

The retinoblastoma protein regulates mutagenic potential via control of UV-induced DNA lesion acquisition, thus revealing novel mechanisms that contribute to its tumor suppressor capabilities.
Abstract
Carcinogenic insult, such as UV light exposure, creates DNA lesions that evolve into mutations if left unrepaired. These resulting mutations can contribute to carcinogenesis and drive malignant phenotypes. Susceptibility to carcinogens (i.e., the propensity to form a carcinogen-induced DNA lesion) is regulated by both genetic and epigenetic factors. Importantly, carcinogen susceptibility is a critical contributor to cancer mutagenesis. It is known that mutations can be prevented by tumor suppressor regulation of DNA damage response pathways; however, their roles carcinogen susceptibility have not yet been reported. In this study, we reveal that the retinoblastoma (RB1) tumor suppressor regulates UV susceptibility across broad regions of the genome. In particular, centromere and telomere-proximal regions exhibit significant increases in UV lesion susceptibility when RB1 is deleted. Several cancer-related genes are located within genomic regions of increased susceptibility, including telomerase reverse transcriptase, TERT, thereby accelerating mutagenic potential in cancers with RB1 pathway alterations. These findings reveal novel genome stability mechanisms of a tumor suppressor and uncover new pathways to accumulate mutations during cancer evolution.
Introduction
Loss of tumor suppressor function is a fundamental characteristic of malignancy. Tumor suppressors regulate proliferation, senescence, and apoptotic pathways through a variety of mechanisms that sense extracellular environments and intracellular stress signals (Hanahan & Weinberg, 2011). The first identified tumor suppressor was the retinoblastoma gene, RB1, which causes pediatric malignancy of the eye after biallelic inactivation (Fung et al, 1987; Lee et al, 1987; Horowitz et al, 1989).
RB1 regulates cell cycle progression through its repression of E2F-responsive genes (Weinberg, 1995). It is active in early G1 phase and inactivated via phosphorylation by CDKs during the G1 to S transition to allow expression of genes, such as S-phase cyclins, that promote DNA synthesis and cell growth. CDK inhibitors CDKN2A/p16 and CDKN1A/p21 can also inhibit phosphorylation of RB1 by binding to and inactivating CDKs, thereby preventing cell cycle progression. This elaborate tumor suppressor-regulated network restricts cell cycle growth and proliferation to favorable environments that promote survival (Weinberg, 1995).
RB1 functions as a transcriptional repressor of cell cycle genes through its association with chromatin modifiers, which effectively inhibits target gene expression (Dick & Rubin, 2013). Specifically, RB1 associates with histone deacetylases, H3K9 and H3K27 methyltransferases, and heterochromatin protein 1 (HP1) to alter the chromatin around target loci (Luo et al, 1998; Nielsen et al, 2001; Morrison et al, 2002; Ishak et al, 2016). Interestingly, RB1 has also been found to regulate constitutive heterochromatin, particularly at centromeres and telomeres, impacting chromatid cohesion and telomere length regulation (García-Cao et al, 2002; Gonzalo et al, 2005; Manning et al, 2010, 2014; Ishak et al, 2016; Gonzalez-Vasconcellos et al, 2017).
Alteration of the RB1 pathway is pervasive in many sporadic cancers (Dyson, 2016). For example, the RB1 pathway is a primary driver of skin cancers, with mutations in RB1, CDKN2A/p16, CDKN1A/p21, CDKN1B/p27, CDKN2B/p15, CCNE1/cyclin E, and CCND1/cyclin D, collectively, in 26.4% of melanomas (Hayward et al, 2017). Loss of tumor suppressor function allows unrestricted proliferation and accumulation of genome instabilities that promote malignant transformation (Hanahan & Weinberg, 2011). This transformation can, expectedly, be accelerated in the presence of carcinogens that initiate mutagenesis. Indeed, alteration of the RB1 pathway in mice leads to increases in tumor formation, particularly after exposure to chemical carcinogens and radiation (Serrano et al, 1996; Krimpenfort et al, 2001; Sharpless et al, 2001). For example, CDKN2A/p16 deletion leads to increased metastatic melanoma after carcinogen exposure (Krimpenfort et al, 2001).
Cancers initiated by carcinogen exposure, such as skin and lung cancers, are among those with the highest frequency of somatic mutation (Alexandrov et al, 2013). Melanoma, in particular, is caused by UV exposure in sunlight, the most ubiquitous environmental carcinogen. The major classes of DNA lesions induced by UV radiation are cyclobutane pyrimidine dimers (CPDs) and 6–4 photoproducts (6–4PPs) (Douki & Cadet, 2001), which are targeted by nucleotide excision repair pathways, transcription-coupled repair and global genomic repair (Hanawalt & Spivak, 2008). CPDs are the most abundant UV-induced DNA lesion and proposed to be the most mutagenic (You et al, 2001; Jans et al, 2005; Pfeifer & Besaratinia, 2012; Brash, 2015). Interestingly, the distribution of UV-induced mutations is not uniform across melanoma genomes and found to be regulated by regional features surrounding mutation sites, such as base composition and replication timing (Wolfe et al, 1989; Stamatoyannopoulos et al, 2009). Additionally, transcriptionally repressive heterochromatic regions accumulate more mutations than transcriptionally active euchromatin (Schuster-Böckler & Lehner, 2012; Polak et al, 2015), which is contributed by lack of transcription-coupled repair in non-genic regions and inefficient global genomic repair in heterochromatic regions (Hu et al, 2015, 2017).
However, mutation frequency can also be influenced by regional propensity to form UV-induced DNA lesions (i.e., susceptibility), which precedes repair. Formation of these UV-induced DNA lesions is the initial step of mutagenesis, and thus, essential for establishing mutagenic potential throughout the genome. Interestingly, published data from our laboratory demonstrates that epigenomic features, such as repeat-rich heterochromatin, influence the abundance of ultraviolet CPD lesion formation across the genomes of primary cells (García-Nieto et al, 2017). In these assays, cells were exposed to brief (less than 10 s) dose of UV light then immediately lysed and purified DNA for immunoprecipitation (IP) with CPD-specific antibodies. Repair of CPD lesions is marginally detectable (<5% of all lesions) within 1 h after UV exposure (Moser et al, 2005; Verbruggen et al, 2014; Adar et al, 2016), thus these assays are designed to measure acute UV-mediated DNA lesion formation rather than repair kinetics.
Surprisingly, we found that DNase I–hypersensitive, gene-dense regions with active transcription, are among the chromatin states that are the least UV susceptible (García-Nieto et al, 2017). Conversely, DNase I–protected heterochromatin with repressed transcription is more susceptible to UV compared with the genome-wide average. Importantly, these trends in UV susceptibility are still observed when corrected for dipyrimidine frequency. Thus, susceptibility is not solely driven by base composition, but can be regulated by epigenetic features. Notably, UV susceptibility in primary cells closely reflects mutation frequency across the genomes of malignant melanomas, thus susceptibility is a significant contributor to mutagenesis during cancer evolution.
Because of its roles in chromatin regulation and tumor suppression, we investigated whether RB1 could also influence UV susceptibility, a mechanism distinct from its previously characterized roles. Given that heterochromatin features positive correlate with UV susceptibility (García-Nieto et al, 2017), we proposed that loss of RB1-regulated heterochromatin would reduce acquisition of UV lesions in corresponding regions of the genome. However, we found that deregulation of heterochromatin at and around telomeres and centromeres in RB1 knockout cells correlates with increased susceptibility. Notably, several cancer driver genes are located in these regions of increased susceptibility, such as TERT, thereby increasing its mutagenic potential. Correspondingly, TERT mutations are significant elevated in melanomas with RB1 pathway alterations.
These results uncover new mechanisms that regulate carcinogen susceptibility. Importantly, this study reveals novel pathways in which tumor suppressors can promote genome stability, and how these pathways can become disrupted to accelerate mutagenesis and malignancy.
Results
After creation of RB1 knockout (hereafter referred to as RB1 KO) via CRISPR-Cas9 editing in IMR90 primary cells (Fig 1A), the CPD lesion IP protocol was repeated as previously described (García-Nieto et al, 2017) and shown in Fig 1B. Importantly, before UV exposure, cells were confluent for 2 d to synchronize cells in G1 phase of the cell cycle and avoid cell cycle effects on susceptibility resulting from reduced RB1 function. FACS analysis determined comparable cell cycle occupancy for both cell lines, specifically 79% G1, 4% S-phase, and 18% G2/M for wild-type; and 81% G1, 3% S-phase, and 16% G2/M for RB1 KO cells.
Figure 1. Deletion of RB1 alters UV susceptibility.

(A) Western blot of RB1 in RB1 knockout (RB1 KO) and wild-type (WT) IMR-90 cells. Tubulin was used as loading control. Molecular weight (MW) in kD is shown next to blot. (B) Illustration of UV lesion mapping method. Both wild-type and RB1 KO cells were treated with 100 J/m2 UVC. Cells were immediately lysed after UV irradiation and DNA was harvested. Purified DNA was then immunoprecipitation with anti-cyclobutane pyrimidine dimer antibody. Immunoprecipitated fragments were then repaired by photolyases prior to library preparation and Illumina sequencing. (C) Annotation of previously published RB1 peaks in proliferating and quiescent IMR-90 cells (Chicas et al, 2010). (D) Expected versus observed RB1 ChIP-seq peaks in top 10% and bottom 10% UV susceptible genomic regions, in both proliferating and quiescent cells. Expected values were generated based on the null hypothesis that susceptibility change has no association with location of RB1 peaks. Chi-squared test (P < 0.001) indicated that there is significant depletion and enrichment of RB1 peaks in top 10% and bottom 10% regions, respectively.
Differences in UV susceptibility between wild-type and RB1 KO cells were first examined at previously published RB1 ChIP-seq binding sites throughout the genome (Chicas et al, 2010). Indicative of its role as a transcription factor of cell cycle genes, RB1 was found to be enriched at promoters in both proliferating (76%) and quiescent (74%) cells (Fig 1C). For both these conditions, the majority of these RB1 binding sites did not overlap with the genomic regions that have significant changes in UV-induced lesions in RB1 KO cells compared with wild-type (Fig 1D). However, there is an enrichment of a minor number of RB1 binding sites in the least (bottom 10%) UV susceptible regions of the genome, and a corresponding reduction of overlap in the most (top 10%) susceptible regions. This is in agreement with previous results demonstrating that gene-rich regions, including those regulated by RB1, are among the most protected against UV-induced damage (García-Nieto et al, 2017); and loss of RB1 function does not significantly increase UV susceptibility in these genic regions.
