

SUMMARY
Expansion of a CAG repeat in ATXN3 causes the dominant polyglutamine disease spinocerebellar ataxia type 3 (SCA3), yet the physiological role of ATXN3 remains unclear. Here, we focus on unveiling the function of Ataxin-3 (ATXN3) in the retina, a neurological organ amenable to morphological and physiological studies. Depletion of Atxn3 in zebrafish and mice causes morphological and functional retinal alterations and, more precisely, photoreceptor cilium and outer segment elongation, cone opsin mislocalization, and cone hyperexcitation. ATXN3 localizes at the basal body and axoneme of the cilium, supporting its role in regulating ciliary length. Abrogation of Atxn3 expression causes decreased levels of the regulatory protein KEAP1 in the retina and delayed phagosome maturation in the retinal pigment epithelium. We propose that ATXN3 regulates two relevant biological processes in the retina, namely, ciliogenesis and phagocytosis, by modulating microtubule polymerization and microtubule-dependent retrograde transport, thus positing ATXN3 as a causative or modifier gene in retinal/macular dystrophies.
In Brief
Toulis et al. show that depletion of Atxn3 causes retinal morphological and electrophysiological alterations, as well as diminished phagocytosis in vivo and in vitro. ATXN3 regulates retinal ciliogenesis and phagocytosis through the KEAP1-SQSTM1-HDAC6-acetylated tubulin pathway by modulating microtubule polymerization and microtubule-dependent retrograde transport.
Graphical Abstract
INTRODUCTION
Ataxin-3 (ATXN3) is a deubiquitinating enzyme (DUB) of the Machado-Joseph disease (MJD) protease family (Nijman et al., 2005). An expanded CAG repeat within the ATXN3 gene encodes a polyglutamine (polyQ) stretch located in the carboxyl terminus of the ATXN3 protein (UniProtKB: 54252) and causes MJD, also known as spinocerebellar ataxia type 3 (SCA3) (OMIM: 109150). MJD/SCA3 is a late-onset autosomal dominant neurodegenerative disorder whose main phenotypic trait is ataxia (Paulson et al., 2017).
ATXN3, a 42-kDa protein, harbors the protease Josephin domain (JD), which requires a cysteine in the catalytic site (C14 residue) to deconjugate ubiquitin (Burnett et al., 2003). ATXN3 participates in ubiquitin-dependent pathways by interacting with ubiquitin E3 ligases, such as parkin (Durcan et al., 2011) and CHIP (C terminus of Hsc70 interacting protein) (Scaglione et al., 2011), and proteasome-associated proteins (Blount et al., 2014; Ristic et al., 2018). Besides, ATXN3 is involved in several other cellular processes, including endoplasmic-reticulum-associated degradation (ERAD) (Zhong and Pittman, 2006), aggresome formation (Wang et al., 2012), cytoskeletal organization (do Carmo Costa et al., 2010; Rodrigues et al., 2010), transcriptional regulation and histone acetylation (Evert et al., 2006), response to different types of cellular and organismal stress (Tsou et al., 2015), DNA repair (Pfeiffer et al., 2017), and autophagy (Ashkenazi et al., 2017).
The vertebrate retina, a structurally complex neuronal tissue covering the inner surface of the eye, consists of six different highly specialized cell types with a layered organization (Hoon et al., 2014; Figure 1A). The light-sensitive photoreceptor cells are responsible for absorption of the light stimuli and the phototransduction cascade (den Hollander et al., 2010). All the proteins involved in photoreception and phototransduction are localized in the outer segment (OS) of photoreceptors, a specialized compartment containing stacked ordered membranous disks. Because the OS is devoid of protein translation machinery, OS components are synthesized in the inner segment (IS) of photoreceptors and transported to the OS through the ciliary gate, a microtubule-based extension known as the connecting cilium (CC) (Khanna, 2015). The tips of photoreceptor OS are physically in contact with the retinal pigment epithelium (RPE), a single layer of pigmented cells, forming the blood-retina barrier (Figure 1A). The RPE participates in many important retinal functions, such as the restoration of visual cycle molecules and in the daily shedding of OS disks through phagocytosis of photoreceptor tips (Sparrow et al., 2010).
Figure 1. Knockdown of ataxin-3 in Zebrafish Embryos Alters Eye Size and Retinal Layer Organization.
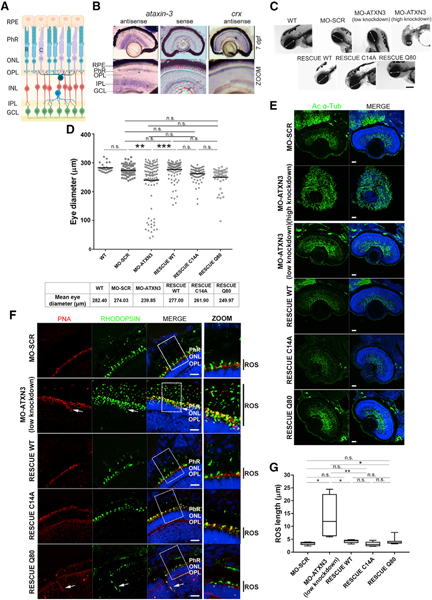
(A) Schematic diagram of the mature vertebrate retina organization, which is similar in humans, mice, and zebrafish. RPE, retinal pigment epithelium; PhRs, photoreceptors; ONL, outer nuclear layer; OPL, outer plexiform layer; INL, inner nuclear layer; IPL, inner plexiform layer; GCL, ganglion cell layer; R, rod; C, cone.
(B) atxn3 is highly expressed in different retinal layers in zebrafish larvae (mRNA in situ hybridization on retinal cryosections at 7 days post-fertilization [dpf]). Sense atxn3 (negative) and antisense crx (positive) riboprobes were used as controls. Magnification, 20×; zoom, 40×.
(C) In vivo imaging of embryos (72 hpf) shows that microinjection of MO-ATXN3 (morpholino against atxn3) compared to MO-SCR (scrambled, negative control) causes alterations in the eye (white double-head arrows indicate the diameter) and head size depending on the level of atxn3 knockdown (low or high). The decrease in eye size was rescued when MO-ATXN3 was co-injected with human ATXN3 WT Q22 mRNA, but not as much with ATXN3 C14A (catalytically inactive mutant) or ATXN3 Q80 (MJD mutant). Scale bar, 50 μm.
(D) Quantification and comparison of eye size in the different embryo groups. A black line indicates the mean eye size in each group (n = 100 independent embryos per group). Mann-Whitney test (**p < 0.01, ***p < 0.001; n.s., non-significant).
(E) Extensive microtubule disorganization and defective formation of the retinal structures are observed in the retinas of high knockdown MO-ATXN3 morphants compared to MO-SCR (control) embryos, as shown by acetylated α-tubulin (green). Phenotypic rescue in retinal structures was observed after co-injecting morpholinos with human ATXN3-derived mRNAs, as indicated. Scale bar, 20 μm.
(F) Knockdown MO-ATXN3 zebrafish embryo retinas show elongation of the PhR outer segment (OS) and mislocalization of opsins compared to MO-SCR retinas (see zoom for detail, black bars on the right side indicate the OS length). This altered rod phenotype in MO-ATXN3 morphants is successfully rescued with co-injection of ATXN3 WT and C14A mRNAs. Rods are detected with rhodopsin (green) and cones with peanut agglutinin (PNA; red). White arrows indicate ectopic expression of opsins in cones and rods. Scale bar, 10 μm. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) (blue).
(G) Rod OS (ROS) length measurements in morphants and rescued embryos (Mann-Whitney test; *p < 0.05; **p < 0.01; n.s., non-significant) (n = 3–4 images per embryo, 5 embryos per group).
Being a complex tissue, the retina requires a rigorous regulation at the transcriptional and protein levels, including post-translational modifications, such as ubiquitination and SUMOylation (Swaroop et al., 2010). Mutations in several ubiquitin-pathway-related genes cause hereditary retinal neurodegeneration, e.g., the genes encoding the E3 ligases TOPORS (Chakarova et al., 2007), KLHL7 (Friedman et al., 2009), TRIM32/BBS11 (Chiang et al., 2006), and the DUB USP45 (Yi et al., 2019). Given the clear implication of ubiquitin pathway genes in retinal development and pathology and the genetic link between cerebellar dysfunction and retinal anomalies in other SCAs (McLaughlin and Dryja, 2002), we aimed to explore the function of ATXN3, whose mutation causes MJD/SCA3, in the retina.
Because humans, mice, and zebrafish show similar mature retinal structures (Figure 1A), here, we combine in vivo zebrafish and mouse models and in vitro cells. We show that ATXN3 abrogation causes retinal dysfunction due to alterations in both the photoreceptor sensory cilium and RPE-mediated phagocytosis, highlighting ATXN3 as a good candidate for genetic retinal diseases.
RESULTS
Knockdown of ataxin-3 Causes Layer Disorganization and Cell-Specific Alterations in Zebrafish Retinas
First, we investigated the role of ATXN3 in retina by using zebrafish (Danio rerio). In early zebrafish embryos, atxn3 transcripts are already detectable from 10 h post-fertilization (hpf) onward (Figure S1A). At 48 hpf, the atxn3 is expressed throughout the whole retinal tissue (Figure S1B), but in the differentiated retina at 7 days post-fertilization (dpf), atxn3 expression is mostly detected in the photoreceptor (PhR), inner plexiform layer (IPL), and ganglion cell layer (GCL) (Figure 1B). This expression pattern of atxn3 in zebrafish is in agreement with that reported for Atxn3 in adult mouse retinas (Esquerdo et al., 2016).
We next performed a knockdown of this gene by morpholino injection in zebrafish embryos. A specific morpholino antisense targeting atxn3 (MO-ATXN3) was designed to interfere with correct splicing—and to ensure degradation of the aberrant mRNAs by non-sense-mediated decay—and the efficiency of atxn3 knockdown was assessed (Figure S1C). Remarkably, in comparison to MO-SCR (scrambled) controls MO-ATXN3 embryos showed smaller brain and eye size, without signs of malformation in the anterior-posterior axis, and with similar survival rates (Figure 1C). The eye size phenotype strongly correlated with the remaining levels of atxn3 expression (Figure S1C). Low levels of knockdown resulted in embryos with moderately reduced eye size (85.9% of total), whereas high atxn3 knockdown values resulted in embryos with very small eyes and heads (14.1%) (Figure 1C). Overall, MO-ATXN3 morphants showed statistically significantly reduced eye size (diameter) compared with that of MO-SCR animals (mean of 239.85 μm and 274.03 μm, respectively) (Figures 1C and D). To validate the observed atxn3-morphant phenotype in embryos, phenotypic rescue experiments were performed by co-injection of MO-ATXN3 with an in-vitro-transcribed mRNA of either human ATXN3 wild type (WT) (polyQ within the normal range, ATXN3Q22), a catalytically inactive form of human ATXN3 (ATXN3 C14A), or human expanded ATXN3 (polyQ within the MJD/SCA3 range, ATXN3 Q80). Co-injection of the human ATXN3 WT mRNA fully rescued the eye size phenotype, whereas co-injection of either the C14A or Q80 ATXN3 mutants slightly increased the eye size but without reaching statistical significance (Figures 1C and 1D).
To assess whether the small eye phenotype in MO-ATXN3-injected embryos was due to retinal disorganization, we immunodetected acetylated α-tubulin (a retinal cytoskeleton marker) in retinal sections of 72 hpf embryos. Compared to controls, MO-ATXN3 morphants, particularly those with high levels of knockdown, showed extensive microtubule disorganization and defective retinal structure formation (Figure 1E). Additionally, immunodetection of cones (peanut agglutinin [PNA]) and rods (rhodopsin) revealed longer rod OS (about 3-fold) in retinas of MO-ATXN3-injected embryos than that of controls (Figures 1F and 1G). Besides, cone L/M opsins were mislocalized in MO-ATXN3 retinas: instead of being solely localized at the OS, ectopic localization at the perinuclear region and axons of cones was clearly detected (Figure S1D). Co-injection of MO-ATXN3 morphants with any of the three human ATXN3-derived mRNAs significantly rescued both the retinal lamination and microtubule organization phenotypes (Figure 1E) and rod OS elongation (Figures 1F and 1G). However, only the human WT ATXN3 mRNA reversed cone opsin mislocalization, indicating that this phenotypic trait is dependent on the preserved DUB activity of a non-expanded polyQ form of ATXN3 (Figure S1D).
