

Abstract
Cellular heterogeneity and an immunosuppressive tumour microenvironment are independent yet synergistic drivers of tumour progression and underlie therapeutic resistance. Recent studies have highlighted the complex interaction between these cell-intrinsic and cell-extrinsic mechanisms. The reciprocal communication between cancer stem cells (CSCs) and infiltrating immune cell populations in the tumour microenvironment is a paradigm for these interactions. In this Perspective, we discuss the signalling programmes that simultaneously induce CSCs and reprogramme the immune response to facilitate tumour immune evasion, metastasis and recurrence. We further highlight biological factors that can impact the nature of CSC–immune cell communication. Finally, we discuss targeting opportunities for simultaneous regulation of the CSC niche and immunosurveillance.
Cellular heterogeneity has been a long-appreciated hallmark of advanced cancers and serves as a framework to understand aggressive and therapeutically resistant cancers. One aspect of this framework is the cancer stem cell (CSC) hypothesis, which refers to treatment-refractory, tumour-initiating cell populations1. While defining a CSC has been challenging for a variety of reasons, including CSC population heterogeneity and patient-to-patient variability, CSC populations have been functionally validated across multiple cancers and have revealed a series of cell-intrinsic and cell-extrinsic molecular mechanisms that underlie tumour growth and therapeutic resistance (BOX 1). CSC signalling has been the focus of therapeutic development efforts that are in the initial stages of clinical evaluation2,3. Another aspect of CSC studies has focused on cell-extrinsic interactions with the surrounding tumour microenvironment that rely on a series of bidirectional cellular mechanisms, such as direct cell contact and ligand–receptor interactions, to drive tumour growth through interaction with resident and infiltrating non-neoplastic tumour cells. These interactions are also being considered in therapeutic development2.
BOX 1 |. Hallmarks of cancer stem cells.
First defined in haematological malignancies, cancer stem cells (CSCs) have since been identified across many solid tumours. While there are no universal markers of CSCs, surface CD133, CD34, CD44, epithelial cell adhesion molecule (EPCAM) and CD24 expression, aldehyde dehydrogenase (AlDH) activity and exclusion of Hoechst 33342 dye have been widely used to isolate these cells in different tumour types1. However, marker expression infers only the CSC state, which needs to be rigorously defined by functional properties of self-renewal and tumour initiation. self-renewal can be determined in many ways, including lineage tracing, but is generally observed using in vitro and in vivo limiting-dilution analysis via sphere formation (in vitro) and tumour initiation (in vivo) assays. generation of heterogeneous tumours upon implantation in immunocompromised mice and serial xenograft transplantation are frequently used to assess tumour-initiation capacity140. CSCs are maintained through a combination of cell-intrinsic signalling pathways and environmental factors147. Collectively, these interactions maintain a CSC state that is defined by self-renewal and tumour initiation and may have enhanced therapeutic resistance and an epithelial–mesenchymal transition programme1,2,140.
Harnessing the immune system to recognize and effectively eliminate tumour cells has transitioned from a theoretical possibility to a viable therapeutic option for many advanced cancers4. Although many of these strategies are based on the systemic targeting of protumorigenic immune cells or the activation of an antitumoural immune response, the molecular mechanisms altering the immune cell function within the tumour microenvironment are not fully elucidated. Furthermore, instructive interactions in the tumour microenvironment may underlie acquired resistance to these therapeutic strategies, as the tumour microenvironment is generally considered to be immunosuppressive5. An additional confounding variable is how immune cell lineages interact with CSCs, as both immune evasion and CSCs are recognized as integral parts of tumour growth and metastatic spread. Moreover, it was recently demonstrated that a high-stemness signature correlates with a poor immunogenic response across 21 solid malignancies, highlighting a potential interaction between these two protumorigenic pathways6. On the basis of these associations, the aim of this Perspective is to evaluate our current understanding of CSC–immune cell interactions in cancer to determine how these interactions drive tumour growth and to identify novel dual CSC targeting and immunotherapy strategies for future therapeutic development efforts. Given the rapid development in each of these seemingly independent areas, CSC biology and immunotherapy, exploring the interface between these fields may provide a new viewpoint and additional insight into each individual field as well as identify key overlapping areas that can be leveraged for additional mechanistic insight and therapeutic development efforts.
To assess CSC–immune cell interactions, we focus on signalling programmes that are common for self-renewal and immune reprogramming and that facilitate immune evasion, tumour growth and metastasis, and therapeutic resistance. In addition, we examine these programmes in the context of individual immune cell lineages that are resident in or infiltrate the tumour microenvironment. We also provide examples of cellular and molecular mechanisms that reciprocally amplify CSC maintenance and immunosuppression. Finally, we integrate these themes to highlight targeting opportunities to concomitantly attenuate CSC maintenance and reduce immunosuppression in the tumour microenvironment.
Myeloid cells
Tumour-infiltrating myeloid cells are a heterogeneous lineage consisting of monocytes, myeloid-derived suppressor cells (MDSCs), granulocytes, macrophages and dendritic cells (DCs) originating from the common myeloid progenitors in the bone marrow, as well as tissue-resident macrophages derived from yolk sac or fetal liver monocytes7. Despite differences in their ontogeny and activation status, these diverse cell types interact with CSCs, albeit the relative contribution of each myeloid population to cancer progression is tumour type dependent. There is growing appreciation of the protumorigenic role of this reciprocal communication axis. These interactions, summarized in FIG. 1, constitute potential therapeutic targets for the reprogramming of the tumour microenvironment across multiple cancers.
FIG. 1 |. A unique set of mediators informs the communication between cancer stem cells and innate immune cell populations.
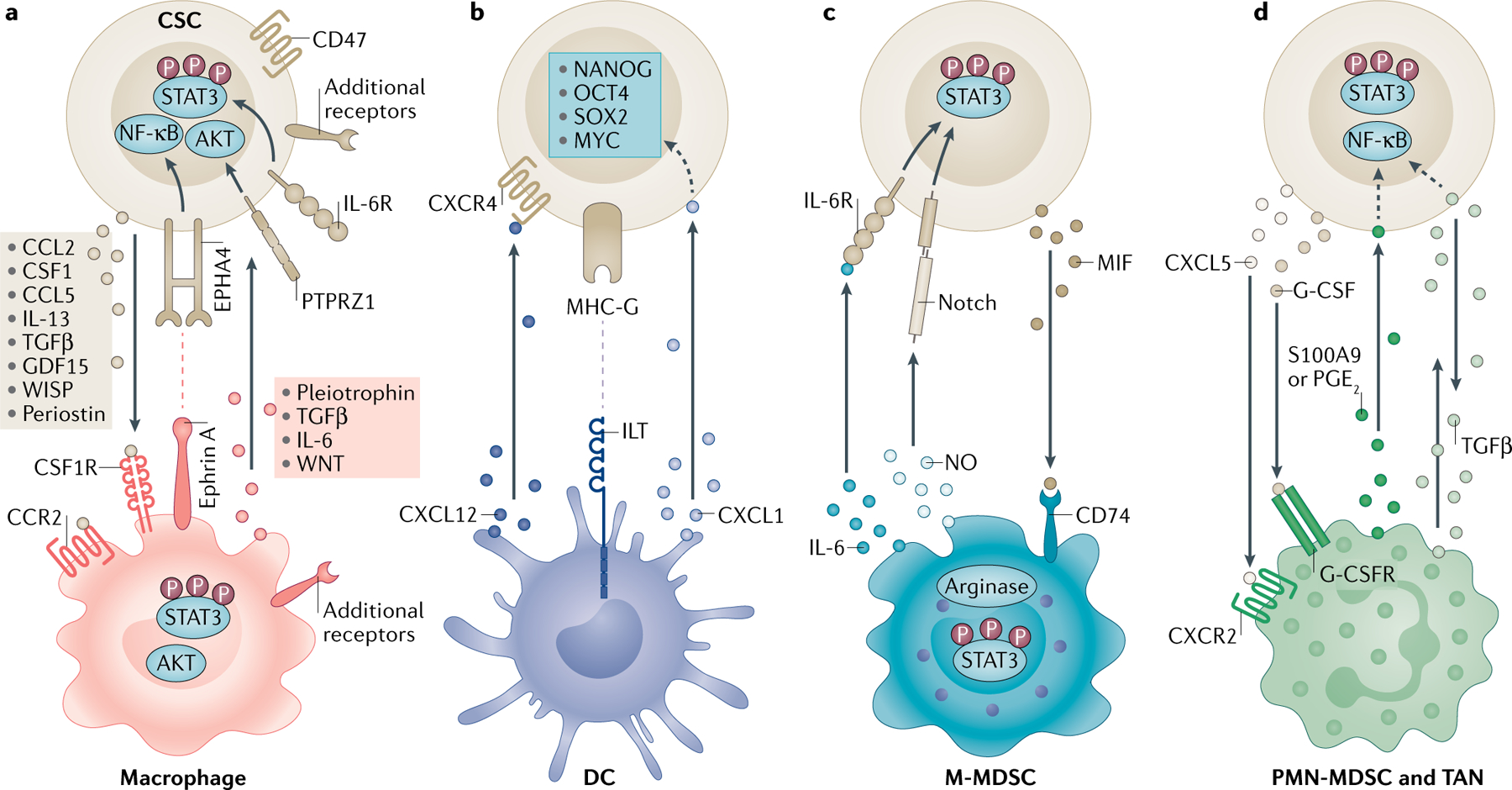
Reciprocal communication between cancer stem cells (CSCs) and myeloid cells through soluble mediators or juxtacrine signalling promotes immunosuppression and stemness. a | CSCs drive monocytes and macrophages via molecules including C-C motif chemokine 2 (CCL2), CCL5, colony-stimulating factor 1 (CSF1), transforming growth factor-β (TGFβ), growth/differentiation factor 15 (GDF15), WNT-induced signalling protein 1 (WISP1) and periostin that bind to surface receptors, including CSF1 receptor (CSF1R) and C-C chemokine receptor type 2 (CCR2). Macrophages, in return, express factors including interleukin-6 (IL-6), IL-13, pleiotrophin and TGFβ that signal through receptors such as receptor-type tyrosine-protein phosphatase-ζ (PTPRZ1) and ephrin type A receptor 4 (EPHA4) to support CSCs. These interactions activate the downstream signal transducer and activator of transcription 3 (STAT3), AKT and nuclear factor-κB (NF-κB) pathways. CSCs also evade macrophage phagocytosis through CD47 expression. b | CSCs interfere with dendritic cell (DC) maturation through major histocompatibility complex class I antigen G (MHC-G)–immunoglobulin-like transcript (ILT) inhibitory receptor interaction. Tolerogenic DCs release C-X-C motif chemokine 12 (CXCL12), which serves as a ligand to C-X-C chemokine receptor type 4 (CXCR4) and CXCL1 to promote CSCs. These interactions lead to the activation of the stem cell transcription factors NANOG, OCT4, SOX2 and MYC. c | Nitric oxide (NO) produced by monocytic myeloid-derived suppressor cells (M-MDSCs) activates Notch on CSCs, and IL-6 signalling increases STAT3 phosphorylation. CSCs recruit M-MDSCs via macrophage migration inhibitory factor (MIF) through CD74. CSCs also promote arginase 1 and STAT3 signalling in M-MDSCs. d | The CXCL5–CXCR2 axis and granulocyte colony-stimulating factor (G-CSF) recruit tumour-associated neutrophils (TANs) and polymorphonuclear MDSCs (PMN-MDSCs). In return, prostaglandin E2 (PGE2) and S100A9 support stemness. TGFβ has a bidirectional role in this communication axis. Dashed arrows indicate indirect associations. G-CSFR, granulocyte colony-stimulating factor receptor; IL-6R, interleukin-6 receptor.
