

Abstract
Bicyclic hydrocarbons, bicyclo[1.1.1]pentanes (BCPs) in particular, play an emerging role as saturated bioisosteres in pharmaceutical, agrochemical, and material chemistry. Taking advantage of strain release strategies, prior synthetic studies have featured the synthesis of bridgehead-substituted (C1, C3) BCPs from [1.1.1] propellane. This work describes an approach to accessing multi-substituted BCPs via a type of intramolecular cyclization. In addition to the C1, C3-disubstituted BCPs, this method also enables the construction of yet under-explored multi-substituted (C1, C2 and C3) BCPs from readily accessible cyclobutanones. The broad generality of this method is also examined through the synthesis of a variety of other caged bicyclic molecules, ranging from [2.1.1] to [3.2.1] scaffolds. The modularity afforded by the pendant bridgehead Bpin generated during the cyclization reaction is demonstrated via several downstream functionalizations, highlighting the ability of this approach to enable the programmed and divergent synthesis of multi-substituted bicyclic hydrocarbons.
Graphical Abstract
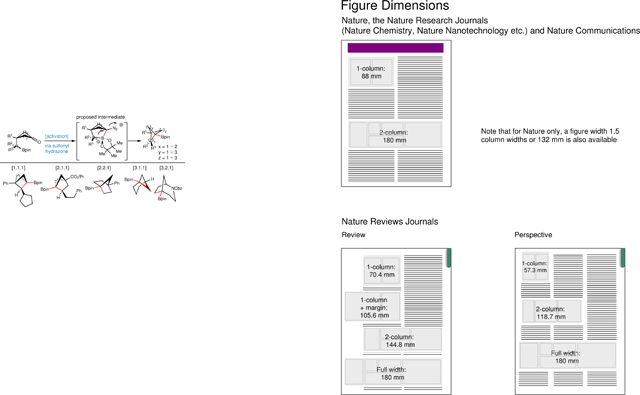
Caged bicyclic molecules that exhibit considerable ring strain have long been the subject of intense study due to their unusual geometries, physical properties and theoretical interest1–3. Recent developments in medicinal chemistry shine a new light on the potential utility of these C(sp3)-rich hydrocarbons4. Owing to their unique physical and chemical properties, bicyclic hydrocarbons exhibit the ability to modulate the pharmacokinetic and physiochemical properties of drug candidates (Figure 1a)5–10. Bicyclo[1.1.1]pentanes (BCPs) containing substitutions at bridgehead positions (C1, C3) are now widely recognized as saturated bioisosteres for para-substituted benzenes11,12. Analogously, related caged scaffolds with differentiated substitutions (Figure 1b) are expected to be ideal bioisosteres of ortho- or meta- substituted benzenes13,14. Currently BCPs are synthesized from the highly strained [1.1.1]propellane (6) (the strain energy of the C–C bond = ~59~65 kcal/mol)15–18, using methodologies pioneered by Wiberg15,19, Michl20, Baran21,22, and others23–41, wherein 6 is transformed to symmetric and asymmetric BCPs using either single- or two-electron transfer pathways (Figure 1c). These efforts have primarily focused on accessing C1 and/or C3- substituted BCPs until few recent reports42–46 disclosed strategies for the systematic functionalization of the backbone (C2) of BCPs. In addition to the strain-release strategy, Wurtz coupling47, Norrish–Yang cyclization48, [2+2] photo-cycloaddition49, ring expansion44–46,50, and ring contraction51 represent other means to access BCPs. However, these methods are often plagued by low yields or limited substrate scope. In light of the aforementioned issues, practical and efficient methodologies to construct multi-substituted (C1/C2/C3) BCPs 8 are highly desirable as they represent elusive bioisosteres of ortho-/meta-substituted benzene rings and would enable access to novel chemical space. Herein we describe an approach for the construction of multi-substituted BCPs via the intramolecular coupling of cyclobutane-tethered sulfonyl hydrazones and boronic esters (Figure 1d). This intramolecular cyclization strategy not only provides a general and operationally simple method for the synthesis of BCPs but can also be expanded to access a wide range of bicyclic alkyl boronates, all of which have the potential to serve as useful benzene bioisosteres. Additionally, the pendant bridgehead alkyl boronate allows for subsequent downstream functionalizations, resulting in a modular and programmable template for the construction of multi-substituted caged bicyclic molecules.
Fig. 1. Bridged hydrocarbons and BCPs syntheses.

(a) Representative drugs bearing ortho- or meta-substituted benzene ring: boscalid and belinostat. (b) Substituted hydrocarbons providing novel chemical space as potential bioisosteres. (c) The state-of-art for BCP synthesis (> 200 reports) from propellane 6 by using strain releasing strategy (left); under-explored C2-substituted BCPs and chiral BCPs (right). (d) Proposed intramolecular cyclization to access strained multi-substituted BCPs from cyclobutanone.
Results and Discussion
Our retrosynthetic analysis to multi-substituted BCPs (strain energy ~71 kcal/mol) relies on cyclization from cyclobutanones 9 (strain energy for cyclobutane ~26 kcal/mol)52. However, previous studies indicated that base-initiated intramolecular substitution proved unsuccessful in BCP formation53, presumably due to the unusual strain energy present in the desired target. Taking inspiration from our54 and other’s prior studies55–63 on base-promoted cross-coupling between alkyl sulfonylhydrazones and boronic acids, we surmised that base-mediated intramolecular coupling of cyclobutane-tethered sulfonyl hydrazones and boronates might enable the formation of a high energy bicyclic [2.1.1] zwitterionic intermediate 10. Furthermore, we hypothesized that this high-energy intermediate might undergo subsequent 1,2-metallate rearrangement to form BCP 11 via extrusion of N2. While the C–B bond in 10 is not perfectly aligned with the leaving group, the loss of N2 could help drive the subsequent 1,2-metallate rearrangement and contraction to the desired BCP scaffold 11.64,65 Alkyl boron pinacol esters (Bpin) have a priori been reported as recalcitrant coupling partners in Barluenga-Valdés coupling55 and its modifications.54 However, from both a practicality and ease of access perspective, alkyl Bpins were identified as ideal starting materials. We envisioned that an intramolecular reaction environment would facilitate the coordination by decreasing the entropic barrier, which might help overcome the poor coordinate reactivity of the Bpin, thereby enabling the intramolecular reaction to occur.
