

Abstract
The ARID1 proteins are mutually exclusive subunits of the BRG1/BRM-associated factor (BAF) complexes that play an important role in chromatin remodeling and regulate many fundamental cell functions. The role of ARID1s is well defined as a tumor-suppressive. The cancer cells evolve different mechanisms to downregulate ARID1s and inactivate their functions. ARID1s are frequently mutated in human cancer. The recent findings of ARID1A/B downregulation at transcriptional and translational levels along with their low levels in human cancers indicate the significance of regulatory mechanisms of ARID1s in cancers. In this review, we present the current knowledge on the regulation and alterations of ARID1 protein expression in human cancers and indicate the importance of regulators of ARID1s as a prognostic marker and in potential therapeutic strategies.
Keywords: : ARID1A, ARID1B, BAF, cancer, chromatin remodelers, post-translational modification, SWI/SNF, transcriptional regulation, tumorigenesis
The ARID1A and ARID1B are paralogs with mutually exclusive properties [1] and are members of the ARID family of DNA-binding proteins [2]. They are components of the ATP-dependent SWItch/sucrose non-fermentable (SWI/SNF) chromatin remodeling complex, which control various processes by modulating the accessibility of DNA to transcriptional regulators resulting in activation or repression of target genes [3,4].
The SWI/SNF complexes have a highly variable protein composition and consist of 12–15 subunits [3]. These subunits are encoded by 29 genes and several of them have mutually exclusive characteristics (Table 1). This extensive diversity of SWI/SNF complexes, generated by different combinations of mutually exclusive subunits, indicates their distinct roles in human tissues [4]. Human SWI/SNF complexes contain one of the two mutually exclusive ATPase subunits (SMARCA2 or SMARCA4) that catalyzes a hydrolysis of ATP; several core subunits that contribute to their catalytic activity; and variable accessory subunits that endow target specificity. Based on their compositions, SWI/SNF complexes are divided into three groups: canonical BRG1/BRM-associated factor (BAF) complexes, polybromo-associated BAF (PBAF) complexes and the most recently discovered noncanonical GLTSCR1/1L-associated BAF (GBAF) [5]. The SWI/SNF complexes frequently localize at enhancers and promoters enriched in acetylation marks such as H3K27ac, which is associated with an open chromatin site and an active transcription. The different subfamilies of SWI/SNF complexes differentially target enhancers and promoters with BAF complexes are enriched at enhancers, while PBAF and GBAF complexes occur most strongly at promoters [6,7].
Table 1. . Summary of the BRG1/BRM-associated factor complex subunits.
| HUGO symbol |
Alternative names | Function | Relationship |
|---|---|---|---|
| Accessory subunits | |||
| ARID1A | BAF250A | Accessory subunit | Mutually exclusive Paralogous |
| ARID1B | BAF250B | Accessory subunit | |
| PHF10 | BAF45A | Accessory subunit | Mutually exclusive Paralogous |
| DPF1 | BAF45B | Accessory subunit | |
| DPF2 | BAF45C | Accessory subunit | |
| DPF3 | BAF45D | Accessory subunit | |
| BCL7A | Accessory subunit | ||
| BCL7B | Accessory subunit | ||
| BCL7C | Accessory subunit | ||
| BCL11A | Accessory subunit | ||
| BCL11B | Accessory subunit | ||
| BRD9 | Accessory subunit | ||
| ATPase module | |||
|---|---|---|---|
| SMARCA2 | BRM | ATPase catalytic subunit | Mutually exclusive Paralogous |
| SMARCA4 | BRG1 | ATPase catalytic subunit | |
| ACTL6A | BAF53A | Actin-related | Mutually exclusive Paralogous |
| ACTL6B | BAF53B | Actin-related | |
| ACTB | β-Actin | Actin-related | |
| SS18/L1 | -/CREST | ||
| Core module | |||
|---|---|---|---|
| SMARCB1 | BAF47, SNF5, INI1 | Core subunit | |
| SMARCC1 | BAF155 | Core subunit | Paralogous |
| SMARCC2 | BAF170 | Core subunit | |
| SMARCD1 | BAF60A | Core subunit | Mutually exclusive Paralogous |
| SMARCD2 | BAF60B | Core subunit | |
| SMARCD3 | BAF60C | Core subunit | |
| SMARCE1 | BAF57 | Core subunit | |
ARID1A and ARID1B associate with the BAF subfamily of SWI/SNF complexes, and their homolog, ARID2, with the PBAF subfamily. These ARID-containing proteins are not specific to the ATPases and associate with either SMARCA2 or SMARCA4; thus, increasing the number of distinct subunit combinations known to be present in cells [1,8]. ARID1 proteins are the largest subunits of the BAF complexes and are essential for their assembly [9,10]. ARID1A and ARID1B are often coexpressed, have a predominantly nuclear subcellular localization and a broad tissue distribution. In cells, the ARID1A/ARID1B ratio is approximately 3.5:1, and SMARCA4 is distributed proportionally between the two ARID1 subunits [1].
The SWI/SNF complexes are frequently disrupted in human diseases, most notably in cancer and neurodevelopmental disorders [3,4,11]. Genes encoding subunits of SWI/SNF complexes are collectively mutated in approximately 20% of all human cancers [11]. Mutually exclusive ARID subunits (ARID1A, ARID1B and ARID2) are among the most frequently mutated subunits in human diseases and they are mutated with different frequencies in different cancer types [2–4,11–14]. Among them, ARID1A is the most frequently mutated in cancer, while ARID1B is the most frequently mutated in intellectual disability disorders. Most mutations in genes encoding ARID1 proteins include nonsense, frameshift and insertion/deletion mutations, and result in the protein loss (Figure 1) [11–16]. Although mutations in ARID1A and ARID1B can occur within the same tumors, recent studies demonstrated that ARID1B has synthetic lethality with ARID1A in cancer cell lines and fibroblasts [3]. The functional basis for the synthetic lethal relationship between ARID1A and ARID1B has not yet been determined.
Figure 1. . Regulation of the ARID1-containing proteins in cancer.

The downregulation of ARID1 proteins in cancer is attributed to both mutational and non-mutational events. Most mutations in genes encoding ARID1 proteins include nonsense, frameshift and insertion/deletion mutations, and result in the protein loss. ARID1 genes are transcriptionally downregulated by their promoter methylation and repressive histone modifications. P-Stat3 transcriptionally represses ARID1B expression by decreasing H3K4Me3 histone modification. The levels of ARID1 proteins is controlled by ubiquitin-proteasome system. In gastric cancer, the DNA-damage repair signaling promotes rapid SCF β-TRCP-dependent ubiquitination and subsequent degradation of ARID1A. The phosphorylation of ARID1A at two serine sites by nuclear kinase ATM is required for its recognition by β-TRCP. In squamous cell carcinoma, ARID1A stability is regulated by E3 ligase TRIM32 and deubiquitinase USP11. ARID1B is not only a subunit of the BAF complexes but also a component of E3 ubiquitin ligase complex. ARID1B-harboring mutations in the BC box are less stable compared with wild-type ARID1B, and undergo autoubiquitination and proteasome-dependent degradation. In CCA-type ovarian cancer cell lines, p-Ser696-ARID1A levels is reduced compared with non-CCA cells possibly because of reduced phosphorylation or downregulation.
PTM: Post-translational modifications.
In tumors with low mutational rates of ARID1 proteins, downregulation of ARID1A and 1B was due to non-mutational events such as epigenetic regulation, transcriptional regulation and post-translational modifications (PTMs) (Figure 1). The regulation of ARID1A and ARID1B in cancer and normal tissues is still poorly investigated. ARID1A is better studied compared with ARID1B. Transcriptional and PTM regulators of ARID1A and 1B levels can be potentially considered as promising therapeutic targets in the clinical treatment of cancer. This review will focus on the regulatory mechanisms that alter ARID1-containing proteins and drive tumor development and progression.