Monocytes and macrophages.
CSC–immune cell interactions are best defined in the context of tumour-associated macrophages (TAMs), which can arise from tissue-resident macrophages or can differentiate from immature monocytes. Tissue-resident macrophages populate organs during embryonic development, diversify to respond to a specific set of environmental cues by establishing distinct expression programmes and maintain organ homeostasis by performing specialized functions depending on their anatomical location8,9. Several studies have identified tissue-resident macrophages as an integral component of adult stem cell niches (BOX 2), raising the possibility that this homeostatic mechanism may be leveraged by CSCs for their maintenance10,11.
BOX 2 |. Interactions between adult stem cells and the immune system.
Several studies have indicated that macrophages and regulatory T cells (Treg cells) are essential components of the stem cell niches across organ systems148. targeting of macrophages through CSF1R blockade was associated with reduced numbers of lgR5+ stem cells in the intestinal crypt149, while depletion of CD169+ macrophages was shown to interfere with retention of haematopoietic stem cells by acting on mesenchymal stem cells within the bone marrow niche150. similarly, mammary gland stem cells failed to repopulate fat pads of mice with the Csf1-null osteopetrotic (op) mutation (Csf1op/op), which have reduced levels of macrophages151. this is a functional stem cell–macrophage interaction with broader implications for organ development, as stem cells isolated from mammary pads of Csf1op/op mice also had decreased clonogenic activity151, and inhibition of CsF1 was previously shown to interfere with mammary gland development152. While this particular crosstalk was mediated through WNT ligand production by NOTCH1+, NOTCH3+ and NOTCH4+ macrophages as a result of interaction with Delta-like 1 (DLL1) expressed on the surface of stem cells153, there is still limited knowledge of the niche factors maintaining the stem cell–macrophage dialogue during homeostasis and injury. Recruitment of macrophages by CCL2 during hair follicle regeneration raises the possibility that this is a dynamic process impacted by environmental stimuli154. Treg cells have also been identified in bone marrow, intestinal and hair follicle stem cell niches155–158. self-renewal of intestinal stem cells and an immune-privileged status of transplanted haematopoietic stem cells are maintained through IL-10 production by Treg cells, and depletion of Treg cells decreased the frequency of stem cells155,157,158. In addition, regeneration of hair follicles required accumulation of Treg cells and Jagged 1 expression, pointing to a potent role for Treg cells in regulating stem cell activity156. these observations also suggest the possibility that CSCs manipulate a host mechanism to form a favourable environment and rely on pre-existing interactions such as Notch or WNT signalling3, especially given the evidence that adult stem cells constitute precursors of CSCs in some cancers116.
While in vitro studies have classified macrophages into dichotomous proinflammatory M1 and immunosuppressive M2 phenotypes8, TAMs can exist in a spectrum of activation states, which is further complicated by spatial and temporal variation12–14. To a certain degree, this variation is determined by the evolving needs of the tumour microenvironment as well as by the source of the protumorigenic macrophage population. TAMs consist of both prenatally derived macrophages, which are retained in some organs during adulthood through local proliferation, and blood-derived macrophages, which contribute to or replace the pool of tissue-resident macrophages15–17. Among the multitude of factors produced by tumour and stromal cells that govern monocyte and macrophage infiltration and survival, C-C motif chemokine 2 (CCL2) and macrophage colony-stimulating factor 1 (CSF1) are the main regulators of this behaviour. Therefore, influx of monocytes in early stage mouse mammary tumours, an essential step for subsequent metastatic spread, is facilitated by the CCL2–C-C chemokine receptor type 2 (CCR2) signalling axis18. CSCs can be imperative to monocyte recruitment across tumour types, as supernatants from patient-derived or mouse cholangiocarcinoma, hepatocellular carcinoma (HCC) or glioblastoma cells grown in CSC-enriching conditions (sphere culture) had elevated levels of protumorigenic macrophage factors, including CCL2, CCL5, CSF1, growth differentiation factor 15 (GDF15), interleukin-13 (IL-13), transforming growth factor-β (TGFβ), periostin or WNT-induced signalling protein 1 (WISP1; also known as CCN4) than their non-CSC counterparts10,19–24 (BOX 3; FIG. 1a). Subsequent treatment of macrophages with conditioned media from these sphere cultures informed expression of markers associated with immunosuppressive function in vitro, suggesting that CSCs may impact the polarization state of TAMs10,19–24.
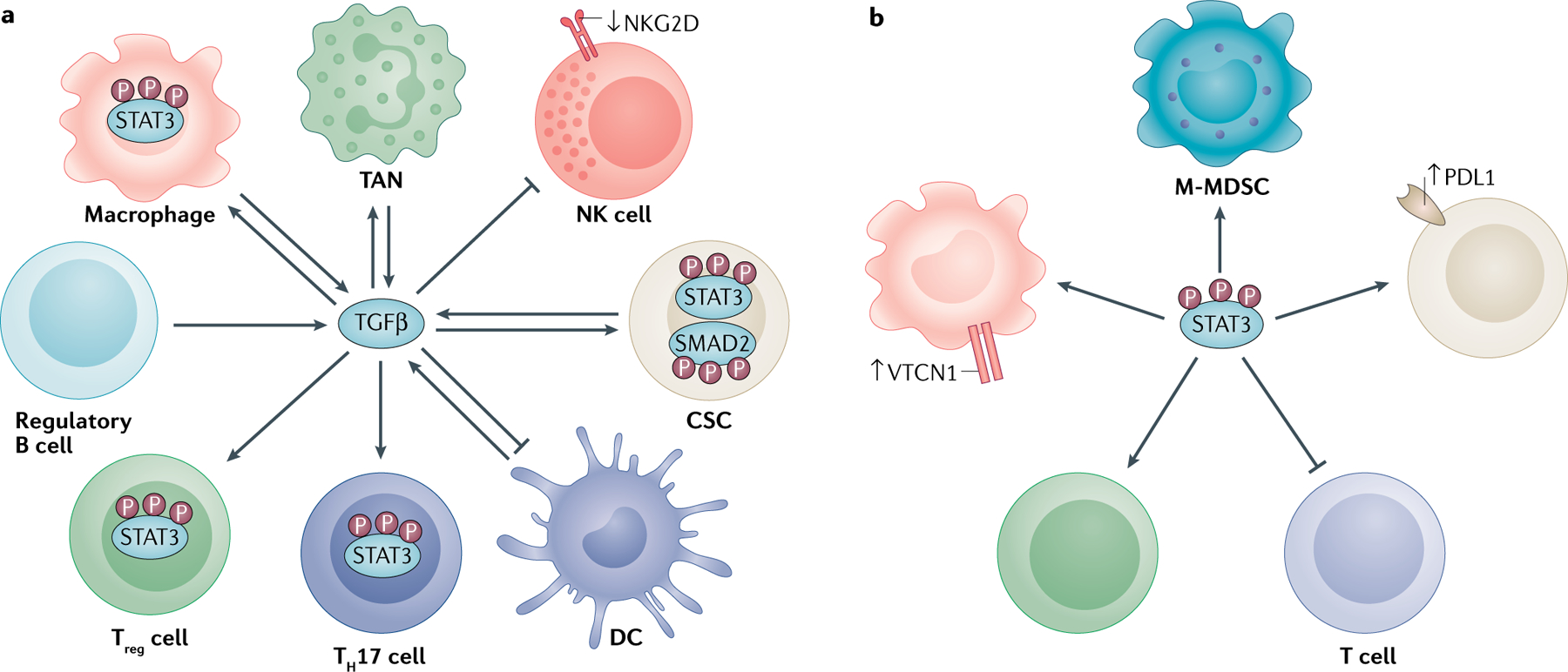
BOX 3 |. TGFβ and STAT3 as master regulators.
Transforming growth factor-β (TGFβ) is a pleiotropic cytokine that lies at the intersection of the pathological immune response and cancer stem cell (CSC) modulation159 (see the figure, part a). As it is produced by and acts on many of the cells involved in CSC–immune cell crosstalk, TGFβ can amplify the impact of the pathogenic signal to drive tumour growth in an autocrine or paracrine manner. TGFβ is an important regulator of the dedifferentiation process that reinforces stem signatures and immune evasion by signalling through SMADs26,30,58,63,71,82,93. the immune effects of CSC-derived TGFβ span the inhibition of natural killer (NK) and cytotoxic T cell activation71, promotion of TANs63, polarization of immunosuppressive macrophages22, and differentiation and recruitment of regulatory T cells (Treg cells)58,99. the close association of TGFβ with other communication axes in the tumour microenvironment, most notably IL-6–Janus kinase 2 (JAK2), can also potentiate downstream signal transducer and activator of transcription 3 (STAT3) activation, further amplifying protumorigenic immune phenotypes and CSC behaviour25,29,54,61,145,146,159. Activation of STAT3 can inform co-inhibitory receptor expression by both macrophages98 and CSCs97, and its inhibition in CSCs further interferes with their ability to promote macrophages and Treg cells and suppress T cell proliferation22,99 (see the figure, part b). Conversely, blockade of STAT3 in immune cells hampers their ability to propagate CSCs53. thereby, STAT3 is a key intracellular pathway that is a therapeutic target for simultaneous reversal of immunosuppression and reprogramming of CSCs90,128,143–145. In this context, TGFβ and STAT3 emerge as master regulators that shape the tumour architecture, warranting testing of combinational approaches targeting these pathways.
DC, dendritic cell; M-MDSC, monocytic myeloid-derived suppressor cell; NKG2D, natural killer cell receptor D; PDL1, programmed cell death 1 ligand 1; TAN, tumour-associated neutrophil; VTCN1, V-set domain-containing T cell activation inhibitor 1.