Reaction optimization.
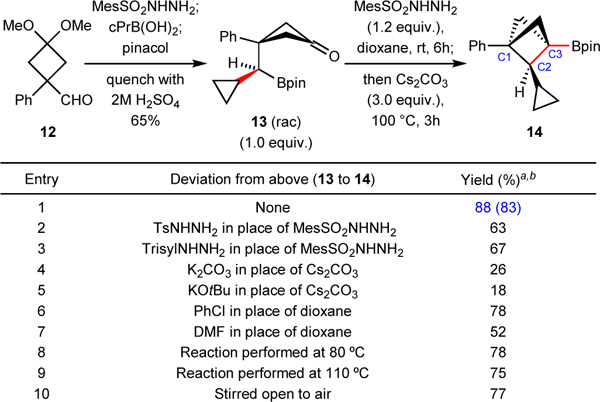
To test this theory, the key intermediate 13 was prepared in one-step using our boron-preserving cross-coupling conditions from cyclobutane aldehyde 12 (Table 1)54. Subjecting 13 to in-situ hydrazone formation followed by our previously reported conditions for intermolecular cross-coupling gratifyingly afforded the desired bridgehead Bpin substituted BCP product 14 in 78% yield (Entry 6). Subsequent optimization of sulfonylhydrazide, base, solvent and temperature (summarized in Table 1; see Page 34 in Supplementary Information for detailed reaction optimizations) resulted in the identification of optimal conditions, employing mesitylsulfonyl hydrazide, cesium carbonate and dioxane to afford the coupling product 14 in 83% isolated yield (88% GC yield) (Entry 1). The use of mesitylsulfonyl hydrazide as the activation reagent was found to be the key for effecting efficient hydrazone condensation and in-situ generation of the diazo intermediate (Entries 2 and 3, starting material 13 is left for these two cases). The selection of base (Entries 4 and 5) and solvent (Entries 6 and 7) were also important for obtaining high yield for this cyclization. Varying the temperatures also afforded the desired product 14 (Entries 8 and 9), albeit in lower yields. It is noteworthy that the reaction does not require inert atmosphere and proceeds smoothly under air, presumably due to the improved stability of the Bpin motif in comparison to its B(OH)2 counterpart (Entry 10).
Table 1.
Intramolecular coupling optimization to access C2-substituted BCPs.
|
Yield determined by GC analysis with trimethoxybenzene as an internal standard.
Yield in parenthesis is the isolated yield. rt, room temperature; Mes, mesityl; cPr, cyclopropyl; Ts, tosyl; Trisyl, triisopropylbenzenesulfonyl; DMF, N,N-dimethylformamide.
Reaction scope for BCPs.
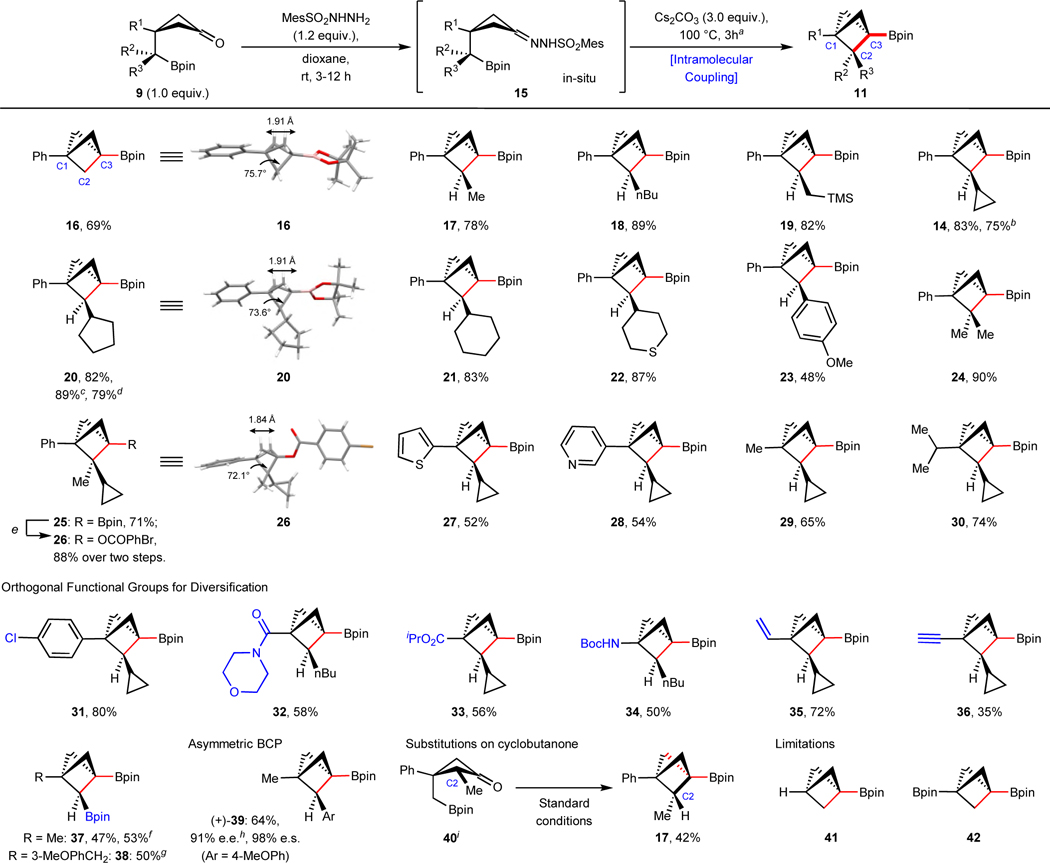
With the optimal conditions in hand, the substrate scope of this intramolecular cyclization to access di-, tri-, and tetra-substituted BCPs was systematically investigated (Table 2). With the hypothesis that this cyclization would be influenced by the conformation of cyclobutane 9, our exploration commenced with a sterically large phenyl group (A value = 3.0) on cyclobutane ring66,67. A-values are the conformational preference of an equatorial compared to an axial substituent in a monosubstituted cyclohexane. The cyclobutanone Bpins 9 were prepared from the corresponding aldehydes, ketones, esters and halides68–73. A primary alkyl Bpin (R2 = H, R3 = H) underwent smooth cyclization to the C1, C3 di-substituted BCP Bpin 16. Starting from secondary alkyl Bpins, a variety of C1, C2 and C3 tri-substituted BCPs, including C2-alkyl (17–22, 14) and C2-aryl substituted (23) BCP Bpins were prepared. Lastly, subjecting tertiary alkyl Bpin starting materials to cyclization condensations afforded BCPs with di-substituted C2 side chains (24, 25). The structures of BCPs 16, 20 and 26 were unambiguously assigned by single crystal X-ray analysis. From this structural data, it is clear that increased C2-substitution reduces the C1–C2–C3 angle, presumably due to the Thorpe–Ingold effect (75.7° in 16, 73.6° in 20, 72.1° in 26). In addition to Ph at C1, other medicinal chemistry relevant motifs such as halogenated aryls (4-chlorophenyl, 31), electron-rich heterocycles (2-thiophene, 27), and Lewis-basic heterocycles (3-pyridyl, 28) were all compatible in this cyclization. Smaller alkyl substitutions, including methyl (A value = 1.7, 29) or isopropyl (A value = 2.15, 30) could also be incorporated at R1 to promote smooth cyclization to the corresponding products. It is noteworthy that the cyclization could also be performed with a variety of functional groups that allow for further downstream functionalizations, including amide (32), isopropyl ester (A value = 1.2, 33), vinyl (A value = 1.35, 35), terminal