

Abstract
Background: At present, the global sever acute respiratory syndrome coronavirus 2 (SARS-CoV-2) situation is still grim, and the risk of local outbreaks caused by imported viruses is high. Therefore, it is necessary to monitor the genomic variation and genetic evolution characteristics of SARS-CoV-2. The main purpose of this study was to detect the entry of different SARS-CoV-2 variants into Jiangsu Province, China.
Methods: First, oropharyngeal swabs were collected from 165 patients (55 locally confirmed cases and 110 imported cases with confirmed and asymptomatic infection) diagnosed with SARS-CoV-2 infection in Jiangsu Province, China between January 2020 and June 2021. Then, whole genome sequencing was used to explore the phylogeny and find potential mutations in genes of the SARS-CoV-2. Last, association analysis among clinical characteristics and SARS-CoV-2 Variant of Concern, pedigree surveillance analysis of SARS-COV-2, and single nucleotide polymorphisms (SNPs) detection in SARS-COV-2 samples was performed.
Results: More men were infected with the SARS-CoV-2 when compared with women. The onset of the SARS-CoV-2 showed a trend of younger age. Moreover, the number of asymptomatic infected patients was large, similar to the number of common patients. Patients infected with Alpha (50%) and Beta (90%) variants were predominantly asymptomatic, while patients infected with Delta (17%) variant presented severe clinical features. A total of 935 SNPs were detected in 165 SARS-COV-2 samples. Among which, missense mutation (58%) was the dominant mutation type. About 56% of SNPs changes occurred in the open reading frame 1ab (ORF1ab) gene. Approximately, 20% of SNP changes occurred in spike glycoprotein (S) gene, such as p.Asp501Tyr, p.Pro681His, and p.Pro681Arg. In total, nine SNPs loci in S gene were significantly correlated with the severity of patients. It is worth mentioning that amino acid substitution of p.Asp614Gly was significantly positively correlated with the clinical severity of patients. The amino acid replacements of p.Ser316Thr and p.Lu484Lys were significantly negatively correlated with the course of disease.
Conclusion: Sever acute respiratory syndrome coronavirus 2 (SARS-CoV-2) may further undergo a variety of mutations in different hosts, countries, and weather conditions. Detecting the entry of different virus variants of SARS-CoV-2 into Jiangsu Province, China may help to monitor the spread of infection and the diversity of eventual recombination or genomic mutations.
Keywords: SARS-CoV-2, pharyngeal swabs, whole genome sequencing, single nucleotide polymorphisms, spike glycoprotein, clinical characteristics, patients severity SARS-CoV-2, patients severity
Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), first detected in Wuhan, China, in December of 2019 (1), has spread throughout the world by travel and community-based contacts (2, 3). The SARS-CoV-2 is a pleomorphic, enveloped, positive-sense, and single-stranded RNA virus (4, 5). It has four structural proteins, such as nucleocapsid (N), membrane “matrix” (M), spike (S), and envelope (E). The assembly of these proteins into the infectious virion results in distinct infectivity of these coronaviruses (5–7). Clinically, the majority of SARS-CoV-2 vaccine candidates are based on the S protein (8), which is the major target of neutralizing antibodies (9, 10). Based on the clinical characteristics, patients with SARS-CoV-2 are classified as mild, moderate, severe, and critical (11). Signs and symptoms of SARS-CoV-2 include fatigue, shortness of breath, cough and chest pain, sore throat, respiratory distress, invasive lung lesions, pneumonia with prominent fever, anosmia, and diarrhea (12–18). For severe SARS-CoV-2 disease, major risk factors include age, male sex, smoking, obesity, and comorbid chronic conditions (such as, hypertension, type 2 diabetes mellitus, and others) (19–21). Now, the global SARS-CoV-2 situation is still grim, and the risk of local outbreaks caused by imported viruses is high. Therefore, it is important to monitor the genomic variation and genetic evolution characteristics of SARS-CoV-2. In this study, we collected oropharyngeal swabs from 165 patients (55 locally confirmed cases and 110 imported cases with confirmed and asymptomatic infection) diagnosed with SARS-CoV-2 infection in Jiangsu Province, China between January 2020 and June 2021 to detect the entry of different SARS-CoV-2 variants into Jiangsu Province. Our study could help to monitor the spread of infection and the diversity of genomic mutations in SARS-CoV-2.