Reciprocally, TAMs can foster CSC phenotypes through soluble mediators, such as IL-6, TGFβ, WNT ligands and pleiotrophin, or via juxtacrine signalling as determined by co-culture experiments25–30 (BOX 3; FIG. 1a). Macrophage signalling to CSCs through receptor-type tyrosine-protein phosphatase-ζ (PTPRZ1) and ephrin type A receptor 4 (EPHA4), among other receptors that have not yet been elucidated, increased self-renewal (as read out by sphere formation and tumour-initiation capacity of CSCs) via activation of nuclear factor-κB (NF-κB), AKT and signal transducer and activator of transcription 3 (STAT3)25,27–29 (BOX 3; FIG. 1a). Importantly, the ability of CSCs to recruit TAMs through the expression of immunomodulatory factors is intertwined with stemness epigenetic programming and CSC transcriptional activity. For example, in human metastatic prostate cell lines, Polycomb repressive complex 1 (PRC1)-dependent chromatin remodelling was necessary for downstream CCL2 expression by CSCs31. In HCC mouse models, CCL2 and CSF1 expression was driven by the activation of the Hippo pathway effector Yes-associated protein (YAP), which is also essential for hepatocyte dedifferentiation and CSC proliferation21.
These observations highlight the complexity of CSC–macrophage crosstalk and underscore its implications for simultaneous targeting of immunosuppression and stemness phenotypes. However, inhibition of the CCL2–CCR2 axis alone was ineffective in reducing CSC frequency in the aforementioned HCC model21, and CSF1 receptor (CSF1R) inhibitors have had variable clinical success32. These limitations are potentially due to the non-specific elimination of antitumorigenic myeloid cells and the presence of compensatory mechanisms that warrant testing of combinational approaches. WISP1 and periostin are among those targets, as their knockdown reduced tumour-infiltrating myeloid cell frequency and delayed tumour growth in preclinical glioblastoma models19,20, but they lack specific inhibitors for clinical use. Pertinently, CSCs not only polarize macrophages to a protumorigenic state but also use protective mechanisms to avoid phagocytosis by macrophages. The CD47 ‘don’t eat me’ signal is upregulated in leukaemia33, HCC34, pancreatic ductal adenocarcinoma (PDAC)35 and lung cancer36 cells expressing phenotypic markers of CSCs. Preclinical studies revealed that blockade of the CD47–signal regulatory protein-α (SIRPα) axis can trigger tumour phagocytosis across disease models33–37, and a recent phase I clinical trial determined that a humanized anti-CD47 antibody is well tolerated in patients with advanced cancers38, suggesting that inhibition of CD47 has translational potential. These observations highlight the complex interplay with functional consequences of enhanced TAM recruitment and polarization as well as increased CSC activity and survival. While the unique set of mediators contributing to this pathological axis might differ depending on the tumour type (TABLE 1), interference with this CSC–TAM communication along with activation of tumour lysis properties of antitumoural macrophages provides the conceptual framework for added therapeutic value.
Table 1 |.
Cellular mechanisms underlying CSC–immune cell communication
| Signalling axis | Mechanism of action | Tumour type | Refs |
|---|---|---|---|
| CCL2–CCR2 | Recruitment of macrophages, MDSCs and Treg cells by CSCs | Glioblastoma | 22,77 |
| HCC | 21 | ||
| Pancreas | 143 | ||
| Prostate | 31 | ||
| CSF1–CSF1R | Polarization of TAMs by CSCs | Breast | 141 |
| Cholangiocarcinoma | 10 | ||
| Colorectal | 141 | ||
| Glioblastoma | 22,77 | ||
| HCC | 21 | ||
| Pancreas | 143 | ||
| Prostate | 31 | ||
| IDO1 | CSC-mediated accumulation of Treg cells | Breast | 84 |
| Glioblastoma | 81,83 | ||
| Prostate | 84 | ||
| IL-1β | CSC phenotype promotion by macrophagesa | Breast | 114 |
| Colorectal | 120 | ||
| Pancreas | 124 | ||
| IL-6 | Upregulation of CSC phenotype, perpetuation of TAM and MDSC function, TH17 cell polarization | Breast | 54 |
| Colorectal | 88 | ||
| Glioblastoma | 98 | ||
| HCC | 29 | ||
| Multiple myeloma | 60 | ||
| NSCLC | 25 | ||
| Ovarian | 144 | ||
| TGFβ | Stimulation of CSC phenotype, promotion of immunosuppressive myeloid cell activity, T cell and NK cell inhibition, Treg cell and TH17 cell polarization | Colorectal | 82 |
| Glioblastoma | 22,71,99 | ||
| HCC | 26,63 | ||
| Melanoma | 58 | ||
| Pancreas | 30 | ||
| STAT3 | CSC maintenance, TAM polarization and function, MDSC polarization and function, cytotoxic T cell suppression, Treg cell generation | Breast | 54 |
| Colorectal | 61 | ||
| Glioblastoma | 11,22,98,99 | ||
| HCC | 29,128 | ||
| HNSCC | 97 | ||
| NSCLC | 25,90 | ||
| Pancreas | 53,143 |
CCL2, C-C motif chemokine 2; CCR2, C-C chemokine receptor type 2; CSC, cancer stem cell; CSF1, colony-stimulating factor 1; CSF1R, colony-stimulating factor 1 receptor; HCC, hepatocellular carcinoma; HNSCC, head and neck squamous cell carcinoma; IDO1, indoleamine 2,3-dioxygenase 1; IL-1β, interleukin-1β; IL-6, interleukin-6; MDSC, myeloid-derived suppressor cell; NK, natural killer; NSCLC, non-small-cell lung carcinoma; STAT3, signal transducer and activator of transcription 3; TAM, tumour associated macrophage; TGFβ, transforming growth factor-β ; TH17 cell, T helper 17 cell; Treg cell, regulatory T cell.
Particular consideration in the context of ageing.
Dendritic cells.
DCs play an essential role in priming the T cell-mediated antitumour immune response by cross-presenting tumour antigens. However, tumours can restrict the antitumorigenic properties of DCs by limiting their trafficking, preventing their maturation and inducing differentiation of tolerogenic subtypes7. These DCs are not devoid of function and can actively support cancer growth as well as metastatic spread39. This is in part mediated by reprogramming of regulatory CD4+ T cell populations by monocyte-derived DCs, suggesting that tumours can inform downstream interactions of DCs with other immune cell populations39,40.
Although there is very limited knowledge of the specific role of CSCs in this process, several recent observations support a reciprocal relationship between CSCs and tumorigenic DCs. For example, cells from human renal cancers expressing CD105 (also known as endoglin), a surrogate marker for CSCs, blocked maturation of monocyte-derived DCs in vitro at a higher rate than CD105− tumour cells, on the basis of the expression of co-stimulatory molecules41. This phenotypic change was attributed to extracellular vesicle-associated expression of the inhibitory receptor major histocompatibility complex class I (MHC-I) antigen G (MHC-G)41 (FIG. 1b). While the precise mechanism of action was not investigated in that study, MHC-G serves as a ligand to immunoglobulin-like transcript (ILT) inhibitory receptors and can arrest DC differentiation by signalling through the IL-6–STAT3 axis42 (BOX 3).
Tumorigenic DCs can also provide prosurvival signals for the maintenance of CSCs in solid and haematological malignancies as a result of an altered secretory phenotype. DCs isolated from patients with colon cancer or conditioned with culture medium from a human colon cancer cell line are characterized by lower IL-12 and higher C-X-C motif chemokine 1 (CXCL1) production43. CXCL1 increases expression of the stem cell signalling network proteins NANOG, OCT4, SOX2 and MYC and drives cell migration43 (FIG. 1b). As CXCL1 also contributes to premetastatic niche formation of colon cancer by recruiting CXCR2+ MDSCs, mediators secreted by dysfunctional or tolerogenic DCs might drive tumour progression through multiple downstream effector functions44. However, it is plausible that the receptor–ligand interaction is tumour specific, as the CXCL12–CXCR4 axis was germane to support provided by follicular DCs to B cell lymphoma stem cells, which were characterized by exclusion of Hoechst 33342 dye, also known as the side population, and increased sphere-forming capacity45 (FIG. 1b).
These early findings highlight the necessity of educating DCs against CSCs for the success of DC vaccine strategies that are currently in clinical development. To this end, DCs pulsed with the lysates of mouse cancer cell lines grown in CSC-enriching sphere conditions or selected for ALDH expression elicited a better immune response than those pulsed with bulk tumour cells in glioblastoma, melanoma and head and neck squamous cell carcinoma (HNSCC) mouse models46,47. Nonetheless, the presence of CSCs in mouse tumour cell lines is a contested subject, and high immunogenicity of these cell lines48 can further complicate accurate representation of human tumours. Thus, identification of additional mechanisms of DC dysregulation in tumours and neoantigens selectively expressed in CSCs as opposed to normal stem cells are essential to improve the success and manage the toxicity of this treatment approach in patients.
Myeloid-derived suppressor cells and tumour-associated neutrophils.
MDSCs are immature bone marrow-derived cells that expand in patients with malignancies and infiltrate tumours. In addition to serving as markers of malignancy, MDSCs suppress the antitumour immune response and limit the efficacy of anticancer therapies49. Monocytic MDSCs (M-MDSCs) and polymorphonuclear MDSCs (PMN-MDSCs) are two subsets of MDSCs that are involved in different steps of tumorigenic growth and metastatic spread50. Tumour-associated neutrophils (TANs) are a potential third cell type closely linked to PMN-MDSCs. Although PMN-MDSCs are widely accepted as a population separate from inflammatory neutrophils on the basis of their functional annotation and transcriptome profile51, the distinction between TANs and PMN-MDSCs is not well defined. The variation between MDSC subtypes and TANs is linked to their complex and distinct roles in the tumour microenvironment but can also depend on the cancer type.
Initial observations suggested that MDSCs co-cultured with patient-derived primary ovarian cancer tissue promote sphere formation and ALDH expression by stimulating miR-101 and blocking carboxy-terminal-binding protein 2 (CTBP2) in tumour cells52. Subsequent studies identified M-MDSCs as the major driver of CSC phenotype in pancreatic and breast cancers53. M-MDSCs, which were detected at high levels in human pancreatic cancer tissue, were capable of increasing the frequency of the fraction of ALDH+ stem cells in mouse tumour cell lines in culture and increasing the in vivo tumour growth rate following implantation of these cells subcutaneously53. Similarly, M-MDSCs constituted the majority of tumour-infiltrating MDSCs in human breast tumour tissue and in mouse breast cancer models50,54. Mechanistic analysis indicated that nitric oxide synthase 2 gene (Nos2) expression and nitric oxide production by M-MDSCs promoted CSC phenotypes via activation of Notch signalling and sustained STAT3 phosphorylation in cancer cells50,54 (BOX 3; FIG. 1c). In mouse breast cancer cell lines, this effect was observed as increased levels of vimentin and phosphorylated STAT3, which was reversed by pharmaceutical inhibition of NOS2, when they were co-cultured with M-MDSCs50. Similarly, culturing of human breast cancer cell lines with MDSCs resulted in an increase in the ALDH+ fraction, and self-renewal, as read out by sphere formation, was blocked by the combination of an NOS2 inhibitor and an anti-IL-6 antibody54. Importantly, this is not a unidirectional relationship, as CSCs recruit MDSCs to constrain T cell activity and create a favourable environment for tumour growth. This can be achieved through a CSC-dependent increase in the levels of STAT3 and arginase, markers of suppressive function, in M-MDSCs through mediators that are not well defined53,55. However, macrophage migration inhibitory factor (MIF) is one such factor secreted by CSCs to promote M-MDSC accumulation and survival in the glioblastoma microenvironment55,56.
