

Abstract
Background
Postpartum iron deficiency anaemia is caused by bleeding or inadequate dietary iron intake/uptake. This condition is defined by iron deficiency accompanied by a lower than normal blood haemoglobin concentration, although this can be affected by factors other than anaemia and must be interpreted in the light of any concurrent symptoms. Symptoms include fatigue, breathlessness, and dizziness. Treatment options include oral or intravenous iron, erythropoietin which stimulates red blood cell production, and substitution by red blood cell transfusion.
Objectives
To assess the efficacy and harms of the available treatment modalities for women with postpartum iron deficiency anaemia.
Search methods
The Cochrane Pregnancy and Childbirth Group’s Trials Register (9 April 2015); the WHO International Clinical Trials Registry Portal (ICTRP), and the Latin‐American and Caribbean Health Sciences Literature database (LILACS) (8 April 2015) and reference lists of retrieved studies.
Selection criteria
We included published, unpublished and ongoing randomised controlled trials that compared a treatment for postpartum iron deficiency anaemia with placebo, no treatment, or another treatment for postpartum iron deficiency anaemia, including trials described in abstracts only. Cluster‐randomised trials were eligible for inclusion. We included both open‐label trials and blinded trials, regardless of who was blinded. The participants were women with a postpartum haemoglobin of 120 g per litre (g/L) or less, for which treatment was initiated within six weeks after childbirth.
Non‐randomised trials, quasi‐randomised trials and trials using a cross‐over design were excluded.
Data collection and analysis
Two review authors independently assessed studies for inclusion, quality, and extracted data. We contacted study authors and pharmaceutical companies for additional information.
Main results
We included 22 randomised controlled trials (2858 women), most of which had high risk of bias in several domains. We performed 13 comparisons. Many comparisons are based on a small number of studies with small sample sizes. No analysis of our primary outcomes contained more than two studies.
Intravenous iron was compared to oral iron in 10 studies (1553 women). Fatigue was reported in two studies and improved significantly favouring the intravenously treated group in one of the studies. Other anaemia symptoms were not reported. One woman died from cardiomyopathy (risk ratio (RR) 2.95; 95% confidence interval (CI) 0.12 to 71.96; two studies; one event; 374 women; low quality evidence). One woman developed arrhythmia. Both cardiac complications occurred in the intravenously treated group. Allergic reactions occurred in three women treated with intravenous iron, not statistically significant (average RR 2.78; 95% CI 0.31 to 24.92; eight studies; 1454 women; I² = 0%; low quality evidence). Gastrointestinal events were less frequent in the intravenously treated group (average RR 0.31; 95% CI 0.20 to 0.47; eight studies; 169 events; 1307 women; I² = 0%; very low quality evidence).
One study evaluated red blood cell transfusion versus non‐intervention. General fatigue improved significantly more in the transfusion group at three days (MD ‐0.80; 95% CI ‐1.53 to ‐0.07; women 388; low quality evidence), but no difference between groups was seen at six weeks. Maternal mortality was not reported.
The remaining comparisons evaluated oral iron (with or without other food substances) versus placebo (three studies), intravenous iron with oral iron versus oral iron (two studies) and erythropoietin (alone or combined with iron) versus placebo or iron (seven studies). These studies did not investigate fatigue. Maternal mortality was rarely reported.
Authors' conclusions
The body of evidence did not allow us to reach a clear conclusion regarding the efficacy of the interventions on postpartum iron deficiency anaemia. The quality of evidence was low.
Clinical outcomes were rarely reported. Laboratory values may not be reliable indicators for efficacy, as they do not always correlate with clinical treatment effects. It remains unclear which treatment modality is most effective in alleviating symptoms of postpartum anaemia.
Intravenous iron was superior regarding gastrointestinal harms, however anaphylaxis and cardiac events occurred and more data are needed to establish whether this was caused by intravenous iron.
The clinical significance of some temporarily improved fatigue scores in women treated with blood transfusion is uncertain and this modest effect should be balanced against known risks, e.g. maternal mortality (not reported) and maternal immunological sensitisation, which can potentially harm future pregnancies.
When comparing oral iron to placebo it remains unknown whether efficacy (relief of anaemia symptoms) outweighs the documented gastrointestinal harms.
We could not draw conclusions regarding erythropoietin treatment due to lack of evidence.
Further research should evaluate treatment effect through clinical outcomes, i.e. presence and severity of anaemia symptoms balanced against harms, i.e. survival and severe morbidity.
Plain language summary
Treatment for women with iron deficiency anaemia after childbirth
Anaemia is a condition where the blood contains less than normal haemoglobin (low blood count), as shown in blood tests. Haemoglobin is the molecule within red blood cells that requires iron to carry oxygen. Insufficient iron intake/uptake and iron loss (bleeding) can cause iron deficiency anaemia. Anaemia symptoms include tiredness, shortness of breath and dizziness. Women may bleed severely at childbirth and many pregnant women already have anaemia, which can worsen as a result of bleeding. Severe anaemia can be linked to maternal deaths. Iron deficiency anaemia after childbirth is more likely to occur in low‐income countries.
Treatment for iron deficiency anaemia includes iron tablets or a solution injected into a vein (intravenously). Another option is to restore red blood cells through transfusion with blood from a blood donor or to boost red blood cell formation with erythropoietin. It is important to investigate if one treatment is better than another in relieving anaemia symptoms, and whether the treatment options are safe.
We included 22 randomised controlled studies with 2858 women and performed 13 comparisons, many of which were based on few studies involving small numbers of women. The overall quality of evidence was low. Most trials were conducted in high‐income countries.
Ten studies, including 1553 women, compared intravenous iron with oral iron. Only one study showed a temporary positive effect on fatigue for intravenous iron. Other anaemia symptoms were not reported. One woman died from heart complications in the intravenous group. Only two studies reported on maternal deaths. Allergic reactions occurred in three women, and heart complications in two women in the intravenous group. Gastrointestinal symptoms were frequent in the oral group and caused some participants to abandon treatment.
One study compared red blood cell transfusion with no transfusion. Some (but not all) fatigue scores temporarily improved in the transfused women. Maternal mortality was not reported.
When comparing oral iron to placebo (three studies), anaemia symptoms were not reported. It remains unknown whether benefits of oral iron outweigh documented gastrointestinal harms.
Other treatment options were compared in other studies, which did not investigate fatigue.
Very few studies reported on relief of anaemia symptoms, although this is perhaps the most important purpose of treatment.
The body of evidence did not allow us to fully evaluate the efficacy of the treatments on iron deficiency anaemia after childbirth and further research is needed.
Summary of findings
Background
Description of the condition
Women who give birth may develop postpartum anaemia, either because of excessive bleeding or pre‐existing conditions in pregnancy. Severe postpartum anaemia can be a serious problem, being linked to possibly 40% of maternal deaths worldwide (WHO 2001). Anaemia also increases the risk of maternal death from other causes such as infections, malnutrition and bleeding (WHO 2012b). For some women, particularly in resource‐poor countries, postpartum anaemia is a major cause of poor health (Bergmann 2010; Gupta 2010; Khan 2006; WHO 2012a).
Anaemia, including postpartum anaemia, is defined by a lower than normal haemoglobin (Hb) value, but clinical symptoms are essential to the evaluation of its importance. Haemoglobin is the molecule contained within red blood cells (RBCs) that is responsible for transporting oxygen around the body. During pregnancy, most women have a physiologically normal reduction in their Hb concentration due to accumulation of fluid (WHO 2001). Postpartum anaemia may be caused or augmented by low dietary iron intake or uptake, blood loss, or infections (e.g. malaria), and the physiological changes during pregnancy, and bleeding associated with childbirth can aggravate the condition (WHO 1999).
Postpartum anaemia can cause symptoms such as breathlessness, palpitations (a sensation of increased heart rate), tiredness, as well as an increased risk of infections. All of these symptoms may impact a woman’s ability to breastfeed and care for her baby in general (Bergmann 2010; Milman 2011).
In pregnancy, the circulating blood volume increases to prepare the woman for blood loss at delivery. Bleeding and/or resorption of excess fluid from body tissues during and after delivery vary in extent between individuals (Milman 2011), which can have a major impact on maternal Hb concentrations. It is generally accepted that a low Hb concentration ‐ usually less than 120 grams per litre (g/L) ‐ is indicative of anaemia in postpartum women, although there is considerable variation in the precise concentration that defines anaemia, and also the time after birth at which this should be measured (Barroso 2011; Bergmann 2010; Bodnar 2005; Breymann 2010; Milman 2011; Richter 1995). Thus, postpartum anaemia is poorly defined and the Hb level in the postpartum period (six weeks after delivery) depends strongly on how long after delivery it is measured (WHO 2012a). It should be emphasised that even though an association between a low Hb and clinical symptoms has been shown in population‐based observational studies, the normal range of Hb is an arbitrarily defined statistical value derived from the population average, and an individual woman’s Hb level does not necessarily reflect the clinical symptoms she may experience (WHO 2001). Since the correlation between the different clinical symptoms and the level of Hb in postpartum anaemia is not well described, the clinical significance of a change in Hb level as a result of any given treatment remains uncertain. This is why a change in the Hb concentration is a surrogate outcome in trials of interventions for anaemia, In clinical practice however, a low Hb level is the most commonly used laboratory test to support or refute the clinical diagnosis of anaemia and it is generally understood that a major drop in the Hb level within a short time frame is likely to correlate with a large blood loss at delivery, which may lead to acute symptoms of anaemia and shock.
During pregnancy, anaemia is also defined by low Hb levels and is stratified into mild, moderate or severe anaemia (WHO 2002). However, there is no clear correlation between the type and severity of anaemia symptoms and these stratifications. Both anaemia in pregnancy (e.g. due to insufficient dietary intake) and haemorrhage during or after birth are strong predictors for postpartum iron deficiency anaemia (Bodnar 2005; Milman 2011; Reveiz 2011). Iron deficiency anaemia is a condition where the low Hb level is caused by an insufficient amount of iron in the body. To our knowledge, only one study has estimated the incidence of postpartum iron deficiency anaemia, at 4.2% among women in the United States examined within the first six months postpartum (Bodnar 2002).
Postpartum iron deficiency anaemia does not have a specific code in the International Classification of Diseases (ICD‐10), but is included in the more general code 099.0 'Anaemia complicating pregnancy, childbirth and the puerperium' (WHO ICD 2010). At this point all definitions of postpartum anaemia rely on Hb values alone, not symptoms. The classification of different stages of severity is also based on Hb values only (Milman 2012). In the postpartum period as well as in pregnancy, it remains unanswered whether any benefits of treating anaemia outweighs the harms of treatment (Reveiz 2011). Known harms depend on the choice of treatment and include e.g. gastrointestinal symptoms and allergic reactions.
Description of the intervention
There is a number of treatment options for women with postpartum anaemia, and the optimal treatment, dose, and balance between benefits and harms may vary depending on the timing and severity of anaemia, clinical symptoms, harms of the intervention, available resources and factors such as geographic location, socioeconomic status and education. The treatment modalities described in this review include iron supplementation administered either orally or directly into a vein or muscle (parenterally), erythropoietin which stimulates RBC production, and substitution of RBCs by blood transfusion.
How the intervention might work
Oral iron therapy
Oral iron therapy has been used for many years as a treatment for iron deficiency anaemia in general (Dudrick 1986), as well as in pregnancy (Pena‐Rosas 2012). Oral iron is often the recommended treatment for mild to moderate iron deficiency anaemia (Bodnar 2005) because of its low cost and ease of use. The body has a limited capacity to absorb iron from the gut, and prolonged treatment over a period of several months is often required to increase the Hb concentration and relieve symptoms of anaemia (Auerbach 2008; Milman 2012; Van Wyck 2007; Westad 2008). Gastrointestinal (GI) adverse effects, such as constipation and nausea are common in oral iron therapy (al‐Momen 1996; Bhandal 2006). This may affect the women's compliance with treatment and consequently prevent correction of the anaemia.
Folate
Folate, also called folic acid and vitamin B9, is a substance found in many foods and is naturally available in especially high concentrations in green vegetables. Folate is involved in the synthesis of DNA, cell division and growth in human cells. Folate deficiency can cause megaloblastic anaemia, not iron deficiency anaemia. However, folate is often added as an adjunct to oral iron because malnutrition often results in a lack of both iron and folate in the body. The long‐term effect of folate supplementation and continuously high levels of folate in the blood have been associated to an increased risk of certain cancers (Almeida 2010). In this review, we will not consider folate supplementation as an independent treatment of anaemia, but accept studies where it is a part of other types of treatment for postpartum iron deficiency anaemia.
Parenteral iron therapy
Parenteral administration of iron has been shown to produce a more rapid increase in the Hb concentration in iron deficiency anaemia during pregnancy (Milman 2012). Parenterally administered iron has been associated with pain and redness (erythema) at the injection site and, rarely, anaphylactic reactions characterised by itching, redness and in severe cases angioedema (swelling), vascular collapse, bronchospasm (constriction of the airways) and shock (Barish 2012; Breymann 2008; Kochhar 2013; Seid 2008; Wysowski 2010). The use of new low‐molecular iron (such as iron sucrose and ferric carboxymaltose) may lower the risk of anaphylactic reactions but these products are expensive compared with oral iron therapy, which does not have these serious harms (Khalafallah 2012; Kochhar 2013).
Erythropoietin
Erythropoietin (EPO) is a hormone produced in the kidneys when blood oxygen levels are low. It acts to stimulate erythropoiesis (blood formation) in the bone marrow (Oster 2012). Initially, EPO was used for anaemia associated with renal (kidney) disease. Later, EPO was used to treat other forms of anaemia and has been used as an alternative to blood transfusion for the treatment of iron deficiency anaemia, including postpartum iron deficiency anaemia (Bergmann 2010; Oster 2012). Adverse effects of EPO treatment include mild flu‐like symptoms such as sore throat, cough, fever, muscle pains and weakness, headache and fatigue. Uncommon but more serious adverse effects include hypertension, thromboembolic complications, seizures, and pure red‐cell aplasia (Dodd 2004; Kliger 2012). Recent research has shown an association with certain haematological cancers, which led to a Food and Drug Agency (FDA) black box warning (label on the product warning against serious or life‐threatening risks). The use of EPO is now restricted to specific patient groups and is rarely used in postpartum anaemic patients (Bunn 2009; Oster 2012).
Blood transfusion
Transfusion of allogeneic blood can be used in the treatment of postpartum anaemia and may be life‐saving in the case of acute or major bleeding at the time of giving birth (Montufar‐Rueda 2013). However, experimentally depleting healthy volunteers to an Hb of 50 g/L in a controlled setting, elicited cardiac compensatory mechanisms, but did not compromise health (Weiskopf 1998). Adverse reactions rather than clinical benefit has been found when transfusing mixed‐patient populations with mild to moderate anaemia (Carson 2012; Rohde 2014; Salpeter 2014). Thus, transfusion is generally not recommended following small to moderate bleedings in patients with a normal physiologic response to anaemia. Transfusion of one unit of RBC usually increases the Hb by 10 g/L in the non‐bleeding, haemodynamically stable patient (Wiesen 1994). There are associated risks, including donor‐transmitted infections (particularly hepatitis and human immunodeficiency virus (HIV)), transfusion‐associated circulatory overload (TACO), and a variety of immunologic reactions such as fever, urticaria (hives), anaphylaxis, transfusion‐related lung injury (TRALI) or antibody formation which may interfere with future pregnancies (Fuller 2010; Hendrickson 2009; SHOT Report 2011; Villanueva 2013). Blood transfusion may rarely cause acute haemolysis (breakdown of RBCs) if incompatible blood is administered by mistake (Fuller 2010). Blood transfusions are expensive, as costs include screening for infection, cross‐matching, storage and sterile and safe administration of blood products (Shander 2010). In low‐income countries or during disasters, blood for transfusion may not be readily available.
Why it is important to do this review
Postpartum anaemia caused by insufficient iron intake and/or bleeding (postpartum iron deficiency anaemia) is a common condition affecting women after childbirth and may be associated with symptoms that can influence survival, health and the ability to care for the baby. The treatment modalities available for postpartum iron deficiency anaemia have harms, some of which are serious. Since all women bleed at delivery, it is a common practice to administer treatment for postpartum iron deficiency anaemia, to enable the women to synthesise new RBCs effectively. Some populations may benefit more than others, and in some populations and categories of disease severity, treatment may be unnecessary, ineffective or even harmful. Women and care givers need reliable estimates of the benefits and harms of the available treatments for postpartum anaemia so that they can be balanced for each individual patient.
This review is an update of an earlier review by Dodd 2004.
Objectives
To assess the efficacy and harms of the available treatment modalities for women with postpartum iron deficiency anaemia. These include oral and parenteral iron, erythropoietin, and blood transfusion.
Methods
Criteria for considering studies for this review
Types of studies
We included published, unpublished and ongoing randomised controlled trials that compared a treatment for postpartum iron deficiency anaemia with placebo, no treatment, or another treatment for postpartum iron deficiency anaemia, including trials described in abstracts only. Cluster‐randomised trials were eligible for inclusion. We included both open‐label trials and blinded trials, regardless of who was blinded. Non‐randomised trials, quasi‐randomised trials and trials using a cross‐over design were excluded.
Types of participants
Women with a postpartum Hb value of 120 g/L (7.4 millimoles per litre) or less, with treatment initiated up to six weeks after giving birth. We distinguished between socioeconomic population groups whenever possible, as this factor may affect the response to treatment, but included all.
Types of interventions
Treatment for postpartum iron deficiency anaemia started within the first six weeks after giving birth compared with placebo, no treatment or another treatment.
Currently, accepted treatment for iron deficiency anaemia includes blood transfusion or iron supplementation administered orally or parenterally, either alone or in combination with folate and/or erythropoietin.
Folate supplementation was not considered as an independent treatment of iron deficiency anaemia, but was accepted as a part of other types of treatment for postpartum iron deficiency anaemia.
New treatment modalities appropriate for iron deficiency anaemia will be included in future updates.
Types of outcome measures
Primary outcomes
Maternal mortality: We considered that no women died only if: a) this was stated explicitly, or b) no dropouts occurred during follow‐up, or c) contact authors provided this information on request. Mortality was considered present only if: a) stated explicitly in a published report or b) contact authors provided this information on request. Mortality was assessed as not reported if a) no mention of dropouts or their causes, b) all dropouts not accounted for, c) dropouts not explicitly reported to be alive at the end of the follow‐up period.
Fatigue: as reported by the women ‐ verbalisation of fatigue or lack of energy and inability to maintain usual routines; measured by a scale or questionnaire; or as defined by the trial authors. Short‐term and long‐term results, thus the minimal and maximal time from baseline.
Secondary outcomes
Persistent anaemia symptoms during treatment. Any of the following symptoms: dyspnoea, tachypnoea, tachycardia, palpitations, orthostatic dizziness, syncopation, paleness.
Psychological well being, including cognitive performance, measured by the 'Blues Questionnaire' (Kennerley 1989), 'Self‐report symptom inventory 90 [SCL‐90‐R]' (Schmitz 1999), 'SF36 [Medical Outcomes Study Short Form]' (Ware 2000) or similar questionnaire; or as defined by the trial authors. Only short‐term results, thus the minimal time from baseline.
Urinary tract infection, endometritis, or other infections (as defined by the trial authors).
Compliance to treatment (as defined by the trial authors).
Breastfeeding (at hospital discharge; six weeks postpartum; six months postpartum).
Length of hospital stay.
Any adverse events during treatment (each type of harm analysed individually, when possible).
Number of RBC transfusions (number of transfused women and number of RBC units per woman).
For outcomes of other psychological well being we did not apply any restrictions regarding follow‐up periods to avoid excluding data on any long‐term benefits or harms. We did not apply language restrictions.
We planned to include the following outcomes in the 'Summary of findings' tables of the review, using the Grade Profiler programme (GRADEpro 2014).
Maternal mortality.
Fatigue.
Constipation (for oral iron substitution).
Allergic reactions (for intravenous iron).
The comparisons included in the 'Summary of findings' tables were chosen based on relevance to current treatment standards according to clinical experts. Therefore we chose not to include treatment with intravenous (IV) erythropoietin (EPO) or yeast extract in the 'Summary of findings' tables, as these methods are no longer practiced. For the treatment‐specific outcomes listed above (constipation and allergic reactions), the results were included in the 'Summary of findings' tables if the specific treatment was present in only one of the study arms.
We chose to include additional outcomes in the 'Summary of findings' tables, which we found important for clinical decision making for each individual treatment modality, when this treatment was present in only one of the study arms. For comparisons with IV iron this outcome was infections. For comparisons with oral iron we included all GI symptoms combined. For comparisons with RBC transfusions we included infections, thromboembolic events and transfusion‐specific adverse events, such as alloantibody formation and transfusion reactions. For comparisons with EPO, thromboembolic events were essential. For all comparisons which met the above mentioned criteria, we found it important to include anaemia symptoms.
Search methods for identification of studies
The following methods section of this review is based on a standard template used by the Cochrane Pregnancy and Childbirth Group.
Electronic searches
We contacted the Trials Search Co‐ordinator to search the Cochrane Pregnancy and Childbirth Group’s Trials Register (9 April 2015).
The Cochrane Pregnancy and Childbirth Group’s Trials Register is maintained by the Trials Search Co‐ordinator and contains trials identified from:
monthly searches of the Cochrane Central Register of Controlled Trials (CENTRAL);
weekly searches of MEDLINE (Ovid);
weekly searches of Embase (Ovid);
monthly searches of CINAHL (EBSCO);
handsearches of 30 journals and the proceedings of major conferences;
weekly current awareness alerts for a further 44 journals plus monthly BioMed Central email alerts.
Details of the search strategies for CENTRAL, MEDLINE, Embase and CINAHL, the list of handsearched journals and conference proceedings, and the list of journals reviewed via the current awareness service can be found in the ‘Specialized Register’ section within the editorial information about the Cochrane Pregnancy and Childbirth Group.
Trials identified through the searching activities described above are each assigned to a review topic (or topics). The Trials Search Co‐ordinator searches the register for each review using the topic list rather than keywords.
In addition to the search carried out by the Trials Search Co‐ordinator, we searched the following trial registers for planned, ongoing or unpublished trials (8 April 2015):
WHO ICTRP (http://apps.who.int/trialsearch/);
LILACS (www.bireme.br).
The search strategy is described in Appendix 1.
Searching other resources
We searched the citation lists of relevant publications, review articles and included studies and contacted manufacturers of parenteral iron pharmaceuticals for knowledge of any ongoing trials.
We did not apply any language or date restrictions.
Data collection and analysis
Selection of studies
Two review authors (Veronika Markova (VM) and Astrid Norgaard (AN)) independently assessed for inclusion all the studies identified. All disagreements were resolved through discussion.
Data extraction and management
We designed a form to extract data. The form contained information on bias assessment, inclusion and exclusion criteria, number of participants and dropouts, demographic data, treatment regimens, and available information on the outcome measures pre‐specified for this review.
For eligible studies, VM and AN extracted the data using the agreed form, blinded to each others results. We resolved discrepancies through discussion or, when required, we consulted Karsten Juhl Jørgensen (KJ) at the Nordic Cochrane Centre. We entered the data into Review Manager software (RevMan 2014) and checked for accuracy. When information regarding any of the above information items was unclear, we attempted to contact authors of the original reports to provide further details. When we identified trials with more than two study arms, we included only the relevant arms in our meta‐analysis. The remaining arm(s) was described and compared with the control arm. If comparisons in the trial could not be included in a meta‐analysis, but the trial otherwise fulfilled our inclusion criteria, we described the results in separate comparisons.
Assessment of risk of bias in included studies
Two review authors (VM and AN) independently assessed risk of bias for each study using the criteria outlined in the Cochrane Handbook for Systematic Reviews of Interventions and a 'Risk of bias' table (Higgins 2011). As per Cochrane standards, we assessed selection bias, performance bias, detection bias, attrition bias, reporting bias, and other bias. Each type of bias was assessed as low, high, or unclear. All disagreements were resolved by discussion, or by involving a third assessor (KJ or Jens Langhoff‐Roos (JLR)).
We made explicit judgements about whether studies were at high risk of bias, according to the criteria given in the Cochrane Handbook (Higgins 2011). Where possible, we assessed the likely magnitude and direction of the bias and whether we considered if it was likely to impact the findings. We explored the impact of bias through Sensitivity analysis.
We used Grade Profiler (GRADEpro 2014) to make 'Summary of findings' tables. We included our primary outcomes, constipation (when treated with oral iron), and allergic reactions (when treated with intravenous (IV) iron). We also included additional outcomes, which we considered important for the decision‐making process.
Measures of treatment effect
Dichotomous data
For dichotomous data, we presented results as a summary risk ratio with 95% confidence intervals from a meta‐analysis, if possible.
Continuous outcome data
For continuous outcome data we used the mean difference if outcomes were measured in the same way between trials. We planned to use the standardised mean difference to combine trial results that measured the same outcome, but used different methods.
Unit of analysis issues
Cluster‐randomised trials
We planned to include cluster‐randomised trials in the analyses along with individually‐randomised trials, but none were found. In future updates, if included, we will adjust the standard error of any cluster‐randomised trials using the methods described in the Cochrane Handbook section 16.3.6, if relevant. We will use an estimate of the intra‐cluster correlation co‐efficient (ICC) derived from the trial (if possible), from a similar trial or from a study of a similar population. If we use ICCs from other sources, we will report this and conduct sensitivity analyses to investigate the effect of variation in the ICC. If we identify both cluster‐randomised trials and individually‐randomised trials, we plan to synthesise the relevant information from both types of studies if there is little heterogeneity between the results from trials using the two types of study design, and if the interaction between the effect of the intervention and the choice of randomisation unit is considered to be unlikely. We also plan to perform a subgroup analysis to investigate the effects of the randomisation unit (cluster or individual).
Dealing with missing data
For each included study, we noted the level of attrition. We planned to explore the impact of including studies with high levels of missing data (more than 10%) in the overall assessment of treatment effect through sensitivity analysis, using our primary outcomes (maternal mortality and fatigue).
For all outcomes, we carried out analyses, as far as possible, on an intention‐to‐treat basis, i.e. we attempted to include all participants randomised to each group in the analyses, and all participants were analysed in the group to which they were allocated, regardless of whether or not they received the allocated intervention. The denominator for each outcome in each trial was the number randomised minus the number of participants whose outcome data were known to be missing.
Assessment of heterogeneity
We assessed statistical heterogeneity in each meta‐analysis using the T², I² and Chi² statistics. We regarded heterogeneity as substantial if an I² was greater than 30% and either a T² was greater than zero, or if there was a low P value (less than 0.10) in the Chi² test for heterogeneity.
Assessment of reporting biases
We planned to investigate reporting biases (such as publication bias) if there were 10 or more studies in a meta‐analysis, using funnel plots and to assess the funnel plot asymmetry visually. In future updates, if asymmetry is suggested by a visual assessment, we will perform exploratory analyses.
Data synthesis
Statistical analysis was performed with Review Manager software (RevMan 2014). We used fixed‐effect meta‐analysis for data where it was reasonable to assume that the studies were estimating the same underlying treatment effect: i.e. where trials were examining the same intervention, and the trials’ populations and methods were judged sufficiently similar.
If clinical heterogeneity was sufficient to expect that the underlying treatment effects differed between trials, we used random‐effects meta‐analysis to produce an overall summary, if an average treatment effect across trials was considered clinically meaningful. The random‐effects summary was treated as the average range of possible treatment effects and we discussed the clinical implications of treatment effects differing between trials. If the estimated treatment effect in the included trials was not clinically meaningful, we did not combine trials.
When we used random‐effects meta‐analyses, the results were presented as the average treatment effect with 95% confidence intervals, and the estimates of T² and I².
Subgroup analysis and investigation of heterogeneity
We planned to investigate heterogeneity (if substantial), using subgroup analyses and sensitivity analyses. In future updates, if more data become available and heterogeneity is identified, we will consider whether an overall summary is meaningful, and if it is, use random effects meta‐analysis to produce it.
We planned to carry out the following subgroup analyses, if necessary.
Study setting: low‐ versus high‐income populations; high‐ versus low‐education status.
Type of intravenous iron therapy: iron sucrose versus iron carboxymaltose.
Dose administered: high versus low dose.
Duration of treatment: four weeks versus longer.
Presence of an adjunct to iron supplementation: folate versus no folate.
Source of funding: public versus corporate.
We planned to assess potential subgroup differences by interaction tests available within RevMan (RevMan 2014) and to report the results of subgroup analyses quoting the χ2 statistic and P value, and the interaction test I² value.
Sensitivity analysis
We planned to carry out a sensitivity analysis based on trial design, thus excluding trials with a high risk of selection, performance, and detection bias.
We also planned to carry out sensitivity analyses to explore the effects of random‐effects analyses for outcomes with statistical heterogeneity and the effects of any assumptions made such as the value of the ICC used for cluster‐randomised trials.
We planned sensitivity analyses only for our primary outcomes (maternal mortality and fatigue). Provided that enough data become available, we will attempt to carry out sensitivity analyses for all comparisons in future updates.
Results
Description of studies
For an individual description of the studies please see Characteristics of included studies, Characteristics of excluded studies, Characteristics of studies awaiting classification, and Characteristics of ongoing studies.
Results of the search
We retrieved 57 (9 April 2015) articles from the Pregnancy and Childbirth Group's Trials Register, 16 (8 April 2015) from the WHO International Clinical Trials Registry Portal (ICTRP), and 154 (8 April 2015) from LILACS. After excluding duplicates, 178 records remained. We screened these records for relevance by title and abstracts, and excluded 118. One discontinued study is awaiting classification and four studies based on five reports are ongoing. The remaining 54 full text articles were assessed for eligibility. Of these, 17 studies based on 19 reports were excluded (Figure 1, Study flow diagram). In addition, we assessed seven studies that were excluded by the previous authors of this review, and agreed with their assessment. Of these, one trial also appeared in our electronic search, resulting in 17 excluded studies.
1.
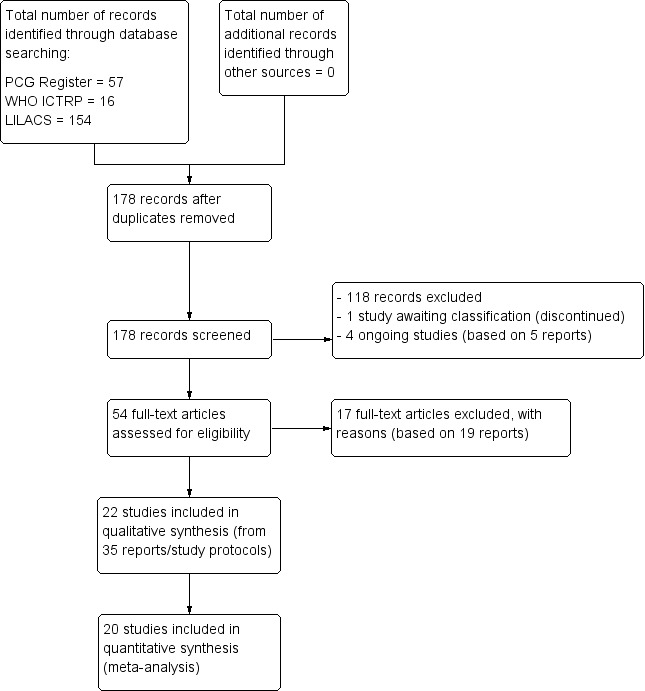
Study flow diagram.
No additional randomised controlled trials (RCTs) were found through screening of the citation lists of relevant publications.
We attempted to contact the trial authors (contact information from articles or of the Internet) for additional information or clarification of methods used for all included trials and trials with an unclear assessment for eligibility. We received the additional information from 10 individual studies (Backe 2009;Daniilidis 2011; Froessler 2013; Giannoulis 2009; Guerra 2012; Krafft 2011; Prick 2014; Van Wyck 2007; Wagstrom 2007; Westad 2008).
The remaining authors did not respond (Bhandal 2006; Breymann 1996; Breymann 2000; Jain 2013; Makrydimas 1998; Mumtaz 2011; Perello 2014; Seid 2008; Tam 2005; Verma 2011), were not possible to contact due to lack of contact information (Beard 2005; Krauss 1972; Lebrecht 1995; Meyer 1995), or did not have resources to provide the requested information (Breymann 2008).
Included studies
Design and sample sizes
We included 22 RCTs with 2858 women (Beard 2005; Bhandal 2006; Breymann 1996; Breymann 2000; Breymann 2008; Froessler 2013; Guerra 2012; Jain 2013; Krafft 2011; Krauss 1972; Lebrecht 1995; Makrydimas 1998; Meyer 1995; Mumtaz 2011; Perello 2014; Prick 2014; Seid 2008; Tam 2005; Van Wyck 2007; Verma 2011; Wagstrom 2007; Westad 2008).
Participants
All the participants were women with postpartum anaemia who received treatment within six weeks postpartum.
Interventions
Itravenous iron versus oral iron
Intravenous (IV) iron (either iron carboxymaltose or iron sucrose) was compared with oral ferrous sulphate in 10 studies including a total of 1553 women (Bhandal 2006; Breymann 2008; Froessler 2013; Guerra 2012; Jain 2013; Mumtaz 2011; Seid 2008; Van Wyck 2007; Verma 2011; Westad 2008). One study added oral iron to those originally assigned to receive IV iron after four weeks (Westad 2008). The follow‐up periods varied from 14 to 84 days between the studies. Socioeconomic status was clearly stated as being low in only one study (Froessler 2013). We did not make assumptions regarding socioeconomic status based on the name of the country.
Red blood cell transfusion
Red blood cell (RBC) transfusion was compared with non‐intervention (standard of care) in one study with 519 women (Prick 2014). The treatment of the non‐intervention arm was decided by the clinicians. This trial reported on all pre‐defined outcomes for this review, except maternal mortality. Follow‐up was six weeks.
Oral iron
Oral iron was compared with either placebo or no treatment in three studies with a total of 315 women (Beard 2005; Krauss 1972; Tam 2005). The preparations used in each trial contained various additives, such as vitamin C, vitamin B, and folic acid. Follow‐up varied from 30 days to nine months among studies. One RCT only included women of low socioeconomic status (Beard 2005). The remaining studies did not specify this. The trial by Krauss 1972 included three study arms. The trial by Tam 2005 was based on two anaemic study groups (one treated and one given placebo) and one non‐anaemic group. The study was included based on intervention, which fulfilled our criteria. However, the majority of the results were combined for both anaemic groups, thus not distinguishing between the treated and untreated group.
