

Abstract
Inactivating mutations in human ecto-nucleotide pyrophosphatase/phosphodiesterase-1 (ENPP1) may result in early-onset osteoporosis (EOOP) in haploinsufficiency and autosomal recessive hypophosphatemic rickets (ARHR2) in homozygous deficiency. ARHR2 patients are frequently treated with phosphate supplementation to ameliorate the rachitic phenotype, but elevating plasma phosphorus concentrations in ARHR2 patients may increase the risk of ectopic calcification without increasing bone mass. To assess the risks and efficacy of conventional ARHR2 therapy, we performed comprehensive evaluations of ARHR2 patients at two academic medical centers and compared their skeletal and renal phenotypes with ENPP1-deficient Enpp1asj/asj mice on an acceleration diet containing high phosphate treated with recombinant murine Enpp1-Fc. ARHR2 patients treated with conventional therapy demonstrated improvements in rickets, but all adults and one adolescent analyzed continued to exhibit low bone mineral density (BMD). In addition, conventional therapy was associated with the development of medullary nephrocalcinosis in half of the treated patients. Similar to Enpp1asj/asj mice on normal chow and to patients with mono- and biallelic ENPP1 mutations, 5-week-old Enpp1asj/asj mice on the high-phosphate diet exhibited lower trabecular bone mass, reduced cortical bone mass, and greater bone fragility. Treating the Enpp1asj/asj mice with recombinant Enpp1-Fc protein between weeks 2 and 5 normalized trabecular bone mass, normalized or improved bone biomechanical properties, and prevented the development of nephrocalcinosis and renal failure. The data suggest that conventional ARHR2 therapy does not address low BMD inherent in ENPP1 deficiency, and that ENPP1 enzyme replacement may be effective for correcting low bone mass in ARHR2 patients without increasing the risk of nephrocalcinosis.
Keywords: ENPP1 MUTATION, OSTEOPOROSIS, AUTOSOMAL RECESSIVE HYPOPHOSPHATEMIC RICKETS (ARHR2), NEPHROCALCINOSIS
Introduction
Generalized arterial calcification of infancy (GACI, OMIM 614473) and autosomal recessive hypophosphatemic rickets type 2 (ARHR2, OMIM 613312) are human diseases associated with biallelic ecto-nucleotide pyrophosphatase/phosphodiesterase-1 (ENPP1) mutations. GACI is characterized by arterial calcifications in the medium and large arteries beginning as early as the late second trimester and culminating in death in about 50% of affected infants around 6 months of age. Hypertension is found in up to a third of patients(1) and can represent a cause of mortality.(2) Although prior data suggest that bisphosphonates might improve survival,(2) a recent large natural history study did not find any statistical difference regarding mortality in those treated with bisphosphonates versus those who remained untreated (unpublished findings). The calcifications arise as a consequence of the generalized deficiency of pyrophosphate,(3) resulting from biallelic mutations that inactivate the genes encoding either the ectoenzyme ENPP1(4) (GACI type 1, about 75% of patients), which hydrolyzes ATP to generate PPi, or ABCC6(5,6) (GACI type 2, about 25% of patients), a transmembrane protein that when deficient is associated with low plasma pyrophosphate (PPi). Affected individuals who reach the age of 6 months are likely to survive and may experience partial spontaneous reversal of vascular calcifications over time.(2,7–9) However, patients with ENPP1 deficiency develop increased plasma levels of the phosphatonin FGF23, which induces renal phosphate (Pi) wasting and ARHR2.(10,11) Although the basis for elevated levels of FGF23 in GACI type 1 is unknown, lowering plasma Pi to reduce the calcification propensity of PPi-deficient individuals may be an adaptive physiologic response, a notion supported by the correlation of FGF23 elevation and Pi wasting with survival in GACI patients.(2) Whatever the cause, the bone disease in ARHR2 can be severe and cause bone pain, fractures of the long bones, rachitic skeletal deformities, and impaired growth. To ameliorate these problems, ARHR2 patients are often treated with oral phosphate and calcitriol, which improve the skeletal symptoms(12) but may cause nephrocalcinosis. Moreover, because of the nearly absent levels of plasma PPi, increasing serum levels of Pi may increase the risk of worsening vascular calcification.(13)
Murine models of ENPP1 deficiency recapitulate essential cardiovascular characteristics of GACI type 1 when the mice are placed on a high phosphate/low magnesium “acceleration diet.”(14) These features include severe aortic, coronary, and cardiac calcifications and are associated with perinatal mortality. We and others have demonstrated that treatment of murine models of ENPP1 deficiency with ENPP1 enzyme replacement therapy prevents the calcifications and mortality,(15) improves cardiac function,(16) and reverses vascular stenosis,(17) all of which represent clinical challenges in the management of GACI patients.
Although ectopic mineralization in the cardiac and vascular tissue of murine models of Enpp1 deficiency reproduces features present in GACI patients, similarities in the skeletal phenotypes of human and mouse ENPP1 deficiency are less apparent. Instead of hypophosphatemic rickets resembling human ARHR2, murine models of Enpp1 deficiency exhibit low bone mass consistent with osteoporosis.(18–22) Questions regarding the relevance of murine skeletal phenotype to human ENPP1 deficiency resolved with the identification of early-onset osteoporosis (EOOP) in several subjects with heterozygous ENPP1 mutations who exhibited findings nearly identical to those in Enpp1asj/asj mice—low plasma Pi, mildly elevated FGF23 levels, low bone mass, and bone microarchitectural defects.(23) Furthermore, bone histomorphometry showed greater accumulation of osteoid by several indices in Enpp1asj/asj mice as well as mineralization defects consistent with skeletal abnormalities observed in rachitic diseases such as ARHR2. The close correspondence between the vascular and skeletal phenotype in ENPP1-deficient humans and mice validates the use of the Enpp1asj/asj model in additional predictive mechanistic and pre-clinical studies.
To understand the efficacy of current ARHR2 treatment regimens, we reviewed the presentation and the clinical course of patients with biallelic ENPP1 mutations treated at two institutions with conventional therapy for hypophosphatemic rickets, comparing the clinical course and treatment response to a murine model of Enpp1 deficiency treated with phosphate supplementation and ENPP1 enzyme replacement therapy (ERT). We focus here on the low bone mass, fracture risk, and microarchitectural defects reflecting the skeletal phenotype in ENPP1 deficiency.
Materials and Methods
Patient evaluation in Bethesda
Nineteen individuals with biallelic ENPP1 mutations were evaluated at the National Institutes of Health Clinical Center under research protocols 76-HG-0238 (“Diagnosis and treatment of patients with inborn errors of metabolism and other genetic disorders,” identifier: NCT00369421) and 18-HG-0064 (“Study of people with generalized arterial calcification of infancy (GACI) or autosomal recessive hypophosphatemic rickets type 2 [ARHR2]),” identifier: NCT03478839). The aforementioned protocols were approved by the National Human Genome Research Institute (NHGRI) Institutional Review Board (IRB). Seventeen of the enrolled individuals underwent renal imaging (ultrasonography and/or computed tomography), whereas three affected adults underwent dual-energy X-ray absorptiometry (DXA) (Hologic Discovery A, Hologic, Inc., Marlborough, MA, USA) with calculation of areal bone mineral density (aBMD). Slides of postmortem renal histopathology from two affected individuals (brothers of patient 1, deceased at 38 and 49 days due to myocardial infarction) were reviewed.
Patient evaluation in Hamburg
We previously reported the clinical phenotype of hypophosphatemic rickets in a family treated at the specialized outpatient clinic (The National Bone Board) of the University Medical Center Hamburg-Eppendorf.(23) Mutation analysis by a custom-designed SureSelect XT gene panel (Agilent, Santa Clara, CA, USA)(24,25) revealed homozygous ENPP1 mutations in two siblings with ARHR2. Here, we present the clinical details of the homozygous son after exposure to conventional therapy (ie, phosphate and calcitriol supplementation adjusted to respective serum levels). This study was performed in accordance with approval of the local ethics committee (PV5364) and the Declaration of Helsinki, and informed consent was obtained.
Fracture history and reoccurring new fractures were assessed via comprehensive medical history. Mineral metabolism and bone turnover were assessed by biochemical analyses. Automated photometric assays were used to measure serum calcium and phosphate (Dimension VISTA 1.500 Analyzer, Siemens, Munich, Germany); automated immunoassays were used to measure parathyroid hormone [PTH(1–84)], 25-OH-D (Centaur, Siemens), bone-specific alkaline phosphatase (bAP), osteocalcin, and 1,25-(OH)2-D (Liaison, Diasorin, Stillwater, MN, USA), as well as DPD (Immulite-XP, Siemens). C-terminal FGF23 was determined by ELISA (Immutopics, Quidel, San Diego, CA, USA).
We assessed aBMD of the lumbar spine, hip, and total body less head (TBLH) (Lunar iDXA, GE Healthcare, Madison, WI, USA) at initial presentation and follow-up, and adjusted the data for height-adjusted Z-score (HAZ).(26,27) All parameters were assessed both before and post-initiation of conventional treatment, consisting of phosphate and calcitriol, as well as 25(OH) D-supplementation.
Enpp1asj/asj mouse model
Animal care and maintenance were provided through Yale University Animal Resource Center (YARC) at Yale University (New Haven, CT, USA). All procedures were approved by the Animal Care and Use Committee of Yale University and complied with the US National Institutes of Health guide for the care and use of laboratory animals. Heterozygous Enpp1asj/+ (genotype C57BL/6J-Enpp1asj/GrsrJ, Jackson Laboratory, Bar Harbor, ME, USA; stock number 012810) breeding pairs were maintained on the “acceleration” high-phosphate diet (TD.00442, Harlan Laboratories, Madison, WI, USA) throughout the entire experiment and food and water were delivered ad libitum. The animal colony was housed in pathogen-free conditions. Pups were weaned at day 21, and the pups were continued on the diet fed to the breeding pairs (so-called in utero acceleration diet). Litters were genotyped on day 8 and weaned at day 21. After weaning, sibling pairs were sequentially divided into cohorts on a rolling basis, meaning we enrolled one wild-type (WT), then one vehicle-treated Enpp1asj/asj, followed by one Enpp1-Fc–treated Enpp1asj/asj mouse before beginning again with a WT animal. Animals were consecutively enrolled in experimental trials without regard to sex, and the following sexes were enrolled in each cohort: WT—8 males and 12 females; vehicle-treated Enpp1asj/asj—8 males and 7 females; Enpp1-Fc–treated Enpp1asj/asj—7 males and 8 females. Enrollment of the experimental cohorts spanned 18 months. Once the enrollment began, both sexes of the appropriate genotype were consecutively enrolled with the exclusion of severely runted animals weighing less than 5.5 grams at 14 days of life. After weaning, all experimental animals were housed with littermates to allow for cooperative grooming and nesting. Investigators were not blinded to experimental groups during the study. Mice were treated from days 21 to 35 with daily subcutaneous doses of either vehicle or 10 mg/Kg of recombinant mouse Enpp1-Fc (mEnpp1-Fc) formulated in vehicle. All animals were terminally bled on day 35 and the skeletal phenotypes were examined.
