

Abstract
Ankyrin scaffolding proteins are critical for membrane domain organization and protein stabilization in many different cell types including neurons. In the cerebellum, Ankyrin-R (AnkR) is highly enriched in Purkinje neurons, granule cells, and in the cerebellar nuclei (CN). Using male and female mice with a floxed allele for Ank1 in combination with Nestin-Cre and Pcp2-Cre mice, we found that ablation of AnkR from Purkinje neurons caused ataxia, regional and progressive neurodegeneration, and altered cerebellar output. We show that AnkR interacts with the cytoskeletal protein β3 spectrin and the potassium channel Kv3.3. Loss of AnkR reduced somatic membrane levels of β3 spectrin and Kv3.3 in Purkinje neurons. Thus, AnkR links Kv3.3 channels to the β3 spectrin-based cytoskeleton. Our results may help explain why mutations in β3 spectrin and Kv3.3 both cause spinocerebellar ataxia.
SIGNIFICANCE STATEMENT Ankyrin scaffolding proteins localize and stabilize ion channels in the membrane by linking them to the spectrin-based cytoskeleton. Here, we show that Ankyrin-R (AnkR) links Kv3.3 K+ channels to the β3 spectrin-based cytoskeleton in Purkinje neurons. Loss of AnkR causes Purkinje neuron degeneration, altered cerebellar physiology, and ataxia, which is consistent with mutations in Kv3.3 and β3 spectrin causing spinocerebellar ataxia.
Keywords: ataxia, cytoskeleton, ion channel, neurodegeneration
Introduction
The mammalian nervous system expresses three ankyrin proteins, Ankyrin-R (AnkR), Ankyrin-B (AnkB), and Ankyrin-G (AnkG), corresponding to the genes Ank1-3, respectively. These large scaffolding proteins function as adaptors between the spectrin-based cytoskeleton and cytoplasmic domains of multiple transmembrane proteins. Furthermore, each ankyrin has unique spectrin and membrane binding partners, and distinct cellular and subcellular distributions. For example, AnkG is highly enriched at nodes of Ranvier and axon initial segments, and its binding partners include β4 spectrin, voltage-gated Na+ (Nav) channels, and Kv7.2/3 K+ channels. AnkB is widely expressed throughout the nervous system and colocalizes and interacts with β2 spectrin and Nav channels (Bréchet et al., 2008; Bennett and Lorenzo, 2013; Nelson and Jenkins, 2017; Stevens and Rasband, 2021). In contrast, much less is known about AnkR's distribution, function, and protein interactions in neurons.
Outside of the nervous system, AnkR is highly expressed in red blood cells where it interacts with β1 spectrin and the bicarbonate-chloride exchanger also known as Band 3 (Bennett, 1982). AnkR mutations cause hereditary spherocytosis, a hemolytic anemia. Intriguingly, some case reports of hereditary spherocytosis because of mutations in AnkR also describe neurologic abnormalities, including cerebellar dysfunction and psychomotor delay (Matheson and Howland, 1974; McCann and Jacob, 1976; Coetzer et al., 1988; Miya et al., 2012). In mice, AnkR was reported in discrete, but largely undefined neuronal populations (Lambert and Bennett, 1993), and aged AnkR hypomorphs (nb/nb mice) have cerebellar Purkinje neuron degeneration and impaired gait (Peters et al., 1991). Like AnkG and AnkB, neuronal AnkR also regulates ion channel localization and density by linking them to the actin cytoskeleton through spectrins. For example, AnkR is required for the normal expression and localization of Kv3.1b K+ channels and β1 spectrin in forebrain fast-spiking parvalbumin-positive (Pv+) GABAergic neurons, and loss of AnkR alters the intrinsic excitability of these cells. Nodal AnkR is sufficient to recruit Kv3.1b K+ channels to nodes of Ranvier (Stevens et al., 2021), and can also function together with β1 spectrin to cluster and maintain Nav channels at nodes (Ho et al., 2014). AnkR also interacts with β3 spectrin (Clarkson et al., 2014). Mutations in β3 spectrin cause spinocerebellar ataxia type 5 (SCA5; Ikeda et al., 2006; Perkins et al., 2010) and spectrin associated autosomal recessive cerebellar ataxia type 1 (SPARCA1; Lise et al., 2012), both of which cause cerebellar degeneration and ataxia. Studies in β3 spectrin-deficient mice suggest these ataxias may result from failure to stabilize membrane protein complexes through AnkR (Clarkson et al., 2014).
Here, we used a new mouse model that permitted us to investigate the function of AnkR in the cerebellum, without confounds because of partial loss of the protein or anemia. Consistent with previous studies (Peters et al., 1991; Lambert and Bennett, 1993; Clarkson et al., 2014), we show that AnkR is highly enriched in cerebellar Purkinje neurons, granule cells, and in the cerebellar nuclei (CN). Deletion of AnkR from the cerebellum or specifically Purkinje neurons resulted in ataxia and progressive Purkinje neuron degeneration predominantly in the anterior zone (AZ) of the cerebellum; this degeneration occurred in parasagittal stripes. We show that AnkR binds directly to Kv3.3 K+ channels, which are highly enriched in the somatodendritic membrane domains of Purkinje neurons, and links them to the β3 spectrin-dependent cytoskeleton; loss of AnkR reduces both Kv3.3 and β3 spectrin in Purkinje neuron somatic membranes, indicating that AnkR is necessary to maintain the high membrane densities of both these proteins. Finally, we show the changes caused by deletion of AnkR from Purkinje neurons decrease the functional output of the CN.
Materials and Methods
Animals
AnkR conditional knock-out mice were previously described (Stevens et al., 2021), and used a similar strategy to that successfully used to create the Ank2 and Ank3 floxed mice where loxP sites flank exons 23/24 and 22/23, respectively (Chang et al., 2014; Ho et al., 2014). In brief, a targeting construct was designed to introduce loxP sites flanking exons 26 and 27 of Ank1. Cre-mediated removal of these exons causes a frame-shift mutation that results in a premature stop codon in exon 28. Forward primer: 5′-GGG AAA CTC CAC AGA GCC TGA CGG GTC AGT-3′, reverse primer: 5′-GGC GTC CCT ATG TTC CAT CCT ATA GAT GAC T-3′. These animals were generated as a service of the University of Rochester Medical Center Transgenic Core Facility. Ank1F/F mice were generated in and subsequently backcrossed to C57BL/6 (IMSR catalog #JAX:000664, RRID:IMSR_JAX:000664) for at least four generations before being crossed to Nestin-Cre transgenic mice (IMSR catalog #JAX:003771, RRID:IMSR_JAX:003771), and Pcp2-Cre transgenic mice (IMSR catalog #JAX:004146, RRID:IMSR_JAX:004146). As germline recombination has been found to occur in these Cre lines (Luo et al., 2020), we used immunostaining with anti-AnkR antibodies as a secondary confirmation of genotype. Both male and female mice were used in our studies. All experiments were conducted in compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Animal Care and Use Committee at Baylor College of Medicine.
Gait analysis
Gait abnormalities were assessed using footprint analysis as previously described (White et al., 2014). In brief, hindfeet were painted with washable, nontoxic paint before mice walked on a strip of white paper fitted to the floor of a 45-cm custom-made Plexiglas tunnel. Runs were repeated to reach a total of 20–30 steps/animal. Three measurements were taken, the distance between one left-foot-step to the next left-foot-step, the straight distance between left and right feet, and the diagonal distance between left and right feet, to obtain lengths for stride, sway, and stance, respectively. Measurements were averaged for each mouse.
Antibodies
The primary antibodies used here include: mouse monoclonal antibodies against AnkR (IF: 1:250, UC Davis/NIH NeuroMab Facility catalog #75-380, RRID:AB_2491109), β1 spectrin (IF: 1:250, UC Davis/NIH NeuroMab Facility catalog #73-374, RRID:AB_2315814), AnkG (IF: 1:500, UC Davis/NIH NeuroMab Facility catalog #73-146, RRID:AB_10697718), Kv3.3 (IF: 1:500, Antibodies-Online catalog #ABIN572016, RRID:AB_10782137), Calbindin (IF: 1:250, UC Davis/NIH NeuroMab Facility catalog #L109/57, RRID:AB_2877197), and Flag-tag or DDDDK-tag (WB: 1:1000, MBL International catalog #M185-3L, RRID:AB_11123930), rabbit polyclonal antibodies against AnkR (IF: 1:500, WB: 1:1000, RRID:AB_2833096), Ank1 (IF: 1:500, WB: 1:1000, Thermo Fisher Scientific catalog #PA5-63372, RRID:AB_2638015), AnkB (IF: 1:250, Santa Cruz Biotechnology catalog #sc-28560, RRID:AB_2242828), β3 spectrin (IF: 1:500, WB: 1:1000, Novus catalog #NB110-58346, RRID:AB_877723), Calbindin D-28k (IF: 1:500, Swant catalog #CB38, RRID:AB_10000340), Kv3.3 (IF: 1:500, WB: 1:1000, Alomone Labs catalog #APC-102, RRID:AB_2040170), and GFP (WB: 1:1000, Thermo Fisher Scientific, catalog #A-11122, RRID:AB_221569). Secondary antibodies were purchased from Jackson ImmunoResearch or Life Technologies (IgG-specific mouse antibodies) and used at a concentration of 1:1000. We encourage researchers to determine the optimal antibody dilutions for themselves as varying tissue treatment can affect staining conditions.
Immunofluorescence
Animals were transcardially saline perfused to reduce red blood cells, then brains were dissected and fixed in 4% paraformaldehyde for 1 h on ice and then immersed in 20% sucrose overnight at 4°C. Tissue was embedded in Tissue-Tek OCT (Sakura Finetek catalog #4583) mounting medium, and frozen on dry ice. Brains were sectioned at 25-μm thickness using a cryostat (Thermo Fisher Scientific Cryostar NX70). Sections were placed on 1% bovine gelatin precoated coverslips (Thermo Fisher Scientific). Sections were blocked with 10% normal goat serum in 0.1 m phosphate buffer (PB) with 0.3% Triton X-100 for 1 h at room temperature. Primary antibodies diluted in the blocking buffer and incubated at room temperature overnight, then sections were washed with blocking buffer. Secondary antibodies were incubated at room temperature for 2 h and washed with 0.1 m PB. Coverslips were mounted using Vectashield mounting media (Vector Laboratories catalog #H-1400, RRID:AB_2336787) and sealed using CoverGrip coverslip sealant (Biotum catalog #23005).
