssey” and the load on the health care system [11]. The impact of a conclusive genetic diagnosis for individuals/families has been investigated in several studies and demonstrates the importance of developing effective diagnostic guidelines [7, 12–14].
Genetic diagnostics have traditionally been initiated at a relatively late stage in the diagnostic pathway and were often performed in a stepwise manner [15, 16]. Since its introduction, high-throughput sequencing has proven to be an effective, rapid but cost-intensive method in genetic diagnostics [17] and has evolved from a research method to a routine diagnostic tool [18]. Next Generation Sequencing (NGS) outperforms traditional genetic diagnostics, as it can achieve a diagnostic yield between 30 and 47% in NDD and epilepsy cohorts combined with speed-up through a single test [16, 19–22]. Thus, the question remains [23] whether first-line exome sequencing (ES) could save costs and time, reduce the risks of obsolete extensive or even invasive diagnostic procedures, and allow families to make earlier reproductive decisions.
To address these questions, we designed a retrospective single-center study focusing on diagnostic procedures and time durations in 111 individuals with NDD and/or epilepsy with NGS based diagnosis. We wanted to determine the extent to which NGS diagnostics influenced the diagnostics duration and to assess the number of diagnostic interventions that might have not been required retrospectively if ES had been implemented earlier in this cohort.
Materials and methods
Study design and patient selection
We retrospectively selected 111 individuals with pediatric-onset NDD and/or epilepsy from a diagnostic cohort of 2128 cases based on a series of filtering and quality control steps (see details in Fig. 1A). All individuals had received a molecular diagnosis using NGS methods at the Institute of Human Genetics at Leipzig University Medical Center (UKL), a tertiary care centre in Germany, between 2017-04-04 and 2020-08-03. Our filtering criteria were intended to ensure that physician letters were available for all individuals to collect data on diagnostic procedures as well as time points of interest. Therefore, we excluded individuals who received no clinical assessments at UKL and those born before the year 2000. We excluded cases where only variants of uncertain significance (VUS) had been reported in the initial diagnostic report. This resulted in a list of 112 individuals for which we collected clinical data. We excluded one individual after variant reevaluation because the reported variant does not sufficiently explain the observed severity (Ind076; for further details see Supplementary File S3).
Fig. 1. Flowchart of participant recruitment and timeline.

A Workflow used to select individuals for this study. Further information on the excluded individual (Ind076) is provided in supplementary files. B Schematic timeline, time points and intervals (compare Supplementary methods for color coding) of the diagnostic trajectory researched from the patient’s medical history.
Data collection and analysis
The UKLs patient information systems were used to curate genetic/clinical information from the individuals’ medical history. This involved all procedures performed from birth to the final molecular report. We classified each procedure by its requirement. Only the final NGS-based investigations leading to diagnosis and their validation was classified as required for genetic analyses. We recorded diagnostic time points (t1–t6) on the structured diagnostic pathway (Fig. 1B). Associated diagnostic costs were determined using a retrospective bottom-up approach by inferring the total health care costs based on individual procedures. Data were compiled in Excel (Microsoft Corporation, Redmond, Washington, USA) and analyzed in RStudio (Version 1.4). See Supplementary methods for details.
Genetic analyses and variants reevaluation
All individuals were diagnosed by NGS-methods such as gene panel, single ES or trio ES and variants were re-evaluated to current standards (see details in Supplementary methods).
Results
Cohort demographics
Of the 111 individuals included, 49 (44.1%) were female and 62 (55.8%) were male (ratio: 1:1.26). Due to the inclusion criteria described earlier (Fig. 1A), all individuals enrolled were under 20 years of age at the time of diagnosis. Details of the sex-specific age distribution are provided in Fig. 2A. Initial clinical diagnosis was NDD in 69 (62.1%) of individuals, epilepsy in 10 (9%), and a combined occurrence of both in 32 (28.8%) (Fig. 2B). In ten individuals with isolated epilepsy the seizure onsets and genetic diagnoses occurred in a very young age, or no medical records were available for the time after genetic diagnosis. We examined further medical letters for possible later onsets of NDD, but no evidence for it was found. Thus, it cannot be excluded that NDD could still develop in these individuals later in life. The onset of the first symptoms associated with the underlying genetic disease developed at a median age of 6.2 months (range: 0–156.6; standard deviation (SD): 20.5) in the individuals. Out of 111 individuals 22 individuals (19.8%) were already symptomatic regarding the molecular diagnosis on their day of birth and 50 individuals (45%) showed symptoms in their first year of life; another 39 individuals (35.1%) became symptomatic in their second year of life or later. A compilation of anonymized individual data is provided in Supplementary File S2.
Fig. 2. Individual and variant features.

A Divergent plot representing the age distribution of individuals. The age determination refers to the date of final establishment of the human genetic diagnosis. The y-axis represents the age classes in years. The x-axis shows the number of associated individuals. B Stacked bar chart indicating the distribution of the phenotypes (epilepsy, NDD and the combination of both) in the 111 included individuals presented in percentages. C Stacked bar chart showing the distribution of all variant types found in the individuals including additional findings seen in no or low association with NDD or epilepsy presented in percentages (n = 127). SNV = Single-nucleotide variant, Indel = Insertion/Deletion Polymorphism, Large CNV = Copy number variation whose alteration affects multiple genes, Small CNV = Copy number variation whose alteration affects a single gene and involves, for example, several exons (D). Stacked bar chart showing the inheritance patterns of those variants assigned to the respective phenotype of epilepsy or NDD (n = 111). homo = homozygous, comphet = compound heterozygous.
