

Abstract
Introduction
Long QT syndrome (LQTS) is a life‐threatening inherited channelopathy, and prolonged QT intervals easily trigger malignant arrhythmias, especially torsades de pointes and ventricular fibrillation.
Materials and methods
The proband with overlapped phenotypes of LQTS and sinoatrial node dysfunction underwent some necessary examinations, including echocardiography, electrocardiogram (ECG), and Holter monitoring. Next, whole‐exome sequencing was performed, and candidate genes were validated by Sanger sequencing. RNA secondary structure and protein physical‐chemical parameter analyses were used to predict the possible structural change of the proteins induced by the mutations.
Results
We identified the digenic heterozygous mutations of KCNH2 p.307_308del (NM_001204798, c.921_923del) and SCN5A p.R1865H (NM_001160160, c.G5594A) in the female and young proband (II: 1) of LQTS and ventricular fibrillation with repeat syncope at rest. Subsequently, she occurred with obvious sinus arrest with persistent ventricular pacing of implantable cardioverter‐defibrillator. The heterozygous SCN5Ap.R1865H was carried by her father and sister but not carried by I:2. II:1 carried with KCNH2 p.307_308del as a de novo mutation, but not existed in other family members. RNA secondary structure of KCNH2 p.307_308del showed a false regional double helix, and its amino acids' hydrophobicity was significantly weakened. For the Nav1.5 protein property, SCN5A p.R1865H slightly increased the molecular weight and aliphatic index but reduced the instability index.
Conclusions
The digenic heterozygous KCNH2 and SCN5A mutations were associated with young early‐onset long QT syndrome and sinoatrial node dysfunction.
Keywords: ion channelopathy, KCNH2, long QT syndrome, SCN5A, whole‐exome sequencing
Abbreviations
- ARVC/D
arrhythmogenic right ventricular cardiomyopathy/dysplasia
- ECG
electrocardiogram
- ICD
implantable cardioverter‐defibrillator
- KCNH2
known as KV11.1, delayed rectifier K+ channels
- LQTS
long QT syndrome
- SCD
sudden cardiac death
- SCN5A
known as Nav1.5, cardiac‐type voltage‐dependent Na+ channel
- WES
whole‐exome sequencing
1. INTRODUCTION
Long QT syndrome (LQTS) is an inherited cardiac electrophysiologic disorder characterized by QT interval prolongation in the electrocardiogram and family history of sudden cardiac death (SCD) (Neira et al., 2019; Wallace et al., 2019). A prolonged QT interval is vulnerable to malignant arrhythmias, including torsade de pointes and ventricular fibrillation, which result in unexplained syncope, seizures, and even sudden death in patients, especially young individuals (Shah et al., 2019). LQTS is caused mainly by functional abnormalities of changed repolarizing ionic channels, including mutant potassium channels, sodium channels, or calcium channels. Sixteen genes and more than 600 mutations are linked to LQTS. Mutations of KCNQ1, KCNH2, SCN5A, and ankyrin‐B are more common, and different types of LQTS were associated with different mutations of the genes (Nakano & Shimizu, 2016). In this study, we examined a young proband characterized as LQTS and sinoatrial node dysfunction. The predisposing genes of potential risk were investigated by whole‐exome sequencing (WES) and validated by Sanger sequencing.
2. MATERIAL AND METHODS
2.1. Study objective
Here, we studied a Chinese family and the proband (II:1), an 18‐year‐old female who suffered from LQTS since five years old. We obtained detailed clinical information including initial ventricular tachycardia manifestation, presentation age, therapeutic process, and family history. The proband underwent echocardiography, electrocardiogram (ECG), Holter monitoring, implantable cardioverter‐defibrillator (ICD) programming, and other necessary examinations. Her family members were asked to accept routine ECG and echocardiography. Their peripheral blood samples were collected to perform the following detection.
This study was approved by the Guangdong Medical Institutional Review Board and Medical Ethics Committees [no. GDREC2016001H (R1)]. Informed written consent was obtained from the family members.
2.2. Diagnosis of long QT syndrome
The clinical diagnostic criteria for LQTS mainly depended on the clinical standard score (Goldenberg & Moss, 2008; Skinner & CSANZ, 2007). (1) Corrected QT interval: when the QT interval ≥0.48 s, the point is 3; 0.46–0.47, 2; 0.45 (in males), 1. (2) when showing torsades de pointes, the points is 2; T‐wave alternans, 1; notched T wave in three leads, 1; and low heart rate for ages, 0.5. (3) Clinical history: when suffering from syncope with stress, the point is 2; syncope without stress, 1; and congenital deafness, 0.5. (4) Family history: family members with definite LQTS, the point is 1; unexplained sudden cardiac death at/under 30‐year old among immediate family member(s), 0.5.
The probability of LQTS is high when scoring ≥4 points; intermediate probability of LQTS when scoring 2 to 3 points; low probability of LQTS when scoring ≤1 point (Goldenberg & Moss, 2008).
2.3. Whole‐exome sequencing, sequence alignment, variation calling, and data annotation
The proband (II: 1) was diagnosed with LQTS and thus selected to conduct WES in this family. Genomic DNA samples were extracted from the peripheral blood of the family members using a standard DNA extraction protocol. The isolated genomic DNA was fragmented into 150–200 bp and then subjected to DNA library preparation Illumina paired‐end protocols. A portion of each library was used to create an equimolar pool. Each pool was amplified to enrich for targets to be sequenced by the Agilent SureSelectXT Target Enrichment System (Agilent Technologies Inc., Santa Clara, CA, USA). Accordingto the manufacturer's protocol, whole‐exome capture was performed with the Agilent SureSelectXT Human All Exon 50 Mb Kit (Agilent Technologies Inc.)
