

Abstract
Gastrointestinal (GI) cancers cause one-third of all cancer-related deaths worldwide. Natural compounds are emerging as alternative or adjuvant cancer therapies given their distinct advantage of manipulating multiple pathways to both suppress tumor growth and alleviate cancer comorbidities; however, concerns regarding efficacy, bioavailability, and safety are barriers to their development for clinical use. Emodin (1,3,8-trihydroxy-6-methylanthraquinone), a Chinese herb-derived anthraquinone, has been shown to exert anti-tumor effects in colon, liver, and pancreatic cancers. While the mechanisms underlying emodin’s tumoricidal effects continue to be unearthed, recent evidence highlights a role for mitochondrial mediated apoptosis, modulated stress and inflammatory signaling pathways, and blunted angiogenesis. The goals of this review are to (1) highlight emodin’s anti-cancer properties within GI cancers, (2) discuss the known anti-cancer mechanisms of action of emodin, (3) address emodin’s potential as a treatment complementary to standard chemotherapeutics, (4) assess the efficacy and bioavailability of emodin derivatives as they relate to cancer, and (5) evaluate the safety of emodin.
Keywords: natural compounds, complementary and alternative medicine, emodin, gastrointestinal cancers, cancer treatments
Introduction
Gastrointestinal (GI) cancers account for a significant proportion of cancer incidence and mortality; indeed, in 2018 total cancer incidence was estimated to be 4.8 million cases worldwide and GI cancers were responsible for 35% of all cancer-related deaths. 1 Currently, the treatment strategies for these cancers may include cytotoxic chemotherapeutics, radiation, immunotherapy, and/or surgery. 2 Although these treatment paradigms have undoubtedly contributed to increased survival, there has been little advancement in counteracting the pervasive off-target effects and acquired drug resistance that occur with these treatments.2-8 The use of natural compounds has shown promise in both suppressing tumor growth and alleviating adverse effects associated with standard chemotherapeutics.9,10 Indeed, many natural compounds boast of very broad pleiotropic effects, making them potentially attractive candidates to target the multiple pathways driving tumorigenesis and treatment side-effects.9,10 As such, natural compounds that increase apoptosis, decrease proliferation, modulate intracellular signaling, and inhibit tumorigenesis have shown the best potential as anti-cancer agents.9,10 The ability of natural compounds to exert anti-cancer and chemoprotective effects may be associated with improved patient tolerability, quality of life, and survival.9,10 Unfortunately, however, their use in clinical oncology remains extremely limited, likely due to inconsistencies in efficacy and toxicity findings across preclinical and clinical studies.
Emodin (1,3,8-trihydroxy-6-methylanthraquinone), isolated from the roots and barks of several plants, is a Chinese herb-derived anthraquinone that has shown promise pre-clinically as a natural anti-cancer agent for the treatment of various cancers including breast, pancreatic, colon, gastric, liver, gallbladder, and lung.5,6,11,12 Recent evidence suggests that emodin exerts its anti-cancer effects through inducing mitochondrial mediated apoptosis, alongside suppressing pathways that promote inflammation, proliferation, angiogenesis, and tumorigenesis (Table 1). Indeed, emodin has been demonstrated to elicit these effects at both low (1-40 mg/kg)13 -20 and higher (50-80 mg/kg)16,18 -22 doses consistently across pre-clinical models of colon, liver, and pancreatic cancer. Given emodin’s multimodal anti-tumor properties, popularity has grown not only in investigating its potential as a stand-alone treatment, but also as a complementary therapy to existent chemotherapeutics as it has been reported to enhance the sensitivity of select anti-cancer drugs.15,21,23,24 Although emodin’s anti-cancer effects have been previously reviewed,11,12,25,26 this review will profile the most recent (2017-2021) advancements in emodin’s efficacy, safety, and mechanisms regulating its anti-cancer properties in GI cancers, including colon, liver, and pancreas. Additionally, emphasis will be placed on emodin dosing as it pertains to the disparity amongst treatment regimens that has contributed to equivocal results and reported toxicities.
Table 1.
Apoptotic and Tumorigenic Effects of Emodin.
| Main effect | Model | Cell line or species | Agent/dose/administration route | Mechanism | Reference |
| Apoptosis | Human HCC | HepaRG | Emodin*; 0-80 µM, 0-48 h IC50 ≈ 40/80 µM at 48/24 h |
Mitochondrial mediated apoptosis by ↑ROS*** and cell cycle arrest (at S and G2/M phases) | Dong et al 30 |
| HepG2 and normal human L02 hepatocytes | Emodin*; 0.14-100 µM, 48 h HepG2 IC50 ≈ 44 µM; L02 IC50 ≈ 22 µM Emodin derivative**; 0-10 µM, 48 h HepG2 IC50 ≈ 5 µM; L02 IC50 ≈ 14 µM |
Mitochondrial mediated apoptosis***; cell arrest at G0/G1 phase (in HepG2)*** | Yang et al 34 | ||
| HepG2 | Emodin*; 1-100 µM, 0-72 h IC50 ≈ 20 µM, 72 h |
MAPK regulation (DEGs association; 20 uM) | Zhou et al 29 | ||
| HepG2 | Emodin; 50 or 100 μM, 0-48 h | Cyp-D↑ regulated by ROS***, p-ERK↓*** | Zhang et al 37 | ||
| In vitro: SMMC-7721 | In vitro: Emodin*; Apoptosis: 0-200 µM, 0-48 h IC50 ≈ 50 µM, 48 h |
↓PI3K (↓p-Akt)***; ↑MAPK (↑p-ERK, ↑p-p38, mild↓JNK)*** | Lin et al 19 | ||
| Human liver fibrosis | LX2 (human hepatic stellate-model for liver fibrosis) | Aloe Emodin; 10 or 20 µM, 72 h | Mitochondrial mediated apoptosis, ↓PI3K signaling pathways, ↓TNF | Cai et al 41 | |
| Normal human liver | L02 | Emodin; 0-80 µM, 0-24 h IC50 ≈ 60-80 µM, 24 h |
↑Autophagy (protective role) by ↓p-PI3K/p-AKT/p-mTOR to inhibit apoptosis | Zheng et al 43 | |
| Human colon cancer | HCT116 | Emodin; 10, 25, and 50 µM, 0-24 h IC50 ≈ 20 µM, 24 h |
Inhibition of fatty acid synthesis (FASN)***, ↓PI3K/Akt (25 uM) signaling pathways | Lee et al 49 | |
| HCT116 | Emodin, 15, 30, 60 μg/ml, 24 h | ↓PI3K, ↓p-AKT, ↓VEGFR2 | Dai et al 20 | ||
| SW620 and HCT116 | Emodin, 10, 20, 40 uM, 24 h | Apoptosis*** | Zhang et al 22 | ||
| CACO-2 | Emodin*; Cell viability: 0-200 µM, 24 h; anti-tumor effects: 15-60 µM) IC50 ≈ 35 µM |
Mitochondrial mediated apoptosis***, ↓PI3K/Akt (phospho-proteins) signaling pathway***, induce G2/M cell cycle arrest*** | Ma et al 28 | ||
| DLD-1 and COLO-20 | Emodin*; cell viability: 0-80 μM, 0-72 h All other experiments: 15-18 μM (IC50), 0-48 h |
Mitochondrial mediated apoptosis via ↓MAPK/JNK, PI3K/AKT, NF-κβ and STAT pathways | Saunders et al 27 | ||
| SW620 and HT29 | Aloe Emodin*; 0-40 μM, 0-72 h IC50 ≈ 40 µM, 72 h |
ER stress by ↑ROS***; mitochondrial mediated apoptosis*** | Cheng and Dong 39 | ||
| HCT116 and LOVO | Emodin; Cell viability: 0-320 µM, 0-48 h All other experiments; 0-40 µM, 24 h IC50 ≈ 40, 48 h |
Mitochondrial mediated apoptosis by autophagy-ROS dependent induction***; mitochondrial dysfunction | Wang et al 46 | ||
| Main effect | Model | Cell line or species | Agent/dose/administration route | Mechanism | Reference |
| HT-29 | Emodin* and silica nanomaterials loaded with emodin, 0-80 μM, 0-48 h | Mitochondrial mediated apoptosis, ↓PI3K/Akt, ↑p-ERK, ↑auophagy (↑LC3A/B) | Janicke et al 48 | ||
| In vitro: LS1034 | Emodin, 0-50 µM, 0-48 h | Mitochondrial mediated apoptosis by ↑ROS | Ma et al 32 | ||
| Human pancreatic cancer | MIAPaCa-2 and PANC-1 | Aloe Emodin*; 0-100 µM, 48 h IC50 ≈ 40 µM |
Mitochondrial mediated apoptosis and autophagy dose-dependently; sub-G1 cell cycle arrest | Du et al 40 | |
| PANC-1 and MiaPaCa2 | Emodin, 10-160 µM, 72 h (apoptosis) Emodin, 40 µM; gemcitabine, 20 µM |
↓Survivin, XIAP, NF-κB, and IKKβ. ↑Caspase-3/9 and IκB-α | Tong et al 31 | ||
| PANC-1 and BxPC-3 | In vitro: emodin*; 0-90 µM, 0-72 h EGFR inhibitor Afatinib*; 20 nM, 0-72 h |
↓STAT3 (emodin with or without afatinib)*** | Wang et al 23 | ||
| 20 BALB/c nude mice (5 mice/group) subcutaneously injected with PANC-1 cells | In vivo: emodin and afatinib, 50 mg/kg, oral, 4 weeks after tumor reached 30-50 mm3. Vehicle: saline | ||||
| ↓Tumorigenesis | Human HCC | In vitro: HepG2 | In vitro: emodin; 10, 20, or 100 nM, 0-36 h | Inhibited VEGFR2-AKT-ERK1/2 signaling pathway*** and ↑miR-34a | Bai et al 13 |
| In vivo: 30 male BALB/c nude mice subcutaneously injected with HepG2 cells | In vivo: emodin; 1 or 10 mg/kg, hypodermic injection, daily/10 days | ||||
| In vitro: HepG2, Hep3B, Huh7, SK-HEP-1, and PLC/PRF5 | In vitro: emodin*; 0-40 μM, 0-72 h optimal: emodin 20 µM and sorafenib*; 2 µM |
↓Cholesterol*** to sensitize cells via ↓Akt*** and STAT3*** by inhibiting sterol regulatory element-binding protein-2 (SREBP-2) | Kim et al 15 | ||
| In vivo: BALB/c-nude mice xenografted with HepG2 or SK-HEP-1 | In vivo: emodin*; 10 mg/kg, i.p. and/or sorafenib*; 5 mg/kg, i.p., daily/3 weeks after tumor reached 250 mm3 | ||||
| HepG2 | Emodin derivative**; 0-1 µM, 48 h | ↓Migration*** (no mechanism) | Yang et al 34 | ||
| In vivo: 15 male BALB/c-nu nude mice (5 mice/group) subcutaneously injected with SMMC-7721 cells | In vivo: Emodin*; 25 or 50 mg/kg, i.p., everyday for 2 weeks after tumor volume 75-100 mm3 | No toxicity or body weight changes ↓ALT, AST, AKP, GGT, Cr and BUN |
Lin et al 19 | ||
| HepG2 | β-dihydroartemisinin-emodin | Mitochondrial mediated apotosis; inhibited G1 to S cell cycle progression; inhibited cell migration by ↓survivin | Li et al 42 | ||
| Main effect | Model | Cell line or species | Agent/dose/administration route | Mechanism | Reference |
| Colon cancer | In vitro: HT29 and RKO (human) | In vitro: emodin; MTT: 0-100 μM, 0-48 h All other experiments: 5, 10, 20 µM IC50 ≈ 40/80 μM 48/24 h |
↓matrix proteins and VEGF***. | Gu et al 17 | |