Inravenous iron and oral iron versus oral iron
Intravenous iron with oral iron was compared with IV placebo and active oral iron treatment in two studies (Breymann 2000; Perello 2014), including a total of 112 women. Follow‐up was two and six weeks, respectively.
Erythropoietin
Erythropoietin and IV iron was compared with IV iron alone in two studies with a total of 100 women (Krafft 2011; Wagstrom 2007). In the trial by Wagstrom 2007, EPO was given subcutaneously (SC) in two different doses in two different EPO groups (total of 40,000 U and 20,000 U). The EPO group with a total dose of 20,000 U was analysed separately. In Krafft 2011, EPO was given IV. Follow‐up was two weeks in both studies.
Erythropoietin combined with IV iron followed by oral iron was compared with IV iron alone followed by oral iron in three studies with a total of 186 women (Breymann 1996; Breymann 2000; Lebrecht 1995). Two of the studies had three study arms (Breymann 1996; Breymann 2000). In one study EPO was given either SC or IV (Breymann 1996), and one study also had a study arm that only received oral iron (Breymann 2000). We compared study arms with similar treatment across studies.
Subcutaneous EPO and oral iron were compared with oral iron in one study with 40 women (Makrydimas 1998). Follow‐up was 40 days.
Intravenous EPO was compared with placebo, without iron supplementation in one study with 71 women (Meyer 1995). Follow‐up was five days.
Outcomes
All of the included publications reported at least one clinical outcome measure that was preplanned for this review. These 22 publications also reported laboratory values such as Hb, ferritin or others.
Of all included studies, six reported on maternal mortality (Breymann 2000; Guerra 2012; Krafft 2011; Lebrecht 1995; Makrydimas 1998; Van Wyck 2007), three on fatigue (Prick 2014; Van Wyck 2007; Westad 2008), three on anaemia symptoms (Perello 2014; Prick 2014; Tam 2005), seven on psychological well being (Beard 2005; Meyer 1995; Perello 2014; Prick 2014; Van Wyck 2007; Wagstrom 2007; Westad 2008), six on infections (Breymann 2008; Guerra 2012; Krafft 2011; Prick 2014; Van Wyck 2007; Wagstrom 2007), nine on compliance (Bhandal 2006; Breymann 2008; Guerra 2012; Jain 2013; Krafft 2011; Prick 2014; Van Wyck 2007; Verma 2011; Westad 2008), four on breastfeeding (Krafft 2011; Makrydimas 1998; Prick 2014; Tam 2005), four on length of hospital stay (Makrydimas 1998; Perello 2014; Prick 2014; Verma 2011), and 20 on adverse events during treatment. The studies that did not report on adverse events were Beard 2005 and Meyer 1995. Eleven studies reported on the use of blood transfusions as a rescue treatment (Bhandal 2006; Breymann 1996; Breymann 2000; Breymann 2008; Froessler 2013; Krafft 2011; Makrydimas 1998; Perello 2014; Prick 2014; Wagstrom 2007; Westad 2008).
We chose not to consider placebo treatment as a type of intervention, based on the lack of evidence for a substantial placebo effect (Hróbjartsson 2010). Groups with inactive placebo were therefore considered comparable with groups not receiving treatment. Also, we chose not to distinguish between SC and IV EPO administration, as we did not expect the effect to be influenced by the route of administration.
This allowed five comparisons based on interventions with more than one study. The rest of the studies and the remaining study arms were analysed separately. Thus, a total of 13 comparisons were conducted in this review.
The included studies are described in detail in the Characteristics of included studies tables. Only our preplanned outcomes chosen for this review were described and analysed.
Excluded studies
We excluded 17 studies. Reasosns for exclusion were inadequate randomisation methods, mixed anaemic and non‐anaemic population without subgroup analysis, summary of two included and one excluded study, analyses based on both antepartum and postpartum anaemia, no definition of the postpartum period (thus including women enrolled more than six weeks postpartum), lack of a control arm, investigation of differences in screening strategies rather than different interventions, and interventions found as not appropriate for treatment of iron deficiency anaemia. For further details, please see Characteristics of excluded studies.
Risk of bias in included studies
The 'Risk of bias' assessment is summarised in Figure 2 and Figure 3.
2.
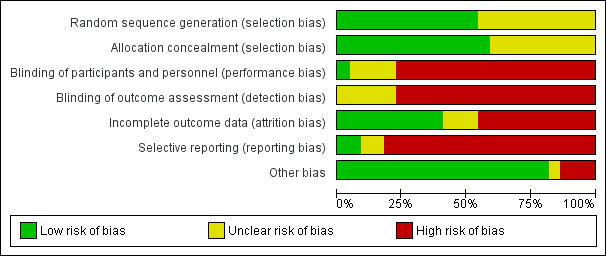
'Risk of bias' graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
3.
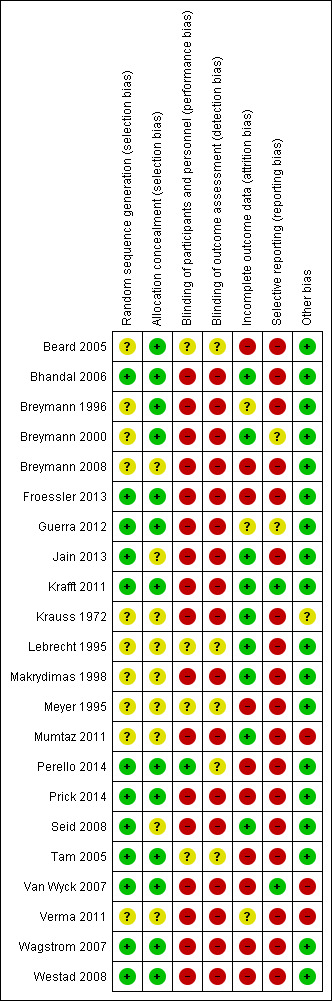
'Risk of bias' summary: review authors' judgements about each risk of bias item for each included study.
Allocation
Random sequence generation
Low risk of bias was found in 12 studies (Bhandal 2006; Froessler 2013; Guerra 2012; Jain 2013; Krafft 2011; Perello 2014; Prick 2014; Seid 2008; Tam 2005; Van Wyck 2007; Wagstrom 2007; Westad 2008). Ten studies had unclear risk of bias, as the random sequence generation method was not described (Beard 2005; Breymann 1996; Breymann 2000; Breymann 2008; Krauss 1972; Lebrecht 1995; Makrydimas 1998; Meyer 1995; Mumtaz 2011; Verma 2011).
Allocation concealment
Allocation concealment was adequately described in 13 studies (Beard 2005; Bhandal 2006; Breymann 1996; Breymann 2000; Froessler 2013; Guerra 2012; Krafft 2011; Perello 2014; Prick 2014; Tam 2005;Van Wyck 2007;Wagstrom 2007; Westad 2008). The method was not described and thus the risk of bias was unclear in the remaining nine studies (Breymann 2008; Jain 2013; Krauss 1972; Lebrecht 1995; Makrydimas 1998; Meyer 1995; Mumtaz 2011; Seid 2008; Verma 2011).
Blinding
Performance bias
In one study, the method of blinding was adequately described and it was clear who was blinded (Perello 2014).
Six studies had an unclear risk of bias: two placebo‐controlled studies described the blinding method as double‐blind, but it was unclear who was blinded (Lebrecht 1995; Meyer 1995). In the study reported by Beard 2005 it was unclear if all treatment components (iron, folate, vitamin C) were prepared in a single tablet and whether this tablet resembled the placebo tablet. In Tam 2005 it was reported that although the trial was double‐blinded, the participants reported stool discolorations when receiving active treatment. The majority of studies were open‐label due to different non‐blinded administration routes and thus considered at high risk (Bhandal 2006; Breymann 1996; Breymann 2000; Breymann 2008; Froessler 2013; Guerra 2012; Jain 2013; Krafft 2011; Krauss 1972; Makrydimas 1998; Mumtaz 2011; Prick 2014; Seid 2008; Van Wyck 2007; Verma 2011; Wagstrom 2007; Westad 2008).
Detection bias
Seven studies were rated as having unclear risk of bias: in two studies described as double‐blinded it was not described who was blinded (Lebrecht 1995; Meyer 1995). In Perello 2014, it was not clear whether personnel who handled self‐rated questionnaires were blinded. In Tam 2005 it was unclear whether the women were able to guess their treatment based on the change in stool colour. The outcomes in this trial were subjective and reported by the women. The risk of detection bias therefore depends on the women's knowledge of the correlation between iron treatment and stool discolouration and the clinician's knowledge of the discolouration at the time when the remaining outcomes were registered. This was not described and the risk of bias was therefore rated as unclear.
In the study reported by Beard 2005, it was not clear from study description who exactly was blinded during the trial and whether the placebo tablet and treatment were sufficiently similar to prevent the patients from guessing the group. Thus the subjective (patient registered) outcome of psychological well being may have been affected by insufficient blinding.
High risk of bias was found in seventeen studies due to open‐label trial design (Bhandal 2006; Breymann 1996; Breymann 2000; Breymann 2008; Froessler 2013; Guerra 2012; Jain 2013; Krafft 2011; Krauss 1972; Makrydimas 1998; Mumtaz 2011; Prick 2014; Seid 2008; Van Wyck 2007; Verma 2011; Wagstrom 2007; Westad 2008). Maternal mortality is one of the few outcome measures which is most probably not affected by a lack of blinding. However, this outcome was rarely reported.
Incomplete outcome data
Information on dropouts and withdrawals after randomisation was reported in 19 studies (Beard 2005; Bhandal 2006; Breymann 2000; Breymann 2008; Froessler 2013; Guerra 2012; Jain 2013; Krafft 2011; Krauss 1972; Lebrecht 1995; Makrydimas 1998; Mumtaz 2011; Perello 2014; Prick 2014; Seid 2008; Tam 2005; Van Wyck 2007; Wagstrom 2007; Westad 2008). Three trial authors provided additional information on dropout rates (Froessler 2013; Van Wyck 2007; Wagstrom 2007).
Dropout varied greatly across studies. Dropout rates after randomisation were lower than 5% in six studies (Bhandal 2006; Breymann 2000; Krafft 2011; Lebrecht 1995; Makrydimas 1998; Seid 2008), between 5% and 9.9% in three studies (Krauss 1972;Mumtaz 2011; Van Wyck 2007), between 10% and 19.9% in six studies (Froessler 2013; Guerra 2012; Jain 2013; Perello 2014; Tam 2005; Wagstrom 2007), and 20% or more in four studies (Beard 2005; Breymann 2008; Prick 2014; Westad 2008). However, for Beard 2005, the missing data were given as lost to follow‐up, not as discontinuation of treatment. The numbers are therefore very high and probably overestimate the actual dropout rate. Three studies did not report sufficient information to calculate the dropout rate after randomisation (Breymann 1996; Meyer 1995; Verma 2011).
Low risk of bias was found in nine studies with a low dropout rate or an equal distribution of dropouts across groups (Bhandal 2006; Breymann 2000; Jain 2013; Krafft 2011; Krauss 1972; Lebrecht 1995; Makrydimas 1998; Mumtaz 2011; Seid 2008).
High risk of bias was found in 10 studies, due to a high dropout rate and/or unequal distribution across groups (Beard 2005; Breymann 2008; Froessler 2013; Meyer 1995; Perello 2014; Prick 2014; Tam 2005; Van Wyck 2007; Wagstrom 2007; Westad 2008).
An unclear risk of attrition bias was found in three studies. In one study it was not possible to assess if the dropouts were in fact not related to the trial (Guerra 2012). In three studies it was not mentioned whether or not any patients dropped out after randomisation (Breymann 1996; Verma 2011).
Selective reporting
We applied strict criteria when evaluating reporting bias because we consider mortality and adverse events as extremely important outcomes, and as per our method section we rated the studies as high risk if the study failed to include results of a key outcome that would have been expected to be reported (i.e. mortality). Therefore, only two studies were rated as having low risk of reporting bias (Krafft 2011; Van Wyck 2007).
Often the trial authors stated that the objectives of their trial were efficacy and safety, but did not specify which preplanned outcome measures were going to be used to evaluate efficacy. Two studies were rated as having unclear risk of bias because no preplanned outcomes were specified (Breymann 2000; Guerra 2012).
High risk of bias was found in 18 studies: 16 of these did not report on adverse events and/or maternal mortality (Beard 2005; Bhandal 2006; Breymann 1996; Breymann 2008; Froessler 2013; Jain 2013; Krauss 1972; Meyer 1995; Mumtaz 2011; Perello 2014; Prick 2014; Seid 2008; Tam 2005; Verma 2011; Wagstrom 2007; Westad 2008), and in two studies there was a lack of data to support their conclusions on quality of life (Lebrecht 1995; Makrydimas 1998). The study by Verma 2011 did not report on the following preplanned outcomes which were stated in the 'aims and objectives' section of the published report: patient satisfaction, quality of life, cost of treatment, length of hospital stay, use of blood transfusion, impact on stress, depression, cognitive function, and breastfeeding.
Other potential sources of bias
One study was found to have unclear risk of bias because the Hb level for inclusion was not stated (Krauss 1972). Three studies were found to have a high risk of bias because of significant errors in the published reports (Mumtaz 2011; Van Wyck 2007; Verma 2011). For further description, please see Characteristics of included studies.
Effects of interventions
See: Table 1; Table 2; Table 3; Table 4; Table 5; Table 6; Table 7; Table 8
Summary of findings for the main comparison. Intravenous iron compared with oral iron for women with postpartum iron deficiency anaemia (Comparison 1).
| Intravenous iron compared with oral iron for women with postpartum iron deficiency anaemia | ||||||
| Patient or population: women with postpartum iron deficiency anaemia Settings: obstetric care units Intervention: intravenous iron Comparison: oral iron | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Oral iron | Intravenous iron | |||||
| Maternal mortality Clinical assessment Follow‐up: mean 42 days | Study population | RR 2.95 (0.12 to 71.96) | 374 (2 studies) | ⊕⊕⊝⊝ low1,2,3,4 | 1 maternal death was reported across the included studies. | |
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| Moderate | ||||||
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| Fatigue at 14, 28, and 42 days Fatigue Linear Analog Scale Assessment. Scale from: 0 to 100. Follow‐up: 14‐42 days | See comment | See comment | Not estimable | 361 (1 study) | ⊕⊝⊝⊝ very low3,5,6 | No statistically significant difference was found at days 14 and 42 days. |
| Persistent anaemia symptoms ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | Not reported. |
| Infections Clinical assessment Follow‐up: mean 41 days | Study population | RR 1.7 (0.58 to 5.03) | 718 (3 studies) | ⊕⊝⊝⊝ very low1,4,7 | ||
| 86 per 1000 | 146 per 1000 (50 to 432) | |||||
| Moderate | ||||||
| 34 per 1000 | 58 per 1000 (20 to 171) | |||||
| Constipation Reported by the women Follow‐up: mean 46 days | Study population | RR 0.21 (0.11 to 0.39) | 1217 (6 studies) | ⊕⊝⊝⊝ very low4,5 | ||
| 114 per 1000 | 24 per 1000 (13 to 44) | |||||
| Moderate | ||||||
| 112 per 1000 | 24 per 1000 (12 to 44) | |||||
| All gastrointestinal symptoms Reported by the women Follow‐up: mean 42 days | Study population | RR 0.31 (0.2 to 0.47) | 1307 (8 studies) | ⊕⊝⊝⊝ very low4,5 | ||
| 216 per 1000 | 67 per 1000 (43 to 102) | |||||
| Moderate | ||||||
| 261 per 1000 | 81 per 1000 (52 to 123) | |||||
| Anaphylaxis or evidence of hypersensitivity Clinical assessment Follow‐up: mean 40 days | Study population | RR 2.78 (0.31 to 24.92) | 1454 (8 studies) | ⊕⊕⊝⊝ low1,2,4 | 3 cases of allergic reactions all occurred in the group treated with intravenous iron. | |
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| Moderate | ||||||
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 The outcome is unlikely to be influenced by risk of bias and so we did not downgrade the evidence for this outcome: open‐label design combined with a objective outcome measure. 2 Downgraded one level due to imprecision: small sample size, few events, broad confidence intervals: likely to lower confidence in effect. 3 Downgraded one level due to risk of bias: at least 1 study suitable for this comparison was terminated by trial sponsors. This trial had fatigue as a pre‐planned outcome. This raises serious concern on the amount of unpublished results which may have been unfavourable to trial sponsors. 4 Downgraded one level due to risk of bias: several studies did not report important harms. 5 Downgraded two levels due to risk of bias: open‐label design combined with a subjective outcome measure. 6 Downgraded one level due to imprecision: broad confidence intervals for raw means and small sample size: likely to lower confidence in effect. 7 Downgraded one level due to inconsistency: significant statistical heterogeneity: I2 = 72%.
Summary of findings 2. Red blood cell transfusion compared with non‐transfusion (Comparison 2).
| Red blood cell transfusion compared with non‐transfusion for postpartum iron deficiency anaemia | ||||||
| Patient or population: patients with postpartum iron deficiency anaemia Settings: obstetric care unit Intervention: red blood cell transfusion Comparison: non‐transfusion | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Non‐transfusion | RBC transfusion | |||||
| Maternal mortality ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | Not reported. |
| Fatigue Multidimensional Fatigue Inventory. Scale from: 4 to 20. Follow‐up: 3‐42 days | See comment | See comment | 519 (1 study) | ⊕⊕⊝⊝ low1 | General fatigue at 3 days was 0.8 lower (1.53 to 0.07) in the transfused group. No statistically significant difference was seen at six weeks. | |
| Persistent anaemia symptoms Reported by the women Follow‐up: mean 42 days | Study population | Not estimable | 519 (1 study) | ⊕⊝⊝⊝ very low1,2 | The outcome was not systematically registered/reported. | |
| See comment | See comment | |||||
| Moderate | ||||||
| Infections Clinical assessment Follow‐up: mean 42 days | Study population | RR 0.93 (0.53 to 1.61) | 519 (1 study) | ⊕⊕⊕⊝ moderate3 | ||
| 92 per 1000 | 86 per 1000 (49 to 148) | |||||
| Moderate | ||||||
| 92 per 1000 | 86 per 1000 (49 to 148) | |||||
| Erythrocyte alloantibody formation Laboratory assessment Follow‐up: mean 42 days | Study population | RR 3.03 (0.12 to 74.15) | 519 (1 study) | ⊕⊝⊝⊝ very low3,4,5 | There was no systematical screening for this outcome in the study population. | |
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| Moderate | ||||||
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| Thromboembolic events Assessment method not described Follow‐up: mean 42 days | Study population | RR 1.01 (0.14 to 7.13) | 519 (1 study) | ⊕⊕⊝⊝ low6,7 | ||
| 8 per 1000 | 8 per 1000 (1 to 55) | |||||
| Moderate | ||||||
| 8 per 1000 | 8 per 1000 (1 to 57) | |||||
| Transfusion reactions Clinical assessment Follow‐up: mean 42 days | Study population | RR 7.08 (0.37 to 136.41) | 519 (1 study) | ⊕⊝⊝⊝ very low3,5 | 3 cases of transfusion reactions occurred in the transfusion group. | |
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| Moderate | ||||||
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Downgraded two levels due to risk of bias: open‐label design combined with a subjective outcome measure. 2 Downgraded one level due to study limitations: the outcome was not systematically registered/reported. 3 The outcome is unlikely to be influenced by risk of bias and so we did not downgrade the evidence for this outcome: open‐label design combined with a objective outcome measure. 4 Downgraded one level due to study limitations: the women were not systematically screened for the presence of antibodies. 5 Downgraded two levels due to imprecision: very broad confidence interval. 6 Downgraded one level due to risk of bias: open‐label study, method for detection not descried. 7 Downgraded one level due to imprecision: broad confidence interval.
Summary of findings 3. Oral iron compared with placebo (Comparison 3).
| Oral iron compared with placebo for women with postpartum iron deficiency anaemia | ||||||
| Patient or population: women with postpartum iron deficiency anaemia Settings: obstetric care units Intervention: oral iron Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Oral iron | |||||
| Maternal mortality ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | Not reported. |
| Fatigue ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | Not reported. |
| Persistent anaemia symptoms Reported by the women Follow‐up: mean 42 days | Study population | Not estimable | (1) | See comment | Symptoms of anaemia were not reported for the anaemic groups separately. | |
| See comment | See comment | |||||
| Moderate | ||||||
| All gastrointestinal symptoms Reported by the patients Follow‐up: mean 30 days | Study population | RR 1 (0.36 to 2.79) | 68 (1 study) | ⊕⊝⊝⊝ very low1,2,3 | ||
| 176 per 1000 | 176 per 1000 (64 to 492) | |||||
| Moderate | ||||||
| 177 per 1000 | 177 per 1000 (64 to 494) | |||||
| Constipation ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | Not reported. |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Downgraded two levels due to risk of bias: open‐label design combined with a subjective outcome measure. 2 Downgraded one level due to imprecision: small sample size, single study ‐ likely to lower confidence in effect. 3 Downgraded one level due to study limitations: adverse events not reported separately.
Summary of findings 4. Intravenous iron with oral iron compared with oral iron (Comparison 6).
| Intravenous iron with oral iron compared with oral iron for women with postpartum iron deficiency anaemia | ||||||
| Patient or population: women with postpartum iron deficiency anaemia Settings: obstetric care unit Intervention: intravenous iron with oral iron Comparison: oral iron | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Oral iron | Intravenous iron with oral iron | |||||
| Maternal mortality | See comment | See comment | Not estimable | ‐ | See comment | In 1 study no maternal deaths were reported. The other study did not report on maternal mortality. |
| Fatigue ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | Not reported. |
| Persistent anaemia symptoms ‐ 1 week Visual Analogue Scale ≥ 7 Follow‐up: mean 7 days | Study population | RR 1.75 (0.56 to 5.46) | 72 (1 study) | ⊕⊝⊝⊝ very low1,2 | ||
| 111 per 1000 | 194 per 1000 (62 to 607) | |||||
| Moderate | ||||||
| 111 per 1000 | 194 per 1000 (62 to 606) | |||||
| Persistent anaemia symptoms ‐ 2 weeks Visual Analogue Scale ≥ 7 Follow‐up: mean 14 days | Study population | RR 0.6 (0.15 to 2.33) | 72 (1 study) | ⊕⊝⊝⊝ very low1,2 | ||
| 139 per 1000 | 83 per 1000 (21 to 324) | |||||
| Moderate | ||||||
| 139 per 1000 | 83 per 1000 (21 to 324) | |||||
| Persistent anaemia symptoms ‐ 6 weeks Visual Analogue Scale ≥ 7 Follow‐up: mean 42 days | Study population | RR 3 (0.33 to 27.5) | 72 (1 study) | ⊕⊝⊝⊝ very low1,2 | ||
| 28 per 1000 | 83 per 1000 (9 to 764) | |||||
| Moderate | ||||||
| 28 per 1000 | 84 per 1000 (9 to 770) | |||||
| Infections ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | Not reported. |
| Anaphylaxis or evidence of hypersensitivity Clinical assessment Follow‐up: mean 28 days | Study population | Not estimable | 0 (1 study) | ⊕⊕⊝⊝ low1 | 1 study reported 0 events, other study pooled adverse events, not reporting allergic reactions separately. Thus the effect was not estimable. | |
| See comment | See comment | |||||
| Moderate | ||||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Downgraded two levels due to risk of bias: the included study had high risk of attrition and reporting bias. 2 Downgraded one level due to imprecision: small sample size, single study.
Summary of findings 5. Erythropoietin (regardless of rout) with intravenous iron compared with intravenous iron (Comparison 7).
| Erythropoietin (regardless of rout) with intravenous iron compared with intravenous iron for women with postpartum iron deficiency anaemia | ||||||
| Patient or population: women with postpartum iron deficiency anaemia Settings: obstetric care units Intervention: erythropoietin (regardless of rout) with intravenous iron Comparison: intravenous iron | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Intravenous iron | EPO (regardless of rout) with IV iron | |||||
| Maternal mortality | See comment | See comment | Not estimable | ‐ | See comment | In 1 study no maternal deaths were reported. The other study did not report on maternal mortality. |
| Fatigue ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | Not reported. |
| Thromboembolic events ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | Not reported. |
| Persistent anaemia symptoms ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | Not reported. |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
Summary of findings 6. Subcutaneous EPO 10,000 U of doses with intravenous iron compared with intravenous iron (Comparison 8).
| Subcutaneous EPO 10,000 U of doses with intravenous iron compared with intravenous iron for women with postpartum iron deficiency anaemia | ||||||
| Patient or population: patients with women with postpartum iron deficiency anaemia Settings: obstetric care unit Intervention: subcutaneous EPO of 2 doses of 10,000 U with intravenous iron Comparison: intravenous iron | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Intravenous iron | Erythropoietin 10,000 U 2 doses with intravenous iron | |||||
| Maternal mortality ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | Not reported. |
| Fatigue ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | Not reported. |
| Persistent anaemia symptoms ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | Not reported. |
| Thromboembolic events ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | Not reported. |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
Summary of findings 7. Subcutaneous EPO with oral iron compared with oral iron (Comparison 10).
| Subcutaneous EPO with oral iron compared with oral iron for women with postpartum iron deficiency anaemia | ||||||
| Patient or population: women with postpartum iron deficiency anaemia Settings: obstetric care unit Intervention: subcutaneous EPO with oral iron Comparison: oral iron | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Oral iron | Subcutaneous EPO with oral iron | |||||
| Maternal mortality | See comment | See comment | Not estimable | 40 (0) | See comment | No maternal deaths were reported. |
| Fatigue ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | Not reported. |
| Persistent anaemia symptoms ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | Not reported. |
| Thromboembolic events ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | Not reported. |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
Summary of findings 8. Subcutaneous EPO with intravenous iron and oral iron compared with intravenous iron with oral iron (Comparison 12).
| Subcutaneous EPO with IV iron and oral iron compared with intravenous iron with oral iron for women with postpartum iron deficiency anaemia | ||||||
| Patient or population: women with postpartum iron deficiency anaemia Settings: obstetric care units Intervention: subcutaneous EPO with intravenous iron and oral iron Comparison: intravenous iron with oral iron | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Intravenous iron + oral iron | Subcutaneous EPO + IV iron + oral iron | |||||
| Maternal mortality ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | Not reported. |
| Fatigue ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | Not reported. |
| Persistent anaemia symptoms ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | Not reported. |
| Thromboembolic events ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | Not reported. |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
Comparison 1: IV iron versus oral iron
Intravenous (IV) iron treatment was compared with oral iron in 10 studies with a total of 1553 women (Bhandal 2006; Breymann 2008; Froessler 2013; Guerra 2012; Jain 2013; Mumtaz 2011; Seid 2008; Van Wyck 2007; Verma 2011; Westad 2008). IV iron was in the form of either iron sucrose (seven studies) or iron‐carboxymaltose (three studies). Doses differed across the trials with a range of 300 mg to 2500 mg in total dose. In several studies doses were individually calculated using the Ganzoni formula, estimating the iron deficit in each patient. Oral iron was given as ferrous sulphate typically using a fixed dose. The content of elemental iron (the dose of the pure iron ion in the iron sulphate tablet) was rarely reported. Treatment regimens differed between studies with regard to doses, number of tablet per day and number of days of treatment. Non‐elemental iron doses ranged from 100 mg to 325 mg per tablet.
Primary outcomes
Maternal mortality
Maternal mortality was only reported by two studies. There was one maternal death in the group receiving IV iron caused by peripartum cardiomyopathy 13 days postpartum, thus it is not clear whether this death was directly caused by the study medication (Van Wyck 2007). A corresponding author from one other study reported that no women died (Guerra 2012) (risk ratio (RR) 2.95; 95% confidence interval (CI) 0.12 to 71.96; two RCTs; one event; 374 women; low quality evidence; Analysis 1.1). In the remaining studies this information was not clear as per our definition in the Primary outcomes section.
1.1. Analysis.
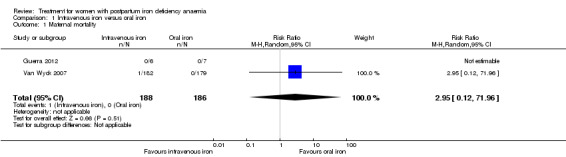
Comparison 1 Intravenous iron versus oral iron, Outcome 1 Maternal mortality.
Fatigue
Fatigue was reported by two studies, and a meta‐analysis was not possible due to lack of data. One study reported a statistically significant improvement in fatigue in the group receiving IV treatment (see below) (Westad 2008). The other study showed no difference in fatigue (see below) (Van Wyck 2007).
Van Wyck 2007 used the Fatigue Linear Analog Scale Assessment (Portenoy 2006) for a mean total fatigue score. Westad 2008 used the Fatigue Score (Chalder 1993), where the scores were reported as mean change from baseline for physical, mental and total fatigue. It was not possible to obtain standard deviations from Westad 2008 (standard deviations were only available for baseline data), and thus we could not perform a meta‐analysis. In both studies the higher score indicated higher level of fatigue. In the trial by Westad 2008, all women received oral iron after four weeks. Results by weeks eight and 12 are described in Comparison 5.
In the published paper by Westad 2008, the authors state that they found a statistically significant improvement in the 'physical fatigue' and 'total fatigue' scores favouring the IV iron group at four weeks with a P value of 0.02 for both scores. They found no between‐group difference in the 'mental fatigue' score.
Van Wyck 2007 provided raw means data on our request. There was no statistically significant differences between groups at 14 days (short term) or 42 days (long term) (very low quality evidence; Analysis 1.2; Analysis 1.3).
1.2. Analysis.
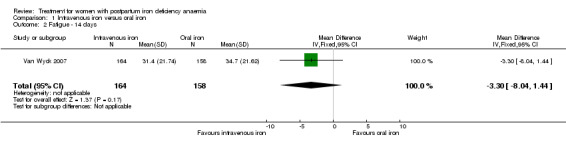
Comparison 1 Intravenous iron versus oral iron, Outcome 2 Fatigue ‐ 14 days.
1.3. Analysis.
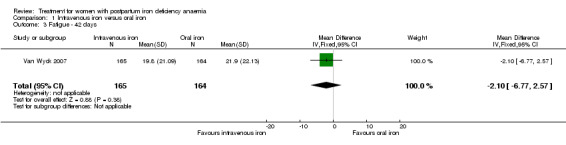
Comparison 1 Intravenous iron versus oral iron, Outcome 3 Fatigue ‐ 42 days.
Secondary outcomes
Anaemia symptoms
Anaemia symptoms (other than fatigue) were not reported.
Psychological well being
Psychological well being was reported by two studies (Van Wyck 2007; Westad 2008). Both used the SF‐36 questionnaire, where higher scores indicate better health state (Ware 2000). There was no overall difference in psychological well being (see below).
It was not possible to carry out a meta‐analyses as standard deviations were only available for baseline data for the study by Westad 2008. This study reported only on four out of eight SF‐36 items. In the published report the authors found no significant between‐group difference at week four.
Van Wyck 2007 provided raw means on all eight items of the SF‐36, thus 'physical function', 'physical role', 'bodily pain', 'social function', 'mental health', 'general health', 'vitality', and 'emotional role'. From the additional data provided, we found no statistically significant difference between the groups at 14 days (Analysis 1.4 to Analysis 1.11).
1.4. Analysis.
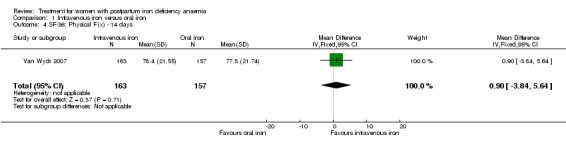
Comparison 1 Intravenous iron versus oral iron, Outcome 4 SF‐36: Physical F(x) ‐ 14 days.
1.11. Analysis.
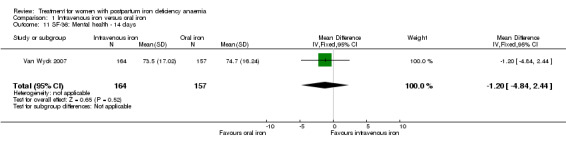
Comparison 1 Intravenous iron versus oral iron, Outcome 11 SF‐36: Mental health ‐ 14 days.
There was no difference in the occurrence of depression (Analysis 1.12).
1.12. Analysis.
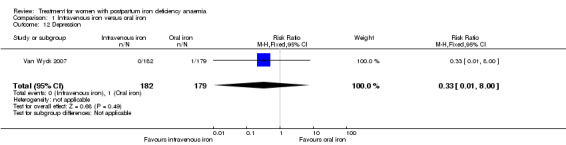
Comparison 1 Intravenous iron versus oral iron, Outcome 12 Depression.
Infections
Infections were analysed as a total for each group based on the assumption that if anaemia can cause immune deficiency, and bioavailable iron can supply microorganisms with nutrition, infections could occur anywhere in body. The results were divergent: In the study by Breymann 2008 infections were more frequent in the IV iron group, whereas there was no difference in the study by Van Wyck 2007. Our analysis found no statistically significant difference in infections (RR 1.70; 95% CI 0.58 to 5.03; three RCTs; 718 women; I² 72 %; T² 0.45; Chi² 3.56; P 0.06; very low quality evidence; Analysis 1.13). We conducted a sensitivity analysis to investigate the high level of heterogeneity in which the difference remained statistically non‐significant after we subtracted the study causing heterogeneity (Analysis 14.1), and when we used fixed‐effect meta‐analysis (Analysis 14.2).
1.13. Analysis.
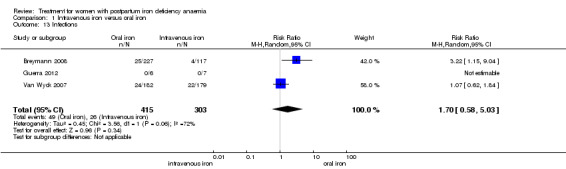
Comparison 1 Intravenous iron versus oral iron, Outcome 13 Infections.