Variant characteristics
In 111 individuals, a total of 127 variants was reported through NGS-based diagnostics. The majority of 87 (68.5%) variants were single-nucleotide variants (SNV), with the remainder composed of 15 (11.8%) copy number variants affecting multiple genes (Large CNV), 13 (10.2%) insertion/deletion variants (indel), ten (7.9%) copy number variation whose alteration affects a single gene and involves several exons (Small CNV), and two complex events (1.6%) summarized as “other” (Fig. 2C). These comprise an unbalanced translocation identified through coverage-based CNV analysis and subsequently confirmed by karyotyping and FISH and a deletion-insertion event in MECP2 (Variant ID: CNV017 and SNV008; Supplementary File S3). Of these 127 variants, 111 variant combinations represented as causative for the phenotypes of NDD/epilepsy in the individuals, whereas the other genetic alterations were related to other concerns or were incidental findings. The origin of most NDD/epilepsy-related variants was recorded as sporadic and de novo in 55 cases (49.5%). The inheritance of eleven variants (9.9%) was recessive and homozygous, ten (9%) were dominant and inherited, another seven (6.3%) variants combinations represented as recessive compound heterozygous variants, six (5.4%) were X-linked recessive and two (1.8%) were postzygotic. For 20 variants (18%) no segregation was performed in the parents (Fig. 2D). A detailed compilation of all identified variants is provided in Supplementary File S3.
Time periods in the diagnostic pathway
The total diagnostic time (first symptoms to final diagnosis) was split into subintervals (Interval 1–5; Fig. 1B) to allow granular analysis of the diagnostic trajectory. The median duration between onset of symptoms associated with the underlying genetic disease and the final genetics report establishing the diagnosis was 34.1 months (range: 0.6–210.5; SD: 57.4). A median of 64.5 days (range: 8–395; SD: 60.8) passed between the initiation of NGS-based diagnostics leading to molecular diagnosis and the report of the molecular diagnosis.
We compared the length of subintervals in individuals with first symptoms before (n = 56) and after (n = 55) the availability of NGS-based diagnostics at UKL in April 2016 (Fig. 3A). While Interval 4 (initiation of first genetic diagnostics to initiation of final genetic diagnostics) dominates in the total diagnostic duration for individuals with symptom onset before the establishment of NGS, Interval 1 (first symptoms to initial diagnostics) emerges prominently for individuals with first symptoms after April 2016. The duration of the total diagnostic trajectory (Interval 1–5) significantly (p < 0.001, Wilcox-Test) differed between individuals with first symptoms before and after 2016-04-01 (Fig. 3B); the median interval length was 106.3 months (range: 21.7–201.5; SD: 50.2) for individuals with first symptoms before establishment of NGS, whereas it was shorter for individuals with first symptoms thereafter, with a median of 9.2 months (range: 0.6–43.3; SD: 12.1). This is expected because recruitment of individuals ended in August 2020. Thus, the total diagnostic time interval length for individuals with first symptoms after the NGS introduction is limited to a maximum of 4 years.
Fig. 3. Impact established NGS-based diagnostics on diagnostic time intervals.

A Stacked bar plot showing the length of each diagnostic subinterval (Interval 1, 2, 3, 4, and 5) per individual, grouped by time of first symptom onset. Cohort is subdivided by availability of NGS-based diagnostics after April 1, 2016. Only individuals for which the duration of all diagnostic intervals could be calculated were included for this figure (n = 84). Colors as in Fig. 1B. B Violin- and scatter-plot of the total diagnostic time interval (Interval 1–5, first symptoms to final diagnosis) grouped by first symptom onset. (p < 0.001, Wilcox-Test). C Violin- and scatter-plot assembling the total diagnostic time interval (Interval 1–5) grouped by phenotype according to the time of onset of first symptoms. Individuals with a combination of both phenotypes were assigned to the phenotype that occurred first. (After Apr16: p ~ 0.002, Wilcox-Test). D Violin- and scatter-plot assembling Interval 1–3 (first symptoms to initiation of first genetic diagnostics) and Interval 4–5 (initiation of first genetic diagnostics to final diagnosis) grouped by onset symptoms. (After Apr16: p < 0.001, Wilcox-Test).
Dividing these subgroups based on initial phenotype (NDD or epilepsy first), no significant differences emerged for individuals with initial symptoms before April 2016. For individuals with first symptoms after 2016, those in the “epilepsy first” group received their molecular diagnosis significantly faster (p ~ 0.002, Wilcox-Test; 3.9 months; range: 0.13–36.53; SD: 10.6) than individuals in the “NDD first” group (15.7 months; range: 0.62–43.3; SD: 12) (Fig. 3C).
To compare the proportion of the total diagnostic interval pertaining to pediatrics with that of clinical genetics, we attributed Interval 1–3 (pediatrics) and 4–5 (genetics) to them. While only a relatively weak significant difference (p ~ 0.03, Wilcox-Test) was found for individuals with first symptoms before April 2016, a highly significant difference (p < 0.001, Wilcox-Test) was found for individuals with first symptoms after 2016. For these individuals, the time allocated to pediatrics (193 days; range: 1–1115; SD: 326.4) was significantly longer than that attributed to genetics (54 days; range: 8–705; SD: 123.2) (Fig. 3D, Supplementary Fig. S3).
