

Abstract
The facility of next‐generation sequencing has led to an explosion of gene catalogs for novel genomes, transcriptomes and metagenomes, which are functionally uncharacterized. Computational inference has emerged as a necessary substitute for first‐hand experimental evidence. PANNZER (Protein ANNotation with Z‐scoRE) is a high‐throughput functional annotation web server that stands out among similar publically accessible web servers in supporting submission of up to 100,000 protein sequences at once and providing both Gene Ontology (GO) annotations and free text description predictions. Here, we demonstrate the use of PANNZER and discuss future plans and challenges. We present two case studies to illustrate problems related to data quality and method evaluation. Some commonly used evaluation metrics and evaluation datasets promote methods that favor unspecific and broad functional classes over more informative and specific classes. We argue that this can bias the development of automated function prediction methods. The PANNZER web server and source code are available at http://ekhidna2.biocenter.helsinki.fi/sanspanz/.
Keywords: evaluation, gene ontology, protein function, web server
1. INTRODUCTION
Climate change, environmental problems, new emerging pathogens and the growing human population have created new critical research questions. Lowering sequencing costs have made it reasonable to study relevant species, linked to these research questions, with next‐generation sequencing projects. Genome and transcriptome projects have sequenced various agricultural plants, microbes used in biochemical processing or biofuel generation, animal and human pathogens, or microbes used in food processing. 1 , 2 , 3 , 4 These genome projects generate massive amounts of sequences. Often the most relevant set of sequences are the predicted genes, extracted from the sequenced genomes. These extracted genes lack any information on what they actually do. Therefore, in order to make this new genome data useful, these genes must be associated with the relevant biological features. Functional annotations can be used to generate hypotheses how the studied agricultural plant reacts to pathogens or what differentiates a multidrug‐resistant bacterium from its relative strains. Annotations could be done manually, but even with one genome, often with 20,000–30,000 genes, it is too overwhelming a task. Therefore, we need Automated Function Prediction methods (AFP methods) that can automatically predict these features.
The goal of the AFP methods is to predict the functions for the studied sequences and communicate these generated predictions to end‐users. This function can be presented in a number of ways: (i) the sequence can have a short descriptive text, (ii) the sequence can have a longer more detailed description, (iii) the sequence can be classified to a sequence family with conserved function, 5 and/or (iv) the sequence can be classified to known signaling or reaction pathways. Short descriptions give a quick summary on the sequence, especially when one is working with FASTA sequence files. They are also required for submitting the sequences to a database. Longer detailed descriptions, presented by databases like SwissProt 6 and RefSeq, 7 are comprehensive descriptions on various sequence functions including literature citations. Sequences can be also classified to various functional classification hierarchies, with Gene Ontology (GO) 8 being the most popular. The classes (also referred to as terms) in GO represent varying levels of information on the gene's function. Still, even the most specific classes often lose some details on the protein function, when compared to a free text description. Classes still have benefits. One sequence can have multiple functions and these can be represented with multiple classes. Furthermore, the classes also allow studies on groups of sequences, where scientists can look for over‐represented functions from the set of sequences. Finally, the function can be presented with various signaling and reaction pathways. These can be used similarly to the aforementioned classes. Here the KEGG (Kyoto Encyclopedia for Genes and Genomes 9 ) pathway database is the most commonly predicted pathway collection. Most of these data types can be predicted with AFP tools.
None of the existing AFP methods invents the function for a novel sequence out of nowhere. They rather use mainly classifiers on various sequence and interaction network derived features, 10 , 11 pre‐annotated protein families 12 , 13 , 14 and/or K‐nearest sequence principles. 15 , 16 Altogether, AFP methods often look for various similarities, between the sequences with known function and the query sequence, and next transfer the function(s) to the query sequence if the similarities are strong enough (Figure 1). This means that even the most advanced AFP methods are only as good as the available training data they use. Most of the existing annotations cumulate to model organisms. Therefore, a more exotic species, with no closely related model organism, is likely to get weakly annotated by all methods. Annotation efficiency is skewed across the different functions and different species. One solution to the lack of data is to use predicted functions also as an input in our predictions. Unreviewed GO annotations are tagged as such, allowing their inclusion or exclusion from the training data. Inclusion has its pros and cons. Over 99% of the current GO annotations for Uniprot are predictions, making our training data significantly larger. The drawback is the inclusion of wrong predictions into the training data. Thus, the repetitive use of predictions could generate a cascade of decisions, where a prediction is used as an input for prediction, which in turn is used as an input for a prediction, leading to error propagation. 17
FIGURE 1.