Overall, these results suggest that a strong depletion of atxn3 causes a considerable reduction in eye size, accompanied by retinal disorganization in the morphants. A milder phenotype (low knockdown) shows rod OS elongation and cone opsin mislocalization. Both severe and milder phenotypes are specifically due to the knockdown of atxn3 because co-injection of MO-ATXN3 morphants with human WT ATXN3 mRNA is able to rescue the eye phenotype. These results in zebrafish prompted us to study the function of ATXN3 in the mammalian mouse retina.
Knockout of Atxn3 in Mice Causes Morphological and Electrophysiological Alterations in PhRs
In agreement with the atxn3 expression pattern in zebrafish (this work) and in mouse retinas (Esquerdo et al., 2016), we detected the ATXN3 protein throughout all mouse retinal layers, but with a stronger signal in the PhR layers, OPLs, IPLs, and GCLs (Figure S2). To understand the outcome of Atxn3 deletion in the retina, we used an existing knockout mouse model (Atxn3 KO) generated by gene trap technology (Reina et al., 2012). Although Atxn3 KO mice do not show any gross alteration, to the best of our knowledge, no comprehensive behavioral and pathological studies have been conducted in these mice. Because expansion of the CAG repeat in the human ATXN3 gene causes MJD/SCA3, mainly characterized by progressive ataxia, we evaluated whether knockout of Atxn3 would lead to motor dysfunction. Assessments of grip strength, gait, balance, and locomotor and exploratory activities in Atxn3 KO and WT littermates were conducted. No differences were observed between WT and KO mouse groups, indicating that ablation of Atxn3 does not impact motor function in ~1-year-old mice (Figure S2). We then focused on characterizing the eye and the visual phenotype.
After confirming that Atxn3 KO mouse retinas showed an absence of Atxn3 transcripts and proteins (Figure S2), we assessed the retinal morphology of 2-year-old Atxn3 KO and WT mice by morphometric measurements of ocular sections at the optic nerve level (Figure S3). We detected a significant enlargement of the retina throughout all layers in the Atxn3 KO compared to WT controls, particularly in the PhR layer (spanning ISs and OSs) and the IPL (Figure S3). Collectively, these findings demonstrate a morphological and functional role for Atxn3 in the mouse retina.
Therefore, we focused in more detail on the role of ATXN3 in PhRs of 2-month-old (fully mature young retinas) and 2-year-old mice (when progressive retinal neurodegeneration, if any, would be observed). No differences in rhodopsin localization in rods were detected between genotypes, whereas the PhR layer was markedly enlarged in 2-month-old Atxn3 KO retinas. The enlargement of this layer was not due to an increase in cell number but instead to a significant elongation of the rod OSs (but not ISs) compared to controls (Figure 2A). This phenotype was consistently observed in retinas from 2-year-old Atxn3 KO mice (Figure S3), with no signs of increased PhR death (at all ages, KO and WT retinas showed an equivalent number of nuclei rows in the ONL; Figures S3B and S4). Similar to rods, Atxn3 KO cones showed longer OSs than those of the WT, independently of age (Figures 2B and S3B). In addition, cone L/M opsins were markedly mislocalized in Atxn3 KO retinas, being detected not only in the OS but also in the IS (Figures 2B and S3B).
Figure 2. Mouse Atxn3 KO Retinas Show Elongation of PhR OSs, Cone Opsin Mislocalization, and Hyperactivation of Cone Response.

(A) Rod OSs (black bars at the right side) in Atxn3 KO (Atxn3−/−) retinas are much longer than those of WT (Atxn3+/+). RHO, rhodopsin (green).
(B) Cone OSs (labeled with PNA, red) in Atxn3 KO retinas are longer and show mislocalization of opsins (L/M opsins, green), which also extend into the inner segment (IS) of cones (black bars at the right side).
(C) GCAP1 (green) is also mislocalized in the IS and ONL of Atxn3 KO cones (PNA, red) compared to WT retinas. Antigen retrieval protocols used to detect GCAP1 partially interfered with PNA staining. Scale bar, 10 μm.
(D) After subretinal microinjection of GFP-ATXN3 WT expression construct in the right eye (R) of Atxn3 KO pups, the mislocalization of L/M cone opsins (red) is partially rescued in transfected cones (green), as L/M opsins localize along the whole length of the cone OS (PNA, magenta). In zoom images, the white arrow indicates a transfected cone where opsins correctly distribute along the OS. The left eye of each animal was not transfected and used as a control eye (L). Nuclei were counterstained with DAPI (blue); scale bar, 10 μm (n = 3 per group). Accompanying 3D reconstructions are in Videos S1 and S2. (A–D) Names of all retinal layers are abbreviated as in Figure 1A.
(E) ERG recordings of WT (black) and Atxn3 KO animals (gray): rod B-wave (scotopic light), photopic (PHOT) B-wave (cones and cone bipolar cells), and combined response to light in dark adapted retinas (DA) A-wave (combined rods and cones) and DA B-wave (bipolar cell response) show that the cone (photopic response) is hyperactivated in the absence of ATXN3 (Mann-Whitney test, *p < 0.05, two eyes of 3–4 animals per group).
(F) Representative ERG recordings of the photopic B-wave in WT and KO retinas and their overlap (bottom) for direct visualization of cone hyperactivation in KO retinas.
We then assessed whether other proteins of the phototransduction cascade were mislocalized in Atxn3 KO retinas. For instance, GCAP1 (involved in the synthesis of cyclic guanosine monophosphate [cGMP] in PhRs) is normally localized in the OS but appeared mislocalized throughout the IS and OS of Atxn3 KO cones (Figure 2C), as well as in the ONL and OPL of rods (Figure S3B). These results were equivalent in young (2-month-old) and aged (2-year-old) mouse retinas (Figures 2 and S3). In contrast, dynamin-2, GRK1, cone-arrestin, and PDE6C were similarly localized in Atxn3 KO retinas compared to controls at all ages (Figure S4).
To confirm that opsin mislocalization was specifically due to Atxn3 ablation, we performed phenotypic rescue assays by transient transgenesis in the retina of Atxn3 KO mice. A GFP-ATXN3 WT expression construct was microinjected into the subretinal space of the right eye of Atxn3 KO P0-P1 newborn pups and electroporated. The left eye of each microinjected animal was used as a control. Control Atxn3 KO animals were transfected with the empty GFP vector. The structure and morphology of GFP-positive PhR cells were analyzed one month post-injection. Because transient transgenesis is detected only in a patch zone close to the injection, the number of animals that we used in these experiments did not provide enough power to evaluate the rescue of the PhR length phenotype. Besides, as rods are present in a markedly higher number than cones in mouse retinas and are difficult to individualize, we centered our rescue analysis on the mislocalization of opsins in individual Atxn3 KO cones (Figure 2D). As anticipated, GFP-positive cones expressing the human GFP-ATXN3 WT showed phenotypic rescue, as cone opsins solely localized along all the OS (Figure 2D). In contrast, the cones of the untreated control eye or those of Atxn3 KO animals microinjected with GFP alone exhibited abnormal opsin localization in the IS and other regions (Videos S1 and S2 for control and Atxn3 KO GFP-ATXN3-WT-microinjected retinas, respectively).
We then explored whether these structural PhR alterations correlated with functional visual alterations in Atxn3 KO mice. We performed electroretinogram recordings (ERGs) in Atxn3 KO and WT littermates under light and dark adaptation conditions to evaluate the affectation of rods (responsible for scotopic vision) and cones (stimulated under photopic conditions). The photopic b-wave response was increased, corresponding to the hyperactivation of cones in Atxn3 KO retinas (Figures 2E and 2F). This photopic hyperexcitation in the Atxn3 KO mice is in accordance with the observed alterations in cones (e.g., protein mislocalization) in Figures 2B and 2C. Although there were no other statistically significant differences between genotypes, a trend for increased response in KO rods to scotopic light (and other ERG measurements) was also observed (Figure 2E).
Collectively, these findings indicate that the ablation of Atxn3 in mice causes an elongation of OSs of PhRs and mislocalization of cone opsins and other specific phototransduction proteins, which are reflected by altered ERG recordings. Our results in the Atxn3 KO mouse retinas are in accordance with those observed in zebrafish embryos and could potentially be explained by an involvement of ATXN3 in the regulation of ciliary formation and/or in protein trafficking from the IS to the OS through the CC. Alternatively, and in a non-mutually exclusive mechanism, ATXN3 could regulate the phagocytosis of the PhR OS tips by the RPE. Hence, we next explored both hypotheses.
ATXN3 Regulates the Length of Sensory and Primary Cilia
The PhR OS is a highly modified sensory cilium that exhibits unique functional properties and requires finely tuned trafficking and localization of phototransduction proteins. As the observed increase in OS length in Atxn3 KO retinas could be due to alterations in the anterograde and/or retrograde ciliary trafficking, we first examined in more detail the structure of PhRs of Atxn3 KO and WT mice. By isolating rods and performing immunofluorescent detection of rhodopsin and acetylated α-tubulin (marking the axoneme), we confirmed the increased length of the OS and also detected elongated cilium axonemes in Atxn3 KO compared to those of the WT (Figure 3A). Measurements from transmission electron microscopy (TEM) ultra-thin sections of retinas from 2-year-old Atxn3 KO and control animals confirmed the significantly increased length of the CC in Atxn3 KO PhRs (mean of 2.066 μm) compared to that of the WT (mean of 1.897 μm) (Figures 3B and 3C). OS membranous stacks of both cones and rods of Atxn3 KO retinas appeared preserved and structurally similar to those of WT PhRs (Figure 3B).
Figure 3. ATXN3 Regulates the Length of the Cilium in PhR Cells.

(A–C) Elongation of the OS (contains the axoneme) and CC (connecting cilium) in Atxn3 KO PhRs, as shown in (A), by rhodopsin (red) and acetylated-α-tubulin (green) detection in isolated rods from 2-year-old WT (Atxn3+/+) and KO (Atxn3−/−) mouse retinas; nuclei were labeled with DAPI (blue) (scale bar, 10 μm); and in (B), by transmission electron microscopy (TEM), where the connecting cilium is indicated by a black line. No other apparent morphological differences were detected (representative images). (C) Mean CC length was 1.897 μm for WT and 2.066 μm for KO (19 TEM pictures, 3 animals per genotype). Two-way ANOVA test (*p < 0.05).
(D–G) The length of primary cilia in ARPE-19 cells is modulated by ATXN3 levels. (D and E) Depletion of endogenous ATXN3 by siATXN3 transfection in human ARPE-19 cells results in longer primary cilium (mean 4.222 μm in siATXN3 cells, n = 41) than that in controls (mean 3.523 μm in siSCR cells, n = 26). Mann-Whitney test (**p < 0.01). (F and G) Starved ARPE-19 cells overexpressing GFP-ATXN3 WT Q22 produce shorter cilia (mean, 3.211 μm) than cells with GFP-ATXN3 C14A mutant (4.208 μm) or the pEGFP empty vector (mean 4.318 μm). Only GFP-positive cells (n > 20–30 per condition) were analyzed (see Figure S5B). Mann-Whitney test (*p < 0.05, **p < 0.01). In (D) and (F), Ciliary microtubules were detected with acetylated α-tubulin (red) and basal bodies with γ-tubulin (green). Scale bar, 5 μm.