The duration of the total diagnostic pathway did not differ between individuals whose initial diagnosis took place at UKL and individuals whose initial diagnosis was performed at another medical institution (Supplementary Fig. S4).
Diagnostic procedures and costs
Before receiving a genetically confirmed diagnosis, the individuals in our cohort underwent various diagnostic procedures (Fig. 4A, Supplementary File S2). All individuals received at least one genetic examination, 101 individuals (91%) had at least one laboratory test other than metabolic testing and 86 individuals (77.5%) received at least one cMRI. Other medical imaging was performed at least once in 78 (70.3%) individuals, medical consults in 75 (67.6%), EEG in 74 (66.7%), metabolic diagnostics in 67 (60.4%), lumbar puncture in 26 (23.4%), electrophysiology in 26 (23.4%), functional tests in 14 (12.6%), ECG in 11 (9.9%). Overall, 95 individuals (85.6%) were in inpatient care at UKL at least once (Supplementary Fig. S5).
Fig. 4. Required and non-required diagnostic costs.
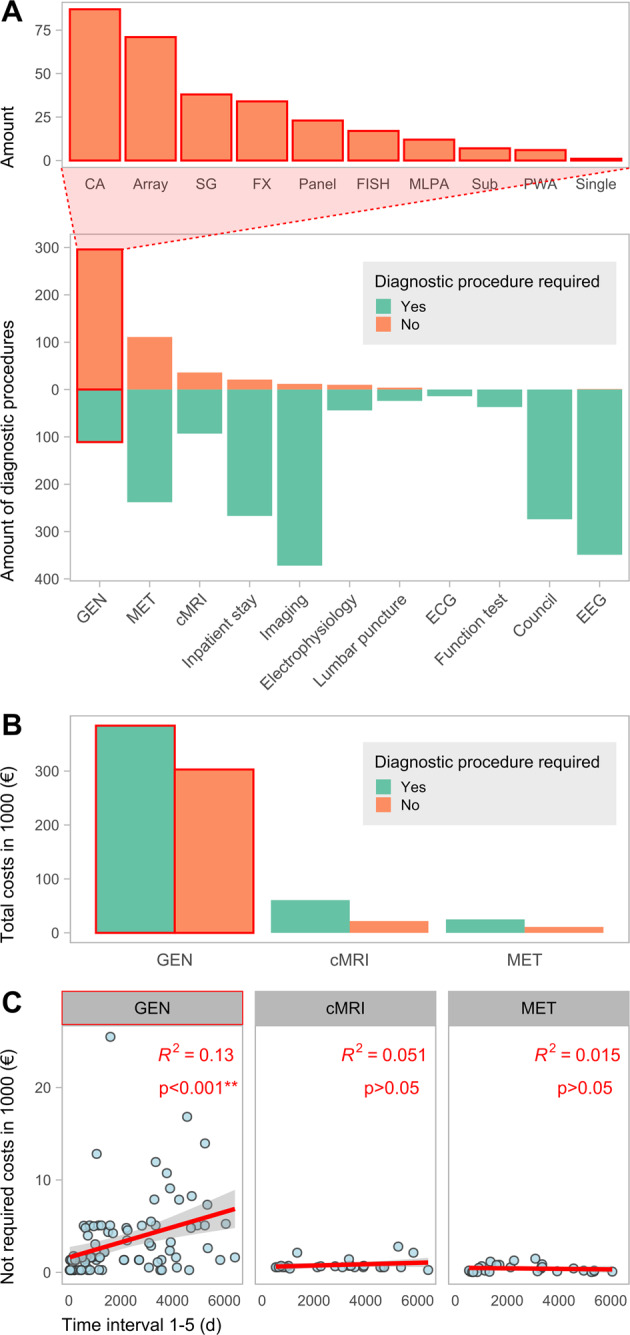
A Divergent plot illustrating the amount of required and not required diagnostics performed on the individuals. GEN = human genetic diagnostics, MET = metabolic laboratory testing, cMRI = cranial magnetic resonance imaging, Imaging = other medical imaging than cMRI, ECG = electrocardiogram, EEG = electroencephalogram. The bar chart above categorizes the non-required genetic diagnostics. CA = Chromosome analysis, SG = Single Gene analysis, FISH = Fluorescence in situ hybridization, MLPA = Multiplex ligation-dependent probe amplification, Sub = Analysis of Subtelomeres, PWA = Prader-Willi syndrome and Angelman syndrome diagnostics, Single = Single Exome Diagnostics. B Bar chart showing the respective costs grouped by top three diagnostic categories (with the highest amount of potential non-required diagnostics) and requirement. Compare Supplementary Fig. S7 for a cost calculation that excludes external costs. C Scatter-plot showing the correlation of the respective unnecessary diagnostic costs with the length of the diagnostic odyssey (Interval 1–5), grouped by diagnostic category. The values for R2 and the p values are given in the plot.