alkyne (A value = 0.41, 36) and amine (34). In addition to the aryl- and alkyl substitutions at C2, productive cyclization of gem-diborylated59,70 precursors provide the di-Bpin substituted BCPs (37 and 38). These substrates open avenues for further diversification. The asymmetric BCP 39 was cyclized from its enantioenriched Bpin precursor in a 69% yield with a high level of chiral fidelity. Besides the above-mentioned substitutions on the cyclobutanone side chain (R2 and/or R3), the cyclobutane ring itself can be prefunctionalized. To that end, the methyl substituted cyclobutanone 40 was cyclized to 17 in 42% yield. This example highlights the possibility of accessing more complicated BCPs via cyclobutanone prefunctionalization.
Table 2.
Substrate scope of BCPs via intramolecular coupling.
|
Starting materials and products are racemic mixtures, unless annotated.
Reaction condition: Cyclobutanone 9 (1.0 equiv., 0.05–1.0 mmol), MesSO2NHNH2 (1.2 equiv.) in dioxane (0.1–0.2 M) stirred at rt for 3–12 h, monitored by TLC; then Cs2CO3 (3.0 equiv.) was added and stirred at 100 °C for another 3 h.
4.8 mmol scale;
3.7 mmol scale;
18 mmol scale;
Reaction condition: 1) NaOAc, H2O2, 0 °C, 1 h; 2) DMAP, 4-bromobenzoyl chloride, DIPEA;
11 mmol scale;
30 mmol scale;
e.e. values were measured after conversion to their alcohol derivatives;
the stereochemistry was assigned based on its derivative; TMS, trimethylsilyl; Boc, tert-butyloxycarbonyl; DMAP, 4-dimethylaminopyridine; DIPEA, N,N-diisopropylethylamine; e.e., enantiomeric excess; e.s., enantiospecificity.
The robustness of this reaction was highlighted by accessing 14 and 20 on gram scale (4.8 mmol and 18 mmol) in a similar yield (75% and 79%) to that on 0.1 mmol scale and under identical conditions. In addition, gram-scale cyclization of gem-diborylated precursors afforded 2.0 g and 6.6 g of di-Bpin substituted BCPs 37 and 38 in 53% and 50% yield, respectively. Consistent with our initial hypothesis that an axial conformation of the Bpin-containing side chain is crucial for the success of this cyclization, small substituents were tolerated (R1 = Me, vinyl and ethynyl) but no reactivity was observed when R1 = H and Bpin (41 and 4274).
Synthetic applications.
As illustrated in Figure 2a, the strategic impact of this methodology shines in its ability to combine the modularity of preparing C2-substituted BCP Bpins (via cross-coupling) and leveraging the plethora of existing transformations for Bpin functionalization for downstream diversification of the BCP bridgehead position. For example, the oxidation of boronic ester 20 led to the alcohol (43) in high yield. Additionally, 20 was subjected to Aggarwal’s arylation protocol75, Zweifel olefination76,77, and Matteson homologation78 to afford C–C bond-forming products 44, 45 and 46, respectively. The Bpin group can also be transformed to the more stable trifluoroborate salt (47), which opens further functionalization opportunities. Radical proto-deborylation79,80 results in C1, C2-disubstitued BCPs (48), and C(sp3)–C(sp2) Pd catalyzed Suzuki cross-coupling conditions81–83 enables arylation at the bridgehead (49–51) (See page 36–37 in Supplementary Information for detailed optimization). Furthermore, cross-coupling of the in-situ-generated boronic acid with sulfonylhydrazone 52 affords the Bpin 53 in 92% yield. Amination with alkyl azides through BF3•Et2O activation of affords amines 54–56 in moderate yields (See page 38–39 in Supplementary Information for detailed optimization).84,85 Therefore, this strategy allows for systematic introduction of substitutions at any position of the BCP, including the bridgeheads (C1 and/or C3) as well as the backbone (C2, mono- and di-). Importantly, this enables the practitioner to access a wide range of substituted BCPs that can serve as bioisosteres for ortho-, meta- or para-substituted benzene rings.
Fig. 2. Derivatization and Synthetic Application of BCP Boronates.

