

Abstract
Phylogenetics is a powerful tool for analyzing protein sequences, by inferring their evolutionary relationships to other proteins. However, phylogenetics analyses can be challenging: they are computationally expensive and must be performed carefully in order to avoid systematic errors and artifacts. Protein Analysis THrough Evolutionary Relationships (PANTHER; http://pantherdb.org) is a publicly available, user‐focused knowledgebase that stores the results of an extensive phylogenetic reconstruction pipeline that includes computational and manual processes and quality control steps. First, fully reconciled phylogenetic trees (including ancestral protein sequences) are reconstructed for a set of “reference” protein sequences obtained from fully sequenced genomes of organisms across the tree of life. Second, the resulting phylogenetic trees are manually reviewed and annotated with function evolution events: inferred gains and losses of protein function along branches of the phylogenetic tree. Here, we describe in detail the current contents of PANTHER, how those contents are generated, and how they can be used in a variety of applications. The PANTHER knowledgebase can be downloaded or accessed via an extensive API. In addition, PANTHER provides software tools to facilitate the application of the knowledgebase to common protein sequence analysis tasks: exploring an annotated genome by gene function; performing “enrichment analysis” of lists of genes; annotating a single sequence or large batch of sequences by homology; and assessing the likelihood that a genetic variant at a particular site in a protein will have deleterious effects.
Keywords: gene ontology, genome analysis, hidden Markov model, molecular evolution, omics data analysis, phylogenetic tree, protein function annotation, protein function evolution
1. INTRODUCTION
Repeated processes (“events”) of speciation, gene duplication and horizontal transfer, for long periods of time, have created families of proteins that are evolutionarily related. Using protein sequences and structures, it is often possible to reconstruct those evolutionary relationships, inferring the phylogenetic tree that identifies the evolutionary events and relates the proteins to each other. However, there are many pitfalls in evolutionary reconstruction that make it challenging, including treating partial or incorrect sequences, ensuring alignment quality, and developing algorithms that can efficiently handle large, diverse protein families. Phylogenetics remains an active field of research. 1 , 2 , 3
The application of phylogenetics to analyze protein function was first proposed by Eisen, 4 who termed it phylogenomics. A phylogenomics analysis begins with the construction of a phylogenetic tree that describes the evolutionary history of a protein‐coding gene family. Knowledge about protein function is then overlaid on the tree to identify how different functions may be partitioned into distinct clades of the tree. Phylogenomics represented an important advance; in previous work, families or groups of related proteins were generally annotated by finding the function that was conserved among the largest number of family members. 5 , 6 Phylogenomics reflected the growing recognition that protein function could diverge during the course of evolution, and that such divergence events could be identified through an analysis of a phylogenetic tree. A phylogenomics analysis can provide a more accurate and precise annotation of protein function, by assigning each protein not just to a particular family, but to a specific subtree (subfamily) within that family.
Protein Analysis THrough Evolutionary Relationships (PANTHER) represented the first knowledgebase to provide access to phylogenomic analyses of thousands of protein families, 7 , 8 and has been continually updated and improved for over 23 years. During this time, incremental update papers have been published, but it may be challenging for users to combine these into a complete description of the current PANTHER knowledgebase content, knowledge generation pipeline, and common applications. Here, we first describe the history of the PANTHER knowledgebase. We then give a description of the contents of the knowledgebase, including how those contents are generated. Finally, we provide an overview of the major applications of PANTHER, before summarizing our planned future directions. We show how PANTHER trees are being applied to the analysis of not only protein function, but also of an increasing number of other characteristics of protein coding genes.
2. HISTORY OF PANTHER: PRE‐GENOME TO POST‐GENOME
Initially established in 1998, PANTHER was the first database of protein phylogenetic trees. The early versions of the knowledgebase (through version 6, released in 2006) were developed during the “pre‐whole genome era”: whole genome sequences were available for very few species, so the compliment of protein‐coding genes for most species was unknown. Protein sequences were available, but the relationship of these sequences to distinct genes was difficult to establish. As a result, in the early versions of PANTHER, protein sequence trees could not be reconciled with the species tree. Expert curators reviewed the trees, and identified clades in the tree in which protein function appeared to be well conserved (based on annotations from NCBI, and particularly the curated annotations from Swiss‐Prot 9 ). These clades were labeled as “subfamilies,” and hidden Markov models (HMMs) were constructed for each subfamily to enable classification of proteins that were not explicitly included in the phylogenetic trees.
Each subfamily was also associated with ontology terms that described gene function, to provide a representation of function that was amenable to computational analysis. Because no ontologies were available when PANTHER was started, we developed our own hierarchical (strictly speaking, it was a directed acyclic graph, as a child term could have more than one parent term) controlled vocabulary of terms, which we called the PANTHER index, or PANTHER/X. 8 However, following the development of the Gene Ontology (GO), 10 we established a collaboration with Michael Ashburner to map PANTHER/X to GO. The collaboration confirmed that the phylogenetic inference, that is, inferring gene function annotation through internal tree branch annotations, was as accurate as manual curation of individual proteins and much more efficient. 11 PANTHER has used GO for function classification ever since that time. PANTHER version 2, released in 2000, included over 2000 families, and was used to analyze the proteins in the first human genome sequence, 7 with both PANTHER/X and GO classifications.
In 2003, we expanded the functional classifications in PANTHER to include pathways, both metabolic and signaling. 12 While GO biological process terms include pathways, computational pathway representations provide additional information not found in traditional GO annotations: the ordering and dependencies of the individual protein functions within the pathway (Figure 1). From 2003 to 2007, pathways were actively curated in PANTHER using the CellDesigner software package, 13 and initially represented using Systems Biology Markup Language. 14 We joined in the effort to create the BioPAX standard for sharing annotated pathway information, 15 and have made PANTHER Pathways available in this format since that time. Graphical representations that are compliant with the Systems Biology Graphical Notation standards 16 are also available for all pathways. PANTHER Pathways 17 are now distributed as part of the PathwayCommons initiative. 18 In addition to PANTHER Pathways, we now import pathways from the Reactome resource 19 , 20 as well. In return, Reactome imports PANTHER orthologs for creating inferred pathways in nonhuman model organisms.
FIGURE 1.
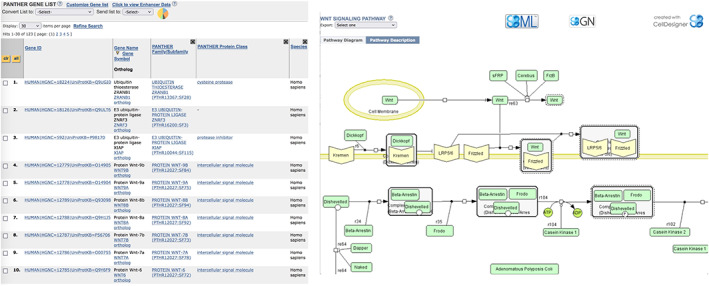
List of unconnected proteins for the GO class (“Wnt signaling pathway” (GO:0016055) (left panel), compared to connected components of the PANTHER Pathway P00057 “Wnt signaling pathway” (right panel). Each pathway component is a generalized protein (a group of one or more specified clades of related proteins in the knowledgebase) that participate in sequential steps/reactions in the pathway
As more whole genomes became available and protein‐coding genes could be robustly identified, it became possible to assemble essentially complete sets of protein‐coding genes for organisms across the tree of life. Between 2007 and 2009, we made a large number of improvements to the PANTHER knowledge generation pipeline to take advantage of these “whole genome era” advancements. Protein sequences were now mapped to protein‐coding genes, and a single representative sequence was selected for each protein coding gene from each genome. Genomes were carefully selected to elucidate genome evolution among the most highly studied genomes (human, mouse, rat, zebrafish, Drosophila, Caenorhabditis elegans, Dictyostelium, fission and budding yeasts, Arabidopsis, and Escherichia coli), which are the source of most knowledge about gene function. The GIGA algorithm was developed for reconstructing reconciled trees, 21 in which each leaf node represents a protein coding gene in an extant organism, and the internal nodes represent identifiable speciation, duplication, and transfer events in evolutionary history. PANTHER version 7 was released in 2010. 22 Since 2010, PANTHER has been continually expanded to cover more families (Table 1) and more species (Supplementary Table).