The diagnostic categories with the highest amounts of potentially non-required procedures are genetic diagnostics, metabolic diagnostics, and cMRI (Fig. 4A). Of 407 genetic examinations performed overall, 296 (72.7%) were categorized as potentially dispensable if an exome-wide analysis would have been performed initially instead. Among those 407 examinations, 314 (77.1%) were performed at UKL and 93 (22.8%) at other hospitals. Based on our study design, all genetic examinations performed outside the UKL were considered as non-essential. The different types of dispensable genetic diagnostics are mainly constituted by chromosomal analyses, arrays, single gene testing, fragile X syndrome diagnostics and small custom gene panels (Fig. 4A, top part). In total 129 cMRI examinations were performed in this cohort, of which 36 (27.9%) were categorized as not required. With one exception, all of those were performed under general anesthesia. Contrast agent was used in 18 (50%) of dispensable imaging procedures. Of 349 metabolic examinations, 111 (31.8%) were categorized as dispensable. Of these 129 cMRI examinations, 94 (72.9%) were performed at UKL and 35 (27.1%) at other hospitals. We categorized 26 cMRI performed at UKL (27.7%) as potentially not required, whereas of 35 cMRI performed elsewhere (28.5%) were considered as dispensable. Only a minority of procedures in the categories of laboratory tests other than metabolic tests, medical imaging except cMRI, electrophysiology, and EEG were classified as non-essential. All consults, ECG, functional tests, EEG examinations were classified as essential. We classified 21 (7.3%) of a total of 288 inpatient stays performed as potentially dispensable. Among these, non-required hospitalizations had a median of two inpatient overnight stays (range: 1–7; SD: 1.9) and were significantly shorter (p ~ 0.016, Wilcox-Test) than hospitalizations classified as indispensable, with a median length of four nights (range: 0–731; SD: 46.7) (Supplementary Fig. S6).
We calculated the costs for the three diagnostic categories with the largest number of non-required diagnostic procedures: genetic diagnostics, metabolic diagnostics, and cMRI. In our cohort, a total of 687,168.02€ was spent on genetic diagnostics. Thereof, 302,947.07€ (44.1%) are associated with dispensable examinations. Of the 82,589.20€ spent on cMRI, 21,903.37€ (26.5%) were considered not required if the final genetic diagnosis would have been known and considered. From 35,980.43€ issued for metabolic examinations, a portion of 10,987.05€ (30.5%) was classified as not required (Fig. 4B). Thus, genetic examinations show the highest cost savings potential with 302,947.07€ (90.2%) out of 335,837.49€.
On average, the total potentially savable cost amounts to 3025.56€ per individual in our study. This corresponds to an average of 2729.25€ for genetic diagnostics, 197.33€ for cMRI examinations and 98.98€ for metabolic testing regarding potential cost savings.
The amount of summed dispensable cost per individual does not correlate with the length of the diagnostic trajectory (Interval 1–5) for metabolic diagnostics and cMRI. In genetic diagnostics, a moderate positive correlation (p < 0.001, Pearson correlation coefficient = 0.367, R2 = 0.13) is observed between the amount of the summed costs of genetic testing per individual and the length of time needed to obtain a diagnosis (Fig. 4C).
Discussion
This retrospective cohort analysis emphasizes the importance of implementing ES as a first line test in the diagnostic pathway for individuals with NDD and/or epilepsy. NGS-based testing ended the hitherto inconclusive diagnostic pathway in all 111 individuals in our study. Therefore, the length of the diagnostic odyssey is significantly shorter in individuals with first symptoms onset after the availability of NGS than in individuals developing first symptoms before this cut-off time. It must be considered that our study included only individuals who received a final molecular diagnosis, and we can thus not assess individuals who had inconclusive NGS-based diagnostics. However, our design is a representative snapshot of the currently achievable diagnostic yield (~31–53%) using ES as a first-tier test [24] in NDD/epilepsy.
Even before the broad availability of NGS in the clinical setting, genetic testing in this cohort was often initiated early by the treating pediatricians. However, the diagnostic odyssey was prolonged by the unavailability of genetic diagnostics covering the considerable heterogeneity observed in NDD and epilepsy (SysID database [25] accessed on 2021-04-24 states 1454 genes associated with NDD whereof 663 were associated with epilepsy). This caused a stepwise evaluation using karyotyping, microarrays, and clinically suspected diagnoses. After the establishment of NGS, this phase receded, while the time interval before initiation of genetic diagnostics became critical for a fast diagnosis. Direct comparison of Interval 1–3 (pediatricians) and 4–5 (medical geneticists) before and after clinical availability of NGS demonstrates a remarkable reduction on the genetics side. This shows the accelerating impact of NGS-based methods on diagnostic time intervals. Thus, further potential for shortening the diagnostics lies in the faster referral of individuals with NDD/epilepsy to genetics testing. We recommend early human genetics consults in the pediatric diagnostic process, which could also be implemented using telemedical methods. A close and coordinated cooperation between pediatricians and human geneticists is essential to achieve a fast diagnosis. These measures are generalizable and will likely improve time to diagnosis in any health care system.
Our data imply that individuals who first presented at other or smaller hospitals had no disadvantages in terms of time to molecular diagnosis. This may be due to good cooperation between institutions or the well-known role of our Center for Rare Diseases in the local area.
Individuals with epilepsy received a quicker diagnosis than those with NDD after establishment of clinical NGS. Thus, the decrease in diagnostic duration is more apparent in individuals with epilepsy. This might be explained by the assumption that pediatricians may be more sensitive to the possible genetic background of epilepsy. The potentially more acute clinical presentation of epileptic seizures could also have led to a more rapid initiation of genetic diagnostics. Furthermore, the focus on epilepsy research in genetics at UKL may have contributed to this. It would be desirable if this could be increasingly established for NDD entities without epilepsy.