Low‐quality reads were filtered, and 3′/5′ adapters were trimmed using the Trim Galore program. Clean reads were mapped to the hg19 version of the human genome using the Burrows‐Wheeler Aligner (BWA) program. Following the exclusion of duplicate reads, insertion–deletions (InDels) and single nucleotide polymorphisms (SNPs) calling, GATK or Sequence Alignment/Map tools (SAM tools) were used. Functional annotation was used to annotate each SNP and InDel, and nonsynonymous SNPs were predicted by SIFT (Kumar et al., 2009) and PolyPhen‐2 (polymorphism phenotyping v2; Adzhubei et al., 2010). For II: 1 patient, there were ~106 variants of candidate genes occurring in coding regions. The criteria of filtering variants were as follows: (1) missense, nonsense and InDel variants; (2) same variants in the WES data; and (3) SNPs with a minor allele frequency not more than 0.01 according to the NCBI‐SNP database (Smigielski et al., 2000; Via et al., 2010).
2.4. Sanger sequencing for candidate genes
The candidate gene sequences were obtained from nucleotide sequence database GenBank (Kuznetsov & Bollin, 2021). The primers (Table 1) were designed using PrimerBank (Wang et al., 2012). These genes from genomic DNA were amplified, and products of amplification were purified using the PCR Purification Kit (Qiagen). They were sequenced directly with the BigDye terminator method (Applied Biosystems, Foster City, CA, USA) on a capillary autosequencer (ABI Prism 3100).
TABLE 1.
Predisposing gene analysis of whole‐exome sequencing
| Name | Sequence (5′–3′) |
|---|---|
| SCN5A exon 27 forward primer | CGCTTATGTCAAGTGGGAGG |
| SCN5A exon 27 reverse primer | AAGGAAGTGGAGGAGATGGAG |
| KCNH2 exon 3 forward primer | GACCTCTGATGCTCGCTCTG |
| KCNH2 exon 3 reverse primer | CCCACCCTTCAGTAGTCCC |
2.5. Conservation analyses
We searched for the candidate protein from the universal protein knowledgebase (UniProt; UniProt Consortium T, 2018). Multiple cross‐species amino acid sequences were selected in the list and extracted into FASTA format. For Multiple Sequence Alignment, T‐Coffee (https://www.ebi.ac.uk/Tools/msa/tcoffee/) was used to compare the conservation of target mutation sites.
2.6. RNA secondary structure prediction
We used the RNAfold WebSever (http://rna.tbi.univie.ac.at/cgi‐bin/RNAWebSuite/RNAfold.cgi) to predict the RNA secondary structure. The difference of the minimum free energy (MFE) in the centroid secondary structure was evaluated between the mutant and wild‐type mRNA.
2.7. Protein physical and chemical parameter prediction
To compare the potential functional influence on the protein physical‐chemical properties, we focused on analyzing the changes of hydrophobicity, transmembrane domain and protein phosphorylation of amino acids induced by mutant and wild‐type SCN5A and KCNH2. The ProtScale (https://web.expasy.org/protscale/) and NetPhos (http://www.cbs.dtu.dk/services/NetPhos/) were used for hydrophobicity, transmembrane domain and phosphorylation analysis. Next, we used ProtParam tool (https://web.expasy.org/protparam/) to predict the theoretical pI, instability index, aliphatic index, grand average of hydropathicity (GRAVY) and so on.
3. RESULT
3.1. Baseline characteristics
The family pedigree and genotype–phenotype correlations are shown in Figure 1a. The female proband II: 1 suffered from repeated convulsions and occasional syncope in April 2008 since five years old. Her ECG monitoring showed torsades de pointes, but the monitoring information did not be saved. The Holter and ECG demonstrated LQTS. The QTc interval was 550 milliseconds (ms) when the heart rate was 79 bpm. It was 560 ms when the heart rate was 131 bpm (Figure 1b, c). The ECGs of her families showed normal QTc intervals (Figure 1d, f). There is no history of syncope, seizures, or unexplained sudden death among her father, mother, and younger sister. The clinical standard score of the proband was 6 points according to the guidelines. Echocardiography, chest radiographs, and thyroid examination presented no significant abnormalities. II: 1 had experienced repeated amaurosis and syncope in May 2012 and been admitted to hospital. Next, she had been treated with ICD. One month later, ICD monitoring showed that the patient experienced multiple ventricular tachycardias (235 bmp) and even ventricular fibrillation. The episodes had triggered ICD to diagnose and release cardioversion and defibrillation treatment (Figure 1g). The charging energy gradually increased from 15J to 36J and achieve successful treatment. Three years later (in Feb 2018), the Holter monitoring presented sinus arrest with persistent ventricular pacing of ICD (Figure 1h). The ICD pacing rhythm accounted for 81.2% (no abnormalities in pacing and sense). Ventricular fusion beat, occasional ventricular premature beats, and long QT intervals were also found in the Holter recording.
FIGURE 1.