Structure of ARID1 proteins & their role in the BAF complex assembly
ARID1 proteins share highly similar primary sequences and are mutually exclusive in the BAF complexes, suggesting that ARID1A- and ARID1B-containing BAF complexes are assembled in a similar manner (Figure 2 & Table 1) [1,10,17]. A recent study using complex affinity purification with gradient fractionation coupled with cross-linking mass spectrometry and mutagenesis provided evidence for the ordered and modular assembly of the BAF complexes [9]. Based on this study, the BAF complex assembly initiates from homo- or heterodimerization of SMARCC1 and/or SMARCC2 subunits, following by incorporation of SMARCD (mutually exclusive SMARCD1, SMARCD2 and SMARCD3), SMARCE1 and SMARCB1 components, building the BAF core module (Figure 2A & Table 1) [9]. Mutually exclusive ARID-containing proteins, ARID1 and ARID2, interact with the fully assembled core module and branch into the canonical BAF and PBAF complexes, respectively. The intermediate ARID1-BAF core module complexes bind DPF (mutually exclusive DPF1, DPF2 and DPF3) accessory subunit and incorporate ATPase module, finalizing the canonical BAF complex assembly (Figure 2A & Table 1) [9]. The ATPase module includes catalytic SMARCA (mutually exclusive SMARCA2 and SMARCA3) subunit, ACTB, ACTL6 (mutually exclusive ACTL6A and ACTL6B), SS18 and BCL7 (mutually exclusive BCL7A, BCL7B and BCL7C) (Figure 2A & Table 1) [9].
Figure 2. . The ARID1-containing proteins are mutually exclusive subunits of the BRG1/BRM-associated factor complexes.
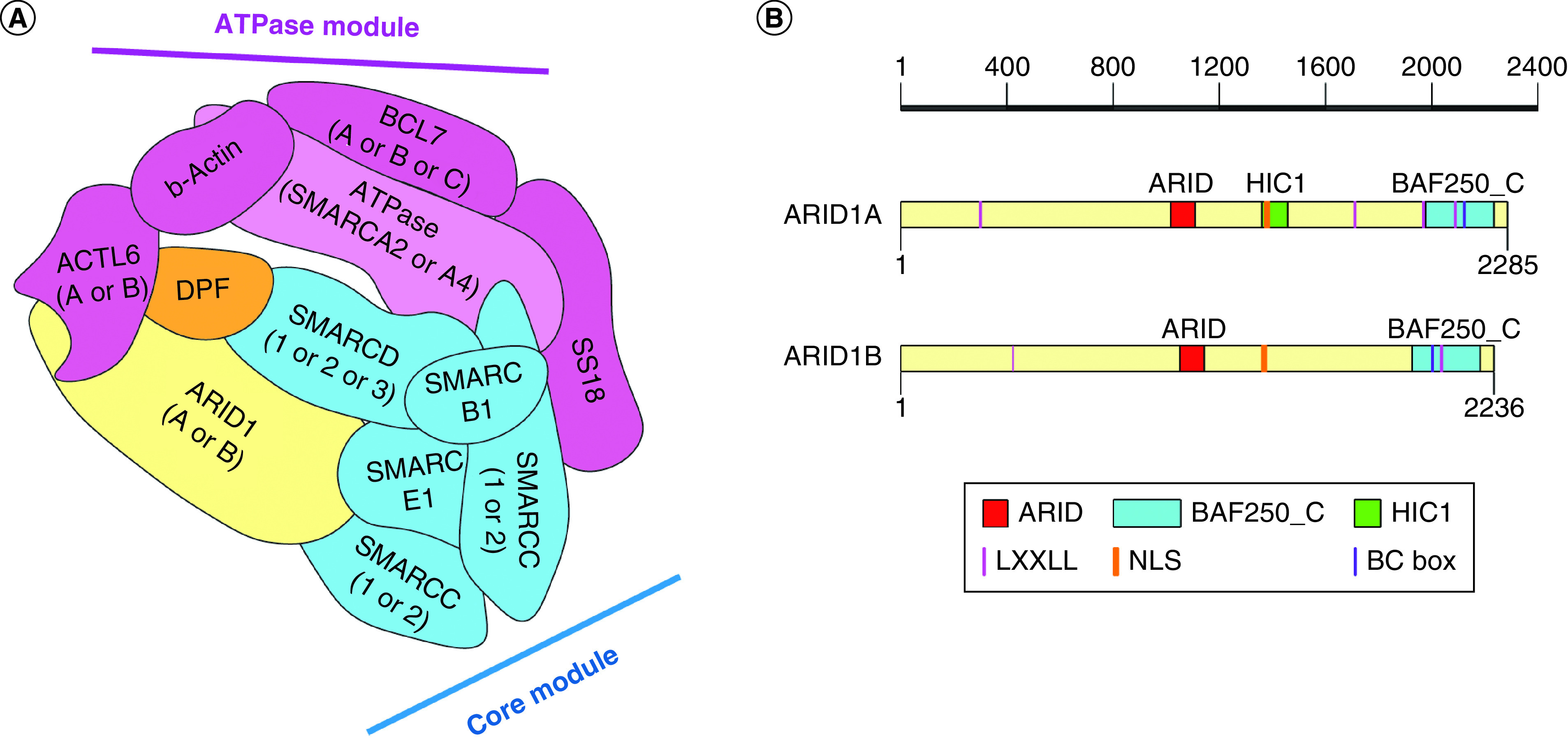
(A) Scheme of the BAF complex based on the model of Mashtalir et al. [9]. Mutually exclusive ARID1 proteins (ARID1A and ARID1B) play important role in the BAF complex assembly. They bind to subunits of the BAF core module and recruit subunits of ATPase module to form fully functioning complex. Subunits of the BAF core module are shown in blue, subunits of the BAF ATPase module are shown in green. ATPase catalytic subunit is either BRG1 (SMARCA4) or BRM (SMARCA2). (B) Schematic domain structure of ARID1 proteins was generated using Illustrator for Biological Sequences tool [82]. Similar motifs and domains are apparent between the amino acid sequences of ARID1A and ARID1B. Red boxes denote the ARID, vertical pink lines indicate LXXLL motifs, vertical orange lines indicate nuclear localizing signal.
BAF: BRG1/BRM-associated factor.
The length of ARID1A and 1B proteins are 2285 and 2236 residues, respectively (Figure 2B). They share approximately 50% sequence identity along the length of the proteins, and approximately 80% sequence identity for their two main functional domains (ARID and BAF250_C) [2,17]. The first 600 N-terminal residues are highly divergent [17].
The DNA-binding ARID domain is a conserved helix-turn-helix-motif-based domain of approximately 100 amino acid residues. Initially, the ARID domain was shown to have DNA-binding preferences for the AT-rich regions, a behavior that prompted the acronym ARID (AT-rich interactive domain). However, recent studies showed that ARID-containing proteins bind DNA through the ARID domain in a nonsequence-specific manner [1,18]. Mutations in the ARID domain can disrupt interaction between ARID1-containing proteins and DNA. For example, mutation of a conserved residue (V1067G) leads to a gross destabilization of the ARID fold and affects its DNA-binding abilities [19]. The BAF250_C domain is required for the interactions of the ARID1-containing proteins with other subunits of the BAF complexes, as well as with other proteins [9]. Thus, mutations in the BAF250_C domain may affect the BAF complex assembly and alter the interaction with different proteins, including transcriptional factors and histone-modifying enzymes [9,19].
Additionally, ARID1 proteins contain multiple LXXLL motifs, elongin-binding BC box motif and nuclear localizing signal (NLS) (Figure 2B) [20,21]. ARID1A has an additional functional domain involved in regulation of transcription, HIC1 binding domain (Figure 2B).
Regulation of ARID1 proteins
Epigenetic & transcriptional regulation
ARID1A and ARID1B genes are located on chromosome 1p36.11 and 6q25.3, respectively. The promoter of ARID1A and ARID1B contains high guanine (G) content and has potential to form alternative secondary structures, called G4 structures. Recent study confirmed the presence of G4 structures in the ARID1A promoter and showed that they increase ARID1A expression, suppress DNA replication and reduce cell proliferation [22].
ARID1 genes are transcriptionally downregulated by their promoter methylation and repressive histone modifications (Figure 1). Low expression levels of ARID1A and ARID1B as a result of promoter hypermethylation have been reported in squamous cell carcinoma (SCC), invasive breast carcinoma and pancreatic cancer [23–25]. Low expression of ARID1A in invasive breast carcinoma was associated with high gene occupancy of H3K27Me3 repressive marks [23].