Vehicle formulation
mEnpp1-Fc was formulated in vehicle as to deliver 16 μL vehicle/g of body weight. Vehicle consisted of AmericanBio (Canton, MA, USA) 10× PBS (stock number AB11072) diluted to 1× with endotoxin-free water and supplemented with 14 μM CaCl2 and 14 μM ZnCl2 (hereafter referred to as PBSPLUS).
mEnpp1-Fc expression construct
Soluble mouse mEnpp1-Fc was purified from CHO cells that had been stably transfected with a cDNA encoding a fusion protein containing the export signal sequence and cytoplasmic domain of human ENPP1 and the transmembrane domain of human ENPP2, similar to the construct previously described for the expression of soluble human ENPP1.(15) The Fc domain of mouse IgG1 was fused to the C-terminus of mEnpp1 and separated by a two-amino acid spacer sequence (GG). The full-length sequence of the transfected protein is shown in Supplemental Fig. S1.
Expression, purification, and characterization of mEnpp1-Fc
Recombinant mEnpp1-Fc was produced and characterized based on previously described methods,(15) with further details provided in Supplemental Materials and Methods.
Evaluation of nephrocalcinosis in mice
A renal pathologist (GM), masked to the identity of the study animal, reviewed each kidney specimen. Multiple sections of renal tissue were evaluated for calcium deposits and scored using a square grid technique. Small squares of a 10 × 10 integrated grid, falling on areas with morphologic features of nonpolarizing calcium deposits in Von Kossa-stained sections were counted. Ten independent fields were counted per kidney (1000 squares per kidney), and the percentage of tissue area with calcium deposits was calculated as percentage of total squares counted.
Quantification of plasma PPi
Blood plasma was prepared and PPi assayed as recently described.(23,28) Data analysis was performed via GraphPad (La Jolla, CA, USA) Prism 7.
Assays for 1,25-dihydroxyvitamin D, PTH(1–84), and FGF-23
1,25-dihydroxyvitamin D enzyme immunoassay (EIA) kits were purchased from Immunodiagnostic Systems Inc. (Boldon, UK; catalog number AC-62F1). Mouse PTH(1–84) ELISA kits were purchased from Quidel Corporation (San Diego, CA, USA; catalog number 60–2305). Mouse/Rat FGF-23 (intact) ELISA kit (catalog number 60–6800) was purchased from Quidel Corporation. A total of 45 μL plasma samples were used for all the above ELISA experiments. Data analysis was performed via GraphPad Prism 7.
Histology, histomorphometry, and micro-CT
Tibias and femurs were stripped of soft tissue, fixed in 70% ethanol, dehydrated, and embedded in methyl-methacrylate before being sectioned and stained with toluidine blue.(29) Histomorphometry measurements were performed on a fixed region 300 to 600 μm below the growth plate corresponding to secondary spongiosa(30) and again at 0 to 600 μm below the growth plate corresponding to primary and secondary spongiosa and analyzed by OsteoMeasure software (OsteoMetrics, Atlanta, GA, USA). For micro-CT, tibias or femurs were stripped of soft tissue and stored in 70% ethanol at 4°C. The bones were scanned using a Scanco μCT-35 (Scanco, Bruttisellen, Switzerland) and analyzed for numerous structural parameters at both the proximal tibia or distal femur just below the growth plate (trabecular bone) and at the tibial or femoral midshaft (cortical bone). The spatial resolution of the micro-CT measurement was 6 μm (isometric voxel size) and was performed to a depth of over 1 mm in each sample. Histomorphometry and micro-CT parameters were evaluated in a sex-specific manner to remove sex as a confounding variable, and reported in the female animals. Similar trends were observed in the male animals.
Bone biomechanical testing
All femurs were loaded to failure. Femurs from mice on the acceleration diet were loaded to failure with three-point bending. All whole bone tests were conducted by loading the femur in the posterior to anterior direction, such that the anterior quadrant was subjected to tensile loads. The widths of the lower and upper supports of the three-point bending apparatus were 7 mm and 3 mm, respectively. Tests were conducted with a deflection rate of 0.05 mm/s using a servo hydraulic testing machine (Instron model 8874; Instron Corp., Norwood, MA, USA). The load and mid-span deflection were acquired directly at a sampling frequency of 200 Hz. Load-deflection curves were analyzed for stiffness, maximum load, and work to fracture. Yield is defined as a 10% reduction in the secant stiffness (load range normalized for deflection range) relative to the initial tangent stiffness. Post-yield deflection, which is defined as the deflection at failure minus the deflection at yield was measured also. Femurs were tested at room temperature and kept moist with phosphate-buffered saline (PBS). Biomechanical measurements were performed in combined male and female animals as sex-specific analysis did not alter the biomechanical findings.
Quantitative backscattered electron imaging (qBEI)
To assess bone mineral density distribution (BMDD), qBEI of mouse tibias in the lateral cortex was performed as previously described.(23) Samples of Enpp1WT mice (3 males, 6 females) and Enpp1asj/asj mice treated with (3 males, 6 females) and without mEnpp1-Fc (5 males, 4 females) were embedded in methyl-methacrylate, polished, and carbon-coated before scanned in a scanning electron microscope (LEO 435, LEO Microscopy Ltd., Cambridge, UK) equipped with a backscattered electron detector (Type 202, K.E. Developments Ltd., Cambridge, UK). Mean calcium weight percentage (CaMean), most frequent calcium weight percentage (CaPeak), and heterogeneity of the calcium content (CaWidth) were evaluated as parameters for BMDD. Mean osteocyte lacunar area (Ot.Lc.Ar) and number of osteocyte lacunae in the cross-sectioned mineralized matrix (N.Ot. Lc/B.Ar) were determined as parameters indicating two-dimensional osteocyte lacunae characteristics.
Raman spectroscopy
Raman spectroscopy (inVia, Renishaw, West Dundee, IL, USA) was performed on the lateral and medial tibia in the central-proximal region in identical planes as assessed with qBEI. Maps of 40 × 200 μm were acquired at 785 nm laser wavelength, 20× objective, and 3-second exposure time with 20 accumulations per step. Data processing was performed using WiRe software (Renishaw) and included cosmic ray removal, automatic polynomial baseline subtraction of fluorescent background, and subtraction of a PMMA reference spectrum acquired near the embedded bone. Using a custom-written Matlab script, peak areas of interest were determined after linear baseline removal below the peaks (phosphate at 963 cm−1, phenylalanine at 1005 cm−1, carbonate at 1075 cm−1, amide III at 1245 cm−1, and amide I between 1615 and 1705 cm−1). Raman ratios were derived based on previously established protocols.(31,32) Sex-specific group comparisons were performed on the mean ratios of each specimen using the nonparametric Kruskal–Wallis independent test at a significance level of α = 0.05.
Statistics
Nephrocalcinosis was statistically compared in ARHR2 patients treated with conventional therapy using a two-tailed Student’s unpaired t test. GraphPad Prism 7 was used to statistically analyze the murine histomorphometry, biomechanical constants, and micro-CT data. Statistical significance was determined using ANOVA comparison of means, stratified as *p < .05, **p < .01, ***p < .001. Raman spectroscopy, sex-specific group comparisons were performed on the mean ratios of each specimen using the nonparametric Kruskal–Wallis independent test at a significance level of α = 0.05.
Results
NIH subject presentation and treatment response
Ten individuals underwent renal imaging after initiation of calcitriol and/or phosphate supplementation for treatment of hypophosphatemic rickets (Table 1). Medullary nephrocalcinosis was not present by ultrasound in any of seven patients who were naïve to treatment at the time of renal imaging, although one patient was found to have a dotted pattern of hyperechogenicity in the renal cortex (Fig. 1C). Five subjects developed bilateral medullary nephrocalcinosis after beginning treatment (Fig. 1A, B), which was significant (p = .04, Student’s two-tailed t test), confirming the association of treatment with nephrocalcinosis. The presence of nephrocalcinosis was not related to age (mean age ± SEM in nephrocalcinosis group 12.7 ± 5.8 years and in group without nephrocalcinosis 11.7 ± 3.5 years, two-tailed p = .88, Student’s unpaired t test). Histo-pathology of autopsy material from two deceased individuals revealed calcification within the cortex of the kidneys in the absence of medullary calcification (Fig. 1D, E).
Table 1.
Relation Between Renal Imaging Findings and Rickets Treatment in NIH Subjects.