To further investigate if ATXN3 is involved in ciliary formation, we used ARPE-19 cells (a retinal cell line derived from human RPE), which produce a primary cilium under serum starvation conditions (Izawa et al., 2015), and induced the knockdown of ATXN3. After testing the efficacy of several ATXN3 small interfering RNAs (siRNAs) and scrambled siRNAs (Figure S5A), we selected siSCR-1 and siATXN3–2 (nearly 90% of ATXN3 knockdown) for subsequent assays (hereafter, siSCR and siATXN3, respectively). We immunodetected the ciliary microtubules or axoneme (acetylated α-tubulin) and the basal body (γ-tubulin) in primary cilia of starved ARPE-19 cells to measure cilium length. In agreement with our observations in mouse retinas, knockdown of endogenous ATXN3 in ARPE19 cells consistently caused primary cilia elongation (Figure 3D). On average, ATXN3-depleted cells showed statistically significantly longer cilia (with a 20% mean increase) than control-transfected cells (4.222 μm and 3.523 μm, respectively) (Figure 3E). On the contrary, cells overexpressing GFP-ATXN3 WT Q22 displayed significantly shortened primary cilia (25% shorter) than controls, whereas the cilium length in cells expressing the ATXN3 C14A mutant were unaffected (Figure 3F). On average, cilium length was 4.318 μm for control, 3.211 μm for ATXN3 WT, and 4.208 μm in cells overexpressing ATXN3 C14A (Figure 3G).
To confirm whether ATXN3 localized in some ciliary components, we performed immunodetections on starved ARPE19 cells. Although non-preferentially, endogenous ATXN3 colocalizes with acetylated α-tubulin at the axoneme of the cilium and γ-tubulin at the basal body (single confocal planes, Figure S5C). We propose that this localization of ATXN3 in ciliary compartments may enable its regulatory activity on ciliary length. Overall, our results support that in PhRs and RPE cells, ATXN3 is a negative regulator of ciliary length in a DUB activity-dependent manner.
KEAP1, a Substrate Adaptor of CUL3, Is Depleted in Atxn3 KO Retinas
To elucidate the role of ATXN3 in ciliogenesis and identify proteins whose abundance in mouse retina is regulated directly or indirectly by this DUB, we compared the proteomes of Atxn3 KO and WT mouse retinas. Protein extracts of neural retinas of 1-year-old Atxn3 KO and WT littermates were pooled for liquid chromatography-mass spectrometry (LC/MS) proteomic assessment. Although not all relevant proteins could be identified in this proteomic approach, over 5,000 proteins were unambiguously identified, quantified, and compared between WT and Atxn3 KO mouse retinas (Table S2, full protein list with fold-change values). As expected, the quantity of Atxn3 was highly over-represented in WT retinas, confirming the sample genotypes (Table S2). The volcano plot in Figure 4A highlights significantly over-represented (88 hits, in green) and under-represented proteins (106 hits, in orange) in the KO versus WT retinas. Gene Ontology (GO) annotation reveals that over-represented proteins in Atxn3 KO retinas are mainly involved in phototransduction (20%) and in endocytosis and vesicular transport pathways (30%) (Table S3), which is in agreement with a corresponding increase of proteins localized at the elongated OS in Atxn3 KO retinas. On the other hand, almost 40% of the under-represented proteins are involved in microtubule formation and organization, in angiogenesis and inflammation, and in the response to stimulus and stress (Table S3), which are of particular interest because many retinal pathologies are associated with alterations of these pathways. Among the under-represented proteins in Atxn3 KO mouse retinas, we selected KEAP1, a Kelch-like ECH-associated protein also related to the ubiquitin signaling pathway, as a good candidate to use to pursue its connection with ATXN3.
Figure 4. Neural Retina Proteomics in Atxn3 KO Mice and Knockdown Assays in Cultured Cells Show That KEAP1 Levels Are Decreased when ATXN3 Expression Is Depleted.

(A) Proteomics volcano plot of neural retinas from 2-year-old Atxn3 KO versus WT mice, plotting the −log10 (p value) against the log2 ratio (fold change). Thresholds were set at a significance of p < 0.01 and a ratio of 50% change (0.5 and 1.5). Over-represented and under-represented proteins in Atxn3 KO retinas are highlighted in green and orange. Some relevant retinal proteins are also indicated. Gene Ontology (GO) classification is in Table S3.
(B) Network generated by RPGenet v2.0 links ATXN3 by KEAP1-SQSTM1 to the HDAC6-acetylated α-tubulin signaling pathway, which controls ciliary length. The direction and strength of the interaction between the connected nodes is respectively indicated by the arrows and width of the connecting edges (red lines, physical interaction; gray lines, genetic interaction). Node size differences are not relevant for this work.
(C) Proposed pathways whereby ATXN3 depletion cause a decrease of KEAP1, which leads to an increase of SQSTM1 (p62), a negative regulator of the HDAC6 deacetylase activity. Inhibition of HDAC6 increases the pool of acetylated α-tubulin, which can then polymerize and increase ciliary length. An asterisk (*) indicates the proteins whose peptides were not identified in the proteomics analyses.
(D) Knockdown of endogenous ATXN3 causes a sharp decrease of KEAP1 levels in ARPE-19 cells, with a concomitant increase of SQSTM1 and acetylated α-tubulin (as well as other proteins of the KEAP1/NR2F2 pathway). Statistical significance was determined by one-sample t test or Wilcoxon rank-sum test (*p < 0.05, **p < 0.01).
(E) The half-life of KEAP1 is clearly decreased after ATXN3 knockdown in HEK293T cells under cycloheximide protein-synthesis inhibition (CHX). Mann-Whitney test, n = 4–6 (*p < 0.05, **p < 0.01).
KEAP1, a substrate adaptor protein of the CUL3 E3 ubiquitin ligase complex, promotes the ubiquitination and proteasome degradation of NRF2 (also known as NFE2L2) (Cullinan et al., 2004) and p62 (also known as SQSTM1) (Johansen and Lamark, 2011), key regulators of cell stress, transcriptional response and autophagy, by the KEAP1-NRF2 signaling pathway. Therefore, a decrease in levels of KEAP1 results in increased levels of NRF2 and p62. To probe for a potential interaction network linking ATXN3 to ciliary formation by KEAP1, we queried an open access webapp, RPGenet v2.0, which allows the visualization of curated network interactions of relevant retinal genes by integrating data from reported literature and high-throughput screenings (Arenas-Galnares et al., 2019). Through this network (Figure 4B), we observed that high levels of p62 not only promote autophagy (Johansen and Lamark, 2011) but also, and of relevance to the Atxn3 KO retinal phenotype, strongly inhibit the deacetylase activity of HDAC6 (Yan et al., 2013). This nuclear and cytosolic deacetylase regulates the length of the cilium by deacetylating α-tubulin TUBA1A, a main component of ciliary microtubules (Ran et al., 2015). The retrieved ATXN3 interactome network allowed us to generate a working model to illustrate how the depletion of ATXN3 might result in the elongation of retinal sensory cilia by ATXN3-KEAP1-SQSTM1-HDAC6-acetylated TUBA1A interactions (Figure 4C).
To confirm this model and to evaluate whether ATXN3 regulates the KEAP1-SQSTM1 signaling pathway, we assessed levels of key proteins in siATXN3-treated ARPE-19 cells. Similar to the Atxn3 KO retina proteome, knockdown of ATXN3 in human cells significantly decreased KEAP1 protein levels (Figure 4D), leading to potentially reduced activity of the KEAP1-CUL3 complex. Our results also confirm that the levels of NRF2 and p62 were increased in ATXN3-knockdown cells compared with those of controls (Figure 4D).
Because KEAP1 seems to be a key player in this potential cascade of events regulating cilium length, we focused on better understanding whether ATXN3 participates in regulating the stability and proteasomal clearance of KEAP1. Indeed, depletion of ATXN3 lead to destabilization of the KEAP1 protein (decreased half-life), further supporting ATXN3 as a regulator of KEAP1 turnover (Figure 4E). Future studies are needed to assess whether ATXN3 directly deubiquitinates KEAP1. Thus, our results suggest that ATXN3 promotes the stability of KEAP1 by slowing down its degradation, although the precise mechanism remains to be elucidated.
ATXN3 Regulates Levels of Extracellular Matrix and Microtubule-Mediated Transport Components in the RPE
In parallel with regulating ciliary length, ATXN3 could regulate the length of the PhR OS by modulation of phagocytosis of the OS tips by the RPE. To assess whether Atxn3 KO mouse RPEs show altered levels of relevant phagocytosis proteins, we defined the proteome in the RPE of the same animals used in the previously described proteomics. The volcano plot in Figure 5A highlights significantly over-represented (7 hits, in green) and under-represented proteins (48 hits, in orange) in the Atxn3 KO versus WT RPEs (full list in Table S2). Although some of the altered proteins in Atxn3 KO versus WT coincide in the neural retina and the RPE proteomes, there were relevant differences due to the intrinsic differential gene expression and protein content in these two tissues. Therefore, Atxn3 ablation seem to differentially affect target proteins or interacting partners in the retina and RPE. GO data analyses showed that several components of the specific extracellular matrix of the retina, namely, the interphotoreceptor matrix (e.g., collagens, fibulin, and laminins; Ishikawa et al., 2015), are over-represented in Atxn3 KO RPE. Increased levels of proteins in the subretinal space also correlate with longer microvilli and other structural proteins that connect the RPE to the PhR OS tips (Bonilha et al., 2006). On the other hand, almost 20% of the specific PhR proteins involved in the visual cycle and phototransduction (e.g., opsins, transducin, recoverin, and peripherin) are under-represented in Atxn3 KO RPE compared to controls, pointing to an alteration of the degradation of PhR proteins by phagocytosis. Proteins involved in the response to stimulus and stress are also diminished, as well as relevant proteins implicated in chromatin structure and transcription regulation that are known to interact with ATXN3 (Table S3). Therefore, our proteomic results support a structural alteration of the subretinal space connecting RPE and PhRs as well as a dysfunction of the phagocytosis of OS tips in Atxn3 KO mice.
Figure 5. ATXN3 Regulates PhR Phagocytosis by Modulating Endosome Maturation and Transport in the RPE.

(A) Proteomics volcano plot of RPEs from Atxn3 KO versus WT mice, the plotting −log10 (p value) against the log2 ratio (fold change). Thresholds and protein highlights are as in Figure 4A.
(B) Network generated by RPGenet v2.0 that links ATXN3 to cytoskeleton and microtubule motor proteins that control phagosome maturation and vesicle transport in RPE cells. Node size is not relevant for this work.
(C) Proposed pathways whereby the depletion of ATXN3 alters phagosome formation and maturation within the RPE cells. An asterisk (*) indicates the proteins whose peptides were not identified in the proteomics analyses.
(D) Ezrin expression (green) is increased in 2-month- and 2-year-old Atxn3 KO RPE compared with controls. The increase of ezrin is visible in the apical zone of the RPE (A), where the microvilli are located, but not in the basal zone (B), see zoom panel at the right. Cones are detected by L/M opsins (magenta) and PNA (red); nuclei are labeled with DAPI (blue). See 3D visualization in Videos S3 and S4. Scale bar, 10 μm. MV, apical microvilli.
(E) Increased ezrin expression is confirmed by an increase in microvilli surface and length (black bar) in the interphotoreceptor matrix of Atxn3 KO retinas in TEM images. The black arrow points to longer MV in Atxn3−/− retinas.
(F–H) The RPE of Atxn3 KO mice displayed delayed phagosome maturation as visualized in (F), a representative TEM image showing more phagosomes in the apical than in the basal zone (white arrows). (G) No apparent difference in the total number of phagosomes per counting area was detected between WT and Atxn3 KO RPEs, but as shown in (H), more phagosomes accumulated in the apical versus the basal zone in Atxn3 KO RPE (51.7% versus 48.3%) compared to WT RPE (33.9% versus 72.1%) (n > 10 images, 3 animals per genotype). Two-way ANOVA test (**p < 0.01).