Transcriptional factors that control expression of ARID1 genes are poorly investigated. In mouse neurofibromas, Arid1b gene is transcriptionally repressed by activated P-Stat3 (Figure 1) [24]. STAT3 is a transcriptional factor with well-known oncogenic properties and its role in regulation of cell-cycle progression and apoptosis. P-Stat3 binds downstream of transcriptional start site (TSS) of the Arid1b gene and represses its transcription by decreasing H3K4Me3 histone modification [24]. In future studies, it will be important to address which signaling pathways and transcriptional factors directly regulate ARID1A and ARID1B transcription.
Post-translational regulation
The detailed mechanisms of the PTMs on ARID1 protein stability and activity remain largely unknown. Among numerous PTMs, phosphorylation and ubiquitination are best studied (Figures 1 & 3).
Figure 3. . Post-translational modification of the ARID1-containing protein in cancer.

In gastric cancer, DNA damage response signaling promotes phosphorylation of 1316 and 1320 serines in DSGXXS canonical degron site for β-TRCP recognition. Phosphorylation of Serines 1316 and 1320 requires ATM kinase activity. SCF (β-TRCP) interacts with phosphorylated recognition site via WD40 domain and results in proteasomal degradation of ARID1A. In squamous cell carcinoma, ARID1A is balanced by TRIM32 and USP11. TRIM32 ubiquitinates ARID1A at lysine 2124 to target for proteasomal degradation, while USP11 deubiquitinates ARID1A at lysine 1961 to stabilize the protein. In CCA-type ovarian cancer cell lines, p-Ser696-ARID1A levels is reduced compared with non-CCA cells possibly because of reduced phosphorylation or downregulation. Cancer-associated G2087R mutation induces ubiquitin-dependent proteasomal degradation of ARID1A.
CCA: Clear cell adenocarcinoma; SCF: Skp, Cullin, F-box containing complex.
Ubiquitination/deubiquitination
The ubiquitin-proteasome system controls ARID1A and ARID1B stability (Figures 1 & 3) [16,20,21,26–28]. Previously, it has been shown that stability of ARID1A is higher in cytosolic than in nuclear subcellular compartment, and that proteasomal degradation of ARID1A occurs mainly in the nucleus [16,20]. ARID1A has both classic NLS and leucine-rich NES motifs that are involved in trafficking of ARID1A from cytosol to the nucleus and vice versa from nucleus to the cytosol, respectively (Figure 2). Leucine-rich NES motif of ARID1A binds XPO1 to form a nuclear export complex, which mediates the transport of large molecules through the nuclear pores. Pharmacological inhibition of XPO1 retained ARID1A in the nucleus resulting in reduced cytosolic ARID1A levels. However, the inhibition of the nuclear export did not increase ARID1A levels in the nucleus due to its rapid proteasomal degradation. Treatment with a proteasome inhibitor MG132 increased the levels of ARID1A in the nucleus but not in the cytosol. Moreover, the ARID1A mutants with a defective NES motif showed reduced protein expression compared with the wild-type ARID1A. These mutants retained in the nucleus with subsequent ubiquitin-dependent proteasomal degradation [16]. In addition, ARID1A harboring a mutated NLS, which is localized in both the cytosol and the nucleus, showed higher stability compared with the solely nuclear localized wild-type ARID1A following proteasomal inhibition with MG132 treatment [20]. These suggest that enzymes, regulating ARID1A through ubiquitin-dependent proteasomal degradation, are localized and function in the nucleus. Trafficking of ARID1A and its proteasomal degradation play an important role in regulation of ARID1A levels in the nucleus, where it functions as chromatin remodeler, transcriptional regulator and tumor suppressor.
Although ubiquitination is a PTM that requires the sequential activity of E1 ubiquitin-activating enzymes, E2 ubiquitin-conjugating enzymes and E3 ubiquitin ligases, the substrate specificity of a ubiquitination is mainly determined by E3 ligases. Two recent studies reported ubiquitination of ARID1A in gastric cancer in response to DNA damage treatment (Figure 3) [26,27]. In gastric cancer, ARID1A protein is rapidly ubiquitinated and subsequently degraded in response to DNA damage. ARID1A is a substrate of SCF β-TRCP E3 ubiquitin ligase complex and interacts with it through WD40 domain of β-TRCP [26]. β-TRCP recognizes a phosphorylated canonical DSGXXS degron site on ARID1A. The phosphorylation of both serine sites (S1316 and S1320) in the DSGXXS degron of ARID1A is required for its recognition by β-TRCP [27].
In SCC, ARID1A is regulated by E3 ligase TRIM32 and deubiquitinase USP11 (Figure 3) [28]. TRIM32 directly ubiquitinates ARID1A mainly at the 2124 lysine site to promote its degradation. USP11 directly deubiquitinates ARID1A at the 1961 lysine site to stabilize ARID1A. It was shown that TRIM32 and USP11 could interact with each other and antagonize for the interaction with ARID1A. In the nucleus of SCC cells, ARID1A protein was negatively correlated with TRIM32 and positively correlated with USP11 [28].
Some disease-associated mutations can affect stability of ARID1 proteins. As previously mentioned, mutations in the NES and NLS motifs affecting nuclear trafficking alter ARID1A protein stability [16,20]. Moreover, it was found that expression of ARID1A with cancer-associated G2087R mutation at C-terminus was significantly lower compare to wild-type ARID1A in HEK293T cells. The G2087R mutation resulted in increased polyubiquitination and decreased stability of the ARID1A protein without affecting its ability to interact with other subunits of the BAF complexes [9]. In addition, the bioinformatic MUpro tool predicted decreased stability of ARID1A with gastric cancer-associated mutation Pro912Thr compared with wild-type ARID1A [29]. The mechanisms underlying the effects of these mutations on ARID1A polyubiquitination and protein stability remain unknown. The possible mechanisms might be associated with changes in the protein configuration that affect the recognition of ARID1A protein by the substrate-specific factor, such as F-box protein, resulting in enhanced loading of ARID1A protein to the E3 ubiquitin ligase. The conformational changes caused by these mutations could possibly enhance the activity of E2 ubiquitin-conjugating enzymes or E3 ubiquitin ligases, which promotes adding up polyubiquitin chain to the ubiquitination site of ARID1A. It is also possible that G2087R and Pro912Thr mutations generate neodegron motifs on ARID1A protein, leading to its enhanced ubiquitination and degradation. Moreover, these mutations could generate a barrier for binding of deubiquitinating enzymes and result in inefficient deubiquitination of ARID1A.
It has been shown that ARID1B is not only a subunit of the BAF complexes but also a component of E3 ubiquitin ligase complex. ARID1B-harboring mutations in the BC box are less stable compared with wild-type ARID1B, and undergo autoubiquitination and proteasome-dependent degradation (Figure 1) [21].
Regulation of ARID1B through PTMs is poorly investigated. It was shown that the treatment of SCC cells with the proteasome inhibitor MG132 increased ARID1A protein levels, however, had no significant effects on ARID1B protein levels [28]. When evaluating ARID1A and ARID1B expression patterns during cell cycle in mouse embryos, a study showed that ARID1A is upregulated in G0 and downregulated throughout the cell cycle, while ARID1B protein levels did not show significant changes throughout the cell cycle [30]. It still remains unknown what mechanisms maintain the levels of ARID1B during the cell cycle. Based on these observations, ARID1B might be more resistant to ubiquitin-dependent proteolysis, or the levels of ARID1B are replenished by increased protein synthesis, or through both [28,30]. ARID1B might be permanently present in the nucleus but at much lower concentrations compared with ARID1A and be able to bind to DNA when ARID1A is downregulated [30].
Phosphorylation
As was mentioned above, phosphorylation of ARID1A at serine residues 1316 and 1320 in the canonical DSGXXS degron site is required for β-TRCP recognition in response to DNA damage response (Figure 3) [27]. It was shown that the phosphorylation of ARID1A is catalyzed by nuclear kinase ATM, which is critical for cellular response to DNA damage treatment [27].
