

Abstract
Ponesimod is a sphingosine 1‐phosphate (S1P) receptor (S1PR) modulator that was recently approved for treating relapsing forms of multiple sclerosis (MS). Three other FDA‐approved S1PR modulators for MS—fingolimod, siponimod, and ozanimod—share peripheral immunological effects via common S1P1 interactions, yet ponesimod may access distinct central nervous system (CNS) mechanisms through its selectivity for the S1P1 receptor. Here, ponesimod was examined for S1PR internalization and binding, human astrocyte signaling and single‐cell RNA‐seq (scRNA‐seq) gene expression, and in vivo using murine cuprizone‐mediated demyelination. Studies confirmed ponesimod’s selectivity for S1P1 without comparable engagement to the other S1PR subtypes (S1P2,3,4,5). Ponesimod showed pharmacological properties of acute agonism followed by chronic functional antagonism of S1P1. A major locus of S1P1 expression in the CNS is on astrocytes, and scRNA‐seq of primary human astrocytes exposed to ponesimod identified a gene ontology relationship of reduced neuroinflammation and reduction in known astrocyte disease‐related genes including those of immediate early astrocytes that have been strongly associated with disease progression in MS animal models. Remarkably, ponesimod prevented cuprizone‐induced demyelination selectively in the cingulum, but not in the corpus callosum. These data support the CNS activities of ponesimod through S1P1, including protective, and likely selective, effects against demyelination in a major connection pathway of the brain, the limbic fibers of the cingulum, lesions of which have been associated with several neurologic impairments including MS fatigue.
Keywords: GPCR, lysophospholipid, multiple sclerosis, neuroinflammation, sphingolipid
Abbreviations
- CC
corpus callosum
- Cg
cingulum
- CIR
compensated interferometric reader
- CNS
central nervous system
- DEGs
differentially expressed genes
- FCM
flow cytometry
- GO
gene ontology
- GPCR
G protein‐coupled receptor
- MS
multiple sclerosis
- MT
metallothionein
- S1P
sphingosine 1‐phosphate
- S1PR
S1P receptor
- scRNA‐seq
single‐cell RNA‐seq
- UMAP
uniform manifold approximation and projection
- UMI
unique molecular identity
1. INTRODUCTION
Sphingosine 1‐phosphate (S1P) is a bioactive lysophospholipid whose effects are mediated by a family of cognate G protein‐coupled receptors (GPCRs). 1 , 2 Five S1P receptor (S1PR) subtypes 3 have been identified—S1P1,2,3,4,5—combinations of which have been successfully targeted for the production of four FDA‐approved oral immune cell trafficking inhibitors to treat relapsing forms of the demyelinating central nervous system (CNS) disease, multiple sclerosis (MS). 2 , 4
Fingolimod (FTY720), 5 approved in 2010, 6 was the first S1PR modulator and oral therapy for MS. It is a pro‐drug that requires endogenous phosphorylation (by sphingosine kinases) to produce fingolimod phosphate (fingolimod‐P) that can then engage four of the five receptor subtypes (all except S1P2) with high affinity. 6 , 7 , 8 , 9 Its receptor mechanism of action is characterized by initial S1PR agonism, followed by functional antagonism with prolonged fingolimod‐P (FTY720‐P) exposure, which effectively eliminates S1PRs, especially S1P1, from the cell surface via irreversible receptor internalization. 6 , 8 , 10 In MS and related animal models, this alters immune cell trafficking whereby disease‐promoting cells, particularly of the adaptive immune system, are sequestered within secondary lymphoid organs to reduce pathogenic entry into the brain. 11 In addition, direct CNS effects have also been implicated, particularly involving astrocytes 12 as well as oligodendrocytes, resident microglia, and likely other cell types that can express one or more S1PR subtypes. 2
Three second‐generation S1PR modulators have more recently been approved for MS and all are distinct from fingolimod in targeting a subset of S1PR subtypes. Siponimod 13 (MAYZENT®, approved in 2019) and ozanimod 14 (ZEPOSIA®, approved in 2020) engage S1P1 and S1P5; siponimod is a direct S1PR modulator whereas ozanimod involves metabolism in humans for receptor engagement. 15 The third approved agent is ponesimod 16 (PONVORY™; approved in 2021) that has been reported to show >10‐fold selectivity for S1P1 as compared to other S1PRs. 17
All four agents met their clinical trial primary endpoint of reducing MS relapses and shared S1P1 engagement. Since ponesimod is only supposed to engage S1P1, it provided an opportunity to examine its mechanistic effects as a mono‐specific S1P1 receptor modulator beyond the previously reported peripheral immunological effects. 18 Other effects included those relevant to ponesimod clinical trial data (OPTIMUM Phase 3 study) 16 that achieved a novel secondary endpoint of reduced MS fatigue. Independent examination of ponesimod was therefore pursued for receptor selectivity, human astrocyte signaling, single‐cell gene expression, and an animal model of cuprizone‐induced demyelination that might elucidate the mechanisms underlying ponesimod efficacy signals related to MS fatigue.
2. MATERIALS AND METHODS
2.1. S1PR modulators
Ponesimod was kindly provided by Janssen Pharmaceuticals Corporation of Johnson & Johnson or obtained from APExBIO (Boston, MA; Cat #B7809). Other S1PR modulators used in this study were S1P (Avanti Polar Lipids, Alabaster, AL; Cat #860429P), fingolimod‐P (Cayman, Ann Arbor, MI; Cat #10008639), and ozanimod and siponimod (Selleck Chemicals, Pittsburgh, PA; Cat #S7952 and S7179, respectively).
2.2. Cells
Primary human astrocytes were obtained from Lonza (Lonza, Basel, Switzerland; Cat #CC‐2565) and cultured with AGM™ Astrocyte Basal Medium (Lonza, Cat #CC‐3187) containing AGM™ SingleQuots Supplements (Lonza, Cat #CC‐4123). HA‐tagged S1PR‐overexpressing C6 glioblastoma cells were established by the methods previously described 19 and maintained in DMEM high glucose (Thermo Fisher Scientific, Waltham, MA; Cat# 11965) containing 10% FBS (Gemini Bio Products, West Sacramento, CA; Cat #100‐500) and penicillin–streptomycin (Thermo Fisher Scientific, Cat #10378016).
2.3. Internalization assay
S1PR‐expressing C6 cells were acutely stimulated with 1 µM S1P or ponesimod for 1 h to test short‐term effects. For wash‐out effect experiments, cells were stimulated with 1 µM S1P, ponesimod, or fingolimod‐P for 4 h, followed by washing out the compounds, medium replacement to 0.1% BSA‐DMEM, and incubation for 18 h. Cells were then stained with primary (anti‐HA antibody, clone 3F10; Millipore Sigma, Burlington, MA; Cat #12158167001) and secondary Abs (phycoerythrin‐labeled anti‐rat IgG secondary Ab; BioLegend, San Diego, CA; Cat #405406), followed by flow cytometry (FCM) analyses using a Novocyte flow cytometer (Agilent, Santa Clara, CA). Human primary astrocytes were transfected with pcDNA3.1 harboring a S1P1‐EGFP fusion construct by Lipofectamine 2000 (Thermo Fisher Scientific, Cat #11668030) and cultured overnight. Cells were stimulated with 1 µM ponesimod or fingolimod‐P for 1 h and fixed with 2% paraformaldehyde. Cells were imaged using a Keyence BZ‐8100 microscope (Osaka, Japan).
2.4. Ca2+ mobilization assay
Human primary astrocytes were plated onto 384 well plates (Greiner, San Diego, CA; Cat #781091) at 10 000 cells/well on day 0. Media were replaced with FreeStyle™ 293 expression medium on day 1 and cultured for 18 h. On day 2, cells were stained with FLIPR Calcium 4 Assay Kit (Molecular Devices, San Jose, CA; Cat #R8141) for 1 h in the presence or absence of S1PR modulators. Intracellular Ca2+ mobilization was monitored with a Hamamatsu FDSS7000.