TABLE 1.
Number of PANTHER families, and proteins in the trees, for different released versions of PANTHER. Version 1 was created in 1998–1999 for testing and validation purposes and not released
| PANTHER version | Year released | Number of families | Number of proteins in family trees |
|---|---|---|---|
| 2 | 2000 | 2,068 | |
| 3.1 | 2002 | 6,155 | 271,779 |
| 4 | 2004 | 6,715 | 262,909 |
| 5 | 2005 | 6,683 | 256,413 |
| 6 | 2006 | 5,546 | 221,609 |
| 7 | 2010 | 8,677 | 407,498 |
| 7.2 | 2012 | 8,677 | 419,652 |
| 8 | 2012 | 7,729 | 642,319 |
| 9 | 2013 | 7,180 | 759,660 |
| 10 | 2014 | 11,928 | 1,026,421 |
| 11 | 2015 | 13,096 | 1,064,054 |
| 12 | 2016 | 14,710 | 995,960 |
| 13 | 2017 | 15,524 | 1,062,191 |
| 14 | 2018 | 15,524 | 1,689,338 |
| 15 | 2019 | 15,702 | 2,065,831 |
| 16 | 2020 | 15,635 | 2,063,337 |
The transition to reconciled trees made it possible to change the PANTHER tree annotation paradigm, which has been accomplished in close collaboration with the GO Consortium. 23 Rather than utilizing a small subset of GO terms for annotation of trees—in essence, representing only the largest leaps in protein function evolution—we developed a method and software infrastructure for annotating the evolutionary gain and loss of a function described by any GO term on any branch in the tree. 24 These annotations are made manually by expert curation, by biocurators in the GO Phylogenetic Annotation project. All experimental GO annotations for proteins in the tree are overlaid on the tree, and an expert uses a variety of information to identify branches where functions were most likely gained and lost; this information includes the phylogenetic tree (especially gene duplication events), taxonomic groups, and presence/absence of protein domains and active site residues. The number of families annotated with these fine‐grained functional evolution events has steadily grown, and is currently over 8,500. In this new paradigm, the previous PANTHER GO‐slim (a subset of GO that mapped to PANTHER/X) was retired, and a new, much larger PANTHER GO‐slim was created using the function gain and loss annotations from the GO Phylogenetic Annotation project. PANTHER/X has now been converted into a simpler, strictly hierarchical “Protein Class” used to classify entire protein families, 25 not functionally distinct subfamilies.
The transition to detailed function annotation also impacts how users can best utilize the PANTHER knowledgebase. Because the functional annotations can now be made to any branch in the phylogenetic tree and not just to “subfamily divergence branches,” PANTHER subfamily HMMs are no longer the best method for classifying protein sequences that were not used to construct the phylogenetic trees. We are aware that many users and data analysis pipelines rely on the subfamily HMMs, and we still construct and publish them with each release. However, we encourage users to transition to the TreeGrafter software, 26 which inserts a query sequence into a PANTHER family phylogenetic tree, resulting in a more precise and informative classification.
3. THE PANTHER KNOWLEDGEBASE
We first present a description of the contents of the PANTHER knowledgebase. We then describe in some detail the processes, both computational and manual, that are used to generate the PANTHER resource and update it on a yearly basis.
3.1. Contents of the KB
An overview of the contents of the PANTHER knowledgebase is shown in Figure 2. The PANTHER knowledgebase contains extensive knowledge about protein families, including how family members are related by evolutionary events (phylogenetic tree) and at the level of individual amino acid sites (multiple sequence alignment). The phylogenetic trees have also been annotated with functional divergence events that have been inferred by overlaying experimental GO annotations onto the tree and manual review by expert biocurators. The protein family knowledge is used to derive knowledge about individual proteins in the knowledgebase. In addition to this family‐derived knowledge, PANTHER imports knowledge from other collaborating resources, in order to facilitate analysis workflows by users of PANTHER tools. A recent addition to the knowledgebase is human enhancer regions, and links between these regions and the genes they may regulate. This knowledge is imported from the PEREGRINE database. 27
FIGURE 2.
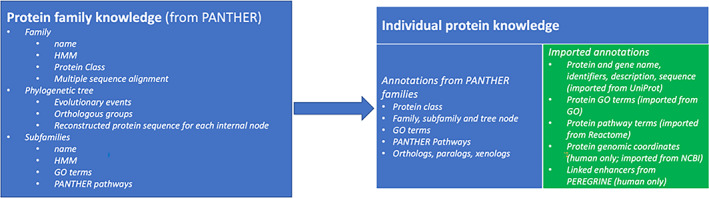
Overview of the content of the Protein Analysis THrough Evolutionary Relationships (PANTHER) KB. Individual proteins (right) are annotated with knowledge derived from the annotated PANTHER families (left), and with knowledge imported from external resources (green). All information is updated yearly, except for imported gene ontology (GO) annotations, which are updated monthly to synchronize with official GO releases
3.1.1. PANTHER family information
Each PANTHER family is given a free‐text name, and assigned to a PANTHER Protein Class. Family names are sometimes curated manually, but more generally, a family is named after the oldest subfamily in the tree (see subfamily naming rules below). If there are multiple subfamilies of the same age (usually due to a gene duplication event at the root of the tree), the one with more extant members is selected, and the word “‐related” is appended to the name. Each family is assigned to a PANTHER Protein Class via manual curation. A family is assigned to a single Protein Class, except in the rare case that its members contain multiple functional domains with distinct functions (generally these are due to domain fusions of enzymes that catalyze multiple reactions in the same pathway).
For each protein family, PANTHER provides a multiple sequence alignment (MSA), a phylogenetic tree, and a family HMM. Details on their construction are provided in the Supplementary Material. Users can also see the exact alignment columns that were used to construct the phylogenetic tree (they are upper case letters and dashes, “‐,” in the alignments; lower case letters and dots, “.,” are masked and not used for phylogenetic reconstruction), in either the downloadable alignment files or the interactive PANTHER TreeViewer tool on the website.