14.1. Analysis.
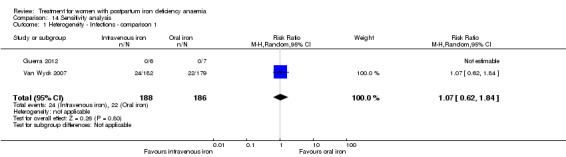
Comparison 14 Sensitivity analysis, Outcome 1 Heterogeneity ‐ Infections ‐ comparison 1.
14.2. Analysis.
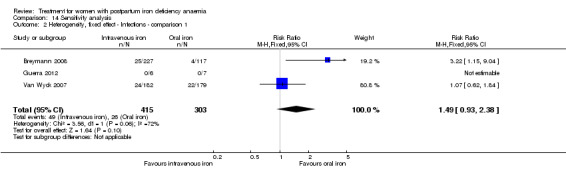
Comparison 14 Sensitivity analysis, Outcome 2 Heterogeneity, fixed effect ‐ Infections ‐ comparison 1.
Compliance
Compliance was reported in seven studies (Bhandal 2006; Breymann 2008; Guerra 2012; Jain 2013; Van Wyck 2007; Verma 2011; Westad 2008). Bhandal 2006 and Guerra 2012 reported 100% compliance in both groups. However, Guerra 2012 did not count the remaining pills. In the study by Breymann 2008, compliance in the group receiving IV iron was 99% and over 90% in the group receiving oral iron. Westad 2008 reported a compliance of 95% specifically for IV injections in the group receiving IV iron. The mean daily intake of oral iron was 99 mg by week four, resulting in 50% compliance in the oral group. Van Wyck 2007 reported a compliance of 98% in the group receiving IV iron and 83.9% in the group receiving oral iron. Jain 2013 reported the group receiving oral iron to have a 100% compliance confirmed by pill count, but compliance for the group receiving IV iron was not specified. Verma 2011 mentioned that compliance was better in the group receiving IV iron than in the group receiving oral iron, but data were not available. Thus, compliance was 95% to 100% for IV iron, and 50% to 100% for oral iron (RR 1.17; 95% CI 1.01 to 1.35; five RCTs; 890 women, I² 90 %; T² 0.02; Chi² 38.44; P < 0.00001; Analysis 1.14).
1.14. Analysis.
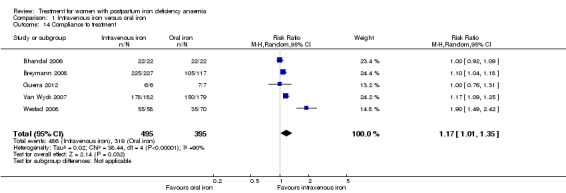
Comparison 1 Intravenous iron versus oral iron, Outcome 14 Compliance to treatment.
Breastfeeding
Breastfeeding was not reported.
Length of hospital stay
Length of hospital stay was generally not reported. Verma 2011 noted that hospital stays were longer in the IV group, but data were not available.
Adverse events during treatment
Three women experienced anaphylaxis or hypersensitivity, all of whom received IV iron. However, there were few events and a reliable absolute risk estimate could therefore not be calculated (RR 2.78; 95% CI 0.31 to 24.92; eight RCTs; three events; 1454 women (767 in the IV arm versus 687 in the oral arm); I² = 0%; low quality evidence; Analysis 1.32.
1.32. Analysis.
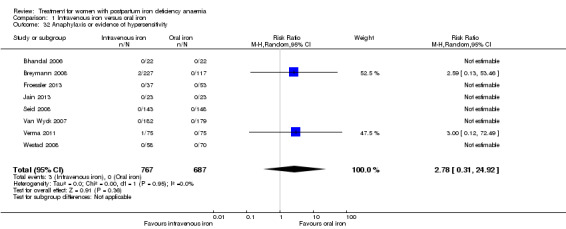
Comparison 1 Intravenous iron versus oral iron, Outcome 32 Anaphylaxis or evidence of hypersensitivity.
One woman who received IV iron developed an arrhythmia during iron infusion (Analysis 1.33).
1.33. Analysis.
Comparison 1 Intravenous iron versus oral iron, Outcome 33 Arythmia.
There was a statistically significant difference in the risk of all combined gastrointestinal (GI) adverse events favouring the group receiving IV iron (RR 0.31; 95% CI 0.20 to 0.47; eight RCTs; 169 events; 1307 women; I² = 0%; very low quality evidence; Analysis 1.15).
1.15. Analysis.
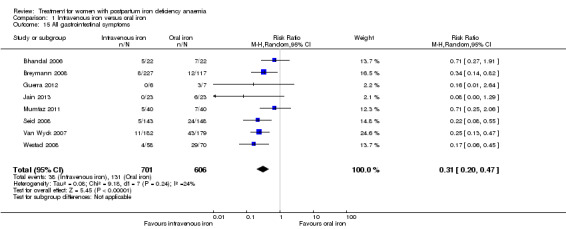
Comparison 1 Intravenous iron versus oral iron, Outcome 15 All gastrointestinal symptoms.
The GI symptoms that were significantly less frequent in the IV iron group were: constipation (RR 0.21; 95% CI 0.11 to 0.39; six RCTs; 74 events; 1217 women; I² = 0%; very low quality evidence; Analysis 1.16), nausea (RR 0.30; 95% CI 0.11 to 0.81; four RCTs; 22 events; 745 women; I² = 0%; Analysis 1.17), GI pain (RR 0.18; 95% CI 0.04 to 0.83; four RCTs; 13 events; 543 women; I² = 0%; Analysis 1.18), and diarrhoea (RR 0.11; 95% CI 0.02 to 0.59; three RCTs; 14 events; 569 women; I² = 0%; Analysis 1.19).
1.16. Analysis.
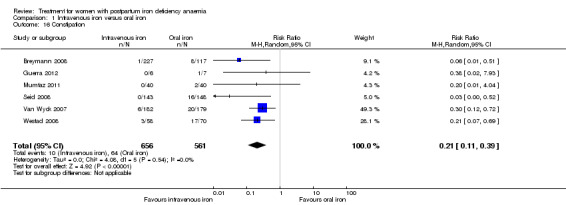
Comparison 1 Intravenous iron versus oral iron, Outcome 16 Constipation.
1.17. Analysis.
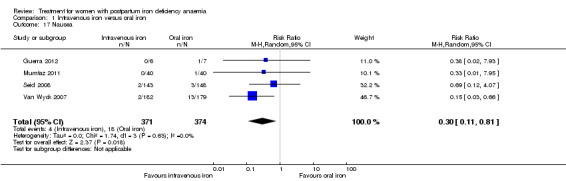
Comparison 1 Intravenous iron versus oral iron, Outcome 17 Nausea.
1.18. Analysis.
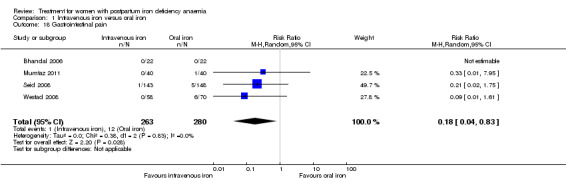
Comparison 1 Intravenous iron versus oral iron, Outcome 18 Gastrointestinal pain.
1.19. Analysis.
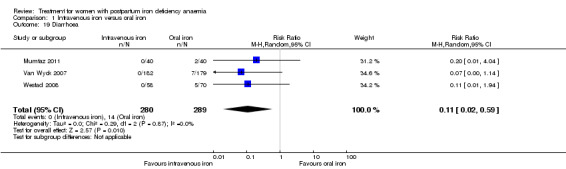
Comparison 1 Intravenous iron versus oral iron, Outcome 19 Diarrhoea.
There was no difference for the occurrence of vomiting (RR 0.40; 95% CI 0.02 to 9.66; one RCT, 128 women; Analysis 1.20) or dyspepsia (RR 0.36; 95% CI 0.04 to 3.20; two RCTs; 93 women; Analysis 1.21).
1.20. Analysis.
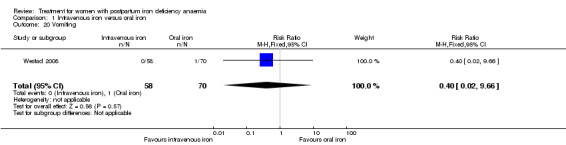
Comparison 1 Intravenous iron versus oral iron, Outcome 20 Vomiting.
1.21. Analysis.
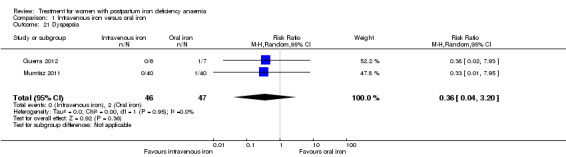
Comparison 1 Intravenous iron versus oral iron, Outcome 21 Dyspepsia.
In the group receiving IV iron we found an increased risk of dysgeusia (distortion of the sense of taste) (RR 7.20; 95% CI 1.63 to 31.76; four RCTs; 13 events; 543 women; I² = 0%; Analysis 1.22), injection site discomfort (RR 4.72; 95% CI 1.03 to 21.54; four RCTs; 10 events; 702 women; I² = 0%; Analysis 1.25), and flush (RR 9.00; 95% CI 1.18 to 68.81; two RCTs, eight events; 124 women; I² = 0%; Analysis 1.28).
1.22. Analysis.
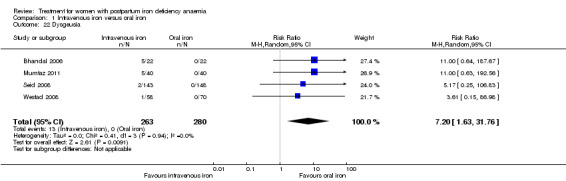
Comparison 1 Intravenous iron versus oral iron, Outcome 22 Dysgeusia.
1.25. Analysis.
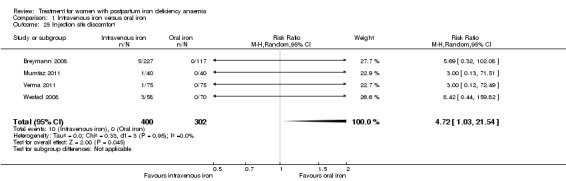
Comparison 1 Intravenous iron versus oral iron, Outcome 25 Injection site discomfort.
1.28. Analysis.
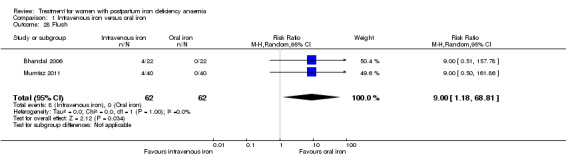
Comparison 1 Intravenous iron versus oral iron, Outcome 28 Flush.
There was no statistically significant difference between IV iron and oral iron regarding other adverse events including headache (RR 1.93; 95% CI 0.87 to 4.29; four RCTs; 1124 women; I² = 0%; Analysis 1.23), skin rash (RR 2.34; 95% CI 0.79 to 6.97; two RCTs; 489 women; I² = 0%; Analysis 1.26), muscle cramps (RR 6.05; 95% CI 0.74 to 49.68; two RCTs, 371 women; I² = 0%; Analysis 1.29), and hepatic involvement (RR 0.45; 95% CI 0.12 to 1.71; three RCTs; 996 women; I² = 51%; T² 0.70; Chi² 4.07; P 0.13; Analysis 1.24). In the analyses for hepatic involvement there was high heterogeneity, therefore we conducted a sensitivity analysis. The difference between groups became statistically significant in favour of the IV group when we removed the study causing heterogeneity (Breymann 2008) (RR 0.22; 95% CI 0.06 to 0.75; two RCTs; 652 women; I² = 0%; Analysis 14.3). The difference was statistically non‐significant when we changed to fixed‐effect meta‐analysis (Analysis 14.4).
1.23. Analysis.
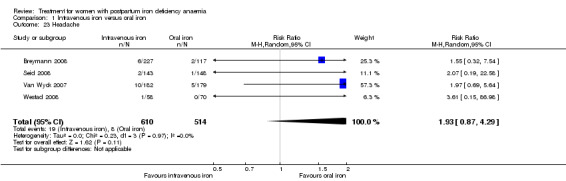
Comparison 1 Intravenous iron versus oral iron, Outcome 23 Headache.
1.26. Analysis.
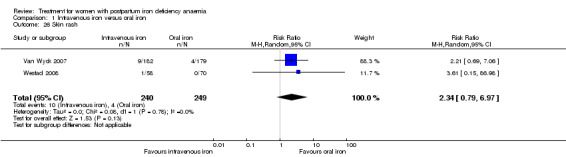
Comparison 1 Intravenous iron versus oral iron, Outcome 26 Skin rash.
1.29. Analysis.
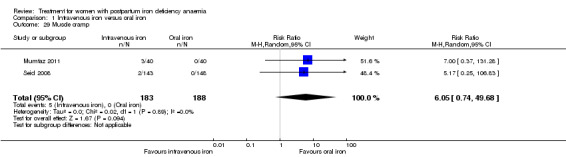
Comparison 1 Intravenous iron versus oral iron, Outcome 29 Muscle cramp.
1.24. Analysis.
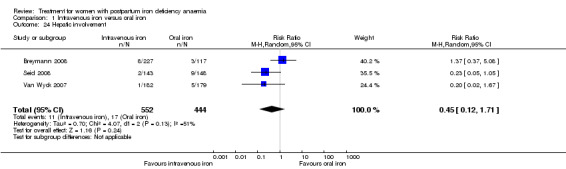
Comparison 1 Intravenous iron versus oral iron, Outcome 24 Hepatic involvement.
14.3. Analysis.
Comparison 14 Sensitivity analysis, Outcome 3 Heterogeneity ‐ Hepatic involvement ‐ comparison 1.
14.4. Analysis.
Comparison 14 Sensitivity analysis, Outcome 4 Heterogeneity, fixed effect ‐ Hepatic involvement ‐ comparison 1.
Some adverse events were rare and reported only by one study. We found no difference in these outcomes, which were urticaria (reported as an isolated symptom and not as part of an allergic reaction) (Analysis 1.27), unspecified pain (Analysis 1.30), and unspecified serious adverse events (Analysis 1.31).
1.27. Analysis.
Comparison 1 Intravenous iron versus oral iron, Outcome 27 Urticaria.
1.30. Analysis.
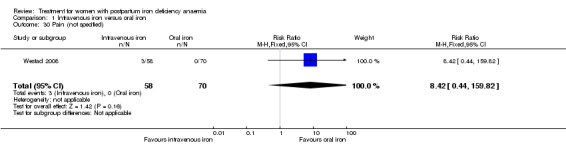
Comparison 1 Intravenous iron versus oral iron, Outcome 30 Pain (not specified).
1.31. Analysis.
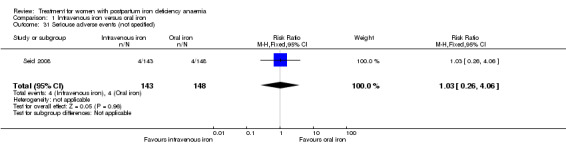
Comparison 1 Intravenous iron versus oral iron, Outcome 31 Seriouse adverse events (not specified).
Red blood cell transfusions
There was no difference between groups the frequency of women receiving blood transfusion as a "rescue treatment" (blood transfusion rates) (RR 0.48; 95% CI 0.19 to 1.23; four RCTs; 18 events; 606 women; I² = 0%; Analysis 1.34). For the trial by Westad 2008, we assumed that the reported number of blood transfusions were received within the first four weeks of treatment, which is clinically most probable. However, this is not specified in the published report. The number of units of RBCs transfused was not reported.
1.34. Analysis.
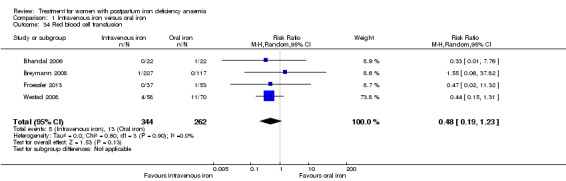
Comparison 1 Intravenous iron versus oral iron, Outcome 34 Red blood cell transfusion.
Discontinued study
One trial by Backe 2009 entitled 'A 6‐week randomised, open comparative, multi‐centre study of IV ferric carboxymaltose (Ferinject) and oral iron (Duroferon) for treatment of post partum anaemia' with the trial identification number NCT00929409 was identified. Based on the type of intervention this trial should have been included in Comparison 1. However, we were informed by the contact person for the trial that "Our controlled trial “A 6‐week randomised, open comparative, multi‐centre study of IV ferric carboxymaltose (Ferinject) and oral iron (Duroferon) for treatment of post partum anemia” was stopped because of slow progress, and the sponsor (Renapharma Vifor) then unfortunately decided to terminate the trial".
We then repeatedly attempted to contract the sponsors for preliminary results and the trial report made after discontinuation. We never received a response from the company. This indicates a high risk of publication bias (Bassler 2010).
Comparison 2: RBC transfusion versus non‐intervention
Prick 2014 was the only trial comparing RBC transfusion to non‐intervention, i.e. other treatment at the clinician's discretion. The trial included 519 women.
Primary outcomes
Maternal mortality
Mortality was not reported.
Fatigue
There was a small and transient, but statistically significant between‐groups difference in fatigue during the first week favouring the group receiving RBC transfusions (see below).
Fatigue was measured by the Multidimentional Fatigue Inventrory (MFI) (Smets 1995). We chose to report only on 'general fatigue', which summarises the remaining domains domains: 'physical fatigue', 'reduced activity', 'reduced motivation', and 'mental fatigue'. High score indicates higher level of fatigue.
The authors provided raw means and standard deviations on our request. However, they pointed out, that it would not be correct to enter the results as raw means while not correcting for baseline differences and mode of delivery. We chose to quote the data from the manuscript, but also import and analyse the data provided by the authors. In the published report's table S1 (data corrected for baseline differences), the authors found a statistically significant between‐groups difference in mean general fatigue at three days. There was no significant difference between groups at six weeks.
The additionally provided data showed that the group receiving RBC transfusions had significantly better scores than the non‐intervention group in general fatigue at three days (mean difference (MD) ‐0.80; 95% CI ‐1.53 to ‐0.07; women 388; low quality evidence; Analysis 2.1), but not at six weeks (low quality evidence; Analysis 2.2).
2.1. Analysis.
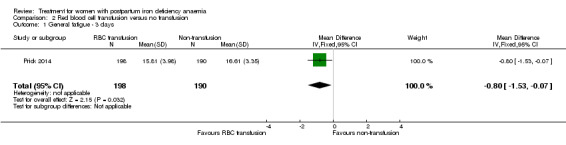
Comparison 2 Red blood cell transfusion versus no transfusion, Outcome 1 General fatigue ‐ 3 days.
2.2. Analysis.
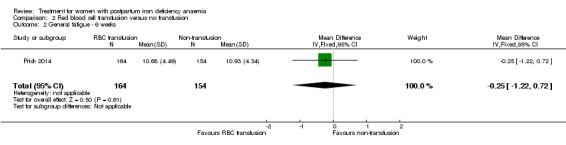
Comparison 2 Red blood cell transfusion versus no transfusion, Outcome 2 General fatigue ‐ 6 weeks.
Secondary outcomes
Anaemia symptoms
Anaemia symptoms eliciting a RBC transfusion occurred in 28 women in the non‐intervention group. However, the frequency of anaemia symptoms (besides fatigue) was not systematically reported for the remaining, non‐transfused members of the non‐intervention group or for the RBC transfusion group (very low quality evidence).
Psychological well being
Psychological well being improved significantly more in the group receiving RBC transfusions (see below).
Psychological well being was registered using the SF‐36 questionnaires (high score indicates better health state). We chose to quote the SF‐36 data from the manuscript, as well as to report the additionally provided data at one week of follow‐up.The published report (Table S1) showed a statistically significant between‐groups difference in 'physical functioning', where scores were 5.5 points lower at one week of follow‐up in the non‐intervention group, thus favouring the group receiving RBC transfusions. When we entered the additionally provided data we found a statistically significant difference in physical functioning favouring the group receiving RBC transfusions at one week (MD 5.67; 95% CI 0.84 to 10.50; 368 women; Analysis 2.3).
2.3. Analysis.

Comparison 2 Red blood cell transfusion versus no transfusion, Outcome 3 SF‐36: Physical functioning ‐ 1 week.
For social function there was a borderline statistically significant difference at one week favouring the group receiving RBC transfusions (MD 5.34; 95% CI 0.11 to 10.57; 369 women; Analysis 2.4). For the remaining items there was no statistically significant difference at one week of follow‐up (Analysis 2.5 to Analysis 2.10). Thus, there was a discrepancy between the reported results and our calculations of additionally provided data, but both sources find effect in favour of RBC transfusions.
2.4. Analysis.
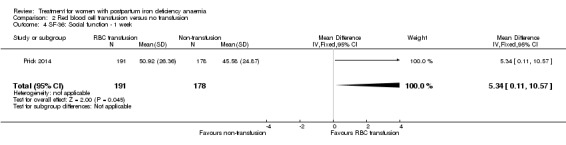
Comparison 2 Red blood cell transfusion versus no transfusion, Outcome 4 SF‐36: Social function ‐ 1 week.
2.5. Analysis.
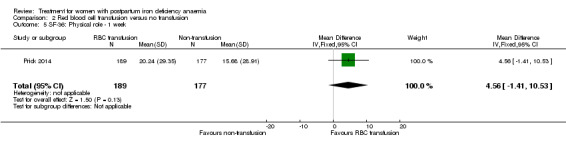
Comparison 2 Red blood cell transfusion versus no transfusion, Outcome 5 SF‐36: Physical role ‐ 1 week.
2.10. Analysis.
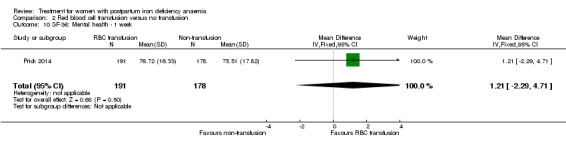
Comparison 2 Red blood cell transfusion versus no transfusion, Outcome 10 SF‐36: Mental health ‐ 1 week.
Infections
Infection rates were similar (RR 0.93; 95% CI 0.53 to 1.61; 519 women; moderate quality evidence; Analysis 2.11).
2.11. Analysis.
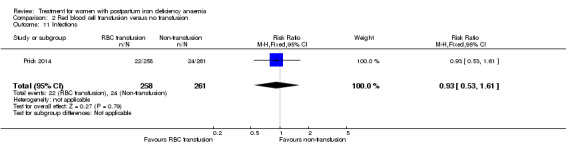
Comparison 2 Red blood cell transfusion versus no transfusion, Outcome 11 Infections.
Compliance
Compliance to treatment was lower in the non‐intervention group, where 33 women did not comply with allocated treatment versus seven in the RBC group (RR 1.11; 95% CI 1.06 to 1.17; 519 women; Analysis 2.12).
2.12. Analysis.
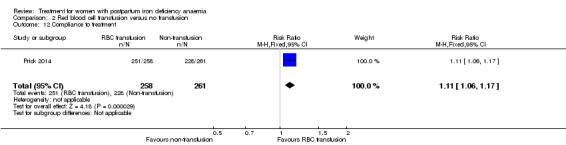
Comparison 2 Red blood cell transfusion versus no transfusion, Outcome 12 Compliance to treatment.
Breastfeeding
Breastfeeding rate at randomisation was 77% in both groups. There was no statistically significant difference in breastfeeding rate between groups at six weeks of follow‐up (RR 0.91; 95% CI 0.78 to 1.07; 297 women; Analysis 2.13).
2.13. Analysis.
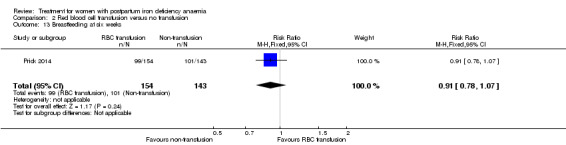
Comparison 2 Red blood cell transfusion versus no transfusion, Outcome 13 Breastfeeding at six weeks.
Length of hospital stay
Length of hospital stay was a median of two days in both groups.
Adverse events during treatment
There was no statistically significant difference in reported adverse events, which were alloantibody formation (very low quality evidence), rash, fever, thromboembolic events (low quality evidence), parenteral iron intolerance, and transfusion reactions (very low quality evidence) (Analysis 2.14 to Analysis 2.19). Transfusion reactions (alloantibodies, fever) only occurred in transfused participants, however, there was no systematical investigation for the presence of new alloantibodies.
2.14. Analysis.
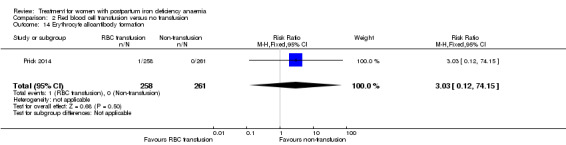
Comparison 2 Red blood cell transfusion versus no transfusion, Outcome 14 Erythrocyte alloantibody formation.
2.19. Analysis.
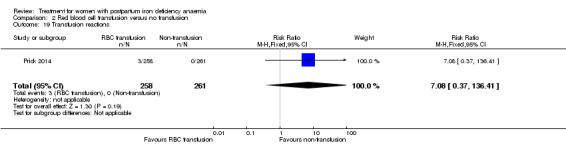
Comparison 2 Red blood cell transfusion versus no transfusion, Outcome 19 Transfusion reactions.
Red blood cell transfusions
In the RBC transfusion group, 251 women received transfusion, seven refused. The total number of RBC units given was 517 (median: 2 units per woman; interquartile range 2‐2). In the non‐intervention group 33 women received RBC transfusion and 88 RBC units were given (median: 0 units per woman; interquartile range 0‐0).
Comparison 3: Oral iron versus placebo
Oral iron was compared with placebo by three studies (Beard 2005; Krauss 1972; Tam 2005). The study by Krauss 1972 had three study arms. For this comparison we chose the study arm that received tablet Eryfer containing ferrous sulphate, ascorbic acid and sodium bicarbonate as the intervention arm (group S) and the placebo arm (empty preparation) as the control arm. The remaining arm received oral iron, magnesium oxide, yeast extract (see Comparison 4). The follow‐up periods for the three studies were 30, 42, and 145 days and the trials did not report results at comparable time points. The trials also reported on different outcomes, as a result it was not possible to perform meta‐analyses for this comparison.
Primary outcomes
None of our primary outcomes were reported.
Secondary outcomes
Anaemia symptoms
Only one study, Tam 2005, reported on persistent anaemia symptoms, but for both study groups combined. These were dyspnoea (n = 6), palpitations (n = 6), chest discomfort (n = 3), dizziness (n = 12), headache (n = 10).
Psychological well being
Psychological well being was significantly better in the placebo group, shown by two different tools (see below).
Psychological well being was reported by Beard 2005. The tools used for the assessment were Digit Symbol Substitution test (high score indicates better cognitive performance) (Hoyer 2004), Edinburgh Postnatal Depression Scale (EPDS), where high scores are associated with depression (Cox 1987), Spielberger State Trait Anxiety Inventory (STAI), where high scores indicate higher anxiety (Marteau 1992), and the Perceived Stress questionnaires, where high scores indicate more stress (Cohen 1983). The Digit Symbol Substitution evaluating cognitive performance showed no difference between groups at 10 weeks (MD 0.0; 95% CI ‐2.76 to 2.76; 51 women, one RCT; Analysis 3.1). The EPDS test did not show any statistical difference between groups at 10 weeks (MD 0.10; 95% CI ‐0.86 to 1.06; 51 women, one RCT; Analysis 3.2). The STAI tool showed no difference at 10 weeks (MD ‐0.40; 95% CI ‐3.18 to 2.38; 51 women, one RCT; Analysis 3.3). The Perceived Stress questionnaire showed a statistically significant difference favouring the placebo group at 10 weeks (MD 4.10; 95% CI 1.70 to 6.50; 51 women, one RCT; Analysis 3.4).
3.1. Analysis.
Comparison 3 Oral iron versus placebo, Outcome 1 Digit Symbol Substitution test ‐ 10 weeks.
3.2. Analysis.
Comparison 3 Oral iron versus placebo, Outcome 2 EPDS ‐ 10 weeks.
3.3. Analysis.
Comparison 3 Oral iron versus placebo, Outcome 3 STAI ‐ 10 weeks.
3.4. Analysis.
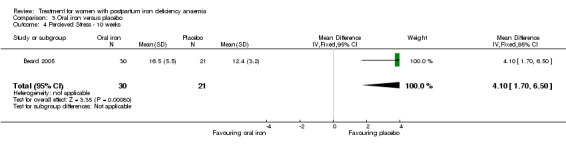
Comparison 3 Oral iron versus placebo, Outcome 4 Percieved Stress ‐ 10 weeks.
In the published report, the authors do not acknowledge the findings listed above.
Infections
Infections were not reported.
Compliance
Compliance was not reported.
Breastfeeding
Tam 2005 reported on breastfeeding rates at two days postpartum, with no statistically significant difference between groups (RR 0.82; 95% CI 0.58 to 1.17; 122 women; one RCT; Analysis 3.5).
3.5. Analysis.
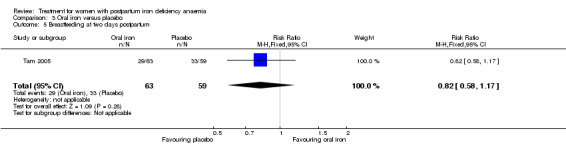
Comparison 3 Oral iron versus placebo, Outcome 5 Breastfeeding at two days postpartum.
Length of hospital stay
Length of hospital stay was not reported.
Adverse events
Adverse events were reported in two studies (Krauss 1972; Tam 2005). Tam 2005 reported only on one type of adverse events (back pain) for each study group individually, with no difference between groups (RR 0.66; 95% CI 0.42 to 1.03; one RCT, 53 events; 150 women; Analysis 3.6). The remaining adverse events were given for both study groups combined. The authors stated that there was no difference regarding nausea, vomiting, or constipation (Tam 2005). Krauss 1972 reported that six women in each group had GI adverse events such as constipation, low appetite, and morning sickness, but the numbers of each adverse event were not reported (very low quality evidence) (Analysis 3.7). No serious adverse events were reported.
3.6. Analysis.
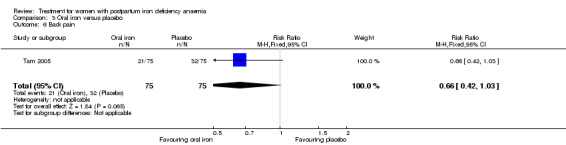
Comparison 3 Oral iron versus placebo, Outcome 6 Back pain.
3.7. Analysis.
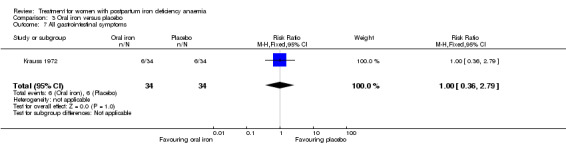
Comparison 3 Oral iron versus placebo, Outcome 7 All gastrointestinal symptoms.
Red blood cell transfusions
Red blood cell transfusions were not reported.
Comparison 4: Oral iron, magnesium oxide and yeast extract versus placebo
One study was included (67 women) in this comparison (Krauss 1972).
Primary outcomes
None of our primary outcomes were reported.
Secondary outcomes
Anaemia symptoms, psychological well being, infections, compliance, breastfeeding, length of hospital stay, blood transfusions
Not reported.
Adverse events
Different types of GI adverse events were not reported separately. Therefore, we analysed all GI symptoms as a whole for each group and found a statistically significant difference favouring the placebo group. Sixteen women in the intervention group had constipation, anorexia, bloating and vomiting. Six women in placebo group had constipation, low appetite and morning sickness (RR 2.75; 95% CI 1.23 to 6.16; one RCT; 22 events; 67 women; Analysis 4.1). No serious adverse events were reported.
4.1. Analysis.
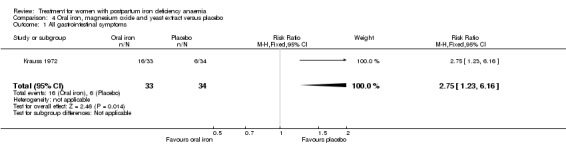
Comparison 4 Oral iron, magnesium oxide and yeast extract versus placebo, Outcome 1 All gastrointestinal symptoms.
Comparison 5: IV iron and oral iron after four weeks versus oral iron (week five to 12)
One study was included (117 women) (Westad 2008). Group A received IV iron immediately after giving birth and started oral iron after four weeks. Group B started receiving oral iron immediately after delivery.
Primary outcomes
Maternal mortality
Maternal mortality was not reported.
Fatigue
In the published report, the authors state that 'physical fatigue' improved significantly more in group A compared to group B. The improvement was seen at week eight (P = 0.02) and at week 12 (P = 0.03). There was no difference between the groups in their 'mental fatigue' score. The 'total fatigue' score was significantly better in group A at eight and 12 weeks (P = 0.02 at both time points). Standard deviations were not available for fatigue at eight and 12 weeks; therefore we could not carry out a statistical analysis.
Secondary outcomes
Anaemia symptoms
Anaemia symptoms were not reported.
Psychological well being
There was no difference between groups in the SF‐36 scores at week eight according to the published report. Standard deviations were not available for analysis.
Infections
Infections were not reported.
Compliance
Compliance to treatment with oral iron was assessed by counting returned pills. Compliance was reported as less than 50% of the recommended dose in both groups.
Breastfeeding
Breastfeeding was not reported.
Length of hospital stay
Length of hospital stay was not reported.
Adverse events
Adverse events did not differ significantly between groups. These were all GI symptoms, GI pain, constipation, diarrhoea, nausea, dysgeusia (distortion of the sense of taste), flatulence, melena, or headache (Analysis 5.1 to Analysis 5.9).
5.1. Analysis.
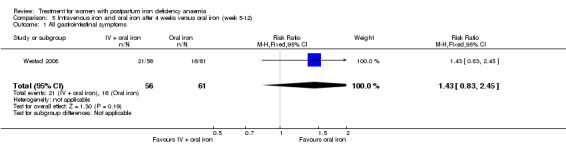
Comparison 5 Intravenous iron and oral iron after 4 weeks versus oral iron (week 5‐12), Outcome 1 All gastrointestinal symptoms.