| Patient | Variant (transcript: NM_006208.2) | Renal parenchymal calcification | Relation to treatment of rickets | Serum phosphatea mmol/L (mg/dL) | TmP/GFRa mmol/L (mg/dL) | TRPa % |
|---|---|---|---|---|---|---|
| 1 | c.1441C > T (p.Arg481Trp) c.2713_2717del (p.Lys905Alafs*16) |
Bilateral medullary nephrocalcinosis at 4 years 0 month | First observed 18 months after initiation of treatment | 1.07 (3.3) | 0.71 (2.19) | 66.3 |
| 2 | c.1412A > G (p.Tyr471Cys) c.1442G > A (p.Arg481Gln) |
None at 25 years 6 months (but bilateral kidney stones) | Received treatment for several years during adolescence | 0.61 (1.9) | 0.44 (1.37) | 72.1 |
| 3 | c.2735 T > C (p.Leu912Ser) 3.4 kb deletion of exon 6 (delIVS5_IVS6) | None at 4 years 2 months | No treatment | 1.29 (4.0) | 1.41 (4.35) | 92.9 |
| 4 | c.1438 T > C (p.Cys480Arg) c.2414G > T (p.Gly805Val) |
Bilateral medullary nephrocalcinosis at 5 years 10 months | First observed 20 months after initiation of treatment | 0.94 (2.9) | 0.75 (2.31) | 84.9 |
| 5 | c.1438 T > C (p.Cys480Arg) c.2414G > T (p.Gly805Val) |
None at 4 years 5 months | Last evaluated 7 months after initiation of treatment | 1.10 (3.4) | 0.90 (2.79) | 82.0 |
| 6 | c.913C > A (p.Pro305Thr) c.1499A > C (p.His500Pro) |
Bilateral medullary nephrocalcinosis at 8 years 3 months | First observed 20 months after initiation of treatment | 1.07 (3.3) | 0.68 (2.12) | 64.1 |
| 7 | c.2320C > T (p.Arg774Cys) c.2662C > T (p.Arg888Trp) |
None at 7 years 10 months | Last evaluated 31 months after initiation of treatment | 0.90 (2.8) | — | — |
| 8 | c.1441C > T (p.Arg481Trp) c.2312–5_2313del |
None at 8 years 2 months | Ultrasound obtained at time of initiation of treatment | 0.71 (2.2) | 0.61 (1.89) | 85.9 |
| 9 | c.1441C > T (p.Arg481Trp) c.2312–5_2313del |
None at 13 years 1 month | Ultrasound obtained at time of initiation of treatment | 0.68 (2.1) | 0.57 (1.76) | 83.9 |
| 10 | c.749C > T (p.Pro250Leu) c.913C > A (p.Pro305Thr) |
None at 26 years 3 months | Receiving treatment since adolescence | 0.74 (2.3) | 0.43 (1.32) | 57.6 |
| 11 | c.913C > A (p.Pro305Thr) c.2662C > T (p.Arg888Trp) |
None at 8 months | Ultrasound obtained at time of initiation of treatment | 1.36 (4.2) | — | — |
| 12 | c.1538A > G (p.Tyr513Cys) c.1538A > G (p.Tyr513Cys) |
Multiple punctate foci of cortical hyperechogenicity bilaterally | No treatment | 0.90 (2.8) | 0.83 (2.58) | 88.9 |
| 13 | c.1652A > G (p.Tyr551Cys) c.1737G > C (p.Leu579Phe) |
None at 2 years 0 month | Ultrasound obtained at time of initiation of treatment | 1.07 (3.3) | 0.91 (2.83) | 85.6 |
| 14 | c.749C > T (p.Pro250Leu) c.749C > T (p.Pro250Leu) |
None at 1 year 8 months | Last evaluated 18 months after initiation of treatment | 0.81 (2.5) | — | — |
| 15 | c.783C > G (p.Tyr261a) c.1756G > A (p.Gly586Arg) |
Bilateral medullary nephrocalcinosis at 9 years 8 months | First observed 2 years after initiation of treatment | 0.87 (2.7) | (2.26) | 84.2 |
| 16 | c.715 + 1G > C c.2376 T > A (p.Asn792Lys) |
Bilateral medullary nephrocalcinosis at 35 years 8 months | Received treatment for 5 years during late childhood and early adolescence | 0.45 (1.4) | 0.38 (1.18) | 85.0 |
| 17 | c.715 + 1G > C c.2376 T > A (p.Asn792Lys) |
None at 38 years 5 months | Received treatment for several years during late childhood/early adolescence | 0.52 (1.6) | - | - |
TmP/GFR = tubular maximum reabsorption of phosphate to glomerular filtration rate; TRP = tubular reabsorption of phosphate.
Obtained at time of renal imaging.
Fig 1.
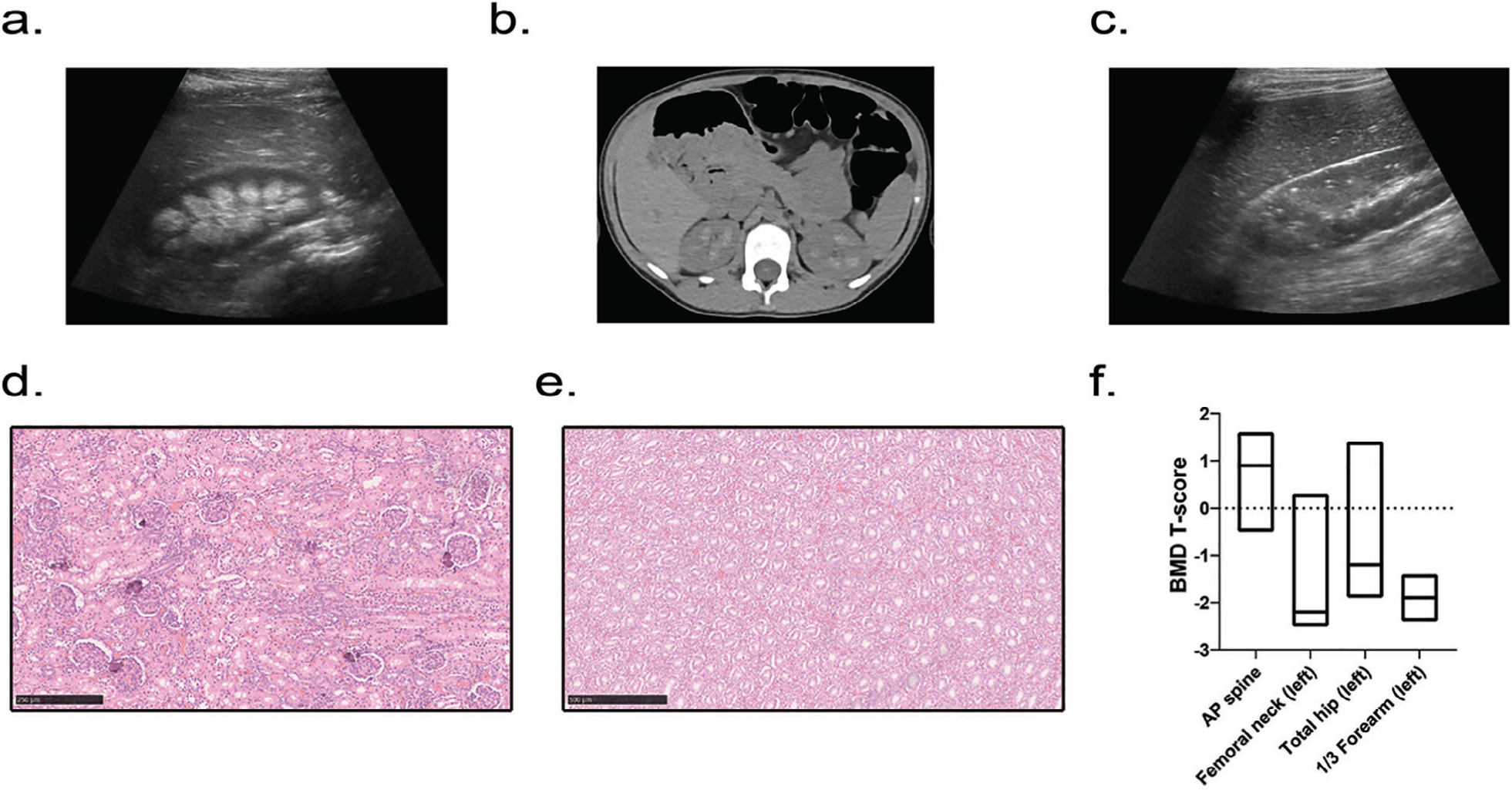
Human phenotype. (A–C) Renal imaging. (A) Renal ultrasound showing medullary nephrocalcinosis (patient 15, right kidney, aged 9 years 8 months). (B) Computed tomography of the abdomen revealing bilateral calcification of the renal pyramids (patient 6, aged 8 years 3 months). (C) Renal ultrasound revealing punctate hyperechogenic foci within the renal cortex (patient 12, right kidney, aged 7 years 11 months). (D, E). Renal histology of patient 1’s deceased sibling revealing foci of calcification within the renal cortex (D), including glomeruli, with absent calcification within the medulla (E). (F) Areal bone mineral density T-scores in adults; data are displayed as box plots denoting median value and interquartile range.
Bone densitometry by DXA evaluation in three adults revealed that they met WHO criteria for osteopenia or osteoporosis in at least one site (Table 2). Two patients were osteopenic (T-score between −1.0 and −2.5) in the distal radius and total hip, and one patient each with osteopenia and osteoporosis (T-score of −2.5 or lower) at the femoral neck (Fig. 1F). Systemic hypertension was identified in 12 of 18 patients (67%).
Table 2.
Bone Mineral Density (Bethesda Patients)
| Bone mineral density (g/cm2) | T-score | Z-score | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient (age; height) | AP spine | Femoral neck (left) | Total hip (left) | Distal radius (left) | AP spine | Femoral neck (left) | Total hip (left) | Distal radius (left) | AP spine | Femoral neck (left) | Total hip (left) | Distal radius (left) |
| Pt. 16 (35 years 8 months; 148 cm) | 1.223 | 0.884 | 1.111 | 0.598 | 1.6 | 0.3 | 1.4 | −1.6 | 1.7 | 0.5 | 1.5 | −1.4 |
| Pt. 17 (38 years 5 months; 149 cm) | 0.987 | 0.577 | 0.790 | 0.552 | −0.5 | −2.5 | −1.2 | −2.4 | −0.4 | −2.2 | −1.1 | −2.1 |
| Pt. 18 (56 years 2 months; 181 cm) | 1.222 | 0.635 | 0.741 | — | 0.9 | −2.2 | −1.9 | — | 1.4 | −1.3 | −1.5 | — |
AP = anteroposterior.
Hamburg subject presentation and treatment response
The male subject (Table 3, age 16 years) had undergone prior surgery for correction of lower limb deformities (ie, left genu varum and right genu valgum) (Fig. 2A, B). Moreover, he suffered multiple vertebral and peripheral traumatic fractures in the context of polytrauma. He was also affected with combined conductive and sensorineural hearing loss and dysplasia of the aortic valve. Upon examination at a follow-up appointment, pronounced skeletal signs of rickets improved (Fig. 2C, D), but ultrasound examination revealed the newly detected nephrocalcinosis.
Table 3.
Overview of Clinical Data Including Sex, Age, GACI Status, History of Symptoms, Mutation Status, Fractures, and Treatment Information (Hamburg Patient)
| Parameter | Patient Hamburg |
|---|---|
| Sex/Age | M/16 yr |
| Height/Z-score | 144.6 cm/−3.5 |
| History of GACI | No |
| Age at diagnosis of rickets | ~2.5 yr |
| Age at initial presentation | 14 yr |
| Age at ARHR2 diagnosis | 14.5 yr |
| ENPP1 mutation status | Homozygous |
| DNA level/protein level | c.755A > G/p.Tyr252Cys |
| Childhood fractures | None |
| Vertebral/nonvertebral fractures | 2a/4a |
| Vitamin D suppl. (IU/d/age at start) | 2000–3000/14 yr |
| Phosphate suppl. (mg/d/age at start) | 1220–1830/14.5 yr |
| Calcitriol suppl. (μg/d/age at start) | 0.5/14.5 yr |
GACI = generalized arterial calcification of infancy; M = male; yr = years; ARHR2 = autosomal recessive hypophosphatemic rickets; ENPP1 = ecto-nucleotide pyrophosphatase/phosphodiesterase-1; suppl. = supplementation.
Traumatic fracture.
Fig 2.
