

Abstract
Unsymmetrical bisacridines (UAs) are a novel potent class of antitumor-active therapeutics. A significant route of phase II drug metabolism is conjugation with glutathione (GSH), which can be non-enzymatic and/or catalyzed by GSH-dependent enzymes. The aim of this work was to investigate the GSH-mediated metabolic pathway of a representative UA, C-2028. GSH-supplemented incubations of C-2028 with rat, but not with human, liver cytosol led to the formation of a single GSH-related metabolite. Interestingly, it was also revealed with rat liver microsomes. Its formation was NADPH-independent and was not inhibited by co-incubation with the cytochrome P450 (CYP450) inhibitor 1-aminobenzotriazole. Therefore, the direct conjugation pathway occurred without the prior CYP450-catalyzed bioactivation of the substrate. In turn, incubations of C-2028 and GSH with human recombinant glutathione S-transferase (GST) P1-1 or with heat-/ethacrynic acid-inactivated liver cytosolic enzymes resulted in the presence or lack of GSH conjugated form, respectively. These findings proved the necessary participation of GST in the initial activation of the GSH thiol group to enable a nucleophilic attack on the substrate molecule. Another C-2028-GSH S-conjugate was also formed during non-enzymatic reaction. Both GSH S-conjugates were characterized by combined liquid chromatography/tandem mass spectrometry. Mechanisms for their formation were proposed. The ability of C-2028 to GST-mediated and/or direct GSH conjugation is suspected to be clinically important. This may affect the patient's drug clearance due to GST activity, loss of GSH, or the interactions with GSH-conjugated drugs. Moreover, GST-mediated depletion of cellular GSH may increase tumor cell exposure to reactive products of UA metabolic transformations.
Keywords: Antitumor agent, Unsymmetrical bisacridine, Metabolic detoxification, Glutathione S-Conjugate, Glutathione S-transferase, Non-enzymatic conjugation
Graphical abstract

Highlights
-
•
We investigated the GSH-mediated metabolic pathway of antitumor bisacridine C-2028.
-
•
Non-enzymatic and GST-catalyzed GSH conjugation of C-2028 was observed.
-
•
The action of human recombinant GSTP1-1 in C-2028 metabolism was proved.
-
•
GSH conjugation occurred without the prior CYP450-mediated activation of C-2028.
-
•
GSH conjugation of C-2028 molecule took place on the system containing nitro group.
1. Introduction
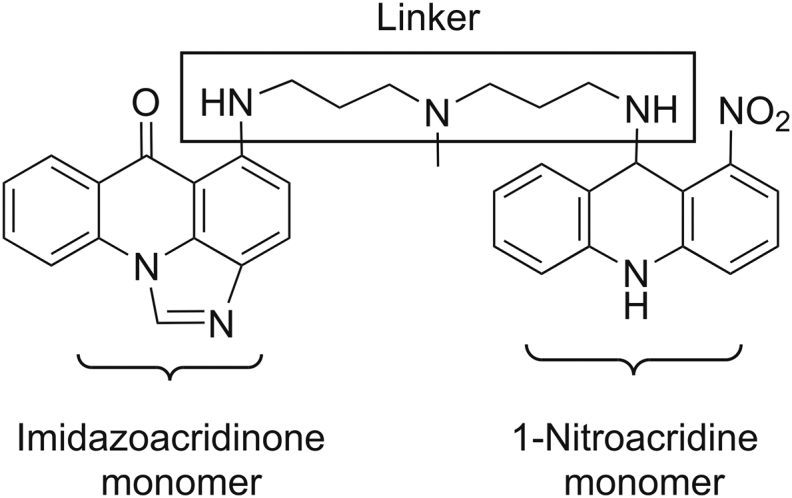
C-2028 (9-{N-[(imidazo[4,5,1-de]-acridin-6-on-5-yl)aminopropyl]-N-methylaminopropylamino}-1′-nitroacridine, Fig. 1) is a representative unsymmetrical bisacridine (UA) developed in our laboratory, patented in Europe [1] and in the United States [2]. This is a completely novel class of potent antitumor agents highly active against many experimental cellular and tumor models (i.e., colon, lung, pancreatic, breast, and prostate) [3,4]. C-2028 molecule consists of imidazoacridinone and 1-nitroacridine monomers, connected by a flexible aminopropyl-N-methylaminopropylamino type linker, which together confer unique physicochemical and biological properties not observed in the case of single monomers [[1], [2], [3], [4]].
Fig. 1.
Schematic representation of the chemical structure of unsymmetrical bisacridine C-2028.
The knowledge about metabolic transformations of a new chemical entity and its affinity to drug-metabolizing enzymes is crucial for explaining its biochemical mechanism of action on the human body [5]. Our previous studies on UA metabolism revealed their slight susceptibility to metabolic transformations with cytochrome P450 (CYP450) isoenzymes (phase I metabolism) both in cell-free and cellular systems [6]. Metabolite profiling of UAs in an electrochemical system indicated that the main metabolism pathway should be the reduction of the nitroaromatic group [7]. However, UA phase II metabolism, which generally serves as a detoxifying step in metabolism of drugs and other xenobiotics as well as endogenous substrates [5], has been studied only to a small extent.