Image analysis
Immunofluorescence labeling was visualized, and images were collected on an AxioImager (Carl Zeiss) fitted with an apotome for optical sectioning, and a digital camera (AxioCam; Carl Zeiss). AxioVision (Carl Zeiss) acquisition software was used for collection of images and molecular layer measurements, three to six measurements/lobule were averaged, and the mean thickness was plotted for each animal. Images were also collected using a Nikon Eclipse Ni-E microscope fitted with a motorized X-Y stage for acquisition of fields. Stitching of images was performed using NIS-Elements (Nikon). In some instances, linear contrast and brightness adjustments were performed using Adobe Photoshop. Fluorescence intensity measurements and cell counts were performed using NIH FIJI/ImageJ. Purkinje neuron density was quantified by dividing Purkinje neuron counts by the distance over which they were measured [NIH FIJI/ImageJ arbitrary unit (AU)], quantifications were from regions in each cerebellar zone and then averaged. Quantification of β1 spectrin, β3 spectrin and Kv3.3 fluorescence intensity at the Purkinje neuron soma was done by measuring the fluorescence intensity of a line across the Purkinje neuron layer (PCL) in each zone, then dividing this by the line length (NIH FIJI/ImageJ AU). Quantification of β1 spectrin and Kv3.3 in the granule cell layer or β3 spectrin in the molecular layer was done by selecting several (three to four) regions for each animal and measuring the fluorescence intensity of the region of that layer then diving it by the area measured (AU). In some instances, quantifications were normalized to controls. No other processing of the images was performed.
Brain lysis at 37°C
Saline perfused mouse brains were homogenized in 20m m HEPES pH 7.4, 2 mm EDTA, and protease inhibitors in a Dounce homogenizer; 1% (v/v) Triton X-100 was added to homogenates and solubilized on a shaker for 30 min at 37°C. Lysates were then centrifuged at 700 × g for 20 min at 4°C to remove nuclei and debris, the supernatants then underwent another centrifugation at 27,200 × g for 60 min at 4°C. Lysates were collected and protein concentrations were measured. The lysates used for immunoprecipitation were prepared by dilution to final protein concentration at 1 mg/ml with lysis buffer [1% (v/v) Triton X-100, 20 mm Tris-HCl pH 8.0, 10 mm EDTA, 150 mm NaCl, 10 mm NaN3 and protease inhibitors]. Lysates were then used for immunoprecipitation.
DSP crosslinking and brain lysis
Saline perfused mouse brains were rapidly dissected and homogenized on ice using a Dounce homogenizer containing ice-cold homogenization and crosslinking buffer: 320 mm sucrose, 5 mm H2NaPO4, pH 7.4, supplemented with 1 mm NaF, 1 mm PMSF, protease inhibitors, and 1 mm dithiobis(succinimidyl propionate) (DSP), Lomant's reagent, ThermoFisher catalog #22585. Homogenates were incubated on ice for 1 h, then DSP was quenched by adding 1 m Tris, pH 7.4 to a final concentration of 20 mm. Homogenates were vortexed and incubated on ice for 10 min. An equal volume of homogenate and 2× radioimmunoprecipitation assay (RIPA) buffer (final 1× concentrations): 1% (v/v) Triton X-100, 0.5% (w/v) sodium deoxycholate, 0.1% (w/v) SDS, 150 mm NaCl, 40 mm HEPES, 10 mm NaN3, 20 mm EDTA with 1 mm PMSF, and protease inhibitor cocktail added before use were incubated on a tube rotator at 4°C for 30 min. Insoluble material was pelleted by centrifugation at 12,000 × g for 15 min at 4°C. Soluble lysates were then used for immunoprecipitation.
Immunoprecipitation
Antibodies (5 µl or 2–4 µg) were added and lysate samples were rotated overnight at 4°C. Protein A (polyclonal antibodies, Thermo Scientific, 20333) or Protein G (monoclonal antibodies, GE Healthcare, 17-0618-01) agarose beads were washed with 1 ml of lysis buffer or RIPA buffer three times and then rotated with the lysates for 1 h at 4°C. The beads were then collected and washed with 1 ml of ice-cold lysis or RIPA buffer seven times and subjected to immunoblotting. For immunoblotting, the samples were resolved by SDS-PAGE, transferred to nitrocellulose membrane, blocked using 4% milk with 150 mm NaCl and 20 mm Tris pH 8.0, then immunoblotted with antibodies diluted in blocking buffer. Immunoblots were imaged using a Licor odyssey Fc system with Image Studio software.
In vivo electrophysiology
Surgery for the awake recordings was performed as described (White et al., 2016). During surgery, a headplate was fixed to the skull and a craniotomy was made at 6.4 mm caudal from bregma and 1.3 mm right-lateral from the midline. These stereotaxic coordinates allowed to record from the interposed nucleus of the cerebellum. Each craniotomy was ∼5 mm in diameter, sealed with antibiotic ointment, and surrounded by a custom-made chamber with screw-top that prevented any particles entering the craniotomy while the mice were recovering from the surgery. All mice were allowed to recover from surgery for at least 2 d during which the mice were closely monitored for pain and distress.
Electrophysiological recordings were performed as previously described in detail (Brown et al., 2020). Briefly, a mouse was placed on a rotating wheel, while head-fixed using its headplate to two horizontal bars. We allowed all animals to acclimate to the electrophysiology rig for at least 1 h before recording to minimalize movement artifacts during our recordings. Electrophysiological recordings were performed using Tungsten electrodes that were controlled from a head stage using a motorized manipulator (MP-225; Sutter Instrument Co). Signals were acquired using an amplifier with bandpass filter (0.3–13 kHz). Analog signals were digitized using a CED Power 1401 and stored using Spike 2 software (CED). Spike 2 software was also used to spike sort the signal from single units.
Electrophysiology data analysis
For our analysis we only included CN cells that were defined as neurons located 2.5–4 mm underneath the cerebellar surface that did not show complex spikes (that are instead characteristic for Purkinje neurons). We analyzed an average of 123.6 ± 8.4 s continuously recorded trace. Spike sorted files were then imported into MATLAB and further analyzed with custom written code (van der Heijden et al., 2021). For this study, we included the following parameters for the firing properties of CN: frequency, covariance (CV) and a second measure of CV (CV2; Holt et al., 1996). Frequency was defined as the total number of spikes observed during the analyzed time frame of the recording. CV was defined as the SD of the interspike intervals (ISIs) divided by the mean of the ISIs. Finally, CV2 was defined with the following formula: CV2 = 2 ·|ISIn+1 – ISIn|/(ISIn+1 + ISIn). CV measures the global ISI variability, whereas CV2 measures the local ISI variability. Statistical significance between groups was determined using a linear mixed model analysis, with genotype as fixed effect and mouse number as random effect.
Plasmids
AnkR-GFP construct was previously described (Ho et al., 2014). The full-length Kv3.3a and 3.3b constructs were gifts from James Trimmer (University of California at Davis). To generate Flag-tagged full-length or truncated Kv3.3a and 3.3b constructs, parts of Kv3.3a and 3.3b were PCR amplified from full-length Kv3.3a and 3.3b, respectively, and then inserted into p3XFLAG-CMV-7.1 vector using In-Fusion cloning reactions (Takara Bio). DNA constructs were verified by sequencing (Genewiz).
Statistical analyses
No statistical methods were used to predetermine sample sizes, but our sample sizes are similar to those previously reported (Stevens et al., 2021). Sets of age-matched conditional knock-out mice and their controls were randomly collected from the same litter or from two litters that had close dates of birth. Data were analyzed using Microsoft Excel and GraphPad Prism. Unless otherwise stated, unpaired, two-tailed Student's t test was used for statistical analysis. Data distributions were assumed to be normal but were not formally tested. All error bars are SEM unless otherwise indicated. Significance values indicated in figures are for post hoc comparisons.
Results
AnkR is enriched in hindbrain nuclei and in the cerebellum
To determine AnkR's cellular and subcellular location, we immunostained the mouse hindbrain using antibodies against AnkR. We found AnkR is highly expressed in many hindbrain nuclei including the CN; consistent with previous reports, AnkR is also enriched at the perisomatic membrane of cerebellar Purkinje neurons, in their dendrites, and granule cells throughout the cerebellum (Fig. 1A–C; Peters et al., 1991; Lambert and Bennett, 1993; Clarkson et al., 2014). To determine the role of AnkR in the cerebellum, we crossed Ank1 floxed mice (Ank1F/F; Stevens et al., 2021) with Nestin-Cre (Ank1F/F;Nestin-Cre) and Pcp2-Cre (Ank1F/F;Pcp2-Cre) mice to remove AnkR from the nervous system or from cerebellar Purkinje neurons, respectively. We confirmed the efficient loss of AnkR by immunostaining of one-month-old Ank1F/F;Nestin-Cre mice. While Ank1F/F;Nestin-Cre mice showed loss of AnkR immunoreactivity in neurons throughout the brain (note the persistent AnkR immunoreactivity in the few isolated red blood cells), including the cerebellum and cortex, Ank1F/F;Pcp2-Cre have selective loss of AnkR in cerebellar Purkinje neurons (Fig. 1D), only ∼60% of Purkinje neurons lose AnkR immunoreactivity at one month of age. We further examined the recombination of AnkR in Purkinje neurons of Ank1F/F;Pcp2-Cre mice at later timepoints and found recombination and progressive loss of AnkR as mice matured; ∼90% of Purkinje neurons lack AnkR immunoreactivity at three months, and by six months of age, AnkR could not be detected in any Purkinje neurons (Fig. 1E). In both Ank1F/F;Nestin-Cre and Ank1F/F;Pcp2-Cre mice, erythrocyte AnkR expression was unaltered, thereby avoiding the potential confound of anemia.
Figure 1.

AnkR is enriched in cerebellar Purkinje neurons, granule cells, and the CN. Immunostaining for AnkR in P56 wild-type, C57BL/6, sagittal mouse (A) hindbrain, scale bar: 500 µm; (B) cerebellar cortex, scale bar: 25 µm; and (C) CN, scale bar: 50 µm. D, Immunostaining for AnkR in one-month Ank1 conditional knock-out mice in cerebellum (top) and cortex (bottom), genotypes indicated. Scale bars: 25 µm. E, Immunostaining for AnkR in three-month (top) and six-month (bottom) Ank1F/F;Pcp2-Cre. Scale bar: 25 µm. ML, molecular layer; PCL, Purkinje neuron layer; GCL, granule cell layer.
Similar to our previous report of loss of AnkR in forebrain neurons, and in contrast to the AnkR's rescue of loss of AnkG at nodes of Ranvier (Ho et al., 2014), AnkG remains highly restricted to the axon initial segment and nodes of Ranvier in Ank1F/F;Nestin-Cre mice (Fig. 2A). Thus, we found no indication AnkG can reciprocally compensate for loss of AnkR. Furthermore, we did not see any apparent changes in AnkB expression levels or patterns in Ank1F/F;Nestin-Cre mice by immunostaining (Fig. 2B). Taken together, these results show AnkR is highly expressed in the hindbrain including in multiple parts of the cerebellar circuit, i.e., cerebellar Purkinje neurons, and we have generated mice to specifically examine the role of AnkR in Purkinje neurons.
Figure 2.