It is still poorly investigated how phosphorylation impacts ARID1 protein stability and functional activity. It was previously shown that clear cell adenocarcinoma (CCA)-type ovarian cancer cell lines have reduced levels of phosphorylated ARID1A at serine 696 (p-Ser696-ARID1a) compared with non-CCA cells (Figure 3). This can be due to p-Ser696-ARID1A downregulation or reduced phosphorylation of ARID1A protein [31].
Functions of ARID1 proteins
As a component of SWI/SNF chromatin remodeling complex, ARID1 proteins play an important role in a lot of fundamental cellular processes, such as transcriptional regulation, DNA replication, DNA-damage repair (DDR), genomic instability, apoptosis, cell proliferation and cell differentiation [3,4,32].
Transcriptional regulation
ARID1 proteins bind to DNA in a nonsequence-dependent manner and sustain chromatin accessibility to transcriptional complexes and regulators [33,34]. They regulate access of RNA polymerase II to TSSs. For example, ARID1A inhibited the recruitment of RNA polymerase II to the TSS of the TERT promoter [35]. They can directly interact with gene-specific transcriptional regulators and be recruited to promoter sites to either coactivate or corepress gene transcription [3,17,33,36–38]. For example, HIC1 directly binds ARID1A through the HIC1-binding domain and recruits it to the promoter sites for a transcriptional repression of some HIC1 target genes, including E2F1 and ATOH1 [17]. Additionally, ARID1A functions as a coactivator of the GATA4/FOXA1 master transcription factor and induces expression of hepatocyte epithelial-differentiation genes [38]. ARID1 proteins have been also linked to steroid hormone receptor-induced transcription [3,36] and expression of cell cycle regulators through association with transcriptional factors, such as E2F [33,37]. ARID1A binds to CDKN1A and SMAD3 promoters and recruits p53 through C-terminus (amino acids 1759–2285) to collaborate in transcriptional activation of these genes [39].
ARID1 proteins collaborate with histone-modifying enzymes to regulate gene expression. They interact with different HTACs and HDACs and recruit them to transcription sites. ARID1A regulates gene expression through interaction with SIN3A HDAC corepressor complex including HDAC1 and HDAC2, while ARID1B interacts with HDAC3 and histone acetyltransferase Tip60 [33,35,40,41]. ARID1A binds to the TERT regulatory element and recruits SIN3A complex to repress TERT transcription [35]. Binding of ARID1A to the TERT regulatory element resulted in increase of transcriptional H3K9me3 repressor marks and decrease of H4H12Ac activation marks [35]. ARID1A inhibits TERT expression and its enzymatic activity resulting in shortening of telomeres. Loss of ARID1A causes reactivation of TERT transcription activity and confers a survival of tumor cells by maintaining their telomeres [35].
Cell proliferation & differentiation
The expression of ARID1 proteins varies during the cell cycle and they play an essential role in cell cycle regulation [33,37]. ARID1A is upregulated in G0 phase and is downregulated throughout the cell cycle, whereas ARID1B is expressed at comparable levels at all phases [30]. In vitro studies showed that ARID1A and ARID1B play distinct roles in the MC3T3-E1 preosteoblasts in regulation of the cell cycle. They produce BAF complexes with antiproliferative (ARID1A) and proproliferative (ARID1B) properties [33]. Both ARID1A- and ARID1B-containing complexes bind to promoters of cell cycle-specific genes, including c-Myc, cdc2, cyclin E and cyclin A [33]. The occupancy of promoter regions of these genes by ARID1A and ARID1B was different during cell cycle. ARID1A binds to the promoter regions during G0 phase and represses expression of cell cycle-specific genes, resulting in cell cycle arrest. ARID1A is required for normal cell cycle arrest in differentiating cells and during DDR process. It has been shown that ARID1A dissociates from promoters of cell cycle-specific genes in proliferating cells [33]. ARID1B was observed at the promoter regions of cell cycle-specific genes at all phases during cell cycle [30]. Inactivation or loss of ARID1A results in suppression of cell differentiation and increased proliferation [33,37]. ARID1A plays an important role in FAS-mediated apoptosis, while ARID1B binds Smad2/3 in response to TGF-β [42].
DNA damage repair
SWI/SNF complexes play an important role in the maintenance of genome integrity and their role in DDR includes modification of chromatin structure around sites of DNA damage and direct recruitment of proteins required for DDR [6]. During DNA damage, SWI/SNF complexes rapidly bind to chromatin surrounding DNA damage through interaction with γ-H2AX [43]. Canonical BAF and PBAF complexes have been shown to be involved in both nonhomologous end joining and homologous recombination repair processes. ARID1 proteins recruit the BAF complexes to DNA damage sites, assist in homologous recombination-mediated DNA repair and nonhomologous end joining at double-strand breaks, and required for cellular resistance to various types of DNA damage [44]. It was shown that ARID1A regulates DNA double-strand break repair through interaction with DNA damage checkpoint kinases ATR and ATM [27,45]. Moreover, ARID1A interacts with TOP2a that resolves newly replicated sister chromatids linked by catenated strands of DNA and prevents DNA entanglements during mitosis [46]. In addition, ARID1A induces expression of a subunit of the cohesion complex STAG1, which plays an important role in telomere cohesion and stabilization [47]. Inactivation of ARID1A results in mitotic defects such as anaphase bridges and chromosomal lagging, leading to genomic instability and polyploidy [46–48]. ARID1A was also found to interact with the mismatch repair (MMR) protein MSH2 and recruit it to chromatin during DNA replication to induce MMR [49].
E3 ubiquitin ligase
ARID1B protein interacts with elongin C via BC box motif and together with cullin 2 and Roc1 form E3 ubiquitin ligase complex that targets histone H2B at lysin 120 for monoubiquitination. H2BK120 monoubiquitination is an upstream event of H3K4 trimethylation associated with gene activation [21]. Although E3 ubiquitin ligase activity was mapped to the ARM-repeat containing BAF250_C domain of both ARID1 proteins, this activity has not been experimentally validated for ARID1A [21].
Despite sharing high similarity in domain architecture, ARID1A and ARID1B of the BAF complexes showed functional distinction. There are some evidences that ARID1A and ARID1B might play opposing roles in osteoblast differentiation [50]. ARID1A and SMARCA2-containing complexes are associates with repression, while ARID1B and SMARCA4-containing complexes with activation of osteocalcin promoter [50]. It is still not known what factors regulate the incorporation of either ARID1A or its paralog ARID1B into the BAF complexes and what specific roles are carried by ARID1A and ARID1B-containing complexes. The functions of ARID1 proteins are context dependent and their detailed mechanisms and physiological roles should be further investigated.
Dysregulation of ARID1 proteins in cancer
Tumorigenesis is a multistep process, involving both genetic mutations and alterations in regulatory mechanisms, including epigenetic regulation, transcriptional regulation and PTMs. Alterations of ARID1 proteins play an important role in cancer development, progression and therapy resistance. The tumor-suppressive function of ARID1A has been well defined [2,11,39,51,52]. As a tumor suppressor, ARID1A decreases cell proliferation [25,39,53,54], promotes differentiation, induces apoptosis [42] and plays an essential role in maintenance of genome integrity [2,26,27,44,45]. As we showed above, ARID1A is required for normal cell cycle arrest and it substantially explains its suppressive effects on proliferation of cancer cells [33,37]. ARID1A regulates the expression of SDC2, a transmembrane heparan sulfate proteoglycan, involved in cell proliferation, cell migration and cell matrix interactions. The depletion of ARID1A decreases the association between Brg1 and the promoter region of SDC2. This dissociation activates SDC2 expression at the transcriptional level through chromatin remodeling-mediated gene activation [28]. ARID1A has been shown to interact and cooperate with other tumor suppressors, such as p53 and HIC1, in suppression of tumor growth [17,39].
ARID1A expression is downregulated or lost in various cancer cell lines [55] and in various human cancers. Its downregulation in cancer is attributed to both mutational and non-mutational events including DNA methylation and ubiquitin-dependent proteasomal degradation (Figure 1). It is noteworthy that although ARID1A is more frequently mutated in cancer, its paralog ARID1B is more frequently mutated in neurodevelopmental disorders, which suggest their differential expression and distinct functions in different cell lineages. Loss of ARID1A has been shown in numerous human malignancies [11,12,15,53,56–59]. ARID1A gene is located at Ch1p36.11, which is also frequently deleted in human cancers (https://www.cbioportal.org/). In cancer, ARID1B expression does not compensate loss of ARID1A. Moreover, ARID1B displays lower expression than ARID1A in most tumor samples. ARID1A is constitutively more highly expressed in the tissues of origin for different cancers as well as in a variety of cancer cell lines, which raises the question as to whether dosage insufficiency upon loss of the dominant paralog, or a unique, paralog-specific functions that cannot be compensated by the remaining paralog, promote tumorigenesis [4,55].