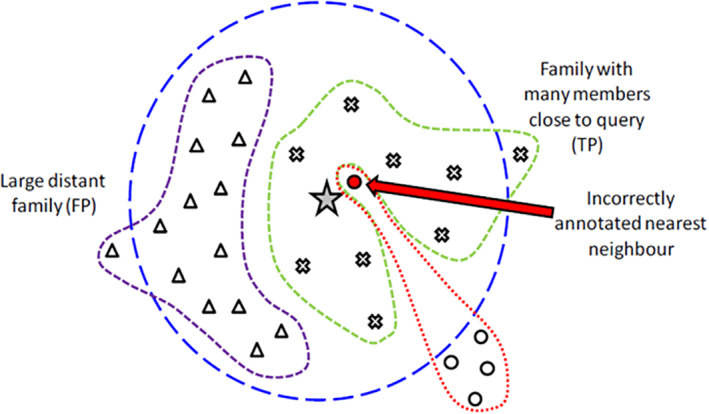
Weighted K‐nearest neighbor approach. A query sequence (star in center) is associated with sequence neighbors. Sequence proximity and database background are taken into account by enrichment statistics. Random mislabels can be rejected—systematic mislabels cannot
The hierarchical structure of the GO means that any sequence, annotated with GO term X, will automatically also get annotated with parent terms of X. For a sequence, predicted to belong to ribosome, the GO hierarchy implies, among others, the following classes: ribosome, translation, protein metabolic process, organic substance metabolic process, cellular metabolic process (http://amigo.geneontology.org/amigo/term/GO:0044391#display-lineage-tab). Notice how information, related to function, decreases as we go forward on the list. These broader classes represent parent terms. This hierarchical GO structure causes less informative GO terms to be more frequent in any training or testing datasets. This in turn causes AFP methods to easily report only unspecific, vague GO terms. Therefore, many AFP methods have been based on enrichment statistics. 15 , 16 Rather than looking what GO classes are frequent in the neighborhood of the query sequence, they look for annotations that are more frequent in the sequence neighborhood than in the whole database. The hierarchical structure of GO also complicates the evaluation of AFP methods. There is ongoing debate how to properly evaluate AFP models. 18 One of the biggest issues with the evaluation metrics is that one can get very good results, with some evaluation metrics, by simply reporting the GO classes in decreasing order of their frequency in the database, for every tested gene. 19 , 20 This baseline model, often referred as the naïve model, 21 constitutes no information to end user.
We have developed PANNZER, a weighted K‐nearest neighbor classifier for protein function prediction. This paper demonstrates the use of PANNZER and recently added features (taxonomic filtering and gene name prediction), future plans and challenges. We present two case studies to illustrate problems related to data quality and AFP method evaluations. We point out that some used evaluation metrics and used evaluation datasets promote methods that favor unspecific and broad classes over more informative and specific classes. We argue that this can bias the development of AFP methods.
2. METHODS
PANNZER is a fast and fully automated tool and web server for the AFP task. PANNZER is one of the few annotation tools that allow genome sized queries (Table 1). PANNZER can predict short sequence description to any species (plants, bacteria, animals). PANNZER outperformed other tools in our comparisons across all datasets (8–45% higher Fmax scores, for example 22 ). In addition, PANNZER has performed well in the international competitions, organized for the AFP methods (ranked overall 3rd in 2013 and frequently between 5th and 7th in 2019 21 , 23 , 24 ) and also in comparisons done by other groups. 25 , 26 , 27
TABLE 1.
Feature comparison between selected annotation servers
| Server schedule | GO prediction | DE prediction | >1,000 query sequences | Open source | Last database update/update schedule |
|---|---|---|---|---|---|
| ARGOT | Yes | No | Yes | No | Nov‐2016 |
| eggNOG | Yes | Yes | Yes | Yes | Jan‐2019 |
| FFpred | Yes | No | No | Yes | Unknown |
| FunFam | Yes | No | Yes (API) | Yes | Daily |
| INGA | Yes | No | No | No | Feb‐2019 |
| NetGO | Yes | No | By request | No | Unknown |
| PANNZER2 | Yes | Yes | Yes | Yes | Jun‐2021/bimonthly |
| PFP | Yes | No | No | No | Sep‐2020 |
Note: DE prediction stands for free text protein descriptions. Last database update is taken from explicit statements on annotation servers (at time of writing 13/07/21). The web servers were accessed at URLs http://www.medcomp.medicina.unipd.it/Argot2‐5/, http://eggnog‐mapper.embl.de/, http://bioinf.cs.ucl.ac.uk/psipred/, http://cathdb.info/search/by_sequence, https://inga.bio.unipd.it/, https://issubmission.sjtu.edu.cn/netgo/, http://ekhidna2.biocenter.helsinki.fi/sanspanz/, https://kiharalab.org/pfp.php.
2.1. Inputs
The inputs are FASTA formatted protein sequences. These sequences can be translated coding sequences from a genome project. Bacterial protein coding genes can be predicted from the genomic sequence using, for example, Prodigal. 28 Eukaryotic gene prediction is more complicated. Gene models can be cross‐mapped from a related genome, guided by homologous proteins or RNA‐seq data, or predicted ab initio. 29 RNA‐seq data should be either mapped to a reference genome, or assembled de novo and translated to peptides using, for example, Trinity and TransDecoder. 30
2.2. Parameters
PANNZER2 uses a fast suffix array neighborhood search (SANSparallel 31 ) to find homologous sequences in the UniProt database. We refer to homology search results as the sequence neighborhood. By default, PANNZER2 uses a maximum of 100 database hits. As we are transferring annotations based on sequence similarity, it is necessary for sequence matches to meet several criteria for inclusion in the sequence neighborhood. Search results must have at least 40% sequence identity, 60% alignment coverage of both the query and target sequences, and a minimum of 100 aligned residues. We refer to this step as sequence filtering. The criteria for sequence filtering can be changed from the Advanced parameters on the web form. Shorter alignment lengths and relaxed coverage requirements can be useful in situations where the query sequences are short fragments or come from an exotic organism.
Output is usually reduced to only one predicted description and to non‐redundant GO terms (see Figure 2). PANNZER ranks GO predictions according to an enrichment statistic, which compares the frequency of a GO class in the sequence neighborhood to its frequency in the database. 15 , 22 The nonredundant subset removes all GO classes that have a higher scoring parent or descendant.
