

Abstract
Deficiency of adenosine deaminase 2 (DADA2) is a unique monogenic autoinflammatory disease caused by autosomal recessive loss-of-function mutations in the CECR1 gene which presents as childhood-onset small- and medium-vessel vasculitis. Previously, many of these patients were misdiagnosed and thought to have clinical features of systemic polyarteritis nodosum, which negatively influenced its outcome, since TNF inhibitors seem to have efficacy on the vasculitic phenotype of DADA2. We present a case of a 28-year-old woman with a lifelong unknown syndrome and unique clinical manifestations recently recognized as DADA2. The first manifestation, at 3 months of age, was an episode of facial paralysis during which renovascular hypertension was diagnosed. Later, she developed episodes of prolonged fever, polyarthritis, Raynaud's phenomenon, gastrointestinal bleeding, and intracerebral hemorrhage. This inflammatory state ultimately led to the development of amyloid A amyloidosis and renal insufficiency.
Keywords: Adenosine deaminase 2 deficiency, Vasculitis, Autoinflammatory disease, Amyloid A amyloidosis, Kidney injury
Introduction
Deficiency of adenosine deaminase 2 (DADA2), first described in 2014, is an autoinflammatory disorder characterized by signs and symptoms of systemic small- and medium-vessel vasculitis and inflammation including early-onset stroke (both, ischemic and hemorrhagic), livedo racemosa, and recurrent fevers [1]. The disease is caused by autosomal recessive loss-of-function mutations in the CECR1 (cat eye syndrome chromosome region, candidate) [1, 2]. Since its initial description, as mimic of systemic polyarteritis nodosa (sPAN), the clinical spectrum of this condition has expanded considerably [3]. Hematological abnormalities, such as hypogammaglobulinaemia, with reduction of memory and terminally differentiated B cells and plasma cells may be present, but a severe clinical picture dominated by cytopenia and lymphoproliferation has been also described [4]. Less common manifestations include nephrological ones, reported only in 14–24% of the cases [5, 6], and glomerular involvement appears to be even more uncommon [6]. Due to this highly variable phenotype, patients with DADA2 may present to different medical subspecialties, which make diagnosis difficult. Given the significant morbidity and mortality, a high index of suspicion is needed for early diagnosis and management [5]. Early initiation of treatment with TNF inhibitors soon after the genetic confirmation of DADA2 results in significant ameliorations of the disease [7].
Here, we describe a case of DADA2 presented at 3 months of age with peripheral facial paralysis and renovascular hypertension, followed in the pediatric department during 16 years, with a presumed diagnosis of sPAN, which ultimately led to nephrotic syndrome and renal insufficiency secondary to amyloid A (AA) amyloidosis development.
Case Report/Case Presentation
A 16-year-old girl began her follow-up in the Nephrology Department for nephrotic proteinuria and worsening renal function.
Her clinical manifestations began at 3 months of age with growth failure and persistent irritability that led to a close follow-up by her pediatrician. At that age, she had an episode of peripheral facial paralysis for 7 days that spontaneously resolved. Further investigations confirmed the presence of renovascular hypertension. She had high blood pressure (BP), renin >250 ng/mL/h (1.9–3.7 ng/mL/h), and aldosterone 1,157 ng/dL (5–80 ng/dL), and initially, BP was difficult to control. Treatment with high-dose atenolol, nifedipine, and enalapril was initiated. Laboratory studies are described in Figure 1. Also, she had persistent anemia (hemoglobin of 9.0 g/dL, MCV 69 fL), high erythrocyte sedimentation rate (ESR) of 32 mm/h, C-reactive protein (CRP) of 2.7 mg/dL, and serum creatinine of 104 μmol/L. Echocardiography was normal, chest radiography revealed cardiomegaly, and technetium-99m dimercaptosuccinic acid scan showed differential kidney function − left kidney 38 and right kidney 62% − with hypoactive areas in the left kidney. Angiographic evaluation completed during her follow-up in the hospitals for sick children showed stenosis and multiple dilatations in the small intrarenal arteries, especially in the left kidney.
Fig. 1.

Evolution of clinical manifestations and laboratory findings through the years.