In RPE cells, early phagosomes (early endosomes) are formed in the apical zone and are transported to the basal zone (late endosomes) by the microtubule motors kinesin-1 and dynein-2 for anterograde and retrograde transport, respectively. Although our proteomic analyses did not identify peptides for all the transport-related proteins (e.g., F-actin or MYO7A were not identified), the interactome analysis of ATXN3 retrieved by RPGenet v2.0 confirmed a close network of interactions with these relevant microtubule transport proteins (Figure 5B). According to a plausible working model, depletion of the ATXN3 protein leads to an increase in F-actin levels, which interacts with the phagosome marker ezrin and the phagosome closure enzyme dynamin-2 (all of them increased), mostly pointing to an increase in phagosome/early endosome formation (Figure 5C). In contrast, the levels of α-tubulin, kinesin-1 subunits (such as KLC1), and the dynein cofactor dynactin-2 were slightly decreased, whereas the levels of dynein-2 were highly increased (Table S2), overall resulting in a delay of the endosome maturation and, consequently, in a decreased number of late endosomes in Atxn3 KO RPE (Figure 5C).
Depletion of ATXN3 Causes a Delay in Phagosome Maturation In Vivo and In Vitro
As predicted by this model, immunodetection in mouse retinal sections confirmed increased ezrin in the apical microvilli of the RPE cells in young (2-month-old) and aged (2-year-old) Atxn3 KO compared to WT mice (Figure 5D; Videos S3 and S4). A more precise visualization by TEM showed an elongation of the apical microvilli in Atxn3 KO RPE compared to that of WT (Figure 5E) and reinforced the implication of RPE dysfunction in the Atxn3 KO retinal phenotype. TEM analysis also revealed a differential distribution of phagosomes in the apical and basal zone of the RPE cells (Figure 5F). Although the WT and Atxn3 KO RPE cells showed a similar number of phagosomes per counting area (Figure 5G), an asymmetrical phagosome distribution between the basal and apical zones was clearly observed in the Atxn3 KO RPE (Figure 5H). The RPE cells of Atxn3 KO retinas showed an increased number of phagosomes in the apical zone and a lower number in the basal zone compared with the WT counterparts (Figures 5F and 5H), thereby indicating a delay in phagosome trafficking from the apical zone (where they are formed) to the basal zone (where fusion to lysosomes occurs) and corroborating our working model. Delayed phagosome maturation in Atxn3 KO RPE could, therefore, result in diminished phagocytosis of the PhR disks, offering an additional explanation for the observed elongation of PhR OS in the retina of these mice.
To confirm the effect of ATXN3 depletion in phagosome maturation, in vitro phagocytosis assays were performed in ARPE19 cells. We induced phagocytosis by adding fluorescent latex beads to the ARPE-19 cell medium (Peng et al., 2017). The formation and maturation of phagosomes containing latex beads in siATXN3 and siSCR-treated ARPE-19 cells were monitored using confocal microscopy. To detect internalization of latex beads within vesicles, cells were immunodetected with early (EEA1) or late (Rab7) endosome markers. In agreement with a delayed maturation of phagosomes in Atxn3 KO RPE cells, ATXN3 knockdown in these human RPE-derived cells caused a consistent and statistically significant increase of early phagocytic endosomes (latex beads within vesicles labeled with EEA1), as well as a significant decrease of late phagocytic endosomes (latex beads in Rab7-positive vesicles) compared to controls (Figures 6A and 6B). Besides, the microtubule network was highly disorganized in cells depleted of ATXN3 (Figures 6C and 6D). In siSCR control cells, dynein-2 localized alongside ordered α-tubulin microtubules, whereas in ATXN3-depleted cells, we observed a marked increase of both F-actin, required to maintain cell shape, and cytoplasmic dynein-2, which did not localize on the disorganized α-tubulin microtubules (Figures 6C and 6D; Videos S5 and S6).
Figure 6. Depletion of ATXN3 in ARPE-19 Cells Causes an Increase of Early Endosomes, Decrease of Late Endosomes, and Alterations in the Cytoskeleton Organization.

(A and B) Immunofluorescent detection of early endosomes (EEA1, red) (A) and late endosomes (RAB7, red) (B) in ATXN3-depleted ARPE-19 cells after incubation with fluorescent latex beads (green) to induce phagocytosis. Quantification of the percentage of the beads within these vesicles shows a significant increase of early endosomes and a decrease of late endosomes. Mann-Whitney test (*p < 0.05, ***p < 0.001, n > 15 cells per group).
(C) Knockdown of ATXN3 in ARPE-19 phagocytic cells alters cytoskeleton organization, as shown by microtubule disorganization (α-tubulin, green), dynein-2 disarray (red), and F-actin increase (phalloidin, gray).
(D) Zoom-in at the central region of images in (C). White arrows point to dynein-2 dots trailing microtubule filaments in control cells (3D visualization in Videos S5 and S6). Nuclei were labeled with DAPI (blue). Scale bar in (A)–(C),10 μm.
Taking together the in vivo and in vitro results, ATXN3 depletion does not affect phagosome formation but rather alters the anterograde/retrograde transport of phagosomes/endosomes, most likely as a result of cytoskeletal network disorganization due to perturbed levels of transport proteins. Thus, ATXN3 seems to function as an arbiter of protein levels of multiple ciliary transport components, plausibly by microtubules, in the vertebrate retina.
DISCUSSION
The vertebrate retinal cup is formed from an evagination of the central nervous system (CNS) during development. Many genes and signaling pathways are, therefore, shared between brain and retina, and pathogenic mutations can both alter specific zones in the brain and cause a retinal or ocular phenotype (e.g., PAX6; Duparc et al., 2006). The retina also shares genetic networks with the cerebellum (Pula et al., 2011), and genes responsible for ataxia are also involved in retinal phenotypes or ciliogenesis. For instance, ATXN7, the protein causing SCA7, interacts with a network of retinal proteins (Kahle et al., 2011), and SCA7 patients show macular degeneration (Hugosson et al., 2009); and ATXN10 (SCA10) or TTBK2 (SCA11) are involved in syndromic ciliopathies that show retinal alterations (van Reeuwijk et al., 2011; Goetz et al., 2012). Although a few studies showed thinning of the nerve fiber layer, GCL, and macular region of MJD/SCA3 patient retinas (Alvarez et al., 2013; Pula et al., 2011; Spina Tensini et.al., 2017), to the best of our knowledge, no investigation has been conducted to comprehensibly assess visual function in MJD/SCA3 patients or to study the function of ATXN3 in retina. Therefore, we sought to characterize the role of ATXN3 in the retina by using genetically amenable animal and cell models.
Focusing on the eye, we found that high knockdown of atxn3 expression in zebrafish embryos caused severe alterations in retinal structure, with no distinguishable layers and disorganization of the microtubule cytoskeleton structure. Mild reduction of Atxn3 protein levels caused a more moderate retinal phenotype in zebrafish, largely characterized by elongation of PhR OSs and mislocalization of cone opsins. Both phenotypes could be rescued by the introduction of WT human ATXN3 mRNA and partially by introduction of the catalytically inactive or the expanded-CAG ATXN3 mutant mRNAs. These results indicate that ATXN3 exerts relevant functional roles both dependently and independently of its DUB activity in the retina. Defining which roles of ATXN3 are DUB dependent or independent deserves further consideration in future studies. The partial rescue by the expanded polyQ protein could be possibly explained by a combination of functional restoration of ATXN3 with concomitant increased cell toxicity due to the longer polyQ tract in the mutant protein. These results are in complete agreement with previous reports showing that both depletion of ATXN3 and overexpression of pathogenic polyQ ATXN3 alleles caused disorganization of cytoskeleton and increased cell death in cultured cells (Neves-Carvalho et al., 2015; Rodrigues et al., 2010).
Particularly, the phenotypic traits observed in the less severe atxn3 knockdown zebrafish embryo/larvae, which affected the OS length and opsin localization in the OS, were highly indicative of alterations in ciliary formation and/or ciliary protein trafficking, respectively (Khanna, 2015; Wheway et al., 2014). The phenotype of lower Atxn3 knockdown in zebrafish is highly consistent with the retinal phenotype of young and aged Atxn3 KO mice. Rigorous assessments of Atxn3 KO and WT retinas by morphometry measurement, confocal microscopy of retinal sections and isolated rods, and TEM showed a consistent elongation of PhR OSs (both in rods and cones), increased length of the CC, and mislocalization of cone (but not rod) opsins in Atxn3 KO retinas compared to controls. The observed morphological phenotype in Atxn3 KO mouse cones reinforced the notion that ATXN3 regulates ciliary microtubule networks and subsequent ciliary protein trafficking. Alteration in ciliary formation and function results in ciliopathies, a generic name that groups syndromic and non-syndromic rare disorders, and the retina is one of the most commonly affected tissues (Bujakowska et al., 2017; Wheway et al., 2014). Syndromic ciliopathies have a very visible multiorgan developmental phenotype, including polydactyly, obesity, heart malformations, and kidney dysfunction, besides neurosensory alterations (Reiter and Leroux, 2017). We have not found any evidence indicating that ATXN3 is involved in syndromic ciliopathies because we have detected only alterations in the cilium of retinal cells (both PhRs and RPE). However, further studies are needed to clarify the precise role of ATXN3 in ciliary formation in other cells/organs.
Additionally, we observed that the increased length of PhR OSs in Atxn3 KO retinas could also indicate phagocytosis dysfunction of the RPE. RPE has an essential physiological role in nursing PhRs by providing nutrients, actively participating in the visual cycle, and daily phagocyting the tips of PhR OSs (Sparrow et al., 2010). Alteration of RPE cells can cause retinal or macular degeneration (Ferrington et al., 2016). Both alterations in RPE phagocytosis and protein trafficking into the PhR OSs may occur in parallel in the retina if microtubule-mediated transport is dysregulated.
Independent proteomic data from neural retina and RPE from Atxn3 KO and control mice highlighted a role for ATXN3 (direct or indirect) in modulating the abundance of proteins involved in the regulation of microtubule formation and microtubule-dependent vesicle-trafficking. The profiles of altered proteins in Atxn3 KO retina and RPE overlapped partially, reflecting a tissue-specific activity of ATXN3. Because DUBs require cofactors and other partners with their own tissue-specific expression pattern in order to recognize their substrates in vivo, the function of ubiquitously expressed ATXN3 appears to be tissue specific. In the neural retina, depletion of this DUB led to decreased levels of KEAP1, which, in turn, resulted in an increase of p62 and potentially subsequent inhibition of HDAC6 activity, leading to an increase of acetylated α-tubulin, the main component of stable microtubules in the cilia. Previous reports showed that ATXN3 interacts with tubulin and microtubules (Rodrigues et al., 2010) and also with HDAC6 and dynein to promote aggresome transport to the microtubule organization center (MTOC) (Burnett and Pittman, 2005). Our in silico analyses of all publicly available data on retinal gene interactomes also support ATXN3 as a regulator of key proteins involved in microtubule polymerization and microtubule-mediated cargo transport. The in vitro assays in human RPE cells and our in vivo observations in mouse and zebrafish models further complement these reports. ATXN3 localizes at ciliary compartments, where it may regulate microtubule polymerization and cilium length. ATXN3 depletion causes an increase in cilium length because there is a decrease in KEAP1 concentration and a subsequent increase in p62, which inactivates HDAC6 deacetylase activity and, in turn, results in increased levels of acetylated α-tubulin and polymerization of ciliary microtubules, thereby altering microtubule-mediated transport. In this context, and likely serving as the main cause of hyperexcitation in the photopic response in the Atxn3 KO mice, altered retrograde ciliary transport in retinal cones could be accounted for the mislocalization of highly expressed photoreception and phototransduction proteins.
In addition, we observed altered OS phagocytosis when ATXN3 was ablated or silenced, with RPE cells showing altered microtubule formation and impaired phagocytic vesicle transport. The number of phagocytic (early) endosomes was normally produced and even enhanced, whereas both the number of late endosomes and levels of the microtubule motor protein KLC1 were decreased in Atxn3 KO retinas, indicating a delay in the microtubule-mediated transport from early to late endosomes. In fact, both Atxn3 KO and Klc1 KO mice display a similar phagosome flux phenotype in the RPE (Jiang et al., 2015). It is intriguing to note that ATXN3 has been recently proposed to transiently regulate autophagosome formation and that ATXN3 depletion also impairs autophagy (Herzog et al., 2019), thereby pointing to a wider role of ATXN3 in regulating vesicle-associated degradation.