FIGURE 2.
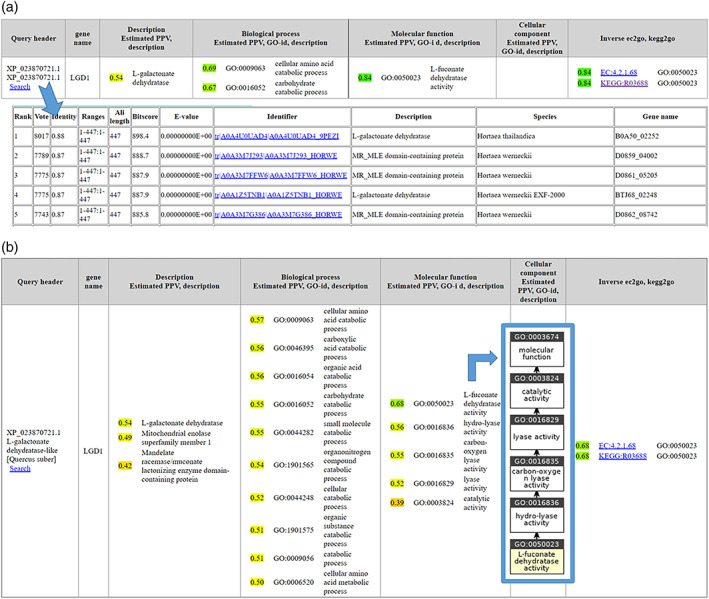
Output of Pannzer web server for test case. This view is for browsing; users can also download the results in a parseable format. Upper part: Nonredundant result obtained using default parameters presents a filtered view. Bottom: sequence neighborhood opens on clicking the “Search” link. Lower part: Result with parameter settings “show only one DE” off and “show only non‐redundant GO terms” off. This view shows alternative predictions ordered by reliability. Inset: GO hierarchy plotted with QuickGO (https://www.ebi.ac.uk/QuickGO/slimming)
Sometimes homology transfer results in biologically implausible annotations such as predicting an organ in plants that only exists in animals. Per a user request, we have implemented a set of taxonomic branch specific GO subsets. These limit the GO predictions only to GO terms occurring in manually curated annotations in fungi, plants, vertebrates, arthropods or bacteria, respectively. This branch specific GO filter is off by default.
2.3. Outputs
Pannzer predicts short descriptions (DE), GO‐terms, gene names, EC classes and KEGG pathways. The web server produces nested tables for browsing (Figure 2) and a tabular format for downloading. PANNZER returns Positive Predictive Value, PPV, as a reliability estimate. PPV estimates the probability of the annotation being correct. Here, correct descriptions are a short TF‐IDF distance (see 15 ) from the most precise terms in the ground truth as given by the Uniprot database. The Uniprot database only reports the most specific terms of a protein, leaving out the implicit parent terms. Pannzer predictions for a query protein are targeted to these biologically most informative GO terms. Note that our definition of correct annotations differs from the probability of class membership of any and all GO classes in the GO hierarchy, which is evaluated, for example, in CAFA. 21 , 23 , 24
2.4. Web server
PANNZER2 provides a fast, publically accessible web server for functional annotation. 22 PANNZER2 uses SANSparallel for high‐performance homology searches, making bulk annotation based on sequence similarity practical. It accepts query sets up to 100,000 protein sequences. Its throughput exceeds 1 M query sequences per day, roughly corresponding to 200 bacterial genomes or 10 eukaryotic transcriptomes. The results can be either downloaded for local post‐processing or browsed via a web application. PANNZER2 outputs both GO and DE predictions. The web server front‐end provides links to homology search results for each query sequence, enabling users to see how predictions were derived.
2.5. Software
PANNZER2 is implemented in a Python framework called SANSPANZ. The SANSPANZ framework frees method developers from data management, as homology searches and fetching metadata can be delegated to remote servers, though it is also possible to install the servers locally. The framework includes “functors” performing computations on data frames. For example, SANSPANZ includes implementations of the scoring functions from ARGOT, 16 BLAST2GO 32 and PANNZER, 15 as well as hypergeometric enrichment and best informative hit. Novel application workflows are implemented by chaining “functors” one after another. PANNZER2 15 and AAI‐profiler for taxonomic profiling 33 are fully integrated into SANSPANZ, while LazyPipe for virus identification 34 is partly built on it. SANSPANZ is open source (http://ekhidna2.biocenter.helsinki.fi/sanspanz/#tab-2).
2.6. Data servers
Both the SANSparallel server and DictServer are maintained by our group. The SANSparallel server takes a protein sequence as input and returns a list of similar sequences from the Uniprot database. Both GO annotations and free text descriptions are gathered for each search result by calling the DictServer. The DictServer is a store of key/value pairs. The servers work locally as a client–server or they can be accessed remotely via CGI. We keep a local copy of the databases used by PANNZER2 (Appendix A). The databases are updated on a regular schedule, ensuring that predictions benefit from new data. Our database update cycle is synchronized with the bimonthly new releases of the Uniprot database. The Uniprot database is mirrored from EBI, indexed and imported to the SANSparallel server. GOA annotations, the GO structure and taxonomy lists are downloaded and imported to the DictServer with recomputed background statistics for DE and GO enrichment analysis.