RB1 regulates UV susceptibility in heterochromatin regions
However, as previously introduced, RB1 not only regulates cell cycle genes but also heterochromatin features at centromeres (Gonzalo et al, 2005; Isaac et al, 2006; Manning et al, 2010) and telomeres (García-Cao et al, 2002). Specifically, loss of RB1 function is associated with changes in the distribution of both constitutive H3K9me3 and facultative H3K27me3 heterochromatin modifications in these regions (Ishak et al, 2016; Gonzalez-Vasconcellos et al, 2017).
Because UV susceptibility is mechanistically linked to H3K9me3-enriched heterochromatin (García-Nieto et al, 2017), the epigenetic features associated with changes in susceptibility upon RB1 loss were explored. Specifically, UV lesion abundances between wild-type and RB1 KO cells were examined in relation to a variety of histone modifications mapped by the Roadmap Epigenomics Project (Roadmap Epigenomics Consortium et al, 2015). Principal component analysis of genome-wide UV lesion abundances in both wild-type and RB1 KO cells cluster closely with constitutive heterochromatin features, such as H3K9me3 and lamin B1, followed by H3K27me3-enriched chromatin, whereas most euchromatic modifications do not cluster with UV susceptibility (Fig 2A).
Figure 2. Genome-wide susceptibility changes in RB1 KO cells are associated with heterochromatin alterations.

(A) Principal component analysis of epigenetic features, replication timing, and UV lesions (UV) in 100 kb bins genome-wide (The ENCODE Project Consortium, 2012; Sadaie et al, 2013; Lund et al, 2014; Roadmap Epigenomics Consortium et al, 2015; Nelson et al, 2016). WT, wild-type; KO, RB1 KO. Variance explained is labelled for principal component 1 (PC1) and PC2. Known euchromatic features are labelled with blue dots, heterochromatic with orange dots, and UV lesions with purple dots. (B) Western blot of H3K9me3 and H3K27me3 in RB1 knockout (RB1 KO) and wild-type (WT) IMR90 cells. Tubulin was used as loading control. Molecular weight (MW) in kD is shown next to blot. (C) Box plots of log2 fold change (FC) of histone modifications (H3K9me3 and H3K27me3) in RB1 KO compared with WT (RB1 KO/WT) in 15 chromatin states from IMR90, as defined in the Roadmap Epigenomics Consortium et al (2015). TSS, transcriptional start site; Biv., Bivalent; Poly., Polycomb. Known heterochromatic states are labelled in red, euchromatic states are labelled in green. Genome-wide median is labelled by the white box and horizontal yellow dotted line. Statistical outliers were omitted. Mann–Whitney U test denotes significantly different (P < 0.001) states from the genome median, indicated by *. Heat maps below box plot depict scaled signal of median H3K9me3 and H3K27me3 ChIP signal in each chromatin state. (D) Scatter plot (100 kb bins) with spearman correlation of log2 FC UV lesion and H3K9me3 and H3K27me3 histone modifications in RB1 KO, WT, and RB1 KO versus WT (RB1 KO/WT). Linear regression line is shown in blue. (E) UV lesion, H3K9me3 and H3K27me3 levels in WT A and B Hi-C compartments, previously determined (Fortin & Hansen, 2015), in RB1 KO, WT, and FC of RB1 KO versus WT (RB1 KO/WT). Each compartment was divided into 50 bins for signal calculation. (A, B) The closed compartments (B) shaded in red, were aligned in the middle with the adjacent open compartments (A) on each side. The mean signal and 95% confidence interval via bootstrapping (R = 1,000) are in black and grey, respectively.
H3K9me3 and H3K27me3 ChIP-seq was then performed in wild-type and RB1 KO cells to identify chromatin changes that may contribute to altered susceptibility. At a cellular level, no significant differences in these histone marks were observed (Fig 2B). However, compared with wild-type cells, RB1 KO cells exhibit reduced H3K9me3 in several bivalent and zinc finger chromatin states (Fig 2C). These bivalent states are enriched in H3K27me3 and H3K9me3 in wild-type cells (Bernstein et al, 2006; Matsumura et al, 2015), yet in RB1 KO cells, levels were reduced. Conversely, more subtle, yet statistically significant, increases in these heterochromatin marks were identified in the “Quiescent” state for H3K9me3 and “Weak Repressed Polycomb” for H3K27me3. A reduction of repressive marks in RB1 KO cells is consistent with RB1’s association with repressive histone modifiers (Dick & Rubin, 2013), and alterations in bivalent domains is in agreement with the association of RB1 with the EZH2 repressor complex that mediates bivalent domain silencing (Ishak et al, 2016).
Across the genomes of wild-type cells we found a positive correlation between UV susceptibility and H3K9me3, whereas H3K27me3 exhibited a modest negative correlation (Fig 2D, and as previously reported [García-Nieto et al, 2017]). The relationship between susceptibility and H3K9me3 and, to a lesser extent, H3K27me3 was reinforced in RB1 KO cells (Fig 2D).
Interestingly, the link between heterochromatic features and UV susceptibility is strong enough to be observed at the level of topologically associated domains (TADs) composed of insulated heterochromatin and euchromatin compartments, and corresponding histone marks and transcriptional activity (Dixon et al, 2016). Specifically, the heterochromatic compartments of TADs acquire more UV lesions than euchromatic compartments, with a sharp transition between the two (Fig 2E). Notably, in RB1 KO cells this pattern of UV susceptibility is exaggerated with heterochromatic compartments accumulating even more UV lesions, and euchromatic compartments less, than wild-type cells. Euchromatic TAD compartments with decreased susceptibility in RB1 KO cells exhibited corresponding decreases in heterochromatin marks. However, in this analysis, increases in UV susceptibility did not correlate with elevated H3K9me3.
The results in Fig 2 demonstrate that RB1 regulates heterochromatin features, which often correlate with UV susceptibility. In particular, decreases in heterochromatin marks in genic regions correspond to reductions in UV susceptibility. However, increases in susceptibility do not obviously appear to be linked to elevated constitutive heterochromatin features in RB1 KO cells.
Loss of RB1 results in elevated UV susceptibility at centromeric and telomeric regions
Because of RB1’s role in modulating the chromatin of centromeric and telomeric repeat regions, UV susceptibility was then investigated in an extensive panel of repeat sequences. Interestingly, a large variation of susceptibility was observed in several repeats (Figs 3A and S1A). Telomeric, centromeric, and satellite repeats, which are enriched around centromeres, exhibited significant increases in susceptibility, whereas L1 repeats exhibited decreased susceptibility.
Figure 3. Centromeric and telomeric regions are more susceptible to UV following RB1 knockout.

(A) Box plots of UV lesion log2 fold change (FC) in RB1 KO cells compared with wild-type (WT) (RB1 KO/WT) in selected families of repeat sequences. (B) Box plots of UV lesion abundance in pericentric or subtelomeric regions, defined as 1 Mb bins next to centromeres or 100 kb next to telomeres, in RB1 KO, WT, and RB1 KO/WT FC. Mann–Whitney U test indicates significant difference between 1,000 groups of random regions and selected regions (ns, not significant, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001). (C) Heat map of scaled median H3K9me3 and H3K27me3 ChIP signal for RB1 KO, WT, and RB1 KO/WT FC in selected families of repeat sequences. For each histone modification, the signal is normalized to genome median for visualization. (D) Box plots of H3K9me3 and H3K27me3 ChIP signal in pericentric and subtelomeric regions, for RB1 KO, WT, and RB1 KO/WT FC. Mann–Whitney U test indicates significant difference between 1,000 groups of random regions and selected regions (ns, not significant, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001). (E) Heat maps of RB1 KO/WT log2 FCs in UV lesion, H3K9me3, and H3K27me3 signal in 100-kb bins along selected chromosomes.
Figure S1. RB1 regulates UV susceptibility in repeat sequences.

(A) Top, box plots of UV lesion log2 fold change (FC) in RB1 KO cells compared with wild type (WT) (RB1 KO/WT) in selected families of repeat sequences. Bottom, heat map of scaled median H3K9me3 and H3K27me3 ChIP signal for RB1 KO, WT, and RB1 KO/WT FC in selected families of repeat sequences. For each histone modification, the signal is normalized to genome median for visualization. (B) Dipyrimidine content of subtelomeric and pericentric regions, defined as 1 Mb bins next to centromeres or 100 kb next to telomeres, compared to random genome control. Mann–Whitney U test indicates significant difference between 1,000 groups of random regions and selected regions (ns, not significant, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001). (C) Abundance of telomere sequences in RB1 KO cells compared to WT using Teltale and Telomere Hunter, as described in Materials and Methods section. (D) Fold change differences in telomere content between WT and RB1 KO cells quantified by qPCR. Cells were sampled at indicated passage number. Two to three biological replicates, and three technical replicates for each biological replicate, were used. Standard error was calculated using error propagation.
Notably, in wild-type cells, centromeric and telomeric-proximal regions are relatively protected from UV compared with a random genomic control (Fig 3B). This protection is particularly unexpected for pericentric regions as TpT frequency, the dipyrimidine with the highest propensity to form a CPD (Douki & Cadet, 2001), is elevated in these regions compared with a random control (Fig S1B). However, in RB1 KO cells, this observed protection for both centromeric and telomeric-proximal regions is significantly disrupted, resulting in an overall increase in UV susceptibility in RB1 KO compared with wild-type (Fig 3B). Notably, increases in telomere length in RB1 KO cells were not observed (Fig S1C and D), thus are not attributed to additional telomeric sequences in mutant cells.
In wild-type cells, these repeat sequences are relatively enriched in heterochromatic H3K9me3 and/or H3K27me3 marks (Figs 3C and S1A). However, in RB1 KO cells, there is a general decrease in these repressive marks at and around these repeats (Fig 3C and D). Some repeats exhibit less H3K27me3, such as L1, whereas centromeric and telomeric repeats exhibit decreases in both H3K27me3 and H3K9me3 (Figs 3C and S1A). Pericentric and subtelomeric regions also display alterations H3K27me3 and/or H3K9me3 (Figs 3D and S2A). Interestingly, these changes in heterochromatin features and UV susceptibility around centromeric and telomeric regions can be observed at a chromosomal level (Figs 3E and S2B). At several telomeric and centromeric regions, changes in H3K9me3 and increases in UV susceptibility can be clearly observed, whereas alterations in H3K27me3 are relatively more dispersed throughout the chromosome.