| In vivo: 30 male BALB/c nude mice (15 mice/group) pretreated with emodin before subcutaneous RKO cell injection into inguinal region, sacrificed after 4 weeks | In vivo: 40 mg/kg, P.O., 7 day pretreament | ↓EMT by Wnt/β-Catenin Pathway inactivation and by ↑E-cadherin and ↓N-cadherin, Snail, and b-catenin (mRNA)*** | |||
| 100 WAG/Rij rats implanted intraperitoneal (ip) or subcutaneous (sc) CC-531 rat colon cancer cells for 28 days then treated | Emodin in 5% polyvinylpyrrolidone (PVP) and 0.9 % sodium chloride; 2.5 or 5 mg/kg; intravenous (iv) port catheter or ip injection, 7 days | N/A | Hohn et al 14 | ||
| 30 male BALB/c nu/nu mice xenografted with LS1034; subcutaneously injected into flank | In vivo: emodin*,40 mg/kg, ip, 1x every 3 days/4 weeks after tumor reached 200 mm3. Vehicle: 1% DMSO | N/A | Ma et al 32 | ||
| HCT116 | Emodin, 20, 40, 80 mg/kg, ip, daily/3 weeks | ↓PI3K, ↓p-AKT, ↓VEGFR2 | Dai et al 20 | ||
| 30 Male BALB/c mice (15/group); AOM/DSS model of colitis-associated intestinal tumorigenesis | Emodin; 50 mg/kg, gavage, 2 or 4 weeks | 14 weeks (in tumor microenvironement): ↓inflammatory cell (i.e. CD11b+ and F4/80+) recruitment, cytokine (i.e. TNFa, IL1a/b, IL6, CCL2, CXCL5) and pro-inflammatory enzymes (i.e. COX-2, NOS2); ↑ CD3+ T cell recruitment. ↓Bleeding and diarrhea | Zhang et al 22 | ||
| HCT116 and LOVO (human) | Emodin; Cell viability: 0-320 μM, 0-48 h All other experiments; 0-40 μM, 24 h IC50 ≈ 40 μM, 48 h. |
Mitochondrial mediated apoptosis by autophagy-ROS dependent induction***; mitochondrial dysfunction | Wang et al 46 | ||
| Human pancreatic cancer | In vitro: AsPC-1, BxPC-3, HPAF-2, MiaPaCa2, and Panc-1 (all human). Cells treated with agents with immediate 6 h hypoxic incubation | In vitro: emodin (0-200 μM) and rhein (0-200 μM) alone, 6 h | HIF1A inhibtion (all)*** by ↓Akt and ERK1/2 pathways*** (in vitro: MiaPaCa2; in vivo) | Hu et al 21 | |
| In vivo: 30 male athymic BALB/c mice (10 mice/group) subcutaneously implanted with MiaPaCa2 cells for 8 weeks | In vivo: emodin (50 mg/kg/day) and/or rhein (50 mg/kg) in PBS; gavage, last 4 weeks (1x/5 days a week) | ||||
| In vitro: PANC-1 and BxPC-3 (human) | In vitro: emodin*; 0-90 µM, 0-72 h Afatinib *; 20 nM, 0-72 h |
↓STAT3 (emodin with or without afatinib)*** | Wang et al 23 | ||
| In vivo: 20 BALB/c nude mice (5 mice/group) subcutaneously injected with PANC-1 cells | In vivo: emodin and EGFR inhibitor afatinib,50 mg/kg, oral, 4 weeks after tumor reached 30-50 mm3. Vehicle: saline | ||||
| 40 female athymic BALB/c nu/nu mice with surgical orthotopic implantation of SW1990 cells (10 mice/group) | Emodin in DMSO; 20, 40, and 80 mg/kg, i.p., 3x a week/3 weeks | Inhibition*** of angiogenesis via TGF-β/Smad signaling pathway (↓TGF-β1, ↓Angptl4, ↑SMAD4). ***↓oncogenic and angiogenesis-associated miR-155 and miR-210; ↑antiangiogenic miR-20b*** | Lin et al 18 | ||
| In vitro: SW1990 (human) | In vitro: emodin; 0-40 μM, 0-48 h | Inhibited EMT and invasion via ↑miR-1271***; inhibited liver metastasis of pancreatic cancer via same mechanism | Li et al 16 | ||
| Human pancreatic liver metastasis | In vivo: SW1990 cells injected into spleens of 45 nude mice (15 mice/group) | In vivo: emodin; 20 or 50 mg/kg; gavaged for about 3 weeks after 8 days of tumor establishement. Sacrificed 6 weeks later. | |||
| Agent dissolved in DMSO* | ((R)-1-(3-(4,5-Dihydroxy-7-methyl-9,10-dioxo-9,10-dihydroanthracen-2-yloxy)propoxy)-1-oxopropan-2-aminium 2,2,2-trifluoroacetate) in DMSO** | Dose-dependent*** |
Emodin’s Beneficial Effects on Tumorigenesis Outcomes
Over the last 2 decades we have witnessed an increase in the investigation of natural compounds, including emodin, for their potential as anti-cancer agents. To put this in perspective, a search of the National Library of Medicine using the term “emodin and cancer” revealed 2 publications in 2000 and 58 in 2020. In this section, we will focus largely on the most recent literature (2017-2021) documenting emodin’s effects on GI cancers; we will first address in vitro studies and then will report the available in vivo findings. Of note, there are emerging reports on (1) emodin’s potential as a complementary therapy to standard anti-cancer therapies and (2) anti-cancer effects of emodin derivatives; these will be addressed in sections IV and V, respectively.
In vitro studies
Several recent in vitro studies have documented emodin’s ability to suppress tumor growth in colon, liver, and pancreatic cell lines. We will address the recent in vitro studies investigating emodin’s effects on these 3 GI cancers. Mechanisms for emodin’s reported anti-cancer effects will be addressed in section III.
With regards to colon cancer, Dai et al 20 reported that emodin (15-60 μg/ml) suppressed the growth, adhesion, and migration of HCT116 cells, a human colon cancer cell line. In another study emodin (15-80 µM) decreased viability of human colon cancer cells (DLD-1 and COLO-20) in a time and dose-dependent manner compared to vehicle-treated control without significantly impacting normal colon epithelial cells (CCD841 CoN). 27 Similarly, Gu et al 17 reported that emodin (5-20 µM) inhibited the invasion and migration abilities of the human colon cancer cell line RKO. Another recent study documented that emodin (15-60 µM) exhibited potent anticancer effects on CACO-2 human colon carcinoma cells. 28 Thus, emodin appears to have anti-tumor efficacy in several colon cancer cell lines.17,20,27,28
The evidence for emodin’s protective effects in liver cancer is just as positive with several in vitro studies documenting beneficial effects of emodin in liver cancer cell lines.13,19,29,30 For example, it was recently reported that emodin can inhibit growth of HepG2 cells in a time- and dose- dependent manner. 29 Using the same cell line, Bai et al 13 reported that emodin (10 and 100 nM, 0-36 hours) time dependently suppressed HepG2 cell proliferation in vitro. Similarly, Dong et al 30 demonstrated that emodin time- and dose- dependently inhibited HepaRG cell growth. Finally, it has been reported that emodin inhibited the proliferation of SMMC-7721 cells, also in a dose- and time-dependent manner 19 These studies support a beneficial effect of emodin as an anti-cancer agent with efficacy against several liver cancer cell lines.13,19,29,30
In pancreatic cancer, emodin significantly inhibited the ability of SW1990 cells to proliferate in a dose- and time-dependent manner. 16 Further, it was reported that emodin increased apoptosis in the pancreatic cell lines, PANC-1 and BxPC-3. 23 Similarly, emodin was found to have efficacy against PANC-1 and MiaPaCa2 given its ability to inhibit cell viability. 31 Although limited, the available studies unequivocally document a beneficial effect of emodin on pancreatic cell lines in vitro.16,23,31
Overall, the available in vitro studies are overwhelmingly positive for a beneficial effect of emodin on colon,17,20,27,28 liver,13,19,29,30 and pancreatic16,23,31 cell lines. However, it must be noted that there remains a dearth of evidence in this area; studies are still parsing out effective dosing and timing for optimal efficacy.
In vivo studies
In vivo studies on emodin as a cancer therapeutic agent for GI cancers are just beginning to emerge in the literature. However, those that are available show great promise for the development of emodin as either a stand-alone anti-cancer therapy or as a combinational therapy (addressed in section IV). We will discuss the most recent available in vivo studies on emodin’s influence on GI cancers.
To our knowledge, prior to 2020, only 2 studies used in vivo models of colon cancer treated with emodin.32,33 The earliest available study (2012) reported that emodin (40 mg/kg administered i.p., twice a week for 4 weeks) effectively suppressed tumor growth in nude mice xenografts bearing LS1034. 32 In contrast, in 2017, Braumann et al 33 reported that emodin (5-10 mg/kg) administered twice a day for a week had no significant tumor suppressive effects in a colon cancer model in which tumor cells were injected in the cecum and back in rats. However, more recently, this same group found that emodin (2.5 and 5 mg/kg) administered to rats for a week inhibited tumorigenesis of established solid colon tumors that were originally implanted intraperitoneally (ip) or subcutaneously (sc). 14 Likewise, emodin (50 mg/kg) prevented colon carcinogenesis in an azoxymethane/dextran sodium sulfate (AOM/DSS) mouse model. 22 Similarly, pretreatment with emodin (40 mg/kg, 1 week) in a xenograft model of colon cancer (RKO) suppressed tumor growth. 17 While the number of studies available remains relatively low, the data consistently showed a beneficial effect of emodin on colon cancer in rodent models.14,17,22
The literature presents fewer studies on the effects of emodin on liver or pancreatic cancer; despite this, the findings are very promising. Indeed, several recent studies have documented emodin’s beneficial effects on liver and pancreatic cancer in vivo (Tables 1 and 2). In 2020, Bai et al 13 reported that daily emodin (1 and 10 mg/kg) administration, initiated 1 day after subcutaneous injection of HepG2 cells, inhibited tumor growth and reduced mortality in a dose-dependent manner. Hu et al 21 implanted MiaPaCa2 cells into athymic mice and reported that emodin (50 mg/kg) inhibited cancer-cell growth when administered as at treatment (i.e. 4 weeks after tumor implantation). Further, emodin (50 mg/kg) also attenuated 2 pathological constituents of cancer cachexia in this model, namely high hepatic gluconeogenesis and skeletal-muscle proteolysis. 21 In a model of pancreatic cancer liver metastasis, emodin (20 and 50 mg/kg) was reported to suppress tumorigenesis. 16 Based on the available studies, emodin (1-50 mg/kg) shows promise as an agent to treat liver cancer and pancreatic cancer.13,16,21
Table 2.