5.9. Analysis.
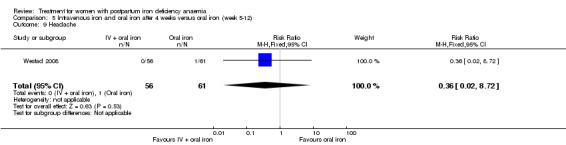
Comparison 5 Intravenous iron and oral iron after 4 weeks versus oral iron (week 5‐12), Outcome 9 Headache.
Red blood cell transfusions
We assumed that the RBC transfusions reported in this study were given within the first four weeks (see Comparison 1).
Comparison 6: IV iron and oral iron versus oral iron
Two studies (112 women) were included (Breymann 2000; Perello 2014). One study administered placebo EPO in the intervention arm (Breymann 2000) and the other study administered placebo IV iron in the comparator arm (Perello 2014). Thus, as per the lack of evidence for a substantial placebo effect (Hróbjartsson 2010) and the similar active treatments in these two studies, we found them comparable. Length of follow‐up was two and six weeks, respectively.
Primary outcomes
None of our primary outcomes were reported.
Secondary outcomes
Anaemia symptoms
One study evaluated anaemia symptoms by the number of patients who scored the severity as being equal to or more than seven on the Visual Analoge Scale (VAS) (higher score indicates higher severity) (Perello 2014). There was no statistically significant difference between groups at any time point (very low quality evidence; Analysis 6.1 to Analysis 6.3).
6.1. Analysis.
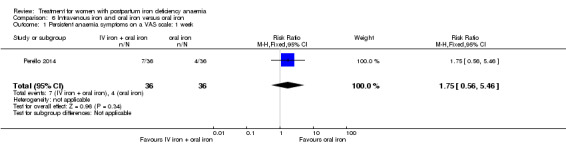
Comparison 6 Intravenous iron and oral iron versus oral iron, Outcome 1 Persistent anaemia symptoms on a VAS scale: 1 week.
6.3. Analysis.
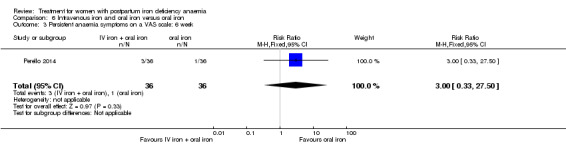
Comparison 6 Intravenous iron and oral iron versus oral iron, Outcome 3 Persistent anaemia symptoms on a VAS scale: 6 week.
Psychological well being
Psychological well being was measured using the EPDS (Cox 1987) and the STAI (Marteau 1992) tools by Perello 2014. A clinically significant EPDS score was defined as being equal to or more than 11. There was no statistical difference between groups at one week of follow‐up (Analysis 6.4).
6.4. Analysis.
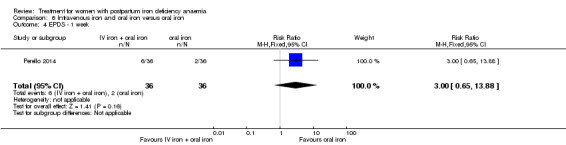
Comparison 6 Intravenous iron and oral iron versus oral iron, Outcome 4 EPDS ‐ 1 week.
Infections
Infections were not reported.
Lenght of hospitalisation
Lenght of hospitalisation was reported by Perello 2014 and did not differ between groups (Analysis 6.5).
6.5. Analysis.
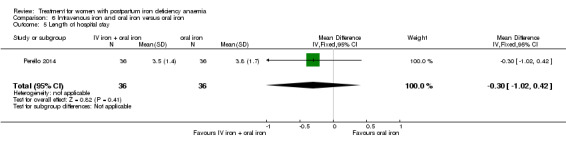
Comparison 6 Intravenous iron and oral iron versus oral iron, Outcome 5 Length of hospital stay.
Adverse events
Adverse events were given as the number of patients who scored severity as being equal to or more than seven on the VAS in one study (Perello 2014). There was no statistically significant difference between groups at any time point (Analysis 6.6 to Analysis 6.8). The other study reported that there were no serious adverse events, including hypersensitivity or thromboembolic events (very low quality evidence). Five cases of dysgeusia, three cases of warm flushes and five cases of GI complaints were reported, but not by group (Breymann 2000).
6.6. Analysis.
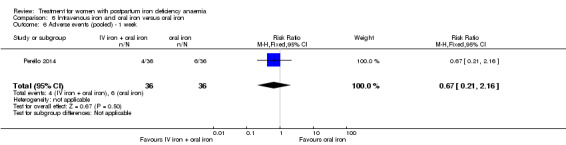
Comparison 6 Intravenous iron and oral iron versus oral iron, Outcome 6 Adverse events (pooled) ‐ 1 week.
6.8. Analysis.
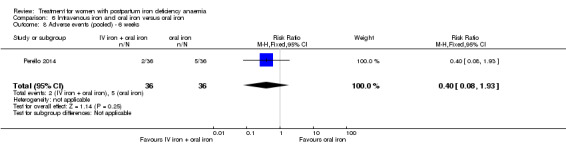
Comparison 6 Intravenous iron and oral iron versus oral iron, Outcome 8 Adverse events (pooled) ‐ 6 weeks.
Red blood cell transfusions
Blood transfusion rates did not differ between groups (Analysis 6.9).
6.9. Analysis.
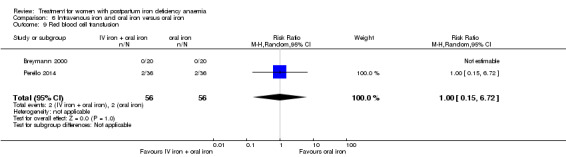
Comparison 6 Intravenous iron and oral iron versus oral iron, Outcome 9 Red blood cell transfusion.
Comparison 7: Erythropoietin (regardless of route) and IV iron versus IV iron
Two studies were included (80 women) (Krafft 2011; Wagstrom 2007). For the study by Wagstrom 2007, which had three study arms, we chose to compare group one (SC EPO 40,000 U + IV iron) and three (IV iron), for optimal resemblance to the study by Krafft 2011. Arm two received SC EPO 20,000 U + IV iron (Comparison 8).
Primary outcomes
None of our primary outcomes were reported.
Secondary outcomes
Anaemia symptoms
Anaemia symptoms were not reported
Psychological well being
One woman developed postpartum depression (RR 0.33; 95% CI 0.01 to 7.72; one RCT; 40 women; Analysis 7.1)
7.1. Analysis.
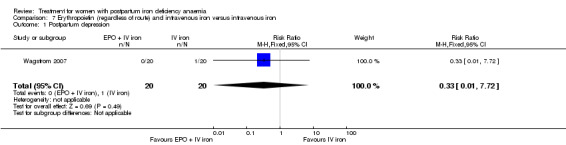
Comparison 7 Erythropoietin (regardless of route) and intravenous iron versus intravenous iron, Outcome 1 Postpartum depression.
Infections
Infection rates were similar (RR 2.00; 95% CI 0.72 to 5.59; two RCTs; 80 women; Analysis 7.2).
7.2. Analysis.
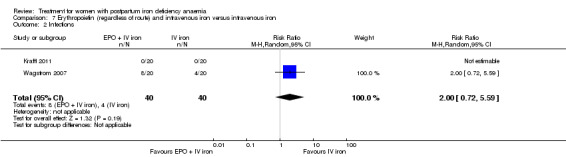
Comparison 7 Erythropoietin (regardless of route) and intravenous iron versus intravenous iron, Outcome 2 Infections.
Compliance
Compliance was reported as 100% in both groups since no women refused injections (Krafft 2011; Analysis 7.3).
7.3. Analysis.
Comparison 7 Erythropoietin (regardless of route) and intravenous iron versus intravenous iron, Outcome 3 Compliance to treatment.
Breastfeeding
All women in both groups of one study breast fed (Analysis 7.4) (Krafft 2011).
7.4. Analysis.
Comparison 7 Erythropoietin (regardless of route) and intravenous iron versus intravenous iron, Outcome 4 Breasfeeding.
Length of hospital stay
Length of hospital stay was not reported.
Adverse events
Adverse events did not differ significantly between groups. These were dysgeusia, flush, diarrhoea, headache, itching including increased liver enzymes, dizziness, or thrombophlebitis (Analysis 7.5 to Analysis 7.11). There were no thromboembolic events.
7.5. Analysis.
Comparison 7 Erythropoietin (regardless of route) and intravenous iron versus intravenous iron, Outcome 5 Dysgeusia.
7.11. Analysis.
Comparison 7 Erythropoietin (regardless of route) and intravenous iron versus intravenous iron, Outcome 11 Thrombophlebitis.
Red blood cell transfusions
There was no difference between groups regarding blood transfusion rate (RR 3.00; 95% CI 0.13 to 69.52; two RCTs; 80 women; Analysis 7.12).
7.12. Analysis.
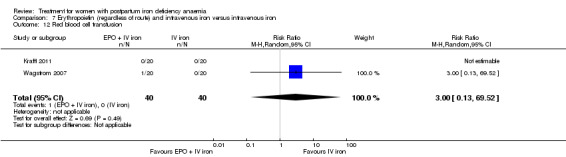
Comparison 7 Erythropoietin (regardless of route) and intravenous iron versus intravenous iron, Outcome 12 Red blood cell transfusion.
Comparison 8: Subcutaneous EPO 10,000 U two doses and IV iron versus IV iron
We included one study (40 women) Wagstrom 2007, with three study arms. For this comparison we used the arm that received SC EPO 20,000 U + IV iron (group two) and IV iron (group three). The last arm received SC EPO 40,000 U + IV iron (Comparison 7).
Primary outcomes
None of our primary outcomes were reported.
Secondary outcomes
Anaemia symptoms
Anaemia symptoms were not reported
Psychological well being
One woman developed postpartum depression (RR 0.33; 95% CI 0.01 to 7.72; one RCT; 40 women; Analysis 8.1)
8.1. Analysis.
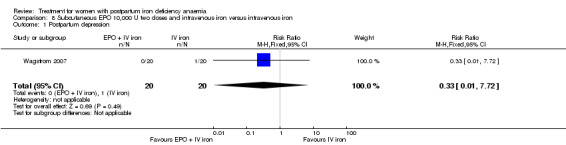
Comparison 8 Subcutaneous EPO 10,000 U two doses and intravenous iron versus intravenous iron, Outcome 1 Postpartum depression.
Infection
Infection rates were similar (RR 0.75; 95% CI 0.19 to 2.93; Analysis 8.2).
8.2. Analysis.
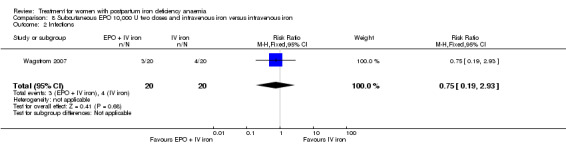
Comparison 8 Subcutaneous EPO 10,000 U two doses and intravenous iron versus intravenous iron, Outcome 2 Infections.
Compliance, breastfeeding, length of hospital stay
Not reported.
Adverse events
There was no between‐group difference for other adverse events including headache, low blood pressure, diarrhoea, dizziness, or itching with increased liver enzymes (Analysis 8.3 to Analysis 8.7). There was no thromboembolic events.
8.3. Analysis.
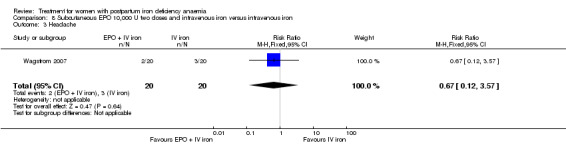
Comparison 8 Subcutaneous EPO 10,000 U two doses and intravenous iron versus intravenous iron, Outcome 3 Headache.
8.7. Analysis.
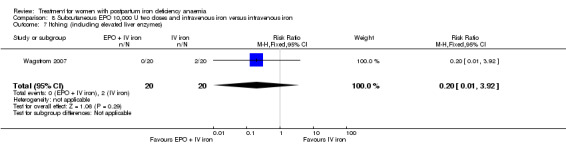
Comparison 8 Subcutaneous EPO 10,000 U two doses and intravenous iron versus intravenous iron, Outcome 7 Itching (including elevated liver enzymes).
Red blood cell transfusions
No women received RBC transfusions (Analysis 8.8).
8.8. Analysis.
Comparison 8 Subcutaneous EPO 10,000 U two doses and intravenous iron versus intravenous iron, Outcome 8 Red blood cell transfusion.
Comparison 9: IV EPO, IV iron and oral iron versus IV iron and oral iron
Three studies were included (Breymann 1996; Breymann 2000; Lebrecht 1995). Two of them had three study arms. For the study by Breymann 1996, we chose the groups receiving IV EPO + IV iron + oral iron (group three) and IV iron + oral iron (group one) for this comparison. The remaining group received SC EPO + IV iron + oral iron (Comparison 12). For the study by Breymann 2000, we chose the group that received IV EPO + IV iron + oral iron (group one) and the group that received IV placebo‐EPO + IV iron + oral iron (group two) for this comparison. The last group received oral iron alone (Comparisons 6 and 13). The follow‐up periods for the three studies were between 14 and 42 days, with no common time point.
Primary outcomes
None of our primary outcomes were reported.
Secondary outcomes
Anaemia symptoms, psychological well being, infections, compliance, breastfeeding, length of hospital stay
Not reported
Adverse events
All three studies reported that there were no serious adverse events such as anaphylactic reactions or thromboembolic events. Leg paraesthesia was the only adverse event reported by group, with no statistically significant difference (RR 0.72; 95% CI 0.08 to 6.65; two RCTs; two events; 76 women; I² = 0%; Analysis 9.1). Two women experienced a warm sensation during iron infusion, dysgeusia was observed in 27 women and 10 women complained of a burning sensation during EPO injection (Breymann 1996). There were five cases of GI adverse events, five cases of dysgeusia and three cases of warm flushes (Breymann 2000). However, these numbers were not given for each group, but were combined, thus it is unknown to which study arm these women were randomised.
9.1. Analysis.
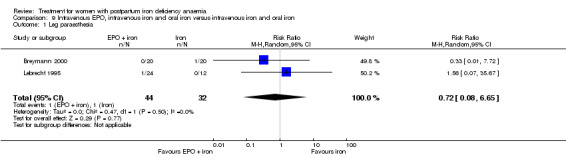
Comparison 9 Intravenous EPO, intravenous iron and oral iron versus intravenous iron and oral iron, Outcome 1 Leg paraesthesia.
Red blood cell transfusions
No women RBC received blood transfusions (Breymann 1996; Breymann 2000; Analysis 9.2).
9.2. Analysis.
Comparison 9 Intravenous EPO, intravenous iron and oral iron versus intravenous iron and oral iron, Outcome 2 Red blood cell transfusion.
Comparison 10: Subcutaneous EPO and oral iron versus oral iron
One study was included (40 women) (Makrydimas 1998).
Primary outcomes
None of our primary outcomes were reported.
Secondary outcomes
Anaemia symptoms, psychological well being, infections, compliance
Not reported.
Breastfeeding
More women were breastfeeding in the EPO group (RR 1.90; 95% CI 1.21 to 2.98; Analysis 10.1). The time point for this observation was not stated.
10.1. Analysis.
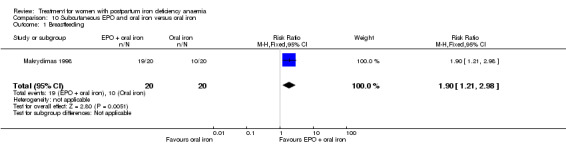
Comparison 10 Subcutaneous EPO and oral iron versus oral iron, Outcome 1 Breastfeeding.
Length of hospital stay
Length of hospital stay was reported as a median with 11 days (range six to 11) for 'EPO + oral iron' group and 14 days (range 11 to 19) for the oral group. The reason for the prolonged hospitalisation was not given. The available data were not sufficient to perform a statistical analysis.
Adverse events
Adverse events including GI symptoms were not reported.
Red blood cell transfusions
Two women in the oral group showed haemodynamic instability and received blood transfusions (RR 0.20; 95% 0.01 to 3.92; Analysis 10.2).
10.2. Analysis.
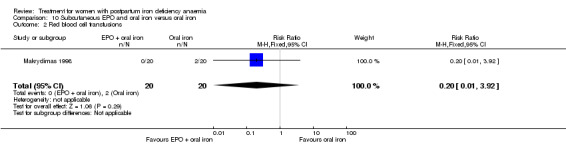
Comparison 10 Subcutaneous EPO and oral iron versus oral iron, Outcome 2 Red blood cell transfusions.
Comparison 11: IV EPO versus IV placebo
One study was included (71 women) (Meyer 1995).
Primary outcomes
None of our primary outcomes were reported.
Secondary outcomes
Anaemia symptoms
Anaemia symptoms were not reported
Psychological well being
According to the study authors, there was no statistically significant difference between the groups regarding psychological well being, which was measured using selected items from the 'Blues Questionnaire' (low score indicates absence of blues) (Kennerley 1989) and the 'Self‐report symptom inventory 90 [SCL‐90‐R]' (high scores indicate high levels of unfavourable symptoms) (Schmitz 1999). Data were not eligible for analysis.
Infections, compliance, breastfeeding, length of hospital stay, adverse events, red blood cell transfusions
Not reported
Comparison 12: Subcutaneous EPO, IV iron and oral iron versus IV iron and oral iron
One study was included (60 women) (Breymann 1996). The study had three study arms. For this comparison, we chose the arm receiving SC EPO + IV iron + oral iron (group two) and IV iron + oral iron (group 1). The remaining study arm received IV EPO + IV iron + oral iron (Comparison 9).
Primary outcomes
None of our primary outcomes were reported.
Secondary outcomes
Anaemia symptoms, psychological well being, infections, compliance, breastfeeding, length of hospital stay
Not reported
Adverse events
The study authors reported that there were no GI events, anaphylactic reactions or serious adverse events. Two women felt a warm sensation during iron infusion, 27 women experienced dysgeusia and 10 women experienced a burning sensation during EPO injection. However, these numbers were not provided per group.
Red blood cell transfusions
No women received blood transfusions.
Comparison 13: IV EPO, IV iron and oral iron versus oral iron
One study was included (40 women) (Breymann 2000). The study had three study arms. For this comparison, we used the arm that received IV EPO + IV iron + oral iron (group one) and oral iron (group three). The remaining study arm received IV placebo‐EPO + IV iron + oral iron (Comparisons 6 and 9).
Primary outcomes
None of our primary outcomes were reported.
Secondary outcomes
Anaemia symptoms, psychological well being, infections, compliance, breastfeeding, length of hospital stay
Not reported
Adverse events
The study reported that there were no serious adverse events, including hypersensitivity or thromboembolic events. Five cases of dysgeusia, three cases of warm flushes and five cases of GI complaints were reported, but not by group.
Red blood cell transfusions
No women received blood transfusions.
Planned analyses
We used random‐effects meta‐analyses for all dichotomous outcomes due to variation in length of treatment and dosages.
We found important statistical heterogeneity in two meta‐analyses in Comparison 1, and therefore we performed a sensitivity analyses for these, although the meta‐analyses were based on secondary outcomes.
Sensitivity analyses regarding trial design could not be performed due to lack of blinding throughout the main comparison and the other comparisons did not include enough studies for a meaningful sensitivity analysis. It was not meaningful to perform subgroup analyses as planned, due to few included studies.
We did not produce a funnel plot as we had a maximum of eight studies in any single meta‐analysis.
'Summary of findings' tables
Please see Table 1; Table 2; Table 3; Table 4; Table 5; Table 6; Table 7; Table 8.
Discussion
Summary of main results
This review included 22 studies with a total of 2858 women. The majority of our analyses are based on a small number of studies. Very few studies report on our primary outcomes of maternal mortality and fatigue. The main results are summarised in the ‘Table 1; Table 2; Table 3; Table 4; Table 5; Table 6; Table 7; Table 8’.
Overview
For intravenous (IV) iron compared with oral iron, fatigue significantly improved in the IV iron group in one of two studies. There were no data on other anaemia symptoms. Psychological well being was not significantly different between groups. There were very little or no data on breastfeeding and length of hospital stay. Infection rates were not significantly different between groups. Maternal mortality was insufficiently reported; however there was one reported death due to cardiomyopathy in the IV iron group. Also, one woman in the IV iron group developed arrhythmia. Allergic reactions only occurred in the IV iron group, but there was no statistically significant difference. Dysgeusia, injection site discomfort, and flushes were only seen in women treated with IV iron. Gastrointestinal symptoms such as constipation, nausea, gastrointestinal (GI) pain and diarrhoea were far more frequent with oral iron. Hepatic involvement was more frequent in the orally treated group, however only after sensitivity analysis. Compliance with treatment was lower in women treated orally, possibly associated with the high frequency of GI symptoms. Blood transfusion rates did not differ significantly.
For red blood cell (RBC) transfusion compared with non‐intervention, there were no data on mortality. For fatigue and psychological well being there was a transient, small but statistically significant improvement favouring the RBC transfused group. There was no difference in infection rates. More women in the RBC transfusion group complied with the allocated treatment. No difference was seen for breastfeeding, length of hospital stay or reported adverse events, including transfusion reactions. One woman developed erythrocyte alloantibodies as a result of transfusion.
For oral iron compared with placebo, none of our primary outcomes were reported. Psychological well being did not improve in the treatment group. There was no difference in breastfeeding or adverse events. No serious events were reported. Treatment with oral iron with magnesium oxide and yeast extract showed significantly more GI adverse events compared with placebo.
For IV iron with oral iron versus oral iron (weeks five to 12), mortality was not reported. Fatigue improved more in the group initially treated with IV iron. Compliance was equally poor in both groups. There was no difference in reported adverse events. No serious events were reported. We assumed that the RBC transfusions took place within the first four weeks of this study.
For IV iron and oral iron versus oral iron, none of our primary outcomes were reported. There were no serious adverse events. Overall, there was no difference between groups.
For erythropoietin (EPO) with IV iron versus IV iron, none of our primary outcomes were reported. There was no difference regarding infections. All women breast fed. There was no difference regarding adverse events. No serious events were reported. No difference was seen regarding blood transfusions.
For IV EPO with IV iron and oral iron versus IV iron with oral iron, none of our primary outcomes were reported. No serious adverse events occurred. There was no difference in other adverse events. No women received transfusions.
For subcutaneous (SC) EPO with oral iron versus oral iron alone, none of our primary outcomes were reported. Significantly more women breast fed in the EPO‐treated group. The duration of hospital stay was quite long in both groups. There was no difference in transfusion rates.
For IV EPO versus IV placebo, none of our primary outcomes were reported. There was no statistically significant differences regarding psychological well being.
For SC EPO, IV iron and oral iron versus IV iron with oral iron, none of our primary outcomes were reported. There were no serious adverse events. No women received transfusions. Some women felt a burning sensation during EPO injection.
For IV EPO, IV iron and oral iron versus oral iron, none of our primary outcomes were reported. There were no serious adverse events. No women received transfusions.
Across all our included studies, data on allergic reactions were available from 1003 women who received IV iron treatment (alone or combined with other treatment). Three of these women developed an anaphylactic reaction.
Interpretation of study methods and results
Main comparison: IV iron versus oral iron
Efficacy
Intravenous iron is perhaps more effective in reducing fatigue than oral iron, but this beneficial effect was only shown in one small study, thus the amount of evidence is very limited and therefore the clinical significance remains uncertain. Other anaemia symptoms were not evaluated. Thus, overall efficacy of treatment can not be concluded upon.
Harms of treatment
One maternal death occurred in the IV group due to peripartum cardiomyopathy. Also, one woman developed arrhythmia during IV iron infusion. Thus, two cases of serious cardiac adverse events were observed. This has not previously been described as an adverse effect of IV iron treatment, and no conclusions can be drawn regarding a causal relationship. However, we suggest that cardiac events be carefully monitored and described in all future studies.
Anaphylaxis was reported in three women, all of whom had received IV iron. Due to the low number of events, and the lack of statistical power, our data do not prove that anaphylaxis or any other serious adverse events are more frequently caused by IV iron than by oral iron. However, a very high number of study participants would be needed to rule out an association between IV iron and anaphylaxis. Since anaphylaxis is a known side effect to IV iron, and a dangerous and potentially fatal reaction, concerns were raised by the French Medicines Agency due to a number of observed cases. The European Medicines Agency therefore issued a safety report (European Medicines Agency 2013) and a risk management recommendation (European Medicines Agency 2013a) regarding allergic reactions to IV iron. It was concluded that treatment with IV iron carries a small risk of allergic reactions which can be life‐threatening.
Urticaria can be a manifestation of an allergic reaction and thus the reported urticarial reactions may be included in the anaphylaxis or evidence of hypersensitivity outcome with the proviso that urticaria also can be caused by other factors.
Taste distortion (dysgeusia), injection site discomfort, and flushing were symptoms associated with IV iron treatment. These reactions are unpleasant but transient and not considered harmful. However, it cannot be excluded that flushing and injection site discomfort could be prodromes heralding more severe hypersensitivity reactions, and they should be observed closely.
Gastrointestinal adverse events were far more frequent with oral iron, both combined and specifically for constipation, nausea, GI pain, and diarrhoea. This corresponds well with the general knowledge and concern among clinicians that GI symptoms are frequent, bothersome and may negatively affect compliance. This means that the patients, who can not handle the adverse effects of treatment are often not treated sufficiently. However, these are the patients who probably are in most need of treatment and it is up to the clinicians to trace this less tolerant subgroup of patients and consider alternative treatment methods. The high compliance for IV iron treatment may be explained by the fact that IV treatment was administered in few doses by clinicians, in contrast to several self‐administered daily doses of oral iron over a period of several weeks.
For hepatic involvement, the results became statistically significant in favour of IV treatment, when we performed a sensitivity analysis. It is surprising that hepatic involvement was shown in the oral group. We could not find any literature that supports the association between oral iron and liver toxicity in humans. However, this effect of oral ferrous sulphate is described in rats (Toblli 2008; Toblli 2013). It should be noted, that the two studies included in the sensitivity analysis for hepatic involvement had an almost five times higher weekly dose of oral iron, than the study which caused heterogeneity. These findings suggest a dose‐response relationship between oral ferrous sulphate and toxic liver effect. This was a rare finding, however it may be more frequent since liver function was not measured in all studies. Also. most studies with an IV iron arm excluded participants with known liver disease. Therefore, there is a population selection favouring women with a healthy liver also in the oral arm of these studies. Further research is needed to investigate the effect of ferrous sulphate on liver function.
We found high heterogeneity for infection suggesting uncertainty of the result. For infections it was not possible to predict with certainty which study was more likely to cause the heterogeneity. However, we know that the study by Breymann 2008 caused heterogeneity in the meta‐analysis for hepatic involvement, and that it had a high level of attrition bias and that the randomisation method was not described. The results for infections remained statistically non‐significant in the sensitivity analysis.
Other treatment modalities
For RBC transfusions compared with non‐intervention data (Comparison 2), a small and transient, but statistically significant improvement was shown in fatigue and SF‐36 data in favour of the RBC treatment. The SF‐36 data provided had very broad standard deviations and our analysis of the raw means differed from what was stated in the published report. However, both sources found a statistically significant effect in favour of the RBC transfusions. Whether the effect of RBC transfusion on fatigue and psychological well being is clinically significant in haemodynamically stable women with no severe anaemia symptoms is debatable, as the small and transient gain in fatigue and psychological well being scores must be balanced against potential severe side effects of RBC transfusions. Also, alleviating relatively mild subjective symptoms with an expensive treatment is not cost‐effective (Prick 2014).
This study did not systematically report all anaemia symptoms in the two groups. However, severe anaemia symptoms led to RBC transfusion in 28 women randomised to the non‐intervention group. This illustrates the importance of registration and reporting of anaemia symptoms as an outcome and the need to evaluate them as an indication for transfusion, because the consequence of these symptoms can trigger a potentially harmful treatment.
The trial was conducted in The Netherlands where the procedure for blood transfusion, including screening and cross‐matching and administration is highly developed and thus safe. The same lack of difference regarding infections, breastfeeding, length of hospital stay, or adverse events may not be the same in a different setting, i.e. in low‐income countries. One woman developed alloantibodies after transfusion. Since there was no systematic post‐transfusion examination for newly formed alloantibodies in all transfused participants, and since these antibodies often have subtle or delayed clinical consequences in the weeks following transfusion, the frequency is most likely underestimated. Generally, the incidence of newly generated alloantibodies is heavily dependent on the patient population, and the precise incidence of antibodies elicited by postpartum transfusion is not known. A recent study showed unexpected alloantibodies occur in 3% of obstetric patients, and approximately one‐third of these cases were associated with haemolytic (breaking down of RBCs) disease of the newborn (Smith 2013). Even though antibodies as a consequence of transfusion may only represent some of these cases, they may potentially harm a future foetus, and thus have significant consequences (Klein 2014).There was low compliance in the non‐intervention group which can be explained by the trial design: If women allocated to the non‐intervention group received transfusion, e.g. due to secondary postpartum haemorrhage, they automatically failed to comply with the allocated treatment, whereas the women allocated to the RBC transfusion group had no obvious reason to refuse transfusion after giving consent to participate in the trial.
When comparing oral iron with placebo, it was not possible to perform a meta‐analysis due to insufficient data. Also, the follow‐up periods were very different and there were no common time points. In assessment of psychological well being one test showed that treatment actually reduced psychological well being compared with placebo. However, the trial authors did not reach the same conclusion. When evaluating measures such as psychological well being it is important to use validated tools for the specific population (i.e. postpartum women).
The women who started their postpartum period with an IV iron infusion and were then supplemented with oral iron at four weeks had significantly less fatigue than women who received oral iron only from the beginning of the postpartum period. However, by the end of this study the dropout rate was very high, and the population was highly exposed to attrition bias, thus potentially selecting the stronger and more healthy patients.
Data from one small study (Makrydimas 1998), indicated that women were able to breastfeed more in the group treated with EPO and oral iron compared with the oral iron group. This effect of EPO has not been reported elsewhere. On the contrary, the study of equal sample size by Krafft 2011 showed that women treated with EPO and IV iron had the same high breastfeeding rate as those treated with IV iron alone. Thus, the difference in lactation rate seen in Makrydimas 1998 was probably coincidental due to small study material.
Handling fatigue and quality of life data
Fatigue and psychological well being are concepts very difficult to define and delimit from one another. Fatigue can be perceived as a uni‐dimensional phenomenon (Visual Analoge Scale (VAS)), it can be subdivided into mental and physical aspects (Fatigue Score), and it may even consist of additional dimensions (reduced motivation and reduced activity), as seen in the Multidimentional Fatigue Inventrory (MFI). These additional aspects of fatigue have certain similarities to items in tools used to measure psychological well being. Psychological well being as measured by SF‐36 may be influenced by a postpartum depression. As defined in the International Classification of Diseases (ICD‐10), one of the core symptoms of depression is 'reduction of energy or increased tiredness', or what is commonly understood as 'fatigue'. In fact, the SF‐36 contains questions specifically targeting disclosure of fatigue symptoms summarised in the 'vitality' item. Thus, although we defined fatigue and psychological well being as separate outcomes, we acknowledge the complexity of these symptoms and the tools developed to measure them. It is therefore in our opinion not advisable to dissect the questionnaire data in an attempt to extract purely fatigue‐related results.
The assessment of health‐related quality of life is highly subjective and the tools developed to measure quality of life merely translate feelings in to numbers which only approximate reality, and statistical significance in calculation of scores does not necessarily equal clinical significance. Therefore, it is perhaps more informative and useful to establish a minimal clinically relevant difference or threshold in scores above or below which a person is confirmed to be experiencing a given condition or emotion (e.g. Perello 2014).
For quality of life outcomes, we reported only the most relevant time points. The intention was to limit the number of analyses to a manageable amount of information and to avoid reporting on time points too close to each other.
For psychological well being, we found the short‐term time points most important when comparing two treatment regiments, as it is clinically relevant how quickly a given treatment can relieve symptoms, and long‐term differences are likely to be affected by an unknown number of confounders and the anaemia would often resolve over time due to physiological factors.
Discrepancy in statistical significance between raw means entered into RevMan 2014 and the findings in the published reports can be explained by the lack of adjustment calculations for additionally provided data (Prick 2014). However, for published data this explanation does not apply (e.g. Beard 2005).
Overall completeness and applicability of evidence
After including 22 studies, we had expected sufficient data to perform a comprehensive meta‐analysis on clinically relevant outcomes for the efficacy of different treatment methods of postpartum iron deficiency anaemia. In terms of treatment efficacy, the available literature provided surprisingly little information on clinical outcomes. Instead, there was focus on surrogate outcomes, such as laboratory values. We were surprised that only three studies investigated fatigue, which we chose as a primary outcome, because of its commonness in anaemic patients.
The identified studies provide an overall acceptable insight on various harmful symptoms, although the reporting was heterogeneous, the symptoms were some times difficult to pool and maternal mortality was rarely reported. We have learned that anaphylaxis, the most feared complications of IV iron, is rare, and we have confirmed that GI adverse events associated with oral iron were common.
It might be difficult to distinguish between symptoms of a condition and adverse effects of a treatment. In theory, anaemia symptoms should decrease over time, while adverse events increase due to drug exposure. Researchers usually did not report the time at which a symptom occurred. It is however important to report the time point at which the patient experiences discomfort of any kind (e.g. headache), to distinguish between symptoms of anaemia and harmful effects of treatment. The studies did not agree on common clinically relevant time points to collect data, which is important in a meta‐analyses.
Surprisingly, only three small studies investigated the effect of oral iron versus placebo, one of which was conducted more than 40 years ago. Cost of different drugs and treatment vary depending on the setting and thus can not be generalised. However, it is certain that IV iron costs substantially more than oral iron (Khalafallah 2012) and infusion often require hospitalisation. When investigating treatment of a condition, which is most likely to occur in low‐income settings where nutritional factors, longer periods of breastfeeding and the number of pregnancies per woman may differ from those of high‐income countries, special attention must be drawn to types of treatment options available in these particular settings, i.e. oral iron.
The majority of trials were conducted in high‐income countries and only in two reports was it specifically stated that the participants derived from a low‐income setting. This questions the external validity of the results and clinicians should bear in mind that women from different settings may respond differently to the same treatment. Women who live in low‐income settings may be more prone to malnutrition and infections. They may have a lower degree of education and according to social standards they may be expected to have more children. These factors may affect compliance, adverse effects of treatment, management of the adverse events (seeking help) and response to treatment.