On the other hand, a combined mutant phenotype in RPE (differential localization of phagosomes and slow endosome maturation) and PhRs (opsin mislocalization with an accumulation in the lower part of the OS and the IS) is also observed in the Myo7a KO mouse, a model of Usher’s syndrome 1B (a human ciliopathy with severe visual and hearing impairment) (Williams and Lopes, 2011). Myosin7a is the motor protein that transports phagosomes from the F-actin filaments at the apical region to the microtubules in the RPE and is located at the CC in the PhRs. The phenotypic similarities of the Myo7a KO and the Atxn3 KO mouse further support a role of ATXN3 associated with vesicular transport.
Notably, Atxn3 KO retinas show increased levels of interphotoreceptor matrix proteins. Indeed, ATXN3 regulates levels of several integrin subunits in muscle, neuronal, and other mammalian cells (do Carmo Costa et al., 2010; Rodrigues et al., 2010; Neves-Carvalho et al., 2015), further supporting a role of ATXN3 in modulating the protein context in the extracellular space. Considering that a correct composition of the interphotoreceptor matrix is key to the proper function of the macula (Ishikawa et al., 2015), alteration of ATXN3 levels may contribute to the eventual dysfunction of the macula, the retinal region with highest concentration of cones in humans. Because the rodent retina does not have a macula, it is difficult to test this hypothesis in mice. Overall, our results highlight ATXN3 as a good candidate for retinal degeneration, particularly for cone/macular affectation and/or ciliary trafficking regulation. Our proposed model for the phenotype observed in the Atxn3 KO retina at the PhR and RPE cell level is summarized in Figure 7.
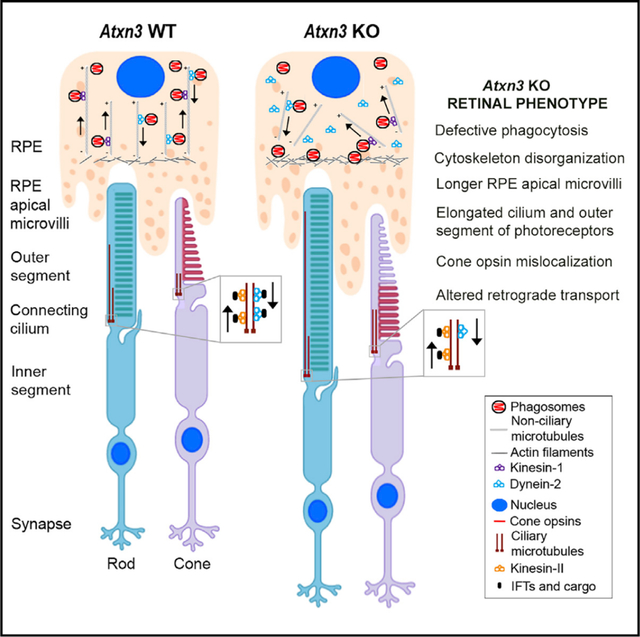
Figure 7. Model for the Molecular Role of ATXN3 in the PhRs and RPE Cells Based on the Phenotype Observed in Atxn3 KO Mouse Retinas.

(A) Rods and cones are PhR cells with a highly specialized sensory cilium, the OS, which contains ordered membranous stacks packed with proteins involved in phototransduction (e.g., rod and cone opsins), which are produced at the IS and must traffic through the ciliary gate. Sensory cilia have two centrioles at the base, from which the corresponding ciliary microtubules grow into the connecting cilium and the axoneme (well within the OS). The intraciliary transport (IFT) of cargo proteins is bidirectional and relies on microtubule-associated motors, kinesins, and dyneins, which are responsible for anterograde and retrograde transport. RPE cells produce microvilli that phagocyte the tips of the OS for renewal of photoreception and phototransduction machinery. Actin filaments, ezrin, and dynamin-2 are involved in RPE phagocytosis. Phagosomes/early endosomes are formed in the apical zone of the RPE, and their maturation process into late endosomes and fusion to the lysosomes at the basal RPE zone involve non-ciliary microtubule-dependent transport (retrograde direction).
(B) Our model proposes that in the retina, ATXN3 modulates the retrograde transport mediated by both ciliary and non-ciliary microtubules by regulation of HDAC6 activity. In the absence or low levels of ATXN3, the following events may occur: (1) the levels of KEAP1 are decreased, which, in turn, increase the levels of p62 and thus inhibits HDAC6; (2) as a result, the pool of acetylated tubulin increases and the polymerization of microtubules is enhanced; (3) the cilium and OS of PhRs increase in length and the retrograde transport is altered, thus resulting in cone opsin mislocalization, phagosome maturation delay, and microtubule disorganization. When overexpressed, ATXN3 localization into the basal body and axoneme increases, where it may enhance microtubule depolymerization and resorption, resulting in shorter cilia (images with biorender.com).
Many studies have been conducted to understand the aggregation properties, stability, and neuronal toxicity of the expanded (CAG)n/polyQ gain-of-function pathogenic alleles of ATXN3. Unfortunately, little is known about the molecular response of cells to ATXN3 loss-of-function mutations. Our data support a diversity of regulatory roles for ATXN3 and indicate a pleiotropic effect of perturbing ATXN3 expression in the retina by altering both PhR ciliary formation and trafficking and PhR OS phagocytosis. In view of the first clinical trials for Huntington disease, based on lowering the expression of the gene in a non-allelic-specific strategy by antisense oligonucleotides (Tabrizi et al., 2019), and the preclinical studies carried out for SCA3 using the same strategy (McLoughlin et al., 2018), it would be prudent to perform additional research to evaluate the impact of reducing the abundance of ATXN3 in several neuronal tissues of animal models before attempting a similar approach in MJD/SCA3 patients.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Gemma Marfany (gmarfany@ub.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
Proteomics data generated in this work is provided in Table S2. The use of RPGenet v2.0 for visualization of interaction networks is of free access (https://compgen.bio.ub.edu/RPGeNet/)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Ethics statement and animal handling
All procedures were performed according to the ARVO statement for the Use of Animals in Ophthalmic and Vision Research, as well as to the regulations of the Animal Care facilities at the University of Barcelona and the University of Michigan. All animal procedures were evaluated and approved by the Animal Research Ethics Committee (CEEA) of the University of Barcelona and the University of Michigan Committee on the Use and Care of Animals (protocols DAAM 6262 and 7185, and PRO00006371, respectively).
Murine retina samples were obtained from 2 month- and 2 year-old Atxn3+/+ and Atxn3−/− mice (mixed sexes, C57BL/6 background) (Reina et al., 2012). Eight to eleven-month-old Atxn3+/+ (n = 4) and Atxn3−/− (n = 4) (mixed sexes) were used for electroretinogram recording. Mice were housed in cages with a maximum number of five animals and maintained at the University of Michigan Unit for Laboratory Animal Medicine in a standard 12-hour light/dark cycle with food and water ad libitum. Animals were anaesthetized with isoflurane and euthanized by decapitation (following the Ethics Committee approved procedures). Zebrafish (Danio rerio) were maintained at 28.5°C on a 14-hour light/10-hour dark cycle at the zebrafish facility of the University of Barcelona. Fertilized eggs were obtained as previously reported (Toulis et al., 2016), and collected at different developmental stages as described in Kimmel et al. (1995). For in vivo imaging, embryos were anaesthetized with MS222, following the approved protocols.
Cell culture
Human ARPE-19 cells (ATCC CRL_2302) were cultured in 1:1 Dulbecco’s Modified Eagle’s Medium (DMEM) (ATCC, Manassas, Virginia) and Ham’s F-12 Nutrient Mix supplemented with 10% Fetal Bovine Serum (FBS) and 1% Penicillin-Streptomycin (all from Life Technologies, Carlsbad, CA) in a 5% CO2 humidified chamber at 37°C. To induce cilia differentiation, ARPE-19 cells were deprived of FBS for 48 h.
METHOD DETAILS
Mouse genotyping by PCR
Mouse Atxn3+/+ and Atxn3−/− genotyping was performed using genomic DNA isolated from tail biopsies at the time of weaning, and genotypes were reconfirmed using DNA extracted from tails collected post-mortem. Total DNA was isolated using the DNeasy Blood and Tissue kit (QIAGEN, Hilden, Germany), following the manufacturer’s instructions with minor modifications. Three primers (Table S1A) were used to genotype WT and knockout alleles in a single PCR reaction.
Tissue dissection and sample preparation
For RNA extraction, zebrafish embryos were collected and preserved in RNAlater® (Ambion/Life Technologies, Carlsbad, CA) at 4°C until processed (maximum 48 hours). For in situ hybridization or immunofluorescence detection, zebrafish embryos were fixed and cryoprotected as reported previously (Toulis et al., 2016). Mouse retinas were dissected and either directly frozen in liquid nitrogen for RNA and protein extraction or fixed and cryoprotected for in situ hybridization and immunofluorescence detection as previously described (Toulis et al., 2016). All the retinas were extracted at the same time point of the day for comparison, as phototransduction proteins shift their localization according to the circadian light-dark cycle.
RNA extraction and RT-PCR
Mouse retinas were homogenized using a Polytron PT1200E homogenizer (Kinematica AG, Lucerne, Switzerland). Total RNA was extracted using the RNeasy® mini kit (QIAGEN, Hilden, Germany) following the manufacturer’s instructions with minor modifications. Reverse transcription of total RNA per sample was performed using the iScript cDNA synthesis kit (Bio-Rad, Hercules, CA) following the manufacturer’s protocol. A pool of 12–20 zebrafish embryos was homogenized using a Polytron PT1200E homogenizer (Kinematica AG, Lucerne, Switzerland). Total RNA was extracted using the High Pure RNA Tissue Kit (Roche Diagnostics, Indianapolis, IN) following the manufacturer’s instructions with minor modifications. Reverse transcription reactions were carried out using the qScript cDNA Synthesis Kit (Quanta BioSciences, Inc., Gaithersburg, MD) following the manufacturer’s protocol. For semiquantitative analysis and riboprobe amplification, the cDNA was amplified according to standard protocols using GoTaq polymerase (Promega, Madison, WI). The expression level quantification and characterization of different mouse isoforms was performed using primers located at the 5′UTR and 3′UTR. Actb (zebrafish) and Gapdh (mouse) genes were used for normalization of gene expression. Primer sequences are listed in Table S1A.
In situ hybridization
In situ hybridizations of zebrafish retinal cryosections and of whole-mount zebrafish embryos were performed as previously described (Thisse and Thisse, 2008; Toulis et al., 2016). In brief, 16–18 μm sections were recovered on commercial Superfrost Plus glass slides (Electron Microscopy Sciences, Hatfield, PA), thawed and rinsed. The name and sequence of all primers are in Table S1A. After overnight hybridization at 55°C with digoxigenin-labeled riboprobes (2 μg/ml) in 50% formamide and 1 mg/ml yeast tRNA in hybridization blocking solution. Sense and antisense riboprobes were generated from cDNA fragments generated by RT-PCR of 400–700 bp cloned into the pGEM-T® Easy Vector (Promega). After hybridization, slides were washed in 50% formamide, 1x SSC, 0.1% Tween-20 at 68°C, equilibrated in MABT (100 mM C4H4O4 pH 7.5, 150 mM NaCl, 0.1% Tween-20) at 37°C, and blocked in Blocking Buffer (100 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% BSA and 0.1% Triton X-100). An anti-digoxigenin-AP conjugate antibody (Roche Diagnostics, Indianapolis, IN) in Blocking Buffer was incubated overnight at 4°C. Sections were washed in MABT prior to adding the BM Purple AP Substrate (Roche Diagnostics, Indianapolis, IN). Antisense and sense ISH staining reactions were processed in parallel. Sections were coverslipped with Fluoprep (Biomérieux, France) and photographed using a Leica DFC Camera connected to MZFLIII (Leica Microsystems, Wetzlar, Germany).