Hepatatoprotective and Hepatotoxic Effects of Emodin and Its Derivatives.
| Main effect | Model | Cell line or species | Agent/dose/administration route | Mechanism | Reference | |
|---|---|---|---|---|---|---|
| Hepatoprotection | In vitro: ↓hepatic oxidative stress and inflammatory response | Hepatic reperfusion (H/R) injury | In vitro: male C57BL/6 primary hepatocytes | In vitro: aloin (C21H22O9)*; 20 μM (pretreatment), 4 h hypoxia | In vitro: ↓TLR4/MyD88/NF-κB signaling pathway to ↓apoptosis and ↓ROS | Du et al 78 |
| In vivo: ↓I/R-induced liver damage | Hepatic ischemia-reperfusion (H I/R) injury | In vivo: 30 male C57BL/6 mice (6 mice/group) pretreated then portal vein and hepatic artery clamped for 1 h; 6 h reperfusion then sacrificed | In vivo: aloin*; 10, 20, 40 mg/kg; i.p., 5 days pretreatment Optimum: 20 mg/kg ↓ALT, AST |
In vivo: ↓TLR4/MyD88/NF-κB signaling pathway to ↓oxidative stress (↑SOD, ↑GSH,↓MDA), ↓hepatocellular apoptosis (↓Bax, ↑Bcl-2), and ↓inflammation (↓IL-6, ↓TNF-α, ↑IL-10) | ||
| ↓Chemically-induced oxidative/liver damage | Arachidonic acid (AA)+iron induced hepatic oxidative stress | In vitro: hepatocytes | In vitro: emodin; 3-30 µM, 1 h pretreatment before AA + iron Optimum: 10 μM |
In vitro: YAP1 inactivation to ↓apoptosis, ↓ROS, and ↓mitochondria damage | Lee et al 47 | |
| Acetaminophen (APAP) induced liver damage | In vivo: C57BL/6 mice | In vivo: emodin in 40% polyethylene glycol (PEG); 10 and 30 mg/kg, pretreated daily/3 days. APAP; 500 mg/kg; gavage, sacrificed after 6 h ↓ALT, ALP, and total bilirubin |
In vivo: LKB1-AMPK pathway activation and Hippo signaling pathway inactivation to ↓apoptosis, ↓antioxidant gene expression, ↓ROS, and ↓mitochondria damage | |||
| In vitro: ↑apototsis | HCC | In vitro: SMMC-7721 | In vitro: emodin*; 0-200 µM, 0-48 h IC50 ≈ 50 µM, 48 h |
In vitro: ↓p-Akt (PI3K), ↑MAPK (↑p-ERK, ↑p-p38, mild ↓JNK) at 100 µM, 0-60 min, time dependent | Lin et al 19 | |
| In vivo: ↓tumorigenesis | In vivo: 15 male BALB/c nude mice (5 mice/group) subcutaneously injected with SMMC-7721 cells | In vivo: emodin*; 25 or 50 mg/kg; i.p., daily/2 wks after tumor volume of 75-100 mm3
No toxicity or body weight changes ↓ALT, AST, AKP, GGT, Cr and BUN |
In vivo: ↑apoptosis | |||
| Protective effect on liver injury and fibrosis | Carbon tetrachloride (CCl4)-induced liver fibrosis | 50 SD rats (10 mice/group) | Emodin in CMC; 0, 10, 20, or 40 mg/kg; subcutaneous injection, daily/12 weeks after 2 ml/kg 40% CCl4 in olive oil; subcutaneous injection, 2x week/12 weeks ↓ALT, AST, ALP, γ-GT |
↓TGF-β1 signaling and EMT (↓p-Smad2, p-Smad3, FN, vimentin, Snail2, twist-related protein 1, ZEB-1; ↑E-cadherin). ↓Inflammatory cell infiltration, ↓hyperplasia, and ↓collagen deposition*** | Liu et al 52 | |
| ↓Hepatic fibrosis (↓HSC activation) | Hepatic fibrosis | HSC-T6 (activated rat hepatic stellate cells) | Emodin; proliferation: 0-300 μM, 0-48 h; All other: 3, 10, 30 μM, 24 h Optimal: 30 μM |
↓TGF-β1 (↓TGF-β receptor I/II) to ↓HSC proliferation and activation (↓α-SMA), ↓MAPK (p38)/Smad4 signaling and EMT genes (↓collagen, ↓FN); Little effect on p-JNK or ERK1/2. | Wang et al 46 | |
| Protective and rescue effect against intrahepatic cholestasis | Up- or down- regulation of farnesoid X receptor (FXR) | In vitro: Normal human hepatocyte line L02 | In vitro: emodin; 0.02, 0.04, and 0.08 µg/ml, 24 h | ↑BSEP, ↑FXR1, and ↑FXR2 (in vitro: regardless of FXR up/down regulation ) • *** | Xiong, Ding et al. (2019)80 | |
| ANIT-induced cholestasis | In vivo: 21 female and 21 male SD rats (6 mice/group) | In vivo: emodin; 20, 40, 80 mg/kg in CMC; gavage, 7 days. ANIT; 50 mg/kg on day 5. Optimum: 40 mg/kg ↓ALT, AST, TBIL, DBIL, ALP, γ-GT, and TBA levels |
||||
| Protective effect on liver lipid metabolism; ameliorated induced hepatic steatosis (lipid accumulation) | FFAs induced lipid accumulation | In vitro: HepG2 (HCC); primary male SD rat hepatocytes | In vitro: emodin; 20,40, 80 μM, 0-24 h, cotreated with 1 mM FFAs | Modulated CaMKK–AMPK–mTOR–p70S6K–SREBP1 signaling pathway (↑p-AMPK,↓SREBP1, ↓fatty acid synthase, ↓p-mTOR, ↓p70S6K); ↓hepatic lipogenesis and ↑fatty acid oxidation*** | Wang, Li et al. (2017) 81 | |
| High-fat diet (HFD)-induced fatty liver | In vivo: 50 male SD rats (10/group) given chow or HFD for 12 weeks. Emodin administered on week 4. | In vivo: emodin in 0.5% CMC; 40, 80, and 160 mg/kg; gavage, 1x daily/8 weeks ↓TC, ALT, AST, ALP, γ-GT, total protein, LDL-c, and MDA |
||||
| AMPK activation | Normal mouse liver | In vitro: primary mouse hepatocytes | In vitro: emodin; 10 µM, 6 h after compound C (AMPK inhibitor) pretreatment; 5 µM, 30 min | In vitro: ↓bsep | Wang et al 65 | |
| Protective against intrahepatic cholestasis liver injury | (ANIT)-induced intrahepatic cholestasis | In vivo (2 models): (1) Male C57BL/6 mice (6-8 mice/4 groups) (2) Male C57BL/6 mice (6-9 mice/4 groups) |
In vivo: (1) Emodin; 150 mg/kg emodin; 10% PEG400 in 0.5% CMC; p.o., daily/7 days. ANIT in corn oil; 50 mg/kg; p.o., 1x on day 5. ↓ALT, ALP, total bilirubin, bile acids (2) Emodin; 30, 90, and 300 mg/kg; gavage, daily/4 or 14 days |
(1) In livers: ↓anti-oxidative expressions (HO-1, Gpx2, Gsta1/2, and Gadd45a); ↓hepatic inflammation (↓IL-1b, IL-6, IL-10, and a-SMA) via ↓NF-kB pathway; ↓neutrophil infiltration, ↓necrosis; little effect on BA accumulation (2) AMPK activation to ↓bsep (time dependent) in livers*** |
||
| ↓LPS induced hepatocyte apoptosis and inflammatory injury activation | LPS-triggered liver cell inflammatory injury model | In vitro: L02 cells and primary hepatocytes (isolated from 4 Wistar rats) | In vitro: emodin; 0-20 µM (cell viability);15 µM (all other) after LPS pretreatment; 5 ug/ml | ↓IL-1B, IL-6, and TNF-a | Xie et al 38 | |
| In vivo: 30 C57BL/6 mice (10 mice/group) | In vivo: emodin; 50 mg/kg; i.p., 12 h pretreatment. LPS; 40 ug/kg; sacrificed 24 h | ↑miR-145, ↓IRAK1, ↓NF-κB pathway | ||||
| Protective against apoptosis | Normal human liver | L02 | Emodin; 0-80 µM, 0-24 h IC50 ≈ 80 µM, 24 h |
Induced autophagy via ↓p-PI3K, ↓p-AKT and ↓p-mTOR (time dependent)*** | Zheng et al 43 | |
| Hepatotoxicity | Aggravated emodin-induced liver injury | Liver injury induced by high dose emodin | In vitro: human primary hepatocytes | In vitro: emodin*; IC50 = 80 µM, 48 h after CYP3A induction (72 h)/inhibition (24 h) or GSH depletion | CYP3A activation and GSH depletion; CYP3A inhibitor with emodin (protective against/rescues liver injury) | Jiang et al. (2018) 82 |
| In vivo: Male SD rats | #In vivo: emodin in 0.5% CMC; 200 mg/kg; gavage, 1x/4 days and 400 mg/kg on day 5 with either CYP3A inhibitor (50 mg/kg) or GSH inhibitor (700 mg/kg); ip, 2 h before last emodin administration | |||||
| ↑Apoptosis | Normal human liver | L02 | Emodin; 50 µM,48 h | Inhibition of mitochondiral respiratory chain complexes (OxPhos), ↑ROS, ↑mitochondrial damage | Lin et al 63 | |
| Liver oxidative damage/injury | Normal liver | 12 Male SD rats (6 rats/group) | Emodin; 150 mg/kg; gavage; daily/4 weeks | ↓Mitochondrial FA b-oxidation, CAC, and ↓OxPhos; ↑ROS and mitochondrial dysfunction/damage; ↓anti-oxidants | Zhang et al 62 | |
| ↑Apoptosis | Normal human liver | L02 | Emodin; 160 µM, 0-24 h | ↑Cleaved cas-3; ↓autophagy via ↓p-PI3K, ↓p-AKT and ↓p-mTOR (time dependent)*** | Zheng et al 43 | |
| Aggravated ANIT-induced cholestatic liver injury | ANIT-induced intrahepatic cholestasis | Male C57BL/6J mice (6–8/ 6 groups) | #Semen cassiae (emodin-containing herb) in saline; 3000 or 10 000mg/kg; p.o., 7 days. ANIT in corn oil; 50 mg/kg; on day 5. ↑total bilirubin, Bas | Bsep inhibition to ↑bile acid accumulation in livers; not due to ↑oxidative stress/inflammation. Activation of AMPK-Fxr crosstalk (↑p-AMPK and ↓FXR in livers). | Wang et al 65 | |
| Liver injury | Normal liver | 48 SD male rats (12/group) | Emodin in saline; 150, 500, 1500 mg/kg; daily/7 weeks # | Inhibition of FADH/NADPH transport to ↑mitochondrial apoptosis | Yang et al 61 | |
| Liver oxidative damage | HCC | In vitro: HepG2; normal human liver HL7702 cells | In vitro: aloe emodin*; 1,15 or 30 μM, 0-48 h | ***Multidrug resistance protein 2 (ABCC2/ MRP2) inhibition via ↑ROS to ↑mitochondrial dysfunction, autophagy, and mitochondrial mediated apoptosis | Liu et al 64 | |
| In vivo: 30 male and female kunming mice (10 mice/group) | In vivo: aloe emodin in 0.5% CMC; 800 or 1600 mg/kg; intragastric, 11 weeks # | |||||
Abbreviations: CMC, sodium carboxymethylcellulose; HO-1, heme oxygenase-1; Gpx2, glutathione peroxidase 2, Gsta1/2: glutathione S-transferases, Gadd45a, growth arrest and DNA-damage-inducible protein.