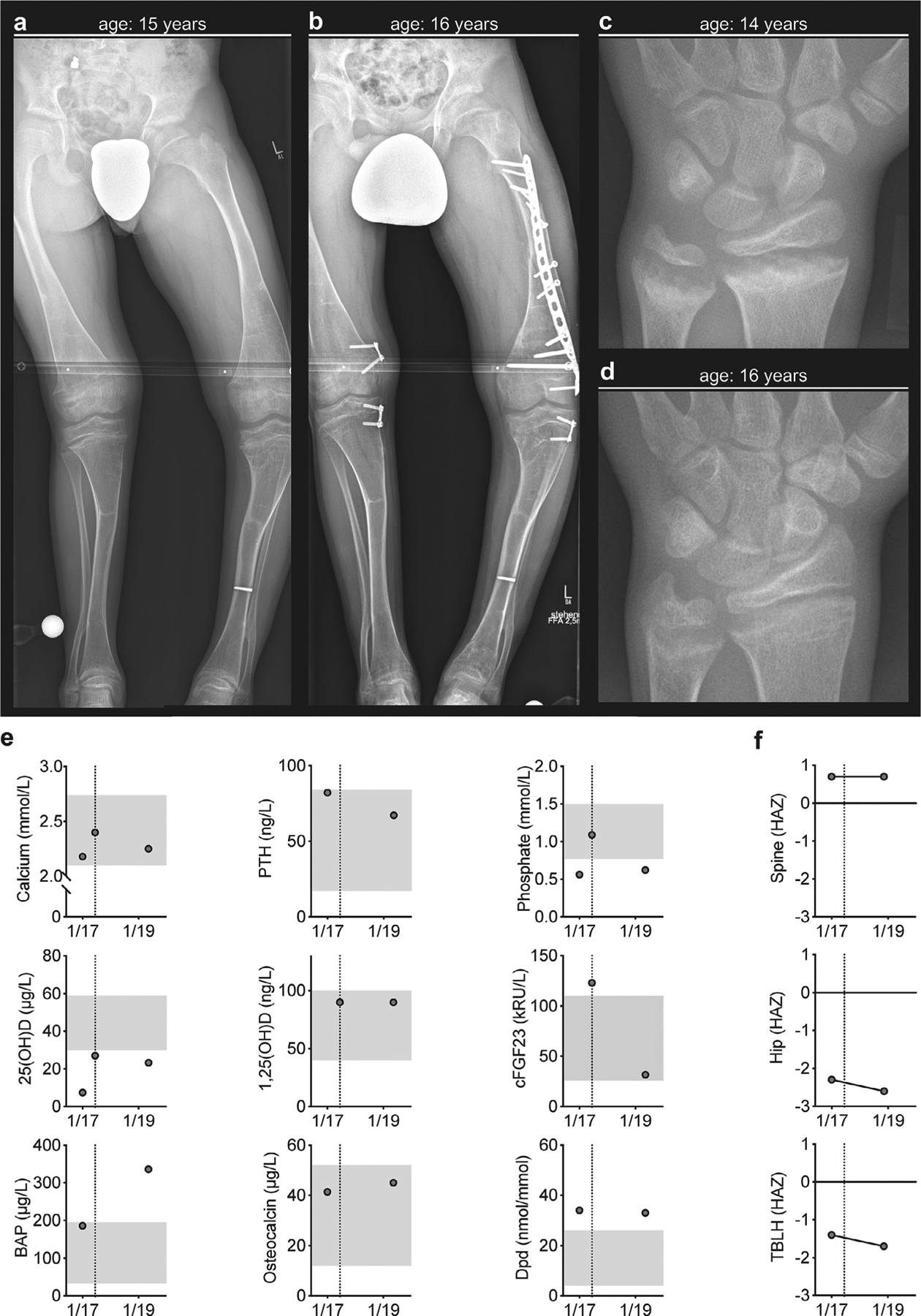
Clinical presentation of an ARHR2 patient with ENPP1−/− and follow-up after initiation of conventional therapy. (A) Skeletal deformities with bending of the long bones of the lower limb displayed in X-ray in context of planning for bilateral hemiepiphyseodesis. (B) Follow-up X-ray after bilateral hemiepiphyseodesis and open reduction internal fixation by a locking compression plate required by traumatic diaphyseal femur fracture. (C) Radiograph of the left hand and distal forearm at the age of 14 years with characteristic signs of rickets at the metaphyses (ie, fraying, spraying, and cupping) at distal radius and ulna. (D) Radiographic improvement at the age of 16 years. (E) Biochemical assessment of calcium and phosphate metabolism as well as bone turnover. Dotted line representing onset of conventional treatment (ie, phosphate and calcitriol). The x axis shows dates of investigation (mo/yr). (F) Assessment of DXA derived bone mineral density Z-score (adjusted for height Z-score, HAZ) is presented for spine, hip, and total body less head (TBLH), revealing further decrease of BMD under conventional treatment. The x axis shows dates of investigation (mo/yr).
Biochemical parameters before treatment initiation revealed levels of calcium within the lower reference range, PTH(1–84) within the upper reference range, and severe vitamin D deficiency (Fig. 2D). Moreover, serum phosphorous levels were below reference values in two of three measurements, accompanied by 1,25(OH)D concentrations in the upper reference range and c-terminal FGF23 levels above the reference range. Markers of bone formation (ie, BAP and osteocalcin) were in the upper reference range, and urinary Dpd excretion was elevated. Follow-up analysis of these parameters under conventional treatment revealed stable values for calcium, phosphorus, 1,25 (OH)2D, osteocalcin, and Dpd but a decrease in PTH(1–84), as well as cFGF23; 25(OH)D increased but remained below the reference range, as well as BAP, which increased above the reference range. Evaluation of aBMD corrected for HAZ revealed stable spine but reduced TBLH and hip mineral densities at follow-up (Fig. 2F).
Murine biochemical phenotype and clinical response to therapy
As previously reported, weight of the Enpp1asj/asj mice on the acceleration diet declines around the fourth week of life, presumably because of poor health induced by vascular and ectopic calcifications.(15) In this study, we noted that 15 of 20 Enpp1asj/asj mice treated with mEnpp1-Fc responded to treatment, exhibiting weights that paralleled WT sibling pairs treated with vehicle (Fig. 3A, B). Nonresponding animals likely developed an immune reaction that either neutralized or increased clearance of the biologic, as has been reported.(17)
Fig 3.
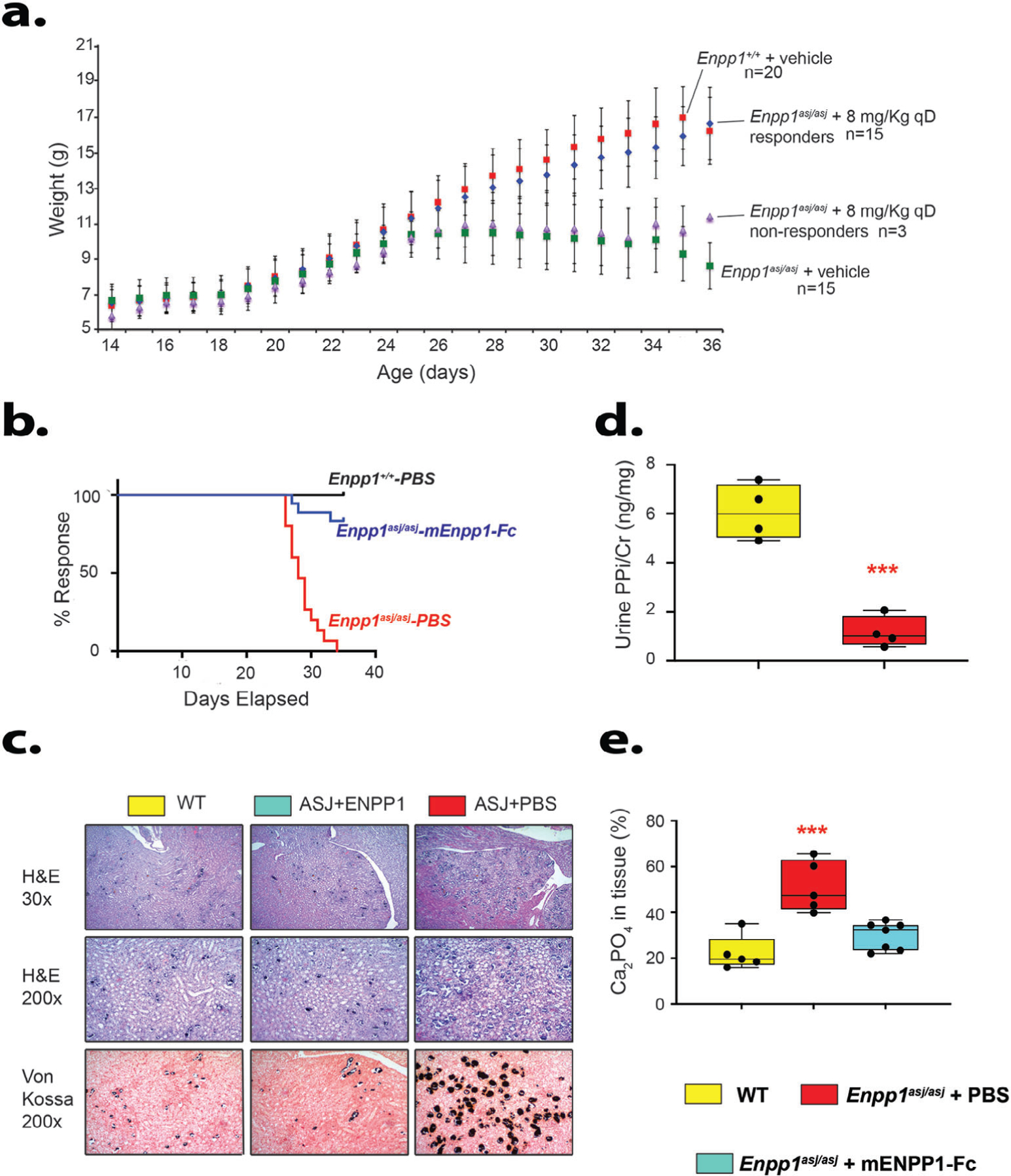
Clinical and renal phenotype of Enpp1asj/asj on high-phosphate diet treated with vehicle or Enpp1-Fc. (A) Daily animal weights. Average daily weights of Enpp1wt and Enpp1asj/asj sibling pairs dosed daily with vehicle (PBSplus) or with mEnpp1-Fc at 8 mg/kg qD. Mean average weights are plotted with standard deviations denoted by error bars. Dosing with mEnpp1-Fc and weighing commenced on day 14 and continued until day 35. Three mEnpp1-Fc-treated Enpp1asj/asj mice did not elevate plasma PPi and were labeled as nonresponders and analyzed separately from the 15 treated Enpp1asj/asj mice that elevated plasma PPi in response to mEnpp1-Fc dosing. (B) Kaplan–Meier plots of WT and Enpp1asj/asj sibling pairs dosed with either vehicle (PBSplus) or with mEnpp1-Fc at 8 mg/kg qD in PBSPLUS. Response was defined as successive weight gain during the experimental period (days 14 to 25 of life). None of the 20 WT mice (8 males, 12 females) experienced weight loss within the experimental period, whereas all Enpp1asj/asj mice dosed with vehicle did (7 males, 8 females). Three of 18 Enpp1asj/asj mice dosed with mEnpp1-Fc were noted to not elevate plasma PPi, presumably due to the presence of anti-drug antibodies, and classified as nonresponders. (C) Renal histology. All experimental cohorts displayed some degree of nephrocalcinosis, which was most pronounced at the corticomedullary junction. Vehicle-treated Enpp1asj/asj mice experienced pronounced nephrocalcinosis throughout the renal parenchyma, including cortex and renal medulla. The calcification was most pronounced in renal tubules near the cortical-medullary junction, as demonstrated by Von Kossa stains (black material within tubules). (D) Comparison of urine pyrophosphate in 8- to 10-week-old WT and Enpp1asj/asj on regular chow revealed that Enpp1asj/asj mice have approximately half the urine PPi levels as WT sibling pairs. Data are displayed as box plots denoting median value and interquartile range, with whiskers denoting minimum and maximum values and individual measurements denoted by circles. ***p < .001, ANOVA comparison of means. (E) Quantitation of nephrocalcinosis and urine pyrophosphate. Nephrocalcinosis was quantitated by a renal pathologist blinded to experimental groups as detailed in Materials and Methods. The quantitation yielded no statistical difference in nephrocalcinosis between the WT and Enpp1asj/asj mice treated with mEnpp1-Fc, whereas vehicle-treated Enpp1asj/asj mice experienced about a twofold increase in nephrocalcinosis when compared with WT or mEnpp1-Fc treated Enpp1asj/asj mice.