Materials and Methods
Collection of Samples
Oropharyngeal swabs were collected from 165 patients (locally confirmed cases and imported cases with confirmed and asymptomatic infection) diagnosed with SARS-CoV-2 infection in Jiangsu Province between January 2020 and June 2021. For imported cases, these patients became ill during the 14-day quarantine period, indicating that they were infected abroad. The relevant clinical characteristics of these patients were recorded. According to the SARS-CoV-2 Diagnosis and Treatment Protocol (Trial Version 7) of the National Health Commission, PRC, for course distribution, patients were divided into the following four groups: (1) mild type: the clinical symptoms were mild, and no pneumonia was observed on imaging; (2) common type: patients with fever, respiratory symptoms, and other imaging manifestations of pneumonia, but no dyspnea or other complications; (3) severe type: patients conformed to any of the following: (1) the occurrence of shortness of breath, breathing rate ≥ 30 times/min; (2) at rest, oxygen saturation ≤ 93%; (3) arterial partial pressure of oxygen (PaO2)/oxygen concentration (FiO2) ≤ 300 mmHg; (4) patients with significant progression > 50% in lung imaging within 24–48 h; (4) asymptomatic type: patients with a positive nucleic acid testing (NAT) result but without any relevant clinical symptoms and radiological changes of the lung during quarantine. All procedures conducted in this study involving human materials were approved by the Ethics Committee of Novel Coronavirus sequence analysis in Jiangsu province (JSJK2021-B016-01). Moreover, all subjects gave written informed consent.
Whole Genome Sequencing
Total viral RNA was extracted from 200 μl oropharyngeal swabs using the QIAamp Viral RNA Mini Kit (Qiagen, Germany) according to the instructions, and stored at −80°C for further use. Genome-wide specific amplification of total viral RNA was performed using ULSEN® Ultra-sensitive Novel Coronavirus Whole-genome capture Kit (Beijing MicroFuture, V-090418-1, China). PCR products were purified using Agencourt AMPure XP kit (Beckman Coulter, CA, USA). DNA sequencing library was constructed according to the steps of NexteraXT DNA Library Preparation Kit (Illumina, CA, USA). The library was diluted into 100 pmol/μl. The whole genome was deeply sequenced using iseq100 sequencer of Illumina Sequencing platform. The original sequencing data were assembled into a complete sequence using 2019nCOV automatic analysis software of Beijing Micro Future Company. Nucleotide variation sites were compared with reference genome SARS-CoV-2 Wuhan-Hu-1 (GenBank: MN908947.3) using CLC Genomic Workbench 12.0. Mafft software was used for multiple sequence comparison analysis of data from this study (165 cases), public database data on 108 cases (from public SARS-CoV-2 database GISAID, 9 clades and 6 continents), and reference sequences. The comparison results were analyzed by using the MEGA-X software. Neighbor-joining algorithm and Bootstrap were used to construct the phylogenetic tree. Ggtree software was used to mark and beautify the phylogenetic tree. Based on the results of the multi-sequence alignment in the previous step, the assembled virus genome sequence was compared with the reference sequence. The annotation information of loci was based on the general feature format (GFF) annotation information of MN908947.3 and the SNPEFF software. Statistical summaries of variation information and annotation information were performed by using custom Python scripts. For each mutation locus, emphasis was placed on the S gene sequence alignment, single nucleotide polymorphism/insertion/deletion (SNP/INDEL) mutation locus detection, and gene function annotation analysis. Clinical disease progression phenotypes of patients with SARS-CoV-2 were analyzed and summarized and referred to the database of outbreak.info.
Results
Basic Clinical Characteristics of the Research Object
Clinical trials of 165 patients with SARS-CoV-2 are shown in Table 1. There were 100 male patients and 65 female patients. Although the number of patients with SARS-CoV-2 decreased in 2021 as compared with that in 2020, there were still more men than women. The onset age of the patients with SARS-CoV-2 in 2021 was 34.55 years, which was younger than that in 2020 (38.55 years). This suggested that the onset of the SARS-CoV-2 showed a trend of younger age. For course distribution, the number of asymptomatic infected patients was large, similar to the number of common patients. The mean infection time of SARS-CoV-2 was 28.22. The infection time in 2021 was 30.2, which was a little longer than that in 2020 (27.5). There were 55 indigenous cases. It is noted that 110 cases came from abroad, such as 49 (49%) cases from Europe and 37 (34%) cases from Asia.
Table 1.
Clinical features of 165 patients with SARS-COV-2.
| Totality | In 2020 | In 2021 | |
|---|---|---|---|
| Gender | 165 | 116 | 49 |
| Male | 100 | 66 | 34 |
| Female | 65 | 50 | 15 |
| Age (years) | |||
| Mean value | 37.36 | 38.55 | 34.55 |
| <20 | 12 | 10 | 2 |
| ≤20 <30 | 51 | 30 | 21 |
| ≤30 <40 | 37 | 25 | 12 |
| ≤40 <50 | 23 | 17 | 6 |
| ≤50 <60 | 23 | 17 | 6 |
| ≥60 | 19 | 17 | 2 |
| Course distribution | |||
| Asymptomatic | 65 | 43 | 22 |
| Mild | 32 | 24 | 8 |
| Common | 67 | 49 | 18 |
| Severe | 1 | 0 | 1 |
| Infection time (days) | |||
| Mean value | 28.22 | 27.5 | 30.2 |
| Source region | |||
| Indigenous cases | 55 | 55 | 0 |
| Imported cases | 110 | 61 | 49 |
| Asia | 37 | 13 | 24 |
| Africa | 4 | 0 | 4 |
| North America | 11 | 6 | 5 |
| South America | 7 | 2 | 5 |
| European | 49 | 40 | 9 |
| Oceania | 2 | 0 | 2 |
Association Between SARS-CoV-2 Variant of Concern and Clinical Characteristics
In addition to GISAID database, there are Pangolin classification A/B main branch, and the descendant branch B.X.X. As shown in Table 2, B.1.1.7 (Alpha), B.1.351 (Beta), and P.1 (Gamma) variants did not appear until March 2021. The B.1.617.2 (Delta) variant did not appear until May 2021. Alpha and Beta variants were mainly from patients in Asia, and Gamma and Delta variants were from patients in South America and Europe. According to the epidemiological characteristics in Table 3, patients infected with Alpha (50%) and Beta (90%) variants were predominantly asymptomatic, while patients infected with Delta (17%) presented severe clinical features.