Agent dissolved in DMSO.
Improved liver and kidney function (↓ ALT, AST, AKP, GGT, Cr and BUN).
Dose-dependent manner.
•More effective than ursodeoxycholic acid (UDCA: anti-cholestasis drug) and dexamethasone (DXM: immunosuppresspressive agent).
#↑ALT, AST.
↓TC, alanine transaminase, aspartate transaminase, alkaline phosphatase, γ-glutamyltransferase, total protein, LDL-c, and MDA.
Overall, the available in vivo studies assessing emodin’s anti-cancer effects in GI cancers are strong; in almost all the reported studies emodin shows promise as a stand-alone therapy in the treatment of colon, liver, or pancreatic cancer.13,14,16,17,21,22 Additional studies to confirm these findings as well as studies that address optimal timing and dosing of emodin administration are needed to support the development of emodin for clinical use. Further, it is worth noting that the majority of studies to date appear to be focused on prevention of tumorigenesis. Thus, pre-clinical evidence to support a therapeutic effect of emodin are warranted.
Mechanisms of Emodin as an Anti-Cancer Therapy
It is clear from section II above that emodin shows promise as an anti-cancer agent for GI cancers.13,14,16,17,21,22 However, a clear understanding of its mechanisms is essential to its development for clinical use. This is complicated by the fact that many natural compounds, like emodin, boast of very broad pleiotropic effects. In the succeeding section we will address the known mechanisms of action of emodin as they relate to GI cancers.
Emodin Targets Mitochondrial Mediated Apoptosis
Direct regulation of mitochondrial mediated apoptosis
It has been well documented that emodin induces apoptosis in GI cancers (Table 1); however, the proposed mechanisms of action have not been well described, resulting in inconsistencies across the literature. While apoptosis can be initiated through several regulatory pathways, recent evidence has emerged showing emodin’s ability to induce apoptosis by targeting mitochondria.27,28,30,34 In order for apoptosis to occur, a family of Cysteine proteases named caspases must become activated to cleave several substrates to result in cellular degradation and death. 35 While mitochondrial mediated apoptosis activates caspases through the release of cytochrome c from the mitochondria, the intracellular signaling mechanisms regulating this process are diverse.35,36 While emodin has been demonstrated to induce mitochondrial mediated apoptosis, these conclusions have been made largely from simple outcome measures with little mechanistic insight. This review will attempt to streamline the findings of several papers on emodin induced apoptosis by examining their reported proteins and apoptotic readouts.
Mitochondrial mediated apoptosis is regulated by maintaining the balance between pro- and anti- apoptotic proteins within the mitochondrial membrane. 36 Interestingly, emodin (50 and 100 µM) has been found to primarily localize in the mitochondria of GI cancer cells. 37 Mitochondrial mediated cell death occurs through the inhibition of the anti-apoptotic B-cell lymphoma 2 (Bcl-2) enabling pro-apoptotic Bax, Bak, and PARP to disrupt the mitochondrial membrane potential (MMP) (Figure 1). 36 The loss of MMP triggers the release of cytochrome c to activate caspase -9 and -3 resulting in apoptosis (Figure 1). 36 To this end, emodin and its derivatives have been shown to reduce Bcl-2, disrupt MMP, and increase Bax, PARP, cytochrome c release, and cleaved caspase -9 and -3 to result in apoptosis of colon,27,28,38,39 pancreatic,23,40 and liver19,30,41-43 cancer cells (Table 1).
Figure 1.
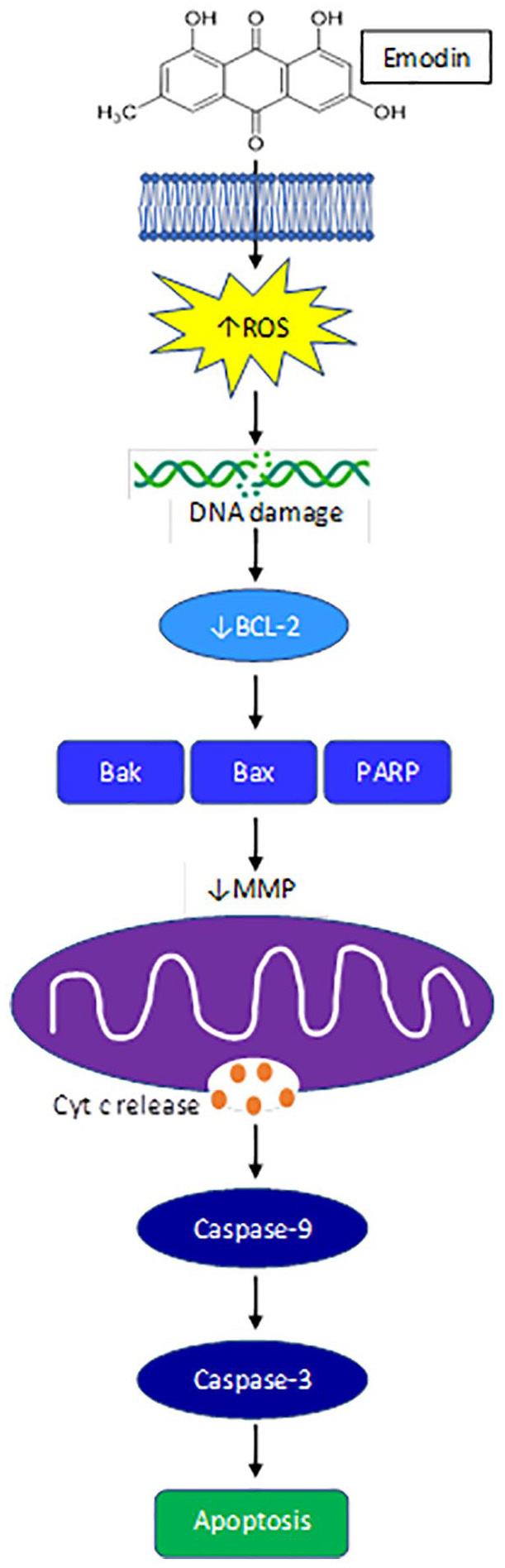
Emodin reduces ROS mitochondrial mediated apoptosis.
While emodin has been shown to modulate several proteins associated with mitochondrial mediated apoptosis in GI cancer cells, the initial cellular stress response required to initiate mitochondrial mediated apoptosis is not as heavily investigated. It has been suggested that the chemical structure of quinones such as emodin, can interact with intracellular molecular oxygen to become superoxide anions, generating reactive oxygen species (ROS).37,44 Although it is possible that emodin could induce other cellular stresses, besides ROS, that are known to result in mitochondrial mediated apoptosis, including ER stress, nutrient deficiencies, hypoxia, etc., 45 this has not been reported for emodin in GI cancers. Emodin has been reported to elicit oxidative stress, primarily via ROS, which was suggested to initiate mitochondrial mediated apoptosis in GI cancers.30,32,37,39,46 Indeed, increased ROS can target structures susceptible to oxidative damage and can disrupt the mitochondrial membrane potential (MMP) and increase cytochrome c release to induce apoptosis. 45 Emodin (20-80 μM) increased intracellular ROS and promoted DNA damage to separately induce mitochondrial dysfunction and subsequent mitochondrial mediated apoptosis, as well as cell cycle arrest through increased p53, p21, and other cyclin E proteins resulting in reduced proliferation in human HepaRG hepatocellular carcinoma (HCC) cells 30 and colon cancer cell lines HCT116 and LS1034 in a dose-dependent manner.32,38 In an in vitro model of HCC, emodin (50 and 100 μM) dose-dependently increased the expression of redox sensitive-mitochondrial matrix protein, cyclophilin D (CypD) that induced MMP loss to further accumulate ROS resulting in mitochondrial-mediated cell death. 37 Emodin (5-40 μM) 46 and aloe-emodin (AE) (10-80 μM) 40 were also shown to promote cell death through inducing autophagy via mitochondrial ROS and the activation of LC3-2. Importantly, emodin does not induce oxidative stress (ROS) nor apoptosis 27 in normal GI cells, 47 or at the very least emodin does not induce ROS in great enough quantities needed to initiate apoptosis as seen in GI cancer cells. An increasing body of evidence supports this notion that the manipulation of ROS by emodin can be selective between cancerous and normal cells. 11 Although not well distinguished in the literature, emodin appears to mediate GI cancer cell death through the promotion of ROS and subsequent mitochondrial damage via the mitochondrial pathway30,32,37,39 (Table 1); however, further work is needed to firmly establish these actions of emodin.
Indirect regulation of mitochondrial mediated apoptosis
The use of natural compounds has the distinct advantage of manipulating numerous aberrant signaling pathways in cancers. Oxidative stress induced by emodin can negatively regulate multiple signaling pathways associated with cell growth, apoptosis, and differentiation to result in GI cancer cell death (Table 1; Figure 1). Emodin at low doses (15-50 μM) was sufficient to induce mitochondrial mediated cell death by suppressing phosphoinositide 3-kinase (PI3K)/Akt,13,19,21,27,28,48,49 mitogen activated protein kinases (MAPK), 27 and extracellular signal-regulated kinase (ERK1/2) signaling (Table 1).13,21,37
The PI3K/Akt and MAPK/ERK signaling pathways are commonly activated in cancers as they promote cancer cell survival, proliferation, and growth. Emodin or its derivatives have been shown to induce liver,13,19 colon,20,27,28,48,49 and pancreatic21,49 cancer cell apoptosis dose dependently at concentrations between 15-200 μM by downregulating PI3K/Akt and AKT-ERK1/2 signaling. Emodin (20-200 μM) also dose dependently decreased the phosphorylation of Akt and Erk1/2 to inhibit pancreatic cancer cell growth and hypoxia, as indicated by decreased HIF-1a. 21 Stress induced activation of these cell regulating pathways, specifically the PI3K/AKT pathway, strains mitochondria to produce energy for survival and provide protection from Redox. 50 This strain eventually results in mitochondrial damage and downregulation of these pathways to promote mitochondrial mediated apoptosis. Altogether, emodin can induce oxidative stress to inhibit numerous signaling pathways which converge in the activation of mitochondrial mediated apoptosis.