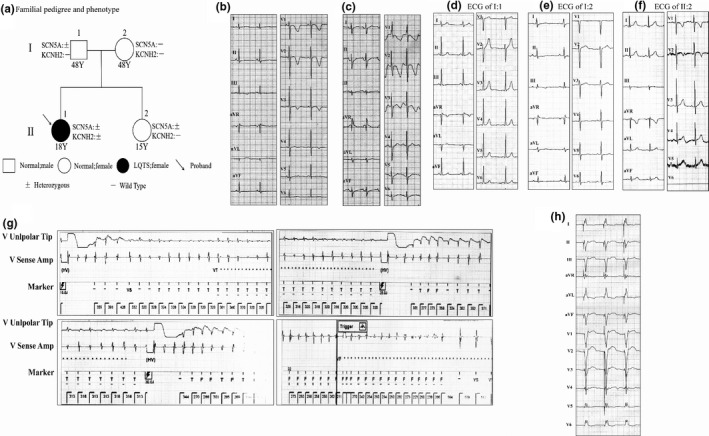
Family pedigree and representative ECGs. (a) The proband diagnosed with LQTS is shown using arrows. II: 1 carried the digenic heterozygous mutations of KCNH2 p.307_308del and SCN5A p.R1865H. I: 1 and II: 2 were heterozygous for SCN5A p.R1865H. Except II: 1, other family members did not carry KCNH2 mutation. (b, c), The ECG images of II: 1 demonstrated LQTS. QTc interval was 550 milliseconds (ms) when the heart rate was 79 bpm. It was 560 ms when the heart rate was 131 bpm. (d–f), Representative ECGs of family members of the proband (the chest lead V6 of II: 2 fell off). (g) The proband experienced multiple ventricular tachycardias and even ventricular fibrillation by ICD monitoring. ICD had released cardioversion and defibrillation treatment. (h) The Holter presented sinus arrest with persistent ventricular pacing of ICD
3.2. WES and predisposing gene analysis
There were 552 variantss in exon and splicing regions by WES. Four potential pathogenic variants, including SCN5A p.R1865H (NM_001160160, c.G5594A), LAMA2 p.A961T (NM_000426, c.G2881A), KCNH2 p.307_308del (NM_001204798, c.921_923del), and DMD p.E1028V (NM_004011, c.A3083T) were involved in the occurrence of arrhythmia and cardiomyopathy (Table 2). In these known and candidate genes, KCNH2 gene encodes voltage‐gated potassium channel activity of cardiomyocytes, which participated in the action potential repolarization. SCN5A gene encodes for voltage‐gated sodium channel subunit as an integral membrane protein, responsible for the initial upstroke of the action potential (obtained from GenBank database). Mutations of KCNH2 and SCN5A genes are closely related to LQTS. The mutations of KCNH2 p.307_308del and SCN5A p.R1865H were found in the proband by WES and validated as positive by Sanger sequencing.
TABLE 2.
Predisposing gene analysis of whole‐exome sequencing
| Chr | Start | Ref | Alt | Gene | Amino acid Change | 1000g2015 | snp142 | SIFT | Polyphen2 | OMIM disease |
|---|---|---|---|---|---|---|---|---|---|---|
| chr3 | 38592170 | C | T | SCN5A | NM_001160160:exon28:c.G5594A:p.R1865H | ‐ | rs370694515 | 0.055(T) | 1 (D) | Ion channelopathy; cardiomyopathy |
| chr6 | 129618854 | G | A | LAMA2 | NM_001098623:exon3:c.A1177G:p.N393D | <0.001 | rs147301872 | 0.005(D) | 0.109 (B) | Muscular dystrophy |
| chr7 | 150648538 | CCA | ‐ | KCNH2 | NM_001204798:exon3:c.921_923del:p.307_308del | ‐ | ‐ | ‐ | ‐ | Short QT syndrome; long QT syndrome |
| chrX | 31854929 | T | A | DMD | NM_004011:exon21:c.A3083T:p.E1028V | ‐ | ‐ | 0.054(T) | 0.999 (D) | Cardiomyopathy; muscular dystrophy |
Chr, chromosome; 1000G, 1000 genomes (2015 version); SNP, single nucleotide polymorphism; B, benign; D, damaging; T, tolerated; –, no report; OMIM, Online Mendelian Inheritance in Man.
Additionally, the heterozygous SCN5A p.R1865H was carried by I: 1 and II: 2, but not carried by I: 2 (Figure 1a). Except II: 1, other family members without cardiac event or cardiac disease did not carry KCNH2 mutation. Moreover, the conservation analyses demonstrated that the mutant sites of amino acid sequences of SCN5A and KCNH2 protein were highly conserved (Figure 2). Therefore, KCNH2 p.307_308del was considered as de novo mutation in II: 1 (Figure 1a and Figure 3).
FIGURE 2.
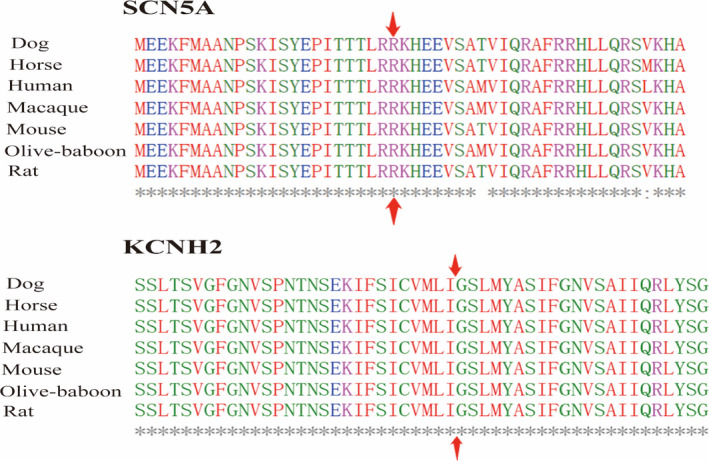
Conservation analyses at the mutant sites of SCN5A and KCNH2 protein. SCN5A p.R1865 and KCNH2 p.307_308 of amino acid sequences were highly conserved across the common species
FIGURE 3.