3.1.2. PANTHER trees and orthologs, paralogs, and xenologs
The phylogenetic trees in PANTHER are properly considered to be gene trees, representing the evolutionary history of each extant protein‐coding gene (leaf) in the tree. The trees in the current version of PANTHER include 142 fully sequenced genomes, covering 19 vertebrates, 15 invertebrates, 14 fungi, 40 plants, 11 other eukaryotes, 8 archaea, and 35 bacteria (full list available at http://aws.pantherdb.org/panther/summaryStats.jsp). The genomes are chosen to sample the tree of life, with deeper sampling surrounding well‐studied organisms that are the source of most experimental functional annotations. PANTHER gene trees are fully reconciled with the species tree, meaning that each internal node in each PANTHER tree is labeled by the evolutionary event type it represents: speciation, gene duplication, or horizontal gene transfer (out of the millions of nodes in PANTHER trees, a handful nodes are labeled “UNK,” for unknown, as we were not able to infer the type of event). The node event type information is available in the downloadable tree files, as well as in the TreeViewer (nodes are colored by their event type). Unlike any other resource of trees of which we are aware, PANTHER maintains stable identifiers for all tree nodes (both leaves and internal nodes), across versions of PANTHER. This feature allows the annotations on the trees to persist across versions. The branch lengths in the trees are expressed in terms of number of amino acid substitutions per site, including the Jukes–Cantor correction 28 to account for reversions.
Pairs of orthologs (genes that descended from the same gene in the last common ancestor genome of two species) are computed directly from the PANTHER trees. Orthologs are defined in a pairwise manner, following Fitch. 29 We first exclude xenologs, which are genes whose lineages include a horizontal transfer event at any point since their divergence. These pairs are labeled as “xenologs” (X). For each remaining pair of genes in the gene tree, we trace to its last common ancestor node. If that node is a speciation node, the genes are orthologs. If it is a gene duplication, they are paralogs. PANTHER reports all ortholog pairs, but only reports paralog pairs if both genes are from the same species (the most common use case for paralogs is to identify related genes in a given genome). Paralog pairs are also labeled with their age (relative to a speciation event). In this way, users can distinguish between paralogs that derive from recent versus more ancient gene duplication events. Users should also note that orthologs can have 1:1, 1:many, or many:many relationships, depending on whether there have been gene duplications in one or more of the lineages following the divergence of the two species. Thus in general, the more distantly related two species are, the greater the chance that gene duplications have occurred since their divergence, and the greater the chance of observing complex (non 1:1) orthology. Because users often want to identify the single “closest” ortholog in the cases of non 1:1 orthology, we use branch lengths following duplication to identify a single “least‐diverged ortholog” (LDO) pair (though we allow multiple LDOs in the rare case that branch lengths are exactly equal). The LDO label is also used for all 1:1 ortholog pairs (as they are closest by definition). Other, non‐LDO ortholog pairs are labeled with O. PANTHER orthologs have been benchmarked using the Quest for Orthologs benchmarking server. 30 The LDOs have high specificity, while the set of all orthologs (LDO plus O) have high sensitivity on these benchmarks.
3.1.3. Tree annotations and their use to annotate proteins using PANTHER
PANTHER trees are annotated with function gain and loss events, by the GO Consortium as described below. The ancestral annotations are propagated through the tree by inheritance, from a node annotated as having gained a function not present in its ancestor, to the leaves. A loss of function annotation will stop propagation. All PANTHER GO‐slim annotations to proteins are inferred from these curated tree annotations; the terms commonly used in tree annotations represent only a subset of all GO terms, and this subset is used to create a slim, or simplified, ontology structure that we call PANTHER GO‐slim. 31 In addition, these ancestral annotations are used to annotate query sequences using the TreeGrafter tool (see below). PANTHER trees are also starting to be used by the UniProt Consortium for annotating other properties of proteins (see below).
3.1.4. Reconstructed ancestral protein sequences
Each internal node in a PANTHER gene tree can also be interpreted as an ancestral gene, specifically a gene that was the common ancestor of two or more extant genes. 32 The PANTHER knowledgebase also contains the reconstructed amino acid sequences for all of these ancestral genes. For each ancestral gene, there are two sequence reconstructions in PANTHER: a simple, discrete reconstruction, and a probabilistic one. The simple one is shown in the TreeViewer on the website, and is a local parsimony‐based reconstruction. Sites that could not be determined are represented as an ‘X' (the standard one‐letter amino acid code for “unknown”). A useful feature of these reconstructions is that we attempt to identify all amino acid residues that were present in each ancestral protein, including sites that may have been deleted in either an ancestor or a descendant and therefore would not be reconstructed under a substitution‐based model. The probabilistic reconstruction is performed under the WAG substitution model 33 using PAML, 34 and is used in the PANTHER PSEP tool described below.
3.1.5. Subfamily information
PANTHER subfamilies are identified automatically from the phylogenetic trees. A subfamily roughly corresponds to a group of least diverged orthologs, in that most members of each subfamily are mutually least diverged orthologs of all other members of the subfamily. There are two exceptions to this rule. First, subfamilies may also contain paralogs that are unique to only one of the 142 species in PANTHER trees, that is, “in‐paralogs.” Second, subfamilies that derive from a duplication at the base of the vertebrates will only span the vertebrates, and not be extended to least diverged orthologs in nonvertebrate organisms. Each subfamily is named after a selected eponymous protein in the subfamily as provided by the UniProt resource. 35 The eponymous protein is selected from a well‐studied model organism whenever possible, preferably human. Subfamilies are annotated with GO terms by propagation/inheritance from the annotated tree nodes.
3.1.6. Protein information
The PANTHER website contains complete family, subfamily and GO annotation for all protein‐coding genes in the 142 genomes in the PANTHER trees. The fraction of protein‐coding genes assigned to PANTHER families varies by organism, from >95% for vertebrate genomes to around 50% for some divergent archaeal genomes, but the coverage is generally very high for both eukaryotic and prokaryotic genomes (see http://pantherdb.org/panther/summaryStats.jsp for details). For each protein‐coding gene in a PANTHER family, PANTHER family and subfamily names are listed, and users can view the gene in a family tree, and see how its protein sequence aligns to other family members. Users can also access the orthologs, xenologs, and within‐species paralogs. In addition to the PANTHER GO‐slim annotations from the GO Phylogenetic Annotation project (labeled with the GO evidence code IBA), all GO annotations (including all GO evidence codes) for each protein‐coding gene are imported from the GO knowledgebase (http://geneontology.org) into PANTHER, including those genes that are not yet in any PANTHER family. As a result, GO enrichment analysis in PANTHER is complete for protein‐coding genes.
In addition to the 142 organisms in PANTHER trees, we provide precalculated PANTHER family/subfamily and GO annotations for over 1,000 other whole reference proteomes from UniProt. These annotations can be used in the PANTHER gene list analysis tools, including the enrichment analysis tools, as described previously. 36 If users cannot find a genome they are interested in, they can request it from us directly using the PANTHER feedback email provided on the website.