Emodin’s Impact on Mechanisms Regulating Tumor Invasion, Metastasis, and Angiogenesis
Epithelial-mesenchymal transition (EMT) and angiogenesis are 2 critical factors that aid in tumor growth, invasion, and metastasis. Tumor EMT is a process in which epithelial cells acquire a mesenchymal, stem-cell-like phenotype possessing increased generation of extracellular (ECM) components and enhanced cell migratory abilities to enhance growth and metastasis. 51 Additionally, sustained growth and metastasis requires the formation of new blood vessels via vascular endothelial growth factor (VEGF)-mediated angiogenesis. 51 Emodin (1-80 mg/kg) has been proposed to inhibit tumor growth through its suppression of angiogenesis and inhibition of EMT.13,16-18,52
The transforming growth factor (TGF)-β/Smad signaling has an established role in tumorigenesis, angiogenesis, and metastasis through ECM remodeling and EMT. 53 Emodin has been shown to reduce TGF-β signaling. 54 Indeed, emodin (20, 40, and 80 mg/kg) was shown to dose dependently inhibit TGF-β1 and upregulate TGF-β1 receptors (TβRI and TβRII) and this was associated with repression of angiogenesis in a pancreatic cancer model (xenograft model using SW1990 human pancreatic cancer cells). 18 Additionally, emodin’s inhibition of TGF-β1 repressed the secretion of Angptl4 by pancreatic tumor cells via Smad4, which is suggested to prevent angiogenesis. 18
Emodin’s inhibition of tumor EMT and promotion of tumor cell migration also has been attributed to its involvement in both Wnt/β-catenin and VEGFR2-AKT-ERK1/2 signaling.13,17 Emodin (40 mg/kg, p.o.) significantly suppressed Wnt/β-catenin signaling in vivo sufficiently to block colon cancer EMT and cell mobility. 17 Emodin (5-20 μM) also reduced colon tumor cell invasion and migration in vitro through reduced VEGF. 17 In addition, it was reported that emodin effectively upregulated the expression of tumor-suppressing miR-34a in cancerous liver tissue, and suppressed VEGFR2-induced activation of AKT, ERK1/2, SMAD2 and SMAD4. 13 Lastly, emodin (20-80 mg/kg, i.p., 3 weeks, 3x/week) suppressed angiogenesis in pancreatic cancer by reducing expression levels of oncogenic and angiogenesis-associated miRNAs, miR-155, and miR-210, and increasing anti-angiogenic miR-20b expression. 18 Drugs that can affect both molecular signaling pathways and miRNAs are of current therapeutic interest as many of them mediate tumorigenesis and cancer progression through the regulation of oncogenes and/or tumor suppressor genes. 55
Emodin as a Complementary Dietary Anti-cancer Therapy
In section II and III above we addressed the effects of emodin as a stand-alone therapy against colon, liver, and pancreatic cancers. Excitingly, there also is an emerging body of literature on emodin’s use as a complementary anti-cancer therapy. In this section we will discuss the available literature on emodin’s efficacy as a complementary therapy in GI cancers along with relevant mechanisms.
Emodin (10-50 mg/kg) has been reported to combat tumorigenesis in HCC and pancreatic cancer models when given in combination with chemotherapeutics.15,21,23,24 Currently, the first-line treatment for advanced HCC is sorafenib, a kinase inhibitor that effectively mitigates tumor growth, angiogenesis, and metastasis. 56 While sorafenib (5 mg/kg, i.p) and emodin (10 mg/kg, i.p.) alone significantly inhibited tumor growth and cell proliferation in HCC cells including HepG2, Hep3B, Huh7, SK-HEP-1, and PLC/PRF5, emodin synergistically increased sorafenib efficacy to result in greater tumor inhibition. 15 Consistently, in the same report, animal models xenografted with HepG2 or SK-HEP-1 cells showed that the combination of emodin and sorafenib, administered following establishment of tumors, was sufficient to inhibit tumor growth suggesting that emodin given as a complementary therapy has the potential to improve efficacy of standard therapies. 15 Mechanistically, the authors associated these benefits of emodin (10 mg/kg, i.p.) with its ability to inhibit sterol regulatory element-binding protein-2 (SREBP-2) transcriptional activity, which suppresses cholesterol biosynthesis and oncogenic protein kinase B (AKT) signaling. 15 Furthermore, attenuated cholesterol synthesis and oncogenic AKT signaling inactivated the oncogenic transcript factor, signal transducer and activator of transcription 3 (STAT3). 15 Additionally, emodin (10 mg/kg, i.p.) synergistically increased cell cycle arrest in the G1 phase and apoptotic cells in the presence of sorafenib (5 mg/kg, i.p). 15
Likewise, to counteract drug resistance of EGFR inhibitors in the treatment of pancreatic cancer, emodin (0-90 μM) was examined in combination with the EGFR inhibitor, afatinib (20 nM), and was reported to reduce proliferation of PANC-1 and BxPC-3 cells. 23 Consistently, the same study reported that the combination of emodin (50 mg/kg) with afatinib (50 mg/kg), administered following tumor establishment, exhibited a synergistic anti-cancer effect in a xenograft pancreatic mouse model. 23 Mechanistically the authors attributed this to emodin’s (50 mg/kg) ability to promote afatinib-induced apoptosis by inhibiting the Stat3 signaling pathway. 23 Several studies have reported benefits when emodin is administered in combination with gemcitabine—a widely utilized first-line drug for advanced pancreatic cancer.24,31 In gemcitabine-resistant PANC-1 pancreatic cancer cell xenografts, the combination of gemcitabine (125 mg/kg) and emodin (40 mg/kg) was shown to significantly reduce xenograft volume and tumor growth in mice compared with treatment with gemcitabine or emodin only. 24 In addition, emodin (40 mg/kg) treatment reduced resistance to gemcitabine, which was attributed to the downregulation of P-glycoprotein, MRP1 and MRP5—drug resistance factors—expression in the group receiving combination treatment. 24 Similarly, in 2 pancreatic cell lines, Panc-1 and MIA-PaCa-2, that were either normal or gemcitabine-resistant, emodin (40 μM) and gemcitabine (20 μM) alone significantly increased the apoptosis rate, but the combination of the 2 drugs further increased the apoptosis rate above the individual treatments in both the normal pancreatic cell lines and the gemcitabine-resistant lines. 31 The authors associated these effects with emodin’s (40 μM) ability to reduce the mRNA and protein expression levels of Survivin, XIAP, NF-κB, and IKKβ, and significantly increase the mRNA and protein expression levels of Caspase-3/9 and IκB-α. 31
Although the data are limited, emodin (10-50 mg/kg) shows promise as a combinational therapy to improve efficacy and reduce resistance of standard anti-cancer drugs.15,23,24,31 This can be attributed to the broad and pleiotropic effects of emodin. Future work in this area should continue to improve our understanding of emodin’s potential as a complementary cancer therapy so that it can be integrated into clinical treatment paradigms.
Anti-cancer Properties of Emodin Derivatives and Formulations
Circumventing the pitfalls of natural compounds like emodin, including poor bioavailability, solubility, and selective cytotoxicity in vivo can be achieved using emodin derivatives and/or formulations. Indeed, there are now several reports of the development of novel emodin-containing drugs to improve its anti-cancer efficacy.34,42,57,58 This section will address the anti-cancer properties of emodin derivatives and/or formulations as they relate to colon, liver, pancreatic cancer.
Several studies have documented improved efficacy of emodin derivatives in liver cancer.34,42,57,58 In one study, a novel series of emodin derivatives were designed and synthesized via the introduction of an amino acid using linkers of varying lengths and composition. 34 In vitro anti-proliferation tests revealed that these derivatives exhibited moderate to potent anti-proliferative activity against HepG2 cells. 34 The authors noted that one specific compound, known as 7a (IC50: 4.95 μM), indicated better selective anti-proliferative activity and specificity than emodin alone. 34 The potential mechanism for this was attributed to a significant effect in inducing cell cycle arrest at G0/G1 phase and inducing cell apoptosis in HepG2 cells via release of cytochrome c and subsequent activation of caspase-9 and caspase-3. 34 In another study, the novel drug compound, β-dihydroartemisinin-emodin (β-DHA-emodin) showed high suppressive activity against the proliferation of HepG2, by inhibiting Ki-67 expression. 42 Further, through modification of emodin’s structure, the derivative emodin succinyl ester (5-20 μM) strongly inhibited HCC cell proliferation and migration in vitro. 58 Another group used emodin-loaded N-acetylaminogalactosyl-poly(lactide-co-glycolide)-succinyl-D-α-tocopherol polyethylene glycol 1000 succinate (GalNAc-PLGA-sTPGS) nanoparticles (EGPTN) to examine efficacy against primary liver cancer; EGPTN was reported to achieve improved anti-cancer efficacy compared to groups without emodin. 57 Similarly, emodin succinyl ester (50-200 mg/kg) dose-dependently suppressed tumorigenicity in xenograft and diethylnitrosamine (DEN)-induced HCC mouse models. 58
With regards to colon cancer and pancreatic cancer fewer reports exist; in fact, we noted just 2 recent reports for colon cancer39,59, and one for pancreatic cancer 40 — all of which used aloe-emodin (AE)—a natural compound extract from Aloe Vera. The first colon cancer study examined the impact of AE on SW620 and HT29 cell lines in vitro; AE (10-40 μM) suppressed cell viability and induced cell apoptosis in SW620 and HT29 cell lines. 39 Furthermore, both cell lines generated ROS when exposed to AE (10-40 μM) supporting its ability to induce apoptosis. 39 The second colon cancer study utilized a genetic model of colorectal cancer, ApcMin/+ mice, treated with or without dextran sodium sulfate (DSS), to induce intestinal mucosa inflammation and multiple neoplasms, that were administered dietary AE. 59 In this study, low dose dietary AE (5 ppm in diet) reduced the number of colorectal tumors in both tumor models. 59 Similarly, AE (0-100 μM) induced dose-dependent cytotoxicity in 2 different pancreatic carcinoma cell lines with MiaPaCa2 cells being more susceptible than PANC-1. 40
In addition to the development of emodin derivatives and formulations to improve efficacy, recent papers also have sought to overcome emodin’s poor solubility observed in water, 10% ethanol, and DMSO. 60 Indeed, a thermoreversible poloxamer gel containing emodin possessed 100-fold greater solubility enhancement and decreased fibroblast cell viability compared to emodin in water or 10% ethanol. 60 In another approach, emodin’s poor solubility may be overcome by using carrier systems such as mesoporous silica nanomaterial (SNM) labeled with fluorochromes. 48 Indeed, nanomaterials loaded with emodin were applied against human colon carcinoma cells HT-29 and found to enter HT-29 cells and release emodin in higher amounts within 48 hours compared to naked emodin. 48 This led to improved bioavailability and selectivity, as well as enhanced autophagy (increased LC3A/B) and mitochondrial mediated apoptosis through increased pro-apoptotic proteins (compared to naked emodin), phosphorylated-AKT downregulation, PI3K/Akt pathway inhibition, and increased phosphorylated-ERK in HT-29 cells at concentrations of 20-80 μM. 48
Overall, there is great potential for emodin derivatives and or formulations to be used in the treatment of GI cancers. In most, if not all cases, the available studies on emodin derivatives and/or formulations show improved efficacy over emodin alone.34,39,40,42,57-59 Further, research is underway to improve the solubility of emodin, which also has resulted in improved efficacy.48,60 These data underscore the potential of this natural compound in the cancer treatment domain.