Table 2.
Genotype (Pangolin) of SARS-CoV-2 patients at different times and in different regions of origin.
| Number | SARS-CoV-2 variant of concern | ||||
|---|---|---|---|---|---|
| Alpha (B.1.1.7) | Beta (B.1.351) | Gamma (P.1) | Delta (B.1.617.2) | ||
| Time | 12 | 10 | 2 | 6 | |
| From January to June in 2020 | 0 | 0 | 0 | 0 | 0 |
| From July to December in 2020 | 0 | 0 | 0 | 0 | 0 |
| From January to February in 2021 | 0 | 0 | 0 | 0 | 0 |
| From March to April in 2021 | 13 | 6 | 7 | 1 | 0 |
| From May to June in 2021 | 12 | 6 | 3 | 1 | 6 |
| Regions of origin | |||||
| Asia | 16 | 6 | 9 | 0 | 1 |
| Africa | 1 | 1 | 0 | 0 | 0 |
| North America | 2 | 1 | 1 | 0 | 0 |
| South America | 3 | 2 | 0 | 1 | 0 |
| Europe | 3 | 2 | 0 | 0 | 1 |
| Oceania | 0 | 0 | 0 | 0 | 0 |
Table 3.
Epidemiological characteristics of patients infected with SARS-CoV-2 Variant of Concern.
| Alpha (B.1.1.7) | Beta (B.1.351) | Gamma (P.1) | Delta (B.1.617.2) | |
|---|---|---|---|---|
| Gender (Male/Female) | 10/2 | 3/7 | 1/1 | 4/2 |
| Age (mean ± standard deviation) | 38.0 ± 12.6 | 34.2 ± 14.2 | 25 | 34.5 ± 4.5 |
| Course distribution | ||||
| Asymptomatic | 6 | 9 | 1 | 1 |
| Mild | 1 | 1 | 0 | 3 |
| Common | 5 | 0 | 1 | 1 |
| Severe | 0 | 0 | 0 | 1 |
| Infection time (days) | 24-59 | 10-48 | 20 | 24-42 |
Pedigree Surveillance of SARS-CoV-2
The phylogenetic tree was constructed to analyze the mutation regularity and transmission mode of virus evolution to provide the theoretical basis for tracing virus origin and determining virus transmission chain. The 165 sequences described in this study were combined with a phylogenetic tree of 108 genomic datasets (9 clades, 6 continents, and selected 2 cases). According to the GISAID classification, phylogenetic analysis showed that the virus samples from 37 patients were located in clade GR and GRY, 15 patients were located in clade V, 10 patients were from clade S, and 6 patients were located in clade L (Figure 1). Other virus samples were scattered in different clades. It is reported that the high predominance of the clades GR is mainly related to the occurrence of the amino acid substitution p.Asp614Gly that improved viral fitness (22, 23). The global prevalence of variant Alpha has generated clade GRY from clade GR.
Figure 1.

Analysis of phylogeny relationship between the severe acute respiratory symptom of coronavirus 2 (SARSCoV-2) genomes of Coronaviridae of GISAID in Jiangsu Province, China. An interactive view of phylogeny was plotted using Interactive Tree Of Life (iTOL).