3.1.7. Example of the information in the PANTHER knowledgebase
Figure 3 shows an example of the kinds of information in PANTHER, for the interleukin‐1 (IL1 gene) family. The family is labeled “IL1 family,” which is assigned to the PANTHER protein class “interleukin superfamily” (which is a subclass of “cytokine,” a subclass of “intercellular signal molecule”). However, despite the fact that it is accurate to say that this is a family of cytokines, the functions of some family members have diverged so that they do not function as cytokines. The GO annotations of the tree capture both the conserved functions, and the diverged functions. Based on the protein sequences, the family can be traced back as far as the common ancestor of vertebrates, and the most parsimonious evolutionary scenario suggests that the common ancestral gene functioned as a cytokine, activating the IL1 receptor, as that function is observed among genes in all descendant lineages. Therefore, the root of the tree (green arrow in Figure 3) is annotated with GO terms such as “cytokine activity,” “cytokine‐mediated signaling pathway” and “inflammatory response.” However, prior to the mammalian common ancestor, an ancestral IL1 family member was duplicated multiple times (orange arrow in Figure 3) to generate four genes now found in many extant mammals, including humans: IL1F10, IL36G, IL36RN, and IL1RN. Two of these duplicate genes still function as cytokines (IL1F10 and IL36G), while two of them (IL36RN and IL1RN) have diverged in function to act as antagonists, rather than agonists, of interleukin receptors. The branches leading to IL1RN and IL36RN (red arrows in Figure 3) have been annotated with a loss of “cytokine activity” and “cytokine‐mediated signaling pathway,” and are annotated with gains of the functions “IL1 receptor antagonist activity” and “negative regulation of cytokine‐mediated signaling pathway.” Thus, the same family contains proteins that have very different GO annotations, due to functional divergence.
FIGURE 3.
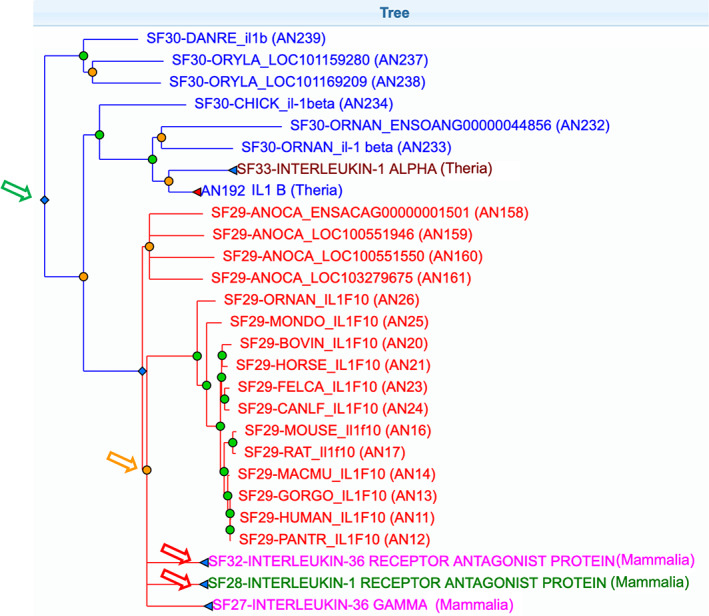
The interleukin 1 family in Protein Analysis THrough Evolutionary Relationships (PANTHER) (PTHR10078). Green circles represent speciation events, while orange circles represent duplication. Diamonds are expanded subfamily nodes, and triangles are collapsed nodes. Most family members are cytokines from the ancestral annotation from the root node of the tree (green arrow), but a phylogenomic analysis suggests that, following gene duplication (orange arrow), two subfamilies of mammalian genes (IL36RN and IL1RN, red arrows) adopted modified functions and now all subfamily members are likely to act as receptor antagonists rather than agonists. Note that we have collapsed some nodes (shown with triangles) here to simplify the diagram by hiding some descendant subtrees. Different colors correspond to different subfamilies. The full family tree can be explored at www.pantherdb.org/treeViewer/treeViewer.jsp?book=PTHR10078
Figure 3 also illustrates PANTHER subfamilies, orthologs and paralogs. The ancestral IL1 gene was duplicated prior to the amniote divergence (orange tree node immediately descending from the root). One of the branches following duplication (namely the branch leading to the clade that includes IL1‐beta in chicken and mammals) is shorter than the other; these leaf genes are therefore in the same subfamily as the fish (e.g., DANRE, Danio rerio) il1b gene, and are also least‐diverged orthologs. The other branch, including IL36, IL10, IL36RN, IL1RN, also contains orthologs of the D. rerio il1b gene, but they are not least diverged (so they are labeled as O in PANTHER, not LDO), and are distinct subfamilies (and therefore colored differently by the PANTHER TreeViewer). Human IL36, IL10, IL36RN, and IL1RN are all paralogs of human IL1, with the age of the duplication dated prior to the amniote common ancestor.
3.2. Creating the KB: The PANTHER pipeline and GO “phylogenetic annotation” project
3.2.1. PANTHER pipeline: From sequences to families and trees
The process for creating the PANTHER knowledgebase is shown in Figure 4. It includes both computational and manual steps, as well as extensive quality assurance (QA). The main inputs to the process are protein sequences, and protein functional annotations. The sequences derive from a selected set of “gene‐centric reference proteomes” from UniProt, 35 each of which is assumed to represent the complete catalog of protein‐coding genes in a given organism. An additional input, necessary to reconstruct reconciled trees (i.e., with annotated evolutionary events, speciation, duplication, and transfer) is the known species tree, which is derived from a meta‐analysis of the literature. 37 The individual PANTHER pipeline steps are described in detail in Supplementary Material.
FIGURE 4.
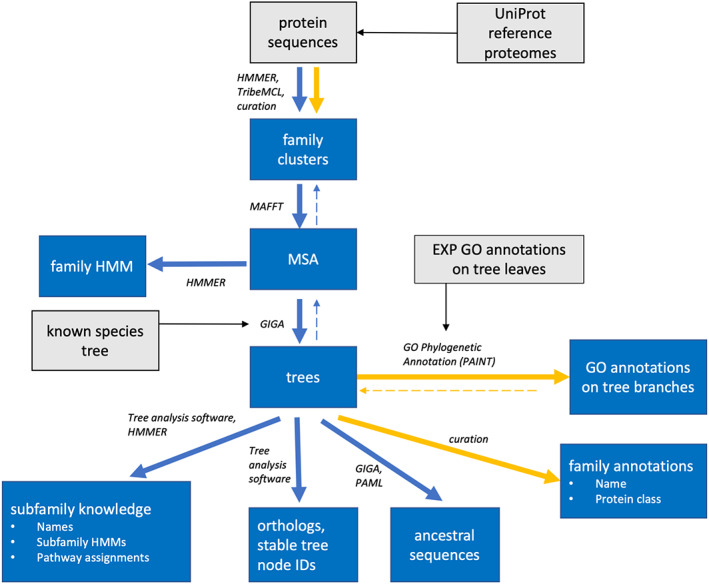
The Protein Analysis THrough Evolutionary Relationships (PANTHER) pipeline transforms raw protein sequence data into knowledge about gene family and function evolution. The major direction of data flow shown with solid arrows. Blue arrows indicate automated computational processes, while orange indicates manually curated processes. QA processes are shown as dashed lines in the reverse direction. Gray boxes and black arrows show external data inputs
The PANTHER pipeline is run yearly, on updated reference proteome sequences. It is designed to be as stable as possible, that is, families should have the same members across versions, and the vast majority of tree nodes, both leaves and internal nodes, should be equivalent across versions. That said, we attempt to improve the PANTHER families in each release, responding to feedback from collaborators and users, as well as our own internal QA processes. Over the past several years, the main contributors of issues and suggestions for improving family boundaries have been the GO Phylogenetic Annotation project (which involves manually reviewing trees, as described below) and the Ensembl Compara project, which uses PANTHER HMMs to define protein families for building gene trees. As a result, ~4,000 protein families have had some change in membership, defined here as a move of at least one protein from one family to another.