Somatic mutations in ARID1A and ARID1B occur in 7.6 and 3.1% of all cancer samples, respectively (https://www.cbioportal.org/). They are disproportionately skewed to the C-terminal and map to both ARID and BAF250_C domains, likely interrupting binding to DNA and interactions with the BAF complex components and other proteins [9,19] (https://www.cbioportal.org/). ARID1A and ARID1B gene mutations affect transcriptional regulation with different consequences, which are determined by the cell type and when during the tumorigenesis the mutation was originated.
Dysregulation of ARID1 proteins & genomic instability
Loss of ARID1 proteins and disruption of SWI/SNF complexes impair one of the critical DDR mechanisms, DNA MMR pathway, leading to high levels of microsatellite instability and high tumor mutational burden, which in turn drive cancer development and can predict responsiveness to immune checkpoint inhibitors [11,15,60–63]. In colorectal carcinoma, ARID1A deficiency led to MMR deficiency due to MLH1 promoter hypermethylation [64]. Depletion of ARID1A is associated with downregulation of DDR pathway in cancer cells [65]. It also has been demonstrated that DDR-inducing reagents downregulate ARID1A in gastric cancer cells [26]. It is well known that checkpoint molecules can pause the cell cycle in response to DDR, which allows to fix errors and maintains genomic stability. G2/M checkpoint is defective in ARID1A-deficient cells exposed to DNA-damaging agents such as ionizing radiation [45].
Although loss of ARID1A is associated with genome instability, some ARID1A-inactivated cancer was shown to be able to maintain genomic stability through negative selection against cells with mitotic defects. ARID1A binds to promoter of STAG1 gene and induces its expression. STAG1, a component of the cohesin complex, is critical for chromatin cohesion at telomeres and maintenance of mitotic integrity [32,47]. It is considered as a tumor suppressor, which is mutated in various malignances and its loss induces genomic instability [32,47]. ARID1A deficiency results in STAG1 loss and is associated with impaired telomere cohesion and telomere loss leading to mitotic defects and genomic instability [32,47]. However, severe mitotic defects in ARID1A-deficient cells were selectively eliminated during mitosis through induction of apoptosis. This can lead to survival of ARID1A-deficient cells lacking genomic instability and increased resistance to mitosis-targeting compounds, such as paclitaxel [32,47].
Dysregulation of ARID1 proteins & treatment resistance
ARID1A deficiency is associated with higher grade tumors, more malignant phenotypes and with high risk or poor outcomes of various cancers [2,25,51,53,58,59,63,66,67]. Loss of ARID1A was correlated with a higher grade of cancer in gastric cancer, breast cancer, bladder cancer and ovarian clear cell carcinoma [53,59]. However, the association between low expression and a poor prognosis is not universal. In some cancer patients, ARID1A was accumulated in primary tumors but not in distant metastatic sites, suggesting that ARID1A can be downregulated through various mechanisms after initiation and play an important role in disease progression [68]. ARID1A and ARID1B mutations have been identified among a few genes that are more frequently mutated in metastatic than in primary human cancers [25,69–73].
ARID1A/B deficiency is associated with treatment resistance in cancer patients [74]. ARID1A increases sensitivity of gastric cancer cells to DNA damage reagents [26]. Depletion of ARID1A is significantly correlated with chemoresistance in ovarian clear cell carcinoma [75]. ARID1A and ARID1B, as well as other BAF components were identified in CRISPR screening to be essential for response of estrogen receptor positive (ER+) breast cancer cells to ER-targeted drugs, tamoxifen and fulvestrant [41]. The ARID1A depletion resulted in drug resistance to both compounds. In contrast, loss of ARID1A sensitized cells to BET inhibitors that disrupt binding of BET proteins to chromatin and subsequently results in transcriptional repression of different oncogenes [41]. It was shown that ARID1A recruits the BAF complex to ER cis-regulatory elements through a pioneer transcriptional factor FOXA1. It facilitates recruitment of HDAC1 that removes acetylation marks resulting in transcriptional repression. Loss of ARID1A prevents HDAC1 binding and facilitates recruitment of BET proteins to ER cis-regulatory elements resulting in induction of target genes that promote tumor growth [41].
Dysregulation of ARID1 proteins & tumor immunity
ARID1A/B plays an important role in regulation of tumor immunity [63,65]. ARID1A deficiency limited chromatin accessibility to interferon (IFN) responsive genes and suppressed their expression in murine cancer models as well as in human cancers [63]. In patients with metastatic urothelial carcinoma, response to immune checkpoint blockade treatment correlated with ARID1A mutations [65]. Moreover, depletion of ARID1A induced expression of IFN responsive genes in malignant cells [65]. Thus, ARID1A loss may enhance the immunogenicity of cancer cells. However, in the recent study, ARID1A had different effect on expression of IFN responsive genes and responsiveness to immune checkpoint blockade. Wild-type ARID1A interacts with EZH2 of PCR2 and antagonizes its function resulting in induction of Th1-type chemokines (CXCL9, CXCL10) and IFN responsive genes expression leading to high CD8+ T-cell tumor infiltration, potent effector T function and high immunotherapy response. In contrast, in case of ARID1A mutation, EZH2 of PRC2 catalyzes methyltransferase reaction converting H3K27 to H3K27me3 on Th1-type chemokine and IFN responsive gene promoters, suppressing their expression, which results in limited CD8+ T-cell tumor infiltration, weak effector T-cell function and low immunotherapy response. Notably, correlation of ARID1A status with immune status, patient survival and responses to immune checkpoint blockade treatment in several types of cancer does not associate with tumor mutational burden [63]. In cancer cells, ARID1A knockdown results in downregulation of angiogenesis pathways [65].
Current state of literature suggests a possibility for protumorigenic roles of ARID1A in certain contexts. In murine liver cancer model, Arid1a promoted tumor initiation by inducing CYP450-mediated oxidative stress [68]. In contrast, in established tumors, ARID1A suppressed tumor progression and metastasis through decrease in chromatin accessibility and reduction of gene expression involved in cancer cell invasion and migration [41]. Although a tumor-suppressive role of ARID1A was well established in colorectal cancer, the presence of ARID1A is crucial for proliferation of KRAS-mutated colorectal cancer cells due to its role in KRAS/AP1-mediated enhancer activity in the MEK/ERK pathway [76]. A number of laboratories reported identical driver mutations in ARID1A in the epithelium of endometrial tissue within the uterus and also in ovarian and extraovarian pelvic endometriosis tissue as well as in ovarian cancers associated with endometriosis (i.e., clear cell and endometrioid type) [77]. The proposed underlying mechanism is that pelvic endometriosis occurs primarily as a result of backward menstruation (through the uterine tubes) and implantation of endometrial tissue fragments in ovarian inclusion cysts or extraovarian peritoneal sites [77]. Thus, intrauterine endometrial epithelial cells with ARID1A mutations may provide a survival and oncogenic potential for the tissue fragments that travel to the ovaries and serve as the origin of cancer [77].
Conclusion
ARID1 proteins are mutually exclusive subunits of the canonical BAF chromatin remodeling complex that are essential for the complex assembly, its binding to DNA and interaction with other proteins. As components of the BAF chromatin remodeling complex, they play an important role in regulation of gene expression and other fundamental cellular processes. ARID1s are frequently mutated in human cancers. In addition to mutations that mostly lead to loss of ARID1s, there are other transcriptional and post-translational regulation mechanisms that downregulate ARID1s in cancers. The ARID1A and ARID1B genes are frequent targets of epigenetic alterations including promoter hypermethylation and other pathways that negatively regulate transcription. The recent studies report that ARID1s are targets of various post-translational modifications, including phosphorylation and ubiquitination that affect protein stability and lead to proteasomal degradation in cancers. Novel discoveries of regulators that affect stability and activity of ARID1s would provide new potent therapeutic strategies to manage cancer and intellectual disability disorders.