(a) Transformations of functional boronates for downstream diversification of the BCP bridgehead position, including oxidation (43), arylation (44), Zweifel olefination (45), Matteson homologation (46), radical proto-deborylation (48), Suzuki cross-coupling (49–51), hydrazone coupling (53) and amination (54–56); (b) Programmable synthesis of C1, C2, C3-trisubstituted BCP 60, a versatile building block amenable for the preparation of hypothetical bioisosteres of an orexin receptor antagonist 57. See “Additional Optimization of Suzuki Cross-Couplings and Aminations with 2-Substituted BCP Boronates” and “Experimental Procedures and Characterization Data of Substrates (compound 43–60)” in Supplementary Information for details.
As illustrated in Figure 2b, compound 57 was developed as an orexin receptor antagonist to treat insomnia86. While this drug possesses a 1,3,4-trisubstituted benzene ring within its structure, previous methods to access functionalized BCPs were not conducive to the preparation of a saturated tri-substituted BCP analogue. In contrast, this methodology provides straightforward and modular access to the intermediate 60 for its higher fraction sp3 (Fsp3) BCP analog 61, via a sequence of 1) cPr installation (59), 2) cyclization (33), 3) Bpin oxidation to alcohol, and 4) alkylation (60).
Reaction scope for other bicyclic scaffolds.
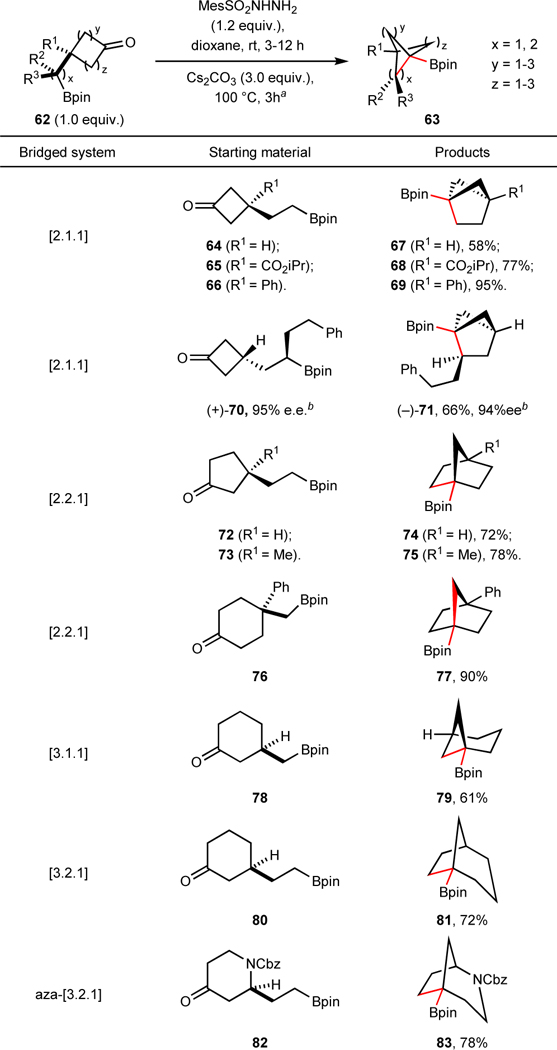
Besides BCPs, other bicyclic scaffolds have also been showcased or proposed as potential saturated bioisosteres. Often the bottleneck in performing SAR (Structure-Activity Relationship) studies on these ring systems, at the bridgehead positions in particular, is the lack of unified synthetic strategies to access suitable diversifiable building blocks. As delineated in Table 3, this cyclization strategy enables the construction of a wide range of bicyclic ring systems with the versatile Bpin preserved at the bridgehead position. Starting from a range of cyclobutanones (64–66, 70), cyclopentanones (72, 73) and cyclohexanones (76, 78, 80), in combination with pendant boronic ester side chains of varying length, bicyclo[2.1.1] (67–69, 71), [2.2.1] (74, 75, 77), [3.1.1] (79) and [3.2.1] (81) systems were successfully prepared using these coupling conditions. Depending on the ease of accessibility of the starting material, [2.2.1] bicycles could be accessed from either cyclopentanones (72, 73) or cyclohexanones (76). Saturated ring systems with a heteroatom embedded in them could also be prepared using this protocol, as demonstrated by the aza-[3.2.1]bicycle (83). Notably, starting from a chiral alkyl Bpin, this protocol allows for complete transfer of stereochemistry into the bicyclic products and enables the asymmetric synthesis of these valuable bioisosteres. For example, the chiral cyclobutanone Bpin 7069 provided chiral [2.1.1] bicycle 71 with no erosion of enantiomeric excess (e.e.).
Table 3.
Intramolecular coupling to access bridged systems.
|
Starting materials and products are racemic mixtures, unless annotated.
Reaction conditions: Cyclic ketone 62 (1.0 equiv.), MesSO2NHNH2 (1.2 equiv.) in dioxane (0.1–0.2 M) stirred at rt for 3–12 h, monitored by TLC; then Cs2CO3 (3.0 equiv.) was added and stirred at 100 °C for another 3 h.
e.e. values were measured after conversion to alcohol derivatives. Cbz, benzyloxycarbonyl.
In conclusion, we have developed an intramolecular cyclization to access C1-, C2-, and C3- substituted BCPs. As showcased in Table 2 and Figure 2, this operationally simple and chemoselective method enables rapid and modular preparation of a variety of synthetically challenging boronate-substituted BCPs. Synergistic application with existing Bpin functionalization strategies allows for rapid diversification and synthesis of complex bioisosteres that are highly desired in drug discovery. In addition, this method was successfully implemented to prepare a range of other pharmaceutically relevant bicyclic bioisosteres (Table 3) that have yet to be fully explored. As a result, we expect this method to have a substantial impact within drug discovery, specifically in how benzene replacements are designed and incorporated into targets of interest.
Methods
Caution:
In the reactions to prepare BCP precursors and the cyclization reactions to afford BCP derivatives, an equivalent of nitrogen is released. In the multi-gram scale procedure, hydrogen and nitrogen are released and the reaction is exothermic. Please handle carefully when conducting the reported reactions in a large scale.
General procedure for synthesis of bicycloalkyl boronates.