A significant route of drug detoxification in many species is the conjugation of xenobiotic with reduced tripeptide glutathione (GSH; L-γ-glutamyl-L-cysteinyl-glycine) [8]. Due to the high nucleophilic reactivity of the GSH thiol group, this process can be non-enzymatic and/or mediated by various GSH-dependent enzymes [9,10]. On the other hand, depending on the properties of the substrate, GSH conjugation may also play an important role in bioactivation reactions as it can generate toxic conjugates [11]. Thus, the study on GSH conjugation is a crucial factor for the determination of drug therapeutic efficacy and potential toxicity. Glutathione S-transferases (GSTs; EC 2.5.1.18) are a family of multigene and multifunctional dimeric enzymes that catalyze the conjugation of reduced GSH to a wide range of exogenous and endogenous substrates, thereby protecting cellular macromolecules (i.e., proteins and nucleic acids) from attack by reactive electrophilic compounds [12,13]. In humans, various GST isoenzymes are present in almost all tissues with the liver exhibiting the highest cytosolic GST activity level, followed by the kidney, lung, and intestines [12]. However, through direct detoxification, GSTs participate in the development of resistance toward chemotherapeutic agents [13]. Hence, the activity and expression levels of GSTs in various tumor tissues are known to be a key element in the success or failure of chemotherapy treatment. The results of initial studies on the pharmacological activity of UAs indicate that their promising clinical usage is particularly seen in the prevention and/or the treatment of human drug-resistant tumors [4]. This observation may suggest an important role of detoxifying transformations in UA metabolism.
Considering all the above, the present study aimed to investigate the specificity of the conjugation of antitumor bisacridine C-2028 with the reduced form of GSH in the presence or absence of various human and rat liver subcellular fractions, the main stores of GSH-dependent enzymes. We carried out detailed investigations to reveal the role of GSTs in enzymatic synthesis of C-2028-GSH S-conjugate. In particular, the possibility to obtain GSH-related product was explored with human recombinant GSTP1-1 (hGSTP1-1), which is the primary GST isoenzyme expressed in a wide range of sensitive and resistant tumor cells [[14], [15], [16]]. The subsequent studies were performed to show whether GSH conjugation of C-2028 proceeded with or without prior CYP450-mediated activation of the parent compound. The structural characterization of the observed products of enzymatic and non-enzymatic GSH conjugation of C-2028 was conducted by using combined liquid chromatography/tandem mass spectrometry (LC/MS/MS). Molecular mechanisms for the formation of specific mono-GSH S-conjugates were proposed and discussed. In order to estimate the sites and the likelihood of reactivity for C-2028 with GSH, we were supported by XenoSite Reactivity Predictor, a computational model that has the ability to predict preferential reactions with macromolecules, compared to common screening traps [17]. The results regarding a novel GSH-mediated metabolic pathway of C-2028 were reported here for the first time. We believe that knowledge presented herein will contribute to the development of an optimal strategy for the treatment with this antitumor-active agent, including achievement of the metabolic balance between therapeutic efficacy and drug resistance.
2. Materials and methods
2.1. Chemicals
The UA C-2028 (Fig. 1) was synthesized and purified in our laboratory according to the method described in the relevant patents [1,2]. 1-Aminobenzotriazole (1-ABT), 1-chloro-2,4-dinitrobenzene (CDNB), ethacrynic acid (EA, Edecrin, Sigma-Aldrich, St. Louis, MO, USA), formic acid (HCOOH), GSH in reduced form, N-(2-hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid) (Hepes), hydrochloric acid (HCl), magnesium chloride (MgCl2), and N-acetylcysteine (NAC) were purchased from Sigma-Aldrich (St. Louis, MO, USA), while ammonium formate (NH4COOH) was ordered from Fisher Scientific (Loughborough, UK). Acetonitrile (CH3CN), methanol (CH3OH) (both of gradient grade quality for LC), β-nicotinamide adenine dinucleotide 2′-phosphate tetrasodium salt (β-NADPH), and sodium hydroxide (NaOH) were obtained from Merck KGaA (Darmstadt, Germany). All other chemicals and solvents were of the highest quality commercially available. Ultrapure water (conductivity 0.056 μS/cm), used in all the experiments, was passed through a Milli-Q water purification system from Merck KGaA (Darmstadt, Germany).
2.2. Enzymes
Human pooled liver cytosol (HuCyt), mixed gender (protein concentration, 10 mg/mL), rat pooled liver cytosol (RCyt) and microsomes (RLMs) from male Sprague-Dawley rats (protein concentration, 10 mg/mL and 20 mg/mL, respectively), and hGSTP1-1 (0.001 mM), expressed in wheat germ cell-free system, were purchased from Tebu-Bio (Le Perray-en-Yvelines, France).
2.3. Experimental procedures
2.3.1. Enzyme catalysis of C-2028 conjugation with GSH
Human/rat liver cytosolic or microsomal incubations (2 mg/mL protein) were performed with 0.05 mM C-2028, 5 mM GSH, with and without the presence of 1.2 mM NADPH in 0.1 M Hepes buffer solution (pH 7.4) at 37 °C for 0, 30, and 60 min, in a total volume of 55 μL. The MgCl2 solution (0.5 mM) was added for enzyme stimulation. C-2028 metabolism with hGSTP1-1 (10-5 mM) was studied using 0.1 M Hepes buffer solution at pH 6.5 to reduce the rate of non-enzymatic reaction. The three types of control incubations were applied for each enzyme system: the first without test compound, the second without NADPH, and the third without GSH. The incubations were terminated by adding ice-cold 1 M HCl to the incubation mixtures (10:90, V/V). The samples were then vortexed, placed in ice for 10 min, and centrifuged at 10,000 g for 10 min. Aliquots of the supernatants (50 μL) in triplicate were then analyzed directly by reversed-phase LC with UV-vis detection and/or diode array and multiple wavelength detection, and monitored by MS(/MS).
To examine the chemical reactivity of C-2028, GSH in RCyt incubations was replaced by 5 mM NAC. In order to demonstrate the GST-mediated conjugation, the enzymes had been properly inactivated. This was accomplished by two methods: 1) heat inactivation – RCyt fraction was incubated at 65 °C for 15 min before adding to the incubation mixture and 2) introduction of EA (0.5 mM), as a potent reversible GST inhibitor [18], to the incubation mixture. Incubation experiments of C-2028 with RLMs were also performed in the presence of 1 mM 1-ABT, a non-specific CYP450 inhibitor [19], to establish whether GSH-mediated conjugation pathway requires the prior substrate bioactivation by CYP450 enzymes.