3. RESULTS
3.1. Case study 1: data conflicts
Database annotations have varying quality. Here, we have deliberately chosen an example, which does not run smooth in Pannzer analysis. The example is a predicted L‐galactonate dehydratase‐like protein from the cork oak (Quercus suber) genome assembly CorkOak1.0. 35 The protein sequence (Appendix B) was submitted to the Pannzer web server (Figure 2). Clicking the “search” link in the web server output calls the SANSparallel server and displays the sequence neighborhood on which the predictions are based. Surprisingly, the sequence neighborhood consists of proteins from unexpected taxa: they are all fungi. This is not an isolated case, either. Taxonomic profiling by PANNZER2's companion tool, AAI‐profiler, 33 shows that 21% of the proteins predicted from the cork oak genome have a closest match in fungi. The proteome is 97% complete with respect to BUSCO's single‐copy ortholog set for Ascomycota and 96% complete with respect to that for eudicots. 36 We conclude that the cork oak genome assembly includes a fungal co‐isolate.
Gene names and descriptions are free text. Pannzer applies spam filters and clustering to identify informative entries with the most support in the data. The gene name is identified as lgd1 (Figure 2, column 2). This gene was first characterized in Trichoderma reesei and shown to convert L‐galactonate to 2‐dehydro‐3‐deoxy‐L‐galactonate and to be required for growth on D‐galacturonate. 37 lgd1 occurs eight times in the sequence neighborhood; all other gene names are unique genomic locus identifiers typified by the use of underscores (these are ignored). Free text descriptions fall into three clusters (Figure 2b, column 3). The alternative descriptions represent different levels of evolutionary classification. Galactonate dehydratase is a subgroup of the mandelate racemase/muconate lactonizing (MR/MLE) family, which in turn belongs to the broad, functionally diverse enolase superfamily. 38 L‐galactonate dehydratase is the preferred description based on sequence similarity weighting. The MR/MLE family and enolase superfamily annotations are based on InterPro profile matches. 5 Phylogenetic analysis confirms that our test example is firmly embedded in the L‐galactonate dehydratase subgroup (Figure 3).
FIGURE 3.
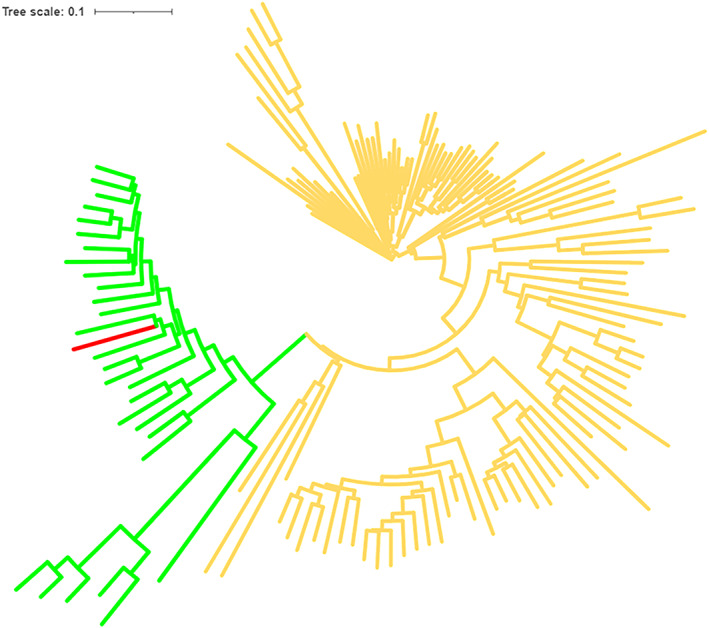
Phylogenetic tree of example test sequence (red) with seed sequences of the L‐galactonate dehydratase (green) and L‐fuconate dehydratase (yellow) subgroups of SFLD. 58 Tree generated by MAFFT, 59 displayed in iToL 60
The second surprise is that, at first glance, the predicted description and predicted GO classification contradict each other (Figure 2, column 3 vs. column 5). L‐galactonate dehydratase activity is a sister class of the predicted L‐fuconate dehydratase activity in GO's molecular function ontology. The GO prediction has high confidence, because 97 of 100 entries in the sequence neighborhood are annotated with L‐fuconate dehydratase activity. Perusal of the literature shows that neither of these predictions is wrong.
L‐galactonate dehydratase (gaaB) and L‐fuconate dehydratase (fucD) are promiscuous, accepting both L‐fuconate and L‐galactonate as substrate. 39 , 40 L‐Galactonate and L‐fuconate have a similar chemical structure; the only difference is that L‐fuconate has no hydroxyl group at the C6. The source material for Pannzer's GO predictions consisted solely of annotations automatically inferred using Interpro2GO. The experimentally characterized proteins fall outside the sequence neighborhood of our test example, which was restricted to 100 hits. The GO annotations for the Lgd1 protein are inferred from direct assay (IDA) as carbonate dehydratase activity and D‐galacturonate catabolic process, a descendant of carboxylic acid catabolic process (cf. Figure 2b, column 4). These annotations have not propagated as far in the database as the gene name or description, which were picked up by Pannzer.
L‐fuconate catabolism has not been demonstrated in fungi. 39 Accordingly, if the taxonomic branch specific GO filter for fungi is activated in Pannzer, L‐fuconate dehydratase activity is not reported and the parent term hydro‐lyase activity becomes the top prediction and also the KEGG pathway prediction (Figure 2, column 7) drops away. Here, the taxonomic filter is set to fungi based on the previous taxonomic observations.