Figure S2. RB1 regulates chromatin and UV susceptibility in centromeric and telomeric regions.

(A) Box plots of H3K9me3 and H3K27me3 ChIP signal in pericentric and subtelomeric regions, for RB1 KO, wild-type (WT), and RB1 KO/WT fold change (FC). Mann–Whitney U test indicates significant difference between 1,000 groups of random regions and selected regions (ns, not significant, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001). (B) Heat maps of RB1 KO/WT log2 FC in UV lesion, H3K9me3, and H3K27me3 signal in 100 kb bins along autosomal chromosomes.
Thus, these repeat-focused analyses differ from the genome-wide overviews presented in Fig 2 and demonstrate that whereas heterochromatin alterations at telomeric and centromeric regions can lead to altered susceptibility, the directionality is not conserved.
Expression of centromere-associated genes is altered after RB1 knockout
Previous studies demonstrate that loss of RB1 and RB family members results in decondensation of telomeres and centromeres, reduced localization of specialized proteins, such as condensin, and chromosomal abnormalities (García-Cao et al, 2002; Gonzalo et al, 2005; Isaac et al, 2006; Coschi et al, 2010, 2014; Longworth & Dyson, 2010; Manning et al, 2010; van Harn et al, 2010). In the case of telomeres specifically, previous studies demonstrate their sensitivity to UV-induced lesion formation (Kruk et al, 1995; Rochette & Brash, 2010), which can persist for days to weeks in cell culture (Mitchell et al, 1999; Bérubé et al, 2018). Interestingly, telomere-associated proteins, such as the Shelterin complex (de Lange, 2005), have been shown to regulate UV susceptibility (Parikh et al, 2015), demonstrating the potential for non-histone, DNA-associated proteins to regulate acquisition of DNA lesions.
To better characterize UV susceptibility defects associated with centromeres and telomeres in RB1 KO cells, RNA-seq was performed to detect changes in the expression of genes associated with centromeres and telomeres (Table S1). Functional annotation analysis of significantly differentially expressed genes identified several down-regulated categories, including “nucleosome assembly,” “cell division,” “chromatid cohesion,” and “kinetochore” (Fig 4), with specific centrosome-associated proteins (CENP) being down-regulated. Up-regulated categories include the “PI3K-Akt signaling pathway,” “pathways in cancer,” and “extracellular matrix” (Fig 4). These results, together with previous publications demonstrating loss of proper centromeric formation upon RB1 depletion (Gonzalo et al, 2005; Isaac et al, 2006; Coschi et al, 2010; Manning et al, 2010), strongly suggest that RB1 regulates centromere assembly to both ensure proper mitotic chromosome function and protect against UV damage formation.
Figure 4. RB1 regulates genes in mitotic pathways.

DAVID functional enrichment analysis (Huang et al, 2009a, 2009b) of significantly differentially expressed genes (upregulated n = 471; downregulated n = 180) from RNA-seq in RB1 KO cells compared to wild-type. Signed enrichment score (−log10 P-value) is displayed for each functional group, with the significance cutoff (P = 0.05) in black. GO, gene ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; IPR, InterPro.
Table S1. Functional enrichment categories of RB1-regulated genes. LSA-2021-01134_TableS1.xlsx (187.9KB, xlsx)
In the case of telomeric regions specifically, expression of related genes, such as Shelterin, were not significantly altered in RB1 KO cells (Table S1). However, previous studies demonstrate that telomeric heterochromatin is regulated by RB1 and related family members (García-Cao et al, 2002; Gonzalo et al, 2005; Gonzalez-Vasconcellos et al, 2017). Thus, general telomere dysfunction, which can alter association of telomere-binding proteins, may be linked to alteration of UV susceptibility.
RB1-regulated pericentric and telomeric regions are more mutable in cancer
Because elevated susceptibility can increase mutagenic potential, we assessed mutation burden in regions of the genome that acquire more UV-induced lesions in RB1 KO cells compared with wild-type. Functional annotation analysis of the genes within the most susceptible (top 5%) genomic regions in RB1 KO cells identified cancer and disease-related categories, including “Chemical Carcinogenesis” and “T-cell lymphoma and Cutaneous Skin Neoplasms” (Fig 5A and Table S2). Categories representing genes in the least susceptible (bottom 5%) genomic regions include several important for essential cell functions, such as “Mitochondrion” and “Lysosome.” “Cell cycle” is also represented, supporting results of Figs 1 and 2, which show that RB1-regulated genes have reduced UV susceptibility and heterochromatin formation. “Endometrial Cancer” is also identified as representing the least susceptible regions of the genome and may indicate some degree of tissue-specific carcinogen susceptibility.
Figure 5. Regions of RB1-regulated susceptibility include highly mutated cancer driver genes.
(A) DAVID functional enrichment analysis (Huang et al, 2009a, 2009b) of genes in the top and bottom 5% UV susceptible regions (top n = 821; bottom n = 2,600) in RB1 KO cells compared with wild-type. Only genes with >10% overlap in top or bottom bin are included in analysis. Signed enrichment score (−log10 P-value) is displayed for each functional group, with the significance cutoff line (P = 0.05) in black. GO, gene ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; GAD, Genetic Association Database; KW, Keyword. (B) Box plots of melanoma mutation frequency (Hoadley et al, 2018) of genes in the top and bottom 10% and 5% susceptible regions compared with the whole genome. Only C>T and G>A mutation were counted. Mutation rates were normalized by cytosine and guanine content of the gene. Additional details provided in the Materials and Methods section. Mann–Whitney U test indicates significant melanoma mutation frequency difference between groups (ns, not significant, *P < 0.05, **P < 0.01, ***P < 0.001). (C) Distance of cancer driver genes to the closest centromere or telomere in base pairs. Genome median is shown as blue dotted line in graph. Genes with oncogenic function are shown in green and genes with tumor suppressor function are shown in pink. (D) Mutational co-occurrence (in red) and mutual exclusivity (in blue) of genes in RB1 pathway, TERT and TPTE. Log2 odds ratio analysis was performed as described in the Materials and Methods section (*q < 0.05, **q < 0.01, ***q < 0.001). (D, E) Percentage of melanoma patients with TERT and TPTE mutations within different genic features from patients with (mutated) and without (not mutated) RB1 pathway mutations for genes shown in (D). Only C>T and G>A mutation were counted. Mutation rates were normalized by cytosine and guanine content of the gene. Additional details provided in the Materials and Methods section (ns, not significant, *P < 0.05, **P < 0.01, ***P < 0.001). (D, F) Proportion of TERT promoter mutations at different ETS sites from patients with (mutated) and without (not mutated) RB1 pathway mutations for genes shown in (D).
Source data are available online for this figure.

Table S2. Functional enrichment analysis of differentially susceptible genes in RB1 KO cells. LSA-2021-01134_TableS2.xlsx (713KB, xlsx)
Interestingly, genes in the most susceptible regions have significantly increased mutation rates in melanoma compared with the least susceptible (Fig 5B). Notably, a few cancer driver genes are found within these regions (Table S2). Among them is telomerase reverse transcriptase (TERT), which is widely mutated in a majority of cancers and ∼10% of melanoma (Hoadley et al, 2018). Somatic mutations in the TERT promoter create de novo ETS binding sites to up-regulate TERT expression during immortalization (Horn et al, 2013; Huang et al, 2013; Chiba et al, 2017). Several of these mutations carry the C>T transition indicative of UV-induced mutagenesis, also known as “solar signature” mutation (Brash, 2015). The increased susceptibility of the TERT promoter was also confirmed using a qPCR interference assay and found to be approximated threefold more in RB1 KO cells compared with wild-type (Fig S3A and B).
Figure S3. Knockout of RB1 increases UV susceptibility at the TERT promoter.
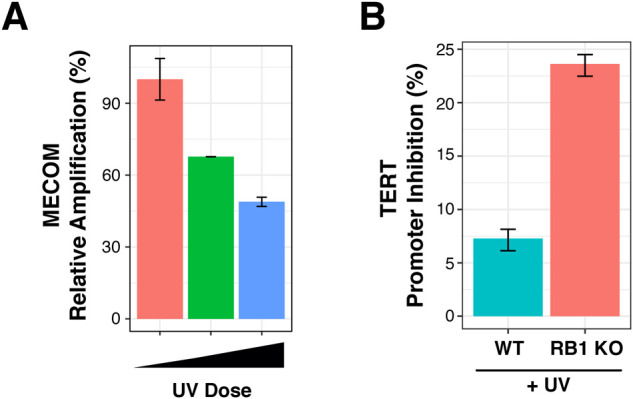
(A) Dose dependent inhibition of qPCR amplification of MECOM locus, a highly susceptible locus, in wild-type (WT) cells (García-Nieto et al, 2017). Relative qPCR amplification is compared with GAPDH, a relatively protected locus in WT cells. UV doses are 0, 100, 200 J/m2 UVB. (B) qPCR inhibition of the TERT promoter in both RB1 KO and WT cells after 100 J/m2 UVC exposure. Primers amplify −224 to −308 of TERT promoter and includes ETS sequences. Three technical replicates were used for each sample. Standard error was calculated using error propagation.
Another cancer-associated gene found to have increased UV susceptibility in RB1 KO cells, and mutation rates in melanoma, is TPTE (Table S2). TPTE is a relatively uncharacterized PTEN-related tyrosine phosphatase that is mutated in 26% of cutaneous melanoma (Hoadley et al, 2018). Interestingly, compared with the genome median, several of these cancer driver genes are located either proximal to a centromere or telomere (Fig 5C). TERT is proximal to the telomere of chromosome 5p, whereas TPTE is proximal to the centromere of chromosome 21p (Fig 5C).
Using sequencing datasets from 1,970 melanomas, we found that cancers with RB1 pathway mutations, including CDKN2A/p16, have significant co-occurrence of mutations in TERT and TPTE (Fig 5D). Mutational co-occurrence between these genes was less prominent in cancers not known to be caused by carcinogen exposure, such as breast and prostate cancer (Fig S4). Further analyses into the specific mutated regions of TERT identified upstream promoter, downstream, and intronic regions as having increased mutation frequency in melanomas that also carry mutations in the RB1 pathway compared with tumors without RB1 pathway alterations (Fig 5E). Site-specific ETS mutations were also found to be differentially enriched in tumors with RB1 pathway mutations (Fig 5F). Significant increases in TPTE mutations were also found in the promoter, downstream, and exonic regions (Fig 5E).