Morpholino injection in zebrafish embryos
To knockdown atxn3 in zebrafish embryos, a morpholino antisense oligonucleotide (MO) that targeted the donor site at the exon 3/intron 3 boundary (MO-ATXN3, 5′-AACGGGTAACTATGACTGACCTGGA-3′) (GeneTools, Philomath, OR) was used to generate aberrant mRNAs by either intron 3 retention or exon 3 skipping. A scrambled (SCR) morpholino was used as a negative control (MO-SCR, 5′-CCTCTTACCTCAGTTACAATTTATA-3′). After optimization of the protocol, we finally injected 65 pL of each MO into 1 to 4-cell embryos as previously described (Toulis et al., 2016), being 72 nmol of MO the final amount per embryo. For phenotypic rescue, the human wild-type ATXN3 plus several mutant cDNAs (the catalytically inactive C14A ATXN3, the MJD mutation Q80 ATXN3) were cloned into the pCS2 expression vector and in vitro transcription was performed using a capped RNA transcription kit (SP6 mMESSAGE mMACHINE®; Ambion/Life Technologies, Carlsbad, CA) following the manufacturer’s instructions. A mixture of mixed MO/mRNA (72 nmol of MO and 30 pg mRNA) was injected as described above. For in vivo eye measurements, anaesthetized embryos at 72 hpf were examined and photographed under the microscope. Eye size measurements from the anterior to the posterior ocular edges were performed using the Fiji software. Significant statistical differences between groups were analyzed by the Mann-Whitney U-test after Bartlett and Shapiro-Wilk tests for equal standard deviation (SD) and normal distribution rendered negative results.
Immunofluorescent detection in retina & cells
For immunofluorescence, 12 μm retinal sections were recovered on commercial Superfrost Plus glass slides (Electron Microscopy Sciences, Hatfield, PA), treated and immunodetected as previously described (Esquerdo et al., 2016). After incubation with the primary and secondary antibodies conjugated to either Alexa Fluor 488, 568 or 647 (Table S1B), sections were mounted in Fluoprep (Biomérieux, Marcy-l’Étoile, France) or ProLong® Gold (Invitrogen, Carlsbad, CA) and analyzed by confocal microscopy (SP2 or SP5, Leica Microsystems, Wetzlar, Germany, or Nikon A1, Tokyo, Japan).
Protein lysates and immunoblotting
Retina lysates from Atxn3+/+ and Atxn3−/− mice were obtained by homogenization and sonication in RIPA lysis buffer (50 mM Tris pH 7.5, 1 mM EDTA, 150 mM NaCl, 0.5% NP40, with protease inhibitors (Complete, Roche Diagnostics, Indianapolis, IN)). Proteins from Atxn3+/+ and Atxn3−/− mouse RPE were extracted following a previously described protocol (Wei et al., 2016). Proteins (30 μg) were resolved in 10%–12.5% SDS-PAGE gels, transferred onto PVDF membranes and blocked with 5%–10% non-fat milk in PBS-T for 1 h, followed by an overnight incubation at 4°C with primary antibodies and a peroxidase-conjugated anti-mouse or anti-rabbit secondary antibody (1:2000) for 1 h. Bands were visualized by treatment either with the ECL-plus reagent (Western Lightning®, PerkinElmer, Waltham, MA) and exposure to autoradiography films or with Lumi-Light Western Blotting Substrate (Roche Diagnostics, Indianapolis, IN) and chemiluminescence detection in a LAS-4000 mini Luminescence Image Analyzer (Fujifilm, Tokyo, Japan). Fiji software (Schindelin et al., 2012) was used to quantify protein band density and α-tubulin or GAPDH were used for normalization. Figure 4 shows representative western blots (n = 4). Uncropped blots are shown in Figure S5D.
Transmission Electron Microscopy (TEM)
Eyes from Atxn3 WT and Atxn3 KO mice were enucleated. The cornea was perforated using a needle to create a small hole, and eyes were immersed in fixative solution (2.5% glutaraldehyde, 2% PFA in 0.1 M cacodylate buffer) for 1 hour at RT, followed by an overnight incubation at 4°C. After several rinses with buffer, eyes were post-fixed in 1% osmium tetroxide, rinsed in double distilled water to remove phosphate salt and then stained en bloc with aqueous 3% uranyl acetate for 1 hour. Eyes were dehydrated in ascending concentrations of ethanol, rinsed two times in propylene oxide, and embedded in epoxy resin. Eye blocks were sectioned into 70 nm ultra-thin sections and stained with uranyl acetate and lead citrate. Negative-stained sections were examined using a JEOL 1400 electron microscope at 80 kV. Images were recorded digitally using a Hamamatsu ORCA-HR digital camera system operated using AMT software (Advanced Microscopy Techniques Corp., Danvers, MA). Fiji software was used for measuring the connecting cilium length of photoreceptors as well as counting phagosomes in the RPE.
Retinal morphometry
Semi-thin sections (0.5 μm) of epoxy-embedded eyes from Atxn3 WT and Atxn3 KO mice (see above) were stained with toluidine blue. Sections of the central retina (containing the optic nerve) were examined and photographed under a stereo microscope (MZFLIII, Leica Microsystems, Wetzlar, Germany). Fiji software was used for measuring the thickness of the retinal layers at 200 μm intervals.
Motor performance evaluation
Groups of Atxn3 KO (n = 5) and Atxn3 WT (n = 8) mice (mixed population, 39–58 weeks of age) were weighed and evaluated for their motor performance in several paradigms blindly to mouse genotype. All groups were generated using littermate male and female mice. Motor function was assessed using tests as previously reported (Costa et al., 2013), including performances on accelerating rotarod, balance beam, and locomotor activity in an open field chamber for 30 min.
Electroretinograms
Electroretinograms (ERGs) were performed in 8–11 month-old WT and ATXN3 KO mouse littermates. Prior to ERG recording, mice were adapted to the dark overnight, anesthetized by intraperitoneal injection of ketamine 100 mg/kg and xylazine 10 mg/kg, and maintained on a heated pad at 37°C. For pupil dilation, 0.5% tropicamide (0.5%) was applied for 10 minutes. Flash-induced ERG responses were recorded from both eyes in response to light stimuli produced with a Ganzfeld stimulator. The recording protocol consisted of dark-adaptation for 20 min, after which scotopic ERG, maximum ERG, and dark-adapted a-wave and b-waves were recorded. The dark-adapted ERG protocol consisted of steps with increasing stimulus strengths. All flashes were presented without background illumination to maintain dark adaptation. Light-adapted ERGs were recorded after light adaptation for 8–10 minutes. The outcome measures were the response amplitudes and implicit time of each ERG component. The ERG recordings complied with International Society for Clinical Electrophysiology of Vision (ISCEV) standards.
Photoreceptor isolation
Retinas from Atxn3 WT and Atxn3 KO mice were dissected as previously described (Toulis et al., 2016). Photoreceptors were isolated using the Neural Tissue Dissociation Kit (MACS, Miltenyi Biotec, Bergisch Gladbach, Germany) following the manufacturer’s instructions with some modifications. Briefly, dissected retinas were washed in PBS, incubated in 1960 μL of enzyme mix 1 at 37°C (20 min) in a water bath, and, after a careful tapping of the tube, the supernatant containing the isolated photoreceptors was placed on commercial Superfrost Plus glass slides (Electron Microscopy Sciences, Hatfield, PA). Photoreceptors were fixed at −20°C (10 min) with cold methanol and assessed for specific protein expression by immunofluorescence as described above.
In vivo mouse subretinal DNA electroporation
pEGFP vector or pEGFP-ATXN3 WT was diluted in PBS (6 μg/μl) and mixed with fast green dye (Fisher Scientific, Waltham, MA) (0.1%). Atxn3 KO neonatal mouse pups (P1) were subjected to subretinal electroporation of the plasmid solutions as described previously (López-Begines et al., 2018). Transient transgenic retinas were collected and processed at postnatal day 25–30 for immunohistochemistry as mentioned above.
Liquid chromatography-mass spectrometry
Retinas and RPE of Atxn3 WT and Atxn3 KO mice (1-year-old, 5 animals per group, 3 females/ 2 males) were separately dissected. Proteins were extracted and quantified as described above. Briefly, protein digestion and isobaric labeling of 2 technical replicate pools of 100 μg of either retinal or RPE protein lysates per mouse group (20 μg/ mouse) was performed using a TMT (tandem mass tag) 10-plex kit (Thermo Fisher Scientific, Waltham, MA) following the manufacturer’s instructions, in preparation for LC-MS/MS analysis as described elsewhere (Tank et al., 2018). In order to obtain superior quantitation accuracy, we employed multinotch-MS3 (McAlister et al., 2014), which minimizes the reporter ion ratio distortion. Orbitrap Fusion (Thermo Fisher Scientific, Waltham, MA) and RSLC Ultimate 3000 nano-UPLC (Dionex, Sunnyvale, CA) was used to acquire the data, and Proteome Discoverer (v2.1; Thermo Fisher, Waltham, MA), for data analysis, as described (Tank et al., 2018). MS2 spectra were searched against SwissProt mouse protein database (release 2017–10-03; 25570 sequences), and only identified proteins and peptides that passed ≤ 1% FDR threshold were retained. Quantitation was performed using high-quality MS3 spectra using the Reporter Ion Quantifier Node of Proteome Discoverer (Average signal-to-noise ratio of 10 and < 30% isolation interference) (Tank et al., 2018). The volcano plot of the proteomic analysis was designed using GraphPad Prism 7.03 (San Diego, CA, USA) by plotting the −log10 (P value) against the log2 ratio (fold change). Gene Ontology (GO) analysis was performed using the web-tools DAVID (https://david.ncifcrf.gov/), AmiGO (http://amigo.geneontology.org/amigo) and PANTHER classification system (http://www.pantherdb.org/).
Transfection and assays in cultured cells
For overexpression experiments, the following constructs were used: pEGFP vector, pEGFP-ATXN3 WT and pEGFP-ATXN3 C14A. For silencing experiments, we used four different siRNAs targeting human ATXN3 (siATXN3: J-012013–05-0002, J-012013–06-0002, J-012013–07-0002, J-012013–08-0002) and two control siRNAs scramble (siSCR) sequences (D-001810–01-05, D-001810–02-05) (all from Dharmacon, Lafayette, CO). For immunohistochemistry and cilia formation imaging, human ARPE-19 cells (1×105/well) were seeded onto poly-L-lysine-coated coverslips and co-transfected with either 1 μg/well of each GFP-ATXN3 constructs for overexpression assays, using Lipotransfectine (Niborlab, Guillena, Spain). Growth medium was replaced 5 hours post-transfection with serum-free medium to induce cilia differentiation. After 48 h of transfection, cells were fixed and used for immunofluorescence assays as described above. For ATXN3 silencing, human ARPE-19 cells (1×105/well) were transfected with 10 nM of each siRNA using Lipofectamine RNAiMAX Transfection Reagent (Invitrogen, Carlsbad, CA) following the manufacturer’s instructions. After 48 h of transfection, cells were either used for immunodetection assays (if grown on L-poly-lysine coverslips) or collected for protein extraction and subsequent immunoblotting analysis. For phagocytosis assays, we used a protocol described elsewhere (Peng et al., 2017). Briefly, ARPE-19 cells were seeded and transfected with either siSCR or siATXN3. After 48 h, 5 × 106 latex beads (Latex beads, amine-modified polystyrene, fluorescent Orange, L9904, Sigma-Aldrich, San Luis, MO) were added per well. After incubation, the coverslips were washed with PBS to remove unbound latex beads. Immunocytochemistry with appropriate antibodies to label early and late endosomes was performed 24 h after addition of latex beads. Human HEK293T cells, acquired from ATCC (CRL_3216), were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) (ATCC, Manassas, Virginia) supplemented with 10% Fetal Bovine Serum (FBS) and 1% Penicillin-Streptomycin (all from Life Technologies, Carlsbad, CA) in a 5% CO2 humidified chamber at 37°C. For protein half-life analysis, cells were treated with 150 μM cycloheximide (CHX, Sigma-Aldrich, San Luis, MO) 36 h after transfection for 0, 1, 2, 3 and 4 h.