2.5. cAMP AlphaScreen assay
Detection of forskolin (FSK)‐induced cAMP inhibition by S1PR compounds in human primary astrocytes was performed using the AlphaScreen cAMP kit (PerkinElmer, Cat #6760635), according to the manufacturer’s instructions. Briefly, primary human astrocytes were harvested and resuspended in stimulation buffer (HBSS, 0.5 mM 3‐isobutyl‐1‐methylxanthine [IBMX; Sigma‐Aldrich, St. Louis, MO; Cat #I5879], 5 mM HEPES, 0.1% BSA) to a final concentration of 1 × 104 cells/µl. Astrocytes and anti‐cAMP AlphaScreen acceptor bead mixes were prepared for a final concentration of 5000 cells/well on a white 384 well microplate (PerkinElmer #6007290). Biotinylated‐cAMP and streptavidin donor bead mixes were prepared in lysis buffer (5 mM HEPES, 0.1% BSA, 0.3% Tween‐20, pH 7.4). Final concentrations of acceptor and donor beads used were according to the manufacturer’s instructions. For Gαi‐coupled agonist mode, S1PR compounds (S1P, ponesimod, FTY720‐P) were added to each well at the indicated final concentrations, along with FSK (Sigma‐Aldrich, Cat #344270; 10 µM final concentration), and incubated for 30 min at room temperature. Following the addition of biotin‐cAMP/streptavidin donor beads in lysis buffer, the mixture was incubated in the dark for 1 h at room temperature. The AlphaScreen signal (light emission at 570 nm) was then measured, followed by excitation at 680 nm using the SpectraMax i3 multi‐modal plate reader (Molecular Devices). To measure functional antagonism, human primary astrocytes were treated with ponesimod and fingolimod‐P (10 µM final concentration) for 18 h prior to stimulation with S1P, as described above. The data were baseline corrected and normalized to the S1P response within each experiment and three parameter concentration‐response curves were fitted using GraphPad Prism (GraphPad Software Inc, San Diego, CA).
2.6. Binding assay
A bioanalytical technique, named compensated interferometric reader (CIR), 19 , 20 was utilized to measure the binding affinity between S1P1 versus ponesimod or siponimod. Briefly, HA‐tagged S1P1‐overexpressing and vector transfected C6 glioblastoma cells were sonicated to prepare S1P1 and vector nanovesicles that were mixed with an increasing concentration of compounds and incubated for 1 h. The assay was analyzed in CIR to measure the binding of compounds to S1P1/vector nanovesicles compared to a reference (no vesicles + compounds) solution. The specific binding was obtained by subtracting the CIR signal of control nanovesicles (non‐specific) from the S1P1 nanovesicles (total).
2.7. Single‐cell RNA‐seq
Human primary astrocytes were plated on 6 cm dishes at 500 000 cells. Cells were stimulated with or without cytokine mixture (25 ng/ml each; TNF‐α, IFN‐γ, IL‐17, IL‐1β, and IL‐6) in the presence or absence of 1 µM ponesimod for 24 or 48 h. Cells were detached and resuspended with PBSE + BSA to obtain around 1000 cells/µl, and the counts were confirmed on a Countess™ II (Thermo Fisher Scientific). Single‐cell capture (target capture of 10 000 cells per sample) and library preparation was conducted using Chromium Single Cell 3′ GEM, Library & Gel Bead Kit v3 (10× Genomics, Pleasanton, CA; Cat #PN‐1000075) and Chromium Chip B Single Cell Kit (10× Genomics, Cat #PN‐1000074) according to the manufacturer’s instructions. Single‐nucleus libraries were sequenced on the Illumina NovaSeq 6000 machine at GENEWIZ, Inc. (La Jolla, CA). Raw FASTQ files were input into the cellranger count pipeline (Cell Ranger V4.0.0, 10× Genomics) to align reads to the GRCh38 human genome. After stringent filtering steps, a total of 49 706 high‐quality cells from six experimental conditions (non‐treated control, ponesimod for 48 h, cytokines for 24 and 48 h, and cytokine + ponesimod for 24 and 48 h). Unique molecular identity (UMI) raw count matrices from individual samples were normalized using a regularized negative binomial regression method 21 to regress out mitochondrial count fractions and then integrated into one combined dataset using Seurat V3.1 22 in R. DEGs (differentially expressed genes; FDR‐adjusted p < .05, log2[fold change] > 1.1) in each cluster were identified by fitting a hurdle model in MAST. 23 Pathway analyses were executed in the Reactome Pathway Database https://reactome.org/ or Metascape https://metascape.org/ and the results are provided in Tables S3 and S5. BAM files, unprocessed gene‐UMI matrices, and cell‐level clustering annotation were deposited into the Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/). The accession number is GSE171684.
2.8. Mice
All animal procedures were conducted in accordance with Institutional Animal Care and Use Committee guidelines of the Sanford Burnham Prebys Medical Discovery Institute. C57BL/6J 8‐week‐old male mice were maintained on a diet of 0.2% cuprizone (Bis(cyclohexanone) oxaldihydrazone 98%; Thermo Fisher Scientific, Cat #370‐81‐0) for 5–6 weeks. A suspension of ponesimod (30 mg/kg) in vehicle (0.25% methylcellulose [400 cp] with 0.05% polysorbate 80) was prepared weekly after stirring overnight at room temperature and aliquoted accordingly to store at 4°C. Vehicle or ponesimod were delivered orally twice a day (8 h apart) by re‐equilibrating the suspension upon stirring for 30 min before the procedure. The ponesimod dose, frequency, delivery, and formulation was based on a prior study published at ACTRIMS.
2.9. Perfusion fixation and cryoprotection
Mice were anesthetized with isoflurane (VetOne, Cat #502017), perfused with 1× PBS and fixed with 10% NBF (Thermo Scientific, Cat #5701) to isolate the brain (cortices) and fixed in 10% NBF for 2 days at room temperature. They were then transferred to 15% sucrose/PBS (2 days), followed by immersion in 30% sucrose (2 days). These fixed and cryoprotected brains were wiped off and embedded in optimum cutting temperature tissue embedding medium Tissue‐Plus™ (Fisher Scientific, Cat #4586) and kept frozen at −20°C.
2.10. Cryosectioning and staining
The coronal sections (30 µm; Leica cryostat CM3050S) were cut at the level of bregma −1.58 mm and beyond. The sections were mounted onto gelatin‐coated slides and black gold II staining was performed (Biosensis, Thebarton, SA, Australia; Cat #TR‐100‐BG) per manufacturer’s instructions to estimate myelination status. Free floating immunofluorescence staining was performed to detect oligodendrocyte progenitor cells (OPCs) and microglia. Briefly, after washing the 30 µm brain sections in 1× PBS, they were incubated with the primary antibody (goat anti‐Olig2 [R&D Systems, Minneapolis, MN; Cat #AF2418], rabbit anti‐Iba1 [Wako, Richmond, VA; Cat #019‐1974]) in 5% normal horse serum (Vector Laboratories, Burlingame, CA; Cat #S‐2000)/0.3% Tx‐100 (Sigma, Cat #T8787), or normal goat serum (Vector Laboratories, Cat #S‐1000) at room temperature, overnight. The tissue was washed thrice in PBS the next day for 5 min each and incubated with the corresponding secondary antibody (Alexa Fluor 568 donkey anti goat Ab, Thermo Fisher Scientific, Cat #A‐11057; Alexa Fluor 488 goat anti rabbit Ab, Thermo Fisher Scientific, Cat #A‐11008) in 0.3% Tx‐100/PBS for 1.5 h at room temperature in the dark. Care was taken to minimize exposure to light from this step onward. After washing thrice in PBS, the sections were mounted onto gelatin‐coated slides and cover slipped with mounting medium containing DAPI (Vectashield, Cat #H‐1500).