3.2.2. Annotation of trees
PANTHER trees are manually annotated, by curators from the GO Phylogenetic Annotation project, 24 with inferred function evolution events. These annotations specify branches in the phylogenetic tree where one or more functions (described by GO terms) have been gained or lost. This (also known as Phylogenetic Annotation and Inference Tool [PAINT] curation) is a manual curation process to infer ancestral functions by reconstructing an explicit model of functional evolution using PANTHER phylogenetic trees. The highly trained curators use the PAINT software (http://wiki.geneontology.org/index.php/Phylogenetic_Annotation_Project) to perform the curation. The tool overlays the GO experimental annotations, MSA and protein information onto the tree. These experimental annotations are GO annotations with an experimental evidence code (http://geneontology.org/docs/guide-go-evidence-codes/), and are used as evidence for any PAINT annotations. Based on the given information, the curators will make the following decisions. First, the curators will determine when a particular function first appeared (was gained) during evolution based on the experimental annotations and the tree topology. The GO term will then be assigned to the ancestral branch (identified by its terminal node identifier) in the tree with the evidence code “Inferred from Biological aspect of Descendant” (IBD). Second, the curators will determine if the function assigned above was subsequently lost in any descendant branches. Losses are often inferred due to one or more of the following conditions: a long branch in the phylogenetic tree, any branch following a duplication node, evidence from a negative experimental GO annotation (identified with the NOT qualifier), mutations of active site or other critical amino acids in the primary sequence, or simply lack of positive experimental annotations in otherwise well‐studied proteins. The PAINT tool also includes automatic taxon constraint checks (using constraints encoded into the GO that restrict the taxa to which some GO terms can be applied 38 ) to ensure all ancestral annotations do not violate these constraints.
3.3. Integration of PANTHER into other resources
PANTHER is part of a larger ecosystem of knowledgebases and resources (Figure 5). As described in the preceding section, PANTHER trees are annotated with GO terms by the GO Consortium, and also by the PhyloGenes project (focusing on plant genes). 39 The UniProt UniRule system 40 is starting to develop PANTHER tree‐based annotations for other protein properties such as protein names. Other resources, including UniProt 35 and the Alliance of Genome Resources, 41 have cross‐references and web links from gene or protein pages to PANTHER subfamilies and trees. Orthologs derived from PANTHER trees are imported by several resources. The Alliance of Genome Resources and the Human Gene Nomenclature Committee 42 projects have developed software that performs meta‐analyses over multiple orthology prediction methods, including PANTHER. Reactome imports PANTHER orthologs to infer pathways in nonhuman model organisms from their curated human pathways. 19 Those pathways, in turn, are imported into the PANTHER knowledgebase for use with PANTHER tools. 20 PANTHER HMMs (and soon also trees) are imported into the InterPro resource, where they are used for classification of UniProt proteins, and redistributed in the widely‐used InterProScan software package 43 for large‐scale protein annotation. PANTHER HMMs and trees are utilized in several steps of the MGnify 44 metagenomics data processing pipeline. The PANTHER HMMs are also used by the TreeFam 45 and Ensembl Compara 46 projects to define protein family boundaries for constructing phylogenetic trees.
FIGURE 5.
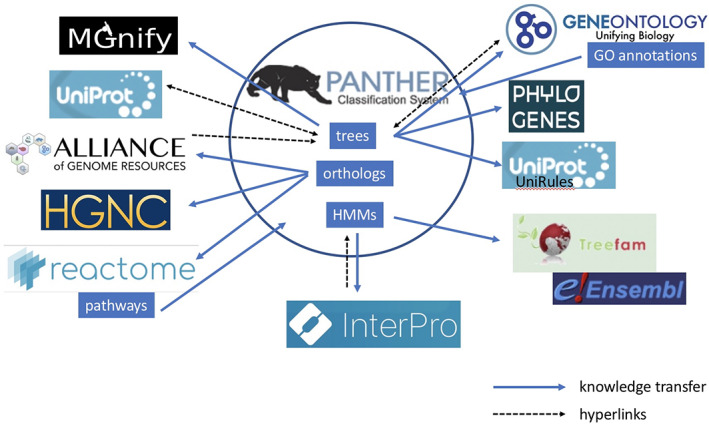
The Protein Analysis THrough Evolutionary Relationships (PANTHER) knowledgebase interoperates with many resources, via regular knowledge transfer and exchange (solid arrows) or hyperlinks (dashed arrows)
4. APPLICATIONS OF THE PANTHER KNOWLEDGEBASE
The main utility of the knowledgebase is that it is applied through a number of protein analysis tools. In this section, we describe several of the most highly used software tools that use the PANTHER knowledgebase. These include tools that can be accessed from the PANTHER website, as well as tools available from third parties.
4.1. Annotating protein sequences at both small and large scales
The PANTHER protein sequence classification tools are designed to take one or more query protein sequences, and provide the following classifications for each of them: protein family and subfamily, GO terms, and pathways. These tools can be run interactively for one sequence at a time on the PANTHER website, or, for large numbers of sequences, users can download the tools and run them via command‐line on a local computer. There are two tools currently supported for this task: PANTHER HMM search, and TreeGrafter. 26 We recommend using TreeGrafter, as it is faster and has been shown to be more accurate, and provides more specific annotations in some cases. The PANTHER HMM search tool uses HMMER3 47 to search the library of ~140,000 family and subfamily HMMs, and reports the best matching HMM and the GO terms associated with it. TreeGrafter first searches the 15,635 family HMMs and then uses RAxML 48 to add the query sequence to the tree, “grafting” it onto the most parsimonious branch of the tree. With the interactive TreeGrafter tool on the PANTHER website, users can view the modified tree that includes the grafted query sequence, while the command‐line tool reports the stable tree node identifier at the end of the graft branch. Both tools can be downloaded from the PANTHER website. In addition, the HMM search tool can also be run using the third‐party InterProScan tool, 49 but efforts are currently underway to replace it with TreeGrafter.
4.2. Browsing whole proteomes or protein families by function
These tools are available for interactive use on the PANTHER website. The whole genome function view tool enables users to select a whole genome, and navigate the set of protein coding genes by function. Any of the different function types can be selected: Protein Class, GO (molecular function [MF], biological process [BP], or cellular component [CC]), or pathways. Users can drill down to more specific function classes, or retrieve a list of the proteins assigned to any selected function class. The PANTHER Prowler (available under the “browse” tab on the homepage) enables users to browse the contents of the knowledgebase, and to combine different classifications to create a set of proteins or protein families that they are interested in. For example, a user could find all proteins in Homo sapiens (NCBI taxonomy) that have “protein kinase activity” (GO) and are in the “Wnt signaling pathway” (PANTHER Pathway).
4.3. Coding variant analysis, and other analyses of individual residues of proteins
The tools described above are applied to entire sequences. However, it is also possible for users to analyze individual protein amino acid sites in PANTHER multiple sequence alignments. PANTHER currently provides two tools of this type. The first is the PANTHER‐PSEP (position‐specific evolutionary preservation) tool, 50 which is commonly used to predict the likely effect of an amino‐acid substitution (“coding variant”) on protein function. PANTHER‐PSEP is available for interactive use (one protein at a time, but multiple variants can be input at the same time), or as a command‐line tool for use on large variant datasets. PANTHER‐PSEP returns, for each variant, the “preservation time” (the length of time the given site has been preserved with no change in the amino acid at that site). Longer times indicate a greater probability of natural selection having acted to prevent change at that site, and therefore a greater chance that a substitution at that site will impact the protein's function. The tool estimates the probability of deleterious impact (Pdeleterious) from the preservation time using an empirical analysis of performance on a curated set of known deleterious and neutral variants. 50 Preservation times are calculated from ancestral sequences reconstructed using PAML. 34
In addition to PANTHER‐PSEP, users can interrogate individual protein sites in multiple different ways. In the interactive PANTHER TreeViewer tool, users can select and view an individual column of a family multiple sequence alignment, by specifying in the URL either the MSA column number, or the site number in a selected single protein sequence. For programmatic access, PANTHER provides an API that can report, for any selected protein and site, the amino acid present at the aligned sites in homologous proteins.