We chose not to suppress a potential neutralizing immune response in the mice with an anti-CD4 T-cell antibody, as we had done previously, to avoid immunosuppressive effects on bone mineralization.
Standard plasma biochemistries in the mEnpp1-treated mice exhibited no significant differences compared with vehicle-treated mice, but other measures that serve as markers of abnormal renal function—BUN, FGF23, and PTH(1–84)—were markedly elevated (Table 4).
Table 4.
Plasma Chemistry, 5-Week-Old Animals on Acceleration Diet
| Analytes | WT (n = 7) | Enpp1asj/asj + PBS (n = 3) | Enpp1asj/asj + ERT (n = 4) |
|---|---|---|---|
| PPi (uM) | 1.68 ± 0.15 | 0.32 ± 0.21** | 2.42 ± 0.61* |
| Pi (mg/dL) | 8.86 ± 1.37 | 8.82 ± 2.95 | 8.46 ± 2.0 |
| PTH (pg/mL) | 586 ± 251 | 1361 ± 607** | 870 ± 461 |
| BUN (mg/dL) | 31 ± 9 | 121 ± 42** | 39 ± 25 |
| Creatinine (mg/dL) | 0.17 ± 0.13 | 0.27 ± 0.10 | 0.18 ± 0.06 |
| FGF23 intact (pg/mL) | 613 ± 297 | 5287 ± 3377** | 1164 ± 334 |
| Na (mmol/L) | 126 ± 2 | 128 ± 9 | 127 ± 3 |
| K (mmol/L) | 4.4 ± 0.5 | 4.6 ± 0.6 | 4.7 ± 0.4 |
| Cl (mmol/l) | 110 ± 3 | 111 ± 3 | 108 ± 1 |
| HCT (%PCV) | 36.6 ± 4.8 | 42.7 ± 5 | 38.7 ± 2.6 |
| Hb (gm/dL) | 12.4 ± 1.6 | 14.5 ± 1.7 | 13.2 ± 0.9 |
| pH | 7.28 ± 0.06 | 7.27 ± 0.04 | 7.28 ± 0.04 |
p = .014.
p < .005.
Murine renal phenotype
To investigate the renal insufficiency further, we examined renal histology and associated analysis in kidneys of all mice. All mice on the acceleration diet exhibited nephrocalcinosis to some degree, with the most pronounced calcification occurring in the medullary tubules at the cortical-medullary junction, although cortical calcification near glomeruli was also observed (Fig. 3C). WT mice exhibited calcifications in 22% of renal area examined, whereas vehicle-treated Enpp1asj/asj mice exhibited calcifications in 51% of examined renal area. Treating Enpp1asj/asj mice with Enpp1-Fc reduced calcifications to WT levels (Fig. 3D). To correlate PPi levels with nephrocalcinosis, we measured urine PPi in 10-week WT and Enpp1asj/asj mice on regular chow, as the urine output in 5-week-old Enpp1asj/asj mice on the acceleration diet was inadequate for measurement. Urinary PPi/creatinine in male Enpp1asj/asj mice was about one-sixth that of WT mice (1.0 versus 6.0 ng/mg [Fig. 3D]), demonstrating an association between urinary PPi and nephrocalcinosis.
Murine skeletal phenotype and treatment response
Bone microarchitecture evaluated by micro-CT in 5-week-old Enpp1asj/asj mice on the acceleration diet exhibited trabecular bone microarchitectural defects similar to 10-week-old Enpp1asj/asj mice on regular chow, namely, decreased trabecular BV/TV and trabecular number at 72% and 83% of WT, respectively, and increased trabecular spacing at 125% of WT. When Enpp1asj/asj mice were treated with Enpp1-Fc, trabecular microarchitecture normalized (Fig. 4A, C). Also, similar to Enpp1asj/asj mice on regular chow, cortical microarchitectural defects were present in 5-week-old Enpp1asj/asj mice on the acceleration diet, with decreased cortical BV/TV, cortical thickness, and apparent density of cortical TV at 90%, 72%, and 76% of WT, respectively. In the case of cortical bone, treatment of Enpp1asj/asj mice on the acceleration diet with Enpp1-Fc improved, but did not normalize, the cortical bone microarchitecture (Fig. 4C).
Fig 4.
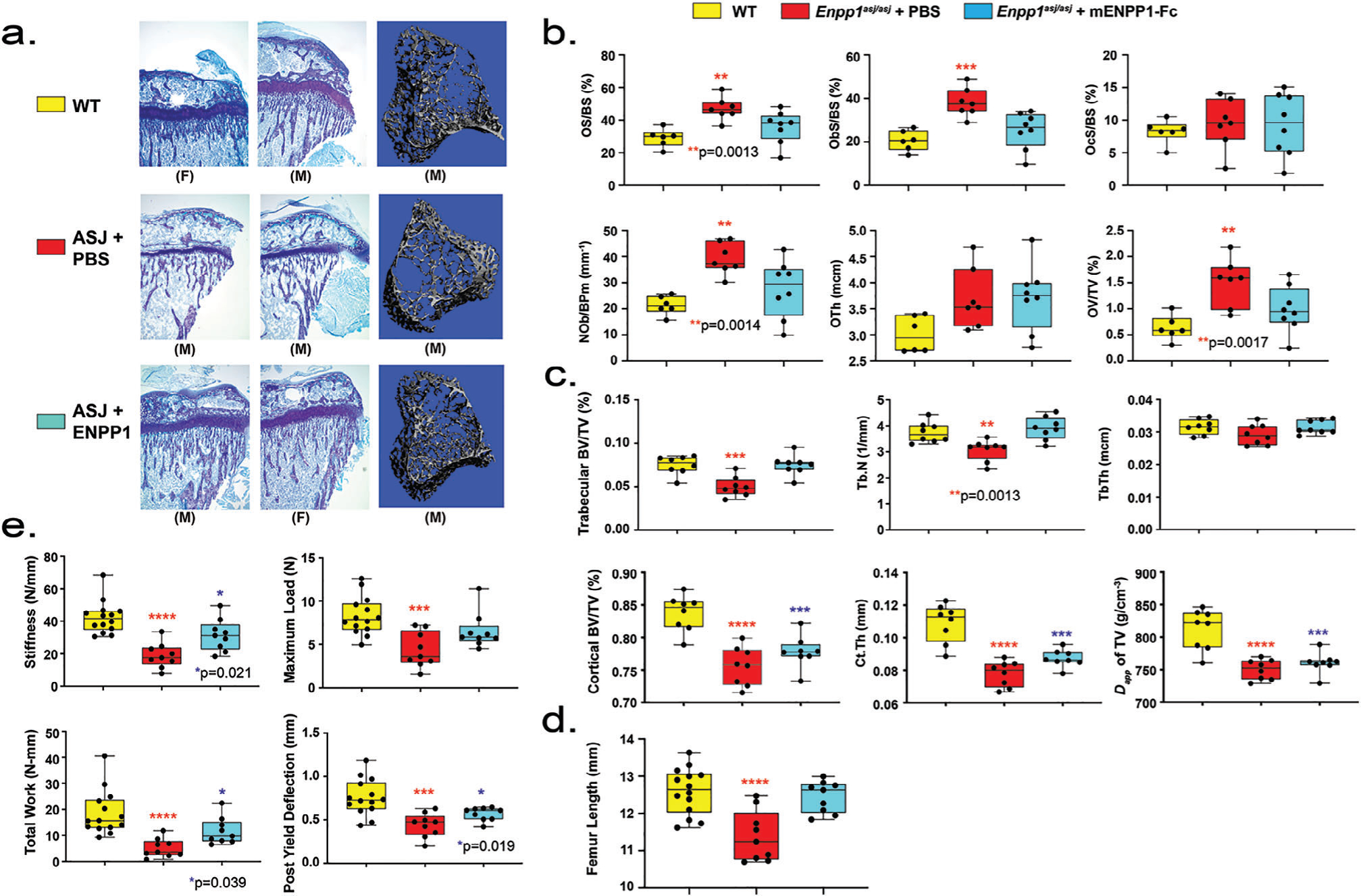
Bone phenotype of Enpp1asj/asj mice on high-phosphate diet treated with vehicle or mEnpp1-Fc. (A) Proximal tibia histology and micro-CT. Histology of the proximal tibias of 5-week-old mice on the high-phosphate diet are displayed from each experimental cohort to evaluate the skeletal phenotype of Enpp1asj/asj mice and the response to mEnpp1-Fc. ENPP1 deficiency resulted in morphologically apparent reductions in trabecular bone and markedly thinner growth plates. Treating Enpp1asj/asj mice with mEnpp1-Fc markedly increased trabecular bone volume and growth plate thickness. (B) Histomorphometry of female mice: osteoid surface area per bone surface area (OS/BS), osteoblast surface per bone surface (ObS/BS), osteoclast surface per bone surface (OcS/BS), osteoblasts number per bone perimeter (NoB/BPm), osteoid thickness, and osteoid volume per total volume (OV/TV). Individual measurements are displayed as circles with bar height representing median and error bars denoting interquartile range (25% to 75%). (C) Micro-CT quantification in female mice of trabecular BV/TV, trabecular number (Tb.N), trabecular thickness (Tb.Th), cortical BV/TV, cortical thickness (Ct.Th), and apparent density of total volume (TV). Data displayed as in panel B. (D) Biomechanical quantification by 3-point bending of femur bone parameters (stiffness—slope of the load versus displacement curve; max load—also known as strength; total work—the energy needed to fracture the bone; and post-yield deflection—the amount of deformation after the yield point) in male and female mice—14 WT mice treated with PBS (5 F and 9 M), 9 Enpp1asj/asj mice treated with PBS (4 F and 5 M), and 9 Enpp1asj/asj treated with mEnpp1-Fc (4 F and 5 M). Data are displayed as box plots denoting median value and interquartile range, with whiskers denoting minimum and maximum values and individual measurements denoted by circles. The p values are explicitly stated when p falls between .05 and .001. ***p < .001, ****p < .0001, ANOVA comparison of means.