Molecular Analysis of SARS-CoV-2 Samples
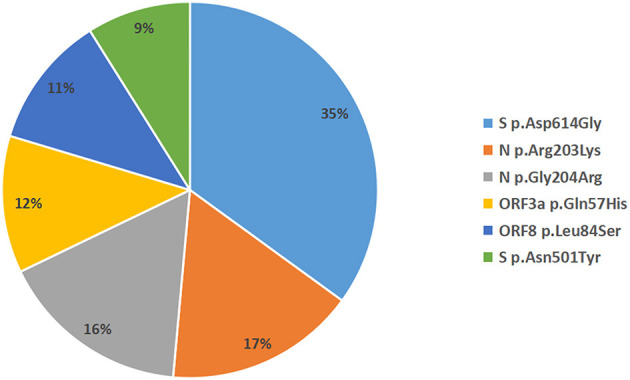
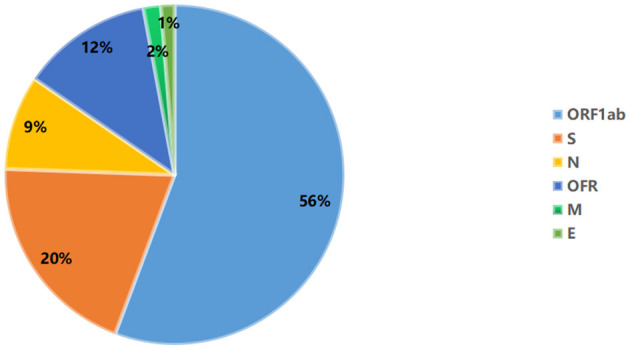
A total of 935 single nucleotide polymorphisms (SNPs) were detected in 165 SARS-COV-2 samples, of which 58% of nucleic acid changes resulted in amino acid substitution (missense mutations) (Supplementary Table 1). In the missense mutation, 303 (56%) SNP changes occurred in the open reading frame 1ab (ORF1ab) gene and 108 (20%) SNP changes occurred in the S gene (Figure 2). As showed in Figure 3, two amino acid substitutions occurred in N gene, such as p.Gli204Arg substitution in 46 SARS-COV-2 samples (16%) and p.Arg203Lys in 46 SARS-COV-2 samples (17%). The p.Glin57His substitution in open reading frame 3a (ORF3a) and the p.Leu84Ser substitution in open reading frame 8 (ORF8) was found in 33 (12%) and 32 (11%) SARS-COV-2 samples, respectively. In S gene, 98 SARS-COV-2 samples (35%) had p.Asp614Gly substitution and 25 SARS-COV-2 samples (9%) had p.Asp501Tyr substitution. In addition to the above mutations, amino acid substitutions of p.Pro681His and p.Pro681Arg were found in the S gene in 27 and 27 SARS-COV-2 samples, respectively (Table 4). In addition, Pearson's correlation coefficient was used to analyze the correlation between SNP changes in the S gene and the severity of patients. Interestingly, 9 SNPs loci in S gene were found to be significantly correlated with severity of patients (Supplementary Table 2). Among which, the amino acid substitution of p.Asp614Gly (cor = 0.29, p < 0.001) was significantly positively correlated with the clinical severity of patients. The amino acid replacements of p.Ser316Thr (cor = −0.16, p = 0.04) and p.Lu484Lys (cor = −0.28, p < 0.001) were significantly negatively correlated with the course of disease.
Figure 2.
Distribution map of missense mutant genes in 165 SARS-COV-2 samples.
Figure 3.
Distribution of amino acid mutation in spike (S), nucleocapsid (N), open reading frame 3a (ORF3a), and open reading frame 3a (ORF8) gene.
Table 4.
Main amino acid mutations in S gene in patients infected with SARS-COV-2.
| Amino acid mutations | Number | Common | Mild | Asymptomatic |
|---|---|---|---|---|
| V70F | 0 | 0 | 0 | 0 |
| A222V | 9 | 1 | 0 | 8 |
| W258L | ||||
| K417N | 10 | 0 | 1 | 9 |
| E484K | 14 | 1 | 1 | 12 |
| E484Q | ||||
| L452R | 9 | 3 | 3 | 1 |
| L452Q | 9 | 3 | 3 | 1 |
| N501Y | 25 | 7 | 2 | 16 |
| D614G | 98 | 32 | 12 | 53 |
| P681H | 27 | 10 | 6 | 10 |
| P681R | 27 | 10 | 6 | 10 |
Discussion
According to the basic clinical characteristics of infected patients in Jiangsu Province, China, we found that the onset age of the patients with SARS-CoV-2 in 2021 was 34.55 years, which was younger than that in 2020 (38.55). This suggested that the onset of the SARS-CoV-2 showed a trend of younger age. Moreover, we found that there were still more male patients than female patients, even though the number of SARS-CoV-2 patients decreased in 2021 compared with that in 2020. A study has reported the association between SARS-COV-2 infection and transmission and gender (24). In addition, Jin et al. found that gender could be an important factor affecting the SARS-COV-2 mortality (25). It is assumed that gender differences in susceptibility toward SARS-CoV-2 may be associated with the immune response (26). The data of 10 European countries on the distribution of SARS-COV-2 cases by gender showed that women of working age outnumbered infected men (27). Interestingly, it is reported that male sex is one of the major risk factors for severe SARS-CoV-2 disease (19–21). This is indicated that men could be more likely to develop SARS-CoV-2 than women. In addition, mean infection time of SARS-CoV-2 was 28.22. The infection time in 2021 was 30.2, which was a little longer than that in 2020 (27.5). Furthermore, the number of asymptomatic infected patients was large, similar to common patients. SARS-CoV-2 causes asymptomatic infections. Asymptomatic infection is defined as an individual with a positive NAT result but without any relevant clinical symptoms and radiological changes of the lung during quarantine. It is found that the number of SARS-CoV-2 infections is still rising rapidly, and asymptomatic infections could play roles in transmission (28). The higher asymptomatic infection of SARS-CoV-2 could be associated with the high replication efficiency of the virus (29, 30). Therefore, it is urgent to timely detect the source of asymptomatic infections to provide scientific data as a reference to fight against SARS-CoV-2. Among all patients, we found that 110 cases came from abroad, such as 49 (49%) cases from Europe and 37 (34%) cases from Asia. Thus, it is also important to strictly control the importation of the virus abroad.