PANNZER results presented on the web page (Figure 2) are hyperlinked to sequences, metadata, and sequence databases with further links to literature. This facilitates tracking down the origins of unexpected results as illustrated here.
3.2. Case Study 2: comparison of results by AFP methods
Our second example represents results from various web servers for a query sequence KAG7012684.1 (Appendix B). This is a ribosomal protein from a recently sequenced pumpkin genome (GenBank date 16‐JUN‐2021). 41 This should be clearly an easy case for AFP methods, as (a) there are several reasonably well annotated plant genomes available and (b) ribosome is well annotated across species. So here we look what different methods report in quite a trivial case. All the compared AFP methods of Table 1 have been among the top 10 in CAFA competitions. In addition, we tested also eggNOG, 14 a popular annotation tool. The results (Table 2) also include the naïve model. The naïve model does not use any information on the sequence. It simply reports the frequency of each GO term in the whole database.
TABLE 2.
Rank of MF predictions by web servers for novel 60S ribosomal protein L5
| GO class | naive | ARGOT | FunFam | INGA | NetGO | PANNZER filtered | PANNZER unfiltered | PFP | Description |
|---|---|---|---|---|---|---|---|---|---|
| GO:0003674 | 1 | 1 | 1 | molecular_function | |||||
| GO:0005198 | 40 | 1 | 3 | 4 | + structural molecule activity | ||||
| GO:0003735 | 49 | 2 | 3 | 1 | 3 | 2 | 3 | 1 | ++ structural constituent of ribosome |
| GO:0005488 | 3 | 1 | 2 | 9 | + binding | ||||
| GO:0005515 | 96 | 1 | 7 | ++ protein binding | |||||
| GO:0097159 | 4 | 1 | 5 | 8 | ++ organic cyclic compound binding | ||||
| GO:1901363 | 5 | 1 | 5 | 7 | 5 | ++ heterocyclic compound binding | |||
| GO:0003676 | 7 | 1 | 8 | 6 | 4 | +++ nucleic acid binding | |||
| GO:0003723 | 37 | 4 | 4 | 1 | 8 | 5 | ++++ RNA binding | ||
| GO:0019843 | 137 | 3 | 1 | 8 | 2 | 2 | +++++ rRNA binding | ||
| GO:0008097 | 1229 | 1 | 2 | 1 | 8 | 1 | 1 | 3 | ++++++ 5S rRNA binding |
Note: The NCBI identifier of the query sequence is KAG7012684.1. Subclasses in GO hierarchy are shown by indentation, padded by “+.” Nonredundant subsets of predictions are bold. Top 10 classes above reliability threshold by any AFP method are included, with the exception of FunFam which had an aberrant profile. Ranks 1–3 are shaded. We show two versions of PANNZER, with and without the filtering of redundant GO terms. FunFam GO terms were ranked by the number of annotations in the top family.
The results show different behavior of methods: PANNZER, 22 ARGOT 16 and PFP 42 report a group of informative GO term as their top predictions. These are linked to ribosomal RNA binding and to ribosome as expected. By default, PANNZER shows a non‐redundant list of GO terms; without the GO filtering, it also shows parent or child terms of the strongest predictions. FunFam 12 maps the query sequence to superfamilies, which include functionally diverse members. FunFam predictions included mRNA 3′/5′‐UTR binding and ubiquitin ligase binding activities, as well as the rather ubiquitous Protein Binding. EggNOG 14 surprisingly did not predict any GO classes. INGA 43 reports a bag of GO classes with the same score. NetGO 10 , 44 , 45 shows different behavior from other methods. NetGO is most confident in predicting the root class and classes close to root resembling, in some ways, the naïve model. Indeed, NetGO uses the naïve model as one of its information sources. This kind of result ranking will improve performance in CAFA competitions, when the evaluation is done with traditional evaluation metrics. These near root classes convey, however, little information to end users. NetGO was actually the clear winner, with some evaluation metrics, in the recent CAFA3 competition. 23 Our discussion covers this topic more in detail. Altogether, these results illustrate how different methods find and emphasize different GO terms.