Between 1 and 6 years, she also had episodes of intermittent rash, recurrent fever, and persistent iron-resistant microcytic anemia (although red blood cell diseases, like thalassemia, were excluded) and subsequently developed nonnephrotic proteinuria, probably due to poorly controlled hypertension. At 5 years, she developed recurrent ataxia and nystagmus that persisted for months. Scintigraphy and cervico-cerebral angiography showed no signs of vasculopathy. Through the years, she also developed gingival hyperplasia, attributed to chronic effects of nifedipine.
At the end of the seventh year of life, she began a prolonged fever syndrome (PFS) for 95 days, abdominal pain, hepatomegaly, splenomegaly, polyarthralgias, livedo racemosa (Fig. 2), and ocular episcleritis. Laboratory tests described in Figure 1 revealed a worsening hemoglobin of 8.5 g/dL, high ESR 115 mm/h, and CRP 20.5 mg/dL. All etiological investigation for infectious prolonged fever was negative; the serum angiotensin converting enzyme (SACE) antineutrophil cytoplasmic antibody was normal or negative; human leukocyte antigen (HLA) class 1 molecule B27 (HLA-B27) was positive; complement C3, C4, and CH50 were normal. One year later, due to a protracted pneumonia, a chest computed tomography was performed that revealed “subsegmental collapse in the middle lobe with incipient cylindrical bronchiectasis and multiple enlarged mesenteric and para-aortic ganglia.” Immunodeficiency investigations disclosed decreased chemotactic activity of neutrophils and deficit of CD15 expression in leukocytes, and studies of phagocytosis and respiratory explosion of granulocytes and monocytes did not show any changes. Serum immunoglobulin concentrations disclosed low IgG (4.6 g/L [N 5.6–13.8]) and IgM levels (0.3 g/L [N 0.47–2.0]) but normal IgA levels (1.0 g/L [N 0.24–2.32]).
Fig. 2.
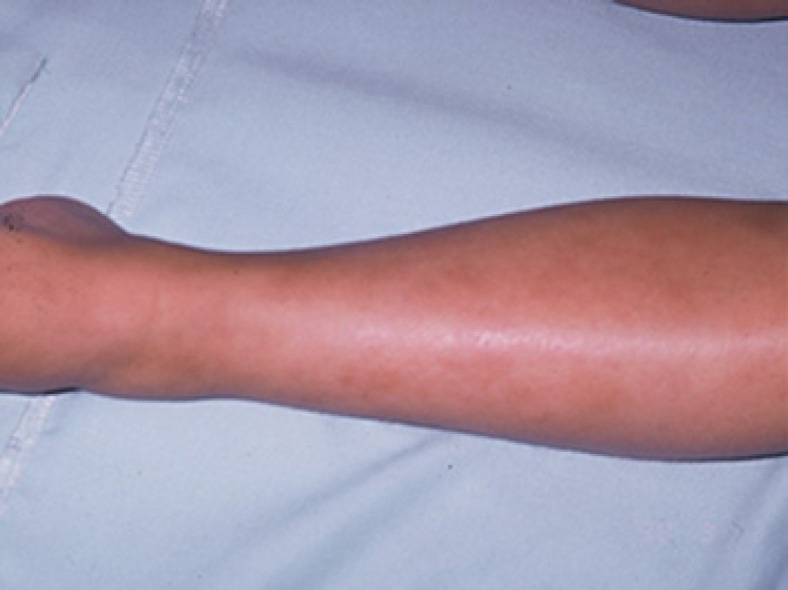
Racemosa livedo reticularis.
At 9 years, she was admitted for abdominal pain, melenas, and severe anemia of 6.6 g/dL. After inconclusive endoscopic study and normal Tc99-m scintigraphy (Meckel scan), she underwent an exploratory laparotomy which only found signs of reactive inflammatory mesenteric lymphadenitis. Lymph node biopsy was negative. After blood transfusion and medical support, she was stabilized. One year later, she was re-admitted for prolonged fever syndrome (for 3 weeks), headaches, and seizures, and a brain CT scan disclosed an intracerebral hemorrhage. Cerebral magnetic resonance imaging showed also a “left posterior parietal brain cavenoma” (probably not related with the hemorrhagic stroke).