Among the randomised studies eligible for inclusion in this review, there was a substantial variation in trial design, and many trials were small. This, along with the lack of clinical outcomes made it very difficult to pool data in meta‐analyses.
Quality of the evidence
The body of evidence in this review does not allow a robust conclusion on clinical efficacy. The conclusion on harms is more comprehensive, however, with serious limitations, especially regarding maternal mortality.
When using the 'Risk of bias' tool, we found that the majority of the included studies had high risk of bias in at least two domains. Also, 11 studies did not describe the randomisation method. In our experience this may disguise a serious selection bias (see Characteristics of excluded studies for examples). Many contact authors did not respond to our letters and potential violation of research ethics remains undisclosed.
Using the GRADE approach, we downgraded the quality level of the body of evidence based on the following reasons. The majority of the studies were open‐label and in some of the blinded studies it was unclear whether the blinding was successful. Clinical outcome measures, such as self‐reported health and most adverse events, are highly subjected to performance and detection bias. Some outcomes are however less or not affected by the subjectivity of the patient, for example infections with a rise in body temperature, elevated liver enzymes and mortality. A large proportion of the included studies had a high dropout rate (especially with oral iron), suggesting an increased risk of attrition bias. Thus, the study populations may have been selected if those who dropped out represented a more vulnerable part of the population. In such cases, it is important to report why the participants dropped out and to perform intention‐to‐treat analyses.
Inconsistency was only substantial for two outcomes (hepatic involvement and infections, Comparison 1) due to a high level of heterogeneity, which was addressed through sensitivity analyses.
We did not experience difficulties regarding indirectness.
Imprecision was however a common problem due to small sample sizes and broad confidence intervals, which naturally lowers our confidence in the effects.
Risk of reporting bias was high. Many studies did not report harmful effects, i.e. maternal mortality or survival even though this must be considered a fundamental knowledge, when testing the safety of drugs. The lack of reporting on harms raises serious concern that unfavourable events may have occurred, but not reported. In several trials there was discrepancy between preplanned and reported outcomes, which also indicates high risk of selective reporting. Also, we are aware of one trial suitable for inclusion, which was terminated prematurely by trial sponsors. This adds to the high risk of reporting bias.
Potential biases in the review process
Our inclusion criteria was based on haemoglobin (Hb) values, although we stated that inclusion criteria should be based on the presence and severely of anaemia symptoms. However, because the included studies chose to include patients based on Hb values as inclusion criteria, we could not use anaemia symptoms as inclusion criteria, as these were almost never systematically screened for.
Due to a lack of details in the method sections of the included studies, we may have pooled partly incompatible data, i.e. data from different time points during follow‐up and different dosages. Also, we could not clearly distinguish adverse events of treatment from anaemia symptoms.
We only reported outcomes prespecified for this review. Thus, the effect of the interventions on laboratory values are not reported.
Some data were provided graphically in the published reports. We consistently requested such data in numerical form along with corresponding standard deviations. When authors did not respond, we read the values from the graphs. However, the lack of data, such as standard deviation, prevented us from carrying out certain analyses.
Agreements and disagreements with other studies or reviews
We only reported outcomes that we found clinically relevant. The original version of this review (Dodd 2004) also reported on laboratory outcomes (Hb and haematocrit (HCT)). The previous review had the limitation that there were very few included studies. In our review we tried to establish the clinical effect of treatment, however this was hampered by the majority of the studies focusing on laboratory values.
We identified several other review articles on treatment of anaemia, including postpartum iron deficiency anaemia (Breymann 2010; Khalafallah 2012; Milman 2011; Milman 2011a; Milman 2012). Overall, the severity of anaemia, evaluation of treatment effect, and the proposed treatments were all based on laboratory values only. The search methods were not clearly described and thus we assumed that the search was not systematic.
We identified one systematic review on safety and efficacy of IV iron therapy, which included some clinical outcome measures in addition to Hb values (Litton 2013). However, this review was based on all types of patients and did not perform a subgroup analysis on postpartum women. Their results may therefore not be applicable for this specific population. The results showed an increased risk of infections in the IV treated group, but no difference in mortality, adverse events, or blood transfusions. The results on infections could not be confirmed by our meta‐analysis, but a similar trend was seen and the lack of significance may be due to the lower number of participants in our analysis.
One systematic review addressed prevention and treatment of maternal anaemia and showed a lack of evidence regarding treatment of anaemia in the postnatal period and poor reporting of clinical outcome measures (Parker 2012).
Authors' conclusions
Implications for practice.
It remains uncertain whether intravenous (IV) iron is superior to oral iron in terms of improving fatigue. Intravenous iron is much more expensive than oral iron and generally requires hospital admission or similar. Intravenous iron appears to carry a high compliance. However, safety of IV iron is not fully disclosed, as maternal mortality was insufficiently reported. Three allergic reactions and two cardiac adverse events were observed, and more data on harms are needed. Due to the sparse amount of evidence on clinical outcomes, it remains unclear whether IV iron therapy is clinically effective in treating the symptoms of postpartum anaemia.
Treatment with red blood cell (RBC) transfusion slightly improved fatigue, but this temporary effect was only shown by one study. Besides the general risks of RBC transfusion for the mother, the risk of antibody formation is particularly important as certain post‐transfusion antibodies may inhibit erythropoiesis and cause haemolytic disease of the newborn in future fetuses. RBC transfusion is expensive and in low‐income countries there may be a greater risk of donor‐transmitted infections and blood group incompatibility. Treatment with RBC transfusions to alleviate mild or symptom‐free anaemia in the stable patient should be weighed against the known risks of blood transfusion.
We found no evidence of advantageous effects of oral iron compared to IV iron or placebo, but oral iron did not have the potentially life‐threatening allergic reactions that IV iron did. Oral iron treatment causes gastrointestinal (GI) adverse effects, which can lead to a reduced quality of life and poor compliance. If compliance is poor, the women may remain untreated and perhaps start their next pregnancy while still being anaemic from the previous one. Clinicians should bear in mind the potentially liver toxic effect of ferrous sulphate, and this should be considered before prescribing oral ferrous sulphate to women with known hepatic disease.
We cannot make conclusions about erythropoietin (EPO) due to lack of evidence but notice that there are alternative treatment options in most cases. For the participants included in this review, we did not find any evidence that questions the safety of EPO, but this analysis is underpowered and the long‐term effects remain unknown.
The above mentioned treatment options have been tested in various combinations in several studies. We did not find evidence that favours one specific combination over others. However, the studies that combined treatments were often standing alone and offered limited information on clinical outcomes.
The 'normal range' for any laboratory test is determined by the background population; some healthy people will fall outside this arbitrary range and there are considerable fluctuations in haemoglobin (Hb) levels during the puerperal period. Laboratory values (i.e. Hb), should be used to confirm the diagnosis of postpartum anaemia in the presence of anaemia symptoms. The value of Hb in the absence of clinical symptoms of postpartum anaemia is uncertain. Once the diagnosis of anaemia has been established, treatment effect should primarily be measured as relief of clinical symptoms.
Implications for research.
After 40 years of research and 22 included studies on the subject, we are still not able to make a clear statement on how we should treat the clinical consequences of postpartum iron deficiency anaemia. The reasons for this are trial quality, the chosen interventions, the chosen outcomes and the many different study designs. Researchers tend to evaluate efficacy trough Hb values. The correlation between Hb levels and anaemia symptoms in postpartum women has not yet been clarified. We strongly encourage authors to choose clinically relevant outcomes, using validated measuring tools. Researchers should distinguish between anaemia symptoms and adverse effects of treatment to evaluate the overall clinical effect. Also, researchers should choose clinically relevant time points during follow‐up. Studies should report on survival and severe morbidity in all study participants. Trials should be designed following the CONSORT Consolidated Standards of Reporting Trials) guidelines in order to minimise sources of bias.
We encourage future researchers to conduct more randomised controlled trials on the treatment for postpartum iron deficiency anaemia focusing on interventions such as oral iron and IV iron treatment, comparing these with each other or placebo. Multicentre trials with large populations are encouraged. Due to the risk of irreversible adverse effects to mother and child, RBC transfusion studies should be reserved for bleeding or severe anaemia, and care should be taken to monitor all adverse effects, including allo‐immunisation. Also, it is of great importance to investigate the long‐term effects of any treatment on both mother and child.
Acknowledgements
We acknowledge the important work of the previous review team (Dodd 2004).
We are grateful for the Pregnancy and Childbirth Group Trials Register search provided by Cochrane Pregnancy and Childbirth's Trials Search Co‐ordinator.
Also, we are very grateful to those corresponding authors and trial investigators who took the time to answer our questions and provided the requested information (Backe 2009;Daniilidis 2011; Froessler 2013; Giannoulis 2009; Guerra 2012; Krafft 2011; Prick 2014; Van Wyck 2007; Wagstrom 2007; Westad 2008).
As part of the pre‐publication editorial process, this review has been commented on by two peers (an editor and referee who is external to the editorial team), a member of the Pregnancy and Childbirth Group's international panel of consumers and the Group's Statistical Adviser.
This project was supported by the National Institute for Health Research, via Cochrane Infrastructure funding to Cochrane Pregnancy and Childbirth. The views and opinions expressed therein are those of the authors and do not necessarily reflect those of the Systematic Reviews Programme, NIHR, NHS or the Department of Health.
Appendices
Appendix 1. Search strategy for WHO ICTRP and LILACS
We searched the WHO ICTRP registry through the advanced search function with the following key words and recruitment status marked as 'All':
#1: Title: Postpartum. Condition: anaemia OR anaemia #2: Title: blank. Condition: Post partum anemia #3: Title: blank. Condition: Postpartum anemia #4: Title: blank. Condition: Postpartum anaemia #5: Title: blank. Condition: Post partum anaemia
The LILACS (www.bireme.br) registry was searched through the advanced search function with the following key words and the filter 'Controlled Clinical Trial': ((postpartum) OR (post partum) OR (postnatal)) AND ((anaemia) OR (anemia)).
Data and analyses
Comparison 1. Intravenous iron versus oral iron.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Maternal mortality | 2 | 374 | Risk Ratio (M‐H, Random, 95% CI) | 2.95 [0.12, 71.96] |
| 2 Fatigue ‐ 14 days | 1 | 322 | Mean Difference (IV, Fixed, 95% CI) | ‐3.30 [‐8.04, 1.44] |
| 3 Fatigue ‐ 42 days | 1 | 329 | Mean Difference (IV, Fixed, 95% CI) | ‐2.10 [‐6.77, 2.57] |
| 4 SF‐36: Physical F(x) ‐ 14 days | 1 | 320 | Mean Difference (IV, Fixed, 95% CI) | 0.90 [‐3.84, 5.64] |
| 5 SF‐36: Physical role ‐ 14 days | 1 | 321 | Mean Difference (IV, Fixed, 95% CI) | 3.50 [‐2.03, 9.03] |
| 6 SF‐36: Bodily pain ‐ day 14 | 1 | 321 | Mean Difference (IV, Fixed, 95% CI) | ‐0.70 [‐6.00, 4.60] |
| 7 SF‐36: General health ‐ 14 days | 1 | 321 | Mean Difference (IV, Fixed, 95% CI) | 0.70 [‐3.09, 4.49] |
| 8 SF‐36: Vitality ‐ 14 days | 1 | 321 | Mean Difference (IV, Fixed, 95% CI) | 0.90 [‐3.64, 5.44] |
| 9 SF‐36: Emotional role ‐ 14 days | 1 | 321 | Mean Difference (IV, Fixed, 95% CI) | 1.10 [‐4.06, 6.26] |
| 10 SF‐36: Social function ‐ 14 days | 1 | 321 | Mean Difference (IV, Fixed, 95% CI) | 1.0 [‐4.08, 6.08] |
| 11 SF‐36: Mental health ‐ 14 days | 1 | 321 | Mean Difference (IV, Fixed, 95% CI) | ‐1.20 [‐4.84, 2.44] |
| 12 Depression | 1 | 361 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.33 [0.01, 8.00] |
| 13 Infections | 3 | 718 | Risk Ratio (M‐H, Random, 95% CI) | 1.70 [0.58, 5.03] |
| 14 Compliance to treatment | 5 | 890 | Risk Ratio (M‐H, Random, 95% CI) | 1.17 [1.01, 1.35] |
| 15 All gastrointestinal symptoms | 8 | 1307 | Risk Ratio (M‐H, Random, 95% CI) | 0.31 [0.20, 0.47] |
| 16 Constipation | 6 | 1217 | Risk Ratio (M‐H, Random, 95% CI) | 0.21 [0.11, 0.39] |
| 17 Nausea | 4 | 745 | Risk Ratio (M‐H, Random, 95% CI) | 0.30 [0.11, 0.81] |
| 18 Gastrointestinal pain | 4 | 543 | Risk Ratio (M‐H, Random, 95% CI) | 0.18 [0.04, 0.83] |
| 19 Diarrhoea | 3 | 569 | Risk Ratio (M‐H, Random, 95% CI) | 0.11 [0.02, 0.59] |
| 20 Vomiting | 1 | 128 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.40 [0.02, 9.66] |
| 21 Dyspepsia | 2 | 93 | Risk Ratio (M‐H, Random, 95% CI) | 0.36 [0.04, 3.20] |
| 22 Dysgeusia | 4 | 543 | Risk Ratio (M‐H, Random, 95% CI) | 7.20 [1.63, 31.76] |
| 23 Headache | 4 | 1124 | Risk Ratio (M‐H, Random, 95% CI) | 1.93 [0.87, 4.29] |
| 24 Hepatic involvement | 3 | 996 | Risk Ratio (M‐H, Random, 95% CI) | 0.45 [0.12, 1.71] |
| 25 Injection site discomfort | 4 | 702 | Risk Ratio (M‐H, Random, 95% CI) | 4.72 [1.03, 21.54] |
| 26 Skin rash | 2 | 489 | Risk Ratio (M‐H, Random, 95% CI) | 2.34 [0.79, 6.97] |
| 27 Urticaria | 1 | 291 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.14 [0.47, 36.59] |
| 28 Flush | 2 | 124 | Risk Ratio (M‐H, Random, 95% CI) | 9.00 [1.18, 68.81] |
| 29 Muscle cramp | 2 | 371 | Risk Ratio (M‐H, Random, 95% CI) | 6.05 [0.74, 49.68] |
| 30 Pain (not specified) | 1 | 128 | Risk Ratio (M‐H, Fixed, 95% CI) | 8.42 [0.44, 159.82] |
| 31 Seriouse adverse events (not specified) | 1 | 291 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.03 [0.26, 4.06] |
| 32 Anaphylaxis or evidence of hypersensitivity | 8 | 1454 | Risk Ratio (M‐H, Random, 95% CI) | 2.78 [0.31, 24.92] |
| 33 Arythmia | 1 | 90 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.26 [0.18, 101.86] |
| 34 Red blood cell transfusion | 4 | 606 | Risk Ratio (M‐H, Random, 95% CI) | 0.48 [0.19, 1.23] |
1.5. Analysis.
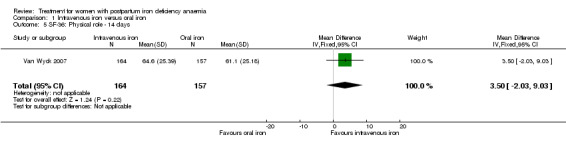
Comparison 1 Intravenous iron versus oral iron, Outcome 5 SF‐36: Physical role ‐ 14 days.
1.6. Analysis.
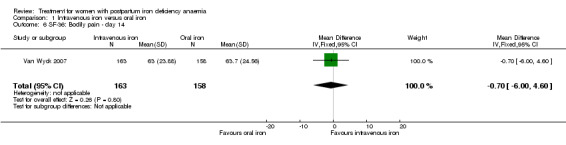
Comparison 1 Intravenous iron versus oral iron, Outcome 6 SF‐36: Bodily pain ‐ day 14.
1.7. Analysis.
Comparison 1 Intravenous iron versus oral iron, Outcome 7 SF‐36: General health ‐ 14 days.
1.8. Analysis.
Comparison 1 Intravenous iron versus oral iron, Outcome 8 SF‐36: Vitality ‐ 14 days.
1.9. Analysis.
Comparison 1 Intravenous iron versus oral iron, Outcome 9 SF‐36: Emotional role ‐ 14 days.
1.10. Analysis.
Comparison 1 Intravenous iron versus oral iron, Outcome 10 SF‐36: Social function ‐ 14 days.
Comparison 2. Red blood cell transfusion versus no transfusion.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 General fatigue ‐ 3 days | 1 | 388 | Mean Difference (IV, Fixed, 95% CI) | ‐0.80 [‐1.53, ‐0.07] |
| 2 General fatigue ‐ 6 weeks | 1 | 318 | Mean Difference (IV, Fixed, 95% CI) | ‐0.25 [‐1.22, 0.72] |
| 3 SF‐36: Physical functioning ‐ 1 week | 1 | 368 | Mean Difference (IV, Fixed, 95% CI) | 5.67 [0.84, 10.50] |
| 4 SF‐36: Social function ‐ 1 week | 1 | 369 | Mean Difference (IV, Fixed, 95% CI) | 5.34 [0.11, 10.57] |
| 5 SF‐36: Physical role ‐ 1 week | 1 | 366 | Mean Difference (IV, Fixed, 95% CI) | 4.56 [‐1.41, 10.53] |
| 6 SF‐36: Bodily pain ‐ 1 week | 1 | 368 | Mean Difference (IV, Fixed, 95% CI) | ‐2.0 [‐5.90, 1.90] |
| 7 SF‐36: General health ‐ 1 week | 1 | 369 | Mean Difference (IV, Fixed, 95% CI) | 2.18 [‐1.47, 5.83] |
| 8 SF‐36: Vitality ‐ 1 week | 1 | 369 | Mean Difference (IV, Fixed, 95% CI) | 1.88 [‐2.01, 5.77] |
| 9 SF‐36: Emotional role ‐ 1 week | 1 | 368 | Mean Difference (IV, Fixed, 95% CI) | 4.37 [‐4.51, 13.25] |
| 10 SF‐36: Mental health ‐ 1 week | 1 | 369 | Mean Difference (IV, Fixed, 95% CI) | 1.21 [‐2.29, 4.71] |
| 11 Infections | 1 | 519 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.93 [0.53, 1.61] |
| 12 Compliance to treatment | 1 | 519 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.11 [1.06, 1.17] |
| 13 Breastfeeding at six weeks | 1 | 297 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.91 [0.78, 1.07] |
| 14 Erythrocyte alloantibody formation | 1 | 519 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.03 [0.12, 74.15] |
| 15 Rash | 1 | 519 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.03 [0.12, 74.15] |
| 16 Fever | 1 | 519 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.06 [0.24, 104.84] |
| 17 Thromboembolic events | 1 | 519 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.01 [0.14, 7.13] |
| 18 Parenteral iron intolerance | 1 | 519 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.34 [0.01, 8.24] |
| 19 Transfusion reactions | 1 | 519 | Risk Ratio (M‐H, Fixed, 95% CI) | 7.08 [0.37, 136.41] |
2.6. Analysis.
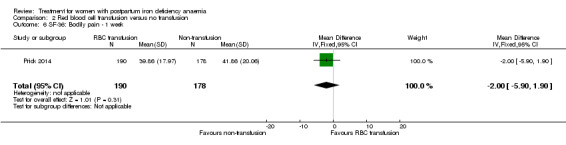
Comparison 2 Red blood cell transfusion versus no transfusion, Outcome 6 SF‐36: Bodily pain ‐ 1 week.
2.7. Analysis.
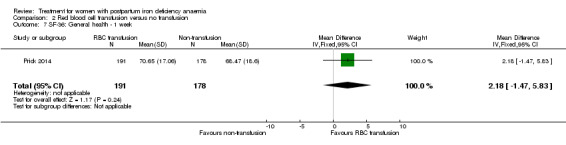
Comparison 2 Red blood cell transfusion versus no transfusion, Outcome 7 SF‐36: General health ‐ 1 week.
2.8. Analysis.
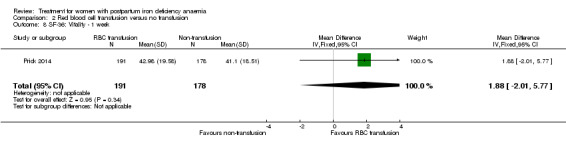
Comparison 2 Red blood cell transfusion versus no transfusion, Outcome 8 SF‐36: Vitality ‐ 1 week.
2.9. Analysis.
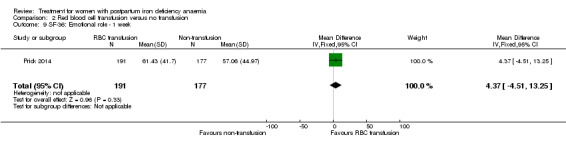
Comparison 2 Red blood cell transfusion versus no transfusion, Outcome 9 SF‐36: Emotional role ‐ 1 week.
2.15. Analysis.
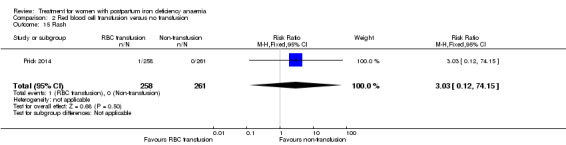
Comparison 2 Red blood cell transfusion versus no transfusion, Outcome 15 Rash.
2.16. Analysis.
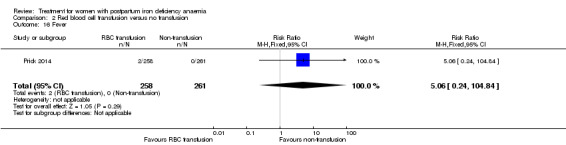
Comparison 2 Red blood cell transfusion versus no transfusion, Outcome 16 Fever.
2.17. Analysis.
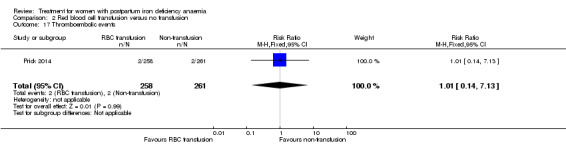
Comparison 2 Red blood cell transfusion versus no transfusion, Outcome 17 Thromboembolic events.
2.18. Analysis.
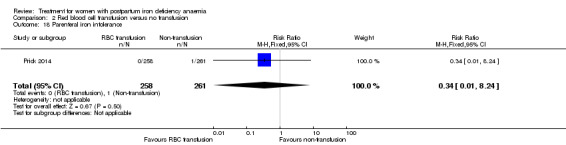
Comparison 2 Red blood cell transfusion versus no transfusion, Outcome 18 Parenteral iron intolerance.
Comparison 3. Oral iron versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Digit Symbol Substitution test ‐ 10 weeks | 1 | 51 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [‐2.76, 2.76] |
| 2 EPDS ‐ 10 weeks | 1 | 51 | Mean Difference (IV, Fixed, 95% CI) | 0.10 [‐0.86, 1.06] |
| 3 STAI ‐ 10 weeks | 1 | 51 | Mean Difference (IV, Fixed, 95% CI) | ‐0.40 [‐3.18, 2.38] |
| 4 Percieved Stress ‐ 10 weeks | 1 | 51 | Mean Difference (IV, Fixed, 95% CI) | 4.1 [1.70, 6.50] |
| 5 Breastfeeding at two days postpartum | 1 | 122 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.82 [0.58, 1.17] |
| 6 Back pain | 1 | 150 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.66 [0.42, 1.03] |
| 7 All gastrointestinal symptoms | 1 | 68 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.36, 2.79] |
Comparison 4. Oral iron, magnesium oxide and yeast extract versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 All gastrointestinal symptoms | 1 | 67 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.75 [1.23, 6.16] |
Comparison 5. Intravenous iron and oral iron after 4 weeks versus oral iron (week 5‐12).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 All gastrointestinal symptoms | 1 | 117 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.43 [0.83, 2.45] |
| 2 Abdominal pain | 1 | 117 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.72 [0.55, 13.48] |
| 3 Constipation | 1 | 117 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.20 [0.55, 2.60] |
| 4 Diarrhoea | 1 | 117 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.27 [0.35, 30.51] |
| 5 Nausea | 1 | 117 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.26 [0.14, 78.49] |
| 6 Dysgeusia | 1 | 117 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.26 [0.14, 78.49] |
| 7 Flatulence | 1 | 117 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.36 [0.02, 8.72] |
| 8 Melaena | 1 | 117 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.36 [0.02, 8.72] |
| 9 Headache | 1 | 117 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.36 [0.02, 8.72] |
5.2. Analysis.
Comparison 5 Intravenous iron and oral iron after 4 weeks versus oral iron (week 5‐12), Outcome 2 Abdominal pain.
5.3. Analysis.
Comparison 5 Intravenous iron and oral iron after 4 weeks versus oral iron (week 5‐12), Outcome 3 Constipation.
5.4. Analysis.
Comparison 5 Intravenous iron and oral iron after 4 weeks versus oral iron (week 5‐12), Outcome 4 Diarrhoea.
5.5. Analysis.
Comparison 5 Intravenous iron and oral iron after 4 weeks versus oral iron (week 5‐12), Outcome 5 Nausea.
5.6. Analysis.
Comparison 5 Intravenous iron and oral iron after 4 weeks versus oral iron (week 5‐12), Outcome 6 Dysgeusia.
5.7. Analysis.
Comparison 5 Intravenous iron and oral iron after 4 weeks versus oral iron (week 5‐12), Outcome 7 Flatulence.
5.8. Analysis.
Comparison 5 Intravenous iron and oral iron after 4 weeks versus oral iron (week 5‐12), Outcome 8 Melaena.
Comparison 6. Intravenous iron and oral iron versus oral iron.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Persistent anaemia symptoms on a VAS scale: 1 week | 1 | 72 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.75 [0.56, 5.46] |
| 2 Persistent anaemia symptoms on a VAS scale: 2 week | 1 | 72 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.6 [0.15, 2.33] |
| 3 Persistent anaemia symptoms on a VAS scale: 6 week | 1 | 72 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.0 [0.33, 27.50] |
| 4 EPDS ‐ 1 week | 1 | 72 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.0 [0.65, 13.88] |
| 5 Length of hospital stay | 1 | 72 | Mean Difference (IV, Fixed, 95% CI) | ‐0.30 [‐1.02, 0.42] |
| 6 Adverse events (pooled) ‐ 1 week | 1 | 72 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.67 [0.21, 2.16] |
| 7 Adverse events (pooled) ‐ 2 weeks | 1 | 72 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.29 [0.06, 1.28] |
| 8 Adverse events (pooled) ‐ 6 weeks | 1 | 72 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.4 [0.08, 1.93] |
| 9 Red blood cell transfusion | 2 | 112 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.15, 6.72] |
| 10 Anaphylaxis or evidence of hypersensitivity | 1 | 40 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
6.2. Analysis.
Comparison 6 Intravenous iron and oral iron versus oral iron, Outcome 2 Persistent anaemia symptoms on a VAS scale: 2 week.
6.7. Analysis.
Comparison 6 Intravenous iron and oral iron versus oral iron, Outcome 7 Adverse events (pooled) ‐ 2 weeks.
6.10. Analysis.
Comparison 6 Intravenous iron and oral iron versus oral iron, Outcome 10 Anaphylaxis or evidence of hypersensitivity.
Comparison 7. Erythropoietin (regardless of route) and intravenous iron versus intravenous iron.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Postpartum depression | 1 | 40 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.33 [0.01, 7.72] |
| 2 Infections | 2 | 80 | Risk Ratio (M‐H, Random, 95% CI) | 2.0 [0.72, 5.59] |
| 3 Compliance to treatment | 1 | 40 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.91, 1.10] |
| 4 Breasfeeding | 1 | 40 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.91, 1.10] |
| 5 Dysgeusia | 1 | 40 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.71 [0.27, 1.88] |
| 6 Flush | 1 | 40 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.0 [0.20, 20.33] |
| 7 Diarrhoea | 1 | 40 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.33 [0.01, 7.72] |
| 8 Headache | 1 | 40 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.14 [0.01, 2.60] |
| 9 Itching (including elevated liver enzymes) | 1 | 40 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.2 [0.01, 3.92] |
| 10 Dizziness | 1 | 40 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.33 [0.01, 7.72] |
| 11 Thrombophlebitis | 1 | 40 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.0 [0.26, 98.00] |
| 12 Red blood cell transfusion | 2 | 80 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.13, 69.52] |
7.6. Analysis.
Comparison 7 Erythropoietin (regardless of route) and intravenous iron versus intravenous iron, Outcome 6 Flush.
7.7. Analysis.
Comparison 7 Erythropoietin (regardless of route) and intravenous iron versus intravenous iron, Outcome 7 Diarrhoea.
7.8. Analysis.
Comparison 7 Erythropoietin (regardless of route) and intravenous iron versus intravenous iron, Outcome 8 Headache.
7.9. Analysis.
Comparison 7 Erythropoietin (regardless of route) and intravenous iron versus intravenous iron, Outcome 9 Itching (including elevated liver enzymes).
7.10. Analysis.
Comparison 7 Erythropoietin (regardless of route) and intravenous iron versus intravenous iron, Outcome 10 Dizziness.
Comparison 8. Subcutaneous EPO 10,000 U two doses and intravenous iron versus intravenous iron.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Postpartum depression | 1 | 40 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.33 [0.01, 7.72] |
| 2 Infections | 1 | 40 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.75 [0.19, 2.93] |
| 3 Headache | 1 | 40 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.67 [0.12, 3.57] |
| 4 Low blood pressure | 1 | 40 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.0 [0.13, 69.52] |
| 5 Diarrhoea | 1 | 40 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.33 [0.01, 7.72] |
| 6 Dizziness | 1 | 40 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.33 [0.01, 7.72] |
| 7 Itching (including elevated liver enzymes) | 1 | 40 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.2 [0.01, 3.92] |
| 8 Red blood cell transfusion | 1 | 40 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
8.4. Analysis.
Comparison 8 Subcutaneous EPO 10,000 U two doses and intravenous iron versus intravenous iron, Outcome 4 Low blood pressure.
8.5. Analysis.
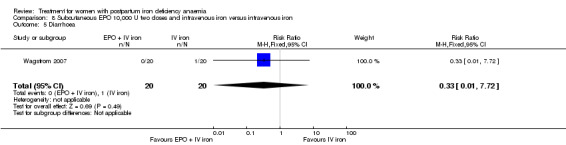
Comparison 8 Subcutaneous EPO 10,000 U two doses and intravenous iron versus intravenous iron, Outcome 5 Diarrhoea.
8.6. Analysis.
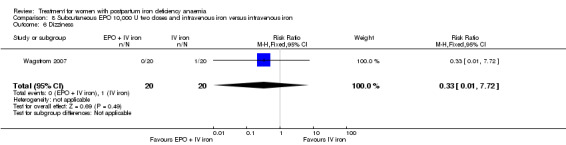
Comparison 8 Subcutaneous EPO 10,000 U two doses and intravenous iron versus intravenous iron, Outcome 6 Dizziness.