Future perspective: ARID1 proteins with potential therapy implications
Uncovering regulatory proteins effectively targeting tumor suppressors and oncogenes provides promising opportunities for the early diagnosis and high efficacy treatment of malignancies. The alterations in PTM can lead to conformational changes of ARID1 proteins and affect their interaction with other proteins, resulting in protein dysfunction. The conformational changes of ARID1 proteins may regulate the chromatin accessibility and transcription of many cancer-related genes. Targeting post-translational modifiers or blocking sites of PTM of ARID1s could be a potential strategy in cancer therapy. While extensive work has been done to dissect ARID1A, future studies on ARID1B could lead to in-depth and entire understanding of the BAF complexes in normal cells and disease status.
Several recent studies reported effects of cancer-associated mutations on stability of ARID1 proteins [9,16,20,29], however, the mechanisms underlying these effects remain unknown. Further validation of the impacts of ARID1A/B mutations in regulating their protein stability, assembly of the BAF complexes, interaction with other proteins, DNA-binding ability and chromatin remodeling function is required. In cancers with ARID1 inactivating mutations, a better understanding of the mechanisms leading to disease progression can help in developing novel disease preventive strategies. Indeed, several recent studies showed that ARID1A alterations, in particularly ARID1A deficiency, may sensitize tumors to drugs targeting ATR [48], EZH2 [78], PI3K, PARP [45], HDAC6 [79] and ARID1B [80]. Thus, promoting ARID1A downregulation by inducing degradation or suppressing stabilization can be beneficial in combination treatment based on synthetic lethal properties of ARID1A.
From PTM angle, we could consider two strategies: downregulating ARID1A for combination therapy using knowledge about synthetic lethality, or stabilizing ARID1A that acts as tumor suppressor. Currently, there are no effective treatments clinically available to selectively fight ARID1A-deficient tumors and there is a high need in development of novel therapeutic strategies [81]. Since ARID1A protein stability is regulated by the ubiquitin-mediated proteasome system in the nucleus, it would be interesting to investigate whether stabilizing nuclear ARID1A with specific ubiquitin-proteasome inhibitors will have antitumor effects. Depletion of TRIM32 in SCC cells led to inhibition of cell proliferation, cell migration, cell invasion and increased their sensitivity to cisplatin (or DDP) treatment through ARID1A stabilization [28]. In contrast, depletion of USP11 significantly promoted SCC cell proliferation, chemoresistance, migration and invasion by promoting ARID1A degradation [28]. No therapy targeting alterations and PTM has been approved yet. It is important to develop specific inhibitor that could prevent etiological signaling-induced proteolysis of ARID1A and ARID1B for therapeutic purposes.
The status of ARID1 proteins in cancer patients can help develop precise therapeutic strategy tailored to a specific patient. Development of antibodies that specifically detect various PTMs of ARID1A and/or ARID1B could potentially be used as diagnostic biomarkers. Future studies evaluating functions of ARID1 proteins and potential therapeutic strategies should consider stage of disease progression, dose and tissue context.
Executive summary.
Structure of the ARID1-containing proteins & their role in the BAF complex assembly
The ARID1A and 1B proteins are the largest mutually exclusive subunits of the canonical BRG1/BRM-associated factor (BAF) chromatin remodeling complexes. They share high sequence similarity and have two main functional domains (ARID and BAF250_C). The ARID domain is required for the DNA binding, while the BAF250_C domain is required for interactions with other subunits of the BAF complexes, as well as with other proteins. ARID1 proteins are essential for the assembly of the BAF complexes.
Regulation of ARID1 proteins
ARID1 genes are transcriptionally downregulated by their promoter methylation and repressive histone modifications. P-Stat3 transcriptionally represses ARID1B expression by decreasing H3K4Me3 histone modification.
The stability of ARID1 proteins is controlled by ubiquitin-proteasome system and can be affected by disease-associated mutations. The proteasomal degradation of ARID1A occurs mainly in the nucleus, where it functions as a chromatin remodeler, transcriptional regulator and a tumor suppressor.
In gastric cancer, the DNA-damage repair (DDR) signaling promotes rapid SCF β-TRCP-dependent ubiquitination and subsequent degradation of ARID1A. The phosphorylation of ARID1A by nuclear kinase ATM is required for its recognition by β-TRCP. In squamous cell carcinoma, ARID1A stability is regulated by TRIM32 and USP11.
Functions of ARID1 proteins
ARID1 proteins regulate chromatin accessibility to RNA polymerase II and transcriptional regulators. They can directly interact with transcriptional factors and histone-modifying enzymes to coactivate or corepress gene transcription.
ARID1A represses expression of cell cycle-specific genes resulting in cell cycle arrest in differentiating cells and during DDR process.
The role of ARID1 proteins in DDR process includes recruitment of the BAF complexes to DNA damage sites and recruitment of other proteins required for DDR.
ARID1B is not only a subunit of the BAF complex but also functions as a substrate recognition module of EloB/C-Cul2-Roc1 E3 ubiquitin ligase complex that monoubiquitinates histone H2B.
Dysregulation of ARID1 proteins in cancer
In cancer, ARID1A functions as a tumor suppressor and is frequently mutated or downregulated due to non-mutational events, such as promoter methylation and ubiquitin-dependent proteasomal degradation.
Loss of ARID1 proteins impairs DDR mechanisms and is associated with genome instability. However, in ARID1A-mutated cancers, the genomic integrity could be preserved by negative selection against cells with severe mitotic defects.
In cancer patients, ARID1A deficiency is associated with higher grade tumors, poor outcomes and treatment resistance.
ARID1A antagonizes EZH2 resulting in increased expression of Th1-type chemokines (CXCL9, CXCL10) that leads to high CD8+ T-cell tumor infiltration and better response to immunotherapy.
ARID1 proteins with potential therapy implications
The understanding of regulatory mechanisms of ARID1 proteins will help to develop novel anticancer therapeutic strategies.
Footnotes
Financial & competing interests disclosure
The work is supported by NIH grants R01CA250110, R01CA202963, R01CA202948. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as: • of interest; •• of considerable interest
- 1.Wang X, Nagl NG, Wilsker D et al. Two related ARID family proteins are alternative subunits of human SWI/SNF complexes. Biochem. J. 383(2), 319–325 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu JN, Roberts CWM. ARID1A mutations in cancer: another epigenetic tumor suppressor? Cancer Discov. 3(1), 35–43 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Masliah-Planchon J, Bièche I, Guinebretière J-M, Bourdeaut F, Delattre O. SWI/SNF chromatin remodeling and human malignancies. Annu. Rev. Pathol. Mech. Dis. 10(1), 145–171 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Pulice JL, Kadoch C. Composition and function of mammalian SWI/SNF chromatin remodeling complexes in human disease. Cold Spring Harb. Symp. Quant. Biol. 81, 53–60 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Wang Y, Hoang L, Ji JX, Huntsman DG. SWI/SNF complex mutations in gynecologic cancers: molecular mechanisms and models. Annu. Rev. Pathol. Mech. Dis. 15(1), 467–492 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang X, Haswell JR, Roberts CWM. Molecular pathways: SWI/SNF (BAF) complexes are frequently mutated in cancer—mechanisms and potential therapeutic insights. Clin. Cancer Res. 20(1), 21–27 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mittal P, Roberts CWM. The SWI/SNF complex in cancer — biology, biomarkers and therapy. Nat. Rev. Clin. Oncol. 17(7 ), 435–448 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nie Z, Xue Y, Yang D et al. A specificity and targeting subunit of a human SWI/SNF family-related chromatin-remodeling complex. Mol. Cell. Biol. 20(23), 8879–8888 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mashtalir N, D’Avino AR, Michel BC et al. Modular organization and assembly of SWI/SNF family chromatin remodeling complexes. Cell 175(5), 1272–1288.e20 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• The first study showing an ordered and modular assembly of mammalian SWI/SNF complexes.