4. DISCUSSION
Managing a comprehensive annotation pipeline involves keeping databases up‐to‐date and ensuring that growing disk space and memory requirements are met. This has made public web servers convenient and popular for annotation. PANNZER can be used via a web server or installed locally with programmatic access to our group's data servers. The benefits of PANNZER include its high throughput, which has allowed users to run a large collection of genomes in pangenome projects and sequence collections from metagenome or transcriptome projects. PANNZER also generates independently DE and GO predictions. Time‐consuming manual curation can focus on contradictory or borderline predictions and checking if they can be confirmed. Current weaknesses include the lack of alternative information sources, like protein–protein interaction data or gene expression data. Notice that these are difficult to apply, when we are studying a novel genome. Large enough collections of interaction data and gene expression data are mostly available for model organisms and other well‐studied organisms. So, the analysis should first map the query sequence to a similar sequence in the interaction database, for example, after which it can process the actual interaction data. Fortunately, there is currently research on this topic. 46 , 47 , 48 Furthermore, various profile alignment based features, like protein domains and sequence motifs, are expected to be useful for function prediction. These two sources are currently missing from PANNZER. We are, however, currently working on these data sources. In addition, the recent surge of structural models 49 , 50 should rekindle research into specificity determining residues. 51 , 52
AFP method development and publishing require that the results from various AFP methods are compared and evaluated. PANNZER selects a more informative level from the GO tree than many competing methods (see Table 2 for example). Comparison of AFP methods also requires that we have established a ground truth, that is, suitable evaluation datasets that contain sequences with known function. The case study illustrated that defining the known function can be a precarious business. The current datasets have practically only knowledge that a protein has a certain function. We can rarely state that a protein does not have a certain function. This is often referred as the positive—unlabeled learning task. This problem is further heightened by some very vague annotations that sequences can have in the databases, like Protein Binding. Any more precise annotation would be assessed as a wrong prediction. 53 Notice that also the biologically correct function predictions will be evaluated as bad predictions, if this function is absent from the evaluation dataset. Other researchers have looked at these research questions in detail. 54 When the evaluation dataset has mostly annotations with very broad unspecific classes, it is better for AFP methods not to predict specific functions but rather broad and vague classes. This is why we have used our own evaluation datasets, with detailed annotations, parallel with the popular CAFA evaluation datasets. 22 Our gold standard has been a set of GO‐annotated sequences, selected from the UniProt database. We especially required that (a) each sequence has at least one manually curated GO annotation, (b) these GO annotations must occur in small informative GO classes, (c) sequences should not have strong sequence similarities (see 22 ). We have been able to show that PANNZER shows good performance especially when the evaluation dataset contains detailed information on functions.
The evaluation metric compares the generated predictions to known correct functions. The selection of this metric for a hierarchical classification like GO is not trivial and it has a strong effect on the results: ranking of the methods varies drastically between different metrics. 20 , 55 This has been shown also outside the bioinformatics field. 56 , 57 Our recent work used simulated data to show that some evaluation metrics fail to separate different amounts of error in the predictions. 19 Furthermore, some used evaluation metrics ranked earlier discussed naïve model, among the best predictions. This will lead to situation where the field is promoting methods that predict quite uninformative results. This can be corrected by using more advanced metrics, like weighted Jaccard correlation (SimGIC), term‐centric AUCPR, etc.; these metrics and further alternatives are explained in Reference 19. We propose that AFP researchers would demonstrate the results on several evaluation datasets and would use mainly evaluation metrics that are insensitive to biases in class sizes.
AUTHOR CONTRIBUTIONS
Petri Toronen: Investigation (equal); methodology (equal); writing – original draft (equal). Liisa Holm: Investigation (equal); methodology (equal); writing – original draft (equal).
CONFLICT OF INTEREST
The authors have no competing interests.
ACKNOWLEDGMENTS
This work was supported by NNF20OC0065157 of the Novo Nordisk Foundation. The Pannzer web server is part of Biocenter Finland's bioinformatics network.
Appendix A. DATA SOURCES USED BY PANNZER2
Appendix B. TEST SEQUENCES USED IN THIS WORK
>KAG7012684.1 60S ribosomal protein L5, partial [Cucurbita argyrosperma subsp. argyrosperma]
MNSPFLELTASMFAFAKAQKTKAYFKRYQVKFKRRREGKTDYRARIRLINQDKNKYNTPKYRIVVRFSNKDITAQIISASIAGDLVLASAYSHELPRYGLEVGLTNYAAAYCTGLLLARRVLKQLEMDDEYEGNVEATGEDYSVEPADTRRPFRALLDVGLLKTTTGNRVFGALKGALDGGLDIPHSDKRFAGFSKDSKQLDADVHRKYIYGGHVAAYMRTLMEDEPEKYQTHFSEYIKKGIEADDIEGLYKKVHAAIRADPSVKKSDKPQPKAHKRYNLKKLTYDERKARLVERLNALNSAANADDDDDDEDDE
>XP_023870721.1 L‐galactonate dehydratase‐like [Quercus suber]
MVLITHATTRDVRFPTSLDKTGSDAMNAAGDYSAAYVMLHSDTSHTGHGMTFTIGRGNEIVCKAISVLAQRVEGKQLEDLVADWGKTWRYLVSDSQLRWIGPEKGVIHLALGAVVNAIWDLWAKVLGKPVWRIVAEMSPQEFVRCIDFRYITDAITPEEAISMLEKEEAGKAQRIKEAEQNRAVPAYTTSAGWLGYGEAKMKGLLEETLAKGYKHFKLKVGTSLEADKQRLAIARDVIGYDNGNVLMVDANQVWSVPEAITYMKELARFKPWFIEEPTSPDDVFGHKAIREALKPYNIGVATGEMCQNRVMFKQLIVQGAIDVCQIDACRIGGVNEVMAVMLIAKKYGVPIVPHSGGVGLPEYTQHLSTIDYVVVSGKLSVLEYVDHLHEHFLHPSIIESGYYVTPTMPGYSVEMKAESMEQYEFPGTEGVSWWRSAQAKGILEGEKI