Figure S4. Mutational co-occurrence of TERT, TPTE, and the RB1 pathway in breast and prostate cancer.
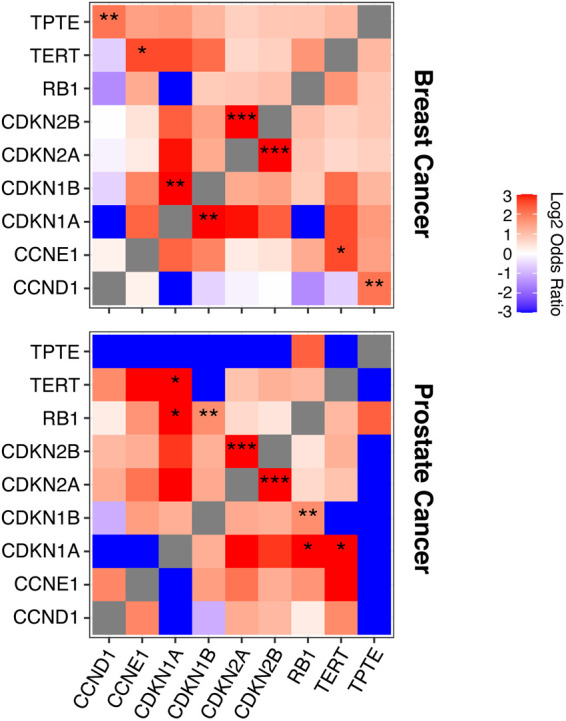
Mutational co-occurrence (in red) and mutual exclusivity (in blue) of genes in RB1 pathway, TERT and TPTE in breast and prostate cancer. Log2 odds ratio analysis was performed as described in the Materials and Methods section (*q < 0.05, **q < 0.01, ***q < 0.001).
These results show that regions of increased UV susceptibility resulting from RB1 loss contain several highly mutated cancer driver genes located proximal to heterochromatic telomeres and centromeres. These increases in mutation frequency can be compounded by reduced repair efficiency, which is observed in pericentric, but not subtelomeric, regions of wild-type cells (Fig S5).
Figure S5. Repair rates of pericentric and subtelomeric regions.

Cumulative XR-seq repair rate at pericentric regions and subtelomeric regions (Hu et al, 2017), compared with random genome control. Mann–Whitney U test indicates significant difference between 1,000 groups of random regions and selected regions (ns, not significant, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001).
Discussion
Collectively, these results demonstrate for the first time that disruption of a tumor suppressor can increase susceptibility to carcinogenic insult, thus accelerating mutagenic potential. This is a significant expansion of our current understanding of tumor suppressors, which are largely known to combat mutagenesis through their participation in DNA damage response pathways (Hanahan & Weinberg, 2011). Specifically, these results demonstrate that tumor suppressors can influence genome maintenance via regulation of carcinogen susceptibility.
Importantly, RB1 loss increases opportunities for mutation of several cancer driver genes, most notably the TERT gene, which plays critical roles in carcinogenesis by sustaining proliferative capacity (Maciejowski & de Lange, 2017). TERT is proximal to a telomere, which, along with centromeric regions, is among the most UV susceptible in RB1 KO cells. This is in line with previous findings demonstrating the important role of RB1 in regulating the proper function and formation of both telomeres and centromeres (García-Cao et al, 2002; Gonzalo et al, 2005; Isaac et al, 2006; Coschi et al, 2010, 2014; Longworth & Dyson, 2010; Manning et al, 2010; van Harn et al, 2010). As expected, increased UV lesions and mutation of centromeric and telomeric sequences can also significantly impair chromosome segregation and increase chromosomal fusions (Lin et al, 2004; Wright & Shay, 2005; Finkel et al, 2007). Indeed, loss of RB1 results in aneuploidy in both retinoblastoma patient-derived fibroblasts and mouse models (Coschi et al, 2014).
Mutagenic potential is, of course, contributed by both susceptibility and repair. Previous studies demonstrate that loss of RB1 alters UV DNA damage responses (Bosco, 2005). Specifically, whereas checkpoint responses are abrogated in mutant cells, repair of UV-induced photoproducts is accelerated. It is hypothesized that this combination of events could lead to propagation of mutations by reducing opportunities for apoptosis if errors are incorporated during accelerated repair. Interestingly, hereditary retinoblastoma patients have increased risk for melanoma development (Moll et al, 1997; Fletcher et al, 2004). Elevated frequency of carcinogen-induced secondary cancers, such as melanoma, in retinoblastoma patients may, indeed, be contributed by both increased susceptibility and altered repair responses.
The specific mechanisms of how RB1 protects telomeric and centromeric regions against carcinogenic insult are still under investigation. Interestingly, although whole genome patterns of UV susceptibility in RB1 KO cells agree with previous results and demonstrate that heterochromatin features positively associate with increased UV susceptibility (García-Nieto et al, 2017), it was not a feature consistent with susceptibility at centromeres and telomeres. Nevertheless, we find that expression of centromere binding proteins, such as CENP proteins, are reduced in RB1 KO cells. In addition, condensin binding to centromeres is disrupted in cells lacking RB1 function (Coschi et al, 2014; Marshall et al, 2019). Collectively, reduced binding of centromere-associated proteins likely contributes to both centromere dysfunction, as well as increased UV susceptibility in RB1 KO cells.
Notably, non-histone DNA binding proteins have previously been shown to regulate UV susceptibility. Namely CTCF and ETS transcription factors alter UV lesion formation at their binding sites (Mao et al, 2016; Fredriksson et al, 2017; Hu et al, 2017). TRF1 of the telomere binding Shelterin complex also protects against UV lesion formation (Parikh et al, 2015). Shelterin provides a protein scaffold that promotes significant DNA compaction important for telomere protection (Bandaria et al, 2016). In addition, heterochromatin plays important roles in the regulation of telomere compaction. Specifically, loss of heterochromatic H3K9me3 in telomeres is associated with decreased heterochromatin protein 1 (HP1) association (García-Cao et al, 2002), which, in turn, impairs telomere cohesion and length maintenance (Canudas et al, 2011). Indeed, H3K9me3 is reduced in telomeric regions of RB1 KO cells, thus binding of heterochromatin proteins would be impaired, which could render associated DNA sequences more vulnerable to UV damage.
These results suggest that disruption of local chromatin structure can alter binding of chromatin-associated proteins that regulate UV susceptibility. These findings have important consequences on genome stability mechanisms that can become disrupted during cancer evolution. Many cancer drivers that are also chromatin regulators, such as RB1, have broad impacts across the genome (Gonzalo et al, 2005; Longworth & Dyson, 2010; Plass et al, 2013; Helin & Minucci, 2017; Valencia & Kadoch, 2019). One specific example is oncogenic Myc overexpression, which is common in cancers, and alters histone modifications globally (Knoepfler et al, 2006; Cotterman et al, 2008; Varlakhanova & Knoepfler, 2009). These global changes in chromatin structure likely impact the association of many DNA binding proteins, which ultimately can alter susceptibility to carcinogens and accelerate mutagenic potential in several regions of the genome.
Findings in this report demonstrate that loss of tumor suppressor function can accelerate mutagenesis via novel genome stability mechanisms that do not involve damage response pathways. These results provide a foundation for additional research on the regulation of carcinogen susceptibility and its disruption in diseases fueled by mutation, such as cancer.
Materials and Methods
Cell culture
Human fetal lung fibroblast IMR90 (Cat. no. CCL-186; ATCC) were obtained from ATCC. Cells were cultured at 37°C and 5% CO2 in DMEM with high glucose, sodium pyruvate, and L-glutamine (Cat. no. SH3024301; Hyclone), supplemented with 15% (vol/vol) FBS (Cat. no. S11150; Atlanta Biologicals) and 1% Pen/Strep (Cat. no. SV30010; Hyclone). HEK293T (Cat. no. CRL-3216; ATCC) cells were obtained from ATCC. Cells were cultured at 37°C and 5% CO2 in DMEM with high glucose, sodium pyruvate, and L-glutamine, supplemented with 10% (vol/vol) FBS and 1% Pen/Strep.
CRISPR-Cas9 knockout cell creation
Guide RNA was selected and designed to target RB1 translational start site using the Broad design tool (Doench et al, 2016; Sanson et al, 2018) (guide RNA sequence: 5′-CACCGGCATGACGCCTTTCCGCGGC-3′ 3′-CCGTACTGCGGAAAGGCGCCGCAAA-5′) and cloned into lentiCRISPR plasmid (Cat. no. 49535; Addgene) and confirmed by Sanger sequencing. The construct was transfected into HEK293T cells with VSV-g, psPAX2 to produce virus to transduce IMR-90 cells. Puromycin resistant cells were pooled together. RB1 disruption was confirmed by Western blot with anti-RB1 antibody (4H1, Cat. no. 9309; Cell Signaling Technology).
CPD IP, library preparation, and sequencing
Wild-type and RB1 KO IMR90 cells were grown to full confluency to eliminate cell cycle effects on UV lesion acquisition. CPD IP was performed as previously described (García-Nieto et al, 2017). Cells were irradiated with 100 J/m2 UVC and were immediately lysed with 1% SDS buffer. Cell lysates were treated with RNase A (Cat. no. NC9729931; Thermo Fisher Scientific) and proteinase K (Cat. no. P2308; Sigma-Aldrich), followed by DNA precipitation with ethanol/sodium acetate. Purified DNA was then sonicated by a Bioruptor (Diagenode) and heated to 99°C to denature. The resulting single-stranded fragments were incubated with anti-CPD antibody (clone TDM-2; Cosmos Bio). Immunoprecipitated DNA fragments were repaired by CPD and 6-4PP photolyases (Selby & Sancar, 2012). Library preparation was performed with NEBNext Ultra II Directional RNA Second Strand Synthesis Module (Cat. no. E7550; NEB) and NEBNext ChIP-seq Library Prep Master Mix Set for Illumina (Cat. no. E6240; NEB) as instructed by the manufacturer. Paired-end sequencing was performed on HiSeq 4000 with read length of 100 bp.