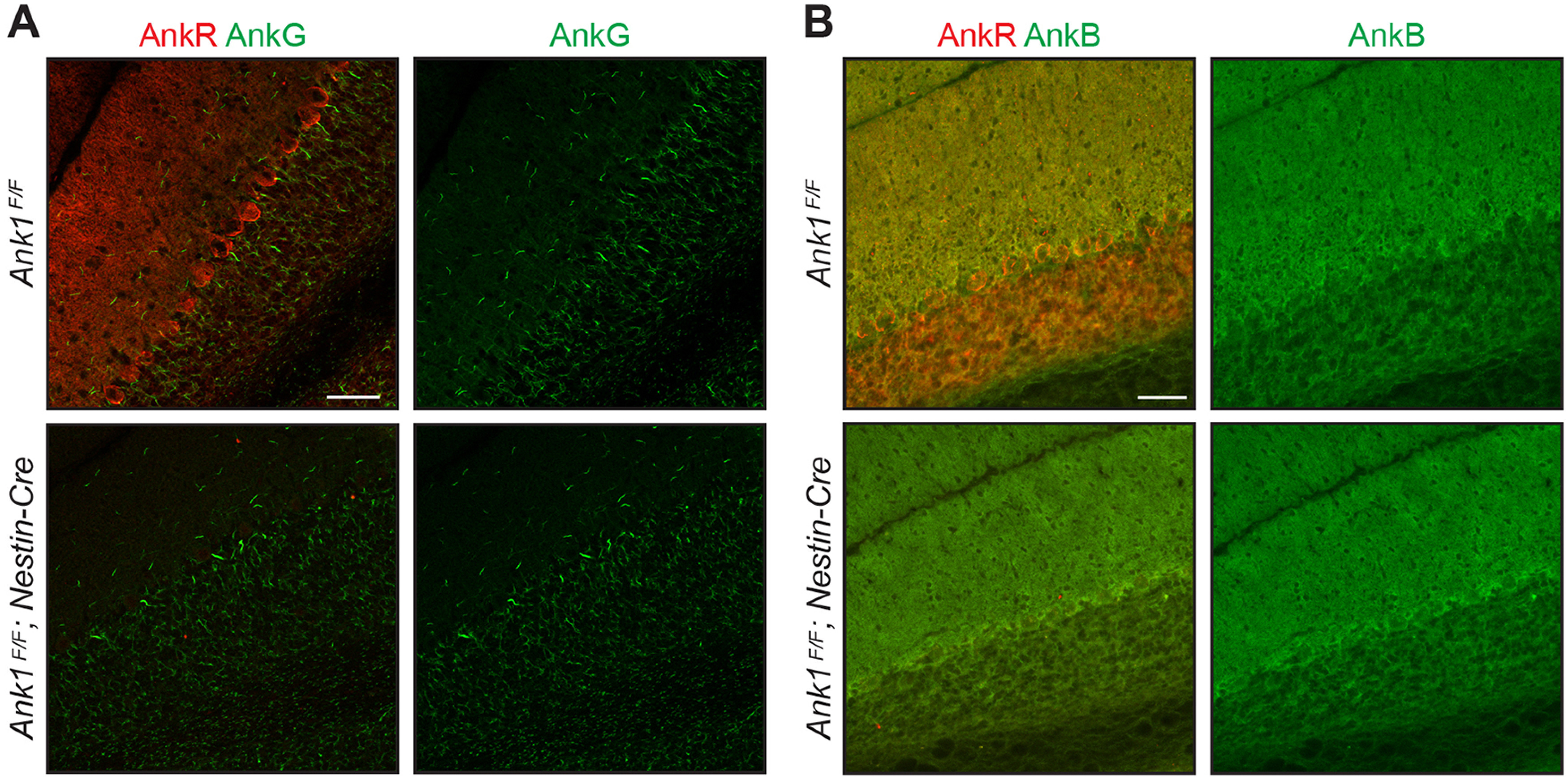
Loss of AnkR does not alter the localization of AnkG or AnkB by immunostaining. A, Immunostaining of Ank1F/F and Ank1F/F;Nestin-Cre sagittal for AnkR (red) and AnkG (green). B, Immunostaining of Ank1F/F and Ank1F/F;Nestin-Cre sagittal cerebellum for AnkR (red) and AnkB (green). Scale bars: 50 µm.
AnkR mutant mice have impaired motor behavior
To determine whether loss of AnkR caused impairments in motor function, we characterized the motor behavior by open field visual inspection and footprint analysis. By open field visual inspection Ank1 mutant mice have a slow, wobbly gait with an apparent tremor during active locomotion. To quantitatively evaluate the extent and severity of this motor impairment we performed gait analysis on control Ank1F/F and mutant Ank1F/F;Nestin-Cre and Ank1F/F;Pcp2-Cre mice at one, six, and 12 months of age. At one month of age, we found Ank1F/F;Nestin-Cre mice had significantly different stride, stance, and sway distances compared with Ank1F/F controls. Similarly, Ank1F/F;Pcp2-Cre mice had significantly different stride and sway distances compared with Ank1F/F; however, Ank1F/F;Pcp2-Cre mice did not significantly differ from controls in stance distances, and compared with Ank1F/F;Nestin-Cre mice the changes observed in stride were moderate (Fig. 3A). We speculate this reflects the partial loss of AnkR at this age (Fig. 1E).
Figure 3.

Loss of AnkR results in ataxia and abnormal gait. Quantification of footprints from gait analysis at (A) one-month-old control (N = 10; stride 5.9 ± 0.17 cm; stance 3.8 ± 0.10 cm; sway 2.3 ± 0.06 cm); Ank1F/F;Nestin-Cre (N = 10; stride 3.8 ± 0.16 cm; stance 3.2 ± 0.06 cm; sway 2.6 ± 0.06 cm); and Ank1F/F;Pcp2-Cre (N = 7; stride 4.9 ± 0.17 cm; stance 3.8 ± 0.09 cm; sway 2.7 ± 0.07 cm); (B) six-month-old control (N = 10; stride 6.9 ± 0.18 cm; stance 4.5 ± 0.10 cm; sway 2.5 ± 0.04 cm); Ank1F/F;Nestin-Cre (N = 9; stride 4.5 ± 0.29 cm; stance 3.8 ± 0.19 cm; sway 3.0 ± 0.11 cm); and Ank1F/F;Pcp2-Cre (N = 7; stride 5.0 ± 0.14 cm; stance 3.8 ± 0.10 cm; sway 2.9 ± 0.08 cm); and (C) 12-month-old control (N = 10; stride 6.5 ± 0.18 cm; stance 4.5 ± 0.10 cm; sway 2.7 ± 0.10 cm); Ank1F/F;Nestin-Cre (N = 8; stride 3.4 ± 0.22 cm; stance 3.7 ± 0.18 cm; sway 3.3 ± 0.12 cm); and Ank1F/F;Pcp2-Cre (N = 7; stride 4.3 ± 0.17 cm; stance 3.7 ± 0.07 cm; sway 3.2 ± 0.06 cm). Error bars indicate mean ± SEM. D, Sample footprints from six-month-old control, Ank1F/F;Nestin-Cre, and Ank1F/F;Pcp2-Cre mice, measurements used for analysis depicted in gray.
At six (Fig. 3B,D) and 12 (Fig. 3C) months of age, when AnkR immunoreactivity is absent from Purkinje neurons in mutant mice, we found both Ank1F/F;Nestin-Cre and Ank1F/F;Pcp2-Cre mice significantly differed from controls in all gait measurements. Furthermore, 12-month-old mutant animals were frequently observed to drag their abdomen and/or tail (Movies 1, 2). These results show that loss of AnkR from the brain (Ank1F/F;Nestin-Cre) impairs motor function. Moreover, loss of AnkR from only Purkinje neurons (Ank1F/F;Pcp2-Cre) results in a similar, albeit slightly delayed, motor impairment.
Gait of 12-month-old Ank1F/F mouse.
Gait of 12-month-old Ank1F/F;Nestin-Cre mouse.
Loss of AnkR induces Purkinje neuron degeneration
Since defects in Purkinje neuron development and survival are known to cause motor impairments (Gerlai et al., 1996; Lalonde and Strazielle, 2007), we used Calbindin staining to determine whether loss of AnkR disrupted Purkinje neuron morphology or survival. At one month of age, Purkinje neurons in all genotypes appeared mostly normal in Ank1F/F;Nestin-Cre and Ank1F/F;Pcp2-Cre mice (Fig. 4A–C). However, axonal torpedoes, a marker of dystrophic axons, were abundant in six-month-old Ank1F/F;Nestin-Cre and Ank1F/F;Pcp2-Cre mice but not control mice (Fig. 4D, arrowheads). Moreover, at six months, we observed disruptions in Purkinje neuron dendritic arbors, with reduced molecular layer thickness in Ank1F/F;Nestin-Cre and Ank1F/F;Pcp2-Cre mice and cell loss in Ank1F/F;Nestin-Cre mice (Fig. 4D–F). This measurement is useful to elucidate whether there are disruptions to Purkinje neuron dendrite size and/or the placement of these normally monolayer cells; altered molecular layer thickness is a common consequence of developmental and diseaseassociated cerebellar mutations (Perkins et al., 2010; Stankewich et al., 2010). These disruptions were progressive and by 12 months of age, significant cell loss was also present in Ank1F/F;Pcp2-Cre mice (Fig. 4G–I), many axonal torpedoes were also visible at this age (Fig. 4G, arrowheads). These observations show that loss of AnkR from Purkinje neurons is sufficient to cause progressive Purkinje neuron degeneration characterized by dystrophic axons, disturbed dendritic arbors, and eventual cell loss.
Figure 4.