Törönen P, Holm L. PANNZER—A practical tool for protein function prediction. Protein Science. 2022;31:118–128. 10.1002/pro.4193
Funding information Novo Nordisk Foundation, Grant/Award Number: NNF20OC0065157
REFERENCES
- 1. Wang B, Lin Z, Li X, et al. Genome‐wide selection and genetic improvement during modern maize breeding. Nat Genet. 2020;52:565–571. [DOI] [PubMed] [Google Scholar]
- 2. Morabito C, Aiese Cigliano R, Maréchal E, Rébeillé F, Amato A. Illumina and PacBio DNA sequencing data, de novo assembly and annotation of the genome of Aurantiochytrium limacinum strain CCAP_4062/1. Data Brief. 2020;31:105729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Simonet C, McNally L. Kin selection explains the evolution of cooperation in the gut microbiota. Proc Natl Acad Sci U S A. 2021;118:e2016046118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Duru IC, Ylinen A, Belanov S, et al. Transcriptomic time‐series analysis of cold‐ and heat‐shock response in psychrotrophic lactic acid bacteria. BMC Genomics. 2021;22:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Blum M, Chang H, Chuguransky S, et al. The InterPro protein families and domains database: 20 years on. Nucleic Acids Res. 2021;49:D344–D354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. The UniProt Consortium . UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021;49:D480–D489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. O'Leary NA, Wright MW, Brister JR. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016;44:D733–D745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ashburner M, Ball C, Blake J, et al. Gene ontology: Tool for the unification of biology. Nat Genet. 2000;25:25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kanehisa M, Furumichi M, Sato Y, Ishiguro‐Watanabe M, Tanabe M. KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 2021;49:D545–D551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yao S, You R, Wang S, et al. NetGO 2.0: Improving large‐scale protein function prediction with massive sequence, text, domain, family and network information. Nucleic Acids Res. 2021;49:W469–W475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cozzetto D, Minneci F, Currant H, et al. FFPred 3: Feature‐based function prediction for all gene ontology domains. Sci Rep. 2016;6:31865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Scheibenreif L, Littmann M, Orengo C, et al. FunFam protein families improve residue level molecular function prediction. BMC Bioinformatics. 2019;20:400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Profiti G, Martelli PL, Casadio R. The Bologna annotation resource (BAR 3.0): Improving protein functional annotation. Nucleic Acids Res. 2017;45:W285–W290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Huerta‐Cepas J, Szklarczyk D, Heller D, et al. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019;47:D309–D314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Koskinen P, Törönen P, Nokso‐Koivisto J, Holm L. PANNZER: High‐throughput functional annotation of uncharacterized proteins in an error‐prone environment. Bioinformatics. 2015;31:1544–1552. [DOI] [PubMed] [Google Scholar]
- 16. Lavezzo E, Falda M, Fontana P, Bianco L, Toppo S. Enhancing protein function prediction with taxonomic constraints – The Argot2.5 web server. Methods. 2016;93:15–23. [DOI] [PubMed] [Google Scholar]
- 17. Schnoes AM, Brown SD, Dodevski I, Babbitt PC. Annotation error in public databases: Misannotation of molecular function in enzyme superfamilies. PLoS Comput Biol. 2009;5:e1000605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Makrodimitris S, van Ham R, Reinders MJT. Automatic gene function prediction in the 2020's. Genes. 2020;11:1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Plyusnin I, Holm L, Törönen P. Novel comparison of evaluation metrics for gene ontology classifiers reveals drastic performance differences. PLoS Comput Biol. 2019;15:e1007419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gillis J, Pavlidis P. Characterizing the state of the art in the computational assignment of gene function: Lessons from the first critical assessment of functional annotation (CAFA). BMC Bioinformatics. 2013;14:S15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Radivojac P, Clark WT, Oron TR, et al. A large‐scale evaluation of computational protein function prediction. Nat Methods. 2013;10:221–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Törönen P, Medlar AJ, Holm L. PANNZER2: A rapid functional annotation web server. Nucleic Acids Res. 2018;46:W84–W88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhou N, Jiang Y, Bergquist TR, et al. The CAFA challenge reports improved protein function prediction and new functional annotations for hundreds of genes through experimental screens. Genome Biol. 2019;20:1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jiang Y, Oron TR, Clark WT, et al. An expanded evaluation of protein function prediction methods shows an improvement in accuracy. Genome Biol. 2016;17:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hippe K, Gbenro S, Cao R, 2020. ProLanGO2: Protein function prediction with ensemble of encoder‐decoder networks. In Proceedings of the 11th ACM international conference on bioinformatics, computational biology and health informatics (BCB '20). Association for Computing Machinery, New York, NY, Article 103, 1–6. DOI: 10.1145/3388440.3414701 [DOI] [Google Scholar]
- 26. Cao R, Freitas C, Chan L, Sun M, Jiang H, Chen Z. ProLanGO: Protein function prediction using neural machine translation based on a recurrent neural network. Molecules. 2017;22:1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wimalanathan K, Friedberg I, Andorf CM, Lawrence‐Dill CJ. Maize GO annotation‐methods, evaluation, and review (maize‐GAMER). Plant Direct. 2018;2:e00052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010;11:119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yandell M, Ence D. A beginner's guide to eukaryotic genome annotation. Nat Rev Genet. 2012;13:329–342. [DOI] [PubMed] [Google Scholar]