Western blotting
Cells were lysed by preheating lysis buffer (1% SDS and 25 mM Tris, pH 8.0) to 99°C and adding to cells. Lysates were then spun down at >18,000g and supernatants were separated on PAGE gels and transferred to polyvinylidene difluoride (PVDF) membranes (Cat. no. 1620177; Bio-Rad) at 100 V at room temperature for 1 h. The membrane was then blocked with 5% BSA (Cat. no. A-420; GoldBio) for 1 h at room temperature and incubated with primary antibody overnight at 4°C. Antibodies used are as follows: anti-H3K9me3, Cat. no. 8898; Abcam; anti-H3K27me3, Cat. no. 6002; Abcam; anti-tubulin Cat. no. sc-53030; Santa Cruz Biotechnology; anti-retinoblastoma 1, Cat. no. 9309; Cell Signaling Technology. The membrane was then incubated with peroxidase-coupled secondary antibody for 1 h at room temperature (donkey anti-rabbit IgG-HRP, Cat. no. NA-934; Cytiva; sheep anti-mouse IgG-HRP, Cat. no. NA-931; Cytiva; goat anti-rat IgG-HRP, NB7115; Novus Biological). Chemiluminescent signal was detected with Advansta Westernbright Quantum-HRP (K-12042). Tubulin was used as loading control.
Histone chromatin IP (ChIP), library preparation, and sequencing
∼2 × 106 confluent cells were fixed with 1% formaldehyde (Cat. no. BP-531; Thermo Fisher Scientific) for 5 min at room temperature under gentle agitation. Formaldehyde was then quenched with the final concentration of 0.125 M glycine. Cells were then washed with ice-cold PBS twice, and washed twice in micrococcal nuclease (MNase) buffer (50 mM Tris–HCl, pH 8.0, 250 mM sucrose, 25 mM KCI, 1 mM MgCl2, 1 mM CaCl2, 0.5 mM 2-mercaptoethanol, and 0.1% NP-40). Nuclei were isolated and DNA was digested with 400 gel units of MNase (Cat. no. M0247; NEB) for 16 min on ice, and digestion was terminated with MNase stop buffer (1% SDS, 50 mM Tris–HCl, pH 8.0, and 20 mM EGTA). Nuclei were then sonicated for 2 min on low with a bioruptor (30 s ON/OFF; Diagenode). Cell debris was removed by centrifugation at 20,000g for 15 min at 4°C. DNA fragment size was verified via bioanalyzer and most fragments (∼90%) were the size of mono or di-nucleosomes (<500 bp). The supernatant was diluted with RIPA buffer without SDS, and 5% of the sample was used as the input for the experiment.
It is important to note that we performed MNase digestion rather than sonication as a method to fragment heterochromatin. This is because when sonication was performed, the immunoprecipitated heterochromatin fragments were greater than 2 kb, whereas total input chromatin displayed a mean fragment size of 250 bp. We reasoned that heterochromatin does not adequately fragment when using sonication. If sonication were used, the vast majority of immunoprecipitated DNA would not be represented in sequencing reads, which requires less than 1 kb DNA fragments.
Antibodies (H3K9me3, Cat. no. 8898; Abcam; H3K27me3, Cat. no. 6002; Abcam, Rabbit IgG, Cat. no. 2729; Cell Signaling Technology) were pre-coupled to Protein G Dynabead (Cat. no. 10-0040D; Thermo Fisher Scientific) in RIPA buffer before IP with the sample. Coupled beads were blocked with 0.1 mg/ml BSA (Fraction V, sterile filtered) for 1 h. Samples were then incubated with antibody-coupled beads overnight at 4°C, followed by elution in elution buffer (1× TE with 1% SDS) at 30°C for 15 min. Eluted DNA–protein complexes were then treated with 10 mg of RNAse A at 37°C for 1 h and proteinase K treatment at 37°C for 2 h. Samples were de-crosslinked overnight at 65°C. De-Crosslinked DNA was purified using Chromatin IP DNA Purification Kit (Cat. no. 58002; Active Motif). Library preparation was performed using NEBNext Ultra II DNA Library Prep Kit for Illumina (Cat. no. E7645S; NEB) and NEBNext Multiplex Oligos for Illumina (Cat. no. 6440S; NEB) as instructed by the manufacturer. Paired-end sequencing was performed on HiSeq 4000 with read length of 100 bp.
ChIP-sequencing data processing
Sequencing data were processed according to ENCODE (phase-3) specification for ChIP-seq, using the ENCODE Transcription Factor and Histone ChIP-Seq processing pipeline (The ENCODE Project Consortium, 2012) (github: https://github.com/ENCODE-DCC/chip-seq-pipeline2). In short, reads were aligned to the reference genome hg19 using Bowtie2 (Langmead & Salzberg, 2012). Aligned reads were then filtered and deduplicated based on their quality. Signal tracks of fold change were generated using MACS2.0 (Zhang et al, 2008) by normalizing IP signals by the input.
RNA-sequencing and analysis
RNA was isolated from confluent cells using Quick-RNA Miniprep plus kit (Cat. no. T1057; Zymo Research). rRNA was then removed using Ribo-Zero Magnetic Kit (Cat. no. 20040526; Illumina), followed by library preparation using NEBNext Ultra II Directional RNA Library Prep Kit for Illumina (Cat. no. E7760S; NEB). RNA quality was examined on Agilent 2100 and all samples had RIN (RNA integrity number) of 10. At least 40 million paired-end reads with read length of 150 bp were sequenced per replicate on an Illumina NovaSeq 6000.
Raw sequencing reads were aligned to GrCh37 with gencode.v19.annotation.gtf using STAR with the following arguments: --readFilesCommand gunzip --outSAMtype BAM SortedByCoordinate --outSAMattributes Standard --quantMode GeneCounts --outFilterMultimapNmax 1 --outFilterMatchNmin 35 --twopassMode Basic (Dobin et al, 2013). Output quantification file was used as input to DESeq2 to identify differentially expressed genes, using cutoff padj = 0.1 (Love et al, 2014).
Repetitive sequence analysis
Raw sequencing reads are aligned to hg19 with Bowtie2 (Langmead & Salzberg, 2012) with default settings. PCR duplicates were then removed with picard MarkDuplicates with --REMOVE_DUPLICATES=TRUE. De-duplicated bam files were then used as input of RepEnrich2 (Criscione et al, 2014) (github: https://github.com/nerettilab/RepEnrich2) to quantify reads aligned to each type of repetitive sequence as annotated by RepeatMasker. Briefly, RepEnrich2 builds a custom genome repeat reference using both the canonical sequence of each repeat and the annotated repetitive sequence of each repeat in RepeatMasker (Smit et al, 2013–2015). Each repeat is assigned to a specific repeat class through unique alignment. Reads that align to multiple families of repeats are divided amongst families.
Repeat enrichment analysis was performed using quantification as input of DESeq2 (Love et al, 2014). For histone modifications, input from RB1 KO and wild-type have different distributions of repeats. Therefore, DESeq2 was used to find significantly different repeats in both input and ChIP. Output log2 fold change value was used to compare the distribution of ChIP versus input via Wilcoxon paired test for each group. Permutation test was performed to assess how likely the difference is obtained by chance, and the result was corrected for multiple testing using Benjamini–Hochberg.
Centromere and telomere analysis
Coordinates of centromeres and telomeres of hg19 were obtained from UCSC genome browser (table browser; assembly: Feb. 2009 (GRCh37/hg19), group = All Tracks, track = gap). Pericentric and subtelomeric regions are defined as the ±1 Mb region around the centromere and 100 kb region proximal to the telomere, respectively. Binned genomic regions of 100 kb that overlapped with the defined pericentric or subtelomeric regions were included in the analysis. The same number of regions were selected at random as control. Mann–Whitney U test indicates significant difference between 1,000 groups of random regions and selected regions.
TAD analysis
A/B compartments of IMR-90 derived from Hi-C were obtained from Fortin and Hansen (2015). Adjacent compartments of the same type were merged to create a dense track for A/B compartments. Each compartment was binned into 50 bins and the median signal for each bin was calculated. The closed domains, shaded in red, were aligned in the middle with its flanking open domains on the side. The mean signal and 95% confidence interval obtained via bootstrapping (R = 1,000) is in black and grey, respectively.
Melanoma mutation frequency analysis
Simple somatic mutation data for melanomas were downloaded from the International Cancer Genome Consortium Data Coordination Centre (Hayward et al, 2017) (https://dcc.icgc.org/). Acral melanoma that are not caused by UV exposure were discarded. Donors were separated into two groups based on their mutation status of RB1-pathway genes (including RB1, CCND1, CCNE1, CDKN1A, CDKN1B, CDKN2A, and CDKN2B). For genic mutational frequency analysis, only C>T and G>A mutation were counted. Mutation rates were normalized by cytosine and guanine content of the gene. Significant differences in mutation rates were detected via Willcox-test.
A set of consolidated cancer driver genes (n = 1,152) was compiled as the union of lists of cancer driver genes from the Catalog of Somatic Mutations (COSMIC) Cancer Gene Census (Tate et al, 2019), the PanCancer Driver Gene list (Bailey et al, 2018), the IntOGen cancer driver database (Gonzalez-Perez et al, 2013), and novel cancer driver genes identified by MutPanning (Dietlein et al, 2020).
Mutation co-occurrence and exclusivity analysis
Eight non-overlapping melanoma studies (n = 1,970 tumors) from cBioPortal for Cancer Genomics were included in this analysis (Berger et al, 2012; Hodis et al, 2012; Krauthammer et al, 2012; Snyder et al, 2014; Van Allen et al, 2014; Akbani et al, 2015). The analysis was performed with the in-house tools of cBioPortal (Cerami et al, 2012; Gao et al, 2013). Briefly, odds ratio tests were performed to identify any mutually exclusive or co-occurring events. Fisher’s exact test was then performed to evaluate the significance of identified events. Two prostate adenocarcinoma studies (n = 2,478) and four breast cancer studies (n = 1,376) from cBioPortal for Cancer Genomics were also included in this analysis (The Oslo Breast Cancer Consortium (OSBREAC) et al, 2012; Banerji et al, 2012; Shah et al, 2012; PCF/SU2C International Prostate Cancer Dream Team et al, 2018; Nguyen et al, 2020). Three additional data sets included in the analysis are: “MSK-IMPACT sequencing of 720 Melanoma tumor samples with matched normals”; “TCGA Skin Cutaneous Melanoma Source data from GDAC Firehose”; and “Breast Invasive Carcinoma Source data from GDAC Firehose” (Cerami et al, 2012; Gao et al, 2013).
qPCR UV interference assay
IMR-90 RB1 KO and wild-type cells were grown to confluency and exposed to indicated doses of UV. Cells were immediately lysed in 1% SDS buffer and DNA purified using Quick-DNA Miniprep Kit (D3024; Zymo Research). GAPDH was used as DNA loading control in qPCR assays. The percentage inhibition was calculated by first subtracting the Cq value for TERT from that of GAPDH within each −UV and +UV sample and cell line. The resulting −UV and +UV values were then subtracted from each other to calculate X, which was used in (1 − (1/2^X)) × 100 to calculate percent inhibition. MECOM primer sequences are: forward 5′-AGCAGGTCTTGATTCGACGTT-3′; reverse 5′-CACAGGTGAGGTCTGCCATA-3′. GAPDH primer sequences are: forward 5′-GACTGAGATTGGCCCGATG-3′; reverse 5′-GACTGAGATTGGCCCGATG-3′. TERT primer sequences are: forward 5′-CTGGAAGGTGAAGGGGCAG-3′; reverse 5′-GGGCTCCCAGTGGATTCG-3′.