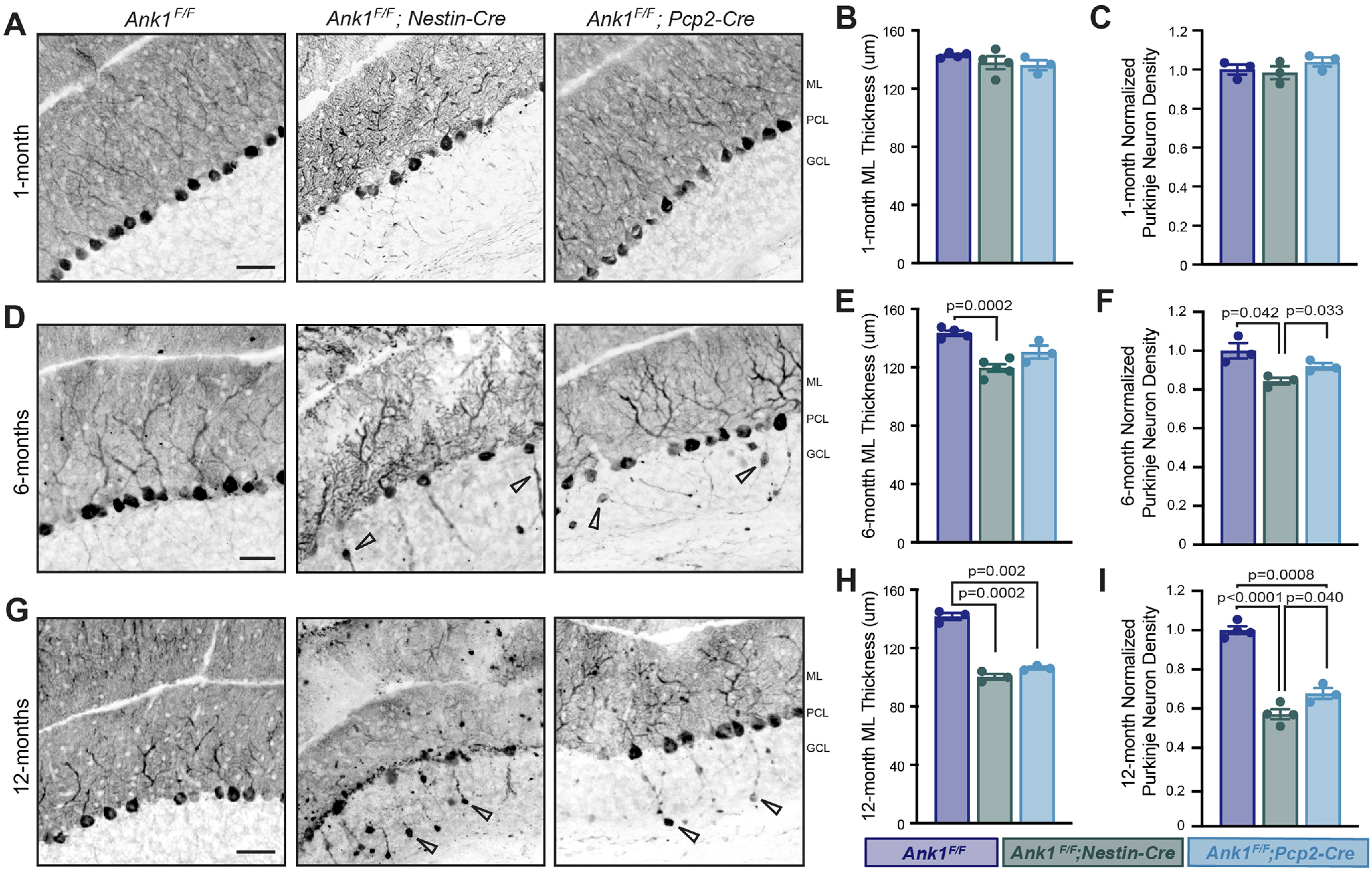
Loss of AnkR results in progressive cerebellar Purkinje neuron degeneration. Immunostaining of Purkinje neurons with Calbindin in the cerebellar cortex of Ank1F/F, Ank1F/F;Nestin-Cre, and Ank1F/F;Pcp2-Cre in (A) one-month-old, (D) six-month-old, and (G) 12-month-old mice of the indicated genotypes. Arrowheads in D, G show examples of axonal torpedoes present in six-month and 12-month Ank1F/F;Nestin-Cre and Ank1F/F;Pcp2-Cre. Scale bars: 50 µm. ML, molecular layer; PCL, Purkinje neuron layer; GCL, granule cell layer. Quantification of molecular layer thickness at (B) one month, Ank1F/F (N = 5; 143.0 ± 0.83 µm), Ank1F/F;Nestin-Cre (N = 4; 137.8 ± 4.43 µm), and Ank1F/F;Pcp2-Cre (N = 3; 136.1 ± 3.44 µm); (E) six months, Ank1F/F (N = 4; 142.1 ± 1.51 µm), Ank1F/F;Nestin-Cre (N = 5; 118.3 ± 2.57 µm), and Ank1F/F;Pcp2-Cre (N = 3; 129.3 ± 4.29 µm); and (H) 12 months, Ank1F/F (N = 3; 141.7 ± 2.58 µm), Ank1F/F;Nestin-Cre (N = 3; 100.5 ± 1.96 µm), and Ank1F/F;Pcp2-Cre (N = 3; 106.44 ± 0.64 µm). Normalized quantification of Purkinje neuron density at (C) one month, Ank1F/F (N = 3; 1.0 ± 0.03), Ank1F/F;Nestin-Cre (N = 3; 0.98 ± 0.03), and Ank1F/F;Pcp2-Cre (N = 3; 1.04 ± 0.02); (F) six months, Ank1F/F (N = 3; 1.0 ± 0.04), Ank1F/F;Nestin-Cre (N = 3; 0.84 ± 0.02), and Ank1F/F;Pcp2-Cre (N = 3; 0.92 ± 0.02); and (I) 12 months, Ank1F/F (N = 4; 1.0 ± 0.02), Ank1F/F;Nestin-Cre (N = 4; 0.57 ± 0.03), and Ank1F/F;Pcp2-Cre (N = 3; 0.68 ± 0.03).
Anterior Purkinje neurons preferentially degenerate
The normal mouse cerebellum consists of 10 major lobules (I–X) that can be divided into four transverse zones: the AZ (lobules I–V), the central zone (CZ; lobules VI–VII), the posterior zone (PZ; lobules VIII–dorsal IX), and the nodular zone (NZ; ventral lobule IX–X; Ozol et al., 1999; Sarna et al., 2006; Fig. 5A, inset). We examined the gross anatomy of the cerebellum using Calbindin staining on sagittal sections to compare all lobules in the same cutting plane in Ank1F/F, Ank1F/F;Nestin-Cre and Ank1F/F;Pcp2-Cre mice at 12 months of age. Both Ank1F/F;Nestin-Cre and Ank1F/F;Pcp2-Cre mice, similar to controls, had all 10 lobules present and their gross size and shape were unaltered, although the Purkinje neuron degeneration in the mutants was more apparent in the anterior lobules (Fig. 5A). To determine whether loss of AnkR caused more subtle disruption of the cerebellum, and to determine whether there were differences between zones, we measured the thickness of the molecular layer by zone. The overall molecular layer thickness from lobules I–X of Ank1F/F;Nestin-Cre and Ank1F/F;Pcp2-Cre mice was significantly reduced compared with controls (Fig. 4H); however, the majority of this reduction was seen in AZ and CZ; however, PZ and NZ were only mildly or not affected (Fig. 5B). We next determined whether there were differences in Purkinje neuron number by zone. Similarly, while the overall cell number was reduced, in Ank1F/F;Nestin-Cre and Ank1F/F;Pcp2-Cre mice (Fig, 4I) only AZ and CZ had significantly fewer Purkinje neurons compared with control mice, and we did not detect differences in PZ and NZ (Fig. 5C).
Figure 5.
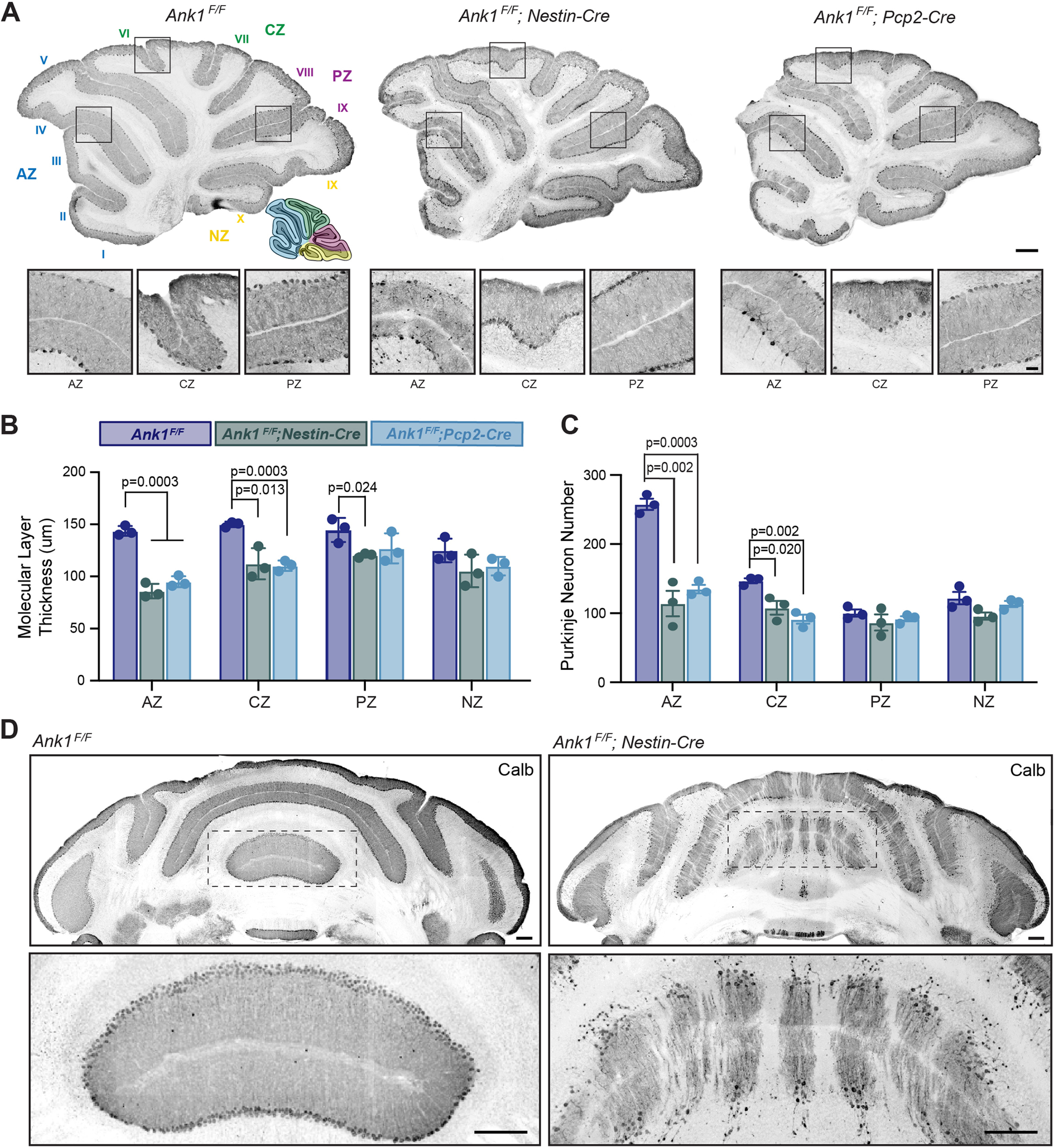
Molecular layer thickness and Purkinje neuron number are dramatically reduced in anterior and central sagittal zones of 12-month-old Ank1 conditional knock-out mice. A, Sample sagittal cerebellum images from 12-month-old Ank1F/F, Ank1F/F;Nestin-Cre, and Ank1F/F;Pcp2-Cre mice immunostained for Calbindin. Cerebellar lobules labeled in Ank1F/F. Cerebellar zones illustrated in Ank1F/F inset. AZ (lobules I–V, blue), CZ (lobules VI–VII, green), PZ (lobules VIII–dorsal IX, purple), and NZ (ventral lobule IX–X, yellow). Scale bars shown in Ank1F/F;Pcp2-Cre, top 250 µm and bottom 50 µm. B, Quantification of molecular layer thickness by zone in 12-month-old mice. Ank1F/F (N = 3; AZ = 144 ± 2.7 µm; CZ = 150 ± 1.5 µm; PZ = 145 ± 6.8 µm; NZ = 125 ± 6.4 µm), Ank1F/F;Nestin-Cre (N = 3; AZ = 86 ± 4.0 µm; CZ = 112 ± 8.4 µm; PZ = 120 ± 1.2 µm; NZ = 105 ± 9.2 µm), and Ank1F/F;Pcp2-Cre (N = 3; AZ = 95 ± 3.0 µm; CZ = 111 ± 3.1 µm; PZ = 127 ± 8.6 µm; NZ = 110 ± 5.1 µm). C, Quantification of Purkinje neuron number using Calbindin by zone in 12-month-old mice. Ank1F/F (N = 3; AZ = 258 ± 8 cells; CZ = 147 ± 4 cells; PZ = 100 ± 5 cells; NZ = 122 ± 9 cells), Ank1F/F;Nestin-Cre (N = 3; AZ = 114 ± 19 cells; CZ = 108 ± 10 cells; PZ = 87 ± 12 cells; NZ = 96 ± 5 cells), and Ank1F/F;Pcp2-Cre (N = 3; AZ = 135 ± 6 cells; CZ = 91 ± 6 cells; PZ = 92 ± 4 cells; NZ = 113 ± 4 cells). D, Immunostaining of Purkinje neurons in coronal cerebellum with Calbindin in 12-month-old Ank1F/F and Ank1F/F;Nestin-Cre mice. Scale bars: 200 µm. Error bars indicate mean ± SEM.