Throughout the years, our patient presented with repeated episodes of fever, abdominal pain, persistent hepatosplenomegaly, and multiple episodes of polyarthralgia associated with Raynaud's phenomenon, as shown in Figure 1. She did not have any sensory or motor deficit and mental development was normal. She maintained follow-up by several medical specialties without reaching a definitive diagnosis.
There was a history of consanguinity and chronic kidney disease requiring hemodialysis. Her mother was diagnosed with juvenile rheumatoid arthritis and Behçet disease at 16 years, together with livedo reticularis, and cerebral aneurysms. She died before the diagnosis of DADA2 on her daughter was made. Thus, plasma ADA2 and/or genetic testing was not performed. However, her parents were also consanguineous.
On her evaluation at the nephrology department's outpatient in 2008, physical examination was only remarkable for the presence of generalized edema, livedo racemosa, Raynaud's phenomenon, and hepatosplenomegaly. Laboratory studies showed renal dysfunction with a serum creatinine of 177 μmol/L, hypoalbuminemia, and nephrotic proteinuria. There was normal blood count but positive inflammatory parameters with negative blood and urine cultures or infection serology. C3, C4, and antistreptolysin test were normal. Autoimmunity study (ANCA, ANA, antiphospholipid antibodies, anti-SSA, and anti-SSB) were all negative. For a better understanding of her renal insufficiency and proteinuria, a kidney biopsy was performed in 2008, leading to the diagnosis of AA amyloidosis, probably secondary to an inflammatory state. The control of kidney disease's progression showed to be increasingly difficult, despite therapeutic optimization and ACE inhibitor as the primary disease leading to this inflammatory state remained unknown. However, there was high suspicion of vasculitis-like syndrome.
The consanguinity of her parents, family history, and early disease onset led to suspicion of DADA2. The absent enzymatic activity of ADA2 confirmed the hypothesis that the causative genetic variant determined the loss of function of the protein. DADA2 was confirmed through mutation analysis of the entire coding region of the CECR1 gene (NM_001282225) that revealed a homozygous pathogenic variant c.1358A>G; p.Tyr453Cys (Fig. 3). This variant was located in exon 9 of this gene and has been previously reported in the literature (1). Her mother was thought to be a heterozygous carrier. Treatment with TNF inhibitors was recommended to the patient after the molecular confirmation of DADA2 as it may have led to significant amelioration in her condition; however, she declined this treatment. At present, her renal function is stable, although with a glomerular filtration rate of 20 mL/min/1.73 m2, CRP is stable, and there is no evidence of progressive neurologic features.
Fig. 3.

Electropherograms showing homozygous c.1358A>G pathogenic variant in the exon 9 of CECR1 gene with a limited reading frame depicting the corresponding amino acid substitution p.Tyr453Cys.
Discussion
Cases of DADA2 diagnosed later in life provide a unique insight into the natural history of the disease and to the wide range of phenotypic variability within its spectrum. Our patient presented early in life with nonspecific findings like fever and growth failure and also with unique characteristics. Involvement of small intrarenal arteries dominated her clinical picture at the beginning, leading to resistant hypertension. Peripheral facial paralysis thought to be secondary to severe renovascular hypertension was one important finding. Facial paralysis can be a rare initial feature of hypertension, although many practitioners may be unaware of this association [8]. As in this case, studies report that approximately 75–80% of children with secondary hypertension have renal abnormalities [8]. However, we now know that facial paralysis could have been an early sign of DADA2, once diverse manifestations of central and peripheral neuropathy have been reported in association with this syndrome, usually with cranial nerve palsies involving mostly III, IV, VI, and VII cranial nerves [9]. Nonnephrotic proteinuria at that time was probably a result of high BP. Her gingival hyperplasia was thought to be drug-induced by calcium channel blockers [10].
The development of recurrent episodes of fever, abdominal pain and polyarthralgias together with livedo racemosa led to the suspicion of a systemic autoinflammatory disease, most likely a necrotizing vasculitis. These events associated with later gastrointestinal and intracerebral hemorrhage that were almost always preceded by an infection lead to the suspicion of sPAN. sPAN is universally accepted to be associated with streptococcal infection [11, 12], and its most frequent findings are general symptoms, neurologic manifestations, skin involvement, abdominal pain, and hypertension [13]. A recent study also reported renal artery microaneurysms in 66.2% of the cases [13]. At that point, our patient met ACR and Chapel Hill diagnostic criteria for PAN [12]. Positive HLA-B27 and low IgM were another clue for an autoinflammatory systemic state [14], although there were no reported associations of these with PAN to our knowledge.