2.11. Imaging and quantitation
Imaging was performed using a Keyence microscope (BZ‐X‐800 series) and signals were quantified using Image J software. A constant region of interest was maintained throughout to quantify the signal in the midline of the corpus callosum or cingulum regions unless otherwise noted.
2.12. Statistical analyses
Results were expressed as means ± SEM or SD. As appropriate, data were analyzed statistically by Student’s t test, one‐way ANOVA, and Kruskal–Wallis test with Dunn’s multiple comparisons test. GraphPad Prism 9 software was used for calculating statistics.
3. RESULTS
3.1. Ponesimod induces sustained S1P1 internalization
The effect of ponesimod on S1PR internalization was analyzed by flow cytometry (FCM) using C6 glioma cell lines that heterologously express HA‐tagged human S1PRs (S1P1, S1P2, S1P3, S1P4, and S1P5‐C6 cells). S1P significantly reduced cell surface expression of S1P1, S1P2, S1P3, and S1P5 (Figure 1A,B). S1P4 and S1P5 were relatively less responsive to S1P‐dependent internalization than other S1PRs. In contrast, acute ponesimod exposure only reduced the cell surface expression of S1P1, and the expression of other S1PRs remained unchanged (Figure 1A,B). These results support previous reports of ponesimod’s selectivity against S1P1. 24 The rapid S1P1 internalization induced by ponesimod and fingolimod‐P was visualized by transfecting a S1P1‐EGFP fusion gene into human primary astrocytes (Figure 1C), which was consistent with prior analyses of fingolimod‐induced S1P1 internalization on primary mouse astrocytes. 12
FIGURE 1.
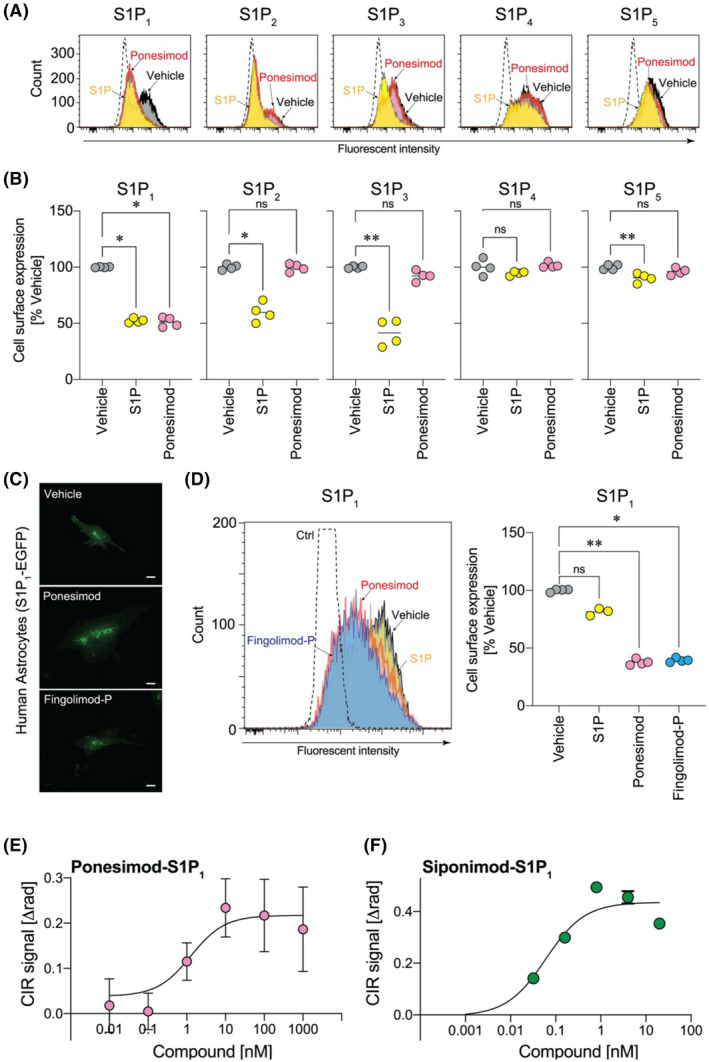
Ponesimod selectively induces S1P1 internalization. (A, B) Short‐term effects of ponesimod exposure in C6 glioma cells. (A) Flow cytometry (FCM) histograms for S1PR‐C6 cells stimulated with S1P (1 µM) or ponesimod (1 µM). Dotted‐line, negative control vector transfected C6 cells. Data are representative of two independent experiments with similar results. (B) Receptor expression levels normalized to vehicle treated group. *p < .05, **p < .01 by one‐way ANOVA with Kruskal–Wallis test. n = 4 pooled from two independent experiments. (C) Short‐term effects of ponesimod (1 µM) exposure in primary human astrocytes transfected with S1P1‐EGFP. Scale bar = 20 µm. (D) Wash‐out effects after short‐term ponesimod exposure in C6 glioma cells. FCM histogram (left) and receptor expression levels (right). *p < .05, **p < .01 by one‐way ANOVA with Kruskal–Wallis test. n = 4. Data are representative of two independent experiments with similar results. Representative specific binding curve against S1P1 for (E) ponesimod (Kd = 2.09 ± 0.27 nM) and (F) siponimod (Kd = 0.80 ± 0.97 nM) from two to six independent experiments. Mean ± SEM, n = 3~7
To test whether ponesimod induced sustained S1P1 internalization and functional antagonism of S1P1 as reported for other S1PR modulators, 2 S1P1‐C6 cells were exposed to S1P, ponesimod, or fingolimod‐P for 4 h, followed by wash‐out and further culturing in fresh, compound‐free media for 18 h before FCM analyses. In this paradigm, internalized S1P1 was recycled to the cell surface after S1P wash‐out (Figure 1D). By contrast, ponesimod reduced cell surface expression of S1P1 for a sustained period of time, which is consistent with known functional antagonism effects of another S1PR modulator, fingolimod‐P (Figure 1D). These results indicated that ponesimod acts as a functional antagonist to S1P1 in primary human astrocytes.
The direct binding of ponesimod to S1P1 was validated by a compensated interferometric reader (CIR)‐based binding assay using nanovesicles in free solution prepared from S1P1‐C6 versus vector‐C6 cells (Figure 1E). 20 Ponesimod bound to S1P1 with a Kd = 2.09 ± 0.27 nM (n = 2, Figure 1E and Table S1). Another S1PR modulator, siponimod that binds to S1P1 and S1P5, bound to S1P1 with a higher affinity (Kd = 0.80 ± 0.97 nM, n = 6) than ponesimod (Figure 1F and Table S1).
3.2. Ponesimod inhibits S1P‐induced intracellular signals in human primary astrocytes
Astrocytes appear to be a major target cell type in the CNS that express S1P1 and may potentially mediate the direct CNS effects of ponesimod. Intracellular signaling was assessed in human primary astrocytes. S1P1 is reported to only couple with Gαi protein but transduces signals to not only cyclic AMP, but also Ca2+, possibly through a coupling of released Gβγ with proximal Gαq protein. 25 S1P induced Ca2+ signals in a concentration‐dependent manner (logEC50 = −7.031 ± 0.063, n = 4; Figure 2A and Table S1). However, none of the S1PR modulators (ponesimod, siponimod, ozanimod, or fingolimod‐P) affected intracellular Ca2+ mobilization under our assay conditions (Figure 2A and Table S1; ponesimod induced a Ca2+ signal only at the highest concentration (30 µM), Figure S1). Functional antagonism was tested by pretreatment with S1PR modulators for 18 h followed by loading with Fluo‐4 in the absence of the compounds for 1 h before the assay. Both ponesimod and ozanimod blocked S1P‐induced Ca2+ mobilization in a concentration‐dependent manner (Figure 2B,C and Table S1), while the inhibitory effects of fingolimod‐P and siponimod were minor as compared to ponesimod and ozanimod (Figure 2B,C and Table S1). These results suggest that ponesimod effectively blocks S1P1‐induced Ca2+ signals as a functional antagonist, likely due to S1P1internalization.