Telomere length quantification
Telomere length estimation was carried out using two separate programs: Teltale from St. Jude children’s research hospital (github: https://stjude.github.io/teltale/) and Telomere Hunter (Feuerbach et al, 2019). ChIP-Seq input from both RB1 KO and wild-type were aligned to hg19, and the resulting bam file was used as the input for both programs. Teltale counts the number of reads in the file consisting of seven or more consecutive occurrences of the telomere motif TTAGGG or its reverse complement. Telomere Hunter estimates the telomere content by counting telomeric reads and normalizing the count with the number of reads with comparable GC content. Unlike Teltale, Telomere Hunter also includes telomere variant repeats for analysis. The analysis was followed with telomere qPCR, as previously described (Cawthon, 2002). The primer sequences were: telo fwd, 5′-CGGTTTGTTTGGGTTTGGGTTTGGGTTTGGGTTTGGGTT-3′; and telo rev, 5′-GGCTTGCCTTACCCTTACCCTTACCCTTACCCTTACCCT-3′; 36B4 fwd, 5′-AGCAAGTGGGAAGGTGTAATCC-3′; and 36B4 rev, 5′-CCCATTCTATCATCAACGGGTACAA-3′. Primer pair teloF/R was used to estimate telomere content, and 36B4F/R was used as control.
XR-seq repair analysis
CPD nucleotide excision repair rates were generated using eXcision Repair-sequencing (XR-seq) and downloaded from Gene Expression Omnibus accession GSE76391 (Adar et al, 2016). Briefly, normal human fibroblasts (NHF1) were irradiated with UVC and were collected 1, 4, 8, 16, 24, and 48 h after irradiation. TFIIH bounded chromatin containing CPD lesions were then immunoprecipitated with anti-TFIIH antibody, followed by IP with anti-CPD antibody. Replicates and strand-specific signals for each time point were averaged for analysis. Cumulative signals for each time point were calculated by summation with all previous time points.
Data Availability
Data sets produced in this study are available on the Gene Expression Omnibus database under: GSE173125, GSE173126, GSE173127, and GSE173128. Computational scripts can be found at https://github.com/orgs/MorrisonLabSU/.
Supplementary Material
Acknowledgements
We thank Aziz Sancar and Chris Selby (University of North Carolina) for the generous gift of photolyases. We also thank Chunlong Chen and Yaqun Liu (Institut Curie) for sharing unpublished datasets. We are grateful for critical feedback from all members of the Morrison Lab. This study was funded by National Institutes of Health (NIH) grants R21CA178529 and R21CA171050 to AJ Morrison.
Author Contributions
KM Wong: data curation, formal analysis, investigation, visualization, methodology, and writing—review and editing.
DA King: formal analysis and methodology.
EK Schwartz: methodology.
RE Herrera: conceptualization and supervision.
AJ Morrison: conceptualization, supervision, funding acquisition, investigation, and writing—original draft, review, and editing.
Conflict of Interest Statement
The authors declare that they have no conflict of interest.
References
- Adar S, Hu J, Lieb JD, Sancar A (2016) Genome-wide kinetics of DNA excision repair in relation to chromatin state and mutagenesis. Proc Natl Acad Sci U S A 113: E2124–E2133. 10.1073/pnas.1603388113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akbani R, Akdemir KC, Aksoy BA, Albert M, Ally A, Amin SB, Arachchi H, Arora A, Auman JT, Ayala B, et al. (2015) Genomic classification of cutaneous melanoma. Cell 161: 1681–1696. 10.1016/j.cell.2015.05.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale AL, et al. (2013) Signatures of mutational processes in human cancer. Nature 500: 415–421. 10.1038/nature12477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey MH, Tokheim C, Porta-Pardo E, Sengupta S, Bertrand D, Weerasinghe A, Colaprico A, Wendl MC, Kim J, Reardon B, et al. (2018) Comprehensive characterization of cancer driver genes and mutations. Cell 173: 371–385.e18. 10.1016/j.cell.2018.02.060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandaria JN, Qin P, Berk V, Chu S, Yildiz A (2016) Shelterin protects chromosome ends by compacting telomeric chromatin. Cell 164: 735–746. 10.1016/j.cell.2016.01.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerji S, Cibulskis K, Rangel-Escareno C, Brown KK, Carter SL, Frederick AM, Lawrence MS, Sivachenko AY, Sougnez C, Zou L, et al. (2012) Sequence analysis of mutations and translocations across breast cancer subtypes. Nature 486: 405–409. 10.1038/nature11154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger MF, Hodis E, Heffernan TP, Deribe YL, Lawrence MS, Protopopov A, Ivanova E, Watson IR, Nickerson E, Ghosh P, et al. (2012) Melanoma genome sequencing reveals frequent PREX2 mutations. Nature 485: 502–506. 10.1038/nature11071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, et al. (2006) A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125: 315–326. 10.1016/j.cell.2006.02.041 [DOI] [PubMed] [Google Scholar]
- Bérubé R, Drigeard Desgarnier M-C, Douki T, Lechasseur A, Rochette PJ (2018) Persistence and tolerance of DNA damage induced by chronic UVB irradiation of the human genome. J Invest Dermatol 138: 405–412. 10.1016/j.jid.2017.08.044 [DOI] [PubMed] [Google Scholar]
- Bosco EE, Knudsen ES (2005) Differential role of RB in response to UV and IR damage. Nucleic Acids Res 33: 1581–1592. 10.1093/nar/gki283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brash DE (2015) UV signature mutations. Photochem Photobiol 91: 15–26. 10.1111/php.12377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canudas S, Houghtaling BR, Bhanot M, Sasa G, Savage SA, Bertuch AA, Smith S (2011) A role for heterochromatin protein 1γ at human telomeres. Genes Dev 25: 1807–1819. 10.1101/gad.17325211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cawthon RM (2002) Telomere measurement by quantitative PCR. Nucleic Acids Res 30: e47. 10.1093/nar/30.10.e47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. (2012) The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov 2: 401–404. 10.1158/2159-8290.CD-12-0095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba K, Lorbeer FK, Shain AH, McSwiggen DT, Schruf E, Oh A, Ryu J, Darzacq X, Bastian BC, Hockemeyer D (2017) Mutations in the promoter of the telomerase gene TERT contribute to tumorigenesis by a two-step mechanism. Science 357: 1416–1420. 10.1126/science.aao0535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chicas A, Wang X, Zhang C, McCurrach M, Zhao Z, Mert O, Dickins RA, Narita M, Zhang M, Lowe SW (2010) Dissecting the unique role of the retinoblastoma tumor suppressor during cellular senescence. Cancer Cell 17: 376–87. 10.1016/j.ccr.2010.01.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coschi CH, Ishak CA, Gallo D, Marshall A, Talluri S, Wang J, Cecchini MJ, Martens AL, Percy V, Welch I, et al. (2014) Haploinsufficiency of an RB-E2F1-Condensin II complex leads to aberrant replication and aneuploidy. Cancer Discov 4: 840–853. 10.1158/2159-8290.CD-14-0215 [DOI] [PubMed] [Google Scholar]
- Coschi CH, Martens AL, Ritchie K, Francis SM, Chakrabarti S, Berube NG, Dick FA (2010) Mitotic chromosome condensation mediated by the retinoblastoma protein is tumor-suppressive. Genes Dev 24: 1351–1363. 10.1101/gad.1917610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotterman R, Jin VX, Krig SR, Lemen JM, Wey A, Farnham PJ, Knoepfler PS (2008) N-Myc regulates a widespread euchromatic program in the human genome partially independent of its role as a classical transcription factor. Cancer Res 68: 9654–9662. 10.1158/0008-5472.CAN-08-1961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criscione SW, Zhang Y, Thompson W, Sedivy JM, Neretti N (2014) Transcriptional landscape of repetitive elements in normal and cancer human cells. BMC Genomics 15: 583. 10.1186/1471-2164-15-583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lange T (2005) Shelterin: The protein complex that shapes and safeguards human telomeres. Genes Dev 19: 2100–2110. 10.1101/gad.1346005 [DOI] [PubMed] [Google Scholar]
- Dick FA, Rubin SM (2013) Molecular mechanisms underlying RB protein function. Nat Rev Mol Cell Biol 14: 297–306. 10.1038/nrm3567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietlein F, Weghorn D, Taylor-Weiner A, Richters A, Reardon B, Liu D, Lander ES, Van Allen EM, Sunyaev SR (2020) Identification of cancer driver genes based on nucleotide context. Nat Genet 52: 208–218. 10.1038/s41588-019-0572-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Gorkin DU, Ren B (2016) Chromatin domains: The unit of chromosome organization. Mol Cell 62: 668–680. 10.1016/j.molcel.2016.05.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR (2013) STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21. 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R, et al. (2016) Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol 34: 184–191. 10.1038/nbt.3437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douki T, Cadet J (2001) Individual determination of the yield of the main UV-induced dimeric pyrimidine photoproducts in DNA suggests a high mutagenicity of CC photolesions. Biochemistry 40: 2495–2501. 10.1021/bi0022543 [DOI] [PubMed] [Google Scholar]
- Dyson NJ (2016) RB1: A prototype tumor suppressor and an enigma. Genes Dev 30: 1492–1502. 10.1101/gad.282145.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuerbach L, Sieverling L, Deeg KI, Ginsbach P, Hutter B, Buchhalter I, Northcott PA, Mughal SS, Chudasama P, Glimm H, et al. (2019) TelomereHunter: In silico estimation of telomere content and composition from cancer genomes. BMC Bioinformatics 20: 272. 10.1186/s12859-019-2851-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel T, Serrano M, Blasco MA (2007) The common biology of cancer and ageing. Nature 448: 767–774. 10.1038/nature05985 [DOI] [PubMed] [Google Scholar]