Safety of Emodin
Evaluation of the safety of dietary compounds is principal to their clinical development and emodin is no exception. Despite the positive effects on tumorigenesis, several studies have reported side effects of emodin that may preclude its development beyond pre-clinical studies. Given the focus of this review on GI cancers, in this section we will address side effects of emodin as related to the GI system. On the contrary, there also is a literature base supporting a protective effect of emodin on the GI system and therefore this will be addressed.
Toxicity of Emodin
There are now a number of reports of hepatotoxic effects of emodin that appear to be dose dependent (Table 2). In one recent study, rats were administered 150, 500, or 1500 mg/kg of emodin daily for 4 weeks and liver damage was assessed. 61 The findings revealed that as the dose of emodin increased, the damage was accelerated, and this was also time dependent. 61 Specifically, inflammation was considered minimal when administered at 150 mg/kg, but was more elevated in the 500 mg/kg group, and was rated as serious in the 1500 mg/kg group and associated with the presence of fat vacuoles. 61 These findings were consistent with alanine transaminase (ALT) and aspartate transaminase (AST) levels supporting a toxic effect of emodin in the liver. 61 In a similar study, rats administered 150 mg/kg of emodin daily for 4 weeks exhibited significant increases in levels of ALT and AST indicating damage to the liver. 62
The mechanisms responsible for emodin-induced hepatotoxicity continue to be unearthed but several studies implicated mitochondria and oxidative stress in playing a role.61 -64 Indeed, 1500 mg/kg daily dose of emodin for 4 weeks inhibited proton transport and subsequently induced the activation of the mitochondrial apoptosis pathway. 61 A role for mitochondria also was implicated in another study, suggesting emodin (160 μM) to induce oxidative stress in the liver by directly targeting acadvl/complex IV and inhibiting fatty acid β-oxidation, citric acid cycle, and oxidative phosphorylation in mitochondria. 62 It is well known that redox homeostasis is maintained by antioxidant production as antioxidants help in degrading free radicals such as ROS. Emodin at very high doses (150-10 000 mg/kg) down-regulates antioxidant proteins thus suppressing antioxidant defenses that may increase susceptibility to prolonged oxidative stress and excessive ROS production.62,64,65 Oxidative stress resulting in a prolonged imbalance between the production of ROS with their elimination by antioxidants in the liver can contribute to liver inflammation leading to fibrosis and HCC. 66 Specifically, high dosage of emodin (150 mg/kg, daily, 4 weeks) induced hepatotoxicity by inhibiting antioxidants within the mitochondria, including peroxiredoxins (Prdx), glutathione peroxidases (Gpx), thioredoxin (Trx), and glutaredoxin (Glrx), all of which help in the degradation of free radicals such as ROS. 62 Emodin’s inhibition of these antioxidants allowed for excessive ROS accumulation that inhibited the activity of all mitochondrial respiratory chain complexes (oxidative phosphorylation) that led to prolonged oxidative stress, mitochondrial damage, chronic inflammation, and liver injury. 62 Similarly, high dose aloe-emodin (800 or 1,600 mg/kg) resulted in oxidative redox imbalance leading to increased accumulation of ROS, mitochondrial dysfunction, and cell death. 64 In summary, the inhibition of mitochondrial pathways along with the downregulation of anti-oxidant proteins helps to explain high dose emodin’s (150-10,000 mg/kg) ability to promote prolonged redox imbalance, excessive ROS accumulation, and oxidative stress resulting in liver damage.61 -64
In addition to reports of hepatoxicity, there have been some studies that have documented laxative effects of emodin.44,67 In one study in mice, treatment with emodin (1, 2, and 3 g/kg) increased the fecal water content in the colon of mice and evaluation index of defecation in a dose-dependent manner; this was associated with upregulation of aquaporin 3—a protein that plays an important role in regulation of water transfer in the colon. 67
While it is clear that emodin does exhibit some toxicity, some perspective is needed when interpreting these findings. The hepatotoxicity and laxative effects that have been documented were following extremely high doses of emodin. For example, the liver damage documented in rats was following a daily dose of emodin 100x the clinical equivalent of rhubarb. 61 Similarly, the laxative effects have been documented following 1-3 g/kg of emodin daily. 67 Of note, this is much higher than the dose range (1-50 mg/kg) that boasts of anti-cancer effects discussed in section II. Finally, in a sub-chronic (12 week) toxicity study using 3 different doses of emodin (~20 mg/kg, 40 mg/kg, and 80 mg/kg) it was reported that emodin did not cause pathophysiological perturbations in major organs nor did it cause an increase in ALT or AST. 68 While assessing toxicity is certainly an important aspect for the development of emodin as an anti-cancer agent; it should not preclude its progress in this regard, given that most every current cancer drug has potent dose-dependent toxicities. Indeed, just as there are reports of toxicity with emodin, there also is a body of evidence that documents a protective role of emodin that is discussed below.
Organ Protective Functions of Emodin
With regard to the liver, based on a review of the literature it is likely that emodin has bidirectional potential; thus, in addition to the reported hepatoxicity described above, liver protection also has been documented (Table 2).68 -77 In a model of alcohol-induced fatty liver injury, emodin ameliorated liver steatosis, lowered ALT and AST, and decreased hepatic oxidative stress. 69 This finding was supported in vitro where emodin (10-100 μM) suppressed ethanol-induced cytotoxicity in Hep2G cells. 70 In another study, the role of emodin (20 mg/kg) in protection against fibrogenesis caused by carbon tetrachloride was examined in rats. 71 Emodin (20 mg/kg), was able to counteract the hepatic fibrosis induced by carbon tetrachloride. 71 Further, levels of ALT and AST and hydroxyproline content were significantly reduced following the carbon tetrachloride insult in rats administered emodin (20 mg/kg). 71 Consistent with this, another study used the same insult (carbon tetrachloride) and administered water extracts from fresh Polygonum multiflorum, containing emodin (3 mg/kg), to rats for 56 days. 72 It was found that the extract containing emodin reduced ALT and AST, decreased inflammatory mediators and malonaldehyde, and rescued glutathione S-transferase and catalase activity. 72 In addition, fatty degeneration and necrosis were found to be reduced following treatment with the emodin containing extract. 72 In a study of acetaminophen-induced toxicity, treatment with emodin (30 and 40 mg/kg, p.o.) conferred hepatoprotective ability. 73 Using a model of concanavalin A (Con A)-induced hepatitis in mice, emodin (12.5-50 mg/kg) was examined for its protective properties. 75 Consistent with other reports, emodin (12.5-50 mg/kg) decreased circulating ALT and AST and reduced liver necrosis. 75 In addition, there was a marked decrease in pro-inflammatory mediators with emodin (12.5-50 mg/kg) and this was consistent with a decrease in activation of p38 MAPK and NF-κB. 75
Studies utilizing lower doses (1-100 mg/kg) overwhelmingly support a hepatoprotective role of emodin following various insults (Table 2). Thus, it is safe to conclude that emodin possesses bidirectional potential with regards to liver toxicity; indeed, low doses of emodin (1-100 mg/kg) offer hepatoprotection whereas higher doses (500-3000 mg/kg) promote toxicity. The mechanisms responsible for emodin-induced hepatoprotection and hepatotoxicity are also bidirectional in their modulation of oxidative stress, ROS, and antioxidants which is dosage dependent (Table 2). At lower doses (10-20 uM in vitro and 10-40 mg/kg in vivo), emodin protects against non-cancerous hypoxic- and chemical- induced liver damage and hepatic oxidative stress by reducing ROS, oxidative stress (↓MDA), and hepatocellular apoptosis as well as upregulating antioxidants (↑SOD and ↑GSH)47,78 (Table 2). While at higher doses (150-10,000 mg/kg), emodin results in liver damage by promoting prolonged redox imbalance, excessive ROS accumulation and oxidative stress, as well as by down-regulating antioxidant proteins to further increase ROS.61 -64 This underscores the importance of integrating dose response studies into the development of natural compounds. Further, it supports the safety of emodin, at doses below 100 mg/kg, for potential use as an anti-cancer agent.
Conclusions
Given the global burden of GI cancers, treatment options that target multi-aspects of tumorigenesis or increase the efficacy of currently approved drugs are continually being sought after. Through the manipulation of multiple pathways, plant-derived agents have been suggested to elicit their tumoricidal effects without displaying the high toxicity exhibited by traditional chemotherapeutics. Emodin is a promising alternative anti-cancer drug that has documented efficacy in pre-clinical models of colon, liver, and pancreatic cancers. While the data continue to accumulate, we can conclude the most, if not all, reports of emodin’s anti-cancer efficacy against GI cancers are positive and using doses that are attainable in humans. Indeed, even at the higher effective pre-clinical dose range of 50-80 mg/kg16,18 -22 the human equivalent dose would be 4-6.5 mg/kg. 79 Evidence suggests that emodin exerts its anti-cancer effects through inducing mitochondrial mediated apoptosis, alongside suppressing pathways that promote inflammation, proliferation, angiogenesis, and tumorigenesis. Equally exciting is the ability of emodin to enhance the efficacy of anti-cancer drugs including sorafenib, afatinib, and gemcitabine, supporting its potential as a complementary treatment to standard chemotherapeutics. As with any natural compound there are concerns surrounding bioavailability; emodin is no exception. To counteract this there are studies that have successfully improved bioavailability of emodin. Finally, there is a body of literature that has addressed toxicity of emodin. Based on a review of the literature emodin possesses bidirectional potential; lower doses of emodin exhibit hepatoprotection whereas higher doses are hepatotoxic. In conclusion, emodin offers great potential as either a stand-alone treatment or a complementary treatment to existing chemotherapeutics that target GI cancers, and should be further developed in this regard.
Footnotes
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) received no financial support for the research, authorship, and/or publication of this article.