FIGURE 2.
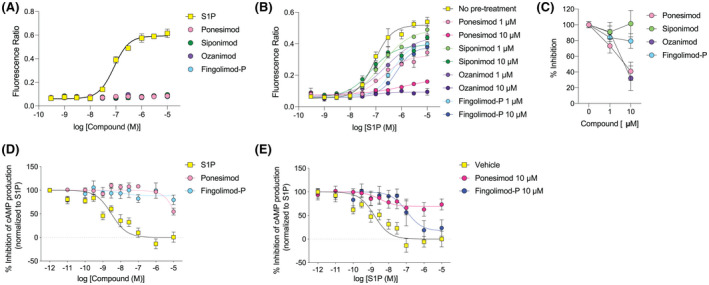
Ponesimod inhibits S1P‐induced Ca2+ and cAMP responses in human primary astrocytes. (A) Intracellular Ca2+ responses for agonistic applications of S1P and sphingosine 1‐phosphate (S1P) receptor (S1PR) modulators. Mean ± SD, n = 2. Data are representative of three independent experiments with similar results. (B) Intracellular Ca2+ responses for S1P that were pretreated with 1 or 10 µM of S1PR modulators overnight. Mean ± SD, n = 2. Data are representative of three independent experiments with similar results. (C) Inhibitory effects of S1PR modulators against 10 µM S1P stimulation. Mean ± SD, n = 4 pooled from three independent experiments. (D) Intracellular cAMP responses for agonistic applications of S1P and S1PR modulators. Mean ± SD, n = 3–5. Data are pooled from three independent experiments with similar results. (E) Intracellular cAMP responses of S1P pretreated with 10 µM of S1PR modulators overnight. Mean ± SD, n = 3–4. Data are pooled from three independent experiments with similar results
Next, in an orthogonal assay, ponesimod’s effect on cAMP signaling was tested in forskolin‐stimulated astrocytes. Agonistic application of S1P inhibited cAMP production in a concentration‐dependent manner (logIC50 = −8.930 ± 0.153, n = 5) (Figure 2D and Table S1). Neither ponesimod nor fingolimod‐P showed any effect on inhibiting cAMP production as agonists, while ponesimod inhibited forskolin‐induced cAMP production at the highest concentration (10 µM). These results were consistent with the Ca2+ signaling data (Figure 2D). Next, functional antagonistic activities on cAMP signaling were evaluated in human astrocytes that were cultured in the presence of ponesimod or fingolimod‐P (only tested for the 10 µM concentration chosen based on the results of Ca2+ assay) for 18 h prior to S1P stimulation (Figure 2E and Table S1). Ponesimod effectively blocked S1P‐induced cAMP inhibition, while fingolimod‐P showed a right shift of cAMP‐inhibitory curves (logIC50 = −6.978 ± 0.318, n = 3) as compared to controls. The corroboration between the Ca2+ and cAMP signaling results indicated that chronic exposure to ponesimod produces S1P1 functional antagonism in human astrocytes.
3.3. Ponesimod blocks astrocytic neuroinflammatory responses
To determine how ponesimod affects neuroinflammatory responses in astrocytes, single‐cell RNA sequencing (scRNA‐seq) was performed in cytokine mixture (IFN‐γ, TNF‐α, IL‐1β, IL‐6, and IL‐17)‐stimulated human primary astrocytes in the presence or absence of ponesimod. Quality control filtering resulted in a total of 49 706 cells with ~16 000 transcripts/cell (Table S2). Cells were clustered into 16 groups (Clusters 1–16) using the Seurat SNN (shared nearest neighbor) algorithm and visualized as Uniform Manifold Approximation and Projection (UMAP) plots (Figure 3A). An isolated, minor cell cluster (~1.5% of total cells) that deviated from the main clusters was considered to be fibroblasts/pericytes based on the high expression of COL1A1 (collagen type I alpha 1 chain), and thus it was excluded from further analyses. The expression of well‐known astrocyte markers including GFAP (glial fibrillary acidic protein) and SLC1A3 (glutamate aspartate transporter 1, GLAST‐1), were limited to Cluster 1 that was considered to be naïve astrocytes. The vast majority of cells expressed NES (nestin), VIM (vimentin), and CDH2 (N‐cadherin) (Figure S2), indicating a reactive phenotype previously observed when using in vitro culture systems. 26
FIGURE 3.
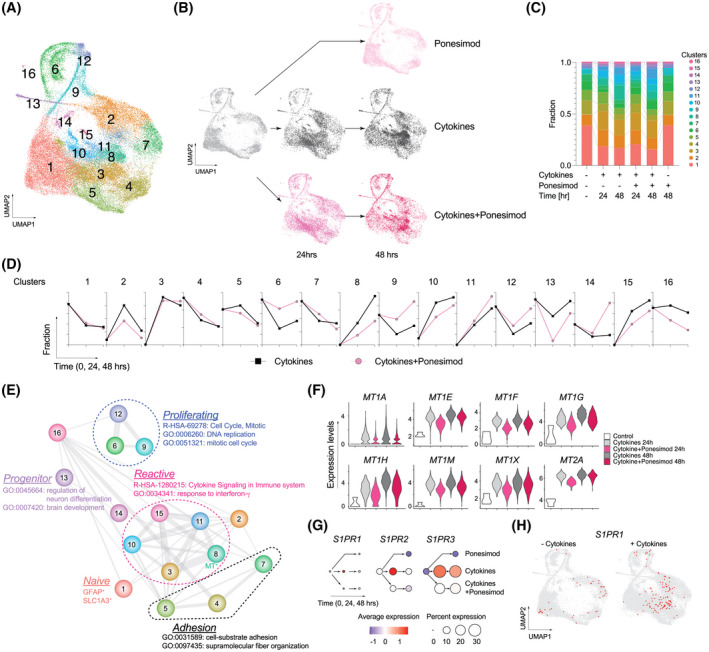
Single‐cell RNA‐seq (scRNA‐seq) of human primary astrocytes and effects of ponesimod. (A) Uniform manifold approximation and projection (UMAP) plots of total 49 706 cells. (B) UMAP plots of individual conditions (non‐treatment control, cytokines, and cytokines +1 µM ponesimod for 24 and 48 h). (C) Fraction of clusters. (D) Time course changes of each fraction. (E) Relationships between clusters. Nodes are clusters. Edges represent similarity between clusters which is weighted by thickness of the line. (F) Violin plots for metallothionein genes in Cluster 8. Statistical data are available in Table S5. (G) Dot plots for sphingosine 1‐phosphate (S1P) receptors (S1PRs). Colors represent standard deviation of expression levels from standardized mean. Dot sizes represent cell distribution rate that are calculated as the proportion of cells that express particular genes. (H) UMAP plots of S1PR1 expression. Red dots represent S1PR1‐expressing cells
Cytokine stimulation altered gene expression profiles that included 4266 differentially expressed genes (DEGs) at 24 h (2809 up‐ vs. 1457 downregulated genes) and 3914 DEGs at 48 h (2440 up‐ vs. 1474 down‐regulated genes) as compared to non‐stimulated controls (Table S3). In total, 3209 and 1780 DEGs were up‐ and downregulated, respectively, by cytokine exposure. Many DEGs were also identified when “cytokines + ponesimod” was compared to “cytokines” (4065 DEGs at 24 h including 34 up‐ and 4031 downregulated genes: 952 DEGs at 48 h including 130 up‐ and 822 downregulated genes). Upregulated DEGs produced by cytokine stimulation were compared to downregulated DEGs following ponesimod treatment, in which 2432 common DEGs were considered as ponesimod‐related genes and pathways (Figure S3). Gene ontology (GO) analyses using the ClueGO plugin of Cytoscape 27 identified that under functional antagonistic conditions, ponesimod significantly regulated IFN signaling, ubiquitin‐related pathways, IL‐1/immunoglobulin secretion, ER‐mediated pathways, and antigen presentation (Figure S3 and Table S4), representing global anti‐inflammatory effects in astrocytes. Importantly, ponesimod significantly inhibited FOS expression (Table S3) in accordance with previously identified ieAstrocytes (immediate early astrocytes that track with increased disease state in EAE and that were reported to be blocked by fingolimod). 28
Next, astrocytic heterogeneity was analyzed to understand ponesimod’s effects on neuroinflammation (Figure 3B–F, Tables S5 and S6). Ponesimod treatment alone showed a highly overlapping cellular distribution as compared to controls (Figure 3B,C), while cytokine exposure produced a major cellular shift within clusters from the outer boundary toward its interior (Clusters 3, 8, 10, 11, and 15) (Figure 3B–C). The cluster similarity was calculated with the top 20 significantly enriched GO terms (Table S6) in each cluster and visualized as a network (Figure 3E). This provided evidence for a strong relationship between newly enriched inside clusters that share similar pathways regulated by NF‐κB and RELA transcription factors (e.g., cytokine signaling in the immune system, R‐HAS‐1280215; and response to interferon‐γ, GO:0034341; Table S6). Previously proposed reactive astrocyte genes (Pan/A1/A2 genes 29 ) were enriched within the interior clusters (Table S5), indicating that the interior clusters presented reactive astrocyte phenotypes. Among reactive astrocyte clusters (Clusters 3, 8, 10, 11, and 15), ponesimod selectively decreased the cellular fractions of Clusters 8, 10, and 15 (Figure 3D). Astrocytes belonging to Cluster 8 were distinctly characterized by expression of metallothionein (MT) genes (MT1A, MT1E, MT1F, MT1G, MT1H, MT1 M, MT1X, and MT2A) that were significantly downregulated by ponesimod treatment (Figure 3F).