- Fletcher O, Easton D, Anderson K, Gilham C, Jay M, Peto J (2004) Lifetime risks of common cancers among retinoblastoma survivors. J Natl Cancer Inst 96: 357–363. 10.1093/jnci/djh058 [DOI] [PubMed] [Google Scholar]
- Fortin JP, Hansen KD (2015) Reconstructing A/B compartments as revealed by Hi-C using long-range correlations in epigenetic data. Genome Biol 16: 180. 10.1186/s13059-015-0741-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredriksson NJ, Elliott K, Filges S, Van den Eynden J, Ståhlberg A, Larsson E (2017) Recurrent promoter mutations in melanoma are defined by an extended context-specific mutational signature. PLoS Genet 13: e1006773. 10.1371/journal.pgen.1006773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung YK, Murphree AL, T’Ang A, Qian J, Hinrichs SH, Benedict WF (1987) Structural evidence for the authenticity of the human retinoblastoma gene. Science 236: 1657–1661. 10.1126/science.2885916 [DOI] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6: pl1. 10.1126/scisignal.2004088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Cao M, Gonzalo S, Dean D, Blasco MA (2002) A role for the Rb family of proteins in controlling telomere length. Nat Genet 32: 415–419. 10.1038/ng1011 [DOI] [PubMed] [Google Scholar]
- García-Nieto PE, Schwartz EK, King DA, Paulsen J, Collas P, Herrera RE, Morrison AJ (2017) Carcinogen susceptibility is regulated by genome architecture and predicts cancer mutagenesis. EMBO J 36: 2829–2843. 10.15252/embj.201796717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Perez A, Perez-Llamas C, Deu-Pons J, Tamborero D, Schroeder MP, Jene-Sanz A, Santos A, Lopez-Bigas N (2013) IntOGen-mutations identifies cancer drivers across tumor types. Nat Methods 10: 1081–1082. 10.1038/nmeth.2642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Vasconcellos I, Schneider R, Anastasov N, Alonso-Rodriguez S, Sanli-Bonazzi B, Fernández JL, Atkinson MJ (2017) The Rb1 tumour suppressor gene modifies telomeric chromatin architecture by regulating TERRA expression. Sci Rep 7: 42056. 10.1038/srep42056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalo S, Blasco MA (2005) Role of Rb family in the epigenetic definition of chromatin. Cell Cycle 4: 752–755. 10.4161/cc.4.6.1720 [DOI] [PubMed] [Google Scholar]
- Gonzalo S, García-Cao M, Fraga MF, Schotta G, Peters AH, Cotter SE, Eguía R, Dean DC, Esteller M, Jenuwein T, et al. (2005) Role of the RB1 family in stabilizing histone methylation at constitutive heterochromatin. Nat Cell Biol 7: 420–428. 10.1038/ncb1235 [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA (2011) Hallmarks of cancer: The next generation. Cell 144: 646–674. 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- Hanawalt PC, Spivak G (2008) Transcription-coupled DNA repair: Two decades of progress and surprises. Nat Rev Mol Cell Biol 9: 958–970. 10.1038/nrm2549 [DOI] [PubMed] [Google Scholar]
- Hayward NK, Wilmott JS, Waddell N, Johansson PA, Field MA, Nones K, Patch AM, Kakavand H, Alexandrov LB, Burke H, et al. (2017) Whole-genome landscapes of major melanoma subtypes. Nature 545: 175–180. 10.1038/nature22071 [DOI] [PubMed] [Google Scholar]
- Helin K, Minucci S (2017) The role of chromatin-associated proteins in cancer. Annu Rev Cancer Biol 1: 355–377. 10.1146/annurev-cancerbio-050216-034422 [DOI] [Google Scholar]
- Hoadley KA, Yau C, Hinoue T, Wolf DM, Lazar AJ, Drill E, Shen R, Taylor AM, Cherniack AD, Thorsson V, et al. (2018) Cell-of-origin patterns dominate the molecular classification of 10,000 tumors from 33 types of cancer. Cell 173: 291–304.e6. 10.1016/j.cell.2018.03.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat J-P, Nickerson E, Auclair D, Li L, Place C, et al. (2012) A landscape of driver mutations in melanoma. Cell 150: 251–263. 10.1016/j.cell.2012.06.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn S, Figl A, Rachakonda PS, Fischer C, Sucker A, Gast A, Kadel S, Moll I, Nagore E, Hemminki K, et al. (2013) TERT promoter mutations in familial and sporadic melanoma. Science 339: 959–961. 10.1126/science.1230062 [DOI] [PubMed] [Google Scholar]
- Horowitz JM, Yandell DW, Park SH, Canning S, Whyte P, Buchkovich K, Harlow E, Weinberg RA, Dryja TP (1989) Point mutational inactivation of the retinoblastoma antioncogene. Science 243: 937–940. 10.1126/science.2521957 [DOI] [PubMed] [Google Scholar]
- Hu J, Adar S, Selby CP, Lieb JD, Sancar A (2015) Genome-wide analysis of human global and transcription-coupled excision repair of UV damage at single-nucleotide resolution. Genes Dev 29: 948–960. 10.1101/gad.261271.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Adebali O, Adar S, Sancar A (2017) Dynamic maps of UV damage formation and repair for the human genome. Proc Natl Acad Sci U S A 114: 6758–6763. 10.1073/pnas.1706522114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang FW, Hodis E, Xu MJ, Kryukov GV, Chin L, Garraway LA (2013) Highly recurrent TERT promoter mutations in human melanoma. Science 339: 957–959. 10.1126/science.1229259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA (2009. a) Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37: 1–13. 10.1093/nar/gkn923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA (2009. b) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57. 10.1038/nprot.2008.211 [DOI] [PubMed] [Google Scholar]
- Isaac CE, Francis SM, Martens AL, Julian LM, Seifried LA, Erdmann N, Binné UK, Harrington L, Sicinski P, Bérubé NG, et al. (2006) The retinoblastoma protein regulates pericentric heterochromatin. Mol Cell Biol 26: 3659–3671. 10.1128/MCB.26.9.3659-3671.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishak CA, Marshall AE, Passos DT, White CR, Kim SJ, Cecchini MJ, Ferwati S, MacDonald WA, Howlett CJ, Welch ID, et al. (2016) An RB-EZH2 complex mediates silencing of repetitive DNA sequences. Mol Cell 64: 1074–1087. 10.1016/j.molcel.2016.10.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jans J, Schul W, Sert Y-G, Rijksen Y, Rebel H, Eker APM, Nakajima S, van Steeg H, de Gruijl FR, Yasui A, et al. (2005) Powerful skin cancer protection by a CPD-photolyase transgene. Curr Biol 15: 105–115. 10.1016/j.cub.2005.01.001 [DOI] [PubMed] [Google Scholar]
- Knoepfler PS, Zhang XY, Cheng PF, Gafken PR, McMahon SB, Eisenman RN (2006) Myc influences global chromatin structure. EMBO J 25: 2723–2734. 10.1038/sj.emboj.7601152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krauthammer M, Kong Y, Ha BH, Evans P, Bacchiocchi A, McCusker JP, Cheng E, Davis MJ, Goh G, Choi M, et al. (2012) Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet 44: 1006–1014. 10.1038/ng.2359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krimpenfort P, Quon KC, Mooi WJ, Loonstra A, Berns A (2001) Loss of p16Ink4a confers susceptibility to metastatic melanoma in mice. Nature 413: 83–86. 10.1038/35092584 [DOI] [PubMed] [Google Scholar]
- Kruk PA, Rampino NJ, Bohr VA (1995) DNA damage and repair in telomeres: Relation to aging. Proc Natl Acad Sci U S A 92: 258–262. 10.1073/pnas.92.1.258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WH, Bookstein R, Hong F, Young LJ, Shew JY, Lee EY (1987) Human retinoblastoma susceptibility gene: Cloning, identification, and sequence. Science 235: 1394–1399. 10.1126/science.3823889 [DOI] [PubMed] [Google Scholar]
- Lin J, Smith DL, Blackburn EH (2004) Mutant telomere sequences lead to impaired chromosome separation and a unique checkpoint response. Mol Biol Cell 15: 1623–1634. 10.1091/mbc.e03-10-0740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longworth MS, Dyson NJ (2010) pRb: A local chromatin organizer with global possibilities. Chromosoma 119: 1–11. 10.1007/s00412-009-0238-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550. 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund E, Oldenburg AR, Collas P (2014) Enriched domain detector: A program for detection of wide genomic enrichment domains robust against local variations. Nucleic Acids Res 42: e92. 10.1093/nar/gku324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo RX, Postigo AA, Dean DC (1998) Rb interacts with histone deacetylase to repress transcription. Cell 92: 463–473. 10.1016/s0092-8674(00)80940-x [DOI] [PubMed] [Google Scholar]
- Maciejowski J, de Lange T (2017) Telomeres in cancer: Tumour suppression and genome instability. Nat Rev Mol Cell Biol 18: 175–186. 10.1038/nrm.2016.171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning AL, Longworth MS, Dyson NJ (2010) Loss of pRB causes centromere dysfunction and chromosomal instability. Genes Dev 24: 1364–1376. 10.1101/gad.1917310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning AL, Yazinski SA, Nicolay B, Bryll A, Zou L, Dyson NJ (2014) Suppression of genome instability in pRB-deficient cells by enhancement of chromosome cohesion. Mol Cell 53: 993–1004. 10.1016/j.molcel.2014.01.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao P, Smerdon MJ, Roberts SA, Wyrick JJ (2016) Chromosomal landscape of UV damage formation and repair at single-nucleotide resolution. Proc Natl Acad Sci U S A 113: 9057–9062. 10.1073/pnas.1606667113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall AE, Ishak CA, Dick FA (2019) An RB-condensin II complex mediates long-range chromosome interactions and influences expression at divergently paired genes. Mol Cell Biol 40: e00452-19. 10.1128/MCB.00452-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumura Y, Nakaki R, Inagaki T, Yoshida A, Kano Y, Kimura H, Tanaka T, Tsutsumi S, Nakao M, Doi T, et al. (2015) H3K4/H3K9me3 bivalent chromatin domains targeted by lineage-specific DNA methylation pauses adipocyte differentiation. Mol Cell 60: 584–596. 10.1016/j.molcel.2015.10.025 [DOI] [PubMed] [Google Scholar]