A screw-capped culture tube was charged with ketone (1.0 equiv.), 2-mesitylenesulfonyl hydrazide (1.2 equiv.) and dried dioxane (0.1 M). The mixture was then stirred at room temperature until the completion of the reaction showed by TLC analysis (usually 3 – 12 h). Cesium carbonate (3.0 equiv.) was added, and the headspace of the tube was purged with a gentle stream of argon for approximately 10 seconds. The system was stirred at 100 °C under argon atmosphere for 3 hours. The suspended solution was then filtered over Celite and washed with diethyl ether. The solvent was removed under high vacuum, and the crude residue was purified by chromatography on silica gel.
Data Availability
Experimental data as well as characterization data for all compounds prepared in the course of these studies are provided in the Supplementary Information of this manuscript. Crystallographic data for the structures reported in this Article have been deposited at the Cambridge Crystallographic Data Centre, under deposition numbers CCDC 2062923 (16), 2062924 (20), 2062925 (26) and 2081087 (40-ester, see X-ray Crystallographic Data in Supplementary Information). Copies of the data can be obtained free of charge via https://www.ccdc.cam.ac.uk/structures/.
Supplementary Material
Acknowledgements
Financial support for this work was provided by Welch Foundation (I-2010-20190330), National Institutes of Health (R01GM141088) and UT Southwestern Eugene McDermott Scholarship. We thank Feng Lin (UTSW) for assistance with NMR spectroscopy; Hamid Baniasadi (UTSW) for HRMS; Vincent Lynch (UT-Austin) for X-ray crystallographic analysis. We thank the Chen, Tambar, Ready, De Brabander, Smith, and Falck groups (UTSW) for generous access to equipment, and helpful discussions. We thank Chuo Chen (UTSW) for theoretical calculation help. We are grateful to Kevin Campos, LC Campeau, Patrick Fier, Cayetana Zarate Saez, and Kyle McClymont (Merck & Co., Inc.) for feedback on this manuscript.
Footnotes
Competing Interests
A provisional patent application naming T.Q., Y.Y., and J.T. as inventors has been filed by the Board of Regents of the University of Texas System, which covers the synthetic method and structural motifs described in this manuscript. The remaining authors declare no competing interests.
References:
- 1.Levin MD, Kaszynski P. & Michl J. Bicyclo[1.1.1]pentanes, [n]Staffanes, [1.1.1]Propellanes, and Tricyclo[2.1.0.02,5]pentanes. Chem. Rev. 100, 169–234 (2000). [DOI] [PubMed] [Google Scholar]
- 2.Dilmaç AM, Spuling E, de Meijere A. & Bräse S. Propellanes—From a Chemical Curiosity to “Explosive” Materials and Natural Products. Angew. Chem. Int. Ed. 56, 5684–5718 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Wiberg KB The Concept of Strain in Organic Chemistry. Angew. Chem. Int. Ed. Engl. 25, 312–322 (1986). [Google Scholar]
- 4.Lovering F, Bikker J. & Humblet C. Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success. J. Med. Chem. 52, 6752–6756 (2009). [DOI] [PubMed] [Google Scholar]
- 5.Mikhailiuk PK et al. Conformationally Rigid Trifluoromethyl‐Substituted α‐Amino Acid Designed for Peptide Structure Analysis by Solid‐State 19F NMR Spectroscopy. Angew. Chem. Int. Ed. 45, 5659–5661 (2006). [DOI] [PubMed] [Google Scholar]
- 6.Stepan AF et al. Application of the Bicyclo[1.1.1]pentane Motif as a Nonclassical Phenyl Ring Bioisostere in the Design of a Potent and Orally Active γ-Secretase Inhibitor. J. Med. Chem. 55, 3414–3424 (2012). [DOI] [PubMed] [Google Scholar]
- 7.Westphal MV, Wolfstaedter BT, Plancher J, Gatfield J. & Carreira EM Evaluation of tert-Butyl Isosteres: Case Studies of Physicochemical and Pharmacokinetic Properties, Efficacies, and Activities. ChemMedChem 10, 461–469 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Costantino G. et al. Synthesis and Biological Evaluation of 2-(3′-(1H-tetrazol-5-yl)bicyclo[1.1.1]pent-1- yl)glycine (S-TBPG), a Novel mGlu1 Receptor Antagonist. Bioorg. Med. Chem. 9, 221–227 (2001). [DOI] [PubMed] [Google Scholar]
- 9.Measom ND et al. Investigation of a Bicyclo[1.1.1]pentane as a Phenyl Replacement within an LpPLA2 Inhibitor. ACS Med. Chem. Lett. 8, 43–48 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Auberson YP et al. Improving Nonspecific Binding and Solubility: Bicycloalkyl Groups and Cubanes as para-Phenyl Bioisosteres. ChemMedChem 12, 590–598 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Talele TT Opportunities for Tapping into Three-Dimensional Chemical Space through a Quaternary Carbon. J. Med. Chem. 63, 13291–13315 (2020). [DOI] [PubMed] [Google Scholar]
- 12.Bauer MR et al. Put a Ring on It: Application of Small Aliphatic Rings in Medicinal Chemistry. RSC Med. Chem. 12, 448–471 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mikhailiuk PK Saturated Bioisoteres of Benzene: Where to Go Next? Org. Biomol. Chem. 17, 2839–2849 (2019). [DOI] [PubMed] [Google Scholar]