Stark et al. [26]. and Tan et al [27] examined the cost-effectiveness of NGS and also considered potential cost savings by omitting other traditional diagnostic procedures. These studies did however not consider whether these diagnostic interventions might nevertheless be indispensable in individual cases. Soden et al. [28] analyzed potentially dispensable medical costs through rapid ES in NDDs, but included diagnostics that would have been performed even if the genetic diagnosis was known in their cost savings model. Therefore, our careful reassessment of each individual case, represents the first study to examine the extent of diagnostics that could retrospectively be replaced by early implementation of ES based on accepted criteria for diagnostics of NDD and epilepsy in a tertiary Center for Rare Diseases.
The greatest potential for cost savings concerns prior genetic diagnostics in our study. Of the genetic tests performed, 72.7% were classified as potentially dispensable representing 90.2% of total savable costs. A reversal from classical genetic diagnostics to an exome first approach [29] would thus have resulted in reduced diagnostic costs in our cohort. Particularly because ES is also increasingly suitable for the detection of CNVs (in our cohort 26 CNVs, including small and complex alternations like translocations, were identified through NGS), which provides an alternative to chromosomal microarrays [22, 30–32]. After inconclusive first tier ES, genetic examinations could be extended accordingly to entities possibly not covered, such as translocations, repeat disorders (fragile X syndrome) or mitochondrial disorders.
The proportion of dispensable metabolic (31.8%) and cMRI examinations (27.9%) is relevant, because genetic testing can compete with, or even surpass them in terms of diagnostic yield [31, 33]. ES prior to performing these diagnostics should therefore be considered. Unlike other studies [34] we did not classify all metabolic diagnostics as dispensable, because they are essential confirmatory diagnostics and used for therapy decisions. Furthermore, metabolic testing currently provides diagnostic results in critical situations faster than genetic testing. Faster genetic results in the future could therefore replace additional metabolic examinations.
Our data suggest that the potential for diagnostic savings through ES lies primarily in genetic diagnostics. This is an effect of the granulated and stepwise approach performed historically. Except for metabolic and cMRI examinations, there was little to no potential for further savings.
A comparison of the savable costs in our cohort with the results of other studies is limited, because of differences in study design and cohort selection. Tan et al. [27] and Stark et al. [26] prospectively designed different diagnostic pathways in the course of ES in an undiagnosed cohort and compared their estimated costs. Based on this, cost savings per additional diagnosis (inflated and currency converted values: 6237.14€ and 1561.58€) were determined. Soden et al. provide the mean costs of prior negative testing in non-acute individuals (inflated and currency converted value: 17,785.18€). Chung et al. [35], Monroe et al. [34], and Vrijenhoek et al. [20] calculated the cost savings per individual resulting from avoidable medical examinations due to early ES implementation (inflated and currency converted values: 110.63€; 3277.66€, and 5090.39€). In some of these publications the costs of potentially unnecessary diagnostics even exceeded costs for ES. In our data, the cost of ES amounts to 3461.45€ while the average cost savings by avoidable diagnostics is 3025.56€ per individual. Thus, the average cost of potentially dispensable diagnostics almost reaches the cost level of an ES examination. The avoidable costs in this study are lower than in some previously publications. This may be an effect of our cautious evaluation of potentially dispensable diagnostics and our focus on solely direct diagnostic costs. Furthermore, previous studies report higher costs for individual parameters of metabolic diagnostics as well as for cMRI which could be explained by different pricing policies of health care systems in other countries.
Hospitalizations that we classified as potentially avoidable were shorter than indispensable inpatient stays. This can be explained by the solely diagnostic purpose of these not required hospitalizations, whereas required hospitalizations were mostly associated with complex therapies, emergency admissions, or a poor general condition of the individuals. While only 7.3% of inpatient stays (with a median of only two nights) were classified as dispensable, their potential omission could have been a major relief for affected families in individual cases [36].
Costs alone should not determine the course of action in rare disease care. Early implementation of ES could reduce diagnostic costs and time, but also prevent exhausting or even risky procedures. In our study, more than a quarter of cMRI examinations were judged to be potentially dispensable and most were performed using anesthesia. Putting a child at risk [37] for imaging with sedation and contrast agent could be influenced by the outcome of prioritized genetic testing since imaging results rarely lead to diagnosis in individuals with NDD [38]. A quick molecular diagnosis can also affect therapy and medical interventions [13, 22, 27], have an impact on family planning [12, 16], and contribute to the psychological well-being of the parents [14].
Moreover, our study demonstrates the importance of reanalyzing and reevaluating exome data (compare Table 1 and Supplementary results) [12, 39]. The reassessment of the SMAD4 variant initially reported as incidental finding in in Ind012 is of decisive clinical importance for the affected family who now do not have to partake invasive colonoscopies anymore. In addition, our reevaluation revealed one individual in which the previously reported genetic diagnosis could not sufficiently explain the observed phenotype because the associated phenotype was too mild. The formal downgrading of five variants is expected and an effect of continuous development and stricter interpretation of the ACMG criteria for variant classification. We considered the five cases as resolved by the plausibility of these “hot VUS”. Future analyses might facilitate a definitive assessment as reevaluation in light of new guidelines and expanding genetic databases may yield new results [40].
Table 1.