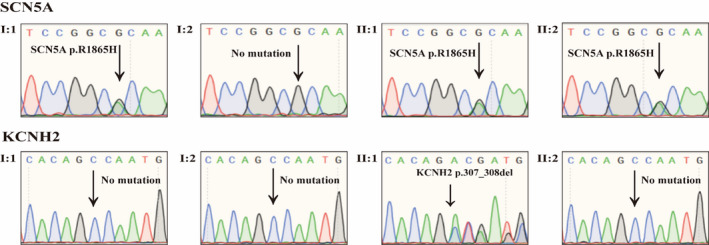
Sanger sequencing for SCN5A and KCNH2 mutations. KCNH2 p.307_308del and SCN5A p.R1865H of the proband were validated as positive by Sanger sequencing. Additionally, I: 1 and II: 2 carried with the heterozygous for SCN5A p.R1865H. Except II: 1, other family members did not carry with the KCNH2 mutation
3.3. RNA secondary structure prediction
The RNA secondary structure differences were presented by the RNAfold WebSever (Figure 4). Compared with wild‐type KCNH2 (Figure 4a), the structure of KCNH2 p.307_308del affected the single‐stranded RNA folding, resulting in a false regional double helix (Figure 4b). The minimum free energy (MFE) of KCNH2 p.307_308del increased, which thus lead to a reduction of structural stability. However, SCN5A p.R1865H showed no significant influence on the RNA structure (Figure 4c,d). The MFE of SCN5A p.R1865H mutation (−178.70 kcal/mol) was approximately similar to that of the wild type (−178.30 kcal/mol), which probably induced no obvious change in the centroid secondary structure.
FIGURE 4.
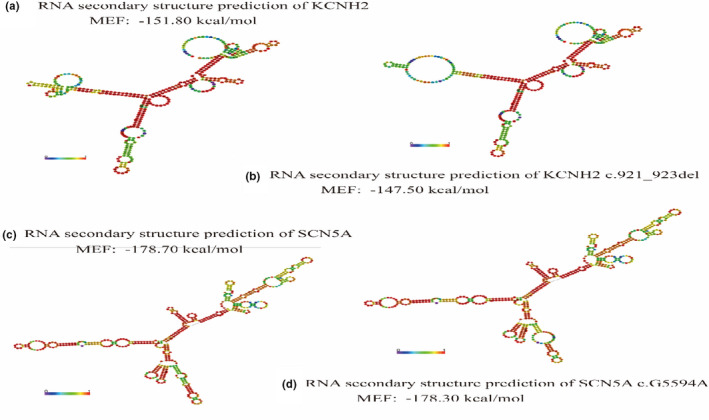
RNA secondary structural prediction. (a, b) Compared with wild‐type KCNH2, the structure of KCNH2 p.307_308del affected the single‐stranded RNA folding, resulting in a false regional double helix. The minimum free energy (MFE) of KCNH2 p.307_308del increased, which thus led to a reduction of structural stability. (c, d) SCN5A p.R1865H showed no significant influence on the RNA structure, and the MFE value of SCN5A p.R1865H mutation was approximately similar to that of the wild type
3.4. Physical and chemical parameter prediction of protein
Compared with the amino acids of wild‐type KCNH2 (Table 3), KCNH2 p.307_308del showed a decreasing trend in molecular weight and increasing instability. However, the prediction of theoretical pI, aliphatic index and GRAVY presented no significant differences. Compared to the Nav1.5 protein properties of wild‐type SCN5A, SCN5A p.R1865H slightly increased its molecular weight and aliphatic index but reduced its instability index. Theoretical pI, aliphatic index, and GRAVY were not affected by SCN5A p.R1865H.
TABLE 3.
Amino acids physical and chemical parameter prediction
| Physical and chemical parameters | KCNH2‐wild type | KCNH2‐p.307_308del | SCN5A‐wild type | SCN5A‐p.R1865H |
|---|---|---|---|---|
| Molecular weight | 14430.15 | 14359.07 | 18856.35 | 18872.39 |
| Theoretical pI | 9.30 | 9.30 | 12.10 | 12.10 |
| Instability index | 30.43 | 30.59 | 95.32 | 91.73 |
| Aliphatic index | 106.54 | 106.59 | 45.00 | 47.22 |
| GRAVY | 0.336 | 0.325 | −0.541 | −0.511 |
Abbreviation: GRAVY, Grand average of hydropathicity.
Next, hydrophobicity analyses for wild‐type and mutant proteins were performed (Table 4, Figure 5). The changed site (position 307) of KCNH2 p.307_308del was located close to the largest hydrophobic region of the protein (Figure 5a). The hydrophobicity of predicted amino acid residues and adjacent sequences was significantly weakened, which probably made the largest hydrophobic domain (position 303) disorganized. The changed site of the SCN5A gene (position 1864) increased the corresponding amino acid residues and nearby sequences hydrophobicity, but the influence was not significant (Figure 5b). Transmembrane structure analysis (Table 4) showed that the mutant site of KCNH2 p.307_308del was located in the protein's transmembrane domain, which suggested that the site may be associated with potential functions, including protein localization, substances transportation, ion channels, and others. Phosphorylation analysis of KCNH2 p.307_308del showed that this mutation caused amino acid residues near position 309 dephosphorylation. The result indicated that KCNH2 p.307_308del might impact the catalytic efficiency of protein. However, there was no significant change in protein phosphorylation for SCN5A p.R1865H (Table 4).
TABLE 4.