CDNB was used as a GST standard substrate to determine enzyme activity in liver matrices. The positive controls of RCyt, HLMs, and RLMs were carried out by incubating 0.1 mM CDNB with 0.5 mM GSH under the aforementioned reaction conditions.
2.3.2. Non-enzymatic synthesis of GSH S-conjugate
The 0.05 mM C-2028 and 5 mM GSH in 0.1 M Hepes buffer solution (pH 7.4 and pH 6.5) were incubated at 37 °C for 0, 1, and 5 h, in a total volume of 55 μL. The incubations were terminated by adding ice-cold 1 M HCl to the incubation mixtures (10:90, V/V). Aliquots of the supernatants (50 μL) in triplicate were analyzed as described above.
2.4. Analytical instrumentation
The reaction mixtures were analyzed directly by reversed-phase LC with electrospray ionization mass spectrometry (ESI-MS). MS/MS mode was used to provide additional structural information for the ions of interest.
LC/MS experiments were carried out on a Nexera-I LC-2040C 3D HPLC system coupled to the LCMS-2020 system through an ESI interface (Shimadzu Corp., Kyoto, Japan). 50 μL of the mixture obtained after enzymatic/non-enzymatic incubations was separated on a reversed-phase Suplex pKb-100 analytical C18 column (Supelco Inc., Bellefonte, PA, USA) with the following dimensions: 250 mm length × 4.6 mm i.d., 5 μm particle size, at room temperature. A gradient at 0.8 mL/min, between solvents A, 0.05 M aqueous NH4COOH buffer (pH 3.4 adjusted with HCOOH), and B, CH3OH, was used for chromatographic separation. A linear gradient from 15% to 85% B in A was kept for 25 min, and then followed by a linear gradient from 80% to 100% B in A for 3 min. A rate of 100% of solvent B was kept for 1.5 min before returning to initial conditions within 0.5 min. These were then kept for 10 min to allow for column equilibration. The eluates were monitored with UV-vis detection and/or diode array and multiple wavelength detection.
LC/MS/MS analyses were performed with an Agilent 6500 Series Accurate-Mass Quadrupole-Time of Flight (Q-TOF) mass spectrometer (Agilent Technologies, Santa Clara, CA, USA) controlled by Agilent MassHunter Workstation software. To ensure accurate mass during the experiment, the mass spectrometer was calibrated daily using a calibration solution (ES-TOF reference mix, Agilent Technologies, Santa Clara, CA, USA). The optimized parameters for the ESI(+)-ion trap MS(/MS) experiments are listed in Table 1.
Table 1.
MS parameters applied in direct LC/MS and LC/Q-TOF-MS/MS experiments for the determination of accurate masses of product ions and getting ion fragmentation patterns.
| Parameter | Value or setting |
|---|---|
| Ion source type | Dual ESI |
| Ion polarity | Positive |
| The range of m/z | 100–1000 |
| MS operating mode | Scan |
| Rate | 1.5 spectra/s |
| Nebulizing gas (N2) pressure | 35 psi |
| Nebulizing gas flow | 1.5 L/min |
| Drying gas (N2) flow | 10 L/min |
| Drying gas temperature | 350 °C |
| Fragmentor | 175 V |
| Skimmer | 45 V |
| OCT 1 RF Vpp | 750 V |
| Capillary voltage | 3500 V |
| MS/MS method | Targeted |
|
|
|
|
|
|
MS: mass spectrometry; LC/MS: liquid chromatography/tandem mass spectrometry; Q-TOF: quadrupole-time of flight; ESI: electrospray ionization; OCT: octopole; RF: radio frequency; Vpp: peak-to-peak value.
2.5. Prediction of chemical reactivity of C-2028 molecule
Simulation of reactivity of C-2028 molecule with GSH (a common trapping agent for electrophiles) was carried out using XenoSite Reactivity Predictor available at https://swami.wustl.edu/xenosite/p/reactivity.
3. Results and discussion
3.1. Enzyme catalysis of C-2028 conjugation with GSH
In this study, antitumor bisacridine C-2028 was profiled for its susceptibility to GSH conjugation in various liver subcellular fractions in the absence or presence of reduced GSH, which was used at a concentration typical for hepatocytes (5 mM). These cell-free incubations were followed by the separation and identification of the metabolic products with the application of an optimized LC/MS(/MS) technique.
C-2028 was initially incubated with the enzymes of HuCyt and RCyt. The representative reversed-phase LC chromatograms in Fig. 2 showed that only a single peak of GSH-related metabolite of C-2028 was detected in GSH-supplemented RCyt incubations but it was not isolated from HuCyt incubations. The peak position of the product eluted at tR = 14.70 min suggested its more polar nature than the parent compound observed at tR = 17.28 min. A high reaction rate of putative GSH S-conjugate formation, which was about 95% of the substrate conversion within 30 min, indicated that this process may be of particular importance in C-2028 metabolism. A small amount of GSH S-conjugate was also formed in the absence of exogenous GSH in control incubations that resulted from the action of GSH naturally present in the commercially available cytosolic fraction. Inter-species variations in the concentrations of drug-metabolizing enzymes can render some metabolic products undetectable [20]. Therefore, no synthesis of GSH S-conjugate of C-2028 in HuCyt may be due to the lack or reduced activity of specific GSH-dependent enzymes involved in conjugation reaction.
Fig. 2.