- 14.Denisenko A, Garbuz P, Shishkina SV, Voloshchuk NM & Mikhailiuk PK Saturated Bioisosteres of ortho-Substituted Benzenes. Angew. Chem. Int. Ed. 59, 20515–20521 (2020). [DOI] [PubMed] [Google Scholar]
- 15.Wiberg KB & Walker FH [1.1.1]Propellane. J. Am. Chem. Soc. 104, 5239–5240 (1982). [Google Scholar]
- 16.Jackson JE & Allen LC The C1-C3 Bond in [1.1.1]Propellane. J. Am. Chem. Soc. 106, 591–599 (1984). [Google Scholar]
- 17.Feller D. & Davidson ER Ab initio Studies of [1.1.1]- and [2.2.2]Propellane. J. Am. Chem. Soc. 109, 4133–4139 (1987). [Google Scholar]
- 18.Wu W, Gu J, Song J, Shaik S. & Hiberty PC The Inverted Bond in [1.1.1]Propellane is a Charge-Shift Bond. Angew. Chem. Int. Ed. 48, 1407–1410 (2009). [DOI] [PubMed] [Google Scholar]
- 19.Wiberg KB, Waddell ST & Laidig K. [1.1.1]Propellane: Reaction with Free Radicals. Tetrahedron Lett. 27, 1553–1556 (1986). [Google Scholar]
- 20.Kaszynki P. & Michl J. A Practical Photochemical Synthesis of Bicyclo[1.1.1]-pentane-1,3-dicarboxylic acid. J. Org. Chem. 53, 4593–4594 (1988). [Google Scholar]
- 21.Gianatassio R. et al. Strain-release Amination. Science, 351, 241–246 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lopchuk JM et al. Strain-Release Heteroatom Functionalization: Development, Scope, and Stereospecificity. J. Am. Chem. Soc. 139, 3209–3226 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ma X, Nhat Pham L. Selective Topics in the Syntheses of Bicyclo[1.1.1]- pentane (BCP) Analogues. Asian J. Org. Chem. 9, 8–22 (2020). [Google Scholar]
- 24.Kanazawa J. & Uchiyama M. Recent Advances in the Synthetic Chemistry of Bicyclo[1.1.1]pentane. Synlett, 30, 1–11 (2019). [Google Scholar]
- 25.Makarov IS, Brocklehurst CE, Karaghiosoff K, Koch G. & Knochel P. Synthesis of Bicyclo[1.1.1]pentane Bioisosteres of Internal Alkynes and para-Disubstituted Benzenes from [1.1.1]- Propellane. Angew. Chem., Int. Ed. 56, 12774–12777 (2017). [DOI] [PubMed] [Google Scholar]
- 26.Kanazawa J, Maeda K. & Uchiyama M. Radical Multicomponent Carboamination of [1.1.1]Propellane. J. Am. Chem. Soc. 139, 17791–17794 (2017). [DOI] [PubMed] [Google Scholar]
- 27.Kondo M. et al. Silaboration of [1.1.1]Propellane: A Storable Feedstock for Bicyclo[1.1.1]pentane Derivatives. Angew. Chem. Int. Ed. 59, 1970–1974 (2020). [DOI] [PubMed] [Google Scholar]
- 28.Caputo DFJ et al. Synthesis and Applications of Highly Functionalized 1-Halo-3-substituted Bicyclo[1.1.1]pentanes. Chem. Sci. 9, 5295–5300 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nugent J. et al. A General Route to Bicyclo[1.1.1]pentanes through Photoredox Catalysis. ACS Catal. 9, 9568–9574 (2019). [Google Scholar]
- 30.Zhang X. et al. Copper-mediated Synthesis of Drug-like Bicyclopentanes. Nature, 580, 220–226 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Trongsiriwat N. et al. Reactions of 2‐Aryl‐1,3‐Dithianes and [1.1.1]Propellane. Angew. Chem. Int. Ed. 58, 13416–13420 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hughes JME, Scarlata DA, Chen AC-Y, Burch J. & Gleason JL Aminoalkylation of [1.1.1]Propellane Enables Direct Access to High-Value 3-Alkylbicyclo[1.1.1]pentan-1-amines. Org. Lett. 21, 6800–6804 (2019). [DOI] [PubMed] [Google Scholar]
- 33.Shelp RA & Walsh PJ Synthesis of BCP Benzylamines From 2‐Azaallyl Anions and [1.1.1]Propellane. Angew. Chem. Int. Ed. 57, 15857–15861 (2018). [DOI] [PubMed] [Google Scholar]
- 34.Yu S, Jing C, Noble A. & Aggarwal VK 1,3-Difunctionalizations of [1.1.1]- Propellane via 1,2-Metalate Rearrangements of Boronate Complexes. Angew. Chem., Int. Ed. 59, 3917–3921 (2020). [DOI] [PubMed] [Google Scholar]
- 35.Kim JH, Ruffoni A, Al-Faiyz YSS, Sheikh N. & Leonori D. Divergent Strain‐Release Amino‐Functionalization of [1.1.1]Propellane with Electrophilic Nitrogen‐Radicals. Angew. Chem. Int. Ed. 59, 8225–8231 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shin S, Lee S, Choi W, Kim N. & Hong S. Visible-Light-Induced 1,3-Aminopyridylation of [1.1.1]Propellane with N Aminopyridinium Salts. Angew. Chem. Int. Ed. 60, 7873–7879 (2021). [DOI] [PubMed] [Google Scholar]
- 37.Toriyama F. et al. Redox-Active Esters in Fe-Catalyzed C-C Coupling. J. Am. Chem. Soc. 138, 11132–11135 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.VanHeyst MD et al. Continuous Flow-Enabled Synthesis of Bench-Stable Bicyclo[1.1.1]pentane Trifluoroborate Salts and Their Utilization in Metallaphotoredox Cross-Couplings. Org. Lett. 22, 1648–1654 (2020). [DOI] [PubMed] [Google Scholar]
- 39.Garlets ZJ et al. Enantioselective C–H Functionalization of Bicyclo[1.1.1]pentanes. Nat. Catal. 3, 351–357 (2020). [Google Scholar]
- 40.Zarate C. et al. Development of Scalable Routes to 1-Bicyclo[1.1.1]pentylpyrazoles. Org. Process Res. Dev. 25, 642–647 (2021). [Google Scholar]