In mouse cervical, prostate and melanoma tumour models, where PMN-MDSCs play a more dominant role, MDSC accumulation is driven by granulocyte colony-stimulating factor (G-CSF), CXCL5 and TGFβ, respectively57–59 (BOX 3; FIG. 1d). Subcutaneous G-CSF-overexpressing human cervical tumours in nude mice were characterized by more MDSCs and ALDH+ tumour cells than were tumours initiated with the parental lines59. In a genetically engineered mouse model of prostate cancer, CXCL5-dependent mobilization of CXCR2+ PMN-MDSCs was linked to YAP hyperactivation57. An increased TGFβ signature in CD133+ mouse melanoma cells (CD133 is also known as PROM1) was associated with increased infiltration of PMN-MDSCs and TAMs in tumours initiated with the CD133+ fraction compared with CD133− counterparts58. These observations point to conserved signalling mechanisms, such as the Hippo–YAP pathway and miR-92 (REFS21,57,58), that drive immunosuppressive myeloid cells into the tumour microenvironment.
Reciprocally, PMN-MDSCs contribute to the stemness of cancer cells via multiple mechanisms. Co-culture of human multiple myeloma cells with PMN-MDSCs increased self-renewal as assessed by tumour sphere formation and increased relative expression of NANOG, OCT4 and SOX2, which was reversed by the PIWI-interacting RNA 823 antagomir or a DNA methyltransferase 3β (DNMT3B) inhibitor60. Similarly, PMN-MDSCs increase STAT3 phosphorylation, CD133 and CD44 expression and sphere formation of mouse and human colorectal cancer cell lines in vitro via transfer of exosomal S100A9 (REF.61). Furthermore, prostaglandin E2 (PGE2) expression by MDSCs was linked to expansion of the ALDH+ fraction in human cervical cancer cell lines in vitro, as this effect was reversed by a PGE2 inhibitor59.
TAN–CSC interactions have been reported in the context of lung cancer and HCC. In multiple mouse models of lung cancer, tumour-derived TGFβ is ascribed as a regulator of protumorigenic TAN polarization62. CSC-derived CXCL5 was also linked to chemotaxis of TANs in mouse models of HCC63 (FIG. 1d). In vitro studies indicated that TANs can also secrete TGFβ along with bone morphogenetic protein 2 (BMP2) to reprogramme cancer cell lines to acquire functional and phenotypical traits of stem cells, including colony formation capacity, through upregulation of NF-κB63, affirming the bidirectional role of TGFβ in the TAN–CSC communication axis (BOX 3). Collectively, these observations reveal that MDSCs and TANs support CSCs and are putative targets for cancer immunotherapy.
Lymphocytes.
T cells and B cells are essential components of immunosurveillance and have a vital role in long-term antigen-specific immune responses. Natural killer (NK) cells and innate lymphoid cells (ILCs) also arise from common lymphoid progenitors but lack antigen-specific receptors. There is limited understanding of how CSCs interact with lymphoid cell populations except that CSCs use multiple mechanisms to escape immunosurveillance. Tumour sphere-forming glioblastoma64, colon cancer65, lung cancer66, ABCB5+ melanoma67 and CD34+ leukaemia68 stem cells are characterized by reduced expression of MHC-I and/or NK cell receptor D (NKG2D) ligand. While lower levels of NKG2D ligand in CD34+ leukaemia stem cells and MHC-I in CD133+ glioblastoma stem cells provided protection against NK cell lysis in vitro68,69, in phenotypically defined lung cancer and colon cancer stem cells, decreased MHC-I expression correlated with susceptibility to in vitro cytotoxic killing66,70. These differences potentially point to variations in NK cell interactions on the basis of tumour type, with the presence of additional factors informing the nature of communication or selective enrichment of NK cell-resistant CSC populations in some cancers. To further add to this complexity, CSCs can inform NK cell activity. CD133+ patient-derived glioblastoma stem cells secrete TGFβ to restrict NKG2D expression on peripheral blood mononuclear cells71 (BOX 3). Given the accumulating evidence for B cell and ILC dysregulation in cancer and its ramifications for antitumour immunity72,73, it is likely that there is underappreciated crosstalk between CSCs and these immune cell populations. The possibility of a direct line of communication between CSCs and ILCs is further suggested by group 3 ILC-dependent STAT3 phosphorylation in intestinal stem cells during tissue regeneration74. While these mechanisms wait to be elucidated, the complex interplay between CSCs and T cell subsets is better described (summarized in FIG. 2).
FIG. 2 |. CSCs interfere with T cell activity directly or through immunosuppressive myeloid cells.
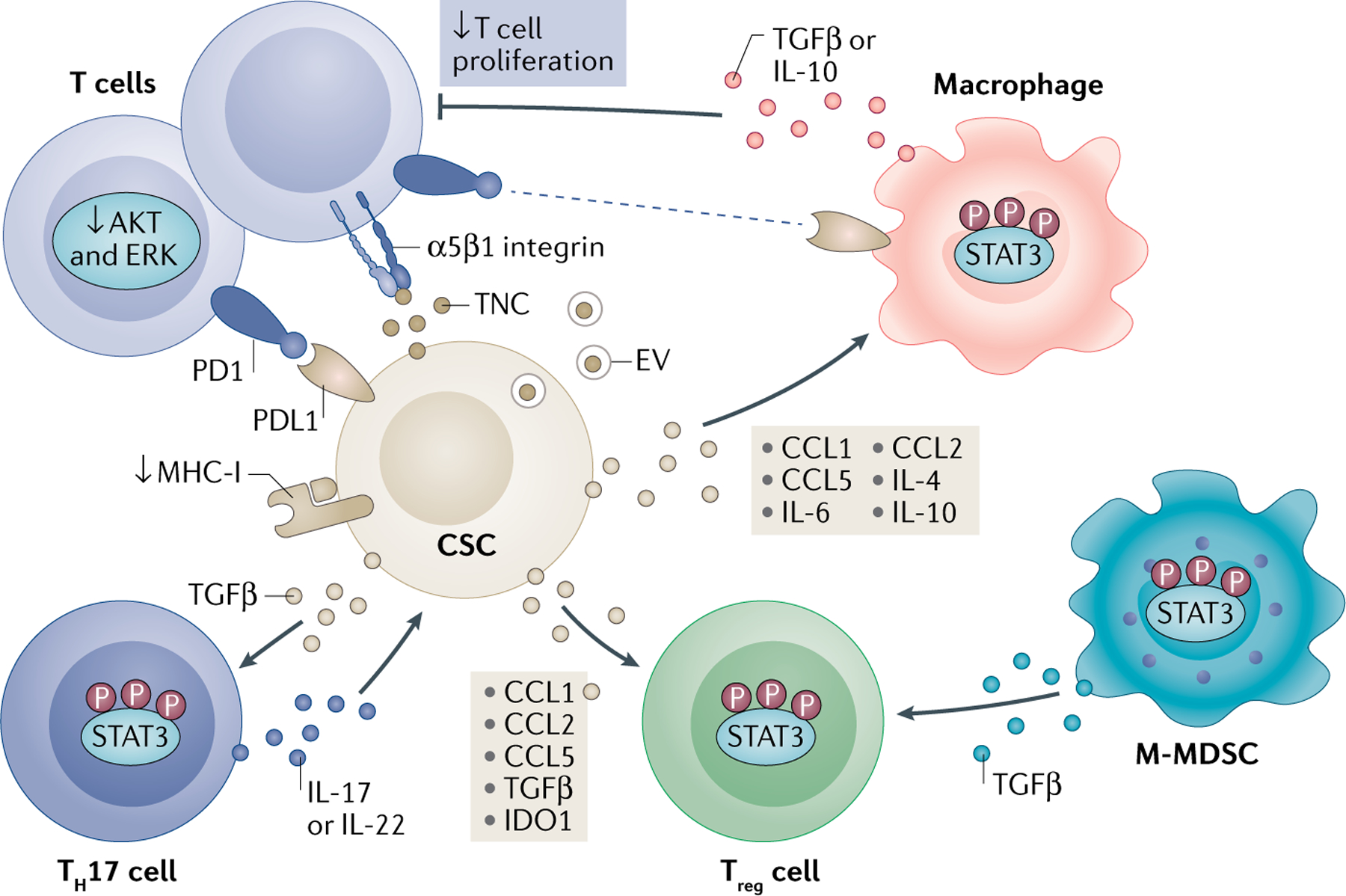
Cancer stem cells (CSCs) suppress or evade antitumorigenic T cells in part by releasing extracellular vesicle (EV)-associated or free tenascin C (TNC) and reducing AKT and ERK signalling. CSCs also induce tumour-promoting regulatory T cells (Treg cells) and T helper 17 (TH17) cells. CSCs downregulate major histocompatibility complex class I (MHC-I) and overexpress checkpoint regulators, including programmed cell death 1 ligand 1 (PDL1). CSCs further drive recruitment and polarization of TH17 cells and Treg cells by the combination of C-C motif chemokine 1 (CCL1), CCL2, CCL5, transforming growth factor-β (TGFβ) and indoleamine 2,3-dioxygenase 1 (IDO1). An additional layer of regulation of T cell activity is mediated indirectly by immunosuppressive myeloid cells, including macrophages and monocytic myeloid-derived suppressor cells (M-MDSCs). This effect partially depends on CCL1, CCL2, CCL5, interleukin-4 (IL-4), IL-6 and IL-10 secreted by CSCs. Myeloid cells produce additional suppressive molecules, such as the checkpoint inhibitor ligand PDL1, to block T cell activation in a contact-dependent manner or by promoting Treg cells. Collectively, these interactions reshape the tumour microenvironment and create a habitat where Treg cells and TH17 cells support CSCs, the latter via IL-17 production. STAT3, signal transducer and activator of transcription 3.
Regulatory T cells and T helper 17 cells.