Cell quantification evaluated by histomorphology in 5-week-old Enpp1asj/asj mice on the acceleration diet exhibited a marked increase in osteoblasts (Ob.S/BS 175% of WT, N.Ob/B.Pm at 193% of WT) without significant alterations in osteoclasts (Fig. 4B). Osteoid accumulation was increased in the mutants (O.Th 117% of WT and OS/BS 150% of WT). Finally, treatment of Enpp1asj/asj mice with Enpp1-Fc normalized both osteoblast quantification and osteoid parameters in the Enpp1-deficient mice with respect to their WT sibling pairs. The histomorphometry parameters were noted to be similar when analyzed in either secondary (Fig. 4C) or primary and secondary spongiosa combined (Supplemental Fig. S4), demonstrating that the differences noted were not artifacts of longitudinal growth or growth plate geometry.
The femurs of Enpp1asj/asj mice were noted to be significantly shorter than WT sibling pairs (Fig. 4D) and when biomechanically evaluated by 3-point bending tests were much less stiff (45% of WT), requiring significantly less total work until fracture (27% of WT) and were able to bear significantly less maximal load (57% of WT) (Fig. 4E). In addition, the bones of Enpp1asj/asj mice on the acceleration diet exhibited significantly reduced ductility (ie, were more brittle), reducing post-yield deflection to 65% of WT. Treating the Enpp1asj/asj mice with Enpp1-Fc increased femur length (Fig.4D), bone stiffness, total work until fracture, and post-yield deflection, and normalized the maximum load, demonstrating a marked biomechanical improvement (Fig. 4E). When normalized for bone length, the biomechanical findings remained unchanged (Supplemental Fig. S5). The data demonstrate that ENPP1 deficiency results in smaller bones with less bone mass, and treating ENPP1-deficient mice with ENPP1 enzyme replacement leads to bigger, stronger bones.
In summary, Enpp1asj/asj mice on the acceleration diet show histomorphometric, microradiographic, and biomechanical defects identical to Enpp1asj/asj mice on regular chow as previously reported, including microarchitectural defects in trabecular bone, increased osteoid accumulation, decreased bone mineral density of both trabecular and cortical bone, and increased propensity to fracture.(23) Treating Enpp1asj/asj mice on the acceleration diet with mEnpp1-Fc prevents many of these defects. We conclude, therefore, that the aberrant skeletal phenotype of Enpp1asj/asj mice on the acceleration diet is not due to the high phosphate content of the diet and that improvements in the skeletal phenotype observed in the treated mice is due to the effects of mEnpp1-Fc.
Assessment of renal osteodystrophy by Raman spectroscopy
Changes in bone matrix composition that may occur due to the mutation or therapy were assessed using Raman spectroscopy (Fig. 5A, B). Although the presence of renal osteodystrophy is expected to accompany higher matrix accumulation of phenylalanine and reductions in phosphate/matrix and carbonate/matrix ratios,(33) we found no difference in phosphate/matrix ratios (phosphate/amide I, phosphate/amide III, phosphate/phenylalanine) and carbonate/amide I ratio between WT mice and Enpp1asj/asj male or female mice whether or not they were treated with mEnpp1-Fc (Fig. 5C–F). Only female mice treated with vehicle showed a trend toward a lower phosphate/phenylalanine ratio. In sum, these results suggest that neither the mutation nor enzyme replacement therapy led to bone matrix composition consistent with renal osteodystrophy.
Fig 5.
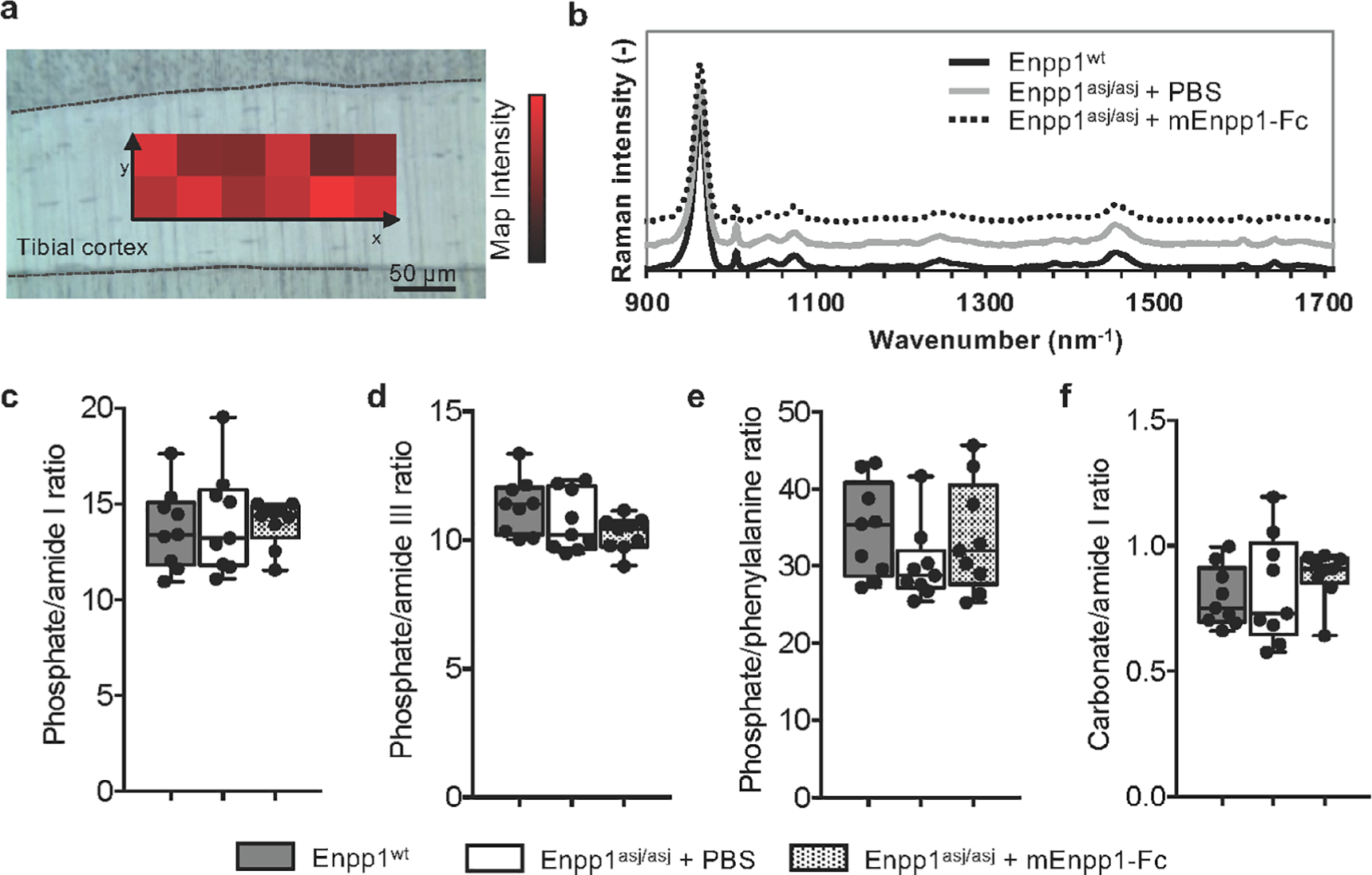
Raman spectroscopy measurements of WT and Enpp1asj/asj mice treated with vehicle or mEnpp1-Fc. (A) Rectangular maps were acquired on the lateral and medial tibial cortex of each specimen. (B) Comparing the normalized average Raman spectra of each group indicates no differences in peak size or location. (C–F) Raman metrics based on the peak areas were assessed in male and female mice and did not show any statistical difference in (C) phosphate/amide I ratio (p = .67 between females, p = .85 between males), (D) phosphate/amide III (p = .22 between females, p = .53 between males), (E) phosphate/phenylalanine (p = .11 between females, p = .91 between males), and (F) carbonate/amide I (p = .76 between females, p = .18 between males), suggesting that neither the mutation nor the therapy significantly affected the bone matrix composition at the tissue level.
Bone mineral density distribution
QBEI analysis of 5-week-old mice on acceleration diet showed only minor differences in matrix mineralization throughout the groups (Fig. 6A–E). In detail, BMDD histograms of Enpp1wt and Enpp1asj/asj treated with or without mEnpp1-Fc are similar, as reflected by CaMean and CaPeak, independent of whether treatment with mEnpp1-Fc was applied. Only CaWidth of male Enpp1asj/asj mice treated with enzyme replacement was reduced compared with WT mice (p = .0248) (Fig. 6E). Hence, in contrast to abnormalities found in 10-week-old animals on normal chow,(23) there is not a strong bone matrix mineralization defect at an age of 5 weeks on the acceleration diet. Further, assessments of Ot.Lc.Ar and N.Ot.Lc/B.Ar (Fig. 6F, G) indicate that neither Enpp1 deficiency nor treatment with enzyme replacement has an effect on two-dimensional osteocyte lacunar characteristics in young male and female mice on acceleration diet.
Fig 6.