The development of nephrotic syndrome secondary to AA amyloidosis reassures the existence of an unknown inflammatory condition. Long-term inflammatory disorders can lead to pathogenic extracellular deposition of misfolded protein of serum amyloid A protein, an acute-phase reactant conducting to organ disfunction [15]. Renal insufficiency progressed despite optimal treatment, hypertension, and proteinuria control because the primary cause was not known. There are few studies reporting glomerular involvement secondary to DADA2 like the one in this case. Batu et al. [16] case series, reported 1 patient with intestinal renal involvement from amyloidosis; also another one reported 1 patient treated with canakinumab for AA amyloidosis secondary to DADA2 with good outcomes [17].
Advances in sequencing technologies and international collaborations led to a better knowledge of mechanisms driving to autoinflammatory diseases, and many new disorders have been reported recently [1]. Initially, described in 2014, DADA2 is recognized as a mimic of PAN with primary features of early-onset strokes, peripheral vasculopathy, and systemic inflammation [2]. Most of the cases had been diagnosed as PAN or undefined vasculitis and autoinflammatory syndrome [1], as in this case. The disease is caused by autosomal recessive loss-of-function mutations in the CERC1 gene, which encodes ADA2 enzyme [18].
Beyond its early description as a monogenic vasculitis, DADA2 is also associated with immunodeficiency and hematologic defects [2], that our patient presented (IgM 0.3 g/L). The onset of disease is usually in childhood with 24% of reported patients presenting before 1 year of age; however, adult onset has also been described [5, 19]. In our case, the symptoms began at age of 3 months. Vasculopathy of small- and medium-sized arteries is the major clinical presentation of DADA2. The skin and central nervous system are the most commonly involved sites, although gastrointestinal, liver, renal, and coronary systems are also affected to various degrees [9]. Skin involvement is reported in 75% of cases, and livedo reticularis is one of the most frequent manifestations [5]. Interestingly, it was one of the most recurrent findings in this case. Stenosis and/or aneurysms of middle sized arteries like renal, mesenteric, hepatic, and celiac arteries are also usual findings both in PAN and DADA2 [1]. Our patient met all of these criteria and also had 2 major hemorrhagic events and gastrointestinal and neurologic (reports describe about 14% and 12% of these events, respectively) [5]. However, as previously said, she had a more uncommon feature since she developed progressive renal dysfunction due to AA amyloidosis secondary to this poorly controlled inflammatory state.
Mortality is high, with up to 8% of patients dying before the age of 30 years and cause of death includes complications of recurrent stroke or infection [5]. Fortunately, until now, there were no other life-threading events such the ones described before.
The highly variable presentation renders early DADA2 diagnosis difficult [9]. However, the presence of early onset stroke, parental consanguinity, or the presence of family history, as well as unresponsiveness to conventional treatments should raise awareness in patients with presumed PAN and should prompt the physician to consider to determine plasma ADA2 activity and genetic analysis to exclude diagnosis of DADA2 [9, 19]. The presence of consanguinity and family history plays an important role in the presumptive diagnosis since it's an autosomal recessive disease [9]. Plasma ADA2 enzyme activity is a well-established test for the diagnosis with 100% sensitivity and 100% specificity in a cohort of Sneddon's syndrome patients [20]. We did not study MEFV genes or any other genes related to familial fevers [21].
Although there is no strong evidence for any treatment strategies until now, immunomodulatory drugs such as TNF inhibitors have been considered [5, 22]. The treatment with biologic TNF-inhibitors was successful in controlling the fever episodes, vasculopathy, and prevention of strokes, in all patients reported [5]. In addition, anti-TNF treatment resulted in positive changes in several key laboratory tests [22].