- 10.He S, Wu Z, Tian Y et al. Structure of nucleosome-bound human BAF complex. Science 367(6480), 875–881 (2020). [DOI] [PubMed] [Google Scholar]
- 11.Kadoch C, Hargreaves DC, Hodges C et al. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat. Genet. 45(6), 592–601 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wiegand KC, Shah SP, Al-Agha OM et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N. Engl. J. Med. 363(16), 1532–1543 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Murakami R, Matsumura N, Brown JB et al. Exome sequencing landscape analysis in ovarian clear cell carcinoma shed light on key chromosomal regions and mutation gene networks. Am. J. Pathol. 187(10), 2246–2258 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Fukumoto T, Magno E, Zhang R. SWI/SNF complexes in ovarian cancer: mechanistic insights and therapeutic implications. Mol. Cancer Res. 16(12), 1819–1825 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jones S, Li M, Parsons DW et al. Somatic mutations in the chromatin remodeling gene ARID1A occur in several tumor types. Hum. Mutat. 33(1), 100–103 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guan B, Gao M, Wu C-H, Wang T-L, Shih I-M. Functional analysis of in-frame indel ARID1A mutations reveals new regulatory mechanisms of its tumor suppressor functions. Neoplasia N.Y.N. 14(10), 986–993 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Van Rechem C, Boulay G, Leprince D. HIC1 interacts with a specific subunit of SWI/SNF complexes, ARID1A/BAF250A. Biochem. Biophys. Res. Commun. 385(4), 586–590 (2009). [DOI] [PubMed] [Google Scholar]
- 18.Wilsker D, Patsialou A, Zumbrun SD et al. The DNA-binding properties of the ARID-containing subunits of yeast and mammalian SWI/SNF complexes. Nucleic Acids Res. 32(4), 1345–1353 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sandhya S, Maulik A, Giri M, Singh M. Domain architecture of BAF250a reveals the ARID and ARM-repeat domains with implication in function and assembly of the BAF remodeling complex. PLoS ONE 13(10), e0205267 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bateman NW, Shoji Y, Conrads KA et al. Identification and functional characterization of a novel bipartite nuclear localization sequence in ARID1A. Biochem. Biophys. Res. Commun. 469(1), 114–119 (2016). [DOI] [PubMed] [Google Scholar]
- 21.Li XS, Trojer P, Matsumura T, Treisman JE, Tanese N. Mammalian SWI/SNF-a subunit BAF250/ARID1 is an E3 ubiquitin ligase that targets histone H2B. Mol. Cell. Biol. 30(7), 1673–1688 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yan T, Zhao B, Wu Q et al. Characterization of G-quadruplex formation in the ARID1A promoter. Int. J. Biol. Macromol. 147, 750–761 (2020). [DOI] [PubMed] [Google Scholar]
- 23.Zhang X, Sun Q, Shan M et al. Promoter hypermethylation of ARID1A gene is responsible for its low mRNA expression in many invasive breast cancers. PLoS ONE 8(1), e53931 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu J, Keng V, Patmore DM et al. Insertional mutagenesis identifies a STAT3/Arid1b/β-catenin pathway driving neurofibroma initiation. Cell Rep. 14(8), 1979–1990 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Luo Q, Wu X, Chang W et al. ARID1A hypermethylation disrupts transcriptional homeostasis to promote squamous cell carcinoma progression. Cancer Res. 80(3), 406–417 (2020). [DOI] [PubMed] [Google Scholar]
- 26.Jiang Z-H, Dong X-W, Shen Y-C et al. DNA damage regulates ARID1A stability via SCF ubiquitin ligase in gastric cancer cells. Eur. Rev. Med. Pharmacol. Sci. 19(17), 3194–3200 (2015). [PubMed] [Google Scholar]
- 27.Jiang Z, Peng T, Qian H, Lu C, Qiu F, Zhang S. DNA damage-induced activation of ATM promotes β-TRCP-mediated ARID1A ubiquitination and destruction in gastric cancer cells. Cancer Cell Int. 19 (2019). https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6567580/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luo Q, Wu X, Nan Y et al. TRIM32/USP11 balances ARID1A stability and the oncogenic/tumor-suppressive status of squamous cell carcinoma. Cell Rep. 30(1), 98–111.e5 (2020). [DOI] [PubMed] [Google Scholar]; •• Shows that the ARID1A stability is controlled by TRIM32 and USP11 in squamous cell carcinoma.
- 29.Qadir J, Majid S, Khan MS et al. AT-rich interaction domain 1A gene variations: genetic associations and susceptibility to gastric cancer risk. Pathol. Oncol. Res. 26(4), 2237–2246 (2020). [DOI] [PubMed] [Google Scholar]
- 30.Flores-Alcantar A, Gonzalez-Sandoval A, Escalante-Alcalde D, Lomelí H. Dynamics of expression of ARID1A and ARID1B subunits in mouse embryos and in cells during the cell cycle. Cell Tissue Res. 345(1), 137–148 (2011). [DOI] [PubMed] [Google Scholar]
- 31.Kimura A, Arakawa N, Hirano H. Mass spectrometric analysis of the phosphorylation levels of the SWI/SNF chromatin remodeling/tumor suppressor proteins ARID1A and Brg1 in ovarian clear cell adenocarcinoma cell lines. J. Proteome Res. 13(11), 4959–4969 (2014). [DOI] [PubMed] [Google Scholar]
- 32.Nacarelli T, Zhao B, Hao X, Zhang R. ARID1A mutation and genomic stability. Mol. Cell. Oncol. 7(3), 1690923 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nagl NG, Wang X, Patsialou A, Van Scoy M, Moran E. Distinct mammalian SWI/SNF chromatin remodeling complexes with opposing roles in cell-cycle control. EMBO J. 26(3), 752–763 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Trizzino M, Barbieri E, Petracovici A et al. The tumor suppressor ARID1A controls global transcription via pausing of RNA polymerase II. Cell Rep. 23(13), 3933–3945 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rahmanto YS, Jung J-G, Wu R-C et al. Inactivating ARID1A tumor suppressor enhances TERT transcription and maintains telomere length in cancer cells. J. Biol. Chem. 291(18), 9690–9699 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Inoue H, Furukawa T, Giannakopoulos S, Zhou S, King DS, Tanese N. Largest subunits of the human SWI/SNF chromatin-remodeling complex promote transcriptional activation by steroid hormone receptors. J. Biol. Chem. 277(44), 41674–41685 (2002). [DOI] [PubMed] [Google Scholar]
- 37.Nagl NG, Patsialou A, Haines DS, Dallas PB, Beck GR, Moran E. The p270 (ARID1A/SMARCF1) subunit of mammalian SWI/SNF-related complexes is essential for normal cell cycle arrest. Cancer Res. 65(20), 9236–9244 (2005). [DOI] [PubMed] [Google Scholar]
- 38.Enane FO, Shuen WH, Gu X et al. GATA4 loss of function in liver cancer impedes precursor to hepatocyte transition. J. Clin. Invest. 127(9), 3527–3542 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guan B, Wang T-L, Shih I-M. ARID1A, a factor that promotes formation of SWI/SNF-mediated chromatin remodeling, is a tumor suppressor in gynecologic cancers. Cancer Res. 71(21), 6718–6727 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim M, Lu F, Zhang Y. Loss of HDAC-mediated repression and gain of NF-κB activation underlie cytokine induction in ARID1A- and PIK3CA-mutation-driven ovarian cancer. Cell Rep. 17(1), 275–288 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nagarajan S, Rao SV, Sutton J et al. ARID1A influences HDAC1/BRD4 activity, intrinsic proliferative capacity and breast cancer treatment response. Nat. Genet. 52(2), 187–197 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• CRISPR screening revealed ARID1 proteins to be involved in treatment response in ER+ breast cancer cells.