Regulatory T cells (Treg cells) have a central role in tumour immunobiology as the main suppressive T cell subset, and were previously recognized as constituents of adult stem cell niches (BOX 2). In general, CSCs secrete the chemokines CCL1, CCL2 and CCL5 to actively recruit Treg cells24,31,75–77 (FIG. 2). In a mouse glioblastoma model, CCL2-mediated Treg cell trafficking was dependent on expression of CCR4, as tumour infiltration was limited in Ccr4-knockout mice77. CD133+ human ovarian cancer cell lines, which had higher levels of CCL5 expression, induced migration of and IL-10 production in Treg cells in a CCR5-dependent manner75. By contrast, Treg cell migration to SOX2+ mouse breast cancer cell lines was induced by CCL1 in transwell assays76, indicating an abundance of chemokines that can recruit Treg cells to the tumour microenvironment. CCL1, CCL2 and CCL5 are also crucial for the trafficking of myeloid cells that are capable of promoting the infiltration of Treg cells31,78–80, suggesting that CSCs drive Treg cell accumulation through a combination of direct and indirect mechanisms. Additionally, self-renewing CSCs from a variety of cancer cell lines exhibit elevated expression of indoleamine 2,3-dioxygenase 1 (IDO1) and TGFβ, two important inducers of Treg cell recruitment and generation58,81–84 (BOX 3; FIG. 2). Through production of these mediators, CSCs can also stimulate Treg cells to enhance CSC marker expression and self-renewal as assessed by increased sphere formation in mouse breast cancer cell lines76.
Treg cells share precursors with T helper 17 (TH17) cells, and these two cell types can transdifferentiate in response to environmental cues85,86. This balance between the two CD4+ T cell subsets is essential for the regulation of self-tolerance versus inflammation. Because of such plasticity, it is not surprising that IDO1 and TGFβ can also impact the pathogenic TH17 cell response in the tumour microenvironment. Whereas IDO1 blocks the conversion of Treg cells to TH17 cells, TGFβ can trigger such a phenotypic switch in collaboration with the IL-6–STAT3 pathway85. These observations further support the composite role of TGFβ in modulation of the antitumour immune response (BOX 3). Although the role of TH17 cells in cancer remains a contested issue87, IL-17 secreted by TH17 cells was shown to augment the self-renewal capacity of CSCs in multiple tumour models, and IL-22, another cytokine produced by these cells, induced the phosphorylation of STAT3 in tumour cells88–92 (FIG. 2). All things considered, the versatile nature of Treg cells and TH17 cells should be extensively interrogated to decipher their means of intercommunication with CSCs and other immune cell populations for effective therapeutic targeting.
Cytotoxic T cells.
Communication of CSCs with CD8+ T cells can be categorized into two areas: evasion of CSCs from T cell-mediated death and inhibition of antitumorigenic properties of T cells. From the perspective of CSCs, effective execution of the first area can occur via limiting the ability of CSCs to present neoantigens to T cells via downregulation of MHC-I expression in the CSCs66,67. Furthermore, single-cell RNA sequencing analysis of mouse epidermal skin squamous cell carcinomas defined an increased stemness signature in tumour cells resistant to adoptive T cell therapy93. Mechanistic studies revealed that these cells expressing markers for CSCs escaped cytotoxic T cell killing by upregulating CD80 in a TGFβ-dependent manner93. While these studies indicate the presence of multiple avoidance mechanisms, our knowledge of therapeutic utility is currently limited.
In contrast, the regulation of T cell activity by CSCs is a more elaborate and better defined process that involves multiple pathways through both direct and indirect contact. One mechanism by which CSCs impair cytotoxic T cell activity is through selective enrichment of inhibitory checkpoint ligands compared with non-CSC tumour cells. While the full array of co-inhibitory receptors expressed by CSCs has not been investigated, several studies revealed that CSCs, which were defined on the basis of expression of phenotypic markers, have higher levels of inhibitory checkpoint receptors. For example, expression of programmed cell death 1 ligand 1 (PDL1) was elevated in CD44+ breast cancer stem cells94,95, CD133+ colon cancer stem cells94,96, and CD44+ HNSCC stem cells97 and expression of V-set domain-containing T cell activation inhibitor 1 (VTCN1) was elevated in CD133+ glioblastoma stem cells98 compared with non-cancer stem cells (FIG. 2). In addition, sphere-forming patient-derived glioblastoma cells or human glioblastoma cell lines64,99,100 and patient-derived colon cancer cells65 and ABCB5+ patient-derived melanoma cells67 have been shown to suppress the proliferation of T cells and limit the expression of co-stimulatory molecules in vitro. Upregulation of phosphorylated STAT3 in T cells co-cultured with CD133+ primary human glioblastoma cells grown in CSC-enriching conditions can account for some of the activity99, but this is not necessarily achieved as a result of direct communication between CSCs and T cells.
It is noteworthy that most of these studies are conducted ex vivo and using artificial culture conditions, which limit their translational potential. Moreover, a portion of the observed effect can be attributed to the presence of myeloid cells that serve as intermediaries. As such, impaired T cell activation is in part mediated by the uptake of extracellular vesicles derived from self-renewing CSCs by myeloid cells in peripheral blood mononuclear cells and subsequent IL-6, IL-10 and IL-1β production100. CSC supernatants are also enriched in IL-4, IL-6 and IL-10, all of which can activate STAT3 signalling in tumour-associated myeloid cells, with outcomes such as induced expression of the checkpoint molecules VTCN1 and PDL1, and production of the immunosuppressive factors IL-10 and TGFβ65,94,98,99,101 (BOX 3; FIG. 2).
Still, the CSC secretome can directly interfere with T cell effector function in the absence of myeloid cells. One of the best documented examples is tenascin C (TNC). Engagement of extracellular vesicle-associated or free TNC with the integrin receptor α5β1 on T cells downregulates AKT and ERK phosphorylation to impair proliferation in co-culture conditions102,103 (FIG. 2). Nevertheless, inhibition of TNC was not sufficient to fully reverse CSC-mediated T cell suppression, warranting further investigation of CSC-derived soluble mediators that can directly impact cytotoxic T cell activity. Importantly, emerging evidence suggests that dysfunctional T cells incapable of lysing CSCs can promote functional and phenotypic stemness traits in a breast cancer cell line through an undefined mechanism104. Gaining mechanistic insight into these interactions will be essential for the design of chimeric antigen receptor T cell strategies directed against CSC antigens105.
Environmental regulators
Although there is accumulating evidence that CSCs and immune cells interact in various types of malignancies, factors impacting the nature of this communication are less well described. A complex interplay within the tumour microenvironment can be influenced by host variation induced by factors including (but not limited to) sex, age and metabolic state. Understanding the contribution of these biological determinants should provide unique insights into altered pathways and additional regulators that sustain the pathological conversation between CSCs and the immune system. These additional factors, while potentially confounding, are likely to inform future therapeutic efforts in the emerging era of personalized medicine.
Sex as a biological variable.
Epidemiologically, male sex is associated with an increased rate of non-reproductive cancers, worse prognosis and higher mortality106. While the impact of biological sex on cancer treatment is under intensive investigation mainly through the use of adjuvant hormone blockers, mechanistic studies focusing on the relative contribution of genetic factors versus hormonal cues are still in their infancy. Early reports indicate that both differences in cellular programming and sex-specific immune interactions can contribute to cancer progression107. As an example, enhanced susceptibility of mouse astrocytes to malignant transformation following inactivation of the tumour suppressors RB1 and p53 has been implicated in the male predominance of glioblastoma108. Importantly, these cells had an increased colony-forming ability, suggesting that they acquire a more stem-like phenotype as compared with female astrocytes lacking RB1 and p53. In parallel, male glioblastoma tumours are infiltrated at higher rates by immunosuppressive myeloid cells, in particular by M-MDSCs109. Although it has been shown that CSCs recruit M-MDSCs to the tumour microenvironment through MIF55,56, any potential correlation between increased SOX2+CD133+ CSC and M-MDSC abundance in male tumours and sexual dimorphism in the expression of immunomodulatory factors by CSCs remains to be investigated. These types of investigations are of particular importance, as sex hormones are capable of altering both immune cell function and CSC behaviour110–114.
Ageing.
Ageing is another risk factor linked to higher cancer prevalence115. Increased accumulation of genetic mutations, reduced immune activity, altered cellular metabolism and decline in sex hormone levels are among the multitude of factors connecting ageing with multiple pathophysiological conditions, including adult cancers. However, there is limited knowledge of how ageing of the tumour microenvironment impacts protumorigenic pathways.
Ontogeny of CSCs is an important consideration for the understanding of their behaviour in the context of ageing. Lineage infidelity characterized by combinational expression of key identity genes distinguishes CSCs from restricted stem cell lineages116, suggesting that deregulation of multipotent cells can give rise to CSCs. By proxy, this observation also indicates that CSCs arising from a pre-existing stem cell pool can inherit age-induced cellular characteristics that provide a survival advantage. With ageing, stem cells acquire a more quiescent state that is associated with downregulation of antigen presentation and enhanced ability to escape from T cell killing117,118, indicating that similar functional changes in CSCs could form the basis of cancer immune evasion and therapeutic resistance.
An opposing point of view is that CSCs emerge from dedifferentiating tumour cells119. Notably, co-existence of these two mechanisms could be the source of CSC heterogeneity observed within tumours. Ageing of the immune system is another factor that could contribute to CSC heterogeneity and abundance since co-culturing cancer cell lines with macrophages, MDSCs or TANs in vitro can trigger tumour cell dedifferentiation as demonstrated by the upregulation of stem cell markers and the acquisition of self-renewal capacity26,29,30,52–54,59,63,120. Importantly, advanced age is accompanied by a heightened inflammation signature. Changes in the frequency of immune cell populations originate from myeloid-biased lineage commitment of aged haematopoietic stem cells121, and epigenetic modification of human immune cells also determines age-dependent immunological phenotypes, such as upregulation of IL-1β122. As a central regulator of the proinflammatory response, IL-1β can affect numerous physiological pathways, including CSC maintenance, especially in inflammation-driven gastrointestinal malignancies. Colon and pancreatic cancers can potentiate macrophages to secrete IL-1β, which upregulates expression of stemness markers in tumour cells and augments sphere-forming ability via activation of NF-κB120,123,124. However, the dynamic role of IL-1β in age-related cancer differences awaits to be elucidated, and understanding the influences of ageing on CSC–immune cell communication axes could inform therapeutic strategies.
Obesity and diet.
Obesity is a risk factor for cancer occurrence125, although the extent of altered metabolic pathways and their ramifications are not fully defined. Obesity and a high-fat diet (HFD) are associated with a chronic low-grade inflammatory state126 that could not only shape the antitumour immune response, such as MDSC accumulation49, but that also has the potential to reshape CSC niches. Studies established that healthy stem cells can change their chromatin accessibility in response to acute inflammatory stimuli to acquire a distinct memory phenotype, which can increase susceptibility to stress-induced lineage infidelity116,127. These observations raise the possibility that stem cells acquire a pro-oncogenic phenotype as a result of a sustained inflammatory response or as part of tissue adaptation mechanisms128. Accordingly, HFD consumption enhanced stem cell function and oncogenic transformation in experimental mouse models of colon cancer as a consequence of altered niche signalling pathways directed by fatty acids129,130.