Cellular fractions in Clusters 1, 6, 9, 12, and 13 decreased after cytokine stimulation (Figure 3D), suggesting an evolution of astrocytic states with cytokine stimulation. A major cellular source of reactive astrocytes was Cluster 1 whose reduction was not affected by ponesimod. However, ponesimod blocked decreases in proliferating astrocyte clusters (Clusters 6, 9, and 12; Figure 3E and Table S6) and augmented reductions in Cluster 13 (progenitor cells). These results suggested that ponesimod prevented conversion of proliferating astrocytes into reactive astrocytes.
Primary human astrocytes express three of the five S1PRs (S1PR1, S1PR2, and S1PR3). Cytokine stimulation enhanced expression of these S1PRs, but their expression was suppressed by ponesimod (Figure 3G). Interestingly, S1PR1 expression was initially distributed along the outer boundary clusters on UMAP plots, followed by internal expression within the reactive astrocyte clusters after cytokine stimulation. This observation strongly implicated the active involvement of S1P1 in astrocytic reactivity as previously proposed. 12 Additionally, S1PR2 and S1PR3 expression was scattered throughout the clusters (Figure S4), suggesting that these receptors might have broader functions related to augmenting S1P1.
3.4. Ponesimod effectively inhibits cuprizone‐induced demyelination
Cuprizone‐induced demyelination, a common animal model for demyelination related to MS, 30 , 31 , 32 , 33 , 34 was used to test in vivo ponesimod efficacy in wild‐type C57BL/6 male mice. Mice kept on 0.2% cuprizone chow for 5 weeks exhibited a decline in weight for the first week, unlike naïve mice which gained weight constantly (Figure S5). Black gold II staining showed demyelination in wild‐type mice on 0.2% cuprizone in both the corpus callosum (CC) and cingulum when compared to controls (Figure S5). After 5 weeks of cuprizone feeding, mice fed on a normal diet for 1 week showed robust remyelination in both the CC and cingulum (Figure S5). These results indicated that the model responded appropriately under our assay conditions.
The preventative effects of ponesimod in cuprizone‐induced demyelination were evaluated by continuous treatment (30 mg/kg; b.i.d.) 35 during the cuprizone chow feeding period (Figure 4A). Ponesimod protected mice from demyelination in the cingulum, (Figure 4B,C). Moreover, ponesimod exposure increased the number of Olig2+ oligodendrocytes in both the CC and cingulum (Figure 4D,E), supporting the protective effects of ponesimod in cuprizone‐induced demyelination, as was observed in EAE model. 35 Ponesimod significantly decreased Iba1+ microglia in the cingulum (Figure 4F,G). GFAP+ astrocytes increased in the CC and tended to decrease in the cingulum following ponesimod treatment (Figure 4H,I). These results showed ponesimod efficacy in preventing demyelination in at least some cuprizone demyelinated fiber tracts and most prominently in the cingulum.
FIGURE 4.
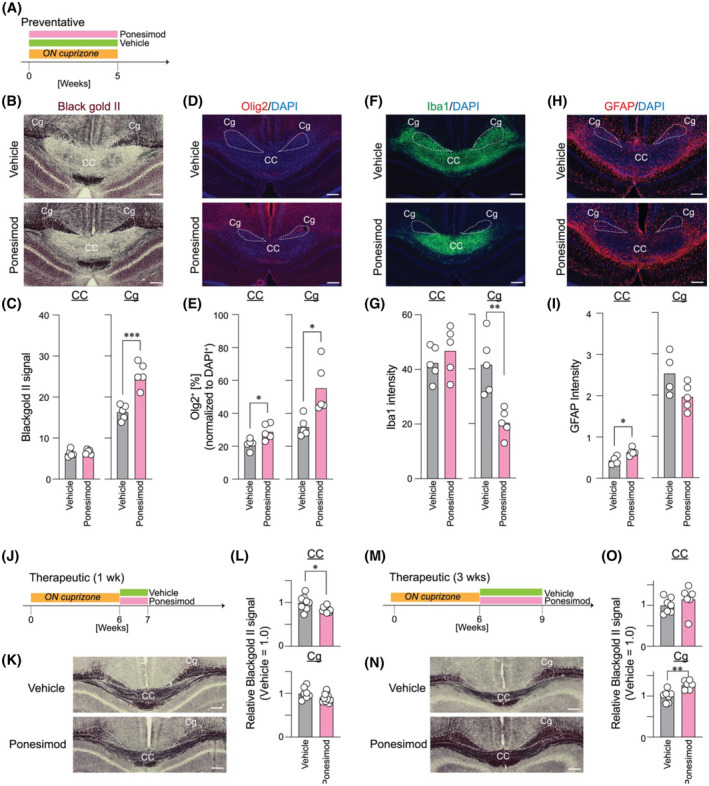
Ponesimod prevents cuprizone‐induced demyelination and improves therapeutic remyelination in the cingulum. Mice were fed a 0.2% cuprizone diet for 5 weeks with concomitant treatment with vehicle or ponesimod (30 mg/kg) or 6 weeks followed by 1 week of similar treatment after returning to a normal diet to identify demyelination or remyelination status, oligodendrocyte progenitor cell population, microglia, and astrocytes. (A) Diagrammatic representation of the preventative paradigm. (B, C) Black gold II staining, ***p < .00005; (D, E) Olig2/DAPI labeling with Olig2+ cell counts normalized to DAPI+ counts, *p < .05; (F, G) Iba1/DAPI labeling, **p < .005; (H, I) GFAP/DAPI labeling, *p < .05; (J–O) Therapeutic effects of ponesimod on remyelination. (J and M) Diagrammatic representation of the therapeutic paradigm; (K and N) Black gold II staining indicating myelination status. (L and O) Quantification of black gold II signals, ***p = .0001, ****p < .0001
Next, therapeutic effects of ponesimod were examined to determine its effect on remyelination following cuprizone diet cessation (Figure 4J–O). Short‐term treatment (1 week) did not show any beneficial effects (Figure 4J–L), whereas prolonged treatment (3 weeks) significantly enhanced remyelination in the cingulum (Figure 4M–O). Together with the preventative experiments, ponesimod may require continuous and long‐term exposure to prevent demyelination and promote remyelination.