Imaging analysis and 3D visualization
Fiji software (Schindelin et al., 2012) was used for cilia length measurements, and Imaris software for 3D visualization and video recording (Oxford Instruments, Zurich, Switzerland). To plot the fluorescence intensity colocalization profiles, single confocal planes centered in either the region of the basal body or the cilium axoneme were used.
Network visualization
To identify new interaction partners of ATXN3 and locate the identified potential substrates of ATXN3 (revealed by proteomics) in a genetic network of interactions, we used the recently updated and versatile webapp of retinal relevant genes, RPGenet v2.0 (https://compgen.bio.ub.edu/RPGeNet/; Arenas-Galnares et al., 2019).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical significance of data, equal standard deviation (SD) and normal distribution were first assessed using Bartlett and Shapiro-Wilk tests. If data followed a normal distribution and showed homogeneity of variance, two-way ANOVA or One-sample t test was used for statistical significance analysis. When data did not follow a normal distribution, non-parametrical Mann-Whitney U-test or Wilcoxon signed-rank test were applied. Analysis was performed using GraphPad Prism 7.03 (San Diego, CA, USA) or Statgraphics Centurion XVII Statistics software (The Plains, VA, USA).
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Rabbit anti-Active Caspase-3 | BD PharMingen (San Jose, CA) | Cat# 559565; RRID:AB_397274 |
| Mouse anti-Acetylated α-tubulin | Sigma Aldrich (San Luis, MO) | Cat# T6793; RRID:AB_477585 |
| Rabbit anti-Acetylated α-tubulin | Abcam (Cambridge, UK) | Cat# ab179484 |
| Lectin PNA | Life Technologies (Carlsbad, CA) | Cat# L32460 |
| Mouse anti-Rhodopsin (1D4) | Abcam (Cambridge, UK) | Cat# ab5417; RRID:AB_304874 |
| Rabbit anti-Red/Green (L/M) Opsin | Merck Millipore (Burlington, MA) | Cat# AB5405; RRID:AB_177456 |
| Rabbit anti-Ataxin-3 (MJD) | Kind gift from Dr. Paulson | N/A |
| Rabbit anti-GCAP1 | Kind gift from Dr. Mendez | N/A |
| Mouse anti-Ezrin | Abcam (Cambridge, UK) | Cat# ab4069; RRID:AB_304261 |
| Rabbit anti-Dynamin-2 | ThermoFisher Scientific (Waltham, MA) | Cat# PA1–661; RRID:AB_2293040 |
| Mouse anti-GRK1 (D11) | Abcam (Cambridge, UK) | Cat# ab2776; RRID:AB_303289 |
| Rabbit anti-Cone Arrestin | Merck Millipore (Burlington, MA) | Cat# AB15282; RRID:AB_1163387 |
| Rabbit anti-PDE6C | Abgent (San Diego, CA) | Cat# AP9728c; RRID:AB_10616160 |
| Mouse anti-HA | BioLegend (San Diego, CA) | Cat# 901502; RRID:AB_2565007 |
| Mouse anti-GAPDH | Abcam (Cambridge, UK) | Cat# ab9484; RRID:AB_307274 |
| Rabbit anti-KEAP1 | Proteintech (Rosemont, IL) | Cat# 10503–2-AP; RRID:AB_2132625 |
| Rabbit anti-CUL3 | Proteintech (Rosemont, IL) | Cat# 11107–1-AP; RRID:AB_2086429 |
| Mouse anti-SQSTM1/p62 | Abcam (Cambridge, UK) | Cat# ab56416; RRID:AB_945626 |
| Mouse anti-α-tubulin | Sigma Aldrich (San Luis, MO) | Cat# T5168; RRID:AB_477579 |
| Rat anti-NRF2 | Merck Millipore (Burlington, MA) | Cat# MABE1799 |
| Rabbit anti-GFP | Abcam (Cambridge, UK) | Cat# ab290; RRID:AB_303395 |
| Rabbit anti-EEA1 | Abcam (Cambridge, UK) | Cat# ab2900; RRID:AB_2262056 |
| Mouse anti-RAB7 | Abcam (Cambridge, UK) | Cat# ab50533; RRID:AB_882241 |
| Rabbit anti-Dynein-2 | Abcam (Cambridge, UK) | Cat# ab122525; RRID:AB_11133051 |
| Alexa Fluor 633 Phalloidin | Life Technologies (Carlsbad, CA) | Cat# A22284 |
| Mouse anti-γ-tubulin | Sigma Aldrich (San Luis, MO) | Cat# T6557; RRID:AB_477584 |
|
| ||
| Experimental Models: Cell Lines | ||
|
| ||
| Human HEK293T | ATCC | Cat# CRL_3216; RRID:CVCL_0063 |
| Human ARPE-19 | ATCC | Cat# CRL_2302; RRID:CVCL_0145 |
|
| ||
| Experimental Models: Organisms/Strains | ||
|
| ||
| Mouse (Mus musculus): Atxn3 KO | Reina et al., 2012 | N/A |
| Zebrafish (Danio rerio): atxn3 WT | This study | N/A |
| Zebrafish (Danio rerio): atxn3 KD | This study | N/A |
| Mouse (Mus musculus): Atxn3 WT | Reina et al., 2012 | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| Morpholino antisense oligonucleotide (MO-ATXN3) | GeneTools (Philomath, OR) | N/A |
| Morpholino antisense oligonucleotide (MO-SCR) | GeneTools (Philomath, OR) | N/A |
| siATXN3 | Dharmacon (Lafayette, CO) | J-012013–05–0002 |
| siATXN3 | Dharmacon (Lafayette, CO) | J-012013–06–0002 |
| siATXN3 | Dharmacon (Lafayette, CO) | J-012013–07–0002 |
| siATXN3 | Dharmacon (Lafayette, CO) | J-012013–08–0002 |
| SiSCR | Dharmacon (Lafayette, CO) | D-001810–01–05 |
| SiSCR | Dharmacon (Lafayette, CO) | D-001810–02–05 |
| Primer DNA oligos | Merck KGaA (Germany) (for sequences, see Table S1) | N/A |
|
| ||
| Software and Algorithms | ||
|
| ||
| GraphPad Prism (7.03) | GraphPad Software (San Diego, CA) | N/A |
| Statgraphics Centurion XVII Statistics | Statgraphics Technologies, Inc. (The Plains, VA) | N/A |
| Fiji software | Schindelin et al., 2012 | N/A |
| Imaris software | Oxford Instruments plc (UK) | N/A |
| Proteome Discoverer (v2.1) | Thermo Fisher (Waltham, MA) | N/A |
Highlights.
Atxn3-depleted zebrafish and mice show elongated photoreceptor outer segments
Atxn3 ablation causes mislocalization of cone opsins and cone hyperexcitation
ATXN3 regulates cilium length and ciliary retrograde transport
ATXN3 controls cytoskeleton organization and phagocytosis in RPE cells
ACKNOWLEDGMENTS
V.T. was a fellow of MINECO (BES-2014-068639, with grants EEBB-I-16-11823 and EEBB-I-17-12664) (Ministerio de Economía, Industria y Competitividad, Spain). This research was supported by grants SAF2013-49069-C2-1-R and SAF2016-80937-R (Ministerio de Economía y Competitividad), 2017 SGR 738 (Generalitat de Catalunya), and La Marató TV3 (Project Marató 201417-30-31-32) to G.M. This research was also supported by Becky Babcox Research Fund/pilot research (award G015617), the Protein Folding Disease Initiative (University of Michigan) - Fund for Proteomics Studies, and University of Michigan discretionary funds to M.d.C.C. The authors thank Henry L. Paulson for providing the Atxn3 KO mice and the anti-MJD antibody; Naila S. Ashraf for technical support; Pennelope Blakely Kunkle and Sasha Meshinchi for TEM assistance; Cagri Besirli, Jungyu Yao, and David Zack for providing access to subretinal injection equipment; Venkatesha Basrur at the Proteomics Resource Facility (Dept. of Pathology, University of Michigan) for the proteomics analysis; and Sergio Castillo-Lara for helpful discussions on network analysis and webapp implementation.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108360.
REFERENCES
- Alvarez G, Rey A, Sanchez-Dalmau FB, Muñoz E, Ríos J, and Adán A (2013). Optical coherence tomography findings in spinocerebellar ataxia-3. Eye (Lond.) 27, 1376–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arenas-Galnares R, Castillo-Lara S, Toulis V, Boloc D, Gonzàlez-Duarte R, Marfany G, and Abril JF (2019). RPGeNet v2.0: expanding the universe of retinal disease gene interactions network. Database (Oxford) 2019, baz120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashkenazi A, Bento CF, Ricketts T, Vicinanza M, Siddiqi F, Pavel M, Squitieri F, Hardenberg MC, Imarisio S, Menzies FM, and Rubinsztein DC (2017). Polyglutamine tracts regulate beclin 1-dependent autophagy. Nature 545, 108–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blount JR, Tsou W-L, Ristic G, Burr AA, Ouyang M, Galante H, Scaglione KM, and Todi SV (2014). Ubiquitin-binding site 2 of ataxin-3 prevents its proteasomal degradation by interacting with Rad23. Nat. Commun. 5, 4638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonilha VL, Rayborn ME, Bhattacharya SK, Gu X, Crabb JS, Crabb JW, and Hollyfield JG (2006). The Retinal Pigment Epithelium Apical Microvilli and Retinal Function. In Retinal Degenerative Diseases (Springer US; ), pp. 519–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bujakowska KM, Liu Q, and Pierce EA (2017). Photoreceptor Cilia and Retinal Ciliopathies. Cold Spring Harb. Perspect. Biol. 9, a028274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnett BG, and Pittman RN (2005). The polyglutamine neurodegenerative protein ataxin 3 regulates aggresome formation. Proc. Natl. Acad. Sci. USA 102, 4330–4335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnett B, Li F, and Pittman RN (2003). The polyglutamine neurodegenerative protein ataxin-3 binds polyubiquitylated proteins and has ubiquitin protease activity. Hum. Mol. Genet. 12, 3195–3205. [DOI] [PubMed] [Google Scholar]
- Chakarova CF, Papaioannou MG, Khanna H, Lopez I, Waseem N, Shah A, Theis T, Friedman J, Maubaret C, Bujakowska K, et al. (2007). Mutations in TOPORS cause autosomal dominant retinitis pigmentosa with perivascular retinal pigment epithelium atrophy. Am. J. Hum. Genet. 81, 1098–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang AP, Beck JS, Yen H-J, Tayeh MK, Scheetz TE, Swiderski RE, Nishimura DY, Braun TA, Kim K-YA, Huang J, et al. (2006). Homozygosity mapping with SNP arrays identifies TRIM32, an E3 ubiquitin ligase, as a Bardet-Biedl syndrome gene (BBS11). Proc. Natl. Acad. Sci. USA 103, 6287–6292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa Mdo.C., Luna-Cancalon K, Fischer S, Ashraf NS, Ouyang M, Dharia RM, Martin-Fishman L, Yang Y, Shakkottai VG, Davidson BL, et al. (2013). Toward RNAi therapy for the polyglutamine disease Machado-Joseph disease. Mol. Ther. 21, 1898–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullinan SB, Gordan JD, Jin J, Harper JW, and Diehl JA (2004). The Keap1-BTB protein is an adaptor that bridges Nrf2 to a Cul3-based E3 ligase: oxidative stress sensing by a Cul3-Keap1 ligase. Mol. Cell. Biol. 24, 8477–8486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Hollander AI, Black A, Bennett J, and Cremers FPM (2010). Lighting a candle in the dark: advances in genetics and gene therapy of recessive retinal dystrophies. J. Clin. Invest. 120, 3042–3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- do Carmo Costa M, Bajanca F, Rodrigues AJ, Tomé RJ, Corthals G, Macedo-Ribeiro S, Paulson HL, Logarinho E, and Maciel P (2010). Ataxin-3 plays a role in mouse myogenic differentiation through regulation of integrin subunit levels. PLoS One 5, e11728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duparc R-H, Boutemmine D, Champagne M-P, Tétreault N, and Bernier G (2006). Pax6 is required for delta-catenin/neurojugin expression during retinal, cerebellar and cortical development in mice. Dev. Biol. 300, 647–655. [DOI] [PubMed] [Google Scholar]