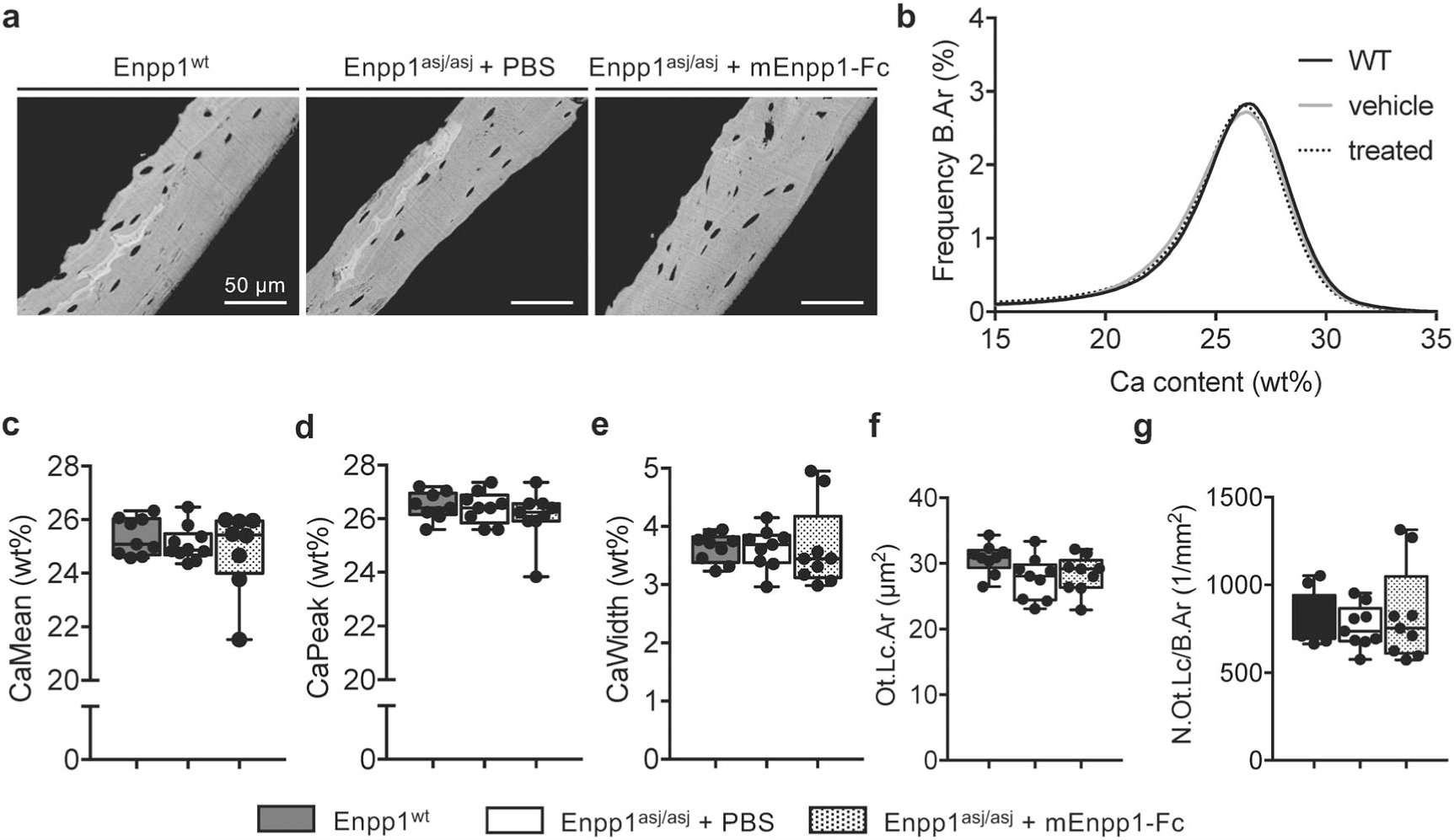
qBEI analysis for BMDD and osteocyte lacunae. (A) qBEI images of Enpp1wt mice and Enpp1asj/asj mice treated with and without mEnpp1-Fc representing the calcium content of mineralized matrix. (B) BMDD curves show no differences between the three groups. (C–E). Determined parameters for mineralization, ie, CaMean, CaPeak and CaWidth, show no significant differences between the groups except reduced CaWidth in male Enpp1asj/asj mice treated with mEnpp1-Fc compared with WT littermates. (F, G) Ot.Lc.Ar and N.Ot.Lc/B.Ar also show no significant differences.
Discussion
In this study, we evaluated the effect of conventional ARHR2 therapy on bone density in patients with biallelic ENPP1 mutations. We found that similar to adults with heterozygous ENPP1 mutations, adults with ARHR2 are likely to exhibit low bone density. Furthermore, half of all individuals who received standard therapy for hypophosphatemic rickets developed nephrocalcinosis as a consequence of this treatment. Enpp1asj/asj mice on a high-phosphate diet recapitulated the skeletal and renal phenotypes of human ENPP1 deficiency, in which renal calcifications and low bone mass are prominent features. Finally, a child with ARHR2 who initially presented with rickets and reduced bone density failed to show improved bone density after 2 years of oral phosphate and calcitriol supplementation, suggesting that conventional therapy for hypophosphatemic rickets might not adequately address bone disease in ARHR2 individuals. We compared the clinical response of ARHR2 subjects with that of Enpp1asj/asj mice maintained on a high-phosphate, low-magnesium diet with or without Enpp1 ERT. We found that Enpp1asj/asj mice on this acceleration diet exhibited osteopenia at 5 weeks comparable to the osteopenia observed at 10 weeks in Enpp1asj/asj mice on regular chow.(23) Treating the Enpp1asj/asj mice with Enpp1 ERT improved bone mineral density, normalized some bone matrix characteristics and cellularity, increased bone strength, and reduced bone fragility, whereas control Enpp1asj/asj mice exhibited renal calcification similar in histologic appearance to that observed in GACI patients at autopsy.
A limitation of our study was the development of renal failure in Enpp1asj/asj mice on the diet used in our model, which is not observed in GACI patients who survive the crucial first 6 months of the disease. The renal insufficiency manifested at 4 weeks in Enpp1asj/asj mice as a decrease in weight, and as we evaluated the skeletal phenotype in 5-week-old animals, uremia was likely present for approximately 1 week. The uremia was induced by extensive nephrocalcinosis and was prevented by treatment with mEnpp1-Fc, likely through the elevation of glomerular/urinary PPi and possibly reduction of phosphate wasting. Although we cannot exclude the possibility that renal osteodystrophy contributed to the skeletal abnormalities present in 5-week-old animals, we did not detect Raman spectroscopic features typical of renal osteodystrophy. Furthermore, the skeletal phenotype of 5-week-old Enpp1asj/asj mice fed an acceleration diet was nearly identical to the skeletal phenotype of 10-week-old Enpp1asj/asj mice fed a normal chow diet, further arguing against assertions that the skeletal phenotype in Enpp1asj/asj mice is a consequence of renal failure. Observations that a skeletal phenotype observed in Enpp1asj/asj mice on either normal chow or a laboratory diet that is corrected by treating the mice on the laboratory diet with Enpp1 ERT strongly supports the notion that Enpp1 ERT addresses the skeletal phenotype induced by ENPP1 deficiency.
We recently investigated the genetic pathways driving the skeletal phenotype of ENPP1 deficiency by comparing gene expression in Enpp1asj/asj mice and WT sibling pairs by RNAseq and qPCR in whole bones and in the liver and kidney by qPCR, directly correlating gene expression with measures of bone microarchitectural and biomechanical phenotypes.(34) The animals were fed a regular chow diet, and we found that ENPP1-deficient mice exhibited reduced gene transcription of Wnt ligands in whole bone and increased transcription of soluble Wnt inhibitors in the liver and kidney, suggestive of multiorgan inhibition of Wnt activity. These findings suggest a genetic mechanism for observations in this study that Enpp1asj/asj mice have smaller bones that are more prone to fracture and that treating the ENPP1-deficient mice with ENPP1 ERT results in bigger and stronger bones in contrast to treating the mice with a high-phosphate diet alone.
Subjects with ARHR2 are often treated with phosphate and calcitriol supplementation, and some patients respond well to this therapy.(12) Nevertheless, there remains the theoretical concern that this therapy will induce progressive or even new vascular calcifications. Treatment of ARHR2 with phosphate and calcitriol supplementation has been reported to induce iatrogenic new-onset calcinosis in at least one ARHR2 patient,(13) with calcifications in the kidneys being especially difficult to reverse. Although nephrocalcinosis has been previously described in individuals with other causes of hyperphosphaturia such as X-linked hypophosphatemia, the concomitant finding of low plasma PPi in patients with ENPP1 deficiency might contribute to mineral deposition in the renal parenchyma and lead to more severe manifestations. Pyrophosphate is the most effective inhibitor of hydroxyapatite precipitation identified in the urine, with concentrations as low as 2.87 μmol/L causing complete inhibition of crystallization for at least 1 hour.(35) Analysis from our cohort of ARHR2 patients showed that medullary nephrocalcinosis was present in half of all patients and occurred as a direct consequence of treatment for rickets within months’ initiation. Our murine findings suggest that ENPP1 ERT may reduce the risk of nephrocalcinosis while also enhancing the beneficial effects of standard therapy. Interestingly, one patient developed the characteristic dotted hyperechogenic cortical pattern typical of patients with pseudoxanthoma elasticum,(36–38) while deceased siblings showed cortical calcification, a finding that has been described in the setting of cortical nephrocalcinosis due to ischemia.(39) In addition, a previously reported infant with GACI exhibited diffuse hyperechogenicity of the renal cortex on ultrasonography before death at 1 month of age.(40) Thus, it appears likely that cortical deposition of calcium is a manifestation of the vascular complications of the disease, whereas medullary calcinosis reflects the increased phosphate and decreased pyrophosphate present in urine.
Human ENPP1 deficiency results in either increased vascular calcification (in GACI), ineffective bone mineralization (in ARHR2), or the asynchronous development of both. For patients with GACI who later develop ARHR2, the association of the identical ENPP1 isoform with vascular calcification and concurrent low bone mass can be referred to as so-called “paradoxical mineralization” to underscore a confusing pathophysiology. A close examination of clinical data may invoke some clarifying principles. GACI is not uniformly fatal, and survival in GACI is associated with elevation of FGF23.(2) FGF23 elevation is also the underlying mechanism of ARHR2, wherein plasma Pi is lowered to the degree that bone mineralization is impaired. The association of phosphate wasting with survival in GACI suggests that elevation of FGF23 may be an adaptive mechanism counteracting potentially lethal ectopic calcifications induced by low plasma PPi through rebalancing plasma Pi, thereby enabling survival at the expense of bone mineralization. The observations also suggest that treatment of ARHR2 with burosomab, a monoclonal antibody against FGF23, may be deleterious.
In summary, treating Enpp1asj/asj mice with daily subcutaneous injections of mEnpp1-Fc improved osteopenia by normalizing trabecular BMD to WT levels, eliminated the osteoblastic proliferation, normalized osteoid volumes, and markedly improved bone strength and bone stiffness. Our findings demonstrate significant and beneficial effects of ENPP1 ERT on the skeletal phenotype of murine ENPP1 deficiency. Our study also reflects that low bone mineral density is common in subjects with biallelic ENPP1 mutations and may indicate osteomalacia and/or osteoporosis.(41) Furthermore, we have demonstrated that the Enpp1asj/asj mouse model recapitulates skeletal and renal manifestations present in human ENPP1 deficiency and that ENPP1 ERT ameliorates these disease phenotypes. Our observations may have relevance for the treatment and management of other so-called paradoxical mineralization disorders exhibiting both increased vascular calcification and decreased BMD, such as chronic kidney disease metabolic bone disorder.