ORCID iD: E. Angela Murphy  https://orcid.org/0000-0002-4803-5822
https://orcid.org/0000-0002-4803-5822
References
- 1. Arnold M, Abnet CC, Neale RE, et al. Global burden of 5 major types of gastrointestinal cancer. Gastroenterology. 2020;159:335-349.e15. doi: 10.1053/j.gastro.2020.02.068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chen C, Di Bartolomeo M, Corallo S, Strickler JH, Goyal L. Overcoming resistance to targeted therapies in gastrointestinal cancers: progress to date and progress to come. Am Soc Clin Oncol Educ Book. 2020;40:161-173. doi: 10.1200/EDBK_280871 [DOI] [PubMed] [Google Scholar]
- 3. VanderVeen BN, Sougiannis AT, Velazquez KT, Carson JA, Fan D, Murphy EA. The acute effects of 5 fluorouracil on skeletal muscle resident and infiltrating immune cells in mice. Front Physiol. 2020;11:593468. doi: 10.3389/fphys.2020.593468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sougiannis AT, VanderVeen BN, Enos RT, et al. Impact of 5 fluorouracil chemotherapy on gut inflammation, functional parameters, and gut microbiota. Brain Behav Immun. 2019;80:44-55. doi: 10.1016/j.bbi.2019.02.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hu N, Liu J, Xue X, Li Y. The effect of emodin on liver disease: comprehensive advances in molecular mechanisms. Eur J Pharmacol. 2020;882:173269. doi: 10.1016/j.ejphar.2020.173269 [DOI] [PubMed] [Google Scholar]
- 6. Iwanowycz S, Wang J, Hodge J, Wang Y, Yu F, Fan D. Emodin inhibits breast cancer growth by blocking the tumor-promoting feedforward loop between cancer cells and macrophages. Mol Cancer Ther. 2016;15:1931-1942. doi: 10.1158/1535-7163.MCT-15-0987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Phillips E, France A, Thatvihane G, Nnaemeka U, Zaidi S. Mucositis and cardiotoxicity due to 5-fluorouracil. Am J Ther. 2018;25:e712-e714. doi: 10.1097/MJT.0000000000000725 [DOI] [PubMed] [Google Scholar]
- 8. Sougiannis AT, VanderVeen BN, Davis JM, Fan D, Murphy EA. Understanding chemotherapy-induced intestinal mucositis and strategies to improve gut resilience. Am J Physiol Gastrointest Liver Physiol. 2021;320:G712-G719. doi: 10.1152/ajpgi.00380.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gezici S, Şekeroğlu N. Current perspectives in the application of medicinal plants against cancer: novel therapeutic agents. Anticancer Agents Med Chem. 2019;19:101-111. doi: 10.2174/1871520619666181224121004 [DOI] [PubMed] [Google Scholar]
- 10. Cragg GM, Pezzuto JM. Natural products as a vital source for the discovery of cancer chemotherapeutic and chemopreventive agents. Med Princ Pract. 2016;25(Suppl 2):41-59. doi: 10.1159/000443404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wei WT, Lin SZ, Liu DL, Wang ZH. The distinct mechanisms of the antitumor activity of emodin in different types of cancer (review). Oncol Rep. 2013;30:2555-2562. doi: 10.3892/or.2013.2741 [DOI] [PubMed] [Google Scholar]
- 12. Hsu SC, Chung JG. Anticancer potential of emodin. BioMedicine. 2012;2:108-116. doi: 10.1016/j.biomed.2012.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bai J, Wu J, Tang R, et al. Emodin, a natural anthraquinone, suppresses liver cancer in vitro and in vivo by regulating VEGFR2 and miR-34a. Investig New Drugs. 2020;38:229-245. doi: 10.1007/s10637-019-00777-5 [DOI] [PubMed] [Google Scholar]
- 14. Höhn P, Braumann C, Freiburger M, Koplin G, Dubiel W, Luu AM. Anti-tumorigenic effects of Emodin and its’ homologue BTB14431 on vascularized colonic cancer in a rat model. Asian Pac J Cancer Prev. 2020;21:205-210. doi: 10.31557/APJCP.2020.21.1.205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kim YS, Lee YM, Oh TI, et al. Emodin sensitizes hepatocellular carcinoma cells to the anti-cancer effect of Sorafenib through suppression of cholesterol metabolism. Int J Mol Sci. 2018;19:E3127. doi: 10.3390/ijms19103127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li N, Wang C, Zhang P, You S. Emodin inhibits pancreatic cancer EMT and invasion by up-regulating microRNA-1271. Mol Med Rep. 2018;18:3366-3374. doi: 10.3892/mmr.2018.9304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gu J, Cui CF, Yang L, Wang L, Jiang XH. Emodin inhibits colon cancer cell invasion and migration by suppressing epithelial-mesenchymal transition via the Wnt/β-Catenin pathway. Oncol Res. 2019;27:193-202. doi: 10.3727/096504018X15150662230295 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 18. Lin SZ, Xu JB, Ji X, et al. Emodin inhibits angiogenesis in pancreatic cancer by regulating the transforming growth factor-β/drosophila mothers against decapentaplegic pathway and angiogenesis-associated microRNAs. Mol Med Rep. 2015;12:5865-5871. doi: 10.3892/mmr.2015.4158 [DOI] [PubMed] [Google Scholar]
- 19. Lin W, Zhong M, Yin H, et al. Emodin induces hepatocellular carcinoma cell apoptosis through MAPK and PI3K/AKT signaling pathways in vitro and in vivo. Oncol Rep. 2016;36:961-967. doi: 10.3892/or.2016.4861 [DOI] [PubMed] [Google Scholar]
- 20. Dai G, Ding K, Cao Q, et al. Emodin suppresses growth and invasion of colorectal cancer cells by inhibiting VEGFR2. Eur J Pharmacol. 2019;859:172525. doi: 10.1016/j.ejphar.2019.172525 [DOI] [PubMed] [Google Scholar]
- 21. Hu L, Cui R, Liu H, Wang F. Emodin and rhein decrease levels of hypoxia-inducible factor-1α in human pancreatic cancer cells and attenuate cancer cachexia in athymic mice carrying these cells. Oncotarget. 2017;8:88008-88020. doi: 10.18632/oncotarget.21330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang Y, Pu W, Bousquenaud M, et al. Emodin inhibits inflammation, carcinogenesis, and cancer progression in the AOM/DSS model of colitis-associated intestinal tumorigenesis. Front Oncol. 2021;10:564674. doi: 10.3389/fonc.2020.564674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang Z, Chen H, Chen J, et al. Emodin sensitizes human pancreatic cancer cells to EGFR inhibitor through suppressing Stat3 signaling pathway. Cancer Manag Res. 2019;11:8463-8473. doi: 10.2147/CMAR.S221877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Guo H, Liu F, Yang S, Xue T. Emodin alleviates gemcitabine resistance in pancreatic cancer by inhibiting MDR1/P-glycoprotein and MRPs expression. Oncol Lett. 2020;20:167. doi: 10.3892/ol.2020.12030 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 25. Shrimali D, Shanmugam MK, Kumar AP, et al. Targeted abrogation of diverse signal transduction cascades by emodin for the treatment of inflammatory disorders and cancer. Cancer Lett. 2013;341:139-149. doi: 10.1016/j.canlet.2013.08.023 [DOI] [PubMed] [Google Scholar]
- 26. Dong X, Fu J, Yin X, et al. Emodin: a review of its pharmacology, toxicity and pharmacokinetics. Phytother Res. 2016;30:1207-1218. doi: 10.1002/ptr.5631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Saunders IT, Mir H, Kapur N, Singh S. Emodin inhibits colon cancer by altering BCL-2 family proteins and cell survival pathways. Cancer Cell Int. 2019;19:98. doi: 10.1186/s12935-019-0820-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ma Q, Ding Y, Wu Z, Li Y. Antitumor effects of emodin in CACO-2 human colon carcinoma cells are mediated via apoptosis, cell cycle arrest and downregulation of PI3K/AKT signalling pathway. J BUON. 2018;23:587-591. [PubMed] [Google Scholar]
- 29. Zhou RS, Wang XW, Sun QF, et al. Anticancer effects of emodin on HepG2 cell: evidence from bioinformatic analysis. Biomed Res Int. 2019;2019:3065818. doi: 10.1155/2019/3065818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dong X, Ni B, Fu J, et al. Emodin induces apoptosis in human hepatocellular carcinoma HepaRG cells via the mitochondrial caspase-dependent pathway. Oncol Rep. 2018;40:1985-1993. doi: 10.3892/or.2018.6620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tong H, Huang Z, Chen H, Zhou B, Liao Y, Wang Z. Emodin reverses gemcitabine resistance of pancreatic cancer cell lines through inhibition of IKKβ/NF-κB signaling pathway. Onco Targets Ther. 2020;13:9839-9848. doi: 10.2147/OTT.S253691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ma YS, Weng SW, Lin MW, et al. Antitumor effects of emodin on LS1034 human colon cancer cells in vitro and in vivo: roles of apoptotic cell death and LS1034 tumor xenografts model. Food Chem Toxicol. 2012;50:1271-1278. doi: 10.1016/j.fct.2012.01.033 [DOI] [PubMed] [Google Scholar]
- 33. Braumann C, Koplin G, Geier C, et al. Dose-dependent role of novel agents emodin and BTB14431 in colonic cancer treatment in rats. Acta Chir Belg. 2017;117:376-384. doi: 10.1080/00015458.2017.1341145 [DOI] [PubMed] [Google Scholar]
- 34. Yang K, Jin MJ, Quan ZS, Piao HR. Design and synthesis of novel anti-proliferative emodin derivatives and studies on their cell cycle arrest, apoptosis pathway and migration. Molecules. 2019;24:E884. doi: 10.3390/molecules24050884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Peter ME. Programmed cell death: apoptosis meets necrosis. Nature. 2011;471:310-312. doi: 10.1038/471310a [DOI] [PubMed] [Google Scholar]
- 36. Leibowitz B, Yu J. Mitochondrial signaling in cell death via the Bcl-2 family. Cancer Biol Ther. 2010;9:417-422. doi: 10.4161/cbt.9.6.11392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhang L, He D, Li K, et al. Emodin targets mitochondrial cyclophilin D to induce apoptosis in HepG2 cells. Biomed Pharmacother. 2017;90:222-228. doi: 10.1016/j.biopha.2017.03.046 [DOI] [PubMed] [Google Scholar]
- 38. Xie MJ, Ma YH, Miao L, et al. Emodin-provoked oxidative stress induces apoptosis in human colon cancer HCT116 cells through a p53-mitochondrial apoptotic pathway. Asian Pac J Cancer Prev. 2014;15:5201-5205. doi: 10.7314/apjcp.2014.15.13.5201 [DOI] [PubMed] [Google Scholar]
- 39. Cheng C, Dong W. Aloe-Emodin induces endoplasmic reticulum stress-dependent apoptosis in colorectal cancer cells. Med Sci Monit. 2018;24:6331-6339. doi: 10.12659/MSM.908400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Y Du, Zhang J, Tao Z, et al. Aloe emodin exerts potent anticancer effects in MIAPaCa-2 and PANC-1 human pancreatic adenocarcinoma cell lines through activation of both apoptotic and autophagic pathways, sub-G1 cell cycle arrest and disruption of mitochondrial membrane potential (ΛΨm). J Buon. 2019;24:746-753. [PubMed] [Google Scholar]
- 41. Cai FF, Bian YQ, Wu R, et al. Yinchenhao decoction suppresses rat liver fibrosis involved in an apoptosis regulation mechanism based on network pharmacology and transcriptomic analysis. Biomed Pharmacother. 2019;114:108863. doi: 10.1016/j.biopha.2019.108863 [DOI] [PubMed] [Google Scholar]