4. DISCUSSION
The entry of S1PR modulators as oral, immune cell trafficking inhibitors for MS has expanded therapeutic choices for relapsing and progressive forms of MS through four FDA‐approved agents: fingolimod, siponimod, ozanimod, and ponesimod. While conceptually they are all S1PR modulators, each is distinct in its chemistry, pharmacology, and receptor subtype engagement. This raises the possibility that meaningful therapeutic differences may be accessed by each agent despite the common effects on immune cell trafficking as a mechanistic explanation for efficacy. 36 In particular, S1PRs that are expressed on multiple CNS cell types 2 , 8 , 12 , 37 , 38 , 39 could contribute to efficacy signals. This possibility is supported by animal studies showing reduced fingolimod efficacy in astrocyte‐specific S1P1‐deficient mice, 12 as well as by clinical studies demonstrating reduced brain atrophy with S1PR modulators, 13 which contrasts with pseudoatrophy produced by natalizumab. 40
Ponesimod was reported to be S1P1 selective, 18 , 24 and an S1PR internalization assay confirmed the selectivity of ponesimod for S1P1. Direct receptor binding as determined here by CIR showed somewhat lower affinities than those published for other agents and identified here for siponimod. 41 Ponesimod showed inhibition of both S1P‐induced intracellular Ca2+ via Gαi‐Gβγ‐Gαq‐mediated signaling 25 and Gαi‐mediated cAMP responses, at least within human primary astrocytes. On the other hand, functional antagonism by fingolimod appeared to be biased for the β‐arrestin pathway because fingolimod‐P induced sustained S1P1 internalization but did not block intracellular signals. Ponesimod blocked G protein signals downstream of S1PRs more effectively than fingolimod in human astrocytes, indicating ponesimod’s unbiased functional antagonism specifically for S1P1, which distinguishes it from other S1PR modulators.
Prior studies identified key roles for astrocytes in MS. 12 , 28 , 29 , 39 , 42 , 43 Using primary human astrocytes challenged with disease‐relevant cytokines, scRNA‐seq identified robust inflammatory gene signatures that were down‐modulated by ponesimod, including a reduction of FOS related to ieAstrocytes that track with disease severity. 28 In addition, high levels of astrocytic metallothionein (MT) genes overlapped with Huntington disease 44 and astrocytomas, 45 suggesting that ponesimod inhibition of MT‐gene expressing astrocytes may have therapeutic utility for treating other diseases. The vast diversity of astrocytes throughout the brain with disease relevance, 26 , 29 , 46 implicates myriad potential effects that could be accessed by ponesimod for neuro‐inflammatory processes and related effects on myelination. 42 , 46 , 47 , 48 , 49 , 50
To assess ponesimod activities on myelination, cuprizone challenge with or without ponesimod was employed in preventative and therapeutic paradigms. Beyond generalized demyelination, a characteristic of MS, clinical studies correlated fatigue with demyelination. 51 , 52 , 53 , 54 , 55 , 56 This particularly involves limbic pathways such as the cingulum. 51 , 57 , 58 , 59 Abnormalities in the cingulum have been associated with several neurological conditions like schizophrenia, 60 attention deficit hyperactivity disorder, 61 depression, 62 post‐traumatic stress disorder, 63 obsessive compulsive disorder (OCD), 64 and autism spectrum disorder. 65 Anterior cingulotomy in patients suffering from OCD, depression, and chronic pain with cingulum lesions improved outcomes. 66 Lesions to the cingulum, among other functions, are clinically associated with MS fatigue in humans. 51 Remarkably, cingulum demyelination has been reported to correlate with the MS symptoms (MS fatigue–cognition). 51 , 57 , 58 , 59 , 67 Cuprizone‐induced demyelination also affects the cingulum, 33 , 34 providing an experimental opportunity to assess ponesimod on this limbic fiber bundle. The precise mechanism through which this prevention occurs is not known but appears to involve increase in oligodendrocytes and reduction in microglia and astrocytes (Figure 4). The mechanisms might likely include non‐cell autonomous effects that involve astrocyte S1P1 engagement, which would be consistent with the documented astrocyte influences on myelination. 46 , 47 , 48 , 49 , 50 Future MS imaging studies on myelin preservation with ponesimod could clarify the effects on the cingulum and other pathways associated with MS.
The experimental data presented here support distinct receptor, gene expression, and myelination effects stimulated by ponesimod, and indicate that an S1P1 mono‐selective receptor modulator can produce positive efficacy signals relevant to MS and possibly other CNS disorders. As all approved S1PR modulators engage S1P1, including singly through ponesimod that exhibited unbiased functional antagonism in human astrocytes, it appears that major benefits accrue through functional antagonism of this receptor subtype. The novel reduction in MS fatigue accessed by ponesimod 16 underscores the benefits, particularly through CNS mechanisms that involve complex cell autonomous and non‐cell autonomous interactions, which may reveal novel therapeutic mechanisms.
DISCLOSURES
J.C. has received consulting fees or research support from Abbott, AbbVie, Amira, Arena Pharmaceuticals, Biogen Idec, BiolineRX, Blade Therapeutics, Brainstorm Cell Therapeutics, Celgene, GlaxoSmithKline, Inception Sciences, Johnson & Johnson, Merck, Mitsubishi Tanabe, Novartis, Ono Pharmaceuticals, Pfizer, SKAI Ventures, and Taisho Pharmaceutical Co. The other authors declare that they have no competing financial interests. Research funding was provided by the National Institute of Neurological Disorders and Stroke of the National Institutes of Health (R01NS103940 to Y.K.) and Janssen Research & Development LLC to J.C.
AUTHOR CONTRIBUTIONS
Yasuyuki Kihara, Deepa Jonnalagadda, Yunjiao Zhu, Manisha Ray, Tony Ngo, Carter Palmer, and Richard Rivera, performed the research and analyzed the data. Yasuyuki Kihara Deepa Jonnalagadda and Jerold Chun wrote the paper. Jerold Chun designed the research.
Supporting information
Fig S1‐S5
Table S1‐S6
ACKNOWLEDGMENTS
The authors thank the Histology Core and Animal Facility at the Sanford Burnham Prebys Medical Discovery Institute, Nyssa Williams and Masami Kachi for technical assistance, and Danielle Jones for editing the manuscript. Research funding was provided by the National Institute of Neurological Disorders and Stroke of the National Institutes of Health (R01NS103940 to Y.K.) and Janssen Research & Development LLC to J.C. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Kihara Y, Jonnalagadda D, Zhu Y, et al. Ponesimod inhibits astrocyte‐mediated neuroinflammation and protects against cingulum demyelination via S1P1‐selective modulation. FASEB J. 2022;36:e22132. doi: 10.1096/fj.202101531R
Yasuyuki Kihara and Deepa Jonnalagadda Joint 1st authors.
Yunjiao Zhu, Manisha Ray and Tony Ngo Joint 2nd authors.