Hydrophobicity, transmembrane domain, and phosphorylation analyses
| KCNH2 | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 302 | 303 | 304 | 305 | 306 | 307 | 308 | 309 | 310 | 311 | 312 | 313 | ||
| Amino acid | WT | I | C | V | M | L | I | G | S | L | M | Y | A |
| p.307_308del | I | C | V | M | L | D | ‐ | S | L | M | Y | A | |
| Hydrophobicity | WT | 2.167 | 3.100(max) | 2.556 | 2.156 | 2.667 | 2.378 | 1.956 | 1.689 | 1.389 | 1.467 | 1.278 | 1.278 |
| p.307_308del | 2.167 | 2.211 | 1.622 | 1.733 | 2.033 | 1.389 | ‐ | 1.311 | 0.756 | 1.044 | 0.933 | 1.278 | |
| Transmembrane | WT | TMhelix | TMhelix | TMhelix | TMhelix | TMhelix | TMhelix | TMhelix | TMhelix | TMhelix | TMhelix | TMhelix | TMhelix |
| p.307_308del | TMhelix | TMhelix | TMhelix | TMhelix | TMhelix | TMhelix | TMhelix | TMhelix | TMhelix | TMhelix | TMhelix | TMhelix | |
| Phosphorylation | WT | Yes | |||||||||||
| p.307_308del | No | ||||||||||||
| SCN5A | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1858 | 1859 | 1860 | 1861 | 1862 | 1864 | 1865 | 1866 | 1867 | 1868 | 1869 | 1870 | ||
| Amino acid | WT | I | T | T | T | L | R | K | H | E | E | V | S |
| p.R1865H | I | T | T | T | L | H | K | H | E | E | V | S | |
| Hydrophobicity | WT | −0.033 | −0.111 | −0.522 | −0.878 | −0.922 | −1.989 | −2.300 | −1.756 | −1.767 | −1.989 | −1.278 | −0.311 |
| p.R1865H | −0.033 | −0.111 | −0.522 | −0.733 | −0.778 | −1.844 | −2.156 | −1.611 | −1.622 | −1.844 | −1.133 | −0.311 | |
| Transmembrane | WT | outside | outside | outside | outside | outside | outside | outside | outside | outside | outside | outside | outside |
| p.R1865H | outside | outside | outside | outside | outside | outside | outside | outside | outside | outside | outside | outside | |
| Phosphorylation | WT | Yes | Yes | ||||||||||
| p.R1865H | Yes | Yes | |||||||||||
FIGURE 5.
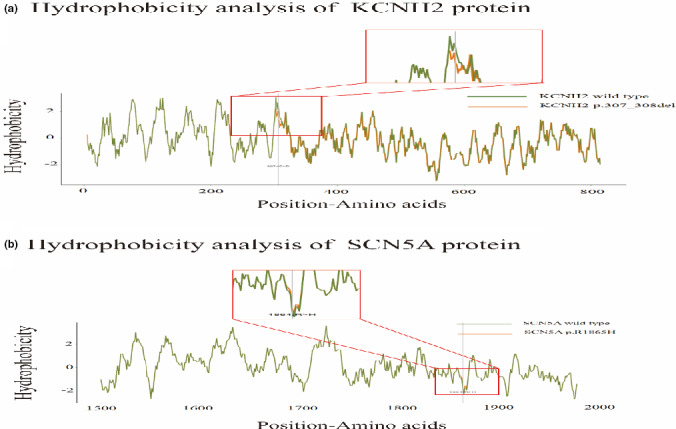
The hydrophobicity prediction of protein. (a) The changed site (position 307) of KCNH2 p.307_308del was located close to the largest hydrophobic region of the protein. The hydrophobicity of predicted amino acid residues and adjacent sequences was significantly weaken. (b) The changed site of SCN5A gene (position 1864) increased the corresponding amino acid residues and nearby sequences hydrophobicity, but the influence was not significant
4. DISCUSSION
In our study, the proband with overlapped phenotypes of young early‐onset LQTS and sinoatrial node dysfunction was first demonstrated to carry with the digenic heterozygous and pathogenic mutations of KCNH2 p.307_308del and SCN5A p.R1865H. KCNH2 p.307_308del induced a regional double helix of the amino acids misfolded and largest hydrophobic domain disorganized, which thus mainly caused LQTS. The SCN5A p.R1865H slightly increased the molecular weight and aliphatic index, but reduced the instability index of Nav1.5 protein property, which potentially induced subsequent sinoatrial node dysfunction.
Genetic screening for LQTS is of great clinical value. The genotype is closely related to gene‐specific epidemiology, risk stratification, and treatment of LQTS. Different channel mutations present their own electrocardiogram signature, typical presentations, and prognosis (Skinner et al., 2019; Wilde & Amin, 2017). KCNH2 gene (HERG) encodes a voltage‐sensitive channel protein mediating the rapid component of delayed rectifier K+ current (Kv11.1) (Butler et al., 2019). The channel protein is expressed mainly in the myocardium and acts as a molecular target for most antiarrhythmic drugs. KCNH2 mutations are associated with inherited arrhythmias involving LQTS, ventricular tachycardia, and SCD (Vandenberg et al., 2012). Especially, loss‐of‐function mutations in KCNH2 are the leading causes of LQTS (type 2) by delaying the ventricular action potential repolarization (Delisle et al., 2004; Ono et al., 2020). In this family, the patient (II: 1) with digenic heterozygous mutations of KCNH2 p.307_308del and SCN5A p.R1865H presented the earliest phenotype of LQTS, and she suffered from syncope, torsades de pointes, and ventricular fibrillation more frequently at rest, whereas the members (I:1 and II:2) without KCNH2 p.307_308del showed normal QT intervals and cardiac function. The changed site of KCNH2 p.307_308 was highly conserved across most species, suggesting p.307_308 of KCNH2 protein playing a significant role in function. The result indicated that the novel genetic background, KCNH2 p.307_308del, may affect and even induce the phenotype of LQTS. The predictions of the RNA secondary structure and physical‐chemical parameters showed KCNH2 p.307_308del affected the single‐stranded RNA folding, and subsequently and significantly weaken the hydrophobicity of mutant amino acid residues, which indicated that KCNH2 mutation probably played a dominant hereditary role in the occurrence of LQTS in this proband.