Comparison 9. Intravenous EPO, intravenous iron and oral iron versus intravenous iron and oral iron.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Leg paraesthesia | 2 | 76 | Risk Ratio (M‐H, Random, 95% CI) | 0.72 [0.08, 6.65] |
| 2 Red blood cell transfusion | 2 | 100 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 10. Subcutaneous EPO and oral iron versus oral iron.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Breastfeeding | 1 | 40 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.9 [1.21, 2.98] |
| 2 Red blood cell transfusions | 1 | 40 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.2 [0.01, 3.92] |
Comparison 14. Sensitivity analysis.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Heterogeneity ‐ Infections ‐ comparison 1 | 2 | 374 | Risk Ratio (M‐H, Random, 95% CI) | 1.07 [0.62, 1.84] |
| 2 Heterogeneity, fixed effect ‐ Infections ‐ comparison 1 | 3 | 718 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.49 [0.93, 2.38] |
| 3 Heterogeneity ‐ Hepatic involvement ‐ comparison 1 | 2 | 652 | Risk Ratio (M‐H, Random, 95% CI) | 0.22 [0.06, 0.75] |
| 4 Heterogeneity, fixed effect ‐ Hepatic involvement ‐ comparison 1 | 3 | 996 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.47 [0.21, 1.07] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Beard 2005.
| Methods | Single‐centre, double‐blind, randomised controlled trial involving iron deficient anaemic mothers and a non‐anaemic control group. Per protocol analysis. Follow‐up was 9 months postpartum. | |
| Participants | 500 puerperal women were screened, and 95 were included. South African population with low socioeconomic status. 21 women were non‐anaemic (control group). 64 anaemic women were randomised to 2 groups:
34 to the intervention group, 30 completed trial;
30 to the comparator group, 21 completed trial. Inclusion criteria: Hb 90 ‐ 115 g/L, and at least 2 of the following: MCV < 80 fL, TSAT < 15%, serum ferritin < 12 µg/L. Age between 18 and 30 years, primary caregivers, breastfeeding for the duration of the study, no chronic diseases, and healthy by physical health screen. Gestation age > 38 weeks, birthweight > 2500 g, no hospitalisation during the neonatal period, and Apgar scores consistent with normal intrauterine growth and development. Exclusion criteria: Hb < 90 g/L. |
|
| Interventions | Intervention: oral ferrous sulphate 125 mg, oral vitamin C 25 mg and 10 μg folic acid daily for 6 months, starting at inclusion 6‐8 weeks postpartum. Total non‐elemental iron dose ≈ 22,500 mg. Comparator: oral vitamin C 25 mg and 10 μg folic acid daily for 6 months. |
|
| Outcomes | Aim was to determine the association between mothers with postpartum iron deficiency anaemia and behavioural changes and present the data on the effect of maternal iron deficiency anaemia on maternal emotions and cognition. Specific preplanned outcome measures were not described. Reported outcomes: Scores on EPDS, STAI, Perceived Stress, Raven’s test, and Digit Symbol. |
|
| Notes | Source of funding: The ILSI Foundation. We only analysed the anaemic women, as per our inclusion criteria. The authors did not respond to the request on additional information. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No method described. |
| Allocation concealment (selection bias) | Low risk | One person who was aware of the code did the allocation. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Study was described as double‐blind. However, it was not clearly described who was blinded. Also, it was unclear whether there was an actual placebo pill, or perhaps if all of the treatment components were dosed in 1 tablet. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Same as for performance bias. If the patients were able to guess their treatment group, this could have influenced the subjective scales used as outcome measures for psychological well being. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | High dropout rate, twice as many in placebo group. Reasons for dropout not described. |
| Selective reporting (reporting bias) | High risk | Intended aim investigated and reported. However, adverse events and maternal mortality were not reported. |
| Other bias | Low risk | None known. |
Bhandal 2006.
| Methods | Single‐centre open‐label randomised controlled trial. Per protocol analysis. Follow‐up period was 40 days. | |
| Participants | 44 puerperal anaemic women from the United Kingdom (socioeconomic conditions not described), were randomised to 2 groups of 22. 1 woman from the comparator group dropped out due to secondary PPH. Inclusion criteria: age > 18 years, Hb < 90 g/L. Exclusion criteria: iron therapy during pregnancy, intolerance to iron derivatives, peripartum blood transfusion, history of asthma, thromboembolism, seizures, alcohol or drug abuse, renal or hepatic dysfunction. |
|
| Interventions | Intervention: IV ferrous sucrose 200 mg (Venofer®) on day 2 and 4 postpartum. Total dose IV iron was 400 mg. Comparator: oral ferrous sulphate 200 mg twice daily for 42 days. Total dose non‐elemental iron was 16,800 mg. |
|
| Outcomes | Preplanned outcomes were laboratory values. Compliance to treatment and adverse events during treatment were reported. | |
| Notes | Source of funding was not stated. Authors did not respond to the request on additional information. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated randomisation. |
| Allocation concealment (selection bias) | Low risk | Opaque, sealed envelopes were prepared and marked with a sequential numerical code by an independent person. After obtaining consent, the next consecutive envelope was opened by the recruiter, who was blinded to the envelope preparation. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial. High risk for subjective outcomes such as adverse events. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label trial. High risk for most outcomes. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Only 1 patient dropped out. However, no report on number of screened and excluded patients prior to randomisation. |
| Selective reporting (reporting bias) | High risk | Intended outcome measures reported. However, maternal mortality was not reported. |
| Other bias | Low risk | None known. |
Breymann 1996.
| Methods | Single‐centre, open‐label, randomised, controlled trial conducted in Switzerland. ITT analysis. Follow‐up was 6 weeks. | |
| Participants | 90 anaemic puerperal women were randomised into 3 groups of 30. Socioeconomic conditions were not described. Inclusion criteria: Hb < 100 g/L 48 to 72 hours after delivery, normal cardiac and renal function, oral iron substitution during pregnancy. Exclusion criteria: anaemia during pregnancy, peripartum infection, peripartum blood transfusion, haematological disease, previous myelosuppressive medication, history of thromboembolism, haemosiderosis, iron intolerance, or rheumatoid polyarthritis. |
|
| Interventions | Intervention referred to rhEPO (intravenously in group 3, subcutaneously in group 2). Group 1 (no EPO): IV ferric carboxymaltose (Ferrum Hausmann®) 100 mg single dose + oral combined tablet containing iron sulphate 160 mg elemental iron and 0.7 mg folic acid daily for 42 days. Total elemental iron dose was 6820 mg (non‐elemental iron dose unknown). Group 2: SC rhEPO (Eprex®) 300 U/kg as a single dose + IV ferric carboxymaltose (Ferrum Hausmann®) 100 mg single dose + oral combined tablet containing iron sulphate 160 mg elemental iron and 0.7 mg folic acid daily for 42 days. Total elemental iron dose was 6820 mg. Total rhEPO dose depended on weight, approximately 20,000 for a person weighing 70 kg. Group 3: IV rhEPO (Eprex®) 300 U/kg as a single dose + IV ferric carboxymaltose (Ferrum Hausmann®) 100 mg single dose + oral combined tablet containing iron sulphate 160 mg elemental iron and 0.7 mg folic acid daily for 42 days. Total elemental iron dose was 6820 mg. Total rhEPO dose depended on weight, approximately 20,000 for a person weighing 70 kg. Treatment was started from 48 to 72 hours after delivery. |
|
| Outcomes | No preplanned outcome measures stated. Adverse events during treatment were reported. | |
| Notes | Source of funding was not stated. Adverse events of iron infusion were reported for the 3 groups combined. Authors did not provide additional information on request. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No method described. |
| Allocation concealment (selection bias) | Low risk | Sealed envelopes. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label ‐ no EPO placebo. High risk for subjective outcomes such as adverse events. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label trial. High risk for most outcomes. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | It was not stated whether any women dropped out. The number of screened and excluded patients prior to randomisation was not reported. |
| Selective reporting (reporting bias) | High risk | No preplanned outcome measures stated. Maternal mortality was not reported. |
| Other bias | Low risk | None known. |
Breymann 2000.
| Methods | Single‐centre, randomised, controlled trial, conducted in Switzerland. ITT analysis. Follow‐up was 14 days. | |
| Participants | 60 anaemic puerperal women (socioeconomic conditions not described) randomised into 3 groups of 20. No women dropped out. Inclusion criteria: postpartum Hb < 100 g/L 24 to 72 hours postpartum. Exclusion criteria: anaemia during pregnancy peripartum blood transfusion, anaemia from causes other than blood loss, history of thromboembolism, signs of infection, history of seizures, alcohol and/or drug abuse, renal or hepatic dysfunction, previous myelosuppressive medication, haemosiderosis, history of iron intolerance, and rheumatoid polyarthritis. |
|
| Interventions | Intervention referred to rhEPO (group 1).
Group 1: IV rhEPO (Eprex®) 300 U/kg daily for 4 days + IV iron sucrose (Venofer®) 200 mg daily for 2 days, followed by oral treatment: tablet (Gynotardiferon ®, Robapharm) containing 80 mg elemental iron and folic acid 0.35 mg daily for 10 days.
Total rhEPO dose depended on weight, 84,000 U for a person weighing 70 kg. Total elemental iron dose was 1200 mg.
Group 2: IV rhEPO placebo (identical administration of physiological saline) + IV Iron sucrose (Venofer®) 200 mg daily for 2 days, followed by oral treatment: tablet iron sulphate (Gynotardiferon ®, Robapharm) containing 80 mg elemental iron and folic acid 0.35 mg daily for 10 days on an empty stomach. Total elemental iron dose was 1200 mg. Group 3: oral iron sulphate (Gynotardiferon ®, Robapharm) containing 80 mg elemental iron and folic acid 0.35 mg daily for 14 days. Total elemental iron dose was 1120 mg. |
|
| Outcomes | No preplanned outcome measures. Incidence and severity of serious or unusual adverse events were recorded. Information on maternal mortality was extrapolated from the numbers of blood tests. | |
| Notes | Financial support from the University of Zurich. Authors did not provide additional information on request. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No method described. |
| Allocation concealment (selection bias) | Low risk | Sealed envelopes. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Low risk for groups 1 and 2 (EPO and placebo), where the patients appear to have been blinded to what drug they received, but high in oral group. High risk for subjective outcomes such as adverse events. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Oral group was not blinded. High risk for most outcomes. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No dropouts. However, the number of screened and excluded patients prior to randomisation was not reported. |
| Selective reporting (reporting bias) | Unclear risk | No preplanned outcome measures mentioned. Data on adverse events were not group specific. |
| Other bias | Low risk | None known. |
Breymann 2008.
| Methods | Multicentre, open‐label, randomised, controlled trial. Trial was conducted from June 2004 to August 2005 in 20 centres in Poland, Romania and Russia. Randomisation ratio was 2:1, stratified by country and severity of anaemia. Efficacy analyses was both ITT and per protocol. Follow‐up was 12 weeks. | |
| Participants | 824 puerperal anaemic women were screened, 349 were randomised:
231 women to the intervention group, where 227 represented the ITT group and 179 represented the per protocol group; 118 women to the comparator group, where 117 represented the ITT group and 89 represented the per protocol group. Socioeconomic conditions were not described. Inclusion criteria: Hb < 105 g/L. Exclusion criteria: anaemia not caused by iron deficiency or haemorrhage. |
|
| Interventions | Intervention referred to IV ferric carboxymaltose. Intervention: IV infusion of ferric carboxymaltose (Ferinject®) at a maximum dose of 1000 mg iron over 15 min (15 mg iron/kg body weight if body weight < 66 kg) on day 1, with subsequent doses at 1‐week intervals until each patient's calculated total iron requirement was reached (up to 3 weekly infusions). Patients' total iron requirement was calculated using the modified formula of Ganzoni. Comparator: oral ferrous sulphate (Plastufer®) 100 mg twice daily for 12 weeks. Total non‐elemental iron dose was 16,800 mg. Treatment was initiated within 7 days postpartum. |
|
| Outcomes | Preplanned outcome measures were laboratory values, and safety of the mother and child. Infections and compliance to treatment were reported. | |
| Notes | This study was supported by an unrestricted scientific grant from Vifor International Inc., Switzerland. Authors did not provide additional information on request. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomised 2:1 ratio, method not described. |
| Allocation concealment (selection bias) | Unclear risk | Method not described. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial. High risk for subjective outcomes such as adverse events. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label trial. High risk for most outcomes. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Unknown reason for consent withdrawal and high dropout rate may have led to selection in the population. |
| Selective reporting (reporting bias) | High risk | Intended outcomes reported. However, maternal mortality was not reported. |
| Other bias | Low risk | None known. |
Froessler 2013.
| Methods | Single‐centre, open‐label, randomised, controlled trial, conducted in Australia from 2009 to 2010. Per protocol analyses. Follow‐up was 6 weeks. | |
| Participants | Both pregnant and puerperal anaemic women. Originally 5950 women were screened. Of the postpartum population 90 women were randomised into 2 groups: 37 to the intervention group, where 31 completed the trial; 53 to the comparator group, where 43 completed the trial. This population came from a very low socioeconomic background (unemployment, teenage pregnancies, (qualitative) nutritional deficiencies, migrants from the Asian Pacific region, Africa and the subcontinent). Inclusion: Hb < 110 g/L and ferritin < 12 μg/L either antepartum or within 72 hours postpartum, following caesarian section and vaginal delivery with at blood loss > 500 mL. Exclusion: other cause of anaemia, acute systemic infection, vitamin B12 or folate deficiency, hepatis, HIV, severe asthma, allergy to iron, pre‐treatment ferritin > 300, multiple pregnancy or high risk of premature birth. |
|
| Interventions | Intervention referred to IV iron sucrose (Venofer®). Intervention group (n = 31): IV iron sucrose 200 mg given twice with a minimum of 24 hours apart + oral folic acid 600 μg daily for 42 days. Total iron dose was 400 mg. Comparator group (n = 43): oral ferrous sulphate 250 mg containing 80 mg elemental iron twice daily + oral folic acid 600 μg daily for 42 days. Total elemental iron dose was 6720 mg. Total non‐elemental iron dose was 21,000 mg. Treatment was initiated between days 1 and 3 postpartum. |
|
| Outcomes | The aim was to determine if treatment could decrease the incidence of severe anaemia, and to measure if an increase in haematological indices was associated with a reduction in the rate of blood transfusions and associated complications, as well as improvement in maternal and neonatal outcomes. Severe adverse events were reported. |
|
| Notes | The source of funding was not stated. The authors declare no conflict of interest. The author of this trial provided additional information on request. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The randomisation was done through a telephone service. |
| Allocation concealment (selection bias) | Low risk | Allocation was conducted by a third party who was blinded to patient data. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial. High risk for subjective outcomes such as adverse events. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label trial. High risk for most outcomes. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | The proportion of dropouts is similar in the 2 groups. However, the numbers are high: 16.2% and 18.9%. The reason for dropout is lost to follow‐up and decline of further participation. |
| Selective reporting (reporting bias) | High risk | Preplanned aim was investigated. However, mild to moderate adverse events and maternal mortality were not reported. |
| Other bias | Low risk | None known. |
Guerra 2012.
| Methods | Single‐centre, comparative, open‐label, randomised trial, conducted in Spain between March 1 and May 31, 2008. ITT analysis. Follow‐up period was 6 weeks. | |
| Participants | 180 puerperal women were screened. 13 women were randomised into 2 groups:
6 women to intervention group A all of whom completed the trial;
7 women to comparator group B, 5 of whom completed the trial. Socioeconomic conditions were not described. Inclusion criteria: age > 18 years, Hb 70 to 100 g/L, and ferritin > 15 µg/L at 24 hours postpartum. Exclusion criteria: iron intolerance, anaemia not caused by iron deficiency, peripartum blood transfusion, severe asthma and atopy, thromboembolism, alcohol or drug abuse, hepatitis B, C or HIV, infection, renal or hepatic dysfunction, no consent. |
|
| Interventions | Intervention referred to IV iron sucrose (Venofer ®). Intervention group A: IV Iron sucrose (Venofer ®) 200 mg on day 2 and 4 after delivery. Total iron dose was 400 mg. Comparator group B: oral ferrous sulphate (Tardyferon®) containing 200 mg twice daily before meals for 42 days. Total dose of non‐elemental iron was 16,800 mg. |
|
| Outcomes | No preplanned outcomes. The aim of the study was to compare efficacy and safety between IV and oral iron treatment. Maternal mortality, infections, compliance to treatment and adverse events during treatment were registered. | |
| Notes | Source of funding not stated. Authors declare no conflict of interest. Trial authors provided unpublished information and corrections to the published text on request. They reported an error in table 2 on page 193, where the values for group A and B are reversed. Table 3 is correct. The article is published in Spanish. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | System based on random distribution of envelopes in 1:1 ratio. |
| Allocation concealment (selection bias) | Low risk | Sealed envelopes. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial. High risk for subjective outcomes such as adverse events. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label trial. High risk for most outcomes. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 2 out of 7 in the comparator group dropped out. Although, the authors state that dropout was unrelated to treatment, it is not specified what the exact reasons were. It is therefore not possible to assess if the dropouts were in fact not related to the trial. |
| Selective reporting (reporting bias) | Unclear risk | No preplanned outcome measures stated. |
| Other bias | Low risk | None known. |
Jain 2013.
| Methods | Single‐centre, open‐label, randomised (block randomisation), controlled trial conducted in India. Per protocol analyses. Follow‐up was 14 days. | |
| Participants | 46 women with postpartum anaemia were randomised into 2 groups: 23 to the intervention group where 21 completed per protocol; 23 to the comparator group where 20 completed per protocol. Socioeconomic conditions were not described. Inclusion criteria: age > 18 years, Hb < 80 g/L within 48 hours postpartum. Exclusion criteria: placenta previa, placental abruption, preeclampsia, clotting disorders, and peripartum blood transfusion. |
|
| Interventions | Intervention referred to IV iron sucrose. Intervention: IV iron sucrose 300–600 mg divided into 3 doses every alternate day for 3 days. Total iron dose was individually calculated. Comparator: oral ferrous fumarate 300 mg daily for 14 days. Each dose contained 99 mg elemental iron. Total elemental iron dose was 1386 mg. Total non‐elemental iron dose was 4200 mg. Treatment was initiated between 24 and 48 hours after delivery. |
|
| Outcomes | No preplanned outcomes stated. The objective was to compare effectiveness of IV iron sucrose vs oral ferrous fumarate in postpartum anaemia. Adverse events were reported. | |
| Notes | Source of funding was not stated. Trial authors did not respond to our request for further details. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated randomisation schedule. |
| Allocation concealment (selection bias) | Unclear risk | No method described. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial. High risk for subjective outcomes such as adverse events. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label trial. High risk for most outcomes. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Low dropout rate. However, reason for dropout not known and number of screened and excluded patients prior to randomisation was not reported. |
| Selective reporting (reporting bias) | High risk | No preplanned outcome measures stated. Maternal mortality was not reported. |
| Other bias | Low risk | None known. |
Krafft 2011.
| Methods | Single‐centre, open‐label, randomised trial conducted in Switzerland. ITT analysis. Follow‐up was 15 days. | |
| Participants | 40 severely anaemic puerperal women were randomised 1:1 to 2 groups. There were no dropouts. Socioeconomic conditions were not described. Inclusion criteria: prepartal Hb > 100 g/L, followed by severe postpartum anaemia, defined by a Hb < 85 g/L 24 to 48 hours after delivery. Exclusion criteria: haematological, chronic inflammatory or malignant disease, cardiac or renal dysfunction, haemosiderosis, history of iron intolerance, peripartum blood transfusion. |
|
| Interventions | Intervention referred to EPO. Group 1 (Comparator): IV iron sucrose (Venofer®) 200 mg for 4 days. Total iron dose was 800 mg. Group 2 (Intervention): IV rhEPO (Eprex®) 10,000 U for 4 days + IV iron sucrose (Venofer®) for 4 days. Total EPO dose was 40,000 U. Total elemental iron dose was 800 mg. Treatment started at the day of delivery. |
|
| Outcomes | Preplanned outcome measures:
primary: proportion of patients not anaemic after 2 weeks; secondary: laboratory values and identification of subgroups which benefit from additional rhEPO treatment. Maternal mortality was extrapolated as lack of dropouts. Infections, compliance to treatment, breastfeeding and adverse events during treatment were also reported. |
|
| Notes | Source of funding was not stated. Trial authors provided unpublished information on request. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Envelopes containing numbers randomly allocated to 1 of the 2 groups. |
| Allocation concealment (selection bias) | Low risk | Sealed envelopes. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial. High risk for subjective outcomes such as adverse events. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label trial. High risk for most outcomes. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | There were no dropouts. However, the number of screened and excluded patients prior to randomisation was not reported. |
| Selective reporting (reporting bias) | Low risk | Preplanned outcomes were reported. |
| Other bias | Low risk | None known. |
Krauss 1972.
| Methods | Single‐centre, single‐blinded, randomised, controlled trial conducted in Germany. For each 3 women with similar parity, pre‐treatment Hb (± 0.3 g%) and age (± 3 years) 1 was allocated to each treatment group. Analysis of laboratory values was done per protocol, analyses on harms was ITT. Follow‐up was 30 days. | |
| Participants | 101 puerperal women were randomised to 3 groups: 34 to intervention group S, 32 completed trial; 34 to intervention group K, 32 completed trial; 34 to control group L, 33 completed trial. Socioeconomic conditions were not described. Inclusion criteria were not described. Exclusion criteria were former iron therapy or transfusion, malabsorption, massive bleeding, GI disease, thyroid disease. |
|
| Interventions | Intervention referred to oral iron. Group S: oral tablet Eryfer® containing 152 mg iron sulphate (elemental 50 mg), 222 mg ascorbic acid and 84 mg sodium bicarbonate twice daily for 30 days. Total non‐elemental iron dose was 9120 mg, total elemental iron dose was 3000 mg. Group K: oral tablet containing 324 mg ferrous sulphate (elemental iron 102 mg), 25 mg magnesium oxide and yeast extract containing vitamin B once daily for 30. Total non‐elemental iron dose was 9720 mg, total elemental iron dose was 3060 mg. Group L: oral tablet empty preparation containing 1 g of milk sugar twice daily for 30 days. |
|
| Outcomes | No preplanned outcomes stated. Adverse events during treatment were reported. | |
| Notes | Source of funding was not stated. Hb values for inclusion in this study were not defined. In table 1 it is shown, that the mean Hb in all groups was < 120 g/L prior to treatment. The table also shows a value of ± s for each Hb measurement. Assuming that "s" is the standard deviation, pre‐treatment Hb plus 2 standard deviations exceeds the value of 120 g/L, which is criteria for this review. However, the population is small and not necessarily normally distributed. Thus, in theory the results can be skewed to the left and not contain any values above 120 g/L. Therefore we chose to include the study. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Stratification based on parity, pre‐treatment Hb (± 0.3 g%) and age (± 3 years). However, the method of this sequence generation was not described. |
| Allocation concealment (selection bias) | Unclear risk | No method described |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | The dose regiment for group K is different than that of group S and L. Thus, the blinding of the patients was inadequate. High risk for subjective outcomes such as adverse events. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | The blinding of the patients was inadequate. High risk for most outcomes. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Few dropouts. Reasons for dropout were reported. However, the number of screened and excluded patients prior to randomisation was not reported. |
| Selective reporting (reporting bias) | High risk | No preplanned outcomes measures stated. However, maternal mortality was not reported. |
| Other bias | Unclear risk | The Hb value as a criteria for inclusion was not stated, thus the study may include non‐anaemic women. |
Lebrecht 1995.
| Methods | Single‐centre, double‐blind, randomised controlled trial conducted in 1992 in Germany. Per protocol analysis. Follow‐up was 4 weeks. | |
| Participants | 36 puerperal anaemic women were randomised to 2 groups:
24 to intervention group, 23 completed the trial, 1 women dropped out due to leg paraesthesia;
12 to comparator group, all of whom completed the trial. Socioeconomic conditions were not described. Inclusion criteria: Hb < 90 g/L on 2nd day postpartum. Anaemia caused by childbirth or pregnancy, healthy baby with gestational age of minimum 38 weeks. Exclusion: other type of anaemia, caesarean section, other surgery, seizures, infections, cardiovascular disease, thromboembolic disease, alcohol or drug abuse, blood transfusions, kidney or liver dysfunction. |
|
| Interventions | Intervention referred to EPO. Intervention: IV rhEPO 20,000 IU single dose + IV iron 400 mg (Ferrum Hausmann®) single dose + oral iron 200 mg (Ferrum Hausman®) + folic acid 1 mg daily for 28 days (starting on second day). Total non‐elemental iron dose was 6000 mg. Comparator: IV placebo EPO (unknown agent) single dose + IV iron 400 mg (Ferrum Hausmann®) + oral iron 200 mg + folic acid 1 mg daily for 28 days (starting on second day). Total non‐elemental iron dose was 6000 mg. |
|
| Outcomes | No preplanned outcome measures stated. Objective was to show if it is enough to use combined oral and IV iron therapy for a quick correction of anaemia, or if it is necessary to supplement with EPO. A brief comment on life quality (unsupported by data), and adverse events were stated. Maternal mortality was extrapolated as lack of lost to follow‐up. |
|
| Notes | Authors did not respond to the request on additional information. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No method described. |
| Allocation concealment (selection bias) | Unclear risk | No method described. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Placebo‐controlled, double‐blinded, however not stated who exactly was blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It is not stated how the personnel were blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Only 1 dropout. However, number of screened and excluded patients prior to randomisation not reported. |
| Selective reporting (reporting bias) | High risk | No statement of preplanned outcomes. The authors comment briefly on quality of life but method is not adequately described and the evaluation was not supported by data. |
| Other bias | Low risk | None known. |
Makrydimas 1998.
| Methods | Single‐centre randomised controlled trial, conducted in Greece. ITT analysis. Follow‐up was 40 days. | |
| Participants | 40 puerperal anaemic women were randomised into 2 groups of 20 on the first day following delivery. There were no dropouts. Socioeconomic conditions were not described. Inclusion criteria: Age 19 to 44 years, Hb < 100 g/L on first day postpartum, no serious illness, no pre‐eclampsia. No exclusion criteria stated. | |
| Interventions | Intervention referred to EPO. Intervention: SC injection rhEPO 200 IU/kg/day for 15 days, oral iron 200 mg/day for 40 days and folic acid 5 mg/day for 40 days. Total rhEPO dose varied according to weight (3000 IU/kg for a person weighing 70 kg). Total non‐elemental iron dose was 8000 mg. Comparator: oral iron 200 mg/day and folic acid 5 mg/day for 40 days. Total non‐elemental iron dose was 8000 mg. |
|
| Outcomes | Preplanned outcomes were subjective symptoms, the ability to lactate and psychological well being. Length of hospital stay was reported. Maternal mortality was extrapolated as lack of dropouts. | |
| Notes | Source of funding was not stated. Improvement in psychological well being was a preplanned measure, however no results were reported. Many of the results were reported as medians. Authors did not respond to the request on additional information. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No method described. |
| Allocation concealment (selection bias) | Unclear risk | No method described. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial, no method of blinding described. High risk for subjective outcomes such as adverse events. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label trial. High risk for most outcomes. However, low risk for maternal mortality, irrespective of blinding. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | There were no dropouts, small study. However, the number of screened patients prior to randomisation was not reported. |
| Selective reporting (reporting bias) | High risk | Psychological well being was planned to be investigated (mentioned in methods), but results are not reported, except an undocumented statement in discussion. |
| Other bias | Low risk | None known. |
Meyer 1995.
| Methods | Multicentre, double‐blind, randomised, controlled trial conducted in 2 German centres from 1991 to 1992. Analysis appears to be per protocol. Follow‐up was 5 days. | |
| Participants | 90 puerperal women were selected, 71 were randomised: 35 to the intervention group and 36 to the placebo group. Socioeconomic conditions were not described. Inclusion criteria: Hb < 100 g/L. Exclusion criteria were not stated. |
|
| Interventions | Intervention referred to EPO. Intervention: IV rhEPO (Eprex) 10,000 U twice with a 24‐hour interval during the first 5 days postpartum. Total EPO dose 20,000 U. Comparator: IV placebo twice with a 24 hour interval during the first 5 days postpartum. |
|
| Outcomes | Preplanned outcome measures were not specified. Objectives were to test the 2 hypotheses:
1) postpartum anaemia implies an additional stress; hence, women with postpartum anaemia suffer more from maternity blues or distress than women with a "normal" postpartum Hb concentration;
2) treatment of postpartum anaemia with rhEPO reduces postpartum blues or distress. Psychologic status was measured using 2 questionnaires; the “Blues Questionnaire” during the first 5 consecutive days postpartum and the “SCL‐90‐R”, used on the 5th day postpartum. |
|
| Notes | Source of funding not stated. The data on psychological well being were not eligible for analysis due to missing standard deviations. Authors did not respond to the request on additional information. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No method described. |
| Allocation concealment (selection bias) | Unclear risk | No method described. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Trial was described as double‐blinded and placebo‐controlled, however it was not stated who exactly was blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It is not stated who of the personnel was blinded. Low risk for the subjective questionnaire since the patients were blinded. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | There was a high dropout rate of more than 20% caused by consent withdrawal or transferal of the child to an intensive care unit. These were excluded prior to randomisation. Unknown if the dropouts would have been equally distributed in intervention and placebo. Number of screened patients prior to randomisation was not reported. |
| Selective reporting (reporting bias) | High risk | Intended objectives were investigated and reported. However, adverse events and maternal mortality were not reported. |
| Other bias | Low risk | None known. |
Mumtaz 2011.
| Methods | Multicentre, open‐label, randomised, controlled trial, conducted in 2009 in 2 centres in Pakistan. Per protocol analysis. Follow‐up was 40 days. | |
| Participants | 86 women were recruited to the trial, 80 were randomised into 2 groups of 40. 76 of the women had a caesarean section. Socioeconomic conditions were not described. Inclusion criteria: Hb < 90 g/L, ferritin < 15 μg/dL at 24 to 48 hours postpartum. Exclusion: intolerance to iron derivatives, peripartum blood transfusion, history of asthma, thromboembolism, seizure, alcohol or drug abuse, infection, renal or hepatic dysfunction. |
|
| Interventions | Intervention referred to IV iron sucrose. Intervention: IV iron sucrose infusion 200 mg on day 2 and 4. Total iron dose was 400 mg. Comparator: the women were advised to take oral ferrous sulphate 200 mg twice daily together with meals for 42 days. Total non‐elemental iron dose was 16,000 mg. |
|
| Outcomes | No preplanned outcome measures stated. The aim was to compare the efficacy of IV ferrous sucrose vs oral ferrous sulphate on postpartum iron deficiency anaemia. Adverse events during treatment were reported. | |
| Notes | Source of funding not stated. Several errors were detected: adverse events were reported as twice as many in the text (page 3) compared to Table 3. Unknown which data are correct. We chose to use the lowest reported. The authors did not respond to our request for further details. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method not described. |
| Allocation concealment (selection bias) | Unclear risk | Method not described. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial. High risk for subjective outcomes such as adverse events. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label trial. High risk for most outcomes. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Low dropout rate. We assume that initial randomisation was 1:1, which would make the 6 dropouts equally distributed among the groups. Reasons for dropout were non‐compliance and complications. Number of screened and excluded patients prior to randomisation was not reported. |
| Selective reporting (reporting bias) | High risk | No preplanned outcome measures stated. Maternal mortality was not reported. |
| Other bias | High risk | Several errors and inconsistencies were detected. |
Perello 2014.
| Methods | Single‐centre, double‐blind, randomised controlled trial conducted from November 2005 to January 2008 in Barcelona, Spain. Per protocol analysis. Follow‐up was 6 weeks. | |
| Participants | 103 puerperal women were screened, 72 of these were randomised into 2 groups of 36. 31 women in the intervention group, and 29 women in the comparator group completed the trial. Inclusion criteria: age > 18 years; postpartum haemorrhage or severe anaemia symptoms and Hb 60 to 80 g/L within the 48 hours after delivery. Written informed consent. Exclusion criteria: antenatal chronic anaemia, infection, asthma, eczema, topical allergy, oral iron intolerance, women with blood transfusion criteria (Hb < 60 g/L or intolerable symptoms of anaemia), anaemia due to other causes than blood loss or iron deficiency, cirrhosis, hepatitis, elevation of liver enzymes, overload or alteration in iron metabolism, hypersensitivity to IV iron, or unwillingness to participate. |
|
| Interventions | Intervention referred to IV iron sucrose. Intervention: IV iron sucrose (Venofer) 200 mg daily for 2 days. Then 2 tablets of ferrous sulphate 525 mg (containing 105 mg of elemental iron per tablet) daily for 30 days. Total elemental iron dose was 6700 mg, total dose of non‐elemental oral iron was 31,500 mg. Comparator: IV NaCl 0,9% equal volume for 2 days. Then 2 tablets of ferrous sulphate 525 mg (containing 105 mg of elemental iron per tablet) daily for 30 days. Total elemental iron dose was 6300 mg, total dose of non‐elemental oral iron was 31,500 mg. |
|
| Outcomes | Preplanned outcome measures: primary: between‐group‐difference in the mean Hb and HCT at 6 weeks postpartum; secondary: ferritin, iron‐binding capacity, reticulocyte count, serum iron, and MCV. Longitudinal progression of Hb and HCT levels within groups. Clinical anaemic signs (pulse and blood pressure), and symptoms (headache, fatigue, tinnitus, dyspnoea, palpitations, tingling, dizziness, nausea, and difficulty in concentration). Levels of depression and anxiety. |
|
| Notes | The trial was partially financed by J Uriach & Co. Information for this description is collected from trial registry and the main report. Authors did not respond to the request on additional information. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated randomisation system. |
| Allocation concealment (selection bias) | Low risk | Sealed envelopes. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | An opaque perfusion system was used in both groups to avoid the identification of the treatment received and maintain the double‐blind nature of the study. Thus, low risk for outcomes evaluated by the patients, such as adverse events. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | It is not stated whether persons who handled patient data were blinded. We are only sure that the patients and the clinicians were blinded. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Lost to follow‐up not equally distributed among groups (14% vs 8%). |
| Selective reporting (reporting bias) | High risk | Intended outcomes (trial register and full report) are reported. However, adverse events are pooled for each group, which makes it impossible to know which adverse events occurred. Mortality is not mentioned and 8 patients were lost to follow‐up. |
| Other bias | Low risk | None known. |
Prick 2014.
| Methods | Multicentre, open‐label, randomised, controlled trial, conducted from May 2004 to February 2011 in 37 centres in the Netherlands. ITT analyses. Follow‐up was 6 weeks. | |
| Participants | 1011 puerperal women were screened, 521 were randomised into 2 groups: 259 to intervention group, 1 did not meet inclusion criteria, 258 represented the ITT population, 251 completed the trial per protocol; 262 to comparator group, 1 did not meet inclusion criteria, 261 represented the ITT population, 228 completed the trial per protocol. The socioeconomic status of the population was considered above average based on education level: non/low (3%), lower/senior secondary vocational education (51% to 56%), higher professional education and university (41% to 46%). Participants were mainly of western ethnic origin (76% to 78%). Inclusion criteria: Hb 48 to 79 g/L 12 to 24 hours postpartum, post partum haemorrhage (> 1000 mL and/or a decrease in Hb > 19 g/L), good knowledge of the Dutch language. Exclusion criteria: dyspnoea, syncope, tachycardia, angina pectoris and/or transient ischaemic attacks, RBC transfusion administered within 12 hours of delivery, severe pre‐eclampsia, severe infection, congenital haemolytic disease, compromised immunological status, malignancy, severe co‐morbidity, and death or critical condition of the neonate. |
|
| Interventions | Intervention referred to RBC transfusion. Intervention: At least 1 unit of RBCs with the aim to a Hb of at least 89 g/L. Comparator: non‐intervention. RBC transfusion was allowed if severe symptoms of anaemia developed or at their physicians’ discretion. Additional use of iron and/or folic acid supplementation according to local protocol was allowed. Iron substitution was administered to 88% of the women in the non‐intervention group. |
|
| Outcomes | Primary outcome: physical fatigue at day 3, measured with the Multidimensional Fatigue Inventory. Secondary outcomes: remaining health related quality of life scores, general and mental fatigue scores (from protocol), number of RBC units transfused, transfusion reactions, length of hospital stay, and physical complications during follow‐up. Data on breastfeeding and compliance were also reported. |
|
| Notes | Source of funding: grants from the Landsteiner Foundation for Blood Transfusion Research (file number 0904) and Stichting Vrienden van de Bloedtransfusie (file number 1201005). Previous funding by Sanquin Blood Supply Foundation, the Netherlands, and the Department of Obstetrics, Erasmus Medical Centre, Rotterdam, the Netherlands. The authors responded to our request for further detail. This trial was registered in ClinicalTrials.gov (NCT00335023) and at the Dutch Trial Register (NTR335). |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Web‐based application for block randomisation with a variable block size of 2 to 8 women. |
| Allocation concealment (selection bias) | Low risk | Randomisation was performed through the study web site (www.studies‐obsgyn.nl/womb); thus allocation was concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial. High risk for subjective outcomes such as adverse events. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label trial. High risk for most outcomes. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Differences in baseline characteristics of questionnaire responders vs non‐responders (western ethnicity in 81% vs 54%, mean age 31 vs 28 years, median blood loss 1500 vs 1150 mL). Big difference in compliance to allocated treatment: 8 vs 34. The design of this trial carries a high risk for selecting the study population. |
| Selective reporting (reporting bias) | High risk | Intended outcomes reported. Maternal mortality was not reported. |
| Other bias | Low risk | None known. |
Seid 2008.