- 42. Li C, Gao S, Yang WS, Jin GZ, Sun S. β-Dihydroartemisinin-Emodin promotes apoptosis by activating extrinsic and intrinsic pathways in human liver cancer cells. Ann Clin Lab Sci. 2019;49:281-290. [PubMed] [Google Scholar]
- 43. Zheng XY, Yang SM, Zhang R, Wang SM, Li GB, Zhou SW. Emodin-induced autophagy against cell apoptosis through the PI3K/AKT/mTOR pathway in human hepatocytes. Drug Des Dev Ther. 2019;13:3171-3180. doi: 10.2147/DDDT.S204958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Srinivas G, Babykutty S, Sathiadevan PP, Srinivas P. Molecular mechanism of emodin action: transition from laxative ingredient to an antitumor agent. Med Res Rev. 2007;27:591-608. doi: 10.1002/med.20095 [DOI] [PubMed] [Google Scholar]
- 45. Redza-Dutordoir M, Averill-Bates DA. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim Biophys Acta. 2016;1863:2977-2992. doi: 10.1016/j.bbamcr.2016.09.012 [DOI] [PubMed] [Google Scholar]
- 46. Wang Y, Luo Q, He X, et al. Emodin induces apoptosis of colon cancer cells via induction of autophagy in a ROS-dependent manner. Oncol Res Featuring Preclin Clin Cancer Ther. 2018;26:889-899. doi: 10.3727/096504017x15009419625178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lee EH, Baek SY, Park JY, Kim YW. Emodin in Rheum undulatum inhibits oxidative stress in the liver via AMPK with Hippo/Yap signalling pathway. Pharm Biol. 2020;58:333-341. doi: 10.1080/13880209.2020.1750658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jänicke P, Lennicke C, Meister A, Seliger B, Wessjohann LA, Kaluđerović GN. Fluorescent spherical mesoporous silica nanoparticles loaded with emodin: synthesis, cellular uptake and anticancer activity. Mater Sci Eng C Mater Biol Appl. 2021;119:111619. doi: 10.1016/j.msec.2020.111619 [DOI] [PubMed] [Google Scholar]
- 49. Lee KH, Lee MS, Cha EY, et al. Inhibitory effect of emodin on fatty acid synthase, colon cancer proliferation and apoptosis. Mol Med Rep. 2017;15:2163-2173. doi: 10.3892/mmr.2017.6254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mikalayeva V, Ceslevičienė I, Sarapinienė I, et al. Fatty acid synthesis and degradation interplay to regulate the oxidative stress in cancer cells. Int J Mol Sci. 2019;20:1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bazzazi H, Isenberg JS, Popel AS. Corrigendum: inhibition of VEGFR2 activation and its downstream signaling to ERK1/2 and calcium by Thrombospondin-1 (TSP1): In silico investigation. Front Physiol. 2017;8:147. doi: 10.3389/fphys.2017.00147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Liu F, Zhang J, Qian J, Wu G, Ma Z. Emodin alleviates CCl4-induced liver fibrosis by suppressing epithelial-mesenchymal transition and transforming growth factor-β1 in rats. Mol Med Rep. 2018;18:3262-3270. doi: 10.3892/mmr.2018.9324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hao Y, Baker D, ten Dijke P. TGF-β-mediated epithelial-mesenchymal transition and cancer metastasis. Int J Mol Sci. 2019;20:2767. doi: 10.3390/ijms20112767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Carver W, Fix E, Fix C, Fan D, Chakrabarti M, Azhar M. Effects of emodin, a plant-derived anthraquinone, on TGF-β1-induced cardiac fibroblast activation and function. J Cell Physiol. 2021;236:7440-7449. doi: 10.1002/jcp.30416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hanna J, Hossain GS, Kocerha J. The potential for microRNA therapeutics and clinical research. Front Genet. 2019;10:478. doi: 10.3389/fgene.2019.00478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Huang A, Yang XR, Chung WY, Dennison AR, Zhou J. Targeted therapy for hepatocellular carcinoma. Signal Transduct Target Ther. 2020;5:146. doi: 10.1038/s41392-020-00264-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Dong H, Wu G, Xu H, et al. N-acetylaminogalactosyl-decorated biodegradable PLGA-TPGS copolymer nanoparticles containing emodin for the active targeting therapy of liver cancer. Artif Cells Nanomed Biotechnol. 2018;46:260-272. doi: 10.1080/21691401.2018.1455055 [DOI] [PubMed] [Google Scholar]
- 58. Khan H, Jia W, Yu Z, et al. Emodin succinyl ester inhibits malignant proliferation and migration of hepatocellular carcinoma by suppressing the interaction of AR and EZH2. Biomed Pharmacother. 2020;128:110244. doi: 10.1016/j.biopha.2020.110244 [DOI] [PubMed] [Google Scholar]
- 59. Shimpo K, Chihara T, Kaneko T, et al. Inhibitory effects of low-dose aloe-emodin on the development of colorectal tumors in min mice. Asian Pac J Cancer Prev. 2014;15:5587-5592. doi: 10.7314/apjcp.2014.15.14.5587 [DOI] [PubMed] [Google Scholar]
- 60. Ban E, Park M, Jeong S, et al. Poloxamer-based thermoreversible gel for topical delivery of Emodin: influence of P407 and P188 on solubility of Emodin and its application in cellular activity screening. Molecules. 2017;22:E246. doi: 10.3390/molecules22020246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Yang X, Zhang Y, Liu Y, Chen C, Xu W, Xiao H. Emodin induces liver injury by inhibiting the key enzymes of FADH/NADPH transport in rat liver. Toxicol Res. 2018;7:888-896. doi: 10.1039/c7tx00307b [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zhang Y, Yang X, Jia Z, et al. Proteomics unravels emodin causes liver oxidative damage elicited by mitochondrial dysfunction. Front Pharmacol. 2020;11:416. doi: 10.3389/fphar.2020.00416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lin L, Liu Y, Fu S, Qu C, Li H, Ni J. Inhibition of mitochondrial complex function-the hepatotoxicity mechanism of Emodin based on quantitative proteomic analyses. Cells. 2019;8:E263. doi: 10.3390/cells8030263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Liu DM, Yang D, Zhou CY, et al. Aloe-emodin induces hepatotoxicity by the inhibition of multidrug resistance protein 2. Phytomedicine. 2020;68:153148. doi: 10.1016/j.phymed.2019.153148 [DOI] [PubMed] [Google Scholar]
- 65. Wang X, Han L, Bi Y, et al. Paradoxical effects of emodin on ANIT-induced intrahepatic cholestasis and herb-induced hepatotoxicity in mice. Toxicol Sci. 2019;168:264-278. doi: 10.1093/toxsci/kfy295 [DOI] [PubMed] [Google Scholar]
- 66. Reyes-Gordillo K, Shah R, Muriel P. Oxidative stress and inflammation in hepatic diseases: current and future therapy. Oxid Med Cell Longev. 2017;2017:3140673. doi: 10.1155/2017/3140673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zheng YF, Liu CF, Lai WF, et al. The laxative effect of emodin is attributable to increased aquaporin 3 expression in the colon of mice and HT-29 cells. Fitoterapia. 2014;96:25-32. doi: 10.1016/j.fitote.2014.04.002 [DOI] [PubMed] [Google Scholar]
- 68. Sougiannis AT, Enos RT, VanderVeen BN, et al. Safety of natural anthraquinone emodin: an assessment in mice. BMC Pharmacol Toxicol. 2021;22:9. doi: 10.1186/s40360-021-00474-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Liu Y, Chen X, Qiu M, Chen W, Zeng Z, Chen Y. Emodin ameliorates ethanol-induced fatty liver injury in mice. Pharmacology. 2014;94:71-77. doi: 10.1159/000363413 [DOI] [PubMed] [Google Scholar]
- 70. Qian ZJ, Zhang C, Li YX, Je JY, Kim SK, Jung WK. Protective effects of emodin and chrysophanol isolated from marine fungus Aspergillus sp. On ethanol-induced toxicity in HepG2/CYP2E1 cells. Evid Based Complement Alternat Med. 2011;2011:452621. doi: 10.1155/2011/452621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Dong MX, Jia Y, Zhang YB, et al. Emodin protects rat liver from CCl(4)-induced fibrogenesis via inhibition of hepatic stellate cells activation. World J Gastroenterol. 2009;15:4753-4762. doi: 10.3748/wjg.15.4753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Lee BH, Huang YY, Duh PD, Wu SC. Hepatoprotection of emodin and polygonum multiflorum against CCl(4)-induced liver injury. Pharm Biol. 2012;50:351-359. doi: 10.3109/13880209.2011.604335 [DOI] [PubMed] [Google Scholar]
- 73. Bhadauria M. Dose-dependent hepatoprotective effect of emodin against acetaminophen-induced acute damage in rats. Exp Toxicol Pathol. 2010;62:627-635. doi: 10.1016/j.etp.2009.08.006 [DOI] [PubMed] [Google Scholar]
- 74. Yin X, Gong X, Jiang R, et al. Emodin ameliorated lipopolysaccharide-induced fulminant hepatic failure by blockade of TLR4/MD2 complex expression in D-galactosamine-sensitized mice. Int Immunopharmacol. 2014;23:66-72. doi: 10.1016/j.intimp.2014.08.018 [DOI] [PubMed] [Google Scholar]
- 75. Xue J, Chen F, Wang J, et al. Emodin protects against concanavalin A-induced hepatitis in mice through inhibiting activation of the p38 MAPK-NF-κB signaling pathway. Cell Physiol Biochem. 2015;35:1557-1570. doi: 10.1159/000373971 [DOI] [PubMed] [Google Scholar]
- 76. Wang J, Zhao Y, Xiao X, et al. Assessment of the renal protection and hepatotoxicity of rhubarb extract in rats. J Ethnopharmacol. 2009;124:18-25. doi: 10.1016/j.jep.2009.04.018 [DOI] [PubMed] [Google Scholar]
- 77. Wang JB, Zhao HP, Zhao YL, et al. Hepatotoxicity or hepatoprotection? Pattern recognition for the paradoxical effect of the Chinese herb Rheum palmatum L. in treating rat liver injury. PLoS One. 2011;6:e24498. doi: 10.1371/journal.pone.0024498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Du Y, Qian B, Gao L, et al. Aloin preconditioning attenuates hepatic ischemia/reperfusion injury via inhibiting TLR4/MyD88/NF-κB signal pathway in vivo and in vitro. Oxid Med Cell Longev. 2019;2019:3765898. doi: 10.1155/2019/3765898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Nair AB, Jacob S. A simple practice guide for dose conversion between animals and human. J Basic Clin Pharm. 2016;7:27-31. doi: 10.4103/0976-0105.177703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Xiong XL, Ding Y, Chen ZL, et al. Emodin rescues intrahepatic cholestasis via stimulating FXR/BSEP pathway in promoting the canalicular export of accumulated bile. Front Pharmacol. 2019;10:522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Wang S, Li X, Guo H, et al. Emodin alleviates hepatic steatosis by inhibiting sterol regulatory element binding protein 1 activity by way of the calcium/calmodulin-dependent kinase kinase-AMP-activated protein kinase-mechanistic target of rapamycin-p70 ribosomal S6 kinase signaling pathway. Hepatol Res. 2017;47(7):683-701. [DOI] [PubMed] [Google Scholar]
- 82. Jiang LL, Jiang Y, Zhao DS, et al. CYP3A activation and glutathione depletion aggravate emodin-induced liver injury. Chem Res Toxicol. 2018;31(10):1052-1060. [DOI] [PubMed] [Google Scholar]