Based on the novel coronavirus genome information currently published worldwide, mutation rate of SARS-CoV-2 is about 1~2 nucleotides/month. On February 25, 2021, the WHO first proposed the definition of SARS-COV-2 Variant of Concern. As of 24 June 2021, 4 SARS-COV-2 Variant of Concern have been defined, such as B.1.1.7 (Alpha), B.1.351 (Beta), P.1 (Gamma), and B.1.617.2 (Delta). Variant of Concern, Alpha, is first detected in southeast England in September 2020 and spread to become the dominant lineage in the United Kingdom. Alpha variant has been spreading to at least 114 countries worldwide. The Alpha variant is characterized by 17 mutations. The Beta variant was isolated from an oropharyngeal swab from a patient in the Ugu district, South Africa in November 2020. The emergence of the Beta variant includes several mutations within the structural and nonstructural proteins (31). It is found that variant Beta is purported to be more transmissible (32). The Gamma variant (acquired 17 mutations) emergence occurred around mid-November 2020 in Brazil and was preceded by a period of faster molecular evolution. Gamma variant is purported to be more transmissible (33). In April 2021, the Delta variant was identified in India and classified on May 11, 2021 as a Variant of Concern due to its fast spreading and potential immune escape (34). The Delta variant has been suggested to be more transmissible than the Alpha variant, which is more transmissible than the wild-type SARS-CoV-2 virus (35, 36). Delta variant has now been detected across the Europe, such as the United Kingdom. In our study, we found that Alpha, Beta, and Gamma variants did not appear until March 2021. Delta variant did not appear until May 2021. Tracing its origin, we found that Alpha and Beta variants were mainly from patients in Asia, and Gamma and Delta variants were from patients in South America and Europe. It is worth mentioning that patients infected with Alpha and Beta variants were predominantly asymptomatic, while patients infected with Delta variant presented severe clinical features. The abovementioned variants have increased transmissibility or harmful changes in epidemic characteristics, increased virulence, or clinical manifestations. Therefore, increased surveillance and strict control of the importation of the virus remain urgent.
The SARS-CoV-2 genome shows the typical structure of coronavirus, 5' Untranslated regions (UTR), genes of ORF1ab, ORF3a, ORF8, N, S, E, Membrane protein (M) and 3' UTR containing poly (A) tail, and several unidentified unstructured ORFs (37). It is noted that the mutation rate is fastest at the ORF1ab, ORF3a, and ORF8, N and S genes (38, 39). In the present study, a total of 935 SNPs were detected in 165 SARS-COV-2 samples in Jiangsu Province, China. Missense mutation (58%) was the dominant mutation type. Among which, 56% of SNP changes occurred in ORF1ab gene. ORF1ab, encodes a large polyprotein pp1ab, is proteolytically cleaved into 16 non-structural proteins critical for viral replication (40). In addition, 20% of SNP changes occurred in the S gene, such as p.Asp501Tyr, p.Pro681His, and p.Pro681Arg. It is worth mentioning that p.Asp614Gly variant was significantly positively correlated with the clinical severity of patients. The p.Ser316Thr and p.Lu484Lys variants were significantly negatively correlated with the course of disease. Around September 2020, genetic variants carrying the p.Asp501Tyr substitution on the S protein of SARS-CoV-2 were first detected in the United Kingdom, and spread to elsewhere globally, such as Brazil, South Africa, and the United States (41–44). The p.Asp501Tyr substitution reduces the neutralization capacity of antibodies generated against wild type virus (45–47). Detection of p.Asp501Tyr mutation flags samples that may harbor mutations associated with immune evasion and increased transmissibility. The p.Pro681His mutation is the characteristic of the new SARS-CoV-2 variants from the United Kingdom and Nigeria. The p.Pro681His mutation has unique and emerging characteristics with a significant exponential increase in worldwide frequency. Previous SARS-CoV-2 studies suggest that the p.Pro681His mutation is associated with enhanced systemic infection and increased membrane fusion (48–50). The p.Pro681Arg mutation, was first identified in Uganda in September 2020, is designated a variant of interest in Africa and with subsequent spread to other countries. The p.Pro681Arg mutation causes a small increase in proteolytic processing that might have an effect on viral replication, transmissibility, or pathogenic properties (51). The variant p.Asp614Gly is associated with increased viral load in patients with SARS-CoV-2 (52–54), which may significantly enhance the infectivity and transmissibility of the virus. The mutation p.Lu484Lys, first identified in March 2020, has now been identified in the United Kingdom fast-spreading variant, prompting fears that the virus is evolving further and could become resistant to vaccines (55). The p.Lu484Lys mutation is associated with the humoral immune response evasion (56), which may affect antibody neutralization. This indicated that abovementioned variants in S gene are associated with poor clinical outcome of patients with SARS-CoV-2.