The representative reversed-phase LC chromatograms (λ = 420 nm) of the reaction mixtures obtained from 30 min incubation of C-2028 (0.05 mM) with GSH (5 mM) in human (inset) and rat pooled liver cytosol (2 mg/mL). The control contained no GSH. GSH: glutathione; RCyt: rat pooled liver cytosol; HuCyt: human pooled liver cytosol.
To examine the chemical reactivity of C-2028 toward nucleophiles, GSH in RCyt incubations was replaced by NAC (5 mM), another common thiol-based trapping agent for electrophiles [21]. However, no conjugation of C-2028 occurred under these conditions (data not shown), which suggested that the observed product was specifically formed only in the reaction with GSH. In order to prove the participation of GSH-dependent enzymes in GSH conjugation reaction, the enzymes of RCyt were properly inactivated in separate experiments. In the presence of heat-inactivated RCyt (65 °C for 15 min) (Fig. 3A) or EA (0.5 mM) (Fig. 3B), a potent reversible GST inhibitor [18], the formation of putative GSH S-conjugate was totally reduced compared to that in control samples. Thus, we demonstrated that cytosolic GSTs mediated in GSH conjugation of C-2028.
Fig. 3.

The representative reversed-phase LC chromatograms (λ = 420 nm) of the reaction mixtures obtained from 30 min incubation of C-2028 (0.05 mM) with GSH (5 mM) in RCyt (2 mg/mL), which was (A) heat-inactivated at 65 °C for 15 min and (B) inactivated by ethacrynic acid (EA, 0.5 mM). The control contained not inactivated cytosolic fraction.
In addition to liver cytosol, the source of GSH-dependent enzymes, including GSTs, is also liver microsomes, which are subcellular fractions derived from the endoplasmic reticulum [9,22]. As shown in Fig. 4A, the product that eluted at the same retention time as in RCyt was also detected in RLMs. Next, purified hGSTP1-1 was also found to catalyze the conjugation reaction (Fig. 4B), but at a much lower level (below 20%) than that observed in the isolated liver subcellular fractions. Nevertheless, this finding supported the role of the GST family of enzymes in the formation of GSH S-conjugate of the studied bisacridine. Similarly, GSTs in RLMs displayed a lower catalytic activity toward GSH S-conjugate formation than enzymes in RCyt (Fig. 5). Finally, we proved that the observed enzymatic GSH conjugation was carried out without prior bioactivation of C-2028 by CYP450s as it was not affected significantly in the presence of NADPH (a cofactor of CYP450 enzymes) as well as the non-selective CYP450 inhibitor 1-ABT [19] (inset in Fig. 5). In both cases, the reaction progress was comparable to that obtained in RLMs, which did not include NADPH or 1-ABT.
Fig. 4.

The representative reversed-phase LC chromatograms (λ = 420 nm) of the reaction mixtures obtained from 30 min incubation of C-2028 (0.05 mM) with GSH (5 mM) (A) in rat liver microsomes (RLMs, 2 mg/mL) and (B) with human recombinant GSTP1-1 (hGSTP1-1) (10-5 mM). The control contained no GSH.
Fig. 5.

Comparison of rates of C-2028-GSH S-conjugate formation in RCyt and RLMs. C-2028 (0.05 mM) was incubated with various rat liver subcellular fractions (2 mg/mL) in the presence of GSH (5 mM) over 0, 30, and 60 min. The inset at the top-left corner shows rates of C-2028-GSH S-conjugate formation in RLMs + GSH incubations supplemented with nicotinamide adenine dinucleotide 2'-phosphate tetrasodium salt (NADPH) (1.2 mM) or 1-aminobenzotriazole (1-ABT) (1 mM). Each bar represents the mean ± standard deviation for three independent experiments.
Studies on drug metabolism play important roles in the optimization of metabolic stability, pharmacokinetic/pharmacodynamic properties, and the safety profiles of drug candidates in drug discovery and development [5]. In this respect, the significance of the above work results from the recognition of novel GSH conjugation reaction of C-2028, which was catalyzed by cytosolic and microsomal GSTs but with no requirement for prior bioactivation by CYP450s. GSTs are extensively distributed in various human tissues, and, in general, GSH conjugation is regarded as a detoxification reaction leading to the formation of chemically less reactive products [8,9]. However, certain human GST-class enzymes exhibit genetic polymorphisms that affect the interpatient variability of the drug clearance [13,23,24]. The polymorphic forms of GSTs are found among specific types of tumors [25,26]. The differential gene expression levels relate, for example, to GSTP1-1 which is reported as the predominant isoform in various human tissues associated with a wide range of sensitive and resistant tumor cells [27]. Thus, the confirmation in our study of the GST-mediated conjugation pathway of C-2028 with GSH is of clinical importance. The key implication will be the dependence of C-2028 clearance of GST activity and GSH stores in the body. In future work, the contributions of specific GST isoforms to UA metabolism will be studied to understand their roles in bisacridine metabolism and disposition in vivo.
3.2. Non-enzymatic synthesis of C-2028-GSH S-conjugate
Non-enzymatic reactions are a widespread and integral part of xenobiotic metabolism [28]. These can occur spontaneously, proceed with the participation of small-molecule compounds or be light-induced. As has been reported for several compounds (i.e., isothiocyanates or catecholamines), the reactive GSH thiol-group, which possesses strong nucleophilic properties, provides the redox potential also for non-enzymatic conjugation [10,29,30].