Supplementary Material
Acknowledgments
These studies were financially supported by Inozyme Pharma and the National Institutes of Health through R01 DK121326 to DTB. Funding to the George M O’Brien Kidney Center at Yale, NIH grant P30DK079310, supported the evaluation of plasma analytes.
Disclosures
PRS and DTB are inventors of patents owned by Yale University for therapeutics treating ENPP1 deficiency. DTB is an equity holder and receives research and consulting support from Inozyme Pharma, Inc. RO received travel reimbursement and honorarium from Inozyme Pharma, Inc.
Footnotes
Additional Supporting Information may be found in the online version of this article.
References
- 1.Chong CR, Hutchins GM. Idiopathic infantile arterial calcification: the spectrum of clinical presentations. Pediatr Dev Pathol. 2008;11(5): 405–415. [DOI] [PubMed] [Google Scholar]
- 2.Rutsch F, Boyer P, Nitschke Y, et al. Hypophosphatemia, hyperphosphaturia, and bisphosphonate treatment are associated with survival beyond infancy in generalized arterial calcification of infancy. Circ Cardiovasc Genet. 2008;1(2):133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rutsch F, Vaingankar S, Johnson K, et al. PC-1 nucleoside triphosphate pyrophosphohydrolase deficiency in idiopathic infantile arterial calcification. Am J Pathol. 2001;158(2):543–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rutsch F, Ruf N, Vaingankar S, et al. Mutations in ENPP1 are associated with ‘idiopathic’ infantile arterial calcification. Nat Genet. 2003; 34(4):379–381. [DOI] [PubMed] [Google Scholar]
- 5.Li Q, Brodsky JL, Conlin LK, et al. Mutations in the ABCC6 gene as a cause of generalized arterial calcification of infancy: genotypic overlap with pseudoxanthoma elasticum. J Invest Dermatol. 2014;134(3): 658–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nitschke Y, Baujat G, Botschen U, et al. Generalized arterial calcification of infancy and pseudoxanthoma elasticum can be caused by mutations in either ENPP1 or ABCC6. Am J Hum Genet. 2012;90(1): 25–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marrott PK, Newcombe KD, Becroft DM, Friedlander DH. Idiopathic infantile arterial calcification with survival to adult life. Pediatr Cardiol. 1984;5(2):119–122. [DOI] [PubMed] [Google Scholar]
- 8.Ciana G, Trappan A, Bembi B, et al. Generalized arterial calcification of infancy: two siblings with prolonged survival. Eur J Pediatr. 2006;165 (4):258–263. [DOI] [PubMed] [Google Scholar]
- 9.Miyai K, Ariyasu D, Numakura C, Yoneda K, Nakazato H, Hasegawa Y. Hypophosphatemic rickets developed after treatment with etidro-nate disodium in patient with generalized arterial calcification in infancy. Bone Rep. 2015;3:57–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Levy-Litan V, Hershkovitz E, Avizov L, et al. Autosomal-recessive hypophosphatemic rickets is associated with an inactivation mutation in the ENPP1 gene. Am J Hum Genet. 2010;86(2):273–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lorenz-Depiereux B, Schnabel D, Tiosano D, Hausler G, Strom TM. Loss-of-function ENPP1 mutations cause both generalized arterial calcification of infancy and autosomal-recessive hypophosphatemic rickets. Am J Hum Genet. 2010;86(2):267–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ferreira CR, Ziegler SG, Gupta A, Groden C, Hsu KS, Gahl WA. Treatment of hypophosphatemic rickets in generalized arterial calcification of infancy (GACI) without worsening of vascular calcification. Am J Med Genet A. 2016;170(5):1308–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Freychet C, Gay C, Lavocat MP, et al. [GACI syndrome: a case report with a neonatal beginning]. Arch Pediatr. 2014;21(6):632–636. [DOI] [PubMed] [Google Scholar]
- 14.Li Q, Guo H, Chou DW, Berndt A, Sundberg JP, Uitto J. Mutant Enpp1asj mice as a model for generalized arterial calcification of infancy. Dis Model Mech. 2013;6(5):1227–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Albright RA, Stabach P, Cao W, et al. ENPP1-Fc prevents mortality and vascular calcifications in rodent model of generalized arterial calcification of infancy. Nat Commun. 2015;6:10006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Khan T, Sinkevicius KW, Vong S, et al. ENPP1 enzyme replacement therapy improves blood pressure and cardiovascular function in a mouse model of generalized arterial calcification of infancy (GACI). Dis Model Mech. 2018;11(10):dmm035691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nitschke Y, Yan Y, Buers I, Kintziger K, Askew K, Rutsch F. ENPP1-Fc prevents neointima formation in generalized arterial calcification of infancy through the generation of AMP. Exp Mol Med. 2018;50 (10):139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kobayashi Y, Goto S, Tanno T, Yamazaki M, Moriya H. Regional variations in the progression of bone loss in two different mouse osteopenia models. Calcif Tissue Int. 1998;62(5):426–436. [DOI] [PubMed] [Google Scholar]
- 19.Okawa A, Goto S, Moriya H. Calcitonin simultaneously regulates both periosteal hyperostosis and trabecular osteopenia in the spinal hyperostotic mouse (twy/twy) in vivo. Calcif Tissue Int. 1999;64(3): 239–247. [DOI] [PubMed] [Google Scholar]
- 20.Hajjawi MO, MacRae VE, Huesa C, et al. Mineralisation of collagen rich soft tissues and osteocyte lacunae in Enpp1(−/−) mice. Bone. 2014; 69:139–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mackenzie NC, Zhu D, Milne EM, et al. Altered bone development and an increase in FGF-23 expression in Enpp1(−/−) mice. PLoS One. 2012;7(2):e32177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Babij P, Roudier M, Graves T, et al. New variants in the Enpp1 and Ptpn6 genes cause low BMD, crystal-related arthropathy, and vascular calcification. J Bone Miner Res. 2009;24(9):1552–1564. [DOI] [PubMed] [Google Scholar]
- 23.Oheim R, Zimmerman K, Maulding ND, et al. Human heterozygous ENPP1 deficiency is associated with early onset osteoporosis, a phenotype recapitulated in a mouse model of Enpp1 deficiency. J Bone Miner Res. 2020;35(3):528–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rolvien T, Koehne T, Kornak U, et al. A novel ANO5 mutation causing gnathodiaphyseal dysplasia with high bone turnover osteosclerosis. J Bone Miner Res. 2017;32(2):277–284. [DOI] [PubMed] [Google Scholar]
- 25.Rolvien T, Sturznickel J, Schmidt FN, et al. Comparison of bone microarchitecture between adult osteogenesis imperfecta and early-onset osteoporosis. Calcif Tissue Int. 2018;103(5):512–521. [DOI] [PubMed] [Google Scholar]
- 26.Fewtrell MS, Gordon I, Biassoni L, Cole TJ. Dual X-ray absorptiometry (DXA) of the lumbar spine in a clinical paediatric setting: does the method of size-adjustment matter? Bone. 2005;37(3):413–419. [DOI] [PubMed] [Google Scholar]
- 27.Rehberg M, Azim M, Martakis K, et al. Bone microarchitecture assessed by trabecular bone score is independent of mobility level or height in pediatric patients with cerebral palsy. J Bone Miner Res. 2020;35(9):1685–1694. [DOI] [PubMed] [Google Scholar]
- 28.Kotwal A, Ferrer A, Kumar R, et al. Clinical and biochemical phenotypes in a family with ENPP1 mutations. J Bone Miner Res. 2020;35 (4):662–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ware CB, Horowitz MC, Renshaw BR, et al. Targeted disruption of the low-affinity leukemia inhibitory factor receptor gene causes placental, skeletal, neural and metabolic defects and results in perinatal death. Development. 1995;121(5):1283–1299. [DOI] [PubMed] [Google Scholar]
- 30.Parfitt AM, Drezner MK, Glorieux FH, et al. Bone histomorphometry: standardization of nomenclature, symbols, and units. Report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res. 1987;2(6):595–610. [DOI] [PubMed] [Google Scholar]
- 31.Fiedler IAK, Casanova M, Keplinger T, Busse B, Muller R. Effect of short-term formaldehyde fixation on Raman spectral parameters of bone quality. J Biomed Opt. 2018;23(11):1–6. [DOI] [PubMed] [Google Scholar]
- 32.Tchanque-Fossuo CN, Gong B, Poushanchi B, et al. Raman spectroscopy demonstrates amifostine induced preservation of bone mineralization patterns in the irradiated murine mandible. Bone. 2013;52 (2):712–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Manciu M, Cardenas M, Bennet KE, et al. Assessment of renal osteodystrophy via computational analysis of label-free Raman detection of multiple biomarkers. Diagnostics (Basel). 2020;10(2):79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maulding ND, Kavanagh D, Zimmerman K, et al. Genetic pathways disrupted by ENPP1 deficiency provide insight into mechanisms of osteoporosis, osteomalacia and paradoxical mineralization. Bone. 2020;142:115656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grases F, Ramis M, Costa-Bauza A. Effects of phytate and pyrophosphate on brushite and hydroxyapatite crystallization. Comparison with the action of other polyphosphates. Urol Res. 2000;28(2):136–140. [DOI] [PubMed] [Google Scholar]
- 36.Seeger H, Mohebbi N. Pseudoxanthoma elasticum and nephrocalcinosis. Kidney Int. 2016;89(6):1407. [DOI] [PubMed] [Google Scholar]
- 37.Vasudevan B, Shijith KP, Bahal A, Raghav V. Pseudoxanthoma elasticum with renal cortical calcification. Med J Armed Forces India. 2010;66(3):272–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Suarez MJ, Garcia JB, Orense M, Raimunde E, Lopez MV, Fernandez O. Sonographic aspects of pseudoxanthoma elasticum. Pediatr Radiol. 1991;21(7):538–539. [DOI] [PubMed] [Google Scholar]
- 39.Schepens D, Verswijvel G, Kuypers D, Vanrenterghem Y. Images in nephrology. Renal cortical nephrocalcinosis. Nephrol Dial Transplant. 2000;15(7):1080–1082. [DOI] [PubMed] [Google Scholar]
- 40.Rosenbaum DM, Blumhagen JD. Sonographic recognition of idiopathic arterial calcification of infancy. AJR Am J Roentgenol. 1986; 146(2):249–250. [DOI] [PubMed] [Google Scholar]
- 41.Jha S, Chapman M, Roszko K. When low bone mineral density and fractures is not osteoporosis. Curr Osteoporos Rep. 2019;17(5): 324–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.