REFERENCES
- 1. Ishii I, Fukushima N, Ye X, Chun J. Lysophospholipid receptors: signaling and biology. Annu Rev Biochem. 2004;73:321‐354. [DOI] [PubMed] [Google Scholar]
- 2. Chun J, Kihara Y, Jonnalagadda D, Blaho VA. Fingolimod: lessons learned and new opportunities for treating multiple sclerosis and other disorders. Annu Rev Pharmacol Toxicol. 2019;59:149‐170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kihara Y, Maceyka M, Spiegel S, Chun J. Lysophospholipid receptor nomenclature review: IUPHAR review 8. Br J Pharmacol. 2014;171:3575‐3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chun J, Giovannoni G, Hunter SF. Sphingosine 1‐phosphate receptor modulator therapy for multiple sclerosis: differential downstream receptor signalling and clinical profile effects. Drugs. 2020;81:1‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cohen JA, Barkhof F, Comi G, et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med. 2010;362:402‐415. [DOI] [PubMed] [Google Scholar]
- 6. Cohen JA, Chun J. Mechanisms of fingolimod’s efficacy and adverse effects in multiple sclerosis. Ann Neurol. 2011;69:759‐777. [DOI] [PubMed] [Google Scholar]
- 7. Chun J, Hartung HP. Mechanism of action of oral fingolimod (FTY720) in multiple sclerosis. Clin Neuropharmacol. 2010;33:91‐101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chun J, Brinkmann V. A mechanistically novel, first oral therapy for multiple sclerosis: the development of fingolimod (FTY720, Gilenya). Discov Med. 2011;12:213‐228. [PMC free article] [PubMed] [Google Scholar]
- 9. Mandala S, Hajdu R, Bergstrom J, et al. Alteration of lymphocyte trafficking by sphingosine‐1‐phosphate receptor agonists. Science. 2002;296:346‐349. [DOI] [PubMed] [Google Scholar]
- 10. Brinkmann V, Davis MD, Heise CE, et al. The immune modulator FTY720 targets sphingosine 1‐phosphate receptors. J Biol Chem. 2002;277:21453‐21457. [DOI] [PubMed] [Google Scholar]
- 11. Matloubian M, Lo CG, Cinamon G, et al. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature. 2004;427:355‐360. [DOI] [PubMed] [Google Scholar]
- 12. Choi JW, Gardell SE, Herr DR, et al. FTY720 (fingolimod) efficacy in an animal model of multiple sclerosis requires astrocyte sphingosine 1‐phosphate receptor 1 (S1P1) modulation. Proc Natl Acad Sci USA. 2011;108:751‐756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kappos L, Bar‐Or A, Cree BAC, et al. Siponimod versus placebo in secondary progressive multiple sclerosis (EXPAND): a double‐blind, randomised, phase 3 study. Lancet. 2018;391:1263‐1273. [DOI] [PubMed] [Google Scholar]
- 14. Cohen JA, Comi G, Selmaj KW, et al. Safety and efficacy of ozanimod versus interferon beta‐1a in relapsing multiple sclerosis (RADIANCE): a multicentre, randomised, 24‐month, phase 3 trial. Lancet Neurol. 2019;18:1021‐1033. [DOI] [PubMed] [Google Scholar]
- 15. Tran JQ, Zhang P, Ghosh A, et al. Single‐dose pharmacokinetics of ozanimod and its major active metabolites alone and in combination with gemfibrozil, itraconazole, or rifampin in healthy subjects: a randomized, parallel‐group, open‐label study. Adv Ther. 2020;37:4381‐4395. [DOI] [PubMed] [Google Scholar]
- 16. Kappos L, Fox RJ, Burcklen M, et al. Ponesimod compared with teriflunomide in patients with relapsing multiple sclerosis in the active‐comparator phase 3 OPTIMUM study: a randomized clinical trial. JAMA Neurol. 2021;78:558‐567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bolli MH, Abele S, Binkert C, et al. 2‐Imino‐thiazolidin‐4‐one derivatives as potent, orally active S1P1 receptor agonists. J Med Chem. 2010;53:4198‐4211. [DOI] [PubMed] [Google Scholar]
- 18. D’Ambrosio D, Freedman MS, Prinz J. Ponesimod, a selective S1P1 receptor modulator: a potential treatment for multiple sclerosis and other immune‐mediated diseases. Ther Adv Chronic Dis. 2016;7:18‐33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mizuno H, Kihara Y, Kussrow A, et al. Lysophospholipid G protein‐coupled receptor binding parameters as determined by backscattering interferometry. J Lipid Res. 2019;60:212‐217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ray M, Nagai K, Kihara Y, et al. Unlabeled lysophosphatidic acid receptor binding in free solution as determined by a compensated interferometric reader. J Lipid Res. 2020;61:1244‐1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hafemeister C, Satija R. Normalization and variance stabilization of single‐cell RNA‐seq data using regularized negative binomial regression. Genome Biol. 2019;20:296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Stuart T, Butler A, Hoffman P, et al. Comprehensive integration of single‐cell data. Cell. 2019;177(1888‐1902):e1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Finak G, McDavid A, Yajima M, et al. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single‐cell RNA sequencing data. Genome Biol. 2015;16:278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Piali L, Froidevaux S, Hess P, et al. The selective sphingosine 1‐phosphate receptor 1 agonist ponesimod protects against lymphocyte‐mediated tissue inflammation. J Pharmacol Exp Ther. 2011;337:547‐556. [DOI] [PubMed] [Google Scholar]
- 25. Pfeil EM, Brands J, Merten N, et al. Heterotrimeric G protein subunit Galphaq is a master switch for Gbetagamma‐mediated calcium mobilization by Gi‐coupled GPCRs. Mol Cell. 2020;80(940‐954):e946. [DOI] [PubMed] [Google Scholar]
- 26. Liddelow SA, Barres BA. Reactive astrocytes: production, function, and therapeutic potential. Immunity. 2017;46:957‐967. [DOI] [PubMed] [Google Scholar]
- 27. Bindea G, Mlecnik B, Hackl H, et al. ClueGO: a Cytoscape plug‐in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics. 2009;25:1091‐1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Groves A, Kihara Y, Jonnalagadda D, et al. A functionally defined in vivo astrocyte population identified by c‐Fos activation in a mouse model of multiple sclerosis modulated by S1P signaling: immediate‐early astrocytes (ieAstrocytes). eNeuro. 2018;5:ENEURO.0239‐18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liddelow SA, Guttenplan KA, Clarke LE, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541:481‐487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tansey FA, Zhang H, Cammer W. Rapid upregulation of the Pi isoform of glutathione‐S‐transferase in mouse brains after withdrawal of the neurotoxicant, cuprizone. Mol Chem Neuropathol. 1997;31:161‐170. [DOI] [PubMed] [Google Scholar]
- 31. Hiremath MM, Saito Y, Knapp GW, Ting JP, Suzuki K, Matsushima GK. Microglial/macrophage accumulation during cuprizone‐induced demyelination in C57BL/6 mice. J Neuroimmunol. 1998;92:38‐49. [DOI] [PubMed] [Google Scholar]
- 32. Morell P, Barrett CV, Mason JL, et al. Gene expression in brain during cuprizone‐induced demyelination and remyelination. Mol Cell Neurosci. 1998;12:220‐227. [DOI] [PubMed] [Google Scholar]
- 33. Schmidt T, Awad H, Slowik A, Beyer C, Kipp M, Clarner T. Regional heterogeneity of cuprizone‐induced demyelination: topographical aspects of the midline of the corpus callosum. J Mol Neurosci. 2013;49:80‐88. [DOI] [PubMed] [Google Scholar]
- 34. Silvestroff L, Bartucci S, Soto E, Gallo V, Pasquini J, Franco P. Cuprizone‐induced demyelination in CNP:GFP transgenic mice. J Comp Neurol. 2010;518:2261‐2283. [DOI] [PubMed] [Google Scholar]