- 41.Shelp RA et al. Strain-release 2-Azaallyl Anion Addition/borylation of [1.1.1]Propellane: Synthesis and Functionalization of Benzylamine Bicyclo[1.1.1]pentyl Boronates. Chem. Sci. 12, 7066–7072 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ma X, Han Y. & Bennett DJ Selective Synthesis of 1-Dialkylamino-2-alkylbicyclo-[1.1.1]pentanes. Org. Lett. 22, 9133–9138 (2020). [DOI] [PubMed] [Google Scholar]
- 43.Zhao J-X et al. 1,2-Difunctionalized bicyclo[1.1.1]pentanes: long-sought-after mimetics for ortho/meta-substituted arenes. Proc. Natl. Acad. Sci. USA 118, e2108881118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bychek RM et al. Difluoro-Substituted Bicyclo[1.1.1]pentanes for Medicinal Chemistry: Design, Synthesis, and Characterization. J. Org. Chem. 84, 15106–15117 (2019). [DOI] [PubMed] [Google Scholar]
- 45.Ma X, Sloman DL, Han Y. & Bennett DJ A Selective Synthesis of 2,2-Difluorobicyclo[1.1.1]pentane Analogues:“BCP-F2”. Org. Lett. 21, 7199–7203 (2019). [DOI] [PubMed] [Google Scholar]
- 46.Kaleta J. et al. Bridge-Chlorinated Bicyclo[1.1.1]pentane-1,3-dicarboxylic Acids. J. Org. Chem. 84, 2448–2461 (2019). [DOI] [PubMed] [Google Scholar]
- 47.Wiberg KB, Connor DS & Lampman GM The Reaction of 3-Bromocyclo-butane-1-methyl Bromide with Sodium: Bicyclo[1. 1. 1]pentane. Tetrahedron Lett. 5, 531–534 (1964). [Google Scholar]
- 48.Padwa A. & Alexander E. Photochemical Formation of a Substituted Bicyclo[1.1.1]pentane. J. Am. Chem. Soc. 89, 6376 (1967). [Google Scholar]
- 49.Srinivasan R. & Carlough KH Mercury(3P1) Photosensitized Internal Cyclo-addition Reactions in 1,4-, 1,5-, and 1,6-dienes. J. Am. Chem. Soc. 89, 4932–4936 (1967). [Google Scholar]
- 50.Applequist DE, Renken TL & Wheeler JW Polar Substituent Effects in 1,3-Disubstituted Bicycle[1.1.1]pentanes. J. Org. Chem. 47, 4985–4995 (1982). [Google Scholar]
- 51.Meinwald J, Szkryalo W. & Dimmel DR Bicyclo[1.1.1]pentane from Mercury Sensitized and Unsensitized Gas Phase Photolyses of Bicyclo[2.1.1]hexan-2-one. Tetrahedron Lett. 8, 731–733 (1967). [Google Scholar]
- 52.Wiberg KB in The Chemistry of Cyclobutanes Ch. 1, 1–15 (Wiley, 2005). [Google Scholar]
- 53.Wiberg KB & Connor DS Bicylo[1.1.1]pentane. J. Am. Chem. Soc. 88, 4437–4441 (1966). [Google Scholar]
- 54.Yang Y. et al. Practical and Modular Construction of C(sp3)–Rich Alkyl Boron Compounds. J. Am. Soc. Chem. 143, 471–480 (2021). [DOI] [PubMed] [Google Scholar]
- 55.Barluenga J, Tomás-Gamasa M, Aznar F. & Valdés C. Metal-Free Carbon–Carbon Bond-Forming Reductive Coupling between Boronic Acids and Tosylhydrazones. Nat. Chem. 1, 494–499 (2009). [DOI] [PubMed] [Google Scholar]
- 56.Préz-Aguilar MC & Valdés C. Olefination of Carbonyl Compounds through Reductive Coupling of Alkenylboronic Acids and Tosylhydrazones. Angew. Chem. Int. Ed. 51, 5953 –5957 (2012). [DOI] [PubMed] [Google Scholar]
- 57.Plaza M. & Valdés C. Stereoselective Domino Carbocyclizations of γ- and δ-Cyano-N-tosylhydrazones with Alkenylboronic Acids with Formation of Two Different C(sp3)–C(sp2) Bonds on a Quaternary Stereocenter. J. Am. Chem. Soc. 138, 12061–12064 (2016). [DOI] [PubMed] [Google Scholar]
- 58.Plaza M, Paraja M, Florentino L. & Valdés C. Domino Synthesis of Benzo-Fused β, γ-Unsaturated Ketones from Alkenylboronic Acids and N-Tosylhydrazone-Tethered Benzonitriles. Org. Lett. 21, 632–635 (2019). [DOI] [PubMed] [Google Scholar]
- 59.Wu G, Deng Y, Wu C, Zhang Y. & Wang J. Synthesis of α- Aryl Esters and Nitriles via Deaminative Coupling of α-Aminoesters and α-Aminoacetonitriles with Arylboronic Acids. Angew. Chem. Int. Ed. 53, 10510–10514 (2014). [DOI] [PubMed] [Google Scholar]
- 60.Merchant RR & Lopez JA A General C(sp3)–C(sp3) Cross-Coupling of Benzyl Sulfonylhydrazones with Alkyl Boronic Acids. Org. Lett. 22, 2271–2275 (2020). [DOI] [PubMed] [Google Scholar]
- 61.Plaza M, Parisotto S. & Valdés C. Heterocyclization and Spirocyclization Processes Based on Domino Reactions of N‐Tosylhydrazones and Boronic Acids Involving Intramolecular Allylborylations of Nitriles. Chem. Eur. J. 24, 14836–14843 (2018). [DOI] [PubMed] [Google Scholar]
- 62.Arunprasath D, Bala BD & Sekar G. Luxury of N-Tosylhydrazones in Transition-Metal-Free Transformations. Adv. Synth. Catal. 361, 1172–1207 (2019). [Google Scholar]
- 63.Li H, Wang L, Zhang Y. & Wang J. Transition‐Metal‐Free Synthesis of Pinacol Alkylboronates from Tosylhydrazones. Angew. Chem. Int. Ed. 51, 2943–2946 (2012). [DOI] [PubMed] [Google Scholar]
- 64.Tani K. & Stoltz BM Synthesis and Structural Analysis of 2-Quinuclidonium Tetrafluoroborate. Nature, 441, 731–734 (2006). [DOI] [PubMed] [Google Scholar]
- 65.Davenport R. et al. Visible Light-Driven Strain-Increase Ring Contraction Allows the Synthesis of Cyclobutyl Boronic Esters. Angew. Chem., Int. Ed. 59, 6525–6528 (2020). [DOI] [PubMed] [Google Scholar]