Alternatively, altered stem cell behaviour could be a secondary effect to diet-induced changes in microbiome composition131. In support of this notion, HFD consumption shifted the commitment of haematopoietic stem cells towards common myeloid progenitors, leading to decreased numbers of common lymphoid progenitors in healthy mice through modulation of the microbiota132. A microbial component, β-glucan, and the associated IL-1β inflammatory response were central to this process132,133. In addition, microbial metabolites are also capable of regulating stem cell behaviour134 and the antitumour immune response135. While microbiome dysbiosis is especially well documented in gastrointestinal malignancies, it has far-reaching consequences for antitumour immunity, tumour growth dynamics and immunotherapy response across malignancies136–139. However, how microbial instructions define the pathological communication between CSCs and immune cells remains an outstanding question.
Therapeutic resistance
Many immunotherapies and CSC pathway inhibitors are currently in clinical development or are being evaluated for solid and non-solid tumours. While these therapeutic strategies (TABLE 1) are likely to improve treatment outcomes, CSCs, which are more resistant to anticancer therapies generally2,140 than the tumour bulk, can alter their intrinsic programming to rewire their mode of interaction with immune cells, and this could hinder therapeutic targeting. For example, chemoresistant CSCs from human breast and colon cancer cell lines were shown to upregulate interferon regulatory factor 5 (IRF5) expression to boost CSF1 production141. These drug-resistant self-renewing cells can enhance the polarization of phenotypically immunosuppressive macrophages from CD14+ peripheral monocytes in a co-transplantation setting, which results in accelerated tumour growth in immunocompromised mice141. As a separate study demonstrated that CSCs undergo chromatin remodelling to acquire tolerance to receptor tyrosine kinase inhibitors and that inhibition of histone deacetylases interferes with CSC sphere formation by overcoming drug resistance, epigenetic modifications emerge as a likely mechanism altering their communication with immune cells142.
TAMs can also drive drug resistance of CSCs in a paracrine fashion. CSCs from colon and lung cancer cell lines prime macrophages to induce secretion of lactadherin and IL-6, which in return activates STAT3 signalling25. In the context of colon cancer, this is accompanied by acquired resistance to cisplatin chemotherapy in vivo. Similar observations were made in mouse PDAC and human ovarian cancer models, where gemcitabine or carboplatin chemotherapy results in an increased tumour-associated myeloid cell frequency in mouse PDAC tumours in vivo and human ovarian heterospheroid cultures in vitro143,144. Inhibition of CSF1R to target myeloid cells was accompanied by reduced ALDH+ PDAC stem cell frequency in vivo and, in combination with gemcitabine, limited tumour growth in part by downregulating STAT3 activation143. Similarly, knockdown of WNT5B in macrophages reduced ALDH activity in ovarian cancer stem cells grown as sphere cultures144. This effect is likely not restricted to the innate immune lineage, and Treg cells can also potentiate chemoresistance of CSCs76. Collectively, these studies suggest that successful treatment strategies will likely need to concomitantly overcome both CSC-dependent and immune-driven resistance mechanisms and that shared signalling pathways can comprise therapeutic vulnerabilities.
Given the cardinal role for STAT3 signalling in CSC maintenance and immunosuppressive cell behaviour (BOX 3), it emerges as a promising candidate for therapeutic targeting. Consequently, there are many ongoing clinical trials testing STAT3 small-molecule inhibitors, oligonucleotides and indirect pathway antagonists as a single agent or in combination with other immunotherapies in cancer145. Similarly, a multimodal role for TGFβ (BOX 3) makes it an attractive therapeutic candidate. Therefore, neutralizing antibodies and small molecules are under clinical investigation, but the central role of TGFβ in tissue remodelling remains a chief safety concern146. Hence, the outcomes of these clinical trials and accompanying biomarker and mechanistic studies will be essential to identify resistance mechanisms and combinational approaches, including simultaneous targeting of TGFβ and STAT3.
Conclusions and perspectives
In this Perspective, we have provided examples of communication of CSCs with individual immune cell lineages, as well as the mechanisms of crosstalk across a variety of cancers. Moving forward, we anticipate that this knowledge will help guide the next generation of therapeutic strategies for simultaneous targeting of CSCs and pathological immune response. However, rapid realization of this will require investment in the interrogation of CSC programmes in immunocompetent mouse models, a topic that is generally overlooked, as the initial CSC observations and models used human tumour tissue in immunocompromised mice. New models leveraging bona fide cell surface markers and/or reporter systems rigorously validated for functional CSC activity are likely to stimulate a new series of in vivo studies investigating the interaction of these cells with the immune system. In parallel, preclinical and clinical immunotherapy studies would benefit from additional assessment of CSCs using a series of functional assays including tumour-sphere formation and an in vivo limiting-dilution assay140. With the rapid advancement in single-cell and spatial analysis techniques, these lines of investigations have tremendous potential to define complex interactions involving multiple immune cell lineages and CSCs. Finally, consideration of biological sex, ageing, and metabolic or microbiome interactions is poised to reveal additional therapeutic opportunities, as these factors individually impact tumour growth and immune response.
Acknowledgements
The authors thank E. Mulkearns-Hubert for editorial assistance. Work in the Lathia laboratory is supported by the Cleveland Clinic, Case Comprehensive Cancer Center, the American Brain Tumor Association, the US National Brain Tumor Society and the NIH (R01 NS109742, R01 NS117104 and P01 CA245705 (J.L.) and F32CA243314 and K99CA248611 (D.B.)).
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Batlle E & Clevers H Cancer stem cells revisited. Nat. Med 23, 1124–1134 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Saygin C, Matei D, Majeti R, Reizes O & Lathia JD Targeting cancer stemness in the clinic: from hype to hope. Cell Stem Cell 24, 2540 (2019). [DOI] [PubMed] [Google Scholar]
- 3.Clara JA, Monge C, Yang Y & Takebe N Targeting signalling pathways and the immune microenvironment of cancer stem cells - a clinical update. Nat. Rev. Clin. Oncol 17, 204–232 (2020). [DOI] [PubMed] [Google Scholar]
- 4.Galluzzi L, Chan TA, Kroemer G, Wolchok JD & Lopez-Soto A The hallmarks of successful anticancer immunotherapy. Sci. Transl. Med 10, eaat7807 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Hinshaw DC & Shevde LA The tumor microenvironment innately modulates cancer progression. Cancer Res 79, 4557–4566 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miranda A et al. Cancer stemness, intratumoral heterogeneity, and immune response across cancers. Proc. Natl Acad. Sci. USA 116, 9020–9029 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Engblom C, Pfirschke C & Pittet MJ The role of myeloid cells in cancer therapies. Nat. Rev. Cancer 16, 447–462 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Davies LC, Jenkins SJ, Allen JE & Taylor PR Tissue-resident macrophages. Nat. Immunol 14, 986–995 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mass E et al. Specification of tissue-resident macrophages during organogenesis. Science 353, aaf4238 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Raggi C et al. Cholangiocarcinoma stem-like subset shapes tumor-initiating niche by educating associated macrophages. J. Hepatol 66, 102–115 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hide T et al. Oligodendrocyte progenitor cells and macrophages/microglia produce glioma stem cell niches at the tumor border. EBioMedicine 30, 94–104 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang YK et al. Macrophage spatial heterogeneity in gastric cancer defined by multiplex immunohistochemistry. Nat. Commun 10, 3928 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bowman RL et al. Macrophage ontogeny underlies differences in tumor-specific education in brain malignancies. Cell Rep 17, 2445–2459 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cassetta L et al. Human tumor-associated macrophage and monocyte transcriptional landscapes reveal cancer-specific reprogramming, biomarkers, and therapeutic targets. Cancer Cell 35, 588–602 e510 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hashimoto D et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity 38, 792–804 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ginhoux F & Guilliams M Tissue-resident macrophage ontogeny and homeostasis. Immunity 44, 439–449 (2016). [DOI] [PubMed] [Google Scholar]
- 17.Laviron M & Boissonnas A Ontogeny of tumor-associated macrophages. Front. Immunol 10, 1799 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qian BZ et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 475, 222–225 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tao W et al. Dual role of WISP1 in maintaining glioma stem cells and tumor-supportive macrophages in glioblastoma. Nat. Commun 11, 3015 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou W et al. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat. Cell Biol 17, 170–182 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guo X et al. Single tumor-initiating cells evade immune clearance by recruiting type II macrophages. Genes Dev 31, 247–259 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu A et al. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro Oncol 12, 1113–1125 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yi L et al. Glioma-initiating cells: a predominant role in microglia/macrophages tropism to glioma. J. Neuroimmunol 232, 75–82 (2011). [DOI] [PubMed] [Google Scholar]
- 24.Guo X, Pan Y & Gutmann DH Genetic and genomic alterations differentially dictate low-grade glioma growth through cancer stem cell-specific chemokine recruitment of T cells and microglia. Neuro Oncol 21, 1250–1262 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jinushi M et al. Tumor-associated macrophages regulate tumorigenicity and anticancer drug responses of cancer stem/initiating cells. Proc. Natl Acad. Sci. USA 108, 12425–12430 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fan QM et al. Tumor-associated macrophages promote cancer stem cell-like properties via transforming growth factor-beta1-induced epithelial-mesenchymal transition in hepatocellular carcinoma. Cancer Lett 352, 160–168 (2014). [DOI] [PubMed] [Google Scholar]
- 27.Lu H et al. A breast cancer stem cell niche supported by juxtacrine signalling from monocytes and macrophages. Nat. Cell Biol 16, 1105–1117 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shi Y et al. Tumour-associated macrophages secrete pleiotrophin to promote PTPRZ1 signalling in glioblastoma stem cells for tumour growth. Nat. Commun 8, 15080 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wan S et al. Tumor-associated macrophages produce interleukin 6 and signal via STAT3 to promote expansion of human hepatocellular carcinoma stem cells. Gastroenterology 147, 1393–1404 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang B et al. Macrophage-expressed CD51 promotes cancer stem cell properties via the TGF-beta1/smad2/3 axis in pancreatic cancer. Cancer Lett 459, 204–215 (2019). [DOI] [PubMed] [Google Scholar]