As illustrated in Fig. 6, chromatographic separation of reaction mixtures obtained after the incubation of C-2028 with reduced GSH under near physiological conditions (at 37 °C and pH 7.4) resulted in the formation of at least two different peaks which intensity increased as the reaction progressed. Their retention time was 18.15 and 19.45 min, respectively. Because they were not observed in GSH-free incubation of C-2028 (control), we considered them to be putative GSH-related products. The products of a non-enzymatic reaction of C-2028 with GSH were detectable just after 1 h incubation and their intensities increased markedly by a change in pH from 6.5 (inset in Fig. 6) to 7.4. It seems that the observed pH-dependent reactivity of C-2028 with GSH could be associated with the changes in protonation states of the UA molecule. Interestingly, no reaction with GSH was detected for C-2045, an analog of C-2028 with a methyl group at para position toward the nitro group (data not shown). Hence, we concluded that a methyl substitution on the aromatic ring could lead to an altered reactivity of the compound with GSH by steric hindrance and/or electron inductive effects.
Fig. 6.

The representative reversed-phase LC chromatograms (λ = 420 nm) of the reaction mixtures obtained from 0, 1, and 5 h incubation of C-2028 (0.05 mM) with GSH (5 mM) at pH 7.4. The control contained no GSH. The inset at the top-left corner shows the LC separation for a mixture of C-2028 (0.05 mM) with GSH (5 mM) after 1 h incubation at pH 6.5.
3.3. Structural characterization of C-2028-GSH S-conjugates
In efforts to know the mechanistic features of GSH-mediated metabolic pathways of antitumor bisacridine C-2028, LC/Q-TOF-MS/MS investigations were performed to characterize the molecular structures of the detected GSH S-conjugates. The results of these analyses are summarized in Fig. 7 and Table 2.
Fig. 7.

The representative ESI(+)-MS/MS fragment spectra of the ions at (A) m/z 846 [M+H]+ and (B) m/z 859 [M+H]+. Proposed structures of the relevant GSH S-conjugates are shown. Dashed lines depict suggested sites of fragmentation (a–f). ESI: electrospray ionization.
Table 2.
Summary of m/z, fragmentations, and retention time for ions attributed to the respective enzymatic and non-enzymatic GSH S-conjugates of C-2028.
| Conjugates | Representative measured m/z [M+H]+ | Molecular formula | Relative mass error a (ppm) | Key MS/MS fragment ions | Retention time (min) |
|---|---|---|---|---|---|
| Enzymatic | 846.3403 | C44H47N9O7S | –1.35 | 212, 347, 485, and 573 | 14.70 |
| Non-enzymatic | 859.3350 | C44H46N10O7S | 0.70 | 586 and 730 | 19.45 |
Exact masses were calculated using Molecular Mass Calculator freeware (v2.02).
The MS/MS spectrum of C-2028-GSH S-conjugate, which was formed in the cytosolic/microsomal fractions and with hGSTP1-1, showed that its accurate mass ([M+H]+ m/z 846.3403) was 260 Da higher than that of C-2028 (theoretical [M+H]+ m/z 586.2561) (Fig. 7A). The observed mass defect suggested an elemental composition of C10H16N2O4S (theoretical molecular weight (MW) 260.0831 Da) consistent with a single molecule of GSH attached to the parent compound lacking the nitro group. Fragmentation of product ion at m/z 846 in positive ion mode gave rise to four fragment ions at m/z 212, 347, 485, and 573. The accurate masses of the proposed structures of these fragments were fully consistent with their calculated masses. The fragment at m/z 573.2430 can be attributed to the structure d with the elemental composition of C34H32N6OS (theoretical [M+H]+ m/z 573.2431), which was the result of the cleavage of the thiol-carbon bond in GSH molecule. In turn, the cleavage of the nitrogen-carbon bond in the acridine moiety led to the formation of the major fragment at m/z 485.1494. For this fragment, we proposed the structure c with the elemental composition of C23H24N4O6S (theoretical [M+H]+ m/z 485.1489). Further fragmentation of the structures c and/or d yielded fragment at m/z 212.0525 with the possible elemental composition of C13H9NS (theoretical [M+H]+ m/z 212.0528). The presence of the above fragment ions indicated that nucleophilic substitution had to occur to the acridine ring; however, mass spectral data did not reveal the exact site of attachment of the GSH molecule. The remaining fragment ion at m/z 347.1874 was characterized as the protonated form of imidazoacridinone monomer with retained aminopropyl-N-methylaminoallyl (-NH(CH2)3N(CH3)CH2CH=CH2) side chain (theoretical [M+H]+ m/z 347.1866). It has been previously identified as a product of C-2028 conversion in an electrochemical system [7].
Among the observed products of non-enzymatic GSH conjugation of C-2028, we have obtained accurate mass and MS/MS ion data only for the peak at tR = 19.45 min. The MS/MS spectrum revealed the product ion at [M+H]+ m/z 859.3350 (Fig. 7B), which was 273 Da higher than that of the substrate. Its fragmentation in positive ion mode led to the major fragments at m/z 586 and 730. On the basis of the accurate mass data, we deduced that these signals might come from structures with the elemental composition of C34H31N7OS (e) and C39H39N9O4S (f), respectively. The ion at m/z 586.2407 (theoretical [M+H]+ m/z 586.2383) was formed due to the fragmentation of the carbon thiol bond of GSH, whereas the fragment ion at m/z 730.2922 (theoretical [M+H]+ m/z 730.2918) was consistent with the neutral loss of anhydroglutamic acid (–129 Da). Thus, we concluded the presence of a single GSH moiety in the molecule of the product ion at m/z 859. Based on the collected values of m/z, the derivative of C-2028 that was conjugated with GSH should have the formula of C34H31N7O (theoretical MW 553.2590 Da). This may correspond to the product with an extra five-membered ring in the molecule of the parent compound. Such a structure was evidenced for a key reactive metabolite of the ledakrin reduction pathway and it was obtained as a result of its incomplete reduction [31].