4.4. Genetic variant annotation
Large‐scale association studies of genetic variants with diseases using whole genome or exome sequencing and GWAS have become methods of choice for identifying genetic variants associated with health traits and diseases. 51 The next step is to develop biological hypotheses about the causal mechanism by which variants act, by predicting the functional consequences of each variant. PANTHER provides a tool to allow users to submit genetic variants (in Variant Call Format) and return functional annotations in two ways. Currently, this functionality is only available for genetic variation in humans, and not in other organisms. First, the tool maps the variant directly to the gene if it falls within the chromosomal location of the gene. 20 Users can specify flanking regions on either side of the gene, so if the variant is outside of the gene region but within the flanking region of the gene, it can be assigned to the gene as well. The idea is that these regions may include cis‐regulatory regions of the gene. Second, the tool will report whether the variant occurs within an enhancer region (as annotated by ENCODE, 52 FANTOM, 53 or VISTA 54 ), and then uses the enhancer‐gene links from the PEREGRINE Project to link the variant to the gene whose expression it might impact. 27 The idea here is that if a variant is within an enhancer that regulates a gene, the variant may impact the regulation of that gene. Since most of the enhancers are in non‐coding regions, this functionality allows users to link variants in those regions to biological functions.
4.5. Gene list analysis
The most highly used tools on the PANTHER website are for analysis of lists of protein‐coding genes. Detailed descriptions of these tools, as well as instructions for using them, can be found elsewhere. 36 , 55 Users upload a list of genes, and can perform four different analyses. The “Annotation Table” (gene list view) displays a table that contains the gene in the first column, and annotations of various types in subsequent columns. The table can be customized to add and remove, or rearrange columns. The “Annotation Chart” displays a pie or bar chart that shows the relative number of genes annotated to different classes. Users can select among multiple classification types, including Protein Class, GO, and pathways. The “overrepresentation test” performs a statistical test for identifying classes that are statistically overrepresented (or underrepresented) in the uploaded list. Finally, there is an “enrichment test,” which requires a user to upload a list that contains, for each gene, a quantitative value. The enrichment test uses the quantitative value to identify classes that have non‐random distributions of values, akin to the well‐known gene set enrichment analysis tool. 56
4.6. Assessing completeness and contamination in MAGs
PANTHER is also used in the third‐party EukCC tool, for assessing the completeness and contamination of metagenome‐assembled genomes (MAGs). 57 This tool utilizes PANTHER trees to identify the most likely taxonomic group to which the MAG belongs, and then uses the PANTHER families and subfamilies that contain members from that group to define the set of genes that would be expected to be present in that clade. This improved sampling of expected genes in a clade has been shown to dramatically improve the estimates of MAG quality.
5. USAGE OF THE PANTHER RESOURCE
PANTHER attracts and supports a large user base. Users access the PANTHER resource interactively at the PANTHER website (http://pantherdb.org), or programmatically using the extensive PANTHER API (http://pantherdb.org/services/openAPISpec.jsp).
5.1. PANTHER website
According to Google Analytics, about 1,300–1,500 unique IP addresses access the PANTHER website daily, and 20,000 monthly. Note that a single IP address can be used by an organization or a university, so this metric underestimates the actual number of users. Typically, there are 500–600k page views monthly. Table 2 shows a breakdown of the types of pages by analysis/information type, with the most highly requested pages at the top. The top three page types represent different tools for analyzing lists of genes based on their annotation information in PANTHER. However, information about individual genes and individual protein families and pathways are also highly accessed.
TABLE 2.
PANTHER website page view statistics (from Google Analytics between July 1 and 31, 2021). Some of the table entries (labeled with *) represent groupings of pages from the same session
| Pages | Total pageviews |
|---|---|
| Overrepresentation test* | 118,775 |
| Pie chart from gene list page* | 85,936 |
| Gene list page* | 42,033 |
| Gene detail page | 10,828 |
| Family detail page | 7,602 |
| Tips/help* | 5,339 |
| Pathways* | 5,229 |
| Pie chart from overrepresentation test | 5,016 |
| GO or protein class detail page | 3,877 |
| Global search | 3,284 |
| Enrichment test graph | 3,246 |
| Coding variant tool (PANTHER‐PSEP) | 2,832 |
| Enrichment test* | 2,416 |
| Sequence search (HMM or TreeGrafter) | 2,004 |
| Tree viewer tool | 1,764 |
| Family list page | 1,424 |
| Prowler | 1,409 |
| Download | 1,300 |
| Pie chart from whole genome view | 1,023 |
| Enhancer | 515 |
Abbreviation: PANTHER, Protein Analysis THrough Evolutionary Relationships.
PANTHER has also been cited regularly in scientific publications by our users. As of August 2021, PANTHER has been cited in over 20,000 papers. Figure 6 shows the total number of citations of PANTHER in Google Scholar in four research areas, gene expression, protein expression, epigenetics, genome‐wide association studies, or GWAS. There is a steady increase in numbers of citations in each of those research areas through the years.
FIGURE 6.
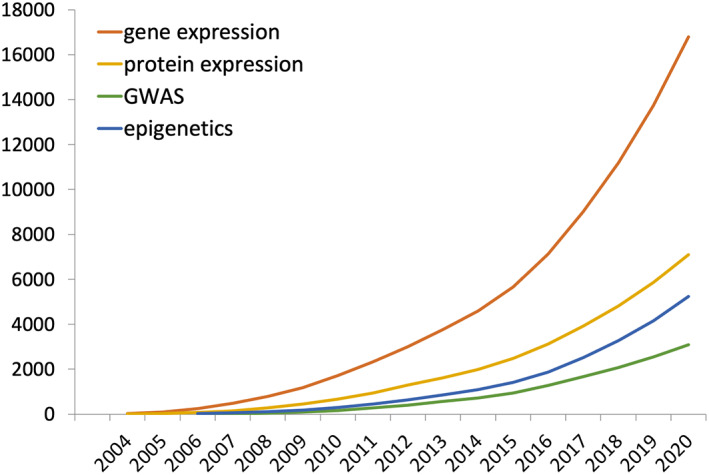
Citations of Protein Analysis THrough Evolutionary Relationships (PANTHER) for analysis of different types of experimental data. With the continued growth in RNA‐seq experiments, the increase in gene expression and epigenomics analysis has increased even more rapidly in the past 5 years or so
5.2. Programmatic access using the PANTHER API
In addition to interactive web pages, PANTHER also allows users to access the knowledgebase and tools programmatically using the PANTHER API. About 160k requests are made to the API monthly. PANTHER has an extensive set of APIs for programmatic access to the knowledgebase and tools. The most highly used APIs are listed in Table 3. PANTHER supports API access to: protein annotation information (“geneinfo” service); the overrepresentation analysis tool; orthologs and residue‐level homology derived from the alignments; families and subfamilies including trees and alignments; the TreeGrafter tool “graftsequence” service); PANTHER Pathway information. The full list of available services is available at http://pantherdb.org/services/openAPISpec.jsp.