Conclusion
Our study provided crucial epidemiological data on the clinical features, variation characteristics of SARS-CoV-2 in Jiangsu Province, China. These findings could help to assess the burden of viral infection in patients. Timely and accurate monitoring of the spreading of infection and the diversity of genomic mutations in SARS-CoV-2 are needed to alleviate the burden of these diseases. There are limitations to our study. First, the small number of samples may bring some errors, therefore, larger numbers of samples are further needed to minimize the errors. Second, potential biological function of variants in genes is further needed to investigate. Anyway, our findings could have certain reference value for the evaluation of patients with SARS-CoV-2 and clinical treatment.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics Statement
All procedures conducted in this study involving human materials were approved by the Ethics Committee of Novel Coronavirus sequence analysis in Jiangsu province (JSJK2021-B016-01). Moreover, all subjects gave written informed consent. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
SW, LZ, and CB: conception and design. LZ, CB, and LC: administrative support. SW, ZL, JF, HF, HY, FD, KZ, and LC: provision of materials and samples. SW, XZ, ZL, JF, HF, HY, FD, HH, and JP: data collection and collation. SW and XZ: data analysis and interpretation. All author read and approved the final manuscript.
Funding
This study was funded by the Social Development Project of Jiangsu Province (BE2021739, provided by LZ).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpubh.2021.791600/full#supplementary-material
References
- 1.Dong E, Du H, Gardner L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect Dis. (2020) 20:533–4. 10.1016/S1473-3099(20)30120-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu JT, Leung K, Leung GM. Nowcasting and forecasting the potential domestic and international spread of the 2019-nCoV outbreak originating in Wuhan, China: a modelling study. Lancet. (2020) 395:689–97. 10.1016/S0140-6736(20)30260-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhu H, Wei L, Niu P. The novel coronavirus outbreak in Wuhan, China. Global Health Res Policy. (2020) 5:6. 10.1186/s41256-020-00135-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.The new scope of virus taxonomy: partitioning the virosphere into 15 hierarchical ranks. Nat Microbiol. (2020) 5:668–74. 10.1038/s41564-020-0709-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lu R, Zhao X, Li J, Niu P, Yang B, Wu H, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. (2020) 395:565–74. 10.1016/S0140-6736(20)30251-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rey FA, Lok SM. Common features of enveloped viruses and implications for immunogen design for next-generation vaccines. Cell. (2018) 172:1319–34. 10.1016/j.cell.2018.02.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kang S, Yang M, Hong Z, Zhang L, Huang Z, Chen X, et al. Crystal structure of SARS-CoV-2 nucleocapsid protein RNA binding domain reveals potential unique drug targeting sites. Acta pharmaceutica Sinica B. (2020) 10:1228–38. 10.1016/j.apsb.2020.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Le TT, Cramer JP, Chen R, Mayhew S. Evolution of the COVID-19 vaccine development landscape. Nat Rev Drug Disc. (2020) 19:667–8. 10.1038/d41573-020-00151-8 [DOI] [PubMed] [Google Scholar]
- 9.Yong SEF, Anderson DE, Wei WE, Pang J, Chia WN, Tan CW, et al. Connecting clusters of COVID-19: an epidemiological and serological investigation. Lancet Infect Dis. (2020) 20:809–15. 10.1016/S1473-3099(20)30273-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zohar T, Alter G. Dissecting antibody-mediated protection against SARS-CoV-2. Nat Rev Immunol. (2020) 20:392–4. 10.1038/s41577-020-0359-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jin A, Yan B, Hua W, Feng D, Xu B, Liang L, et al. Clinical characteristics of patients diagnosed with COVID-19 in Beijing. Biosafety Health. (2020) 2:104–11. 10.1016/j.bsheal.2020.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hung LS. The SARS epidemic in Hong Kong: what lessons have we learned? J Royal Soc Med. (2003) 96:374–8. 10.1258/jrsm.96.8.374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen J, Qi T, Liu L, Ling Y, Qian Z, Li T, et al. Clinical progression of patients with COVID-19 in Shanghai, China. J Infect. (2020) 80:e1–6. 10.1016/j.jinf.2020.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guo YR, Cao QD, Hong ZS, Tan YY, Chen SD, Jin HJ, et al. The origin, transmission and clinical therapies on coronavirus disease 2019 (COVID-19) outbreak - an update on the status. Military Med Res. (2020) 7:11. 10.1186/s40779-020-00240-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li Q, Guan X, Wu P, Wang X, Zhou L, Tong Y, et al. Early transmission dynamics in Wuhan, China, of novel coronavirus-infected pneumonia. N Engl J Med. (2020) 382:1199–207. 10.1056/NEJMoa2001316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chan JF, Yuan S, Kok KH, To KK, Chu H, Yang J, et al. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: a study of a family cluster. Lancet. (2020) 395:514–23. 10.1016/S0140-6736(20)30154-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brann DH, Tsukahara T. Non-neuronal expression of SARS-CoV-2 entry genes in the olfactory system suggests mechanisms underlying COVID-19-associated anosmia. Sci Adv. (2020) 6:eabc5801. 10.1126/sciadv.abc5801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tay MZ, Poh CM, Rénia L. The trinity of COVID-19: immunity, inflammation and intervention. Nat Rev Immunol. (2020) 20:363–74. 10.1038/s41577-020-0311-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu D, Yang XO. TH17 responses in cytokine storm of COVID-19: an emerging target of JAK2 inhibitor Fedratinib. J Microbiol Immunol Infect. (2020) 53:368–70. 10.1016/j.jmii.2020.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. (2020) 395:1054–62. 10.1016/S0140-6736(20)30566-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garibaldi BT, Fiksel J. Patient trajectories among persons hospitalized for COVID-19: a cohort study. Ann Int Med. (2021) 174:33–41. 10.7326/M20-3905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brufsky A. Hyperglycemia, hydroxychloroquine, and the COVID-19 pandemic. J Med Virol. (2020) 92:770–5. 10.1002/jmv.25887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Plante JA, Liu Y, Liu J, Xia H. Spike mutation D614G alters SARS-CoV-2 fitness. Nature. (2021) 592:116–21. 10.1038/s41586-020-2895-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saghazadeh A, Rezaei N. Immune-epidemiological parameters of the novel coronavirus - a perspective. Exp Rev Clin Immunol. (2020) 16:465–70. 10.1080/1744666X.2020.1750954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jin JM, Bai P, He W, Wu F, Liu XF, Han DM, et al. Gender differences in patients with COVID-19: focus on severity and mortality. Front Public Health. (2020) 8:152. 10.3389/fpubh.2020.00152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aleksova A, Gagno G, Sinagra G, Beltrami AP. Effects of SARS-CoV-2 on cardiovascular system: the dual role of angiotensin-converting enzyme 2 (ACE2) as the virus receptor and homeostasis regulator-review. Int J Mol Sci. (2021) 22:4526. 10.3390/ijms22094526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shoaib MH, Ahmed FR, Sikandar M, Yousuf RI, Saleem MT. A journey from SARS-CoV-2 to COVID-19 and beyond: a comprehensive insight of epidemiology, diagnosis, pathogenesis, and overview of the progress into its therapeutic management. Front Pharmacol. (2021) 12:576448. 10.3389/fphar.2021.576448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Al-Tawfiq JA. Asymptomatic coronavirus infection: MERS-CoV and SARS-CoV-2 (COVID-19). Travel Med Infect Dis. (2020) 35:101608. 10.1016/j.tmaid.2020.101608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.V'Kovski P, Kratzel A, Steiner S, Stalder H. Coronavirus biology and replication: implications for SARS-CoV-2. Nat Rev Microbiol. (2021) 19:155–70. 10.1038/s41579-020-00468-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rossi GA, Sacco O, Mancino E, Cristiani L, Midulla F. Differences and similarities between SARS-CoV and SARS-CoV-2: spike receptor-binding domain recognition and host cell infection with support of cellular serine proteases. Infection. (2020) 48:665–9. 10.1007/s15010-020-01486-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tegally H, Wilkinson E, Giovanetti M, Iranzadeh A, Oliveira TD. Emergence and rapid spread of a new severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) lineage with multiple spike mutations in South Africa. mSystems. (2021) 6:e0035321. 10.1101/2020.12.21.2024864034128696 [DOI] [Google Scholar]
- 32.Mwenda M, Saasa N, Sinyange N, Busby G, Chipimo PJ, Hendry J, et al. Detection of B.1.351 SARS-CoV-2 variant strain - Zambia, December 2020. MMWR Morbid Mortal Week Rep. (2021) 70:280–2. 10.15585/mmwr.mm7008e2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Francisco RDS, Jr., Benites LF, Lamarca AP, de Almeida LGP, Hansen AW, Gularte JS, et al. Pervasive transmission of E484K and emergence of VUI-NP13L with evidence of SARS-CoV-2 co-infection events by two different lineages in Rio Grande do Sul, Brazil. Virus Res. (2021) 296:198345. 10.1016/j.virusres.2021.198345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Singh J, Rahman SA, Ehtesham NZ, Hira S, Hasnain SE. SARS-CoV-2 variants of concern are emerging in India. Nat Med. (2021) 27:1131–3. 10.1038/s41591-021-01397-4 [DOI] [PubMed] [Google Scholar]