| Methods | Multicentre, open‐label, randomised controlled trial, conducted from May 9, 2006 to December 27, 2006 in 28 centres in USA. Participants were stratified based on Hb, ferritin levels, and mode of delivery. ITT analyses. Follow‐up was 6 weeks. | |
| Participants | 291 anaemic puerperal women were randomised to 2 groups:
143 to the intervention group,where 138 competed the study, modified ITT population was 139 (72.7% were Caucasian, 10.8% Hispanic, 15.8% African American, 0% Asian, 0.7% other); 148 to comparator group,where, 144 competed the study, modified ITT population was 147 (65.3% were Caucasian, 13.6% Hispanic, 18.4% African American, 2% Asian, 0.7% other). Socioeconomic conditions were not described, study population were of mixed ethnic origin. Inclusion criteria: healthy women, Hb < 100 g/L 10 days or less postpartum on 2 or more laboratory tests conducted at least 12 hours apart. Exclusion criteria: estimated blood loss > 1 litre 24 hours prior to randomisation, history of anaemia other than iron deficiency anaemia or peripartum bleeding, current treatment with myelosuppressive therapy or asthma therapy, recent blood transfusions, or EPO treatment within 3 months prior to screening. |
|
| Interventions | Intervention referred IV ferric carboxymaltose. Intervention: IV ferric carboxymaltose (brand unknown) given weekly until individual calculated cumulative dose was reached or a maximum of 2500 mg was administered. The maximum single weekly dose was 15 mg/kg, not to exceed 1000 mg per dose. Comparator: oral ferrous sulphate 325 mg (65 mg elemental iron) 3 times daily for 6 weeks. Total dose of elemental iron was 8190 mg. Total dose of non‐elemental iron was 40,950 mg. |
|
| Outcomes | Preplanned outcomes were laboratory values and adverse events. Adverse events for participants who were randomised to ferric carboxymaltose and withdrew from the study early were reported for 28 days after the last treatment. | |
| Notes | Trial funded by research grants from American Regent, Inc. Authors provided additional information on request. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Centralised computer randomisation system. |
| Allocation concealment (selection bias) | Unclear risk | No method described. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial. High risk for subjective outcomes such as adverse events. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label trial. High risk for most outcomes. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Low dropout rate, detailed flowchart which accounts for all dropouts. However, reason for voluntary dropouts was not stated and the number of screened and excluded patients prior to randomisation was not reported. |
| Selective reporting (reporting bias) | High risk | Intended outcome measures reported. However, maternal mortality was not reported. |
| Other bias | Low risk | None known. |
Tam 2005.
| Methods | Single‐centre, double‐blind, randomised, controlled trial, conducted from August 1998 to July 1999 in China. Per protocol analysis. Follow‐up was 6 weeks. | |
| Participants | 170 puerperal anaemic women were screened, 150 were included and randomised into 2 groups:
75 to the intervention group, 63 completed the trial per protocol, 75 to the comparator group, 59 completed the trial per protocol. Socioeconomic conditions were not described, ethnic origin was 76% to 81% Chinese, 19% to 24% Filippino. Inclusion criteria: Hb 80 to 99 g/L 2 days postpartum. Exclusion criteria: MCV < 80 fL, significant anaemia symptoms (tachycardia, severe dizziness, and shortness of breath), estimated blood loss > 500 mL. |
|
| Interventions | Intervention referred to oral ferrous sulphate. Intervention: oral ferrous sulphate 200 mg (65 mg elemental iron) 3 times daily for 42 days. Total dose elemental iron was 8200 mg. Total dose non‐elemental iron was 25,200 mg. Comparator: placebo tablets containing lactose and drug binder 3 times daily for 42 days. |
|
| Outcomes | No preplanned outcome measures stated. Aim was to determine effects of mild postpartum anaemia and iron supplementation in women. Laboratory values, subjective evaluation of general well being score on 4‐point scale, anaemia symptoms, ability to lactate and adverse events during treatment were reported. | |
| Notes | Source of funding was not stated. Placebo tablets contained lactose. Majority of Asian people are lactose intolerant. This may have influenced GI adverse events. In this trial anaemia symptoms in the anaemic group were compared to that of the non‐anaemic group, which did not describe the effect of treatment of the anaemic women. Authors did not respond to the request on additional information. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated randomisation table. |
| Allocation concealment (selection bias) | Low risk | Pharmacy responsible for randomisation, identical tablets. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Both patient and clinicians were blinded to the given treatment. However, intervention group's stool turned black. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Assuming all parties involved in the trial were unaware of treatment, risk of bias is low for all outcomes. However, women in the intervention group may have been able to guess their allocation, as it was reported that their stool turned black due to iron supplementation. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | High dropout rate. Reason for dropout after randomisation was not described. |
| Selective reporting (reporting bias) | High risk | No preplanned outcome measures stated. Aim was to determine effects of mild postpartum anaemia and iron supplementation in women. Maternal mortality was not reported. |
| Other bias | Low risk | None known. |
Van Wyck 2007.
| Methods | Multicentre, open‐label, randomised, controlled trial conducted from February 8, 2005 to November 11, 2005 at 43 sites, including 40 in USA and 3 in Mexico. Per protocol analysis. Follow‐up was 6 weeks. | |
| Participants | 660 women were screened, 361 were randomised to 2 groups: 182 to intervention group 168 completed the trial per protocol. 179 to comparator group, 169 completed the trial per protocol. Socioeconomic conditions were not described, ethnic origin of the population was 46% to 52% Caucasian, 26% to 30% Hispanic, 19% to 22% African American and 2% to 3% other. Inclusion criteria: Hb ≤ 100 g/L, use of acceptable contraception, enrolment within 10 days after delivery. Exclusion criteria: previous non‐adherence to oral iron therapy, history of anaemia from other causes than iron deficiency or blood loss secondary to pregnancy or delivery, estimated blood loss > 100 mL 24 hours before randomisation, active severe infection, TSAT > 50 %, serum ferritin > 500 ng/mL, serum creatinine > 2.0 mg/dL, serum transaminases > 1.5 times upper limit, untreated B12 or folate deficiency; erythropoiesis‐stimulating treatment within 3 months before screening, history of myelosuppressive therapy, asthma under treatment, hepatitis, HIV, or hematologic disorder other than iron deficiency. |
|
| Interventions | Intervention referred to IV ferric carboxymaltose.
Intervention: IV ferric carboxymaltose (Injectafer®) was administrated with a maximum dose of 15 mg/kg in a single day, not to exceed 1000 mg. If the total calculated dose exceeded 1000 mg, subsequent doses were administered weekly until the total dose was received, up to a maximal total dose of 2500 mg. The total dose was calculated using Ganzoni formula. The mean total iron dose was 1403.1 mg. Comparator: oral ferrous sulphate 325 mg (65 mg elemental iron) 3 times daily for 42 days. Total elemental iron dose was approximately 8190 mg. Total dose of non‐elemental iron was 40,950 mg. |
|
| Outcomes | Preplanned outcome measures were the proportion of patients with improved quality of life. Maternal mortality, fatigue, psychological well being, infections, compliance and adverse events during treatment were reported. | |
| Notes | Supported by American Regent, Inc, the human drug division of Luitpold Pharmaceuticals, Shirley, New York. The authors provided unpublished information and corrections to the published text on request. They reported 3 errors in figure 1: ‐ ITT population in the IV iron group was corrected to 168; ‐ ITT population in the oral iron group was 169; ‐ In oral iron group 2 women had a Hb not less than 110 g/L at baseline. Errors detected: Figure 1, figure 2C, text page 270. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computerised random number generation, blocked randomisation, interactive voice response system. |
| Allocation concealment (selection bias) | Low risk | Computerised system. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial. High risk for subjective outcomes such as adverse events. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label trial. High risk for most outcomes. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Different frequency of dropouts prior to dosing in the 2 groups. Discrepancy between groups in reasons for dropout. |
| Selective reporting (reporting bias) | Low risk | Preplanned outcome measures reported. |
| Other bias | High risk | Several errors detected in publication. |
Verma 2011.
| Methods | Multicentre, randomised trial, conducted from January 2010 to July 2010 in 2 Indian centres. ITT analysis. Follow‐up was 30 days. | |
| Participants | 150 puerperal anaemic women were randomised to 2 groups of 75. No dropouts were reported. Socioeconomic conditions were not described in detail. 93.3% of the women came from rural areas. Inclusion criteria: Hb < 80 g/L 24 hours after delivery. Exclusion criteria: anaemia from other cause than nutritional deficiency during pregnancy, immuno‐compromised patients, terminal illness, severe cardiac, hepatic, renal, cerebrovascular, malignant, or chronic uncontrolled systemic disease, other serious medical illness, allergy/reaction to iron complex and unwillingness to participate. |
|
| Interventions | Intervention referred to IV iron sucrose.
Intervention: IV iron sucrose 200 mg on day 1, 3 and 5. The total iron dose was 600 mg, calculated by following formula: Weight (target Hb–actual Hb) 0.24 + 500 mg. Comparator: oral ferrous sulphate 200 mg twice daily for 1 month. Total non‐elemental was approximately 12,000 mg. |
|
| Outcomes | Preplanned outcomes were laboratory values, quality of life, patient satisfaction, impact on cost and hospital stay, blood transfusion frequency, impact on stress, depression and cognitive function, impact on breastfeeding compared to oral iron therapy and recommendation of iron sucrose to postpartum anaemic patients. Compliance and adverse events during treatment were also reported. | |
| Notes | Source of funding was not reported. Two errors were detected in figure 2: first Hb value of oral iron does not correspond to text on page 68 (Haemoglobin Response); last Hb value for oral iron does not correspond to the graph, decimal error. Total IV iron dose was listed both as a fixed dose of 600 mg, and as a weight‐dependant dose calculated by the Ganzoni formula. On page 68 the authors state: "In oral group this mean rise of Hb was noted from 9.65 ± 0.88 gm/dl to 11.02 ± 1.02 gm/dl (p < 0.0001) in 30 days (Fig. 2)." These values cannot be found in figure 2. Adverse events in oral group stated but their rate not given. Authors did not respond to the request on additional information. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Study claimed to be randomised in abstract, but not elsewhere, and no method was described. |
| Allocation concealment (selection bias) | Unclear risk | No method described. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial. High risk for subjective outcomes such as adverse events. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label trial. High risk for most outcomes. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Unclear description of participants, no mention of dropouts. Number of screened and excluded patients prior to randomisation was not reported. |
| Selective reporting (reporting bias) | High risk | No report on the following preplanned outcomes: patient satisfaction, quality of life, cost of treatment, length of hospital stay, use of blood transfusion, impact on stress, depression and cognitive function, lactation. Maternal mortality was not reported. |
| Other bias | High risk | Study offers limited amount of information and is difficult to evaluate. Several errors detected. |
Wagstrom 2007.
| Methods | Multicentre, open‐label, randomised, controlled trial, conducted from November 1999 to May 2001 in 2 Swedish centres. Per protocol analysis. Follow‐up was 14 days. | |
| Participants | 60 puerperal anaemic women were randomised to 3 equal groups of 20: intervention: group 1 (20,000 U rhEPO), 15 completed the trial; intervention: group 2 (10,000 U rhEPO), 19 completed the trial; comparator: group 3, 16 completed trial. Socioeconomic conditions were not described. Inclusion criteria: age > 18 years, Hb < 80 g/L within 72 hours after delivery. All women had complicated deliveries: emergency caesarean, vacuum extractions, uterine explorations, lacerations or uterine atony. Exclusion criteria: malignant, infectious, epileptic, hypertensive, haematological, or cardiac disease, rheumatoid arthritis, diseases treated with cytostatic drugs. |
|
| Interventions | Intervention referred to EPO.
Intervention group 1: SC rhEPO (NeoRecormon®) 20,000 U on day 0 and 3 + IV iron sucrose (Venofer) 250 mg on day 0 and 200 mg on day 3. Total rhEPO dose was 40,000 U. Total IV iron dose was 450 mg. Intervention group 2: SC rhEPO (NeoRecormon®) 10,000 U on day 0 and 3 + IV iron sucrose (Venofer) 250 mg on day 0 and 200 mg on day 3. Total rhEPO dose was 20,000 U. Total iron IV dose was 450 mg. Comparator group: IV iron sucrose (Venofer) 250 mg on day 0 and 200 mg on day 3. Total IV iron dose was 450 mg. All women were advised to take supplementary iron, 100 mg daily, after 1 week. Doses were not registered, thus total iron dose is not known. |
|
| Outcomes | The primary objective was to evaluate laboratory values. Infections and adverse events during treatment were reported. | |
| Notes | Source of funding: Roche AB, Stockholm, Sweden and the Swedish Research Council, Karolinska Institutet. Trial authors provided additional data on request. We included the discontinued patients in the analysis of adverse events. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Sequentially numbered envelopes. |
| Allocation concealment (selection bias) | Low risk | Sealed envelopes unknown to recruiter. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial. High risk for subjective outcomes such as adverse events. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label trial. High risk for most outcomes. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | The patients who dropped out had significantly lower Hb than the rest. Dropout rate was high. Authors provided reasons for dropout according to treatment group. Infections (endometritis) were the most frequent reason for dropout, potentially selecting the population. The number of screened and excluded patients prior to randomisation was not reported. |
| Selective reporting (reporting bias) | High risk | Outcomes for intended objectives were reported. In the original published paper reason for dropout and serious complications was not given by group. However this information was provided by trial author on request. Maternal mortality was not reported. |
| Other bias | Low risk | None known. |
Westad 2008.
| Methods | Multicentre, open‐label, randomised, controlled trial, conducted from June 2004 to September 2006 in 5 Norwegian centres. Randomisasion according to the minimization method, controlling for age and Hb at inclusion, parity and iron treatment during the third trimester. ITT analysis. Follow‐up was 12 weeks. | |
| Participants | 128 puerperal women were randomised to 2 groups:
58 to intervention group, 56 completed by 4 weeks, 45 completed by 12 weeks;
70 to comparator group, 61 completed by 4 weeks, 48 completed by 12 weeks. Totally, 117 women completed the first 4 weeks and 93 completed 12 weeks per protocol. Socioeconomic conditions were not described. Inclusion criteria: age 18 to 45, Hb 65 to 85 g/L. Exclusion criteria: inability to read and understand the Norwegian language, prior commencement of postpartum iron supplementation, clinically significant disease, serum creatinine > 130 µmol/L or contraindications for Venofer® or Duroferon®. |
|
| Interventions | Interventions referred to IV ferrous sucrose. Intervention: IV iron sucrose (Venofer®) 200 mg daily over 3 consecutive days. Total iron dose given IV was 600 mg. After 4 weeks the women were given ferrous sulphate tablets containing 100 mg elemental iron twice daily from week 4 to week 12 postpartum. Total dose of elemental iron after 12 weeks was 11,800 mg. Comparator: oral ferrous sulphate (Duroferon®) containing 100 mg elemental iron twice daily from inclusion until 12 weeks. Total dose of elemental iron was 5600 mg after 4 weeks and 16,800 mg after 12 weeks. Treatment was initiated within 48 hours after delivery. |
|
| Outcomes | Preplanned primary outcomes were laboratory values. Secondary outcomes were quality of life measured by SF‐36 and the Fatigue Score after 4, 8 and 12 weeks of treatment. Compliance to treatment and adverse events during treatment were reported. |
|
| Notes | The trial was sponsored by Renapharma AB, the Swedish representative of the manufacturer of iron sucrose (Venofer®). Trial authors provided unpublished information on request. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Internet‐based central randomisation. |
| Allocation concealment (selection bias) | Low risk | Internet‐based. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial. High risk for subjective outcomes such as adverse events. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Open‐label trial. High risk for most outcomes. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | There was a big difference in number of dropouts in the 2 groups at 4 weeks (2 vs 9) and thereby high risk of selection of population regarding all outcomes. Dropout rate was very high 27% by the end of the trial. Number of screened and excluded patients prior to randomisation was not reported. |
| Selective reporting (reporting bias) | High risk | Intended outcomes reported. Reason for choosing 4 out of 8 scales from the SF‐36 was not explained. Maternal mortality was not reported. |
| Other bias | Low risk | None known. |
dl: decilitre EPDS: Edinburgh Postnatal Depression Scale EPO: erythropoietin fL: femtolitres g: grams g/L: grams per litre GI: gastrointestinal Hb: haemoglobin HCT: haematocrit HIV: human immunodeficiency virus ITT: intention‐to‐treat IU: international units IV: intravenous kg: kilograms MCV: mean corpuscular volume mg: milligrams mL: millilitres n: number NaCl: Sodium chloride PPH: postpartum haemorrhage RBC: red blood cell rhEPO: recombinant human erythropoietin SC: subcutaneous SCL‐90‐R: Symptom Checklist‐90‐Revised SF‐36: Short Form 36 STAI: State‐Trait Anxiety Inventory TSAT: transferrin saturation U: units vs: versus µg/L: micrograms per litre
Characteristics of excluded studies [author‐defined order]
| Study | Reason for exclusion |
|---|---|
| Breymann 2007 | An observational cohort study, not randomised controlled trial. |
| Casparis 1996 | Population consists of both women with anaemia during pregnancy and postpartum anaemia, with no subgroup analyses. |
| Daniilidis 2011 | Not a randomised trial. This was clear from authors response on requested method description: "The patients were selected and placed to each group only according to their consent since from 135 women only 109 agreed to be treated with IV iron and the rest who refused intravenous treatment were treated with oral supplements". |
| Danko 1990 | It was not stated in the text that the women were randomised. Therefore, we do not consider this trial a randomised controlled trial. |
| Dede 2005 | It was not stated in the text that the women were randomised. Therefore, we do not consider this trial a randomised controlled trial. |
| Giannoulis 2009 | Not randomised controlled trial. This was clear from authors reply on requested method description. Authors used same allocation method as in the trial by Daniilidis 2011. |
| Haidar 2005 | Study design did not define the postpartum period and the period for enrolment, most probably including women who were enrolled more than 6 weeks postpartum, which conflicts with the review author’s inclusion criteria. |
| Hashmi 2006 | The study population consists of both pregnant and postpartum patients and subgroup analyses were not reported. |
| Huch 1992 | Quasi‐randomised trial. The women were assigned treatment based on their name in alphabetical order. |
| Mara 1999 | It was not stated in the text of the translation that the women were randomised. Therefore, we do not consider this trial a randomised controlled trial. Trial ID: 00264270 and 00413969. |
| Mara 2001 | Population reported in the study included non‐anaemic women. Trial ID 00328429 and 00324138. |
| Mitra 2012 | This study has a non‐anaemic control group which is therefore not relevant according to our predefined criteria. The remaining 2 groups received the exact same treatment and are therefore not comparable by intervention. The study investigates differences in screening strategies. It does not compare the effect of different treatment regiments as we predefined for this review. |
| Osmond 1953 | The intervention focuses on crude liver extract given intramuscularly, an intervention not accepted for treatment of postpartum iron deficiency anaemia in current time. |
| Picha 1975 | The study assessed the usefulness of iron therapy in prevention, not treatment, of postpartum anaemia. |
| Zimmermann 1995 | This report summarises three trials: Breymann 1996 (included), Huch 1992 and Zimmermann 1994 (excluded). |
| Zimmermann 1994 | This trial does not have a control arm and is therefor not a randomised controlled trial. |
| Van Der Woude 2014 | This study compares oral iron with folate supplementation with oral iron alone. In our review we did not consider folate as an independent treatment of iron deficiency anaemia. In this study, folate is the only difference between the two treatments and thus the study does not evaluate the effect of iron treatment. |
Characteristics of studies awaiting assessment [ordered by study ID]
Backe 2009.
| Methods | Multicentre, open‐label, randomised controlled trial, conducted in Norway. Randomisation was performed by use of opaque envelopes. Laboratory analyses were provided by a recognised Swedish biochemical laboratory. |
| Participants | 200 patients with postpartum anaemia, included within 48 postpartum. Hb 65 to 85 g/L. |
| Interventions | Intervention: IV iron carboxymaltose (Ferinject) in a dose calculated by the Ganzoni formula. Comparator: oral iron sulphate (Duroferon) 100 mg twice daily. |
| Outcomes | Preplanned outcomes were laboratory values, fatigue (Fatigue Scale), quality of life (SF‐36), post partum depression, (Edinburgh Post Partum Depression Scale). |
| Notes | The trial was initiated, partially conducted and then terminated by sponsor (Renapharma Vifor) because of slow progress (citation B. Backe). Trial registered in ClinicalTrials.gov (Trial ID: NCT00929409). Renapharma was contacted March 7th and March 27th 2013 for a report of the trial and reasons for discontinuation. However, no‐one responded to our request. Contact person for this trial intends to publish a paper based on the trial report and the results from the incomplete study, which will be shared with the review authors in the future. |
g/L: grams per litre Hb: haemoglobin IV: intravenous mg: milligrams SF‐36: Short Form 36
Characteristics of ongoing studies [ordered by study ID]
Chaudhuri 2013.
| Trial name or title | Public Title of Study: Comparison of beneficial effects of IV iron with oral iron in treating anemia following childbirth. Scientific Title of Study: IV iron‐sucrose complex versus oral iron in the treatment of postpartum anemia. |
| Methods | Computer‐generated randomisation. Sequentially‐numbered, sealed, opaque envelopes. Open‐label trial. |
| Participants | Women with anaemia in the postnatal period. Sample size: 100 Inclusion criteria: postpartum women between 18 and 45 year of age, irrespective of mode of delivery who are haemodynamically stable with moderate iron deficiency anaemia (Hb 60‐80 g/L) and serum ferritin < 15 μg/L at 24‐48 hours after delivery. Exclusion criteria: thalassaemic trait, peripartum blood transfusion, active PPH, allergy or intolerance to iron preparation previously, evidence of sepsis, hepatic, cardiovascular, renal, thromboembolic disorder. |
| Interventions | Intervention: IV iron‐sucrose injection 200 mg every alternate days until the total calculated dose is given (method not described). Comparator: oral ferrous sulphate tablet 200 mg (60 mg elemental iron) 3 times a day for 6 weeks. |
| Outcomes | Primary outcomes: Hb, HCT, red cell indices, serum ferritin.
Secondary outcomes: side effects and complications. Time points: 0, 7, 14, 40 days. |
| Starting date | Date of first enrolment 02/10/2012. |
| Contact information | Dr. Picklu Chaudhuri, associate professor, N.R.S Medical College, Kolkata Phone: 9432277443 Fax: 22658179 Email: picklu.chaudhuri@gmail.com |
| User defined 1 | CTRI Number: CTRI/2013/05/003624 [Registered on: 09/05/2013] Trial registered retrospectively. |
| Notes |
Holm 2015.
| Trial name or title | Intravenous iron isomaltoside 1000 administered by high single‐dose infusions or standard medical care for the treatment of fatigue in women after postpartum haemorrhage: study protocol for a randomised controlled trial. Name in trial register: A randomized comparative, open‐label study of IV iron isomaltoside 1000 (Monofer®) administered by high single dose in‐fusions or RBC transfusion in women with severe postpartum iron deficiency anaemia ‐ P‐Monofer‐PP‐02 |
| Methods | Single‐centre, open‐label, randomised comparative study. |
| Participants | Estimated enrolment: 200 Inclusion criteria: PPH > 1000 mL, Hb 55 to 80 g/L, and signed informed consent. Exclusion criteria: age < 18 years, multiple births, peripartum RBC transfusion, iron overload or disturbances in utilisation of iron (e.g. haemochromatosis and haemosiderosis), hypersensitivity to parenteral iron or any excipients in the investigational drug products, history of active asthma within the last 5 years or a history of multiple allergies, decompensated liver cirrhosis and active hepatitis, HELLP syndrome, active acute infection assessed by clinical judgement, active rheumatoid arthritis, history of anaemia caused by e.g. thalassaemia, hypersplenism or haemolytic anaemia (known haematologic disorder other than iron deficiency), not able to read, speak and understand the Danish language, participation in any other clinical study where the study drug has not passed 5 half‐lives prior to the baseline, any other medical condition that, in the opinion of the investigator, may cause the patient to be unsuitable for completion of the study or place the patient at potential risk from being in the study. |
| Interventions | Intervention arm: IV iron isomaltoside 1000 (Monofer®) given as a single dose of 1200 mg. Comparator arm: standard medical care. Standard medical Care is most often to recommend women with PPH to continue oral iron supplementation as recommended during pregnancy or to advise the participant to take 100 mg oral iron 1‐2 times a day. |
| Outcomes | Primary outcome measures: physical fatigue. The primary objective of this study is to compare efficacy of IV high single dose infusion of iron isomaltoside 1000 to standard medical care in women with PPH evaluated as physical fatigue.
Secondary outcome measures: changes in fatigue symptoms and postpartum depression symptoms; changes in concentrations of Hb, plasma ferritin, plasma iron, plasma transferrin, transferrin saturation, reticulocyte count, mean reticulocyte haemoglobin content, and haematology parameters; breastfeeding, RBC transfusions, adverse drug reactions. Other outcome measures: change in anaemia symptoms, change in GI symptoms. Time frame for all outcome measures: from exposure to day 3, week 1, 3, 8 and 12 post‐exposure. |
| Starting date | Study start date: June 2013. Estimated study completion date: Febuary 2015. |
| Contact information | Correspondence: charlotteholm@dadlnet.dk Department of Obstetrics, Juliane Marie Centre, Copenhagen University Hospital, Rigshospitalet, Blegdamsvej 9, DK‐2100 Kbh Ø Copenhagen, Denmark Pharmacosmos A/S Clinical R & D Telephone: +4559485959 Fax: +4559485962 Email: llt@pharmacosmos.com |
| User defined 1 | Trial ID: NCT01895218; 2012‐005782‐12 |
| Notes |
Hossain 2013.
| Trial name or title | Use of Iron Isomaltoside 1000 (Monofer) in Postpartum Anemia. |
| Methods | Open‐label, randomised controlled trial. |
| Participants | Estimated enrolment: 300 women. Inclusion criteria: Hb < 100 g/L 24 to 48 hours after delivery. Exclusion criteria: history of PPH, or significant blood loss in last 24 hours, history of allergy to iron preparation, Hb < 70 g/L, sign and symptoms of cardiac failure, blood transfusion in last 3 months, chronic liver diseases, increased creatinine. |
| Interventions | Intervention: IV infusion of isomaltoside 1000 (Monofer), calculated according to Ganzoni formula. Active comparator: oral ferrous sulphate 200 mg twice daily. |
| Outcomes | Primary: to see the rise in Hb concentration of 2 g/dL or more, measured at day 14 and at 3 months. Secondary: time required for rise in haemoglobin concentration. Both groups will be compared in terms of time interval, to see the rise in Hb concentration. |
| Starting date | May 2012. |
| Contact information | Nazli Hossain, Dow University of Health Sciences. |
| User defined 1 | Trial ID: NCT01628770. |
| Notes |
Suneja 2014.
| Trial name or title | Public title: A clinical trial to compare oral iron ferrous sulfate with newer intravenous iron (ferric carboxymaltose) injection in patients of iron deficiency anemia in post delivery period Scientific title: Comparison of ferric carboxymaltose injection with oral iron in treatment of postpartum iron deficiency anemia ‐ a randomized controlled clinical trial |
| Methods | Open‐label, randomised controlled trial. Computerised sequence generation for randomisation. |
| Participants | Target sample size: 140 women. Age: 20 to 40 years. Inclusion criteria: Women between 20 and 40 years of age, within 10 days of normal delivery with a Hb between 7 and 10 g% and iron deficiency measured by PCV < 36%, MCV < 80 fl, MCH < 27 pg and MCHC < 33 g/dL) with negative NESTROF test. Exclusion criteria: Weight < 35 kg, puerperal pyrexia, known drug allergy or intolerance to iron therapy, history of chronic medical illness (tuberculosis, asthma, liver diseases, kidney diseases, diabetes mellitus, hypertension, HIV infection), other anaemia treatment (blood transfusion, erythropoietin) within the last three months. |
| Interventions | Intervention: Injection ferric carboxymaltose, calculated by dose not exceeding 1000 mg per infusion. Comparator: Ferrous sulphate tablet containing 60 mg elemental iron thrice a day for 6 weeks. |
| Outcomes | Primary outcomes: Percentage of patients achieving Hb rise 3 g/dL from baseline at 3 and 6 weeks. Secondary outcomes: Percentage of patients achieving Hb 12 g/dL at 3 and 6 weeks. Rise in Hb from baseline to 3 and 6 weeks. Change in red cell indices and serum iron parameters from baseline to 6 weeks. Recording the side effects in both the groups. |
| Starting date | 1 November 2012 |
| Contact information | Dr Amita Suneja Department of obstetrics and gynaecology, 110095 East, DELHI, India Telephone: 9868399728 Email: amita_suneja@yahoo.co.in Affiliation: UCMS & GTB Hospital Dr Shikha Dilshad Garden, 110095, Thiruvananthapuram, DELHI, India Telephone: 9868399728 Email: shikha.pmch@gmail.com Affiliation: UCMS & GTB Hospital |
| User defined 1 | Trial ID: CTRI/2014/10/005099 |
| Notes |
fL: femtolitres GI: gastrointestinal g/dL: grams per decilitre g/L: grams per litre Hb: haemoglobin HCT: haematocrit (= PCV) HELLP: haemolysis elevated liver enzymes and low platelets IV: intravenous kg: kilograms MCH: mean corpuscular haemoglobin MCHC: mean corpuscular haemoglobin concentration MCV: mean corpuscular volume mg: milligrams NESTROF: Naked Eye Single Tube Red Cell Osmotic Fragility Test PPH: postpartum haemorrhage PCV: packed cell volume (= HCT) pg: picograms RBC: red blood cell µg/L: micrograms per litre
Differences between protocol and review
Section: Objectives
Protocol
To assess the efficacy and safety of the available treatment modalities for women with postpartum iron deficiency. These include oral and parenteral iron supplementation, folate, erythropoietin, and blood transfusion.
Review
To assess the efficacy and harms of the available treatment modalities for women with postpartum iron deficiency anaemia. These include oral and parenteral iron, erythropoietin, and blood transfusion.
Comment
We have now learned that the term "safety" is mostly used by intervention trials and indicates a positive tone. We find the term "harms" more appropriate, as we report on registered adverse events of treatment and thus on lack of safety. Also, it is not appropriate to list folate as a treatment for iron deficiency anaemia.
Section: Types of interventions
Protocol
Iron supplementation administered orally or parenterally, either alone or in combination with folate, erythropoietin or blood transfusion started within the first six weeks after giving birth and compared with placebo, another treatment, or no treatment.
Review
Treatment for postpartum iron deficiency anaemia started within the first six weeks after giving birth compared with placebo, no treatment or another treatment.
Currently, accepted treatment for iron deficiency anaemia includes blood transfusion or iron supplementation administered orally or parenterally, either alone or in combination with folate and/or erythropoietin.
Folat supplementation was not considered as an independent treatment of iron deficiency anaemia, but was accepted as a part of other types of treatment for postpartum iron deficiency anaemia.
New treatment modalities appropriate for iron deficiency anaemia will be included in future updates.
Comment
This to ensure inclusion of any new treatments appropriate for iron deficiency anaemia that will be investigated in the future.
Section: Outcomes
Protocol
Primary outcomes
Maternal mortality.
Fatigue (as reported by the women ‐ verbalisation of fatigue or lack of energy and inability to maintain usual routines; measured by a scale or questionnaire; or as defined by the trial authors).
Secondary outcomes
Persistent anaemia symptoms during treatment. Any of the following symptoms: dyspnoea, tachypnoea, tachycardia, palpitations, orthostatic dizziness, syncopation, paleness.
Psychological well being (including cognitive performance); measured by the ’Blues Questionnaire’ (Kennerley 1989), ’Selfreport symptom inventory 90 [SCL‐90‐R]’ (Schmitz 1999), ’SF36 [Medical Outcomes Study Short Form]’ (Ware 2000), or similar questionnaire; or as defined by the trial authors).
Urinary tract infection, endometritis, or other infections (as defined by the trial authors).
Compliance to treatment (as defined by the trial authors).
Breastfeeding (at hospital discharge; six weeks postpartum; six months postpartum).
Length of hospital stay.
Any adverse events during treatment (each type of harm analysed individually, when possible).
Number of red blood cell transfusions (number of transfused women and number of red blood cell units per woman).
We will not apply any restrictions regarding follow‐up periods, to avoid excluding data on any long‐term benefits or harms. Studies of included interventions that do not report any of the above mentioned outcomes will be described in the ’Characteristics of included studies’ section, but will not be included in any meta‐analyses.
We plan to include the following outcomes in the ’Summary of findings’ tables of the review, using the Grade Profiler programme (GRADEpro).
Maternal mortality
Fatigue
Constipation (when treatment was oral iron substitution)
Allergic reactions (when treatment was intravenous iron)
Review
Primary outcomes
Maternal mortality: We considered that no women died only if: a) this was stated explicitly, or b) no dropouts occurred during follow‐up, or c) contact authors provided this information on request. Mortality was considered present only if: a) stated explicitly in published report or b) contact authors provided this information on request. Mortality was assessed as not reported if a) no mention of dropouts or their causes, b) all dropouts not accounted for, c) dropouts not explicitly reported to be alive at the end of the follow‐up period.
Fatigue: as reported by the women ‐ verbalisation of fatigue or lack of energy and inability to maintain usual routines; measured by a scale or questionnaire; or as defined by the trial authors. Short‐term and long‐term results, thus the minimal and maximal time from baseline.
Secondary outcomes
Persistent anaemia symptoms during treatment. Any of the following symptoms: dyspnoea, tachypnoea, tachycardia, palpitations, orthostatic dizziness, syncopation, paleness.
Psychological well being, including cognitive performance, measured by the 'Blues Questionnaire' (Kennerley 1989), 'Self‐report symptom inventory 90 [SCL‐90‐R]' (Schmitz 1999), 'SF36 [Medical Outcomes Study Short Form]' (Ware 2000) or similar questionnaire; or as defined by the trial authors. Only short‐term results, thus the minimal time from baseline.