Each transverse zone can be further subdivided into parasagittal stripes aligned with mossy fiber and climbing fiber terminal fields; these stripes are reproducibly marked by antibodies against zebrinII. The expression pattern is characterized as narrow zebrinII-positive stripes separated by large zebrinII-negative stripes in AZ, roughly equal zebrinII-positive and zebrinII-negative stripes in PZ, and uniformly zebrinII-positive CZ and NZ (Sillitoe and Hawkes, 2002; Cerminara et al., 2015). Conversely, phospholipase C β4 (PLCβ4), roughly expressed in zebrinII-negative Purkinje neurons, is highly expressed in AZ but only expressed in some Purkinje neurons in CZ and PZ (Sarna et al., 2006). Interestingly, Calbindin staining in coronal cerebellar sections shows Purkinje neuron loss in Ank1F/F;Nestin-Cre mice occurs in a striped pattern (Fig. 5D).
AnkR loss from Purkinje neurons reduces CN firing rate
Synapses from Purkinje neurons converge onto neurons in the CN, which in turn modulate motor function by sending their information to brain regions essential for movement, including the thalamus, brainstem, and spinal cord (Beckinghausen and Sillitoe, 2019). CN signals are altered in mouse models of ataxia and tremor (Brown et al., 2020; Palarz et al., 2021), and deep brain stimulation of the CN can alleviate the motor defects (Brown et al., 2020; Miterko et al., 2021). This raises the possibility that Purkinje neuron degeneration in the Ank1F/F;Pcp2-Cre mice likely results in ataxia and tremor through changes in the output of the CN. To test this hypothesis, we measured the activity of neurons in the interposed CN (Fig. 6A), as this region controls ongoing movements (Bracha et al., 1999; Low et al., 2018; Becker and Person, 2019). We found that the firing frequency of the CN in awake, head-fixed mice was significantly lower in Ank1F/F;Pcp2-Cre mice compared with littermate controls (Fig. 6B). However, the average global (CV) and local (CV2) in Ank1F/F;Pcp2-Cre mice, was not significantly different (Fig. 6C,D). Nevertheless, the change in firing activity are similar to the previously reported reduction in firing rate in mice with tremor (van der Heijden et al., 2021) and lower firing rates are also reported in a mouse model for spinocerebellar ataxia (Palarz et al., 2021). In summary, these data suggest that the abnormal firing output of the CN may promote the motor phenotypes that we observed in Ank1F/F;Pcp2-Cre mice.
Figure 6.

Loss of AnkR from Purkinje neurons results in reduced cerebellar output. A, Example traces of electrophysiological recordings from nuclei cell from control and Ank1F/F;Pcp2-Cre mice. B, Firing frequency of CN cells recorded in control (frequency = 68.0 ± 4.0 Hz) and Ank1F/F;Pcp2-Cre (frequency = 41.1 ± 5.2 Hz) mice. C, CV (global variability of ISIs) of CN cells recorded in control (CV = 5.1 ± 0.04) and Ank1F/F;Pcp2-Cre (CV = 0.66 ± 0.06) mice. D, CV2 (local variability of ISIs) of CN cells recorded in control (CV2 = 0.40 ± 0.02) and Ank1F/F;Pcp2-Cre (CV2 = 0.47 ± 0.03) mice. Small, filled circles are the firing frequency of individually recorded cells. Large, open circles represent average firing frequency of individual animals. Each vertical column of datapoints represents an individual animal. 11- to 12-month-old control (N = 4 mice, n = 24 cells) and Ank1F/F;Pcp2-Cre (N = 3 mice, n = 14 cells) animals. Error bars indicate mean ± SEM of all recorded cells for each genotype. Statistical significance was determined using a linear mixed model with genotype as a fixed variable and mouse number as a random variable.
AnkR interacts with β3 spectrin in the cerebellum
Both β1 and β3 spectrin are expressed in the cerebellum but in largely complementary patterns. β1 spectrin is expressed in granule cells but weakly in Purkinje neuron dendrites; β3 spectrin is highly expressed in the Purkinje neuron dendrites, extending throughout the main dendrite, smooth dendrites, and spiny dendrites, but is weakly expressed in granule cells. Both β1 and β3 spectrin are expressed in the soma of Purkinje neurons (Fig. 7A,B; Peters et al., 1991; Stankewich et al., 2010). To determine whether loss of AnkR altered β1 or β3 spectrin protein amounts, we immunostained three-month-old Ank1F/F and Ank1F/F;Nestin-Cre sagittal cerebellum for β1 spectrin (Fig. 7C) and β3 spectrin (Fig. 7D). However, compared with controls, we found no difference in β1 spectrin fluorescence intensity in the granule or Purkinje cell layers (Fig. 7E,F), or β3 spectrin fluorescence intensity in the molecular layer (Fig. 7G). In contrast, β3 spectrin fluorescence intensity was significantly reduced in Purkinje neuron somas (Fig. 7H). We previously showed AnkR and β1 spectrin are binding partners in the brain (Stevens et al., 2021), and consistent with previous reports (Clarkson et al., 2014), reciprocal coimmunoprecipitation also shows that β3 spectrin and AnkR are binding partners (Fig. 7I). Taken together, these experiments indicate that AnkR interacts with and maintains β3 spectrin in cerebellar Purkinje neurons.
Figure 7.
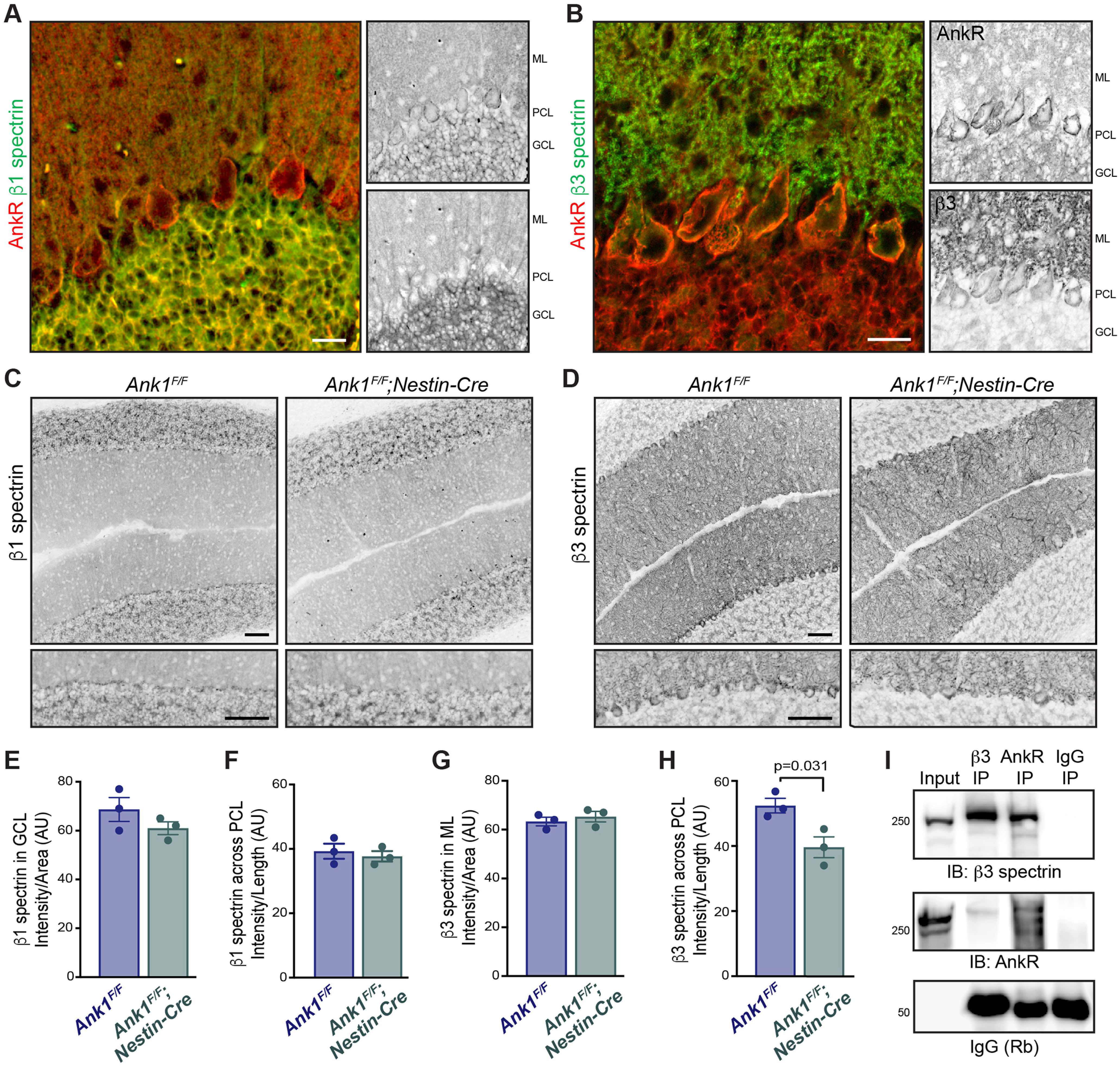
AnkR-spectrin interactions in the cerebellum. A, Immunostaining of sagittal cerebellum for AnkR (polyclonal, red) and β1 spectrin (monoclonal, green). Scale bars: 20 µm. B, Immunostaining of sagittal cerebellum for AnkR (monoclonal, red) and β3 spectrin (polyclonal, green). Scale bars: 20 µm. C, Immunostaining of sagittal cerebellum for β1 spectrin in three-month Ank1F/F and Ank1F/F;Nestin-Cre. Scale bar: 50 µm. D, Immunostaining of sagittal cerebellum for β3 spectrin in three-month Ank1F/F and Ank1F/F;Nestin-Cre. Scale bar: 50 µm. E, Quantification of β1 spectrin fluorescence intensity in the granule cell layer (GCL) of three-month Ank1F/F (N = 3; 68 ± 6.1 AU) and Ank1F/F;Nestin-Cre (N = 3; 61 ± 3.8 AU). F, Quantification of β1 spectrin fluorescence intensity at the Purkinje neuron soma by measuring the fluorescence intensity of a line across the PCL and dividing by length in three-month sagittal cerebellum of Ank1F/F (N = 3; 39 ± 2.4 AU) and Ank1F/F;Nestin-Cre (N = 3; 38 ± 1.6 AU). Error bars indicate mean ± SEM. G, Quantification of β3 spectrin fluorescence intensity in the molecular layer (ML) of three-month Ank1F/F (N = 3; 63 ± 2.2 AU) and Ank1F/F;Nestin-Cre (N = 3; 66 ± 3.1 AU). H, Quantification of β3 spectrin fluorescence intensity at the Purkinje neuron soma by measuring the fluorescence intensity of a line across the PCL and dividing by length in three-month sagittal cerebellum of Ank1F/F (N = 3; 52 ± 4.4 AU) and Ank1F/F;Nestin-Cre (N = 3; 40 ± 3.9 AU). Error bars indicate mean ± SEM. I, Immunoblots of β3 spectrin, AnkR, and IgG immunoprecipitation (IP) reactions using antibodies against AnkR and β3 spectrin.