UV-vis spectroscopic analysis of C-2028-GSH S-conjugates (data not shown) demonstrated that both products absorbed light from 250 to 500 nm and, in contrast to the UV-vis spectrum of C-2028, their first absorbance maximum at about 350 nm was shifted toward higher wavelength values. This may indicate structural changes within one of the chromophore systems while the structure of the dimer has been retained.
Fig. 8 presents the proposed metabolic pathways of enzymatic and non-enzymatic GSH conjugation of C-2028. The sites and the likelihood of reactivity for C-2028 with GSH were estimated by the XenoSite Reactivity Predictor. Accordingly, the 1-nitroacridine moiety seems to be the likely site of GSH conjugation, mainly due to the presence of electronegative nitrogen atoms of the amino and nitro functional groups that decrease the electron density in the aromatic ring. In particular, the redox properties of the latter are usually responsible for the pharmacological and/or toxicological properties of many compounds of biological significance [32].
Fig. 8.

Proposed GSH-mediated metabolic pathways of C-2028. The frame contains C-2028 structure with an indication of sites susceptible to conjugation with GSH. Simulation of molecule reactivity was carried out using the XenoSite Reactivity Predictor available at https://swami.wustl.edu/xenosite/p/reactivity. GST: glutathione S-transferase.
For the enzymatic synthesis of GSH S-conjugate of C-2028, we postulate the GST-catalyzed nucleophilic aromatic substitution with GSH. This reaction pathway is quite common for many GST substrates [[33], [34], [35]]. It is also interesting to note that the mechanisms of GST-mediated conjugations and reductions are extremely variable and highly depend on the GST class and on the substrate [9]. Biochemical and structural analyses have shown that the proteins of GST family, including GSTP1-1 class, exist as dimers [22]. Each subunit of the dimer contains two ligand binding sites: the GSH binding site (G-site), which is highly specific for GSH, and a hydrophobic substrate binding site, which can accept a wide variety of hydrophobic compounds. The reaction mechanism reported by Oakley et al. [36] assumes that two independent structures of the same compound may interact with different enzyme subunits. In view of the above, C-2028 is likely to bind to the one of GST active sites with the imidazoacridinone monomer, whereas the second 1-nitroacridine monomer is simultaneously donated to the other GST active site located in the other subunit of the enzyme. Of the two binding sites of GST enzyme, the high substrate specificity of the G-site indicates that this site is a suitable binding site for GST inhibitors [22]. Our early findings revealed that C-1311 (5-diethylaminoethylamino-8-hydroxyimidazoacridinone) acts as a potent inhibitor of GST (unpublished data). If this were the case for UAs, it would mean the possibility to enhance the effect of these agents in antitumor therapy. The first step of the proposed reaction mechanism seems to be GST-mediated activation of the GSH molecule to the reactive thiolate anion (GS-), which performs a nucleophilic attack on the aromatic moiety of C-2028 that contains the nitro group. This creates Meisenheimer complex as a transition state (with the negative charge delocalized over the entire acridine ring), from which the nitro group may be easily removed in the form of nitrite anion (NO2-) [37,38]. Therefore, this did not require an earlier reduction of the nitro group, which would explain the observed lack of CYP450 involvement in the process. Moreover, the results of our studies also showed that 1-nitroacridine did not give any enzymatic GSH S-conjugate, while in the case of 1-nitro-9-aminoacridine and 1-nitro-9-methylaminoacridine, such products were detected (data not shown). Thus, the presence of a nitrogen atom from an amino group may be crucial here. Nucleophilic substitution of the nitroaromatic group by GSH can be facilitated because of the supportive role of amino acid moieties located in the active site of the GST enzyme and relatively low bond dissociation energy which is 298.0 kJ/mol [39].
For non-enzymatic GSH S-conjugate of C-2028, we considered the reductive modification of the parent molecule that gave rise to a five-membered ring at positions 1 and 9 of the acridine core. The proximity of nitrogen atoms at these positions seems to enable fast intramolecular cyclization, through N-hydroxy intermediate, to adopt such a final structure, which promoted further GSH conjugation via a thiol linkage. The substitution of electron-donating and sterically hindered methyl group at para position toward the nitro group (in bisacridine C-2045) produced an enhancement of the push-pull effect in the molecule and thus blocked the spontaneous conjugation reaction. Based on this observation, we conclude that ortho position toward the nitro group may be the most potential site of the nucleophilic attack of the GSH molecule.
Although we demonstrated the existence of GSH S-conjugates of C-2028 and GST-catalyzed reaction, these findings do not suggest a general reactivity of 1-nitroacridine monomer towards biomacromolecules. Further studies will be required to gain insights into the structural features of UAs that render them susceptible to GSH conjugation.
4. Conclusions
The studies conducted in this work were aimed, for the first time, to investigate the GSH-mediated metabolic pathway of antitumor bisacridine C-2028. For this purpose, we treated the substrate drug with GSH in the presence or absence of human/rat liver subcellular fractions, which are the main resources of GSH-dependent enzymes in the body. Our results showed that C-2028 was conjugated with GSH either non-enzymatically or through the catalytic action of cytosolic and microsomal GSTs to give two various GSH-related products. In addition, C-2028 was also proved to be a substrate for hGSTP1-1. Furthermore, we excluded prior CYP450-mediated activation of the parent compound for the formation of GSH S-conjugate. Finally, we proposed molecular mechanisms leading to the observed GSH S-conjugates of C-2028. We conclude that the modifications of the C-2028 molecule were located in the ring system containing the nitro group. The findings regarding a novel GSH-mediated metabolic pathway of C-2028 may contribute to the development of an optimal treatment strategy with this antitumor agent, including achievement of the metabolic balance between therapeutic efficacy and drug resistance. These also provide the background for future research that should include investigations of UA metabolism in tumor cell lines with differentiated GSH concentration or activity of various GSTs.