- Mitchell DL, Greinert R, de Gruijl FR, Guikers KL, Breitbart EW, Byrom M, Gallmeier MM, Lowery MG, Volkmer B (1999) Effects of chronic low-dose ultraviolet B radiation on DNA damage and repair in mouse skin. Cancer Res 59: 2875–2884. https://cancerres.aacrjournals.org/content/59/12/2875.long [PubMed] [Google Scholar]
- Moll AC, Imhof SM, Bouter LM, Tan KE (1997) Second primary tumors in patients with retinoblastoma. A review of the literature. Ophthalmic Genet 18: 27–34. 10.3109/13816819709057880 [DOI] [PubMed] [Google Scholar]
- Morrison AJ, Sardet C, Herrera RE (2002) Retinoblastoma protein transcriptional repression through histone deacetylation of a single nucleosome. Mol Cell Biol 22: 856–865. 10.1128/mcb.22.3.856-865.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser J, Volker M, Kool H, Alekseev S, Vrieling H, Yasui A, van Zeeland AA, Mullenders LH (2005) The UV-damaged DNA binding protein mediates efficient targeting of the nucleotide excision repair complex to UV-induced photo lesions. DNA Repair (Amst) 4: 571–582. 10.1016/j.dnarep.2005.01.001 [DOI] [PubMed] [Google Scholar]
- Nelson DM, Jaber-Hijazi F, Cole JJ, Robertson NA, Pawlikowski JS, Norris KT, Criscione SW, Pchelintsev NA, Piscitello D, Stong N, et al. (2016) Mapping H4K20me3 onto the chromatin landscape of senescent cells indicates a function in control of cell senescence and tumor suppression through preservation of genetic and epigenetic stability. Genome Biol 17: 158. 10.1186/s13059-016-1017-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen B, Mota JM, Nandakumar S, Stopsack KH, Weg E, Rathkopf D, Morris MJ, Scher HI, Kantoff PW, Gopalan A, et al. (2020) Pan-cancer analysis of CDK12 alterations identifies a subset of prostate cancers with distinct genomic and clinical characteristics. Eur Urol 78: 671–679. 10.1016/j.eururo.2020.03.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen SJ, Schneider R, Bauer UM, Bannister AJ, Morrison A, O’Carroll D, Firestein R, Cleary M, Jenuwein T, Herrera RE, et al. (2001) Rb targets histone H3 methylation and HP1 to promoters. Nature 412: 561–565. 10.1038/35087620 [DOI] [PubMed] [Google Scholar]
- Parikh D, Fouquerel E, Murphy CT, Wang H, Opresko PL (2015) Telomeres are partly shielded from ultraviolet-induced damage and proficient for nucleotide excision repair of photoproducts. Nat Commun 6: 8214. 10.1038/ncomms9214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- PCF/SU2C International Prostate Cancer Dream Team, Armenia J, Wankowicz SAM, Liu D, Gao J, Kundra R, Reznik E, Chatila WK, Chakravarty D, Han GC, et al. (2018) The long tail of oncogenic drivers in prostate cancer. Nat Genet 50: 645–651. 10.1038/s41588-018-0078-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeifer GP, Besaratinia A (2012) UV wavelength-dependent DNA damage and human non-melanoma and melanoma skin cancer. Photochem Photobiol Sci 11: 90–97. 10.1039/c1pp05144j [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plass C, Pfister SM, Lindroth AM, Bogatyrova O, Claus R, Lichter P (2013) Mutations in regulators of the epigenome and their connections to global chromatin patterns in cancer. Nat Rev Genet 14: 765–780. 10.1038/nrg3554 [DOI] [PubMed] [Google Scholar]
- Polak P, Karlić R, Koren A, Thurman R, Sandstrom R, Lawrence M, Reynolds A, Rynes E, Vlahoviček K, Stamatoyannopoulos JA, et al. (2015) Cell-of-origin chromatin organization shapes the mutational landscape of cancer. Nature 518: 360–364. 10.1038/nature14221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roadmap Epigenomics Consortium, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, et al. (2015) Integrative analysis of 111 reference human epigenomes. Nature 518: 317–330. 10.1038/nature14248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rochette PJ, Brash DE (2010) Human telomeres are hypersensitive to UV-induced DNA damage and refractory to repair. PLoS Genet 6: e1000926. 10.1371/journal.pgen.1000926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadaie M, Salama R, Carroll T, Tomimatsu K, Chandra T, Young AR, Narita M, Pérez-Mancera PA, Bennett DC, Chong H, et al. (2013) Redistribution of the Lamin B1 genomic binding profile affects rearrangement of heterochromatic domains and SAHF formation during senescence. Genes Dev 27: 1800–1808. 10.1101/gad.217281.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanson KR, Hanna RE, Hegde M, Donovan KF, Strand C, Sullender ME, Vaimberg EW, Goodale A, Root DE, Piccioni F, et al. (2018) Optimized libraries for CRISPR-Cas9 genetic screens with multiple modalities. Nat Commun 9: 5416. 10.1038/s41467-018-07901-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster-Böckler B, Lehner B (2012) Chromatin organization is a major influence on regional mutation rates in human cancer cells. Nature 488: 504–507. 10.1038/nature11273 [DOI] [PubMed] [Google Scholar]
- Selby CP, Sancar A (2012) The second chromophore in Drosophila photolyase/cryptochrome family photoreceptors. Biochemistry 51: 167–171. 10.1021/bi201536w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano M, Lee H-W, Chin L, Cordon-Cardo C, Beach D, DePinho RA (1996) Role of the INK4a locus in tumor suppression and cell mortality. Cell 85: 27–37. 10.1016/s0092-8674(00)81079-x [DOI] [PubMed] [Google Scholar]
- Shah SP, Roth A, Goya R, Oloumi A, Ha G, Zhao Y, Turashvili G, Ding J, Tse K, Haffari G, et al. (2012) The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature 486: 395–399. 10.1038/nature10933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpless NE, Bardeesy N, Lee K-H, Carrasco D, Castrillon DH, Aguirre AJ, Wu EA, Horner JW, DePinho RA (2001) Loss of p16Ink4a with retention of p19Arf predisposes mice to tumorigenesis. Nature 413: 86–91. 10.1038/35092592 [DOI] [PubMed] [Google Scholar]
- Smit A, Hubley R, Green P (2013–2015) RepeatMasker Open-4.0.
- Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, Walsh LA, Postow MA, Wong P, Ho TS, et al. (2014) Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med 371: 2189–2199. 10.1056/NEJMoa1406498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatoyannopoulos JA, Adzhubei I, Thurman RE, Kryukov GV, Mirkin SM, Sunyaev SR (2009) Human mutation rate associated with DNA replication timing. Nat Genet 41: 393–395. 10.1038/ng.363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, Boutselakis H, Cole CG, Creatore C, Dawson E, et al. (2019) COSMIC: The catalogue of somatic mutations in cancer. Nucleic Acids Res 47: D941–D947. 10.1093/nar/gky1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The ENCODE Project Consortium (2012) An integrated encyclopedia of DNA elements in the human genome. Nature 489: 57–74. 10.1038/nature11247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Oslo Breast Cancer Consortium (OSBREAC), Stephens PJ, Tarpey PS, Davies H, Van Loo P, Greenman C, Wedge DC, Nik-Zainal S, Martin S, Varela I, et al. (2012) The landscape of cancer genes and mutational processes in breast cancer. Nature 486: 400–404. 10.1038/nature11017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valencia AM, Kadoch C (2019) Chromatin regulatory mechanisms and therapeutic opportunities in cancer. Nat Cell Biol 21: 152–161. 10.1038/s41556-018-0258-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Allen EM, Wagle N, Sucker A, Treacy DJ, Johannessen CM, Goetz EM, Place CS, Taylor-Weiner A, Whittaker S, Kryukov GV, et al. (2014) The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov 4: 94–109. 10.1158/2159-8290.CD-13-0617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Harn T, Foijer F, van Vugt M, Banerjee R, Yang F, Oostra A, Joenje H, te Riele H (2010) Loss of Rb proteins causes genomic instability in the absence of mitogenic signaling. Genes Dev 24: 1377–1388. 10.1101/gad.580710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varlakhanova NV, Knoepfler PS (2009) Acting locally and globally: Myc's ever-expanding roles on chromatin. Cancer Res 69: 7487–7490. 10.1158/0008-5472.CAN-08-4832 [DOI] [PubMed] [Google Scholar]
- Verbruggen P, Heinemann T, Manders E, von Bornstaedt G, van Driel R, Höfer T (2014) Robustness of DNA repair through collective rate control. PLoS Comput Biol 10: e1003438. 10.1371/journal.pcbi.1003438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg RA (1995) The retinoblastoma protein and cell cycle control. Cell 81: 323–330. 10.1016/0092-8674(95)90385-2 [DOI] [PubMed] [Google Scholar]
- Wolfe KH, Sharp PM, Li W-H (1989) Mutation rates differ among regions of the mammalian genome. Nature 337: 283–285. 10.1038/337283a0 [DOI] [PubMed] [Google Scholar]
- Wright WE, Shay JW (2005) Telomere biology in aging and cancer. J Am Geriatr Soc 53: S292–S294. 10.1111/j.1532-5415.2005.53492.x [DOI] [PubMed] [Google Scholar]
- You Y-H, Lee D-H, Yoon J-H, Nakajima S, Yasui A, Pfeifer GP (2001) Cyclobutane pyrimidine dimers are responsible for the vast majority of mutations induced by UVB irradiation in mammalian cells. J Biol Chem 276: 44688–44694. 10.1074/jbc.M107696200 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008) Model-based analysis of ChIP-seq (MACS). Genome Biol 9: R137. 10.1186/gb-2008-9-9-r137 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Functional enrichment categories of RB1-regulated genes. LSA-2021-01134_TableS1.xlsx (187.9KB, xlsx)
Table S2. Functional enrichment analysis of differentially susceptible genes in RB1 KO cells. LSA-2021-01134_TableS2.xlsx (713KB, xlsx)
Source Data for Figure 5LSA-2021-01134_SdataF5.xlsx (1.4MB, xlsx)
Data Availability Statement
Data sets produced in this study are available on the Gene Expression Omnibus database under: GSE173125, GSE173126, GSE173127, and GSE173128. Computational scripts can be found at https://github.com/orgs/MorrisonLabSU/.