AnkR maintains Kv3.3 K+ channels at the somatic membrane
Ankyrin scaffolding proteins interact with, cluster, and maintain ion channels in neuronal membranes. We previously reported that AnkR interacts with Kv3.1b in forebrain Pv+ neurons, and that loss of AnkR results in both a dramatic reduction of Kv3.1b protein and altered intrinsic excitability in these cells (Stevens et al., 2021). Furthermore, we discovered that AnkR and Kv3.1b interact using a 6-aa binding motif (EDCPHI) in Kv3.1b. A nearly identical motif is found in Kv3.3, but the motif is not present in Kv3.2 or Kv3.4. Kv3-type potassium channels are highly expressed in the cerebellum; they are found in Purkinje neurons (Kv3.3 and Kv3.4), granule cells (Kv3.1 and Kv3.3), and in the CN (Kv3.1, Kv3.2, Kv3.3, and Kv3.4; Hurlock et al., 2009). The fast activation and deactivation of these channels is essential to generate and sustain the brief action potentials and high frequency firing rates observed in cerebellar neurons (Joho and Hurlock, 2009). Based on these observations, we considered Kv3.3 to be a good candidate to interact with AnkR in Purkinje neurons.
Immunostaining of control three-month-old mice showed AnkR and Kv3.3 are highly colocalized in Purkinje neurons and granule cells (Fig. 8A). To determine whether AnkR and Kv3.3 interact we used crosslinked reciprocal immunoprecipitation of brain homogenates and found Kv3.3 coimmunoprecipitates AnkR (Fig. 8B). To further define this interaction and determine whether AnkR and Kv3.3 interact using the predicted binding motif (EDCPAI), we cotransfected HEK cells with AnkR-GFP and full-length Flag-tagged Kv3.3a, full-length Flag-tagged Kv3.3b, or Flag-tagged Kv3.3a and Kv3.3b lacking the predicted AnkR binding motif. AnkR-GFP efficiently coprecipitated full-length Flag-Kv3.3a and full-length Flag-Kv3.3b, however loss of the binding motif precluded the interaction with AnkR (Fig. 8C). Taken together, these results show that AnkR binds Kv3.3 through the EDCPAI binding motif.
Figure 8.

AnkR binds to Kv3.3 K+ channels and is critical for its proper membrane localization in Purkinje neurons. A, Immunostaining of AnkR (red) and Kv3.3 (green) in sagittal cerebellum of Ank1F/F. Scale bar: 25 µm. B, Immunoblots of urea sample buffer (USB) input, reducing sample buffer (RSB) input, Kv3.3, and AnkR DSP-crosslinked immunoprecipitation reactions using antibodies against AnkR and Kv3.3. C, Immunoblots of AnkR-GFP immunoprecipitations in HEK293T cells coexpressing AnkR-GFP with full-length Flag-tagged Kv3.3a, truncated (ΔEDCPAI) Flag-tagged Kv3.3a, full-length Flag-tagged Kv3.3b, or truncated (ΔEDCPAI) Flag-tagged Kv3.3b. D, Immunostaining of Calbindin (red) and Kv3.3 (green) in sagittal cerebellum of three-month-old Ank1F/F and Ank1F/F;Nestin-Cre. Scale bar: 25 µm. E, Normalized quantification of Kv3.3 expression at the Purkinje neuron soma by measuring the fluorescence intensity of a line across the PCL and dividing by length in three-month sagittal cerebellum of Ank1F/F (N = 3; 1.00 ± 0.04 AU), and Ank1F/F;Nestin-Cre (N = 3; 0.76 ± 0.004 AU). F, Normalized quantification of Kv3.3 expression in the granule cell layer Ank1F/F (N = 3; 1 ± 0.05 AU), and Ank1F/F;Nestin-Cre (N = 3; 0.68 ± 0.05 AU). Error bars indicate mean ± SEM. ML, molecular layer; PCL, Purkinje neuron layer; GCL, granule cell layer.
To determine whether loss of AnkR impacts Kv3.3 K+ channels, we immunostained three-month-old cerebella and found AnkR-deficient Ank1F/F;Nestin-Cre mice have reduced amounts of Kv3.3 K+ channels in Purkinje neuron somas and in granule cells (Fig. 8D–F). We measured the reduction in Kv3.3 fluorescence intensity in Purkinje neuron somas and found that Ank1F/F;Nestin-Cre mice have a similar reduction in Kv3.3 across all cerebellar zones (Ank1F/F;Nestin-Cre AZ = 0.77 ± 0.01; CZ = 0.73 ± 0.02; PZ+NZ = 0.78 ± 0.004, normalized to Ank1F/F = 1.0). Thus, similar to AnkR and β spectrins, these results show AnkR has multiple K+ channel binding partners in the brain and cerebellum, which vary depending on both cell type and subcellular location.
Discussion
We used Ank1 conditional knock-out mice to investigate the function of AnkR in the cerebellum and in Purkinje neurons. This is an important technical advance since previous loss of function studies used AnkR hypomorphs (nb/nb) or whole-body knock-outs (Ank1pale/pale; Peters et al., 1991; Ho et al., 2014). Ank1pale/pale mice die within a week of birth, and nb/nb mice develop motor deficits by six months of age. Both nb/nb and Ank1pale/pale mice have severe erythrocyte dysfunction and hemolytic anemia, and the most likely cause of perinatal death in Ank1pale/pale mice is the severe anemia since bone marrow transplants into neonates rescues their viability (data not shown). The development of Ank1F/F;Nestin-Cre and Ank1F/F;Pcp2-Cre mice enabled us to explore the role of AnkR in the cerebellum, while avoiding the confound of anemia and other non-neuronal effects of AnkR deficiency.
We found that both Ank1F/F;Nestin-Cre and Ank1F/F;Pcp2-Cre mice had similar ataxic phenotypes. In addition, Purkinje neurons in the AZ and CZ of the cerebellum were more likely to degenerate without AnkR, indicating they are either more susceptible or less resistant to degeneration. Several molecularly distinct populations of Purkinje neurons have been described in the cerebellum, and parasagittal stripes of Purkinje neurons, reproduced across individuals and species, have been shown using markers like zebrinII (Sarna and Hawkes, 2003). Interestingly, multiple studies have found specific populations of Purkinje neurons preferentially degenerate depending on the genetic model (Sarna et al., 2003; Sarna and Hawkes, 2011). In general, sagittal sections with more degeneration in the AZ (lobules I–V), as shown in the Ank1 mutant mice reported here, are often associated with degeneration occurring in the zebrinII-negative population of Purkinje neurons (Sarna and Hawkes, 2003). However, these patterns of degeneration can shift slightly away from the strict zebrin code, as shown in the ataxic “sticky” mouse, which was found to have striped, patterned degeneration that was difficult to characterize into one Purkinje neuron subtype (Sarna and Hawkes, 2011). It will be of interest to further characterize the patterned degeneration occurring in Ank1 mutant animals, and to determine whether the Purkinje neuron degeneration reported in β3 spectrin knock-out mice is similar (Clarkson et al., 2014).
Pathogenic variants of SPTBN2 (β3 spectrin) and KCNC3 (Kv3.3) cause spinocerebellar ataxia types 5 (SCA5) and 13 (SCA13), respectively (Ikeda et al., 2006; Hsieh et al., 2020). As shown here, AnkR interacts with both β3 spectrin and Kv3.3. AnkR conditional knock-out mice have multiple similarities to β3 spectrin knock-out mice including altered gait, with an increase in sway or base width distance, decreased molecular layer thickness in mature animals, and progressive Purkinje neuron degeneration (Perkins et al., 2010; Stankewich et al., 2010). Interestingly, β3 spectrin knock-out mice were shown to have a reduction in vivo of Purkinje neuron simple spikes (Perkins et al., 2010), this same reduction has also been shown in Kv3.3 knock-out mice (Hurlock et al., 2008). In the AnkR-deficient Purkinje neurons, we found reductions in both β3 spectrin and Kv3.3. Although altered glutamatergic transmission has been proposed as a pathomechanism in β3 spectrin knock-out animals (Perkins et al., 2010; Stankewich et al., 2010), reduced AnkR was also reported in their Purkinje neurons (Clarkson et al., 2014). Thus, altered Kv3.3 could be an additional pathomechanism involved in SCA5. Mice lacking the K+ channels Kv3.1 or Kv3.3 have only small behavioral deficits (Porcello et al., 2002; Hurlock et al., 2008; Zagha et al., 2010), but simultaneous deletion of both Kv3.1 and Kv3.3 causes a much more severe phenotype including ataxia and tremor (Espinosa et al., 2001, 2004). However, mice lacking Kv3.3 do not have any reported Purkinje neuron degeneration (Zhang and Kaczmarek, 2016). Taken together, our results suggest that AnkR may function as a scaffold on which multiple SCA proteins converge to perform their functions in Purkinje neurons. We speculate that proteins associated with other spinocerebellar ataxias may also work together with AnkR. Nevertheless, the precise mechanisms underlying the dysfunction and degeneration of Purkinje neurons remains poorly understood and could reflect structural changes because of loss of cytoskeletal integrity, excitotoxicity because of altered membrane properties and Ca2+ handling, or other pathomechanisms (Huang and Verbeek, 2019).