- Durcan TM, Kontogiannea M, Thorarinsdottir T, Fallon L, Williams AJ, Djarmati A, Fantaneanu T, Paulson HL, and Fon EA (2011). The Machado-Joseph disease-associated mutant form of ataxin-3 regulates parkin ubiquitination and stability. Hum. Mol. Genet. 20, 141–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esquerdo M, Grau-Bové X, Garanto A, Toulis V, Garcia-Monclús S, Millo E, López-Iniesta MJ, Abad-Morales V, Ruiz-Trillo I, and Marfany G (2016). Expression Atlas of the Deubiquitinating Enzymes in the Adult Mouse Retina, Their Evolutionary Diversification and Phenotypic Roles. PLoS One 11, e0150364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evert BO, Araujo J, Vieira-Saecker AM, de Vos RAI, Harendza S, Klockgether T, and Wüllner U (2006). Ataxin-3 represses transcription via chromatin binding, interaction with histone deacetylase 3, and histone deacetylation. J. Neurosci. 26, 11474–11486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrington DA, Sinha D, and Kaarniranta K (2016). Defects in retinal pigment epithelial cell proteolysis and the pathology associated with age-related macular degeneration. Prog. Retin. Eye Res. 51, 69–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman JS, Ray JW, Waseem N, Johnson K, Brooks MJ, Hugosson T, Breuer D, Branham KE, Krauth DS, Bowne SJ, et al. (2009). Mutations in a BTB-Kelch protein, KLHL7, cause autosomal-dominant retinitis pigmentosa. Am. J. Hum. Genet. 84, 792–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetz SC, Liem KF Jr., and Anderson KV (2012). The spinocerebellar ataxia-associated gene Tau tubulin kinase 2 controls the initiation of ciliogenesis. Cell 151, 847–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog LK, Kevei E, Marchante R, Böttcher C, Bindesbøll C, Lystad AH, Pfeiffer A, Gierisch ME, Salomons FA, Simonsen A, et al. (2019). The Machado–Joseph disease deubiquitylase ataxin-3 interacts with LC3C/GABARAP and promotes autophagy. Aging Cell 19, e13051. 10.1111/acel.13051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoon M, Okawa H, Della Santina L, and Wong ROL (2014). Functional architecture of the retina: development and disease. Prog. Retin. Eye Res. 42, 44–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugosson T, Gränse L, Ponjavic V, and Andréasson S (2009). Macular dysfunction and morphology in spinocerebellar ataxia type 7 (SCA 7). Ophthalmic Genet. 30, 1–6. [DOI] [PubMed] [Google Scholar]
- Ishikawa M, Sawada Y, and Yoshitomi T (2015). Structure and function of the interphotoreceptor matrix surrounding retinal photoreceptor cells. Exp. Eye Res. 133, 3–18. [DOI] [PubMed] [Google Scholar]
- Izawa I, Goto H, Kasahara K, and Inagaki M (2015). Current topics of functional links between primary cilia and cell cycle. Cilia 4, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M, Esteve-Rudd J, Lopes VS, Diemer T, Lillo C, Rump A, and Williams DS (2015). Microtubule motors transport phagosomes in the RPE, and lack of KLC1 leads to AMD-like pathogenesis. J. Cell Biol. 210, 595–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen T, and Lamark T (2011). Selective autophagy mediated by autophagic adapter proteins. Autophagy 7, 279–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahle JJ, Gulbahce N, Shaw CA, Lim J, Hill DE, Barabási A-L, and Zoghbi HY (2011). Comparison of an expanded ataxia interactome with patient medical records reveals a relationship between macular degeneration and ataxia. Hum. Mol. Genet. 20, 510–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna H (2015). Photoreceptor Sensory Cilium: Traversing the Ciliary Gate. Cells 4, 674–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, and Schilling TF (1995). Stages of embryonic development of the zebrafish. Dev. Dyn. 203, 253–310. [DOI] [PubMed] [Google Scholar]
- López-Begines S, Plana-Bonamaisó A, and Méndez A (2018). Molecular determinants of Guanylate Cyclase Activating Protein subcellular distribution in photoreceptor cells of the retina. Sci. Rep. 8, 2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlister GC, Nusinow DP, Jedrychowski MP, Wühr M, Huttlin EL, Erickson BK, Rad R, Haas W, and Gygi SP (2014). MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal. Chem. 86, 7150–7158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin ME, and Dryja TP (2002). Ocular findings in spinocerebellar ataxia 7. Arch. Ophthalmol. 120, 655–659. [PubMed] [Google Scholar]
- McLoughlin HS, Moore LR, Chopra R, Komlo R, McKenzie M, Blumenstein KG, Zhao H, Kordasiewicz HB, Shakkottai VG, and Paulson HL (2018). Oligonucleotide therapy mitigates disease in spinocerebellar ataxia type 3 mice. Ann. Neurol. 84, 64–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neves-Carvalho A, Logarinho E, Freitas A, Duarte-Silva S, do Carmo Costa M, Silva-Fernandes A, Martins M, Serra SC, Lopes AT, Paulson HL, et al. (2015). Dominant negative effect of polyglutamine expansion perturbs normal function of ataxin-3 in neuronal cells. Hum. Mol. Genet. 24, 100–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijman SMB, Luna-Vargas MPA, Velds A, Brummelkamp TR, Dirac AMG, Sixma TK, and Bernards R (2005). A genomic and functional inventory of deubiquitinating enzymes. Cell 123, 773–786. [DOI] [PubMed] [Google Scholar]
- Paulson HL, Shakkottai VG, Clark HB, and Orr HT (2017). Polyglutamine spinocerebellar ataxias - from genes to potential treatments. Nat. Rev. Neurosci. 18, 613–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng RM, Hong J, Jin Y, Sun YZ, Sun YQ, and Zhang P (2017). Mertk gene expression and photoreceptor outer segment phagocytosis by cultured rat bone marrow mesenchymal stem cells. Mol. Vis. 23, 8–19. [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer A, Luijsterburg MS, Acs K, Wiegant WW, Helfricht A, Herzog LK, Minoia M, Böttcher C, Salomons FA, van Attikum H, and Dantuma NP (2017). Ataxin-3 consolidates the MDC1-dependent DNA double-strand break response by counteracting the SUMO-targeted ubiquitin ligase RNF4. EMBO J. 36, 1066–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pula JH, Towle VL, Staszak VM, Cao D, Bernard JT, and Gomez CM (2011). Retinal Nerve Fibre Layer and Macular Thinning in Spinocerebellar Ataxia and Cerebellar Multisystem Atrophy. Neuroophthalmology 35, 108–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran J, Yang Y, Li D, Liu M, and Zhou J (2015). Deacetylation of α-tubulin and cortactin is required for HDAC6 to trigger ciliary disassembly. Sci. Rep. 5, 12917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reina CP, Nabet BY, Young PD, and Pittman RN (2012). Basal and stress-induced Hsp70 are modulated by ataxin-3. Cell Stress Chaperones 17, 729–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter JF, and Leroux MR (2017). Genes and molecular pathways underpinning ciliopathies. Nat. Rev. Mol. Cell Biol. 18, 533–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ristic G, Sutton JR, Libohova K, and Todi SV (2018). Toxicity and aggregation of the polyglutamine disease protein, ataxin-3 is regulated by its binding to VCP/p97 in Drosophila melanogaster. Neurobiol. Dis. 116, 78–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues A-J, do Carmo Costa M, Silva T-L, Ferreira D, Bajanca F, Logarinho E, and Maciel P (2010). Absence of ataxin-3 leads to cytoskeletal disorganization and increased cell death. Biochim. Biophys. Acta 1803, 1154–1163. [DOI] [PubMed] [Google Scholar]
- Scaglione KM, Zavodszky E, Todi SV, Patury S, Xu P, Rodríguez-Lebrón E, Fischer S, Konen J, Djarmati A, Peng J, et al. (2011). Ube2w and ataxin-3 coordinately regulate the ubiquitin ligase CHIP. Mol. Cell 43, 599–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparrow JR, Hicks D, and Hamel CP (2010). The retinal pigment epithelium in health and disease. Curr. Mol. Med. 10, 802–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spina Tensini F, Sato MT, Shiokawa N, Ashizawa T, and Teive HAG (2017). A Comparative Optical Coherence Tomography Study of Spinocerebellar Ataxia Types 3 and 10. Cerebellum 16, 797–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaroop A, Kim D, and Forrest D (2010). Transcriptional regulation of photoreceptor development and homeostasis in the mammalian retina. Nat. Rev. Neurosci. 11, 563–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabrizi SJ, Ghosh R, and Leavitt BR (2019). Huntingtin Lowering Strategies for Disease Modification in Huntington’s Disease. Neuron 101, 801–819. [DOI] [PubMed] [Google Scholar]
- Tank EM, Figueroa-Romero C, Hinder LM, Bedi K, Archbold HC, Li X, Weskamp K, Safren N, Paez-Colasante X, Pacut C, et al. (2018). Abnormal RNA stability in amyotrophic lateral sclerosis. Nat. Commun. 9, 2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thisse C, and Thisse B (2008). High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat. Protoc. 3, 59–69. [DOI] [PubMed] [Google Scholar]
- Toulis V, Garanto A, and Marfany G (2016). Combining Zebrafish and Mouse Models to Test the Function of Deubiquitinating Enzyme (Dubs) Genes in Development: Role of USP45 in the Retina. Methods Mol. Biol. 1449, 85–101. [DOI] [PubMed] [Google Scholar]
- Tsou W-L, Ouyang M, Hosking RR, Sutton JR, Blount JR, Burr AA, and Todi SV (2015). The deubiquitinase ataxin-3 requires Rad23 and DnaJ-1 for its neuroprotective role in Drosophila melanogaster. Neurobiol. Dis. 82, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Reeuwijk J, Arts HH, and Roepman R (2011). Scrutinizing ciliopathies by unraveling ciliary interaction networks. Hum. Mol. Genet. 20, R149–R157. [DOI] [PubMed] [Google Scholar]
- Wang H, Ying Z, and Wang G (2012). Ataxin-3 regulates aggresome formation of copper-zinc superoxide dismutase (SOD1) by editing K63-linked polyubiquitin chains. J. Biol. Chem. 287, 28576–28585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei H, Xun Z, Granado H, Wu A, and Handa JT (2016). An easy, rapid method to isolate RPE cell protein from the mouse eye. Exp. Eye Res. 145, 450–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheway G, Parry DA, and Johnson CA (2014). The role of primary cilia in the development and disease of the retina. Organogenesis 10, 69–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams DS, and Lopes VS (2011). The many different cellular functions of MYO7A in the retina. Biochem. Soc. Trans. 39, 1207–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J, Seibenhener ML, Calderilla-Barbosa L, Diaz-Meco M-T, Moscat J, Jiang J, Wooten MW, and Wooten MC (2013). SQSTM1/p62 interacts with HDAC6 and regulates deacetylase activity. PLoS One 8, e76016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi Z, Ouyang J, Sun W, Xiao X, Li S, Jia X, Wang P, and Zhang Q (2019). Biallelic mutations in USP45, encoding a deubiquitinating enzyme, are associated with Leber congenital amaurosis. J. Med. Genet. 56, 325–331. [DOI] [PubMed] [Google Scholar]
- Zhong X, and Pittman RN (2006). Ataxin-3 binds VCP/p97 and regulates retrotranslocation of ERAD substrates. Hum. Mol. Genet. 15, 2409–2420. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Proteomics data generated in this work is provided in Table S2. The use of RPGenet v2.0 for visualization of interaction networks is of free access (https://compgen.bio.ub.edu/RPGeNet/)