- 42.Luo B, Cheung HW, Subramanian A et al. Highly parallel identification of essential genes in cancer cells. Proc. Natl Acad. Sci. USA 105(51), 20380–20385 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Park J-H, Park E-J, Lee H-S et al. Mammalian SWI/SNF complexes facilitate DNA double-strand break repair by promoting γ-H2AX induction. EMBO J. 25(17), 3986–3997 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Watanabe R, Ui A, Kanno S et al. SWI/SNF factors required for cellular resistance to DNA damage include ARID1A and ARID1B and show interdependent protein stability. Cancer Res. 74(9), 2465–2475 (2014). [DOI] [PubMed] [Google Scholar]
- 45.Shen J, Peng Y, Wei L et al. ARID1A deficiency impairs the DNA damage checkpoint and sensitizes cells to PARP inhibitors. Cancer Discov. 5(7), 752–767 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dykhuizen EC, Hargreaves DC, Miller EL et al. BAF complexes facilitate decatenation of DNA by topoisomerase II[alpha]. Nature 497(7451), 624 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhao B, Lin J, Rong L et al. ARID1A promotes genomic stability through protecting telomere cohesion. Nat. Commun. 10(1), 4067 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Shows that ARID1A induces the expression of telomere cohesion subunit STAG1 and its loss causes mitotic defects and leads to genomic instability.
- 48.Williamson CT, Miller R, Pemberton HN et al. ATR inhibitors as a synthetic lethal therapy for tumours deficient in ARID1A. Nat. Commun. 7(1), 13837 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shen J, Ju Z, Zhao W et al. ARID1A deficiency promotes mutability and potentiates therapeutic antitumor immunity unleashed by immune checkpoint blockade. Nat. Med. 24(5), 556–562 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Flowers S, Nagl NG, Beck GR, Moran E. Antagonistic roles for BRM and BRG1 SWI/SNF complexes in differentiation. J. Biol. Chem. 284(15), 10067–10075 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mamo A, Cavallone L, Tuzmen S et al. An integrated genomic approach identifies ARID1A as a candidate tumor-suppressor gene in breast cancer. Oncogene 31(16), 2090–2100 (2012). [DOI] [PubMed] [Google Scholar]
- 52.Wu R-C, Wang T-L, Shih I-M. The emerging roles of ARID1A in tumor suppression. Cancer Biol. Ther. 15(6), 655–664 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang D, Chen Y, Pan K et al. Decreased expression of the ARID1A gene is associated with poor prognosis in primary gastric cancer. PLoS ONE 7(7), e40364 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Guo X, Zhang Y, Mayakonda A et al. ARID1A and CEBPα cooperatively inhibit UCA1 transcription in breast cancer. Oncogene 37(45), 5939–5951 (2018). [DOI] [PubMed] [Google Scholar]
- 55.Wang X, Nagl NG, Flowers S, Zweitzig D, Dallas PB, Moran E. Expression of p270 (ARID1A), a component of human SWI/SNF complexes, in human tumors. Int. J. Cancer 112(4), 636 (2004). [DOI] [PubMed] [Google Scholar]
- 56.Jones S, Wang T-L, Shih I-M et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science 330(6001), 228–231 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Guan B, Mao T-L, Panuganti PK et al. Mutation and loss of expression of ARID1A in uterine low-grade endometrioid carcinoma. Am. J. Surg. Pathol. 35(5), 625–632 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gui Y, Guo G, Huang Y et al. Frequent mutations of chromatin remodeling genes in transitional cell carcinoma of the bladder. Nat. Genet. 43(9), 875–878 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang X, Zhang Y, Yang Y et al. Frequent low expression of chromatin remodeling gene ARID1A in breast cancer and its clinical significance. Cancer Epidemiol. 36(3), 288–293 (2012). [DOI] [PubMed] [Google Scholar]
- 60.Okamura R, Kato S, Lee S, Jimenez RE, Sicklick JK, Kurzrock R. ARID1A alterations function as a biomarker for longer progression-free survival after anti-PD-1/PD-L1 immunotherapy. J. Immunother. Cancer 8(1), (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pasanen A, Loukovaara M, Bützow R. Clinicopathological significance of deficient DNA mismatch repair and MLH1 promoter methylation in endometrioid endometrial carcinoma. Mod. Pathol. 33(7), 1443–1452 (2020). [DOI] [PubMed] [Google Scholar]
- 62.Zhu G, Pei L, Li Y, Gou X. EP300 mutation is associated with tumor mutation burden and promotes antitumor immunity in bladder cancer patients. Aging 12(3), 2132–2141 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li J, Wang W, Zhang Y et al. Epigenetic driver mutations in ARID1A shape cancer immune phenotype and immunotherapy. J. Clin. Invest. 130(5), 2712–2726 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Villatoro TM, Ma C, Pai RK. Switch/sucrose nonfermenting nucleosome complex-deficient colorectal carcinomas have distinct clinicopathologic features. Hum. Pathol. 99, 53–61 (2020). [DOI] [PubMed] [Google Scholar]
- 65.Goswami S, Chen Y, Anandhan S et al. ARID1A mutation plus CXCL13 expression act as combinatorial biomarkers to predict responses to immune checkpoint therapy in mUCC. Sci. Transl. Med. 12(548), (2020). https://stm.sciencemag.org/content/12/548/eabc4220 [DOI] [PubMed] [Google Scholar]
- 66.Yang Y, Wang X, Yang J et al. Loss of ARID1A promotes proliferation, migration and invasion via the Akt signaling pathway in NPC. Cancer Manag. Res. 11, 4931–4946 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Han X, Chen W, Chen P et al. Aberration of ARID1A is associated with the tumorigenesis and prognosis of sporadic nonfunctional pancreatic neuroendocrine tumors. Pancreas 49(4), 514–523 (2020). [DOI] [PubMed] [Google Scholar]
- 68.Sun X, Wang SC, Wei Y et al. Arid1a has context-dependent oncogenic and tumor suppressor functions in liver cancer. Cancer Cell. 33(1), 151–152 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Stein MK, Williard FW, Xiu J et al. Comprehensive tumor profiling reveals unique molecular differences between peritoneal metastases and primary colorectal adenocarcinoma. J. Surg. Oncol. 121(8), 1320 –1328 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gibson WJ, Hoivik EA, Halle MK et al. The genomic landscape and evolution of endometrial carcinoma progression and abdominopelvic metastasis. Nat. Genet. 48(8), 848–855 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.He F, Li J, Xu J et al. Decreased expression of ARID1A associates with poor prognosis and promotes metastases of hepatocellular carcinoma. J. Exp. Clin. Cancer Res. CR. 34, 47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zehir A, Benayed R, Shah RH et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 23(6), 703–713 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yates LR, Knappskog S, Wedge D et al. Genomic evolution of breast cancer metastasis and relapse. Cancer Cell. 32(2), 169–184.e7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Katagiri A, Nakayama K, Rahman MT et al. Loss of ARID1A expression is related to shorter progression-free survival and chemoresistance in ovarian clear cell carcinoma. Mod. Pathol. 25(2), 282–288 (2012). [DOI] [PubMed] [Google Scholar]
- 75.Luo Q, Wu X, Zhang Y et al. ARID1A ablation leads to multiple drug resistance in ovarian cancer via transcriptional activation of MRP2. Cancer Lett. 427, 9–17 (2018). [DOI] [PubMed] [Google Scholar]
- 76.Sen M, Wang X, Hamdan FH et al. ARID1A facilitates KRAS signaling-regulated enhancer activity in an AP1-dependent manner in colorectal cancer cells. Clin. Epigenetics. 11(1), 92 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bulun SE, Wan Y, Matei D. Epithelial mutations in endometriosis: link to ovarian cancer. Endocrinology 160(3), 626–638 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bitler BG, Aird KM, Garipov A et al. Synthetic lethality by targeting EZH2 methyltransferase activity in ARID1A-mutated cancers. Nat. Med. 21(3), 231–238 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Demonstrated synthetic lethality between EZH2 and ARID1A.
- 79.Bitler BG, Wu S, Park PH et al. ARID1A-mutated ovarian cancers depend on HDAC6 activity. Nat. Cell Biol. 19(8), 962–973 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Shows that loss of ARID1A suppresses p53-mediated apoptosis by upregulating HDAC6.
- 80.Helming KC, Wang X, Wilson BG et al. ARID1B is a specific vulnerability in ARID1A-mutant cancers. Nat. Med. 20(3), 251–254 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bitler BG, Fatkhutdinov N, Zhang R. Potential therapeutic targets in ARID1A-mutated cancers. Expert Opin. Ther. Targets. 19(11), 1419–1422 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Liu W, Xie Y, Ma J et al. IBS: an illustrator for the presentation and visualization of biological sequences. Bioinforma. Oxf. Engl. 31(20), 3359–3361 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]