- 30. Grabherr MG, Haas BJ, Yassour M, et al. Full‐length transcriptome assembly from RNA‐seq data without a reference genome. Nat Biotechnol. 2011;29:644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Somervuo P, Holm L. SANSparallel: Interactive homology search against Uniprot. Nucleic Acids Res. 2015;43:W24–W29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Götz S, Garcia‐Gomez JM, Terol J, et al. High‐throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008;36:3420–3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Medlar AJ, Törönen P, Holm L. AAI‐profiler: Fast proteome‐wide exploratory analysis reveals taxonomic identity, misclassification and contamination. Nucleic Acids Res. 2018;46:W479–W485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Plyusnin I, Kant R, Jääskeläinen AJ, et al. Novel NGS pipeline for virus discovery from a wide spectrum of hosts and sample types. Virus Evol. 2020;6:veaa091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ramos AM, Usié A, Barbosa P, et al. The draft genome sequence of cork oak. Sci Data. 2017;5:180069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. BUSCO: assessing genome assembly and annotation completeness with single‐copy orthologs. Bioinformatics. 2015;31:3210–3212. 10.1093/bioinformatics/btv351. [DOI] [PubMed] [Google Scholar]
- 37. Kuorelahti S, Jouhten P, Maaheimo H, Penttila M, Richard P. L‐galactonate dehydratase is part of the fungal path for D‐galacturonic acid catabolism. Mol Microbiol. 2006;61:1060–1068. [DOI] [PubMed] [Google Scholar]
- 38. Babbitt PC, Hasson MS, Wedekind JE, et al. The enolase superfamily: A general strategy for enzyme‐catalyzed abstraction of the alpha‐protons of carboxylic acids. Biochemistry. 1996;35:16489–16501. [DOI] [PubMed] [Google Scholar]
- 39. Motter FA, Kuivanen J, Keränen H, Hilditch S, Penttilä M, Richard P. Categorisation of sugar acid dehydratases in Aspergillus niger . Fungal Genet Biol. 2014;64:67–72. [DOI] [PubMed] [Google Scholar]
- 40. Yew WS, Fedorov AA, Fedorov EV, et al. Evolution of enzymatic activities in the enolase superfamily: l‐fuconate dehydratase from Xanthomonas campestris . Biochemistry. 2006;45:14582–14597. [DOI] [PubMed] [Google Scholar]
- 41. Barrera‐Redondo J, Ibarra‐Laclette E, Vázquez‐Lobo A, et al. The fenome of Cucurbita argyrosperma (Silver‐Seed Gourd) reveals faster rates of protein‐coding gene and long noncoding RNA turnover and neofunctionalization within cucurbita. Mol Plant. 2019;12:506–520. [DOI] [PubMed] [Google Scholar]
- 42. Hawkins T, Chitale M, Luban S, Kihara D. PFP: Automated prediction of gene ontology functional annotations with confidence scores using protein sequence data. Proteins. 2009;74:566–582. [DOI] [PubMed] [Google Scholar]
- 43. Piovesan D, Tosatto S. INGA 2.0: Improving protein function prediction for the dark proteome. Nucleic Acids Res. 2019;47:W373–W378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. You R, Yao S, Xiong Y, et al. NetGO: Improving large‐scale protein function prediction with massive network information. Nucleic Acids Res. 2019;47:W379–W387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. You R, Zhang Z, Xiong Y, Sun F, Mamitsuka H, Zhu S. GOLabeler: Improving sequence‐based large‐scale protein function prediction by learning to rank. Bioinformatics. 2018;34:2465–2473. [DOI] [PubMed] [Google Scholar]
- 46. You R, Yao S, Mamitsuka H, Zhu S. DeepGraphGO: Graph neural network for large‐scale, multispecies protein function prediction. Bioinformatics. 2021;37:i262–i271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fan K, Guan Y, Zhang Y. Graph2GO: A multi‐modal attributed network embedding method for inferring protein functions. GigaScience. 2020;9:giaa081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Barot M, Gligorijević V, Cho K, Bonneau R. NetQuilt: Deep multispecies network‐based protein function prediction using homology‐informed network similarity. Bioinformatics. 2021;37:2414–2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jumper J, Evans R, Pritzel A, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Baek M, DiMaio F, Anishchenko I, et al. Accurate prediction of protein structures and interactions using a three‐track neural network. Science. 2021;373:871–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Marttinen P, Corander J, Törönen P, Holm L. Bayesian search of functionally divergent protein subgroups and their function specific residues. Bioinformatics. 2006;22:2466–2474. [DOI] [PubMed] [Google Scholar]
- 52. Ward RM, Venner E, Daines B, et al. Evolutionary trace annotation server: Automated enzyme function prediction in protein structures using 3D templates. Bioinformatics. 2009;25:1426–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Clark WT, Radivojac P. Information‐theoretic evaluation of predicted ontological annotations. Bioinformatics. 2013;29:i53–i61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Warwick Vesztrocy A, Dessimoz C. Benchmarking gene ontology function predictions using negative annotations. Bioinformatics. 2020;36:i210–i218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kahanda I, Funk CS, Ullah F, Verspoor KM, Ben‐Hur A. A close look at protein function prediction evaluation protocols. GigaScience. 2015;4:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Valverde‐Albacete FJ, Peláez‐Moreno C. 100% classification accuracy considered harmful: The normalized information transfer factor explains the accuracy paradox. PloS one. 2014;9:e84217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ferri C, Hernández‐Orallo J, Modroiu R. An experimental comparison of performance measures for classification. Pattern Recognit Lett. 2009;30:27–38. [Google Scholar]
- 58. Akiva E, Brown S, Almonacid DE, et al. The structure‐function linkage database. Nucleic Acids Res. 2014;42:D521–D530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Katoh K, Rozewicki J, Yamada KD. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinformatics. 2019;20:1160–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Letunic I, Bork P. Interactive tree of life (iTOL) v3: An online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016;44:W242–W245. [DOI] [PMC free article] [PubMed] [Google Scholar]