Information of the five individuals with “hot VUS” variants after reevaluation.
| Variant ID | Individual ID | Gene | Variant | Variant classification | Classification rules | Zygosity | Origin |
|---|---|---|---|---|---|---|---|
| SNV_002 | Ind002 | TUBB2B | c.611 A > T, p.(Asn204Ile) | VUS | PM2_Supporting, PP2_Supporting, PP3_Supporting, PP4_Supporting | Heterozygous | Maternal |
| SNV_022 | Ind019 | NPRL3 | c.434 T > C, p.(Leu145Pro) | VUS | PS2_Moderate, PM2_Supporting, PP3_Supporting | Heterozygous | de novo (confirmed) |
| SNV_027 | Ind025 | DYNC1H1 | c.8945 G > A, p.(Arg2982His) | VUS | PS2_Moderate, PM2_Supporting, PP2_Supporting, PP3_Supporting | Heterozygous | de novo (confirmed) |
| SNV_067 | Ind069 | SCN1A | c.4581 + 18 A > G, p.? | VUS | PS2_Moderate, PM2_Supporting, PP3_Supporting | Heterozygous | de novo (confirmed) |
| SNV_073 | Ind077 | NBEA | c.4662 + 1 G > C, p.Thr1519_Val1554del | VUS | PVS1_Moderate, PS2_Moderate, PM2_Supporting | Heterozygous | de novo (confirmed) |
Limitations of this study include the relatively small cohort, the lack of control group by focusing on solved cases only and the retrospective approach. Our study design included data on examinations from hospitals other than UKL to increase the number of available data for these important categories. We used the GOÄ billing system for consistent cost calculation to minimize potential bias through this approach. Also, only a part of dispensable costs was determined, so the extent of these may potentially be higher. Overall, some of the study design choices were imposed by the billing system and decentralized medical system in Germany.
Further studies on this topic should involve larger cohorts in a prospective setting. Collaboration among multiple rare disease centers and more complete collection of individuals’ medical data across the boundaries of single medical institutions would benefit for this research aim. However, individuals with rare NDD and/or epilepsy entities will surely benefit from continued development and research into rapid and effective diagnostic pathways. Therefore, close and informed collaboration between different medical specialties, such as pediatrics and human genetics, is essential. Both early consideration of a genetic differential diagnosis and quick performance of ES can contribute to reduce diagnostic time, costs and exhausting medical procedures and enable a sooner reproductive choice in the families.
Supplementary information
Acknowledgements
We thank all involved families for participating in this study. We appreciate the support of our colleagues Julia Herrmann, Uwe Herrmann, Dr Julia Hentschel, Anja Heinze and Marcel Frömming. This paper is part of a doctoral thesis of JK.
Author contributions
JRL and SB conceived the initial study concept. JK and SB collected, curated and classified the clinical data. RAJ and MR provided the genetic data and performed database submission, with updates by JK, JK, HO, MR and BP re-evaluated all variants. JK, SB, and BP cleaned, standardized and analyzed all clinical and genetic data. JK and BP created Figs. 1, 2, 3 and 4 and Supplementary materials. JK, JRL, and BP wrote and edited the paper. All authors reviewed and commented on the final paper draft.
Funding
BP is supported by the Deutsche Forschungsgemeinschaft (DFG) through grant PO2366/2–1. Open Access funding enabled and organized by Projekt DEAL.
Data availability
All data generated or analyzed during this study are included in this published paper and its supplementary information files.
Competing interests
The authors declare no competing interests.
Ethical approval
The project was approved by the ethic committee of the University of Leipzig, Germany (224/16-ek and 402/16-ek) and was conducted in concordance to the declaration of Helsinki. Individuals or their parents or legal guardians consented to genetic testing.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Skadi Beblo, Bernt Popp
Supplementary information
The online version contains supplementary material available at 10.1038/s41431-021-00981-z.
References
- 1.Kumar P, Radhakrishnan J, Chowdhary MA, Giampietro PF. Prevalence and Patterns of Presentation of Genetic Disorders in a Pediatric Emergency Department. Mayo Clin Proc. 2001;76:777–83. doi: 10.4065/76.8.777. [DOI] [PubMed] [Google Scholar]
- 2.Yoon PW. Contribution of Birth Defects and Genetic Diseases to Pediatric Hospitalizations: a Population-Based Study. Arch Pediatr Adolesc Med. 1997;151:1096. doi: 10.1001/archpedi.1997.02170480026004. [DOI] [PubMed] [Google Scholar]
- 3.Gilissen C, Hehir-Kwa JY, Thung DT, van de Vorst M, van Bon BWM, Willemsen MH, et al. Genome sequencing identifies major causes of severe intellectual disability. Nature. 2014;511:344–7. doi: 10.1038/nature13394. [DOI] [PubMed] [Google Scholar]
- 4.Aaberg KM, Gunnes N, Bakken IJ, Lund Søraas C, Berntsen A, Magnus P, et al. Incidence and Prevalence of Childhood Epilepsy: a Nationwide Cohort Study. Pediatrics. 2017;139:e20163908. doi: 10.1542/peds.2016-3908. [DOI] [PubMed] [Google Scholar]