- 31.Su W et al. The Polycomb repressor complex 1 drives double-negative prostate cancer metastasis by coordinating stemness and immune suppression. Cancer Cell 36, 139–155 e110 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cannarile MA et al. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J. Immunother. Cancer 5, 53 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Theocharides AP et al. Disruption of SIRPalpha signaling in macrophages eliminates human acute myeloid leukemia stem cells in xenografts. J. Exp. Med 209, 1883–1899 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee TK et al. Blockade of CD47-mediated cathepsin S/protease-activated receptor 2 signaling provides a therapeutic target for hepatocellular carcinoma. Hepatology 60, 179–191 (2014). [DOI] [PubMed] [Google Scholar]
- 35.Cioffi M et al. Inhibition of CD47 effectively targets pancreatic cancer stem cells via dual mechanisms. Clin. Cancer Res 21, 2325–2337 (2015). [DOI] [PubMed] [Google Scholar]
- 36.Liu L et al. Anti-CD47 antibody as a targeted therapeutic agent for human lung cancer and cancer stem cells. Front. Immunol 8, 404 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hutter G et al. Microglia are effector cells of CD47-SIRPalpha antiphagocytic axis disruption against glioblastoma. Proc. Natl Acad. Sci. USA 116, 997–1006 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sikic BI et al. First-in-human, first-in-class phase I trial of the anti-CD47 antibody Hu5F9-G4 in patients with advanced cancers. J. Clin. Oncol 37, 946–953 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kenkel JA et al. An immunosuppressive dendritic cell subset accumulates at secondary sites and promotes metastasis in pancreatic cancer. Cancer Res 77, 4158–4170 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barilla RM et al. Specialized dendritic cells induce tumor-promoting IL-10+IL-17+ FoxP3neg regulatory CD4+ T cells in pancreatic carcinoma. Nat. Commun 10, 1424 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grange C et al. Role of HLA-G and extracellular vesicles in renal cancer stem cell-induced inhibition of dendritic cell differentiation. BMC Cancer 15, 1009 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liang S et al. Modulation of dendritic cell differentiation by HLA-G and ILT4 requires the IL-6–STAT3 signaling pathway. Proc. Natl Acad. Sci. USA 105, 8357–8362 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hsu YL et al. Interaction between tumor-associated dendritic cells and colon cancer cells contributes to tumor progression via CXCL1. Int. J. Mol. Sci 10.3390/ijms19082427 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang D, Sun H, Wei J, Cen B & DuBois RN CXCL1 is critical for premetastatic niche formation and metastasis in colorectal cancer. Cancer Res 77, 3655–3665 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee CG et al. A rare fraction of drug-resistant follicular lymphoma cancer stem cells interacts with follicular dendritic cells to maintain tumourigenic potential. Br. J. Haematol 158, 79–90 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pellegatta S et al. Neurospheres enriched in cancer stem-like cells are highly effective in eliciting a dendritic cell-mediated immune response against malignant gliomas. Cancer Res 66, 10247–10252 (2006). [DOI] [PubMed] [Google Scholar]
- 47.Ning N et al. Cancer stem cell vaccination confers significant antitumor immunity. Cancer Res 72, 1853–1864 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lechner MG et al. Immunogenicity of murine solid tumor models as a defining feature of in vivo behavior and response to immunotherapy. J. Immunother 36, 477–489 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Veglia F, Sanseviero E & Gabrilovich DI Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat. Rev. Immunol 10.1038/s41577-020-00490-y (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ouzounova M et al. Monocytic and granulocytic myeloid derived suppressor cells differentially regulate spatiotemporal tumour plasticity during metastatic cascade. Nat. Commun 8, 14979 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhou J, Nefedova Y, Lei A & Gabrilovich D Neutrophils and PMN-MDSC: their biological role and interaction with stromal cells. Semin. Immunol 35, 19–28 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cui TX et al. Myeloid-derived suppressor cells enhance stemness of cancer cells by inducing microRNA101 and suppressing the corepressor CtBP2. Immunity 39, 611–621 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Panni RZ et al. Tumor-induced STAT3 activation in monocytic myeloid-derived suppressor cells enhances stemness and mesenchymal properties in human pancreatic cancer. Cancer Immunol. Immunother 63, 513–528 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Peng D et al. Myeloid-derived suppressor cells endow stem-like qualities to breast cancer cells through IL6/STAT3 and NO/NOTCH cross-talk signaling. Cancer Res 76, 3156–3165 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Otvos B et al. Cancer stem cell-secreted macrophage migration inhibitory factor stimulates myeloid derived suppressor cell function and facilitates glioblastoma immune evasion. Stem Cell 34, 2026–2039 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Alban TJ et al. Glioblastoma myeloid-derived suppressor cell subsets express differential macrophage migration inhibitory factor receptor profiles that can be targeted to reduce immune suppression. Front. Immunol 11, 1191 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang G et al. Targeting YAP-dependent MDSC infiltration impairs tumor progression. Cancer Discov 6, 80–95 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shidal C, Singh NP, Nagarkatti P & Nagarkatti M MicroRNA-92 expression in CD133+ melanoma stem cells regulates immunosuppression in the tumor microenvironment via integrin-dependent activation of TGFbeta. Cancer Res 79, 3622–3635 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kuroda H et al. Prostaglandin E2 produced by myeloid-derived suppressive cells induces cancer stem cells in uterine cervical cancer. Oncotarget 9, 36317–36330 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ai L et al. Myeloid-derived suppressor cells endow stem-like qualities to multiple myeloma cells by inducing piRNA-823 expression and DNMT3B activation. Mol. Cancer 18, 88 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang Y et al. Granulocytic myeloid-derived suppressor cells promote the stemness of colorectal cancer cells through exosomal S100A9. Adv. Sci 6, 1901278 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fridlender ZG et al. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 16, 183–194 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhou SL et al. A positive feedback loop between cancer stem-like cells and tumor-associated neutrophils controls hepatocellular carcinoma progression. Hepatology 70, 1214–1230 (2019). [DOI] [PubMed] [Google Scholar]
- 64.Di Tomaso T et al. Immunobiological characterization of cancer stem cells isolated from glioblastoma patients. Clin. Cancer Res 16, 800–813 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Volonte A et al. Cancer-initiating cells from colorectal cancer patients escape from T cell-mediated immunosurveillance in vitro through membrane-bound IL-4. J. Immunol 192, 523–532 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Morrison BJ, Steel JC & Morris JC Reduction of MHC-I expression limits T-lymphocyte-mediated killing of cancer-initiating cells. BMC Cancer 18, 469 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schatton T et al. Modulation of T-cell activation by malignant melanoma initiating cells. Cancer Res 70, 697–708 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Paczulla AM et al. Absence of NKG2D ligands defines leukaemia stem cells and mediates their immune evasion. Nature 572, 254–259 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wu A et al. Expression of MHC I and NK ligands on human CD133+ glioma cells: possible targets of immunotherapy. J. Neurooncol 83, 121–131 (2007). [DOI] [PubMed] [Google Scholar]
- 70.Tallerico R et al. Human NK cells selective targeting of colon cancer-initiating cells: a role for natural cytotoxicity receptors and MHC class I molecules. J. Immunol 190, 2381–2390 (2013). [DOI] [PubMed] [Google Scholar]
- 71.Beier CP et al. The cancer stem cell subtype determines immune infiltration of glioblastoma. Stem Cell Dev 21, 2753–2761 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sharonov GV, Serebrovskaya EO, Yuzhakova DV, Britanova OV & Chudakov DM B cells, plasma cells and antibody repertoires in the tumour microenvironment. Nat. Rev. Immunol 20, 294–307 (2020). [DOI] [PubMed] [Google Scholar]
- 73.Bruchard M & Ghiringhelli F Deciphering the roles of innate lymphoid cells in cancer. Front. Immunol 10, 656 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lindemans CA et al. Interleukin-22 promotes intestinal-stem-cell-mediated epithelial regeneration. Nature 528, 560–564 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.You Y et al. Ovarian cancer stem cells promote tumour immune privilege and invasion via CCL5 and regulatory T cells. Clin. Exp. Immunol 191, 60–73 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Xu Y et al. Sox2 communicates with Tregs through CCL1 to promote the stemness property of breast cancer cells. Stem Cell 35, 2351–2365 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chang AL et al. CCL2 produced by the glioma microenvironment is essential for the recruitment of regulatory T cells and myeloid-derived suppressor cells. Cancer Res 76, 5671–5682 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ban Y et al. Targeting autocrine CCL5-CCR5 axis reprograms immunosuppressive myeloid cells and reinvigorates antitumor immunity. Cancer Res 77, 2857–2868 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Eruslanov E et al. Expansion of CCR8+ inflammatory myeloid cells in cancer patients with urothelial and renal carcinomas. Clin. Cancer Res 19, 1670–1680 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Curiel TJ et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med 10, 942–949 (2004). [DOI] [PubMed] [Google Scholar]
- 81.Wainwright DA et al. IDO expression in brain tumors increases the recruitment of regulatory T cells and negatively impacts survival. Clin. Cancer Res 18, 6110–6121 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nakano M et al. Dedifferentiation process driven by TGF-beta signaling enhances stem cell properties in human colorectal cancer. Oncogene 38, 780–793 (2019). [DOI] [PubMed] [Google Scholar]
- 83.Ozawa Y et al. Indoleamine 2,3-dioxygenase 1 is highly expressed in glioma stem cells. Biochem. Biophys. Res. Commun 524, 723–729 (2020). [DOI] [PubMed] [Google Scholar]
- 84.Stapelberg M et al. Indoleamine-2,3-dioxygenase elevated in tumor-initiating cells is suppressed by mitocans. Free Radic. Biol. Med 67, 41–50 (2014). [DOI] [PubMed] [Google Scholar]
- 85.Sharma MD et al. Indoleamine 2,3-dioxygenase controls conversion of Foxp3+ Tregs to TH17-like cells in tumor-draining lymph nodes. Blood 113, 6102–6111 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gagliani N et al. Th17 cells transdifferentiate into regulatory T cells during resolution of inflammation. Nature 523, 221–225 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Martin F, Apetoh L & Ghiringhelli F Controversies on the role of Th17 in cancer: a TGF-beta-dependent immunosuppressive activity? Trends Mol. Med 18, 742–749 (2012). [DOI] [PubMed] [Google Scholar]
- 88.Yang S et al. Foxp3+IL-17+ T cells promote development of cancer-initiating cells in colorectal cancer. J. Leukoc. Biol 89, 85–91 (2011). [DOI] [PubMed] [Google Scholar]
- 89.Zhang Y et al. Immune cell production of interleukin 17 induces stem cell features of pancreatic intraepithelial neoplasia cells. Gastroenterology 155, 210–223 e213 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wang R et al. Th17 cell-derived IL-17A promoted tumor progression via STAT3/NF-kappaB/Notch1 signaling in non-small cell lung cancer. Oncoimmunology 7, e1461303 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.He W et al. IL22RA1/STAT3 signaling promotes stemness and tumorigenicity in pancreatic cancer. Cancer Res 78, 3293–3305 (2018). [DOI] [PubMed] [Google Scholar]