Conclusion
In conclusion, cases of DADA2 diagnosed later in life provide a unique opportunity to understand the natural history of the disease and demonstrate the potential for these patients to develop life-threatening sequelae if left untreated. Furthermore, this case highlight the need for greater awareness about DADA2 among nephrologists and rheumatologists, when evaluating cases of suspected PAN, and neurologists when evaluating adults with unexplained neurologic features [7]. Early recognition is essential for the management and prevention of major complications. We suggest screening ADA2 among children with vasculitic rash or unexplained early-onset central nervous system disease with systemic inflammation and in cases of unknown cause of renal amyloidosis, since it could enable an earlier diagnosis of DADA2 and treatment. More studies are required, especially in the pediatric population, for a better understanding of its pathophysiology and future treatment research.
Statement of Ethics
The patient had provided written informed consent to publish her case (including publication of images). The study is exempt from ethical committee approval once the article just reports the clinical case, and written patient consent was obtained.
Conflict of Interest Statement
The authors have no conflicts of interest to declare.
Funding Sources
There were no funding sources.
Author Contributions
Research idea and study design: C.P., L.E., and G.S.; data acquisition: L.E., G.S., J.R., and M.S.; data analysis/interpretation: M.R.A.; supervision or mentorship: R.A., G.S., M.S., C.P., G.S., L.E., M.A., J.R., R.A., and M.S. takes responsibility that this study has been reported honestly, accurately and transparently, and accepts accountability for the overall work by ensuring that questions pertaining to the accuracy or integrity of any portion of the work are appropriately investigated and resolved.
References
- 1.Çakan M, Aktay-Ayaz N, Karadağ ŞG, Tahir-Turanlı E, Stafstrom K, Bainter W, et al. Atypical phenotype of an old disease or typical phenotype of a new disease: deficiency of adenosine deaminase 2. Turk J Pediatr. 2019;61((3)):413–7. doi: 10.24953/turkjped.2019.03.014. [DOI] [PubMed] [Google Scholar]
- 2.Lee PY. Vasculopathy, immunodeficiency, and bone marrow failure: the intriguing syndrome caused by deficiency of adenosine deaminase 2. Front Pediatr. 2018 Oct 18;6:282. doi: 10.3389/fped.2018.00282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee PY, Kellner ES, Huang Y, Furutani E, Huang Z, Bainter W, et al. Genotype and functional correlates of disease phenotype in deficiency of adenosine deaminase 2 (DADA2) J Allergy Clin Immunol. 2020;145((6)):1664–72.e10. doi: 10.1016/j.jaci.2019.12.908. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0091674920300300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Caorsi R, Penco F, Grossi A, Insalaco A, Omenetti A, Alessio M, et al. ADA2 deficiency (DADA2) as an unrecognised cause of early onset polyarteritis nodosa and stroke: a multicentre national study. Ann Rheum Dis. 2017;76((10)):1648–56. doi: 10.1136/annrheumdis-2016-210802. [DOI] [PubMed] [Google Scholar]
- 5.Meyts I, Aksentijevich I. Deficiency of adenosine deaminase 2 (DADA2): Updates on the phenotype, genetics, pathogenesis, and treatment. J Clin Immunol. 2018;38((5)):569–78. doi: 10.1007/s10875-018-0525-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fayand A, Sarrabay G, Belot A, Hentgen V, Kone-Paut I, Grateau G, et al. Multiple facets of ADA2 deficiency: Vasculitis, auto-inflammatory disease and immunodeficiency: a literature review of 135 cases from literature. Rev Med Interne. 2018;39((4)):297–306. doi: 10.1016/j.revmed.2017.11.006. [DOI] [PubMed] [Google Scholar]