- 35.Davies NG, Abbott S. Estimated transmissibility and impact of SARS-CoV-2 lineage B.1.1.7 in England. Science. (2021) 372:eabg3055. 10.1126/science.abg3055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leung K, Shum MH, Leung GM, Lam TT, Wu JT. Early transmissibility assessment of the N501Y mutant strains of SARS-CoV-2 in the United Kingdom, October to November 2020. Euro Surveil. (2021) 26:2002106. 10.2807/1560-7917.ES.2020.26.1.2002106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weiss SR, Navas-Martin S. Coronavirus pathogenesis and the emerging pathogen severe acute respiratory syndrome coronavirus. Microbiol Mol Biol Rev. (2005) 69:635–64. 10.1128/MMBR.69.4.635-664.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Laha S, Chakraborty J, Das S, Manna SK, Biswas S, Chatterjee R. Characterizations of SARS-CoV-2 mutational profile, spike protein stability and viral transmission. Infect Genet Evol. (2020) 85:104445. 10.1016/j.meegid.2020.104445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Popa A, Genger JW. Genomic epidemiology of superspreading events in Austria reveals mutational dynamics and transmission properties of SARS-CoV-2. Sci Transl Med. (2020) 12:eabe2555. 10.1126/scitranslmed.abe2555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chan JF, Kok KH. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg Microbes Infect. (2020) 9:221–36. 10.1080/22221751.2020.1719902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tang JW, Tambyah PA, Hui DS. Emergence of a new SARS-CoV-2 variant in the UK. J Infect. (2021) 82:e27–8. 10.1016/j.jinf.2020.12.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tang JW, Toovey OTR, Harvey KN, Hui DDS. Introduction of the South African SARS-CoV-2 variant 501Y.V2 into the UK. J Infect. (2021) 82:e8–10. 10.1016/j.jinf.2021.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Claro IM, da Silva Sales FC, Ramundo MS, Candido DS, Silva CAM, de Jesus JG, et al. Local transmission of SARS-CoV-2 Lineage B.1.1.7, Brazil, December 2020. Emerg Infect Dis. (2021) 27:970–2. 10.3201/eid2703.210038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Galloway SE, Paul P, MacCannell DR, Johansson MA, Brooks JT, MacNeil A, et al. Emergence of SARS-CoV-2 B.1.1.7 lineage - United States, December 29, 2020-January 12, 2021. MMWR Morbid Mortal Weekly Rep. (2021) 70:95–9. 10.15585/mmwr.mm7003e2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mahase E. Covid-19: Novavax vaccine efficacy is 86% against UK variant and 60% against South African variant. BMJ. (2021) 372:n296. 10.1136/bmj.n296 [DOI] [PubMed] [Google Scholar]
- 46.Wu K, Werner AP, Moliva JI, Koch M, Choi A, Stewart-Jones GBE, et al. mRNA-1273 vaccine induces neutralizing antibodies against spike mutants from global SARS-CoV-2 variants. bioRxiv [Preprint]. (2021). 10.1101/2021.01.25.427948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Starr TN, Greaney AJ. Prospective mapping of viral mutations that escape antibodies used to treat COVID-19. bioRxiv. (2021) 371:850–4. 10.1126/science.abf9302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huang Y, Yang C, Xu XF, Xu W, Liu SW. Structural and functional properties of SARS-CoV-2 spike protein: potential antivirus drug development for COVID-19. Acta pharmacologica Sinica. (2020) 41:1141–9. 10.1038/s41401-020-0485-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang Q, Qiu Y, Li JY, Zhou ZJ, Liao CH, Ge XY. A unique protease cleavage site predicted in the spike protein of the novel pneumonia coronavirus (2019-nCoV). potentially related to viral transmissibility. Virologica Sinica. (2020) 35:337–9. 10.1007/s12250-020-00212-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yarmarkovich M, Warrington JM, Farrel A, Maris JM. Identification of SARS-CoV-2 vaccine epitopes predicted to induce long-term population-scale immunity. Cell Rep Med. (2020) 1:100036. 10.1016/j.xcrm.2020.100036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tada T, Zhou H, Dcosta BM, Samanovic MI, Landau NR. The Spike Proteins of SARS-CoV-2 B.1.617 and B.1.618 variants identified in india provide partial resistance to vaccine-elicited and therapeutic monoclonal antibodies. bioRxiv [Preprint]. (2021). 10.1101/2021.05.14.444076 [DOI] [Google Scholar]
- 52.Korber B, Fischer WM, Gnanakaran S, Yoon H, Theiler J, Abfalterer W, et al. Tracking changes in SARS-CoV-2 Spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell. (2020) 182:812–27.e19. 10.1016/j.cell.2020.06.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McNamara RP, Caro-Vegas C, Landis JT, Moorad R, Pluta LJ, Eason AB, et al. High-density amplicon sequencing identifies community spread and ongoing evolution of SARS-CoV-2 in the Southern United States. Cell Rep. (2020) 33:108352. 10.1016/j.celrep.2020.108352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Volz E, Hill V, McCrone JT, Price A, Jorgensen D, O'Toole Á, et al. Evaluating the effects of SARS-CoV-2 spike mutation D614G on transmissibility and pathogenicity. Cell. (2021) 184:64–75.e11. 10.1016/j.cell.2020.11.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wise J. Covid-19: The E484K mutation and the risks it poses. BMJ. (2021) 372:n359. 10.1136/bmj.n359 [DOI] [PubMed] [Google Scholar]
- 56.Xie X, Liu Y, Liu J, Zhang X, Zou J, Fontes-Garfias CR. Neutralization of SARS-CoV-2 spike 69/70 deletion, E484K and N501Y variants by BNT162b2 vaccine-elicited sera. Nat Med. (2021) 27:620–1. 10.1038/s41591-021-01270-4 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.