- 92.Jiang R et al. IL-22 is related to development of human colon cancer by activation of STAT3. BMC Cancer 13, 59 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Miao Y et al. Adaptive immune resistance emerges from tumor-initiating stem cells. Cell 177, 1172–1186 e1114 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wu Y et al. Increased PD-L1 expression in breast and colon cancer stem cells. Clin. Exp. Pharmacol. Physiol 44, 602–604 (2017). [DOI] [PubMed] [Google Scholar]
- 95.Hsu JM et al. STT3-dependent PD-L1 accumulation on cancer stem cells promotes immune evasion. Nat. Commun 9, 1908 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhi Y et al. B7H1 expression and epithelial-to-mesenchymal transition phenotypes on colorectal cancer stem-like cells. PLoS ONE 10, e0135528 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lee Y et al. CD44+ cells in head and neck squamous cell carcinoma suppress T-cell-mediated immunity by selective constitutive and inducible expression of PD-L1. Clin. Cancer Res 22, 3571–3581 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yao Y et al. B7-H4(B7x)-mediated cross-talk between glioma-initiating cells and macrophages via the IL6/JAK/STAT3 pathway lead to poor prognosis in glioma patients. Clin. Cancer Res 22, 2778–2790 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wei J et al. Glioblastoma cancer-initiating cells inhibit T-cell proliferation and effector responses by the signal transducers and activators of transcription 3 pathway. Mol. Cancer Ther 9, 67–78 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Domenis R et al. Systemic T cells immunosuppression of glioma stem cell-derived exosomes is mediated by monocytic myeloid-derived suppressor cells. PLoS ONE 12, e0169932 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gabrusiewicz K et al. Glioblastoma stem cell-derived exosomes induce M2 macrophages and PD-L1 expression on human monocytes. Oncoimmunology 7, e1412909 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mirzaei R et al. Brain tumor-initiating cells export tenascin-C associated with exosomes to suppress T cell activity. Oncoimmunology 7, e1478647 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Jachetti E et al. Tenascin-C protects cancer stem-like cells from immune surveillance by arresting T-cell activation. Cancer Res 75, 2095–2108 (2015). [DOI] [PubMed] [Google Scholar]
- 104.Stein RG et al. Cognate nonlytic interactions between CD8+ T cells and breast cancer cells induce cancer stem cell-like properties. Cancer Res 79, 1507–1519 (2019). [DOI] [PubMed] [Google Scholar]
- 105.Wang D et al. CRISPR screening of CAR T cells and cancer stem cells reveals critical dependencies for cell-based therapies. Cancer Discov 10.1158/2159-8290.CD-20-1243 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Clocchiatti A, Cora E, Zhang Y & Dotto GP Sexual dimorphism in cancer. Nat. Rev. Cancer 16, 330–339 (2016). [DOI] [PubMed] [Google Scholar]
- 107.Klein SL & Flanagan KL Sex differences in immune responses. Nat. Rev. Immunol 16, 626–638 (2016). [DOI] [PubMed] [Google Scholar]
- 108.Sun T et al. Sexually dimorphic RB inactivation underlies mesenchymal glioblastoma prevalence in males. J. Clin. Invest 124, 4123–4133 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Bayik D et al. Myeloid-derived suppressor cell subsets drive glioblastoma growth in a sex-specific manner. Cancer Discov 10, 1210–1225 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Fillmore CM et al. Estrogen expands breast cancer stem-like cells through paracrine FGF/Tbx3 signaling. Proc. Natl Acad. Sci. USA 107, 21737–21742 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sun Y et al. Estrogen promotes stemness and invasiveness of ER-positive breast cancer cells through Gli1 activation. Mol. Cancer 13, 137 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Svoronos N et al. Tumor cell-independent estrogen signaling drives disease progression through mobilization of myeloid-derived suppressor cells. Cancer Discov 7, 72–85 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Generali D et al. Immunomodulation of FOXP3+ regulatory T cells by the aromatase inhibitor letrozole in breast cancer patients. Clin. Cancer Res 15, 1046–1051 (2009). [DOI] [PubMed] [Google Scholar]
- 114.Sarmiento-Castro A et al. Increased expression of interleukin-1 receptor characterizes anti-estrogen-resistant ALDH+ breast cancer stem cells. Stem Cell Rep 15, 307–316 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.White MC et al. Age and cancer risk: a potentially modifiable relationship. Am. J. Prev. Med 46, S7–15 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ge Y et al. Stem cell lineage infidelity drives wound repair and cancer. Cell 169, 636–650 e614 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kalamakis G et al. Quiescence modulates stem cell maintenance and regenerative capacity in the aging brain. Cell 176, 1407–1419 e1414 (2019). [DOI] [PubMed] [Google Scholar]
- 118.Agudo J et al. Quiescent tissue stem cells evade immune surveillance. Immunity 48, 271–285 e275 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Bocci F et al. Toward understanding cancer stem cell heterogeneity in the tumor microenvironment. Proc. Natl Acad. Sci. USA 116, 148–157 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kaler P, Godasi BN, Augenlicht L & Klampfer L The NF-kappaB/AKT-dependent induction of Wnt signaling in colon cancer cells by macrophages and IL-1beta. Cancer Microenviron 2, 69–80 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Pang WW et al. Human bone marrow hematopoietic stem cells are increased in frequency and myeloid-biased with age. Proc. Natl Acad. Sci. USA 108, 20012–20017 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Marquez EJ et al. Sexual-dimorphism in human immune system aging. Nat. Commun 11, 751 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Li Y, Wang L, Pappan L, Galliher-Beckley A & Shi J IL-1beta promotes stemness and invasiveness of colon cancer cells through Zeb1 activation. Mol. Cancer 11, 87 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Nomura A et al. NFkappaB-mediated invasiveness in CD133+ pancreatic TICs is regulated by autocrine and paracrine activation of IL1 signaling. Mol. Cancer Res 16, 162–172 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Lauby-Secretan B et al. Body fatness and cancer–viewpoint of the IARC Working Group. N. Engl. J. Med 375, 794–798 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Duan Y et al. Inflammatory links between high fat diets and diseases. Front. Immunol 9, 2649 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Naik S et al. Inflammatory memory sensitizes skin epithelial stem cells to tissue damage. Nature 550, 475–480 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Li XF et al. Chronic inflammation-elicited liver progenitor cell conversion to liver cancer stem cell with clinical significance. Hepatology 66, 1934–1951 (2017). [DOI] [PubMed] [Google Scholar]
- 129.Beyaz S et al. High-fat diet enhances stemness and tumorigenicity of intestinal progenitors. Nature 531, 53–58 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Yoshimoto S et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 499, 97–101 (2013). [DOI] [PubMed] [Google Scholar]
- 131.David LA et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 505, 559–563 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Luo Y et al. Microbiota from obese mice regulate hematopoietic stem cell differentiation by altering the bone niche. Cell Metab 22, 886–894 (2015). [DOI] [PubMed] [Google Scholar]
- 133.Mitroulis I et al. Modulation of myelopoiesis progenitors is an integral component of trained immunity. Cell 172, 147–161 e112 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kaiko GE et al. The colonic crypt protects stem cells from microbiota-derived metabolites. Cell 165, 1708–1720 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Mager LF et al. Microbiome-derived inosine modulates response to checkpoint inhibitor immunotherapy. Science 369, 1481–1489 (2020). [DOI] [PubMed] [Google Scholar]
- 136.Gopalakrishnan V et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 359, 97–103 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Matson V et al. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science 359, 104–108 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Routy B et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 359, 91–97 (2018). [DOI] [PubMed] [Google Scholar]
- 139.Riquelme E et al. Tumor microbiome diversity and composition influence pancreatic cancer outcomes. Cell 178, 795–806 e712 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Lathia JD, Mack SC, Mulkearns-Hubert EE, Valentim CL & Rich JN Cancer stem cells in glioblastoma. Genes Dev 29, 1203–1217 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Yamashina T et al. Cancer stem-like cells derived from chemoresistant tumors have a unique capacity to prime tumorigenic myeloid cells. Cancer Res 74, 2698–2709 (2014). [DOI] [PubMed] [Google Scholar]
- 142.Sharma SV et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 141, 69–80 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Mitchem JB et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res 73, 1128–1141 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Raghavan S, Mehta P, Xie Y, Lei YL & Mehta G Ovarian cancer stem cells and macrophages reciprocally interact through the WNT pathway to promote pro-tumoral and malignant phenotypes in 3D engineered microenvironments. J. Immunother. Cancer 7, 190 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Zou S et al. Targeting STAT3 in cancer immunotherapy. Mol. Cancer 19, 145 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Ciardiello D, Elez E, Tabernero J & Seoane J Clinical development of therapies targeting TGFbeta: current knowledge and future perspectives. Ann. Oncol 31, 1336–1349 (2020). [DOI] [PubMed] [Google Scholar]
- 147.Laplane L & Solary E Towards a classification of stem cells. eLife 10.7554/eLife.46563 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Naik S, Larsen SB, Cowley CJ & Fuchs E Two to Tango: dialog between immunity and stem cells in health and disease. Cell 175, 908–920 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Sehgal A et al. The role of CSF1R-dependent macrophages in control of the intestinal stem-cell niche. Nat. Commun 9, 1272 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Chow A et al. Bone marrow CD169+ macrophages promote the retention of hematopoietic stem and progenitor cells in the mesenchymal stem cell niche. J. Exp. Med 208, 261–271 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Gyorki DE, Asselin-Labat ML, van Rooijen N, Lindeman GJ & Visvader JE Resident macrophages influence stem cell activity in the mammary gland. Breast Cancer Res 11, R62 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Van Nguyen A & Pollard JW Colony stimulating factor-1 is required to recruit macrophages into the mammary gland to facilitate mammary ductal outgrowth. Dev. Biol 247, 11–25 (2002). [DOI] [PubMed] [Google Scholar]
- 153.Chakrabarti R et al. Notch ligand Dll1 mediates cross-talk between mammary stem cells and the macrophageal niche. Science 10.1126/science.aan4153 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Chen CC et al. Organ-level quorum sensing directs regeneration in hair stem cell populations. Cell 161, 277–290 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Fujisaki J et al. In vivo imaging of Treg cells providing immune privilege to the haematopoietic stem-cell niche. Nature 474, 216–219 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Ali N et al. Regulatory T cells in skin facilitate epithelial stem cell differentiation. Cell 169, 1119–1129 e1111 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Hirata Y et al. CD150high bone marrow Tregs maintain hematopoietic stem cell quiescence and immune privilege via adenosine. Cell Stem Cell 22, 445–453 e445 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Biton M et al. T helper cell cytokines modulate intestinal stem cell renewal and differentiation. Cell 175, 1307–1320 e1322 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Bellomo C, Caja L & Moustakas A Transforming growth factor beta as regulator of cancer stemness and metastasis. Br. J. Cancer 115, 761–769 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]