SCN5A gene is located in chromosome 3p22.2, encoding the alpha subunit of the voltage‐gated sodium channel (Nav1.5) in human cardiomyocytes (Li et al., 2018). Nav1.5 channel was described first in 1955 to be responsible for inward sodium current, which participated in the rapid flow of Na+ during the depolarization phase in cardiomyocytes (George et al., 1995). The protein contains four homologous domains (DI–DIV), and each includes six transmembrane spanning segments (S1–S6). SCN5A mutations may influence the expression and function of the Nav1.5 channel. Therefore, the pathogenic mutations of SCN5A probably caused distinct clinical diseases, including LQTS, Brugada syndrome, sick sinus syndrome, and even fatal arrhythmias (Benson et al., 2003; Remme et al., 2006; Wilde & Amin, 2018). Over 300 gain‐of‐function mutations were associated with type 3 of LQTS (Flaim et al., 2007; Jenewein et al., 2017; Remme, 2013; Wilde et al., 2016). Our study firstly identified SCN5A p.R1865H in the patient with overlapped phenotypes of LQTS and sinoatrial node dysfunction. The RNA secondary structure predictions of SCN5A p.R1865H slightly affected its RNA folding. In our study, the proband was diagnosed with the earliest LQTS and subsequently experienced sinoatrial node dysfunction, such as sinus arrest with persistent ventricular pacing of ICD. It was worth mentioning that SCN5A p.R1865H was found in one patient with arrhythmogenic right ventricular cardiomyopathy/dysplasia (ARVC/D) (Te Riele et al., 2017) and demonstrated a close association with reduced sodium current. Loss‐of‐function mutations of SCN5A were commonly related to sinoatrial node dysfunction. Therefore, we speculated that SCN5A p.R1865H, a loss‐of‐function mutation, led to subsequent sinus arrest in the proband by reducing sodium channel current. As mentioned above, some family members carrying heterozygous SCN5A p.R1865H mutation showed no evidence of cardiac events or cardiac diseases. The reason may be as follows: First, I:1 and II:2 who carried with the heterozygous SCN5A p.R1865H presented no clinical syndromes because of incomplete penetrance or delayed onset. Moreover, gain‐of‐function mutation of SCN5A commonly induced LQTS, while loss‐of‐function mutation of SCN5A ordinary led to sinoatrial node dysfunction, atrioventricular block, atrial fibrillation and cardiomyopathy (e.g., ARVC/D; Blana et al., 2010; Han et al., 2018). Therefore, in this study, SCN5A p.R1865H may be the main cause of sinoatrial node dysfunction, whereas KCNH2 p.307_308del only carried by II: 1 may potentially induce the phenotype of LQTS. However, it was hard to determine whether the coexisting interactions of KCNH2 p.307_308del and SCN5A p.R1865H increased the risk of young and early‐onset LQTS, or whether KCNH2 mutation was only associated with LQTS, while SCN5A mutation was only associated with sinoatrial node dysfunction.
5. CONCLUSIONS
We firstly identified the novel digenic heterozygous mutations by WES, KCNH2 p.307_308del and SCN5A p.R1865H, which resulted in LQTS with repeat syncope, torsades de pointes, ventricular fibrillation, and sinoatrial node dysfunction. KCNH2 p.307_308del may affect the function of Kv11.1 channel in cardiomyocytes by inducing a regional double helix of the amino acids misfolded and largest hydrophobic domain disorganized. SCN5A p.R1865H reduced the instability index of Nav1.5 protein and sodium current. All of these were closely related to young early‐onset LQTS and sinoatrial node dysfunction.
6. LIMITATIONS
Our study was performed only in the statistical field on KCNH2 p.307_308del and SCN5A p.R1865H by WES and predisposing genes analyses. More cellular and animal research is needed to further investigate whether the coexisting interaction of KCNH2 p.307_308del and SCN5A p.R1865H increases the risk of the early‐onset LQTS and sinoatrial node dysfunction.
7. CONSENT FOR PUBLICATION
Not applicable.
CONFLICT OF INTEREST
The authors declare that they have no competing interest.
AUTHOR CONTRIBUTIONS
ZY and YBL contributed to pedigree analysis and writing; YTM contributed to bioinformatics analysis; JNH, WLZ, and LC contributed to Sanger sequencing; JZX, YH, LBW, and FW made case collection and follow‐up; XFL and YBL made quality control of clinical data and clinical design.
ACKNOWLEDGMENTS
We thank Professor Shulin Wu, Yumei Xue, and Fang Rao from Guangdong Cardiovascular Institute. We appreciate Jinsheng Tao and Zhipeng Cao for assistance in data analysis and visualization.
Yang, Z. , Ma, Y. , Huang, J. , Zhang, Z. , Xian, J. , Huang, Y. , Wu, L. , Zhu, W. , Wang, F. , Chen, L. , Lin, X. , & Lin, Y. (2022). Digenic heterozygous mutations of KCNH2 and SCN5A induced young and early‐onset long QT syndrome and sinoatrial node dysfunction. Annals of Noninvasive Electrocardiology, 27, e12889. 10.1111/anec.12889
Funding information
This study was supported by the following funding: The Foundation Project of Medical Science and Technology Research in Guangdong Province (A2018043); Science and Technology Project of Zhuhai (20191210E030072); Natural Science Foundation of Guangdong (2016A030313279); and the Construction Foundation of the Key Clinical Specialties in Zhuhai (ZHLCZD011)
Contributor Information
Xiufang Lin, Email: linxiufang_126@126.com.