- 7.Springer JM, Gierer SA, Jiang H, Kleiner D, Deuitch N, Ombrello AK, et al. Deficiency of adenosine deaminase 2 in adult siblings: many years of a misdiagnosed disease with severe consequences. Front Immunol. 2018 Jun;9:1361. doi: 10.3389/fimmu.2018.01361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aynaci FM, Sen Y. Peripheral facial paralysis as initial manifestation of hypertension in a child. 2002:73–5. [PubMed] [Google Scholar]
- 9.Tanatar A, Karadağ ŞG, Sözeri B, Sönmez HE, Çakan M, Kendir Demirkol Y, et al. ADA2 deficiency: case series of five patients with varying phenotypes. J Clin Immunol. 2019;40((2)):253–8. doi: 10.1007/s10875-019-00734-0. [DOI] [PubMed] [Google Scholar]
- 10.Lawrence DB, Weart CW, Laro JJ, Neville BW. Calcium channel blocker-induced gingival hyperplasia: case report and review of this iatrogenic disease. J Fam Pract. 1994;39((5)):483–8. [PubMed] [Google Scholar]
- 11.Ozen S. The changing face of polyarteritis nodosa and necrotizing vasculitis. Nat Rev Rheumatol. 2017 Jun;13((6)):381–6. doi: 10.1038/nrrheum.2017.68. [DOI] [PubMed] [Google Scholar]
- 12.Hernández-Rodríguez J, Alba MA, Prieto-González S, Cid MC. Diagnosis and classification of polyarteritis nodosa. J Autoimmun. 2014;48–49:84–9. doi: 10.1016/j.jaut.2014.01.029. [DOI] [PubMed] [Google Scholar]
- 13.Pagnoux C, Seror R, Henegar C, Mahr A, Cohen P, Le Guern V, et al. Clinical features and outcomes in 348 patients with polyarteritis nodosa: a systematic retrospective study of patients diagnosed between 1963 and 2005 and entered into the French Vasculitis Study Group Database. Arthritis Rheum. 2010;62((2)):616–26. doi: 10.1002/art.27240. [DOI] [PubMed] [Google Scholar]
- 14.Boiwness P. HLA-B27. Annu Immunol. 2015;33:29–48. doi: 10.1146/annurev-immunol-032414-112110. [DOI] [PubMed] [Google Scholar]
- 15.Papa R, Lachmann HJ. Secondary, AA, Amyloidosis. Rheum Dis Clin North Am. 2018;44((4)):585–603. doi: 10.1016/j.rdc.2018.06.004. [DOI] [PubMed] [Google Scholar]
- 16.Batu ED, Karadag O, Taskiran EZ, Kalyoncu U, Aksentijevich I, Alikasifoglu M, et al. A case series of adenosine deaminase 2-deficient patients emphasizing treatment and genotype-phenotype correlations. J Rheumatol. 2015;42((8)):1532–4. doi: 10.3899/jrheum.150024. [DOI] [PubMed] [Google Scholar]
- 17.Ekinci RMK, Balci S, Bisgin A, Hershfield M, Atmis B, Dogruel D, et al. Renal amyloidosis in deficiency of adenosine deaminase 2: successful experience with canakinumab. Pediatrics. 2018;142((5)):e20180948. doi: 10.1542/peds.2018-0948. [DOI] [PubMed] [Google Scholar]
- 18.Ozen S, Bilginer Y, Batu ED, Taşkıran E, Özkara HA, Ünal Ş, et al. A monogenic disease with a variety of phenotypes: deficiency of adenosine deaminase 2 subtitle: deficiency of adenosine deaminase 2. J Rheumatol. 2019 doi: 10.3899/jrheum.181384. [DOI] [PubMed] [Google Scholar]
- 19.Goldenberg I, Kutyifa V, Moss AJ. Survival with cardiac-resynchronization therapy. N Engl J Med. 2014;371((5)):477–8. doi: 10.1056/NEJMc1407182. [DOI] [PubMed] [Google Scholar]
- 20.Santo GC, Baldeiras I, Guerreiro R, Ribeiro JA, Cunha R, Youngstein T, et al. Adenosine deaminase two and immunoglobulin M accurately differentiate adult sneddon's syndrome of unknown cause. Cerebrovasc Dis. 2019;46((5–6)):257–64. doi: 10.1159/000495794. [DOI] [PubMed] [Google Scholar]
- 21.Accetturo M, D'Uggento AM, Portincasa P, Stella A. Improvement of MEFV gene variants classification to aid treatment decision making in familial Mediterranean fever. Rheumatol. 2020;59((4)):754–61. doi: 10.1093/rheumatology/kez332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Receptor SU, Kidney C, Stents PDC, Dyspepsia F, Regression T. Correspondence treatment of advanced renal-cell carcinoma. 2016:888–90. [Google Scholar]