The observed decrease in CN firing rate after loss of Purkinje neurons in Ank1F/F;Pcp2-Cre mice may be counterintuitive given the inhibitory nature of Purkinje neurons and their predominant output onto CN neurons. However, the lack of an expected negative correlation between the spontaneous firing rate in individual pairs of connected Purkinje neurons and CN neurons has been reported (McDevitt et al., 1987; Lang and Blenkinsop, 2011). Purkinje neurons also send convergent output to the CN (Person and Raman, 2011), with circuit level responses that drive the canonical Purkinje neuron-CN inhibitory relationship typically evident at the level of ensemble activity within the functional landscape of zonal modules (Witter et al., 2013). However, in vivo single-unit recordings from CN neurons show that their firing rate is unchanged in awake mice where GABAergic neurotransmission from Purkinje neurons is permanently blocked (Brown, 2020). One recent study suggests that at the single cell level, the lack of an inverse relationship between CN and Purkinje neuron firing rates results from adaptation of the CN firing rates to global changes in Purkinje neuron firing rate (Khan et al., 2021). These data indicate that adaptive mechanisms may be in place to protect the CN against excitotoxicity that could result from Purkinje neuron silencing or degeneration. Thus, the relationship between CN and Purkinje neuron firing rates cannot be strictly predicted based on the known point-to-point wiring diagram of the fundamental cerebellar circuit. Instead, CN responses are context specific. In the case of degeneration, specifically when it occurs in a pattern, functional changes may reflect cumulative loss of connectivity and compensatory change in downstream neurons. While further studies are needed to determine how progressive Purkinje neuron degeneration leads to specific abnormalities in CN firing rate and pattern, our results underscore the pathophysiological complexity of how Purkinje neurons modulate CN function in the context of patterned neurodegenerative diseases.
In addition to the cerebellum, AnkR is also found in the forebrain where it is highly enriched in GABAergic, fast-spiking Pv+ interneurons. There, AnkR interacts with β1 spectrin, Kv3.1b K+ channels, and the cell adhesion molecules (CAMs) NrCAM and PlexinA4 (Stevens et al., 2021). These AnkR-interacting CAMs function to recruit and stabilize perineuronal nets surrounding these interneurons; the nets consist of chondroitin-sulfate proteoglycans and other extracellular matrix molecules. Loss of AnkR significantly reduces and disrupts perineuronal nets, and reduces the amount of both β1 spectrin and Kv3.1b in the perisomatic neuronal membranes. The reduction in Kv3.1b is accompanied by changes to the intrinsic excitability and firing properties of Pv+ forebrain interneurons. Purkinje neurons and Pv+ forebrain interneurons are both fast-spiking GABAergic neurons, and after loss of AnkR both cell types have reduced Kv3 K+ channels and their β spectrin binding partners. Despite these similarities and unlike Purkinje neurons, AnkR-deficient forebrain Pv+ interneurons do not degenerate. One major difference between the two cell types is the presence of the perineuronal net. Although the nets are less compact and reduced in density in AnkR-deficient forebrain Pv+ interneurons, they are not completely lost. Some studies suggest that perineuronal nets are neuroprotective and may function to buffer ion concentrations (Cabungcal et al., 2013; Suttkus et al., 2016; Fawcett et al., 2019). We speculate the profound degeneration of AnkR-deficient Purkinje neurons may reflect a lack of neuroprotective functions provided by the perineuronal nets surrounding other fast-spiking GABAergic neurons.
In conclusion, although prior evidence supported a role for AnkR in the cerebellum (Peters et al., 1991; Clarkson et al., 2014), until now genetic tools did not exist to allow a clear determination of AnkR's function. Our experiments reveal that AnkR is a cytoskeletal scaffolding protein required to link Kv3.3 K+ channels to the β3 spectrin-based cytoskeleton in Purkinje neurons. Loss of AnkR from Purkinje neurons results in ataxia, neurodegeneration, and altered cerebellar output. Moreover, our results, combined with studies of Kv3 channels in Pv+ forebrain interneurons (Stevens et al., 2021), demonstrate that AnkR is the critical scaffold regulating the Kv3 subfamily of K+ channels in the nervous system.
Footnotes
This work was supported by National Institutes of Health Grants R35 NS122073 (to M.N.R.); R01NS089664, R01NS1000874, R01NS119301, and U54HD083092 (to R.V.S.), Dystonia Medical Research Foundation (DMRF; M.E.v.d.H.), and F31 NS100300 (to S.R.S.). This work was also supported by the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation (M.N.R.).
The authors declare no competing financial interests.
References
- Becker MI, Person AL (2019) Cerebellar control of reach kinematics for endpoint precision. Neuron 103:335–348.e5. 10.1016/j.neuron.2019.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckinghausen J, Sillitoe RV (2019) Insights into cerebellar development and connectivity. Neurosci Lett 688:2–13. 10.1016/j.neulet.2018.05.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett V (1982) The molecular basis for membrane - cytoskeleton association in human erythrocytes. J Cell Biochem 18:49–65. 10.1002/jcb.1982.240180106 [DOI] [PubMed] [Google Scholar]
- Bennett V, Lorenzo DN (2013) Spectrin- and ankyrin-based membrane domains and the evolution of vertebrates. Curr Top Membr 72:1–37. 10.1016/B978-0-12-417027-8.00001-5 [DOI] [PubMed] [Google Scholar]
- Bracha V, Kolb FP, Irwin KB, Bloedel JR (1999) Inactivation of interposed nuclei in the cat: classically conditioned withdrawal reflexes, voluntary limb movements and the action primitive hypothesis. Exp Brain Res 126:77–92. 10.1007/s002210050718 [DOI] [PubMed] [Google Scholar]
- Bréchet A, Fache MP, Brachet A, Ferracci G, Baude A, Irondelle M, Pereira S, Leterrier C, Dargent B (2008) Protein kinase CK2 contributes to the organization of sodium channels in axonal membranes by regulating their interactions with ankyrin G. J Cell Biol 183:1101–1114. 10.1083/jcb.200805169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AM, White JJ, van der Heijden ME, Zhou J, Lin T, Sillitoe RV (2020) Purkinje cell misfiring generates high-amplitude action tremors that are corrected by cerebellar deep brain stimulation. Elife 9:e51928. 10.7554/eLife.51928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabungcal JH, Steullet P, Morishita H, Kraftsik R, Cuenod M, Hensch TK, Do KQ (2013) Perineuronal nets protect fast-spiking interneurons against oxidative stress. Proc Natl Acad Sci USA 110:9130–9135. 10.1073/pnas.1300454110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerminara NL, Lang EJ, Sillitoe RV, Apps R (2015) Redefining the cerebellar cortex as an assembly of non-uniform Purkinje cell microcircuits. Nat Rev Neurosci 16:79–93. 10.1038/nrn3886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang KJ, Zollinger DR, Susuki K, Sherman DL, Makara MA, Brophy PJ, Cooper EC, Bennett V, Mohler PJ, Rasband MN (2014) Glial ankyrins facilitate paranodal axoglial junction assembly. Nat Neurosci 17:1673–1681. 10.1038/nn.3858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarkson YL, Perkins EM, Cairncross CJ, Lyndon AR, Skehel PA, Jackson M (2014) β-III spectrin underpins ankyrin R function in Purkinje cell dendritic trees: protein complex critical for sodium channel activity is impaired by SCA5-associated mutations. Hum Mol Genet 23:3875–3882. 10.1093/hmg/ddu103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coetzer TL, Lawler J, Liu SC, Prchal JT, Gualtieri RJ, Brain MC, Dacie JV, Palek J (1988) Partial ankyrin and spectrin deficiency in severe, atypical hereditary spherocytosis. N Engl J Med 318:230–234. 10.1056/NEJM198801283180407 [DOI] [PubMed] [Google Scholar]
- Espinosa F, McMahon A, Chan E, Wang S, Ho CS, Heintz N, Joho RH (2001) Alcohol hypersensitivity, increased locomotion, and spontaneous myoclonus in mice lacking the potassium channels Kv3.1 and Kv3.3. J Neurosci 21:6657–6665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinosa F, Marks G, Heintz N, Joho RH (2004) Increased motor drive and sleep loss in mice lacking Kv3-type potassium channels. Genes Brain Behav 3:90–100. 10.1046/j.1601-183x.2003.00054.x [DOI] [PubMed] [Google Scholar]
- Fawcett JW, Oohashi T, Pizzorusso T (2019) The roles of perineuronal nets and the perinodal extracellular matrix in neuronal function. Nat Rev Neurosci 20:451–465. 10.1038/s41583-019-0196-3 [DOI] [PubMed] [Google Scholar]
- Gerlai R, Millen KJ, Herrup K, Fabien K, Joyner AL, Roder J (1996) Impaired motor learning performance in cerebellar En-2 mutant mice. Behav Neurosci 110:126–133. 10.1037//0735-7044.110.1.126 [DOI] [PubMed] [Google Scholar]
- Ho TS, Zollinger DR, Chang KJ, Xu M, Cooper EC, Stankewich MC, Bennett V, Rasband MN (2014) A hierarchy of ankyrin-spectrin complexes clusters sodium channels at nodes of Ranvier. Nat Neurosci 17:1664–1672. 10.1038/nn.3859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt GR, Softky WR, Koch C, Douglas RJ (1996) Comparison of discharge variability in vitro and in vivo in cat visual cortex neurons. J Neurophysiol 75:1806–1814. 10.1152/jn.1996.75.5.1806 [DOI] [PubMed] [Google Scholar]
- Hsieh JY, Ulrich BN, Issa FA, Lin MA, Brown B, Papazian DM (2020) Infant and adult SCA13 mutations differentially affect Purkinje cell excitability, maturation, and viability in vivo. Elife 9:e57358. 10.7554/eLife.57358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang M, Verbeek DS (2019) Why do so many genetic insults lead to Purkinje Cell degeneration and spinocerebellar ataxia? Neurosci Lett 688:49–57. 10.1016/j.neulet.2018.02.004 [DOI] [PubMed] [Google Scholar]