Yubi Lin, Email: linyubi88@qq.com.
DATA AVAILABILITY STATEMENT
The data used in this study are not publicly available, but it might be available from the corresponding author upon reasonable request and permission from relevant Chinese Authorities.
REFERENCES
- Adzhubei, I. A. , Schmidt, S. , Peshkin, L. , Ramensky, V. E. , Gerasimova, A. , Bork, P. , Kondrashov, A. S. , & Sunyaev, S. R. (2010). A method and server for predicting damaging missense mutations. Nature Methods, 7(4), 248–249. 10.1038/nmeth0410-248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson, D. W. , Wang, D. W. , Dyment, M. , Knilans, T. K. , Fish, F. A. , Strieper, M. J. , Rhodes, T. H. , & George, A. L. (2003). Congenital sick sinus syndrome caused by recessive mutations in the cardiac sodium channel gene (SCN5A). Journal of Clinical Investigation, 112(7), 1019–1028. 10.1172/JCI200318062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blana, A. , Kaese, S. , Fortmüller, L. , Laakmann, Sandra , Damke, D. , van Bragt, K. , Eckstein, J. , Piccini, I. , Kirchhefer, U. , Nattel, S. , Breithardt, G. , Carmeliet, P. , Carmeliet, E. , Schotten, U. , Verheule, S. , Kirchhof, P. , & Fabritz, L. (2010). Knock‐in gain‐of‐function sodium channel mutation prolongs atrial action potentials and alters atrial vulnerability. Heart Rhythm, 7(12), 1862–1869. 10.1016/j.hrthm.2010.08.016. [DOI] [PubMed] [Google Scholar]
- Butler, A. , Helliwell, M. V. , Zhang, Y. , Hancox, J. C. , & Dempsey, C. E. (2019). An update on the structure of hERG. Frontiers in Pharmacology, 10, 1572. 10.3389/fphar.2019.01572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delisle, B. P. , Anson, B. D. , Rajamani, S. , & January, C. T. (2004). Biology of cardiac arrhythmias: ion channel protein trafficking. Circulation Research, 94(11), 1418–1428. 10.1161/01.RES.0000128561.28701.ea. [DOI] [PubMed] [Google Scholar]
- Flaim, S. N. , Giles, W. R. , & McCulloch, A. D. (2007). Arrhythmogenic consequences of Na+ channel mutations in the transmurally heterogeneous mammalian left ventricle: analysis of the I1768V SCN5A mutation. Heart Rhythm, 4(6), 768–778. 10.1016/j.hrthm.2007.02.009. [DOI] [PubMed] [Google Scholar]
- George Jr, A. L. , Varkony, T. A. , Drabkin, H. A. , Han, J. , Knops, J. F. , Finley, W. H. , Brown, G. B. , Ward, D. C. , & Haas, M. (1995). Assignment of the human heart tetrodotoxin‐resistant voltage‐gated Na+ channel alpha‐subunit gene (SCN5A) to band 3p21. Cytogenetics and Cell Genetics, 68(1–2), 67–70. [DOI] [PubMed] [Google Scholar]
- Goldenberg, I. , & Moss, A. J. (2008). Long QT syndrome. Journal of the American College of Cardiology, 51(24), 2291–2300. 10.1016/j.jacc.2008.02.068. [DOI] [PubMed] [Google Scholar]
- Han, D. , Tan, H. , Sun, C. , & Li, G. (2018). Dysfunctional Nav1.5 channels due to SCN5A mutations. Exp Biol Med (Maywood), 243(10), 852–863. 10.1177/1535370218777972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenewein, T. , Beckmann, B. M. , Rose, S. , Osterhues, Hh , Schmidt, U. , Wolpert, C. , Miny, P. , Marschall, C. , Alders, M. , Bezzina, Cr , Wilde, Aam , Kääb, S. , & Kauferstein, S. (2017). Genotype‐phenotype dilemma in a case of sudden cardiac death with the E1053K mutation and a deletion in the SCN5A gene. Forensic Science International, 275, 187–194. 10.1016/j.forsciint.2017.02.038. [DOI] [PubMed] [Google Scholar]
- Kumar, P. , Henikoff, S. , & Ng, P. C. (2009). Predicting the effects of coding non‐synonymous variants on protein function using the SIFT algorithm. Nature Protocols, 4(7), 1073–1081. 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- Kuznetsov, A. , & Bollin, C. J. (2021). NCBI Genome Workbench: Desktop Software for Comparative Genomics, Visualization, and GenBank Data Submission. Methods in Molecular Biology, 2231, 261–295. [DOI] [PubMed] [Google Scholar]
- Li, W. , Yin, L. , Shen, C. , Hu, K. , Ge, J. , & Sun, A. (2018). SCN5A variants: Association with cardiac disorders. Frontiers in Physiology, 9, 1372. 10.3389/fphys.2018.01372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano, Y. , & Shimizu, W. (2016). Genetics of long‐QT syndrome. Journal of Human Genetics, 61(1), 51–55. 10.1038/jhg.2015.74. [DOI] [PubMed] [Google Scholar]
- Neira, V. , Enriquez, A. , Simpson, C. , & Baranchuk, A. (2019). Update on long QT syndrome. Journal of Cardiovascular Electrophysiology, 30(12), 3068–3078. 10.1111/jce.14227. [DOI] [PubMed] [Google Scholar]