TABLE 3.
PANTHER API access statistics (from Google Analytics between July 1 and 31, 2021)
| API events | Total events |
|---|---|
| Overrepresentation | 83,752 |
| Ortholog information | 51,652 |
| Gene list annotation (geneinfo) | 23,074 |
| Family information | 741 |
| TreeGrafter (graftsequence) | 82 |
Abbreviation: PANTHER, Protein Analysis THrough Evolutionary Relationships.
6. FUTURE DEVELOPMENTS
We expect that PANTHER will continue to be improved with every release at an even more rapid rate, due to ongoing feedback from a growing number of users. The quality of the raw protein sequences that are used as an input into any evolutionary reconstruction method has always been an important consideration in constructing accurate trees. Identification of potentially incorrect or fragmented sequences is a critical step in the PANTHER knowledge generation pipeline, but it inevitably results in some information loss. To address this problem, improvements are required in protein sequence annotation from genome assemblies, a process known as gene structure annotation. Even with improved protein sequences, there are several improvements that can be made to the PANTHER pipeline. One such improvement would be treatment of gene fusion events; in the current version of PANTHER, a gene can be assigned to only one family, so fused genes cannot appear in all the families to which their constituent parts belong. Another improvement would be to perform a full maximum‐likelihood reconstruction of all ancestral node sequences in the tree at each release. Another would be to provide bootstrap values on tree branches, so users can distinguish between parts of a tree that are well‐supported by the sequence data, and those that are not. We also plan to look for potential sources of systematic error in the PANTHER tree reconstruction process, via a thorough comparison to other computational methods that have been submitted to the Quest for Orthologs benchmarking service.
Finally, we note that we are currently working with the UniProt automatic annotation team to enable UniProt curators to construct evolutionary models of other properties of proteins, in addition to the GO function terms that are currently being modeled by the GO Consortium. The first properties this project will treat are protein name, and Enzyme Commission number. Already the feedback from this project has resulted in additional QA steps that have been added to the PANTHER pipeline.
AUTHOR CONTRIBUTIONS
Paul D. Thomas: Conceptualization (lead); data curation (supporting); funding acquisition (lead); investigation (supporting); methodology (lead); project administration (equal); software (supporting); validation (equal); writing – original draft (lead); writing – review and editing (lead). Dustin Ebert: Investigation (supporting); software (equal); validation (supporting). Anushya Muruganujan: Investigation (supporting); software (equal). Tremayne Mushayahama: Software (supporting). Laurent‐Philippe Albou: Software (supporting). Huaiyu Mi: Data curation (lead); funding acquisition (supporting); investigation (lead); methodology (supporting); project administration (equal); software (equal); validation (equal); writing – original draft (supporting); writing – review and editing (supporting).
Supporting information
Appendix S1: Supporting Information
ACKNOWLEDGMENTS
Funding was provided by the National Human Genome Research Institute of the National Institutes of Health (grant U41HG002273) and the National Science Foundation (grant number 1917302). The authors would like to thank the collaborating groups who have provided critical feedback and guidance on the PANTHER knowledgebase: the GO Phylogenetic Annotation team (Marc Feuermann, Pascale Gaudet, Michael Kesling, Karen Christie, Donghui Li, Suzanna Lewis); the InterPro team (Rob Finn, Alex Bateman, Lorna Richardson, Alex Mitchell, Tiago Grego, Gift Nuka); the UniProt UniRule team (Maria Martin, Hermann Zellner, ThankGod Ebenezer, Alistair MacDougall); the TreeFam/Ensembl Compara team (David Thybert, Matthieu Muffato, Mateus Patricio); the Quest for Orthologs benchmarking team (Christophe Dessimoz, Adrian Altenhoff, Brigitte Boeckmann, Salvador Capella‐Gutierrez, Laura Portell Silva); the PhyloGenes team (Peifen Zhang, Tanya Berardini, Qian Li, Trilok Prithvi, Leonore Reiser, Swapnil Sawant, Eva Huala); the MEROPS team (Neil Rawlings); the Reactome team (Peter D'Eustachio, Robin Haw, Guanming Wu); and the CellDesigner and Garuda teams (Hiroaki Kitano, Yukiko Matsuoka, Akira Funahashi).
Thomas PD, Ebert D, Muruganujan A, Mushayahama T, Albou L‐P, Mi H. PANTHER: Making genome‐scale phylogenetics accessible to all. Protein Science. 2022;31:8–22. 10.1002/pro.4218
Protein Analysis THrough Evolutionary Relationships (PANTHER) is a publicly accessible resource of information about the evolution of proteins and protein families, represented as phylogenetic trees; and protein function, derived from curated models of how evolutionarily related proteins have conserved or diverged in function. Information in the PANTHER knowledgebase can be searched, browsed, downloaded, and applied to numerous problems in protein research using publicly available software tools.
Funding information National Human Genome Research Institute, Grant/Award Number: U41HG002273; National Science Foundation, Grant/Award Number: 1917302
REFERENCES
- 1. Legried B, Molloy EK, Warnow T, Roch S. Polynomial‐time statistical estimation of species trees under gene duplication and loss. J Comput Biol. 2021;28:452–468. [DOI] [PubMed] [Google Scholar]
- 2. Glover N, Dessimoz C, Ebersberger I, et al. Advances and applications in the quest for orthologs. Mol Biol Evol. 2019;36:2157–2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stolzer M, Siewert K, Lai H, Xu M, Durand D. Event inference in multidomain families with phylogenetic reconciliation. BMC Bioinformatics. 2015;16(Suppl 14):S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Eisen JA. A phylogenomic study of the muts family of proteins. Nucleic Acids Res. 1998;26:4291–4300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mistry J, Chuguransky S, Williams L, et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021;49:D412–D419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tatusov RL, Koonin EV, Lipman DJ. A genomic perspective on protein families. Science. 1997;1997(278):631–637. [DOI] [PubMed] [Google Scholar]
- 7. Venter JC, Adams MD, Myers EW, et al. The sequence of the human genome. Science. 2001;291:1304–1351. [DOI] [PubMed] [Google Scholar]
- 8. Thomas PD, Campbell MJ, Kejariwal A, et al. PANTHER: A library of protein families and subfamilies indexed by function. Genome Res. 2003;13:2129–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bairoch A, Boeckmann B. The SWISS‐PROT protein sequence data bank. Nucleic Acids Res. 1991;19(suppl):2247–2249. 10.1093/nar/19.suppl.2247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ashburner M, Ball CA, Blake JA, et al. Gene ontology: Tool for the unification of biology. The gene ontology consortium. Nat Genet. 2000;25:25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mi H, Vandergriff J, Campbell M, et al. Assessment of genome‐wide protein function classification for drosophila melanogaster. Genome Res. 2003;13:2118–2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mi H, Lazareva‐Ulitsky B, Loo R, et al. The PANTHER database of protein families, subfamilies, functions and pathways. Nucleic Acids Res. 2005;33:D284–D288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Funahashi A, Tanimura N, Morohashi M, Kitano H. Celldesigner: A process diagram editor for gene‐regulatory and biochemical networks. BIOSILICO. 2003;1:159–162. [Google Scholar]
- 14. Keating SM, Waltemath D, König M, et al. Sbml level 3: An extensible format for the exchange and reuse of biological models. Mol Syst Biol. 2020;16:e9110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Demir E, Cary MP, Paley S, et al. The biopax community standard for pathway data sharing. Nat Biotechnol. 2010;28:935–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Le Novère N, Hucka M, Mi H, et al. The systems biology graphical notation. Nat Biotechnol. 2009;27:735–741. [DOI] [PubMed] [Google Scholar]