- Hurlock EC, McMahon A, Joho RH (2008) Purkinje-cell-restricted restoration of Kv3.3 function restores complex spikes and rescues motor coordination in Kcnc3 mutants. J Neurosci 28:4640–4648. 10.1523/JNEUROSCI.5486-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurlock EC, Bose M, Pierce G, Joho RH (2009) Rescue of motor coordination by Purkinje cell-targeted restoration of Kv3.3 channels in Kcnc3-null mice requires Kcnc1. J Neurosci 29:15735–15744. 10.1523/JNEUROSCI.4048-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda Y, Dick KA, Weatherspoon MR, Gincel D, Armbrust KR, Dalton JC, Stevanin G, Dürr A, Zühlke C, Bürk K, Clark HB, Brice A, Rothstein JD, Schut LJ, Day JW, Ranum LP (2006) Spectrin mutations cause spinocerebellar ataxia type 5. Nat Genet 38:184–190. 10.1038/ng1728 [DOI] [PubMed] [Google Scholar]
- Joho RH, Hurlock EC (2009) The role of Kv3-type potassium channels in cerebellar physiology and behavior. Cerebellum 8:323–333. 10.1007/s12311-009-0098-4 [DOI] [PubMed] [Google Scholar]
- Khan MM, Chen CH, Regehr WG (2021) Unusually slow spike frequency adaptation in deep cerebellar nuclei neurons preserves linear transformations on the sub-second time scale. bioRxiv 2021.2008.2010.455865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalonde R, Strazielle C (2007) Brain regions and genes affecting postural control. Prog Neurobiol 81:45–60. 10.1016/j.pneurobio.2006.11.005 [DOI] [PubMed] [Google Scholar]
- Lambert S, Bennett V (1993) Postmitotic expression of ankyrinR and beta R-spectrin in discrete neuronal populations of the rat brain. J Neurosci 13:3725–3735. 10.1523/JNEUROSCI.13-09-03725.1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang EJ, Blenkinsop TA (2011) Control of cerebellar nuclear cells: a direct role for complex spikes? Cerebellum 10:694–701. 10.1007/s12311-011-0261-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lise S, Clarkson Y, Perkins E, Kwasniewska A, Sadighi Akha E, Schnekenberg RP, Suminaite D, Hope J, Baker I, Gregory L, Green A, Allan C, Lamble S, Jayawant S, Quaghebeur G, Cader MZ, Hughes S, Armstrong RJE, Kanapin A, Rimmer A, et al. (2012) Recessive mutations in SPTBN2 implicate β-III spectrin in both cognitive and motor development. PLoS Genet 8:e1003074. 10.1371/journal.pgen.1003074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low AYT, Thanawalla AR, Yip AKK, Kim J, Wong KLL, Tantra M, Augustine GJ, Chen AI (2018) Precision of discrete and rhythmic forelimb movements requires a distinct neuronal subpopulation in the interposed anterior nucleus. Cell Rep 22:2322–2333. 10.1016/j.celrep.2018.02.017 [DOI] [PubMed] [Google Scholar]
- Luo L, Ambrozkiewicz MC, Benseler F, Chen C, Dumontier E, Falkner S, Furlanis E, Gomez AM, Hoshina N, Huang WH, Hutchison MA, Itoh-Maruoka Y, Lavery LA, Li W, Maruo T, Motohashi J, Pai ELL, Pelkey KA, Pereira A, Philips T, et al. (2020) Optimizing nervous system-specific gene targeting with Cre driver lines: prevalence of germline recombination and influencing factors. Neuron 106:37–65.e5. 10.1016/j.neuron.2020.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matheson DW, Howland JL (1974) Erythrocyte deformation in human muscular dystrophy. Science 184:165–166. 10.1126/science.184.4133.165 [DOI] [PubMed] [Google Scholar]
- McCann SR, Jacob HS (1976) Spinal cord disease in hereditary spherocytosis: report of two cases with a hypothesized common mechanism for neurologic and red cell abnormalities. Blood 48:259–263. [PubMed] [Google Scholar]
- McDevitt CJ, Ebner TJ, Bloedel JR (1987) Relationships between simultaneously recorded Purkinje cells and nuclear neurons. Brain Res 425:1–13. 10.1016/0006-8993(87)90477-x [DOI] [PubMed] [Google Scholar]
- Miterko LN, Lin T, Zhou J, van der Heijden ME, Beckinghausen J, White JJ, Sillitoe RV (2021) Neuromodulation of the cerebellum rescues movement in a mouse model of ataxia. Nat Commun 12:1295. 10.1038/s41467-021-21417-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miya K, Shimojima K, Sugawara M, Shimada S, Tsuri H, Harai-Tanaka T, Nakaoka S, Kanegane H, Miyawaki T, Yamamoto T (2012) A de novo interstitial deletion of 8p11.2 including ANK1 identified in a patient with spherocytosis, psychomotor developmental delay, and distinctive facial features. Gene 506:146–149. 10.1016/j.gene.2012.06.086 [DOI] [PubMed] [Google Scholar]
- Nelson AD, Jenkins PM (2017) Axonal membranes and their domains: assembly and function of the axon initial segment and node of Ranvier. Front Cell Neurosci 11:136. 10.3389/fncel.2017.00136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozol K, Hayden JM, Oberdick J, Hawkes R (1999) Transverse zones in the vermis of the mouse cerebellum. J Comp Neurol 412:95–111. [DOI] [PubMed] [Google Scholar]
- Palarz K, Neves-Carvalho A, Duarte-Silva S, Maciel P, Khodakhah K (2021) Cerebellar neuronal dysfunction accompanies early motor symptoms in spinocerebellar ataxia type 3 and is partially alleviated upon chronic citalopram treatment. bioRxiv 2021.2004.2029.441865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins EM, Clarkson YL, Sabatier N, Longhurst DM, Millward CP, Jack J, Toraiwa J, Watanabe M, Rothstein JD, Lyndon AR, Wyllie DJ, Dutia MB, Jackson M (2010) Loss of beta-III spectrin leads to Purkinje cell dysfunction recapitulating the behavior and neuropathology of spinocerebellar ataxia type 5 in humans. J Neurosci 30:4857–4867. 10.1523/JNEUROSCI.6065-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Person AL, Raman IM (2011) Purkinje neuron synchrony elicits time-locked spiking in the cerebellar nuclei. Nature 481:502–505. 10.1038/nature10732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters LL, Birkenmeier CS, Bronson RT, White RA, Lux SE, Otto E, Bennett V, Higgins A, Barker JE (1991) Purkinje cell degeneration associated with erythroid ankyrin deficiency in nb/nb mice. J Cell Biol 114:1233–1241. 10.1083/jcb.114.6.1233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porcello DM, Ho CS, Joho RH, Huguenard JR (2002) Resilient RTN fast spiking in Kv3.1 null mice suggests redundancy in the action potential repolarization mechanism. J Neurophysiol 87:1303–1310. 10.1152/jn.00556.2001 [DOI] [PubMed] [Google Scholar]
- Sarna JR, Hawkes R (2003) Patterned Purkinje cell death in the cerebellum. Prog Neurobiol 70:473–507. 10.1016/s0301-0082(03)00114-x [DOI] [PubMed] [Google Scholar]
- Sarna JR, Hawkes R (2011) Patterned Purkinje cell loss in the ataxic sticky mouse. Eur J Neurosci 34:79–86. 10.1111/j.1460-9568.2011.07725.x [DOI] [PubMed] [Google Scholar]
- Sarna JR, Larouche M, Marzban H, Sillitoe RV, Rancourt DE, Hawkes R (2003) Patterned Purkinje cell degeneration in mouse models of Niemann-Pick type C disease. J Comp Neurol 456:279–291. 10.1002/cne.10522 [DOI] [PubMed] [Google Scholar]
- Sarna JR, Marzban H, Watanabe M, Hawkes R (2006) Complementary stripes of phospholipase Cbeta3 and Cbeta4 expression by Purkinje cell subsets in the mouse cerebellum. J Comp Neurol 496:303–313. 10.1002/cne.20912 [DOI] [PubMed] [Google Scholar]
- Sillitoe RV, Hawkes R (2002) Whole-mount immunohistochemistry: a high-throughput screen for patterning defects in the mouse cerebellum. J Histochem Cytochem 50:235–244. 10.1177/002215540205000211 [DOI] [PubMed] [Google Scholar]
- Stankewich MC, Gwynn B, Ardito T, Ji L, Kim J, Robledo RF, Lux SE, Peters LL, Morrow JS (2010) Targeted deletion of betaIII spectrin impairs synaptogenesis and generates ataxic and seizure phenotypes. Proc Natl Acad Sci USA 107:6022–6027. 10.1073/pnas.1001522107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens SR, Rasband MN (2021) Ankyrins and neurological disease. Curr Opin Neurobiol 69:51–57. 10.1016/j.conb.2021.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens SR, Longley CM, Ogawa Y, Teliska LH, Arumanayagam AS, Nair S, Oses-Prieto JA, Burlingame AL, Cykowski MD, Xue M, Rasband MN (2021) Ankyrin-R regulates fast-spiking interneuron excitability through perineuronal nets and Kv3.1b K(+) channels. Elife 10:e66491. 10.7554/eLife.66491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suttkus A, Morawski M, Arendt T (2016) Protective properties of neural extracellular matrix. Mol Neurobiol 53:73–82. 10.1007/s12035-014-8990-4 [DOI] [PubMed] [Google Scholar]
- van der Heijden ME, Kizek DJ, Perez R, Ruff EK, Ehrlich ME, Sillitoe RV (2021) Abnormal cerebellar function and tremor in a mouse model for non-manifesting partially penetrant dystonia type 6. J Physiol 599:2037–2054. 10.1113/JP280978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JJ, Arancillo M, Stay TL, George-Jones NA, Levy SL, Heck DH, Sillitoe RV (2014) Cerebellar zonal patterning relies on Purkinje cell neurotransmission. J Neurosci 34:8231–8245. 10.1523/JNEUROSCI.0122-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JJ, Lin T, Brown AM, Arancillo M, Lackey EP, Stay TL, Sillitoe RV (2016) An optimized surgical approach for obtaining stable extracellular single-unit recordings from the cerebellum of head-fixed behaving mice. J Neurosci Methods 262:21–31. 10.1016/j.jneumeth.2016.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witter L, Canto CB, Hoogland TM, de Gruijl JR, De Zeeuw CI (2013) Strength and timing of motor responses mediated by rebound firing in the cerebellar nuclei after Purkinje cell activation. Front Neural Circuits 7:133. 10.3389/fncir.2013.00133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagha E, Manita S, Ross WN, Rudy B (2010) Dendritic Kv3.3 potassium channels in cerebellar purkinje cells regulate generation and spatial dynamics of dendritic Ca2+ spikes. J Neurophysiol 103:3516–3525. 10.1152/jn.00982.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Kaczmarek LK (2016) Kv3.3 potassium channels and spinocerebellar ataxia. J Physiol 594:4677–4684. 10.1113/JP271343 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Gait of 12-month-old Ank1F/F mouse.
Gait of 12-month-old Ank1F/F;Nestin-Cre mouse.