- Ono, M. , Burgess, D. E. , Schroder, E. A. , Elayi, C. S. , Anderson, C. L. , January, C. T. , Sun, B. , Immadisetty, K. , Kekenes‐Huskey, P. M. , & Delisle, B. P. (2020). Long QT Syndrome Type 2: Emerging strategies for correcting class 2 KCNH2 (hERG) mutations and identifying new patients. Biomolecules, 10(8), 1144. 10.3390/biom10081144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remme, C. A. (2013). Cardiac sodium channelopathy associated with SCN5A mutations: electrophysiological, molecular and genetic aspects. Journal of Physiology, 591(17), 4099–4116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remme, C. A. , Verkerk, A. O. , Nuyens, D. , van Ginneken, A. C. G. , van Brunschot, S. , Belterman, C. N. W. , Wilders, R. , van Roon, M. A. , Tan, H. L. , Wilde, A. A. M. , Carmeliet, P. , de Bakker, J. M. T. , Veldkamp, M. W. , & Bezzina, C. R. (2006). Overlap syndrome of cardiac sodium channel disease in mice carrying the equivalent mutation of human SCN5A‐1795insD. Circulation, 114(24), 2584–2594. [DOI] [PubMed] [Google Scholar]
- Shah, S. R. , Park, K. , & Alweis, R. (2019). Long QT syndrome: A comprehensive review of the literature and current evidence. Current Problems in Cardiology, 44(3), 92–106. 10.1016/j.cpcardiol.2018.04.002. [DOI] [PubMed] [Google Scholar]
- Skinner, J. R. , & CSANZ Cardiovascular Genetics Working Group (2007). Guidelines for the diagnosis and management of familial long QT syndrome. Heart, Lung & Circulation, 16(1), 22–24. 10.1016/j.hlc.2006.10.021. [DOI] [PubMed] [Google Scholar]
- Skinner, J. R. , Winbo, A. , Abrams, D. , Vohra, J. , & Wilde, A. A. (2019). Channelopathies that lead to sudden cardiac death: Clinical and genetic aspects. Heart, Lung & Circulation, 28(1), 22–30. 10.1016/j.hlc.2018.09.007. [DOI] [PubMed] [Google Scholar]
- Smigielski, E. M. , Sirotkin, K. , Ward, M. , & Sherry, S. T. (2000). dbSNP: A database of single nucleotide polymorphisms. Nucleic Acids Research, 28(1), 352–355. 10.1093/nar/28.1.352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Te Riele, A. S. , Agullo‐Pascual, E. , James, C. A. , Leo‐Macias, A. , Cerrone, M. , Zhang, M. , Lin, X. , Lin, B. , Sobreira, N. L. , Amat‐Alarcon, N. , Marsman, R. F. , Murray, B. , Tichnell, C. , van der Heijden, J. F. , Dooijes, D. , van Veen, T. A. B. , Tandri, H. , Fowler, S. J. , Hauer, R. N. W. , … Judge, D. P. (2017). Multilevel analyses of SCN5A mutations in arrhythmogenic right ventricular dysplasia/cardiomyopathy suggest non‐canonical mechanisms for disease pathogenesis. Cardiovascular Research, 113(1), 102–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UniProt Consortium T (2018). UniProt: the universal protein knowledgebase. Nucleic Acids Research, 46(5), 2699. 10.1093/nar/gky092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenberg, J. I. , Perry, M. D. , Perrin, M. J. , Mann, S. A. , Ke, Y. , & Hill, A. P. (2012). hERG K(+) channels: structure, function, and clinical significance. Physiological Reviews, 92(3), 1393–1478. [DOI] [PubMed] [Google Scholar]
- Via, M. , Gignoux, C. , & Burchard, E. G. (2010). The 1000 Genomes Project: New opportunities for research and social challenges. Genome Medicine, 2(1), 3. 10.1186/gm124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace, E. , Howard, L. , Liu, M. , O’Brien, T. , Ward, D. , Shen, S. , & Prendiville, T. (2019). Long QT syndrome: Genetics and future perspective. Pediatric Cardiology, 40(7), 1419–1430. 10.1007/s00246-019-02151-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X. , Spandidos, A. , Wang, H. , & Seed, B. (2012). PrimerBank: a PCR primer database for quantitative gene expression analysis, 2012 update. Nucleic Acids Research, 40(Database issue), D1144–D1149. 10.1093/nar/gkr1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilde, A. , & Amin, A. (2017). Channelopathies, genetic testing and risk stratification. International Journal of Cardiology, 237, 53–55. 10.1016/j.ijcard.2017.03.063. [DOI] [PubMed] [Google Scholar]
- Wilde, A. , & Amin, A. S. (2018). Clinical spectrum of SCN5A mutations: Long QT Syndrome, Brugada Syndrome, and Cardiomyopathy. JACC: Clinical Electrophysiology, 4(5), 569–579. 10.1016/j.jacep.2018.03.006. [DOI] [PubMed] [Google Scholar]
- Wilde, A. A. , Moss, A. J. , Kaufman, E. S. , Shimizu, W. , Peterson, D. R. , Benhorin, J. , Lopes, C. , Towbin, J. A. , Spazzolini, C. , Crotti, L. , Zareba, W. , Goldenberg, I. , Kanters, J. K. , Robinson, J. L. , Qi, M. , Hofman, N. , Tester, D. J. , Bezzina, C. R. , Alders, M. , Aiba, T. , … Ackerman, M. J. (2016). Clinical Aspects of Type 3 Long‐QT Syndrome: An International Multicenter Study. Circulation, 134(12), 872–882. 10.1161/CIRCULATIONAHA.116.021823. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data used in this study are not publicly available, but it might be available from the corresponding author upon reasonable request and permission from relevant Chinese Authorities.