- 66.Hirsch JA Table of Conformational Energies. Topics in Stereochemistry 1, 199–222 (1967). [Google Scholar]
- 67.Eliel EL, Wilen SH & Doyle MP in Basic Organic Stereochemistry p. 443 (Wiley, 2001). [Google Scholar]
- 68.Yang C-T et al. Alkylboronic Esters from Copper‐Catalyzed Borylation of Primary and Secondary Alkyl Halides and Pseudohalides. Angew. Chem. Int. Ed. 51, 528–532 (2012). [DOI] [PubMed] [Google Scholar]
- 69.Stymiest JL, Dutheuil G, Mahmood A. & Aggarwal VK Lithiated Carbamates: Chiral Carbenoids for Iterative Homologation of Boranes and Boronic Esters. Angew. Chem. Int. Ed. 46, 7491–7494 (2007). [DOI] [PubMed] [Google Scholar]
- 70.Li H. et al. Formal Carbon Insertion of N-Tosylhydrazone into B–B and B–Si Bonds: gem-Diborylation and gem-Silylborylation of sp3 Carbon. Org. Lett. 16, 448–451 (2014). [DOI] [PubMed] [Google Scholar]
- 71.Bonet A, Pubill-Ulldemolins C, Bo C, Gulyás H. & Fernández E. Transition‐Metal‐Free Diboration Reaction by Activation of Diboron Compounds with Simple Lewis Bases. Angew. Chem. Int. Ed. 50, 7158–7161 (2011). [DOI] [PubMed] [Google Scholar]
- 72.Kisan S, Krishnakumar V. & Gunanathan C. Ruthenium-Catalyzed Anti-Markovnikov Selective Hydroboration of Olefins. ACS Catal. 7, 5950–5954 (2017). [Google Scholar]
- 73.Yamamoto Y, Fujikawa R, Umemoto T. & Miyaura N. Iridium-catalyzed Hydro-boration of Alkenes with Pinacolborane. Tetrahedron 60, 10695–10700 (2004). [Google Scholar]
- 74.Fasano V. et al. How Big is the Pinacol Boronic Ester as a Substituent? Angew. Chem. Int. Ed. 59, 22403–22407 (2020). [DOI] [PubMed] [Google Scholar]
- 75.Odachowski M. et al. Development of Enantiospecific Coupling of Secondary and Tertiary Boronic Esters with Aromatic Compounds. J. Am. Chem. Soc. 138, 9521–9532 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zweifel G, Arzoumanian H. & Whitney CC A Convenient Stereoselective Synthesis of Substituted Alkenes via Hydroboration-iodination of Alkynes. J. Am. Chem. Soc. 89, 3652–3653 (1967). [Google Scholar]
- 77.Armstrong RJ & Aggarwal VK 50 Years of Zweifel Olefination: A Transition-Metal-Free Coupling. Synthesis 49, 3323–3336 (2017). [Google Scholar]
- 78.Sadhu KM, & Matteson DS (Chloromethyl)lithium: Efficient Generation and Capture by Boronic Esters and a Simple Preparation of Diisopropyl (chloromethyl)boronate. Organometallics 4, 1687–1689 (1985). [Google Scholar]
- 79.Pozzi D, Scanlan EM & Renaud P. A Mild Radical Procedure for the Reduction of B-Alkylcatecholboranes to Alkanes. J. Am. Chem. Soc. 127, 14204–14205 (2005). [DOI] [PubMed] [Google Scholar]
- 80.André-Joyaux E, Kuzovlev A, Tappin NDC & Renaud P. A General Approach to Deboronative Radical Chain Reactions with Pinacol Alkylboronic Esters. Angew. Chem. Int. Ed. 59, 13859–13864 (2020). [DOI] [PubMed] [Google Scholar]
- 81.Littke AF, Dai C. & Fu GC Versatile Catalysts for the Suzuki Cross-Coupling of Arylboronic Acids with Aryl and Vinyl Halides and Triflates under Mild Conditions. J. Am. Chem. Soc. 122, 4020–4028 (2000). [Google Scholar]
- 82.Dreher SD, Dormer PG, Sandrock DL & Molander GA Efficient Cross-Coupling of Secondary Alkyltrifluoroborates with Aryl Chlorides–Reaction Discovery Using Parallel Microscale Experimentation. J. Am. Chem. Soc. 130, 9257–9259 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Li L, Zhao S, Joshi-Pangu A, Diane M. & Biscoe MR Stereospecific Pd-Catalyzed Cross-Coupling Reactions of Secondary Alkylboron Nucleophiles and Aryl Chlorides. J. Am. Chem. Soc. 136, 14027–14030 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Brown HC, Midland MM, & Levy AB, Facile reaction of alkyl-and aryldichloroboranes with organic azides. General stereospecific synthesis of secondary amines. J. Am. Chem. Soc. 95, 2394 – 2396 (1973). [Google Scholar]
- 85.Bagutski V, Elford TG, & Aggarwal VK, Synthesis of Highly Enantioenriched C‐Tertiary Amines From Boronic Esters: Application to the Synthesis of Igmesine. Angew. Chem., Int. Ed. 50, 1080–1083 (2011). [DOI] [PubMed] [Google Scholar]
- 86.Kuduk SD & Skudlarek JW 2-Pyridyloxy-3-substituted-4-nitrile Orexin Receptor Antagonists. WO2014066196A1 (2014). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Experimental data as well as characterization data for all compounds prepared in the course of these studies are provided in the Supplementary Information of this manuscript. Crystallographic data for the structures reported in this Article have been deposited at the Cambridge Crystallographic Data Centre, under deposition numbers CCDC 2062923 (16), 2062924 (20), 2062925 (26) and 2081087 (40-ester, see X-ray Crystallographic Data in Supplementary Information). Copies of the data can be obtained free of charge via https://www.ccdc.cam.ac.uk/structures/.