- 5.Wang J, Lin Z-J, Liu L, Xu H-Q, Shi Y-W, Yi Y-H, et al. Epilepsy-associated genes. Seizure. 2017;44:11–20. doi: 10.1016/j.seizure.2016.11.030. [DOI] [PubMed] [Google Scholar]
- 6.Hanly C, Shah H, Au PYB, Murias K. Description of neurodevelopmental phenotypes associated with 10 genetic neurodevelopmental disorders: a scoping review. Clin Genet. 2021;99:335–46. doi: 10.1111/cge.13882. [DOI] [PubMed] [Google Scholar]
- 7.Carmichael N, Tsipis J, Windmueller G, Mandel L, Estrella E. “Is it Going to Hurt?”: the Impact of the Diagnostic Odyssey on Children and Their Families. J Genet Couns. 2015;24:325–35. doi: 10.1007/s10897-014-9773-9. [DOI] [PubMed] [Google Scholar]
- 8.Gonzaludo N, Belmont JW, Gainullin VG, Taft RJ. Estimating the burden and economic impact of pediatric genetic disease. Genet Med. 2019;21:1781–9. doi: 10.1038/s41436-018-0398-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McCandless SE, Brunger JW, Cassidy SB. The Burden of Genetic Disease on Inpatient Care in a Children’s Hospital. Am J Hum Genet. 2004;74:121–7. doi: 10.1086/381053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arim RG, Miller AR, Guèvremont A, Lach LM, Brehaut JC, Kohen DE. Children with neurodevelopmental disorders and disabilities: a population-based study of healthcare service utilization using administrative data. Dev Med Child Neurol. 2017;59:1284–90. doi: 10.1111/dmcn.13557. [DOI] [PubMed] [Google Scholar]
- 11.Strzelczyk A, Reese JP, Dodel R, Hamer HM. Cost of epilepsy: a systematic review. PharmacoEconomics. 2008;26:463–76. doi: 10.2165/00019053-200826060-00002. [DOI] [PubMed] [Google Scholar]
- 12.Schofield D, Rynehart L, Shresthra R, White SM, Stark Z. Long-term economic impacts of exome sequencing for suspected monogenic disorders: diagnosis, management, and reproductive outcomes. Genet Med. 2019;21:2586–93. doi: 10.1038/s41436-019-0534-x. [DOI] [PubMed] [Google Scholar]
- 13.Dixon-Salazar TJ, Silhavy JL, Udpa N, Schroth J, Bielas S, Schaffer AE, et al. Exome Sequencing Can Improve Diagnosis and Alter Patient Management. Sci Transl Med. 2012;4:138ra78–138ra78. doi: 10.1126/scitranslmed.3003544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Graungaard AH, Skov L. Why do we need a diagnosis? A qualitative study of parents? experiences, coping and needs, when the newborn child is severely disabled. Child Care Health Dev. 2007;33:296–307. doi: 10.1111/j.1365-2214.2006.00666.x. [DOI] [PubMed] [Google Scholar]
- 15.Iglesias A, Anyane-Yeboa K, Wynn J, Wilson A, Truitt Cho M, Guzman E, et al. The usefulness of whole-exome sequencing in routine clinical practice. Genet Med. 2014;16:922–31. doi: 10.1038/gim.2014.58. [DOI] [PubMed] [Google Scholar]
- 16.Srivastava S, Cohen JS, Vernon H, Barañano K, McClellan R, Jamal L, et al. Clinical whole exome sequencing in child neurology practice: WES in Child Neurology. Ann Neurol. 2014;76:473–83. doi: 10.1002/ana.24251. [DOI] [PubMed] [Google Scholar]
- 17.Rump P, Jazayeri O, van Dijk-Bos KK, Johansson LF, van Essen AJ, Verheij JBGM, et al. Whole-exome sequencing is a powerful approach for establishing the etiological diagnosis in patients with intellectual disability and microcephaly. BMC Med Genom. 2015;9:7. doi: 10.1186/s12920-016-0167-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fernandez-Marmiesse A, Gouveia S, Couce ML. NGS Technologies as a Turning Point in Rare Disease Research, Diagnosis and Treatment. CMC. 2018;25:404–32. doi: 10.2174/0929867324666170718101946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Demos M, Guella I, DeGuzman C, McKenzie MB, Buerki SE, Evans DM, et al. Diagnostic Yield and Treatment Impact of Targeted Exome Sequencing in Early-Onset Epilepsy. Front Neurol. 2019;10:434. doi: 10.3389/fneur.2019.00434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vrijenhoek T, Middelburg EM, Monroe GR, van Gassen KLI, Geenen JW, Hövels AM, et al. Whole-exome sequencing in intellectual disability; cost before and after a diagnosis. Eur J Hum Genet. 2018;26:1566–71. doi: 10.1038/s41431-018-0203-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dillon OJ, Lunke S, Stark Z, Yeung A, Thorne N, Gaff C, et al. Exome sequencing has higher diagnostic yield compared to simulated disease-specific panels in children with suspected monogenic disorders. Eur J Hum Genet. 2018;26:644–51. doi: 10.1038/s41431-018-0099-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zacher P, Mayer T, Brandhoff F, Bartolomaeus T, Le Duc D, Finzel M, et al. The genetic landscape of intellectual disability and epilepsy in adults and the elderly: a systematic genetic work-up of 150 individuals. Genet Med. 2021. 10.1038/s41436-021-01153-6. [DOI] [PMC free article] [PubMed]
- 23.Lemke JR. High-Throughput Sequencing as First-Tier Diagnostics in Congenital and Early-Onset Disorders. JAMA Pediatr. 2017;171:833. doi: 10.1001/jamapediatrics.2017.1970. [DOI] [PubMed] [Google Scholar]