- 17. Mi H, Thomas P. PANTHER pathway: An ontology‐based pathway database coupled with data analysis tools. Methods Mol Biol. 2009;563:123–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rodchenkov I, Babur O, Luna A, et al. Pathway commons 2019 update: Integration, analysis and exploration of pathway data. Nucleic Acids Res. 2020;48:D489–D497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jassal B, Matthews L, Viteri G, et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2020;48:D498–D503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mi H, Huang X, Muruganujan A, et al. PANTHER version 11: Expanded annotation data from gene ontology and reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. 2017;45:D183–D189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Thomas PD. GIGA: A simple, efficient algorithm for gene tree inference in the genomic age. BMC Bioinformatics. 2010;11:312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mi H, Dong Q, Muruganujan A, Gaudet P, Lewis S, Thomas PD. PANTHER version 7: Improved phylogenetic trees, orthologs and collaboration with the gene ontology consortium. Nucleic Acids Res. 2010;38:D204–D210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Consortium RGGotGO . The gene ontology's reference genome project: A unified framework for functional annotation across species. PLoS Comput Biol. 2009;5:e1000431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gaudet P, Livstone MS, Lewis SE, Thomas PD. Phylogenetic‐based propagation of functional annotations within the gene ontology consortium. Brief Bioinform. 2011;12:449–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mi H, Ebert D, Muruganujan A, et al. PANTHER version 16: A revised family classification, tree‐based classification tool, enhancer regions and extensive api. Nucleic Acids Res. 2021;49:D394–D403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tang H, Finn RD, Thomas PD. Treegrafter: Phylogenetic tree‐based annotation of proteins with gene. Bioinformatics. 2019;35:518–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mills C, Muruganujan A, Ebert D, et al. Peregrine: A genome‐wide prediction of enhancer to gene relationships supported by experimental evidence. PLoS One. 2020;15:e0243791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jukes TH, Cantor CR. Evolution of protein molecules. New York, NY: Academic Press, 1969. [Google Scholar]
- 29. Fitch WM. Distinguishing homologous from analogous proteins. Syst Zool. 1970;19:99–113. [PubMed] [Google Scholar]
- 30. Altenhoff AM, Garrayo‐Ventas J, Cosentino S, et al. The quest for orthologs benchmark service and consensus calls in 2020. Nucleic Acids Res. 2020;48:W538–W545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mi H, Muruganujan A, Ebert D, Huang X, Thomas PD. PANTHER version 14: More genomes, a new PANTHER GO‐slim and improvements. Nucleic Acids Res. 2019;47:D419–D426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Huang X, Albou LP, Mushayahama T, Muruganujan A, Tang H, Thomas PD. Ancestral genomes: A resource for reconstructed ancestral genes and. Nucleic Acids Res. 2019;47:D271–D279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Whelan S, Goldman N. A general empirical model of protein evolution derived from multiple protein families using a maximum‐likelihood approach. Mol Biol Evol. 2001;18:691–699. [DOI] [PubMed] [Google Scholar]
- 34. Yang Z. Paml: A program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci. 1997;13:555–556. [DOI] [PubMed] [Google Scholar]
- 35. UniProt Consortium . Uniprot: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019;47:D506–D515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mi H, Muruganujan A, Huang X, et al. Protocol update for large‐scale genome and gene function analysis with the PANTHER classification system (v.14.0). Nat Protoc. 2019;14:703–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Boeckmann B, Marcet‐Houben M, Rees JA, et al. Quest for orthologs entails quest for tree of life: In search of the gene stream. Genome Biol Evol. 2015;7:1988–1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Deegan Née Clark JI, Dimmer EC, Mungall CJ. Formalization of taxon‐based constraints to detect inconsistencies in annotation and ontology development. BMC Bioinformatics. 2010;11:530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhang P, Berardini TZ, Ebert D, et al. Phylogenes: An online phylogenetics and functional genomics resource for plant gene function inference. Plant Direct. 2020;4:e00293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. MacDougall A, Volynkin V, Saidi R, et al. Unirule: A unified rule resource for automatic annotation in the uniprot knowledgebase. Bioinformatics. 2020;36:4643–4648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Consortium AoGR . The alliance of genome resources: Building a modern data ecosystem for model organism databases. Genetics. 2019;213:1189–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yates B, Gray KA, Jones TEM, Bruford EA. Updates to hcop: The hgnc comparison of orthology predictions tool. Brief Bioinform. 2021;22:bbab155. 10.1093/bib/bbab155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Blum M, Chang HY, Chuguransky S, et al. The interpro protein families and domains database: 20 years on. Nucleic Acids Res. 2021;49:D344–D354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mitchell AL, Almeida A, Beracochea M, et al. Mgnify: The microbiome analysis resource in 2020. Nucleic Acids Res. 2020;48:D570–D578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Schreiber F, Patricio M, Muffato M, Pignatelli M, Bateman A. Treefam v9: A new website, more species and orthology‐on‐the‐fly. Nucleic Acids Res. 2014;42:D922–D925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hubbard TJ, Aken BL, Beal K, et al. Ensembl 2007. Nucleic Acids Res. 2007;35:D610–D617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Eddy SR. A new generation of homology search tools based on probabilistic inference. Genome Inform. 2009;23:205–211. [PubMed] [Google Scholar]
- 48. Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post‐analysis of large phylogenies. Bioinformatics. 2014;30:1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jones P, Binns D, Chang HY, et al. Interproscan 5: Genome‐scale protein function classification. Bioinformatics. 2014;30:1236–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tang H, Thomas PD. PANTHER‐PSEP: Predicting disease‐causing genetic variants using position‐specific evolutionary preservation. Bioinformatics. 2016;32:2230–2232. [DOI] [PubMed] [Google Scholar]
- 51. Westra HJ, Franke L. From genome to function by studying eqtls. Biochim Biophys Acta. 2014;1842:1896–1902. [DOI] [PubMed] [Google Scholar]
- 52. Moore JE, Purcaro MJ, Pratt HE, et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature. 2020;583:699–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Abugessaisa I, Ramilowski JA, Lizio M, et al. Fantom enters 20th year: Expansion of transcriptomic atlases and functional annotation of non‐coding RNAs. Nucleic Acids Res. 2021;49:D892–D898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Visel A, Minovitsky S, Dubchak I, Pennacchio LA. Vista enhancer browser—A database of tissue‐specific human enhancers. Nucleic Acids Res. 2007;35:D88–D92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mi H, Muruganujan A, Casagrande JT, Thomas PD. Large‐scale gene function analysis with the PANTHER classification system. Nat Protoc. 2013;8:1551–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: A knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Saary P, Mitchell AL, Finn RD. Estimating the quality of eukaryotic genomes recovered from metagenomic analysis with eukcc. Genome Biol. 2020;21:244. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supporting Information