- 35. Fourgeaud L, Le M, Needham A, et al. A Central Effect of Ponesimod on Neuroinflammation in a Pre‐clinical Model of Multiple Sclerosis. ACTRIMS; 2021. [Google Scholar]
- 36. Brinkmann V. Sphingosine 1‐phosphate receptors in health and disease: mechanistic insights from gene deletion studies and reverse pharmacology. Pharmacol Ther. 2007;115:84‐105. [DOI] [PubMed] [Google Scholar]
- 37. Lee CW, Choi JW, Chun J. Neurological S1P signaling as an emerging mechanism of action of oral FTY720 (fingolimod) in multiple sclerosis. Arch Pharm Res. 2010;33:1567‐1574. [DOI] [PubMed] [Google Scholar]
- 38. Soliven B, Miron V, Chun J. The neurobiology of sphingosine 1‐phosphate signaling and sphingosine 1‐phosphate receptor modulators. Neurology. 2011;76:S9‐S14. [DOI] [PubMed] [Google Scholar]
- 39. Groves A, Kihara Y, Chun J. Fingolimod: direct CNS effects of sphingosine 1‐phosphate (S1P) receptor modulation and implications in multiple sclerosis therapy. J Neurol Sci. 2013;328:9‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vidal‐Jordana A, Sastre‐Garriga J, Perez‐Miralles F, et al. Early brain pseudoatrophy while on natalizumab therapy is due to white matter volume changes. Mult Scler. 2013;19:1175‐1181. [DOI] [PubMed] [Google Scholar]
- 41. Gergely P, Nuesslein‐Hildesheim B, Guerini D, et al. The selective sphingosine 1‐phosphate receptor modulator BAF312 redirects lymphocyte distribution and has species‐specific effects on heart rate. Br J Pharmacol. 2012;167:1035‐1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lee SC, Moore GR, Golenwsky G, Raine CS. Multiple sclerosis: a role for astroglia in active demyelination suggested by class II MHC expression and ultrastructural study. J Neuropathol Exp Neurol. 1990;49:122‐136. [DOI] [PubMed] [Google Scholar]
- 43. De Keyser J, Zeinstra E, Frohman E. Are astrocytes central players in the pathophysiology of multiple sclerosis? Arch Neurol. 2003;60:132‐136. [DOI] [PubMed] [Google Scholar]
- 44. Al‐Dalahmah O, Sosunov AA, Shaik A, et al. Single‐nucleus RNA‐seq identifies Huntington disease astrocyte states. Acta Neuropathol Commun. 2020;8:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Masiulionyte B, Valiulyte I, Tamasauskas A, Skiriute D. Metallothionein genes are highly expressed in malignant astrocytomas and associated with patient survival. Sci Rep. 2019;9:5406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Werkman IL, Dubbelaar ML, van der Vlies P, de Boer‐Bergsma JJ, Eggen BJL, Baron W. Transcriptional heterogeneity between primary adult grey and white matter astrocytes underlie differences in modulation of in vitro myelination. J Neuroinflamma. 2020;17:373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sorensen A, Moffat K, Thomson C, Barnett SC. Astrocytes, but not olfactory ensheathing cells or Schwann cells, promote myelination of CNS axons in vitro. Glia. 2008;56:750‐763. [DOI] [PubMed] [Google Scholar]
- 48. Nash B, Thomson CE, Linington C, et al. Functional duality of astrocytes in myelination. J Neurosci. 2011;31:13028‐13038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kiray H, Lindsay SL, Hosseinzadeh S, Barnett SC. The multifaceted role of astrocytes in regulating myelination. Exp Neurol. 2016;283:541‐549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Molina‐Gonzalez I, Miron VE. Astrocytes in myelination and remyelination. Neurosci Lett. 2019;713:e134532. [DOI] [PubMed] [Google Scholar]
- 51. Sepulcre J, Masdeu JC, Goni J, et al. Fatigue in multiple sclerosis is associated with the disruption of frontal and parietal pathways. Mult Scler. 2009;15:337‐344. [DOI] [PubMed] [Google Scholar]
- 52. Basu N, Murray AD, Jones GT, Reid DM, Macfarlane GJ, Waiter GD. Fatigue‐related brain white matter changes in granulomatosis with polyangiitis. Rheumatology (Oxford). 2013;52:1429‐1434. [DOI] [PubMed] [Google Scholar]
- 53. Grandjean J, Azzinnari D, Seuwen A, et al. Chronic psychosocial stress in mice leads to changes in brain functional connectivity and metabolite levels comparable to human depression. NeuroImage. 2016;142:544‐552. [DOI] [PubMed] [Google Scholar]
- 54. Clark AL, Delano‐Wood L, Sorg SF, Werhane ML, Hanson KL, Schiehser DM. Cognitive fatigue is associated with reduced anterior internal capsule integrity in veterans with history of mild to moderate traumatic brain injury. Brain Imaging Behav. 2017;11:1548‐1554. [DOI] [PubMed] [Google Scholar]
- 55. Kang SY, Bang M, Hong JY, et al. Neural and dopaminergic correlates of fatigue in Parkinson’s disease. J Neural Transm (Vienna). 2020;127:301‐309. [DOI] [PubMed] [Google Scholar]
- 56. Provenzano D, Washington SD, Rao YJ, Loew M, Baraniuk J. Machine learning detects pattern of differences in functional magnetic resonance imaging (fMRI) data between chronic fatigue syndrome (CFS) and gulf war illness (GWI). Brain Sci. 2020;10:456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Pardini M, Bonzano L, Bergamino M, et al. Cingulum bundle alterations underlie subjective fatigue in multiple sclerosis. Mult Scler. 2015;21:442‐447. [DOI] [PubMed] [Google Scholar]
- 58. Bisecco A, Caiazzo G, d’Ambrosio A, et al. Fatigue in multiple sclerosis: the contribution of occult white matter damage. Mult Scler. 2016;22:1676‐1684. [DOI] [PubMed] [Google Scholar]
- 59. Novo AM, Batista S, Alves C, et al. The neural basis of fatigue in multiple sclerosis: a multimodal MRI approach. Neurol Clin Pract. 2018;8:492‐500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Shepherd AM, Laurens KR, Matheson SL, Carr VJ, Green MJ. Systematic meta‐review and quality assessment of the structural brain alterations in schizophrenia. Neurosci Biobehav Rev. 2012;36:1342‐1356. [DOI] [PubMed] [Google Scholar]
- 61. van Ewijk H, Heslenfeld DJ, Zwiers MP, Buitelaar JK, Oosterlaan J. Diffusion tensor imaging in attention deficit/hyperactivity disorder: a systematic review and meta‐analysis. Neurosci Biobehav Rev. 2012;36:1093‐1106. [DOI] [PubMed] [Google Scholar]
- 62. Bracht T, Linden D, Keedwell P. A review of white matter microstructure alterations of pathways of the reward circuit in depression. J Affect Disord. 2015;187:45‐53. [DOI] [PubMed] [Google Scholar]
- 63. Daniels JK, Lamke JP, Gaebler M, Walter H, Scheel M. White matter integrity and its relationship to PTSD and childhood trauma–a systematic review and meta‐analysis. Depress Anxiety. 2013;30:207‐216. [DOI] [PubMed] [Google Scholar]
- 64. Radua J, Grau M, van den Heuvel OA, et al. Multimodal voxel‐based meta‐analysis of white matter abnormalities in obsessive‐compulsive disorder. Neuropsychopharmacology. 2014;39:1547‐1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Travers BG, Adluru N, Ennis C, et al. Diffusion tensor imaging in autism spectrum disorder: a review. Autism Res. 2012;5:289‐313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Bubb EJ, Metzler‐Baddeley C, Aggleton JP. The cingulum bundle: anatomy, function, and dysfunction. Neurosci Biobehav Rev. 2018;92:104‐127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Koenig KA, Sakaie KE, Lowe MJ, et al. The relationship between cognitive function and high‐resolution diffusion tensor MRI of the cingulum bundle in multiple sclerosis. Mult Scler. 2015;21:1794‐1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S5
Table S1‐S6