- 24.Srivastava S, Love-Nichols JA, Dies KA, Ledbetter DH, Martin CL, Chung WK, et al. Meta-analysis and multidisciplinary consensus statement: exomesequencing is a first-tier clinical diagnostic test for individuals withneurodevelopmental disorders. Genet Med. 2019;21:2413–21. doi: 10.1038/s41436-019-0554-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kochinke K, Zweier C, Nijhof B, Fenckova M, Cizek P, Honti F, et al. Systematic Phenomics Analysis Deconvolutes Genes Mutated in Intellectual Disability into Biologically Coherent Modules. Am J Hum Genet. 2016;98:149–64. doi: 10.1016/j.ajhg.2015.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stark Z, Schofield D, Alam K, Wilson W, Mupfeki N, Macciocca I, et al. Prospective comparison of the cost-effectiveness of clinical whole-exome sequencing with that of usual care overwhelmingly supports early use and reimbursement. Genet Med. 2017;19:867–74. doi: 10.1038/gim.2016.221. [DOI] [PubMed] [Google Scholar]
- 27.Tan TY, Dillon OJ, Stark Z, Schofield D, Alam K, Shrestha R, et al. Diagnostic Impact and Cost-effectiveness of Whole-Exome Sequencing for Ambulant Children With Suspected Monogenic Conditions. JAMA Pediatr. 2017;171:855. doi: 10.1001/jamapediatrics.2017.1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Soden SE, Saunders CJ, Willig LK, Farrow EG, Smith LD, Petrikin JE, et al. Effectiveness of exome and genome sequencing guided by acuity of illness for diagnosis of neurodevelopmental disorders. Sci Transl Med. 2014;6:265ra168–265ra168. doi: 10.1126/scitranslmed.3010076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stark Z, Tan TY, Chong B, Brett GR, Yap P, Walsh M, et al. A prospective evaluation of whole-exome sequencing as a first-tier molecular test in infants with suspected monogenic disorders. Genet Med. 2016;18:1090–6. doi: 10.1038/gim.2016.1. [DOI] [PubMed] [Google Scholar]
- 30.Quenez O, Cassinari K, Coutant S, Lecoquierre F, Le Guennec K, Rousseau S, et al. Detection of copy-number variations from NGS data using read depth information: a diagnostic performance evaluation. Eur J Hum Genet. 2021;29:99–109. doi: 10.1038/s41431-020-0672-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berg AT, Coryell J, Saneto RP, Grinspan ZM, Alexander JJ, Kekis M, et al. Early-Life Epilepsies and the Emerging Role of Genetic Testing. JAMA Pediatr. 2017;171:863. doi: 10.1001/jamapediatrics.2017.1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miyatake S, Koshimizu E, Fujita A, Fukai R, Imagawa E, Ohba C, et al. Detecting copy-number variations in whole-exome sequencing data using the eXome Hidden Markov Model: an ‘exome-first’ approach. J Hum Genet. 2015;60:175–82. doi: 10.1038/jhg.2014.124. [DOI] [PubMed] [Google Scholar]
- 33.Yubero D, Brandi N, Ormazabal A, Garcia-Cazorla À, Pérez-Dueñas B, Campistol J, et al. Targeted Next Generation Sequencing in Patients with Inborn Errors of Metabolism. PLoS ONE. 2016;11:e0156359. doi: 10.1371/journal.pone.0156359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Monroe GR, Frederix GW, Savelberg SMC, de Vries TI, Duran KJ, van der Smagt JJ, et al. Effectiveness of whole-exome sequencing and costs of the traditional diagnostic trajectory in children with intellectual disability. Genet Med. 2016;18:949–56. doi: 10.1038/gim.2015.200. [DOI] [PubMed] [Google Scholar]
- 35.Chung CCY, Leung GKC, Mak CCY, Fung JLF, Lee M, Pei SLC, et al. Rapid whole-exome sequencing facilitates precision medicine in paediatric rare disease patients and reduces healthcare costs. Lancet Regional Health - West Pac. 2020;1:100001. doi: 10.1016/j.lanwpc.2020.100001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Seliner B, Latal B, Spirig R. When children with profound multiple disabilities are hospitalized: A cross-sectional survey of parental burden of care, quality of life of parents and their hospitalized children, and satisfaction with family-centered care: Parents of hospitalized children with disabilities. J Spec Pediatr Nurs. 2016;21:147–57. doi: 10.1111/jspn.12150. [DOI] [PubMed] [Google Scholar]
- 37.Salerno S, Granata C, Trapenese M, Cannata V, Curione D, Rossi Espagnet MC, et al. Is MRI imaging in pediatric age totally safe? A critical reprisal. Radio Med. 2018;123:695–702. doi: 10.1007/s11547-018-0896-1. [DOI] [PubMed] [Google Scholar]
- 38.Murias K, Moir A, Myers KA, Liu I, Wei X-C. Systematic review of MRI findings in children with developmental delay or cognitive impairment. Brain Dev. 2017;39:644–55. doi: 10.1016/j.braindev.2017.04.006. [DOI] [PubMed] [Google Scholar]
- 39.Fung JLF, Yu MHC, Huang S, Chung CCY, Chan MCY, Pajusalu S, et al. A three-year follow-up study evaluating clinical utility of exome sequencing and diagnostic potential of reanalysis. Npj Genom Med. 2020;5:37. doi: 10.1038/s41525-020-00144-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li J, Gao K, Yan H, Xiangwei W, Liu N, Wang T, et al. Reanalysis of whole exome sequencing data in patients with epilepsy and intellectual disability/mental retardation. Gene. 2019;700:168–75. doi: 10.1016/j.gene.2019.03.037. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this published paper and its supplementary information files.