Declaration of competing interest
The authors declare that there are no conflicts of interest.
Acknowledgments
The authors wish to thank Dr. Ewa Paluszkiewicz (Department of Pharmaceutical Technology and Biochemistry, Faculty of Chemistry, Gdańsk University of Technology, Gdańsk, Poland) for the synthesis of the C-2028 compound performed according to the procedures described in patents. We are grateful to Dr. Weronika Hewelt-Belka (Department of Analytical Chemistry, Faculty of Chemistry, Gdańsk University of Technology, Gdańsk, Poland) for her help in the field of MS(/MS) analysis. Acknowledgments are also given to the Shim-pol A.M. Borzymowski Company (Warsaw, Poland) for supplying the Nexera-I LC-2040C 3D and LCMS-2020 systems, and for their technical assistance.
Footnotes
Parts of this work were presented at the 3rd Congress of Polish Biosciences BIO2018 (Gdańsk, Poland, 2018).
Peer review under responsibility of Xi’an Jiaotong University.
References
- 1.J.K. Konopa, B. Horowska, E.M. Paluszkiewicz, et al., Inventors; Asymmetric bis-acridines with antitumour activity and use thereof, European patent: EP 3070078 A1. 4 October 2017.
- 2.J.K. Konopa, B. Horowska, E.M. Paluszkiewicz, et al., Asymmetric bis-acridines with antitumour activity and their uses, United States patent: US10202349B2. 2 December 2019.
- 3.Pilch J., Matysiak-Brynda E., Kowalczyk A., et al. New unsymmetrical bisacridine derivatives non-covalently attached to quaternary quantum dots improve cancer therapy via enhancing cytotoxicity towards cancer cells and protecting normal cells. ACS Appl. Mater. Interfaces. 2020;12:17276–17289. doi: 10.1021/acsami.0c02621. [DOI] [PubMed] [Google Scholar]
- 4.Paluszkiewicz E., Horowska B., Borowa-Mazgaj B., et al. Design, synthesis and high antitumor potential of new unsymmetrical bisacridine derivatives towards human solid tumors, specifically pancreatic cancers and their unique ability to stabilize DNA G-quadruplexes. Eur. J. Med. Chem. 2020;204:112599. doi: 10.1016/j.ejmech.2020.112599. [DOI] [PubMed] [Google Scholar]
- 5.Zhang Z., Tang W. Drug metabolism in drug discovery and development. Acta Pharm. Sin. B. 2018;8:721–732. doi: 10.1016/j.apsb.2018.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mieszkowska A., Nowicka A.M., Kowalczyk A., et al. Metabolic profiles of new unsymmetrical bisacridine antitumor agents in electrochemical and enzymatic noncellular systems and in tumor cells. Pharmaceuticals (Basel) 2021;14:317. doi: 10.3390/ph14040317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Potęga A., Paczkowski S., Paluszkiewicz E., et al. Electrochemical simulation of metabolic reduction and conjugation reactions of unsymmetrical bisacridine antitumor agents, C-2028 and C-2053. J. Pharm. Biomed. Anal. 2021;179:113970. doi: 10.1016/j.jpba.2021.113970. [DOI] [PubMed] [Google Scholar]
- 8.Lushchak V.I. Glutathione homeostasis and functions: potential targets for medical interventions. J. Amino Acids. 2012;2012 doi: 10.1155/2012/736837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deponte M. Glutathione catalysis and the reaction mechanisms of glutathione-dependent enzymes. Biochim. Biophys. Acta. 2013;1830:3217–3266. doi: 10.1016/j.bbagen.2012.09.018. [DOI] [PubMed] [Google Scholar]
- 10.Romański M., Główka F.K. In vitro study of the enzymatic and nonenzymatic conjugation of treosulfan with glutathione. Eur. J. Drug Metab. Pharmacokinet. 2019;44:653–657. doi: 10.1007/s13318-019-00555-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van Bladere P.J. Glutathione conjugation as a bioactivation reaction. Chem. Biol. Interact. 2000;129:61–76. doi: 10.1016/s0009-2797(00)00214-3. [DOI] [PubMed] [Google Scholar]
- 12.Hayes J.D., Flanagan J.U., Jowsey I.R. Glutathione transferases. Annu. Rev. Pharmacol. Toxicol. 2005;45:51–88. doi: 10.1146/annurev.pharmtox.45.120403.095857. [DOI] [PubMed] [Google Scholar]
- 13.Di Pietro G., Magno L.A., Rios-Santos F. Glutathione S-transferases: an overview in cancer research. Expet Opin. Drug Metabol. Toxicol. 2010;6:153–170. doi: 10.1517/17425250903427980. [DOI] [PubMed] [Google Scholar]
- 14.Tew K.D., Monks A., Barone L., et al. Glutathione-associated enzymes in the human cell lines of the national cancer institute drug screening program. Mol. Pharmacol. 1996;50:149–159. [PubMed] [Google Scholar]
- 15.Bulus H., Oguztuzun S., Güler Simsek G., et al. Expression of CYP and GST in human normal and colon tumor tissues. Biotech. Histochem. 2019;94:1–9. doi: 10.1080/10520295.2018.1493220. [DOI] [PubMed] [Google Scholar]
- 16.Kural C., Kaya Kocdogan A., Simsek G.G., et al. Glutathione S-transferases and cytochrome P450 enzyme expression in patients with intracranial tumors: preliminary report of 55 patients. Med. Princ. Pract. 2019;28:56–62. doi: 10.1159/000494496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hughes T.B., Miller G.P., Swamidass S.J. Site of reactivity models predict molecular reactivity of diverse chemicals with glutathione. Chem. Res. Toxicol. 2015;28:797–809. doi: 10.1021/acs.chemrestox.5b00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ploemen J.H., van Ommen B., van Bladeren P.J. Inhibition of rat and human glutathione S-transferase isoenzymes by ethacrynic acid and its glutathione conjugate. Biochem. Pharmacol. 1990;40:1631–1635. doi: 10.1016/0006-2952(90)90465-w. [DOI] [PubMed] [Google Scholar]
- 19.Emoto C., Murase S., Sawada Y., et al. In vitro inhibitory effect of 1-aminobenzotriazole on drug oxidations catalyzed by human cytochrome P450 enzymes: a comparison with SKF-525A and ketoconazole. Drug Metab. Pharmacokinet. 2003;18:287–295. doi: 10.2133/dmpk.18.287. [DOI] [PubMed] [Google Scholar]
- 20.Issa N.T., Wathieu H., Ojo A., et al. Drug metabolism in preclinical drug development: a survey of the discovery process, toxicology, and computational tools. Curr. Drug Metabol. 2017;18:556–565. doi: 10.2174/1389200218666170316093301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aldini G., Altomare A., Baron G., et al. N-Acetylcysteine as an antioxidant and disulphide breaking agent: the reasons why. Free Radic. Res. 2018;52:751–762. doi: 10.1080/10715762.2018.1468564. [DOI] [PubMed] [Google Scholar]
- 22.Allocati N., Masulli M., Di Ilio C., et al. Glutathione transferases: substrates, inhibitors and pro-drugs in cancer and neurodegenerative diseases. Oncogenesis. 2018;7:8. doi: 10.1038/s41389-017-0025-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McIlwain C.C., Townsend D.M., Tew K.D. Glutathione S-transferase polymorphisms: cancer incidence and therapy. Oncogene. 2006;25:1639–1648. doi: 10.1038/sj.onc.1209373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.den Braver M.W., Zhang Y., Venkataraman H., et al. Simulation of interindividual differences in inactivation of reactive para-benzoquinone imine metabolites of diclofenac by glutathione S-transferases in human liver cytosol. Toxicol. Lett. 2016;255:52–62. doi: 10.1016/j.toxlet.2016.05.015. [DOI] [PubMed] [Google Scholar]
- 25.Hayes J.D., Strange R.C. Glutathione S-transferase polymorphisms and their biological consequences. Pharmacology. 2000;61:154–166. doi: 10.1159/000028396. [DOI] [PubMed] [Google Scholar]
- 26.Okat Z. Clinical importance of glutathione-S-transferase enzyme polymorphisms in cancer. Int. Phys. Med. Rehab. J. 2018;3:491–493. [Google Scholar]
- 27.Bocedi A., Noce A., Marrone G., et al. Glutathione transferase P1-1 an enzyme useful in biomedicine and as biomarker in clinical practice and in environmental pollution. Nutrients. 2019;11:1741. doi: 10.3390/nu11081741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Keller M.A., Piedrafita G., Ralser M. The widespread role of non-enzymatic reactions in cellular metabolism. Curr. Opin. Biotechnol. 2015;34:153–161. doi: 10.1016/j.copbio.2014.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kolm R.H., Danielson U.H., Zhang Y., et al. Isothiocyanates as substrates for human glutathione transferases: structure-activity studies. Biochem. J. 1995;311:453–459. doi: 10.1042/bj3110453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baez S., Segura-Aguilar J., Widersten M., et al. Glutathione transferases catalyse the detoxication of oxidized metabolites (o-quinones) of catecholamines and may serve as an antioxidant system preventing degenerative cellular processes. Biochem. J. 1997;324:25–28. doi: 10.1042/bj3240025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gorlewska K., Mazerska Z., Sowiński P., et al. Products of metabolic activation of the antitumor drug Ledakrin (Nitracrine) in vitro. Chem. Res. Toxicol. 2001;14:1–10. doi: 10.1021/tx000081c. [DOI] [PubMed] [Google Scholar]
- 32.Olender D., Żwawiak J., Zaprutko L. Multidirectional efficacy of biologically active nitro compounds included in medicines. Pharmaceuticals (Basel) 2018;11:54. doi: 10.3390/ph11020054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oakley A. Glutathione transferases: a structural perspective. Drug Metab. Rev. 2011;43:138–151. doi: 10.3109/03602532.2011.558093. [DOI] [PubMed] [Google Scholar]
- 34.Angelucci F., Baiocco P., Brunori M., et al. Insights into the catalytic mechanism of glutathione S-transferase: the lesson from Schistosoma haematobium. Structure. 2005;13:1241–1246. doi: 10.1016/j.str.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 35.Ji X., Pal A., Kalathur R., et al. Structure-based design of anticancer prodrug PABA/NO, Drug Des. Devel. Ther. 2008;2:123–130. doi: 10.2147/dddt.s3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oakley A.J., Lo Bello M., Nuccetelli M., et al. The ligandin (non-substrate) binding site of human pi class glutathione transferase is located in the electrophile binding site (H-site) J. Mol. Biol. 1999;291:913–926. doi: 10.1006/jmbi.1999.3029. [DOI] [PubMed] [Google Scholar]
- 37.Rickert D.E. Metabolism of nitroaromatic compounds. Drug Metab. Rev. 1987;18:23–53. doi: 10.3109/03602538708998299. [DOI] [PubMed] [Google Scholar]
- 38.Rusinov V.L., Sapozhnikova I.M., Ulomskii E.N., et al. Nucleophilic substitution of nitro group in nitrotriazolotriazines as a model of potential interaction with cysteine-containing proteins. Chem. Heterocycl. Compd. (N. Y.) 2015;51:275–280. [Google Scholar]
- 39.Vlasov V.M. Nucleophilic substitution of the nitro group, fluorine and chlorine in aromatic compounds. Russ. Chem. Rev. 2003;72:681–703. [Google Scholar]