Urinary tract infection, endometritis, or other infections (as defined by the trial authors).
Compliance to treatment (as defined by the trial authors).
Breastfeeding (at hospital discharge; six weeks postpartum; six months postpartum).
Length of hospital stay.
Any adverse events during treatment (each type of harm analysed individually, when possible).
Number of RBC transfusions (number of transfused women and number of RBC units per woman).
For outcomes other than psychological well being, we did not apply any restrictions regarding follow‐up periods to avoid excluding data on any long‐term benefits or harms. We did not apply language restrictions.
We planned to include the following outcomes in the 'Summary of findings' tables of the review, using the Grade Profiler programme (GRADEpro 2014).
Maternal mortality
Fatigue
Constipation (for oral iron substitution)
Allergic reactions (for intravenous iron)
The comparisons included in a 'Summary of findings' tables were chosen based on relevance to current treatment standards according to clinical experts. Therefore we chose not to include treatment with IV EPO or yeast extract in a 'Summary of findings' table, as these methods are no longer practiced. For the treatment‐specific outcomes listed above (constipation and allergic reactions,) the results were included in a 'Summary of findings' table if the specific treatment was present in only one of the study arms.
We chose to include additional outcomes in the 'Summary of findings' tables, which we found important for clinical decision making for each individual treatment modality, when this treatment was present in only one of the study arms. For comparisons with IV iron, this outcome was infections. For comparisons with oral iron we included all GI symptoms combined. For comparisons with RBC transfusions we included infections, thromboembolic events and transfusion‐specific adverse events, such as alloantibody formation and transfusion reactions. For comparisons with EPO, thromboembolic events were essential. For all comparisons which met the above mentioned criteria, we found it important to include anaemia symptoms.
Comment
Mortality is an important primary outcome, and it should be clear how we interpreted the data.
Quality of life outcomes (fatigue and psychological well being) were reported in a manner that produced a very large amount of data. This was due to reporting on multiple domains at multiple different time points. We had to rationally restrict this vast amount of analyses to a manageable and amount of information.
The additional outcomes in the 'Summary of findings' tables are important for clinical decision making.
Section: Sensitivity analysis
Protocol
We plan to carry out a sensitivity analysis based on trial design involving trials with a low risk of bias in all bias domains of the ’Risk of bias’ tool, thus removing trials with a high or unclear risk of bias in any domain. We will also carry out sensitivity analyses to explore the effects of random‐effects analyses for outcomes with statistical heterogeneity and the effects of any assumptions made such as the value of the ICC used for cluster‐randomised trials. We will use our primary outcomes only (maternal mortality and fatigue) in the sensitivity analyses.
Review
We planned to carry out a sensitivity analysis based on trial design, thus excluding trials with a high risk of selection, performance, and detection bias.
We also planned to carry out sensitivity analyses to explore the effects of random‐effects analyses for outcomes with statistical heterogeneity and the effects of any assumptions made such as the value of the ICC used for cluster‐randomised trials. We planned sensitivity analyses only for our primary outcomes (maternal mortality and fatigue). Provided that enough data become available, we will attempt to carry out sensitivity analyses for all comparisons in future updates.
Comment
The original phrase was far too restrictive, as it is practically impossible to find a trial with low risk of bias in all domains, and we would never be able to make sensitivity analyses. Sensitivity analyses on the above mentioned domains will allow us to investigate the effect of trial design, thus factors directly controlled by the trial authors.
Section: Assessment of risk of bias in included studies
Protocol
Standart text listing all seven bias domains (please see the protocol for this review).
Review
Two review authors (VM and AN) independently assessed risk of bias for each study using the criteria outlined in the Cochrane Handbook for Systematic Reviews of Interventions and a 'Risk of bias' table (Higgins 2011). As per Cochrane standards, we assessed selection bias, performance bias, detection bias, attrition bias, reporting bias, and other bias. Each type of bias was assessed as low, high, or unclear. All disagreements were resolved by discussion, or by involving a third assessor (KJ or Jens Langhoff‐Roos (JLR)).
We made explicit judgements about whether studies were at high risk of bias, according to the criteria given in the Cochrane Handbook (Higgins 2011). Where possible, we assessed the likely magnitude and direction of the bias and whether we considered if it was likely to impact the findings. We explored the impact of bias through Sensitivity analysis.
We used Grade Profiler (GRADEpro 2014) to make 'Summary of findings' tables. We included our primary outcomes, constipation (when treated with oral iron), and allergic reactions (when treated with intravenous (IV) iron). We also included additional outcomes, which we considered important for the decision‐making process.
Comment
We received permission to simply refer to the Cochrane Handbook in the 'Assessment of risk of bias in included studies' section, instead of copying the standard text along with the seven listed bias domains. We are aware that traditionally this complete text is written in Pregnancy and Childbirth Group reviews. However, it is fully described in the easily accessible Cochrane Handbook and refraining from citing the whole text saves a lot of space.
Contributions of authors
Veronika Markova wrote the protocol update and developed this version of the review, searched the references for the background section, adjusted the methodology section, determining the outcomes and types of analyses and conducted the search in the WHO ICTRP and LILACS registries.
Veronika Markova and Astrid Norgaard independently screened the literature for inclusion, assessed risk of bias and extracted data from relevant trials. Veronika Markova entered data into Review Manager 5.3 (proofread by Astrid Norgaard), carried out the statistical analyses, produced the GRADE 'Summary of findings' tables with the support from Karsten Juhl Jørgensen and wrote review drafts.
Astrid Norgaard also provided expert knowledge on current trends in anaemia treatment options and outcomes.
Karsten Juhl Jørgensen provided expert knowledge regarding the methods. He assisted with the statistical analyses and the 'Summary of findings' tables.
Jens Langhoff‐Roos provided expert clinical knowledge on current treatment regiments for postpartum anaemia. He took part in initiating the project of this Cochrane review.
All authors reviewed all manuscript drafts and contributed to the final preparation of the review.
Sources of support
Internal sources
This was a non‐profit project and the co‐authors did not receive financial support for their efforts, Denmark.
External sources
No sources of support supplied
Declarations of interest
Jens Langhoff‐Roos and Astrid Norgaard are supervisors of an ongoing PhD study (Holm 2015) at University of Copenhagen by Charlotte Holm. The PhD study (EUCTR2012‐005783‐10‐DK) is partly financed by Pharmacosmos which supplies IV iron for the studies. Jens Langhoff‐Roos has no financial interest in this or other pharmaceutical companies. Astrid Norgaard is the principal investigator of one clinical trial and the sponsor of another clinical trial, both partly financed by Pharmacosmos (EudraCT Number 2012‐001529‐28 and 2013‐004979‐13) ‐ neither of these trials would be potentially eligible for inclusion in this review. Astrid Norgaard has no financial interest in this or other pharmaceutical companies.
Veronika Markova: none known
Karsten Juhl Jørgensen: none known
New
References
References to studies included in this review
Beard 2005 {published data only}
- Beard JL, Hendricks MK, Perez EM, Murray‐Kolb LE, Berg A, Vernon‐Feagans L, et al. Maternal iron deficiency anemia affects postpartum emotions and cognition. Journal of Nutrition 2005;135(2):267‐72. [DOI] [PubMed] [Google Scholar]
- Murray‐Kolb LE, Beard JL. Iron deficiency and child and maternal health. American Journal of Clinical Nutrition 2009;89(3):946S‐950S. [DOI] [PubMed] [Google Scholar]
- Perez EM, Hendricks MK, Beard JL, Murray‐Kolb LE, Berg A, Tomlinson M, et al. Mother‐infant interactions and infant development are altered by maternal iron deficiency anemia. Journal of Nutrition 2005;135(4):850‐5. [DOI] [PubMed] [Google Scholar]
Bhandal 2006 {published data only}
- Bhandal N, Russell R. Intravenous versus oral iron therapy for postpartum anaemia. BJOG: an International Journal of Obstetrics and Gynaecology 2006;113(11):1248‐52. [DOI] [PubMed] [Google Scholar]
- Bhandal N, Russell R. Intravenous versus oral iron therapy for postpartum anaemia. International Journal of Obstetric Anesthesia 2004;13(3):S7. [DOI] [PubMed] [Google Scholar]
Breymann 1996 {published data only}
- Breymann C, Zimmermann R, Huch R, Huch A. Use of recombinant human erythropoietin in combination with parenteral iron in the treatment of postpartum anaemia. European Journal of Clinical Investigation 1996;26(2):123‐30. [DOI] [PubMed] [Google Scholar]
- Krafft A, Breymann C, Huttner C, Huch R, Huch A. Erythropoietic quality of maternal milk. Lancet 1999;354(9180):778. [DOI] [PubMed] [Google Scholar]
Breymann 2000 {published data only}
- Breymann C, Richter C, Huttner C, Huch R, Huch A. Effectiveness of recombinant erythropoietin and iron sucrose vs Iron therapy only, in patients with postpartum anaemia and blunted erythropoiesis. European Journal of Clinical Investigation 2000;30(2):154‐61. [DOI] [PubMed] [Google Scholar]
Breymann 2008 {published data only}
- Breymann C, Gliga F, Bejenariu C, Strizhova N. Comparative efficacy and safety of intravenous ferric carboxymaltose in the treatment of postpartum iron deficiency anemia. International Journal of Gynecology & Obstetrics 2008;101(1):67‐73. [DOI] [PubMed] [Google Scholar]
Froessler 2013 {published and unpublished data}
- Froessler B, Cocchiaro C, Saadat‐Gilani K, Hodyl N, Dekker G. Intravenous iron sucrose versus oral iron ferrous sulfate for antenatal and postpartum iron deficiency anemia: a randomized trial. Journal of Maternal‐Fetal & Neonatal Medicine 2013;26(7):654‐9. [DOI] [PubMed] [Google Scholar]
Guerra 2012 {published and unpublished data}
- Guerra S, Lopez A, Munoz H, Marin JM, Lete I, Aizpuru F. Randomized clinical trial to evaluate the effectiveness of two routes of iron administration, oral and intravenous, in the treatment of postpartum iron deficiency anemia [Spanish] [Ensayo clinico aleatorizado para evaluar la efectividad de dos vias de administracion de hierro, oral e intravenosa, en el tratamiento de la anemia ferropenica posparto]. Clinica e Investigacion en Ginecologia y Obstetricia 2012;39(5):190‐5. [Google Scholar]
Jain 2013 {published data only}
- Jain G, Palaria U, Jha SK. Intravenous iron in postpartum anemia. Journal of Obstetrics and Gynecology of India 2013;63(1):45‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Krafft 2011 {published and unpublished data}
- Krafft A, Breymann C. Iron sucrose with and without recombinant erythropoietin for the treatment of severe postpartum anemia: a prospective, randomized, open‐label study. Journal of Obstetrics and Gynaecology Research 2011;37(2):119‐24. [DOI] [PubMed] [Google Scholar]
Krauss 1972 {published data only}
- Krauss V, Nowotny D, Scharifzadeh AH. Effect and tolerance of oral iron therapy in the puerperium. Munchener Medizinische Wochenschrift 1972;114(43):1877‐9. [PubMed] [Google Scholar]
Lebrecht 1995 {published data only}
- Lebrecht A, Haberlin F, Eberhard J. Postpartum anaemia: Intravenous iron supplementation renders therapy with rHuEPO redundant [Anamie im Wochenbett; parenterale Eisensubstitution macht Erythropoetin‐Therapie entbehrlich]. Geburtshilfe und Frauenheilkunde 1995;55(3):167‐70. [DOI] [PubMed] [Google Scholar]
Makrydimas 1998 {published data only}
- Makrydimas G, Lolis D, Lialios G, Tsiara S, Georgiou I, Bourantas KL. Recombinant human erythropoietin treatment of postpartum anemia Preliminary results. European Journal of Obstetrics & Gynecology and Reproductive Biology 1998;81(1):27‐31. [DOI] [PubMed] [Google Scholar]
Meyer 1995 {published data only}
- Meyer JW, Eichhorn KH, Vetter K, Christen S, Schleusner E, Klos A, et al. Does recombinant human erythropoietin not only treat anemia but reduce postpartum (emotional) distress as well?. Journal of Perinatal Medicine 1995;23:99‐109. [DOI] [PubMed] [Google Scholar]
Mumtaz 2011 {published data only}
- Mumtaz A, Farooq F. Comparison for effects of intravenous versus oral iron therapy for postpartum anemia. Pakistan Journal of Medical and Health Sciences 2011;5(1):116‐20. [Google Scholar]
Perello 2014 {published data only}
- Palacio M. Intravenous iron versus oral iron for severe postpartum anemia randomized trial. ClinicalTrials.gov (accessed 5 June 2012) 2007.
- Perello MF, Coloma JL, Masoller N, Esteve J, Palacio M. Intravenous ferrous sucrose versus placebo in addition to oral iron therapy for the treatment of severe postpartum anaemia: a randomised controlled trial. BJOG: an International Journal of Obstetrics & Gynaecology 2014;121(6):706‐14. [DOI] [PubMed] [Google Scholar]
Prick 2014 {published and unpublished data}
- Jansen AJG, Duvekot JJ, Essink‐Bot ML, Hop WCJ, Rhenen DJ. Multicentre clinical study into the optimal blood transfusion policy in patients with postpartum haemorrhage: The 'Wellbeing of obstetric patients on minimal blood transfusions' (WOMB) study [Een klinisch multicentrisch onderzoek naar het optimale bloedtransfusiebeleid bij patienten met een fluxus post partum: De 'Wellbeing of obstetric patients on minimal blood transfusions'(WOMB)‐studie]. Nederlands Tijdschrift voor Geneeskunde 2007;151(39):2170‐2. [PubMed] [Google Scholar]
- Prick B, Jansen A, Steegers E, Hop W, Essink‐Bot M, Uyl‐de Groot C, et al. Transfusion policy after severe postpartum haemorrhage: a randomised non‐inferiority trial. BJOG: an International Journal of Obstetrics and Gynaecology 2014;121(8):1005‐14. [DOI] [PubMed] [Google Scholar]
- Prick BW, Duvekot JJ, Moer PE, Gemund N, Salm PC, Jansen AJ, et al. Cost‐effectiveness of red blood cell transfusion vs. non‐intervention in women with acute anaemia after postpartum haemorrhage. Vox Sanguinis 2014;107(4):381‐8. [DOI] [PubMed] [Google Scholar]
- Prick BW, Jansen AJG, Steegers EAP, Hop WCJ, Essink‐Bot ML, Uyl‐de Groot CA, et al. RBC transfusion leads to an improvement of physical fatigue in women with acute postpartum anemia: the WOMB study (NCT00335023). American Journal of Obstetrics and Gynecology 2012;206(Suppl 1):S41‐2. [Google Scholar]
- Prick BW, Jansen AJG, Steegers EAP, Hop WCJ, Essink‐Bot ML, Uyl‐Groot CA, et al. Health related quality of life in patients with acute anemia after primary postpartum hemorrhage: A randomized controlled trial of red blood cell transfusion vs expectant management: The womb study. Vox Sanguinis 2012;103(Suppl 1):35. [Google Scholar]
- Prick BW, Steegers EAP, Jansen AG, Hop WCJ, Essink‐Bot M‐L, Peters NCJ, et al. Well being of obstetric patients on minimal blood transfusions (WOMB trial). BMC Pregnancy and Childbirth 2010;10:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhenen DJ. Wellbeing of obstetric patients on minimal bloodtranfusions study ‐ WOMB study. Netherlands Trial Register (http://www.trialregister.nl) (accessed 1 November 2005)) 2005.
Seid 2008 {published data only}
- Seid MH, Derman RJ, Baker JB, Banach W, Goldberg C, Rogers R. Ferric carboxymaltose injection in the treatment of postpartum iron deficiency anemia: a randomized controlled clinical trial. American Journal of Obstetrics and Gynecology 2008;199(4):435.e‐1‐435.e7. [DOI] [PubMed] [Google Scholar]
- Seid MH, Rogers R, Dinh Q. Treating postpartum anemia with intravenous ferric carboxymaltose in a randomized controlled study. American Journal of Obstetrics and Gynecology 2007;197(6 Suppl 1):S26, Abstract no: 55. [Google Scholar]
Tam 2005 {published data only}
- Tam KF, Lee CP, Pun TC. Mild postnatal anemia: is it a problem?. American Journal of Perinatology 2005;22(7):345‐9. [DOI] [PubMed] [Google Scholar]
Van Wyck 2007 {published and unpublished data}
- Wyck DB, Martens MG, Seid MH, Baker JB, Mangione A. Intravenous ferric carboxymaltose compared with oral iron in the treatment of postpartum anemia: a randomized controlled trial. Obstetrics & Gynecology 2007;110(2 Pt 1):267‐78. [DOI] [PubMed] [Google Scholar]
Verma 2011 {published data only}
- Verma S, Inamdar SA, Malhotra N. Intravenous iron therapy versus oral iron in postpartum patients in rural area. Journal of SAFOG 2011;3(2):67‐70. [Google Scholar]
Wagstrom 2007 {published and unpublished data}
- Wagstrom E, Akesson A, Rooijen M, Larson B, Bremme K. Erythropoietin and intravenous iron therapy in postpartum anaemia. Acta Obstetricia et Gynecologica Scandinavica 2007;86(8):957‐62. [DOI] [PubMed] [Google Scholar]
Westad 2008 {published and unpublished data}
- Westad S, Backe B, Salvesen K, Nakling J, Okland I, Borthen I, et al. A 12‐week randomised, multi‐centre study comparing intravenous iron sucrose versus oral ferrous sulphate for treatment of postpartum anaemia. International Journal of Gynecology & Obstetrics 2009;107(Suppl 2):S377. [Google Scholar]
- Westad S, Backe B, Salvesen KA, Nakling J, Okland I, Borthen I, et al. A 12‐week randomised study comparing intravenous iron sucrose versus oral ferrous sulphate for treatment of postpartum anemia. Acta Obstetricia et Gynecologica Scandinavica 2008;87(9):916‐23. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Breymann 2007 {published data only}
- Breymann C, Seefried B, Stahel M, Geisser P, Canclini C. Milk iron content in breast‐feeding mothers after administration of intravenous iron sucrose complex. Journal of Perinatal Medicine 2007;35(2):115‐8. [DOI] [PubMed] [Google Scholar]
Casparis 1996 {published data only}
- Casparis D, Carlo P, Branconi F, Grossi A, Merante D, Gafforio L. Effectiveness and tolerance of oral doses of liquid ferrous gluconate in iron‐deficiency anaemia during pregnancy and in the immediate post‐natal period: comparison with other liquid or solid formulations containing bivalent or trivalent iron. Minerva Ginecologica 1996;48:511‐8. [PubMed] [Google Scholar]
Daniilidis 2011 {published and unpublished data}
- Daniilidis A, Giannoulis C, Pantelis A, Tantanasis T, Dinas K. Total infusion of low molecular weight iron‐dextran for treating postpartum anemia. Clinical and Experimental Obstetrics and Gynecology 2011;38(2):159‐61. [PubMed] [Google Scholar]
Danko 1990 {published data only}
- Danko J, Huch R, Huch A. Epoetin alfa for treatment of postpartum anaemia. Lancet 1990;335(8691):737‐8. [DOI] [PubMed] [Google Scholar]
Dede 2005 {published data only}
- Dede A, Uygur D, Yilmaz B, Mungan T, Ugur M. Intravenous iron sucrose complex vs. oral ferrous sulfate for postpartum iron deficiency anemia. International Journal of Gynecology & Obstetrics 2005;90(3):238‐9. [DOI] [PubMed] [Google Scholar]
Giannoulis 2009 {published and unpublished data}
- Giannoulis C. Intravenous administration of iron sucrose for treating anemia in postpartum women. Hippokratia 2009;13(1):38‐40. [PMC free article] [PubMed] [Google Scholar]
Haidar 2005 {published data only}
- Haidar J, Umeta M, Kogi‐Makau W. Effect of iron supplementation on serum zinc status of lactating women in Addis Ababa, Ethiopia. East African Medical Journal 2005;82(7):349‐52. [PubMed] [Google Scholar]
Hashmi 2006 {published data only}
- Hashmi Z, Bashir G, Azeem P, Shah S. Effectiveness of intra‐venous iron sucrose complex versus intra‐muscular iron sorbitol in iron deficiency anemia. Annals of Pakistan Institute of Medical Sciences 2006;2(3):188‐91. [Google Scholar]
Huch 1992 {published data only}
- Huch A, Eichhorn KH, Danko J, Lauener PA, Huch R. Recombinant human erythropoietin in the treatment of postpartum anemia. Obstetrics & Gynecology 1992;80(1):127‐31. [PubMed] [Google Scholar]
- Huch A, Huch R. EPO (rHuEPO) use in obstetrics. Proceedings of Ross Laboratories Special Conference, Hot Topics '90 in Neonatology; 1990 Dec 9‐11; Washington DC, USA. 1990:79‐80.
Mara 1999 {published data only}
- Mara M, Eretova V, Zivny J, Kvasnicka J, Umlaufova A, Marova E. Anemia and its treatment with peroral anti‐anemia agents in women during the postpartum period. [Czech]. Ceska Gynekologie 1999;64:153‐8. [PubMed] [Google Scholar]
Mara 2001 {published data only}
- Mara M, Zivny J, Eretova V, Kvasnicka J, Kuzel D, Umlaufova A, et al. Changes in markers of anemia and iron metabolism and how they are influenced in the postpartum period. Acta Obstetricia et Gynecologica Scandinavica 2001;80:142‐8. [PubMed] [Google Scholar]
Mitra 2012 {published data only}
- Mitra AK, Khoury AJ. Universal iron supplementation: a simple and effective strategy to reduce anaemia among low‐income, postpartum women. Public Health Nutrition 2012;15(3):546‐53. [DOI] [PubMed] [Google Scholar]
Osmond 1953 {published data only}
- Osmond TG. Post‐partum anaemia ‐ a practical treatment. Practitioner 1953;171:77‐80. [PubMed] [Google Scholar]
Picha 1975 {published data only}
- Picha E. Iron treatment by effervescent tablets. Geburtshilfe und Frauenheilkunde 1975;35(10):792‐5. [PubMed] [Google Scholar]
Van Der Woude 2014 {published data only}
- Woude DAA, Vries J, Wijk EM, Verzijl JM, Pijnenborg JMA. A randomized controlled trial examining the addition of folic acid to iron supplementation in the treatment of postpartum anemia. International Journal of Gynaecology and Obstetrics 2014;126:101‐5. [DOI] [PubMed] [Google Scholar]
- Woude D. Iron and folic acid v.s. iron solely in the treatment of post partum anaemia; effects on haemoglobin and health status. Netherlands Trial Register (http://www.trialregister.nl) (accessed 8 July 2011) 2011.
Zimmermann 1994 {published data only}
- Zimmermann R, Breymann C, Huch R, Huch A. rHuEPO in the treatment of postpartum anemia: subcutaneous versus intravenous administration. Clinical Investigator 1994;72(6 Suppl):S25‐30. [PubMed] [Google Scholar]
Zimmermann 1995 {published data only}
- Zimmermann R, Breymann C, Richter C, Huch R, Huch A. rhEPO treatment of postpartum anemia. Journal of Perinatal Medicine 1995;23:111‐7. [DOI] [PubMed] [Google Scholar]
References to studies awaiting assessment
Backe 2009 {published data only}
- Backe B. A 6‐week randomised, open comparative, multi‐centre study of intravenous ferric carboxymaltose (ferinject) and oral iron (duroferon) for treatment of post partum anemia. http://clinicaltrials.gov/ct2/show/record/NCT00929409 [accessed 5 June 2012] 2009.
References to ongoing studies
Chaudhuri 2013 {published data only}
- Chaudhuri P. Intravenous iron‐sucrose complex versus oral iron in the treatment of postpartum anemia. Clinical Trials Registry ‐ India [accessed 12 November 2013] 2013.
Holm 2015 {published data only}
- EUCTR2012‐005783‐10‐DK. A randomized comparative, open‐label study of intravenous iron isomaltoside 1000 (Monofer®) administered by high single dose in‐fusions or red blood cell transfusion in women with severe postpartum iron deficiency anaemia. EU Clinical Trials Register [accessed 13 November 2013] 2012.
- Holm C, Thomsen LL, Norgaard A, Langhoff‐Roos J. Intravenous iron isomaltoside 1000 administered by high single‐dose infusions or standard medical care for the treatment of fatigue in women after postpartum haemorrhage: study protocol for a randomised controlled trial. Trials 2015;16(1):5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hossain 2013 {published data only}
- Hossain N. Use of iron isomaltoside 1000 (Monofer) in postpartum anemia. ClinicalTrials.gov [accessed 12 November 2013] 2013.
Suneja 2014 {published data only}
- Suneja A. A clinical trial to compare oral iron ferrous sulfate with newer intravenous iron (ferric carboxymaltose) injection in patients of iron deficiency anemia in post delivery period. Clinical Trials Registry ‐ India (http://ctri.nic.in) [accessed 14 October 2014] 2014.
Additional references
al‐Momen 1996
- al‐Momen AK, al‐Meshari A, al‐Nuaim L, Saddique A, Abotalib Z, Khashogji T, et al. Intravenous iron sucrose complex in the treatment of iron deficiency anemia during pregnancy. European Journal of Obstetrics, Gynecology, and Reproductive Biology 1996;69(2):121‐4. [DOI] [PubMed] [Google Scholar]
Almeida 2010
- Almeida LC, Cardoso MA. Recommendations for folate intake in women: implications for public health strategies. Cadernos de Saude Publica 2010;26(11):2011‐26. [PUBMED: 21180975] [DOI] [PubMed] [Google Scholar]
Auerbach 2008
- Auerbach M, Goodnough LT, Picard D, Maniatis A. The role of intravenous iron in anemia management and transfusion avoidance. Transfusion 2008;48(5):988‐1000. [DOI] [PubMed] [Google Scholar]
Barish 2012
- Barish CF, Koch T, Butcher A, Morris D, Bregman DB. Safety and efficacy of intravenous ferric carboxymaltose (750 mg) in the treatment of iron deficiency anemia: two randomized, controlled trials. Anemia 2012;2012:172104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Barroso 2011
- Barroso F, Allard S, Kahan BC, Connolly C, Smethurst H, Choo L, et al. Prevalence of maternal anaemia and its predictors: a multi‐centre study. European Journal of Obstetrics, Gynecology, and Reproductive Biology 2011;159(1):99‐105. [DOI] [PubMed] [Google Scholar]
Bassler 2010
- Bassler D, Briel M, Montori VM, Lane M, Glasziou P, Zhou Q, et al. Stopping randomized trials early for benefit and estimation of treatment effects: systematic review and meta‐regression analysis. JAMA 2010;303(12):1180‐7. [PUBMED: 20332404] [DOI] [PubMed] [Google Scholar]
Bergmann 2010
- Bergmann RL, Richter R, Bergmann KE, Dudenhausen JW. Prevalence and risk factors for early postpartum anemia. European Journal of Obstetrics, Gynecology, and Reproductive Biology 2010;150(2):126‐31. [DOI] [PubMed] [Google Scholar]
Bodnar 2002
- Bodnar LM, Cogswell ME, Scanlon KS. Low income postpartum women are at risk of iron deficiency. Journal of Nutrition 2002;132(8):2298‐302. [DOI] [PubMed] [Google Scholar]
Bodnar 2005
- Bodnar LM, Cogswell ME, McDonald T. Have we forgotten the significance of postpartum iron deficiency?. American Journal of Obstetrics and Gynecology 2005;193(1):36‐44. [DOI] [PubMed] [Google Scholar]
Breymann 2010
- Breymann C, Honegger C, Holzgreve W, Surbek D. Diagnosis and treatment of iron‐deficiency anaemia during pregnancy and postpartum. Archives of Gynecology and Obstetrics 2010;282(5):577‐80. [DOI] [PubMed] [Google Scholar]
Bunn 2009
Carson 2012
- Carson JL, Carless PA, Hebert PC. Transfusion thresholds and other strategies for guiding allogeneic red blood cell transfusion. Cochrane Database of Systematic Reviews 2012, Issue 4. [DOI: 10.1002/14651858.CD002042.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Chalder 1993
- Chalder T, Berelowitz G, Pawlikowska T, Watts L, Wessely S, Wright D, et al. Development of a fatigue scale. Journal of Psychosomatic Research 1993;37(2):147‐53. [PUBMED: 8463991] [DOI] [PubMed] [Google Scholar]
Cohen 1983
- Cohen S, Kamarck T, Mermelstein R. A global measure of perceived stress. Journal of Health and Social Behavior 1983;24(4):385‐96. [PUBMED: 6668417] [PubMed] [Google Scholar]
Cox 1987
- Cox JL, Holden JM, Sagovsky R. Detection of postnatal depression. Development of the 10‐item Edinburgh Postnatal Depression Scale. British Journal of Psychiatry 1987;150(6):782‐6. [PUBMED: 3651732] [DOI] [PubMed] [Google Scholar]
Dudrick 1986
- Dudrick SJ, O'Donnell JJ, Matheny RG, Unkel SP, Raleigh DP. Stimulation of hematopoiesis as an alternative to transfusion. Southern Medical Journal 1986;79(6):669‐73. [DOI] [PubMed] [Google Scholar]
European Medicines Agency 2013
- European Medicines Agency. Assessment report for: Iron containing intravenous (IV) medicinal products. http://www.ema.europa.eu/docs/en_GB/document_library/Referrals_document/IV_iron_31/WC500150771.pdf 2013.
European Medicines Agency 2013a
- European Medicines Agency. New recommendations to manage risk of allergic reactions with intravenous iron‐containing medicines. http://www.ema.europa.eu/docs/en_GB/document_library/Referrals_document/IV_iron_31/WC500151308.pdf 2013.
Fuller 2010
- Fuller AJ, Bucklin BA. Blood product replacement for postpartum hemorrhage. Clinical Obstetrics and Gynecology 2010;53(1):196‐208. [DOI] [PubMed] [Google Scholar]
GRADEpro 2014 [Computer program]
- McMaster University. GRADEpro. [Computer program on www.gradepro.org]. Version 2014. McMaster University, 2014.
Gupta 2010
- Gupta SD, Khanna A, Gupta R, Sharma NK, Sharma ND. Maternal mortality ratio and predictors of maternal deaths in selected desert districts in Rajasthan a community‐based survey and case control study. Womens Health Issues 2010;20(1):80‐5. [DOI] [PubMed] [Google Scholar]
Hendrickson 2009
- Hendrickson JE, Hillyer CD. Noninfectious serious hazards of transfusion. Anesthesia and Analgesia 2009;108(3):759‐69. [DOI] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S, editors. Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Hoyer 2004
- Hoyer WJ, Stawski RS, Wasylyshyn C, Verhaeghen P. Adult age and digit symbol substitution performance: a meta‐analysis. Psychology and Aging 2004;19(1):211‐4. [PUBMED: 15065945] [DOI] [PubMed] [Google Scholar]
Hróbjartsson 2010
- Hróbjartsson A, Gotzsche PC. Placebo interventions for all clinical conditions. Cochrane Database of Systematic Reviews 2010, Issue 1. [DOI: 10.1002/14651858.CD003974.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Kennerley 1989
- Kennerley H, Gath D. Maternity blues. I. Detection and measurement by questionnaire. British Journal of Psychiatry 1989;155:356‐62. [PubMed] [Google Scholar]
Khalafallah 2012
- Khalafallah AA, Dennis AE. Iron deficiency anaemia in pregnancy and postpartum: pathophysiology and effect of oral versus intravenous iron therapy. Journal of Pregnancy 2012;2012:630519. [DOI] [PMC free article] [PubMed] [Google Scholar]
Khan 2006
- Khan KS, Wojdyla D, Say L, Gulmezoglu AM, Look PF. WHO analysis of causes of maternal death: a systematic review. Lancet 2006;367(9516):1066‐74. [DOI] [PubMed] [Google Scholar]
Klein 2014
- Klein HG, Anstee DJ. Mollison’s Blood Transfusion in Clinical Medicine. 12th Edition. ISBN:978‐1‐4051‐9940‐7, Wiley, 2014. [Google Scholar]
Kliger 2012
- Kliger AS, Fishbane S, Finkelstein FO. Erythropoietic stimulating agents and quality of a patient's life: individualizing anemia treatment. Clinical Journal of the American Society of Nephrology: CJASN 2012;7(2):354‐7. [DOI] [PubMed] [Google Scholar]
Kochhar 2013
- Kochhar PK, Kaundal A, Ghosh P. Intravenous iron sucrose versus oral iron in treatment of iron deficiency anemia in pregnancy: a randomized clinical trial. Journal of Obstetrics and Gynaecology Research 2013;39(2):504‐10. [DOI] [PubMed] [Google Scholar]
Litton 2013
- Litton E, Xiao J, Ho KM. Safety and efficacy of intravenous iron therapy in reducing requirement for allogeneic blood transfusion: systematic review and meta‐analysis of randomised clinical trials. BMJ (Clinical research ed.) 2013;347:f4822. [PUBMED: 23950195] [DOI] [PMC free article] [PubMed] [Google Scholar]
Marteau 1992
- Marteau TM, Bekker H. The development of a six‐item short‐form of the state scale of the Spielberger State‐Trait Anxiety Inventory (STAI). British Journal of Clinical Psychology / the British Psychological Society 1992;31 ( Pt 3):301‐6. [PUBMED: 1393159] [DOI] [PubMed] [Google Scholar]
Milman 2011
- Milman N. Postpartum anemia I: definition, prevalence, causes, and consequences. Annals of Hematology 2011;90(11):1247‐53. [DOI] [PubMed] [Google Scholar]
Milman 2011a
- Milman N. Anemia‐‐still a major health problem in many parts of the world!. Annals of Hematology 2011;90(4):369‐77. [PUBMED: 21221586] [DOI] [PubMed] [Google Scholar]
Milman 2012
- Milman N. Postpartum anemia II: prevention and treatment. Annals of Hematology 2012;91(2):143‐54. [DOI] [PubMed] [Google Scholar]
Montufar‐Rueda 2013
- Montufar‐Rueda C, Rodriguez L, Jarquin JD, Barboza A, Bustillo MC, Marin F, et al. Severe postpartum hemorrhage from uterine atony: a multicentric study. Journal of pregnancy 2013;2013:525914. [PUBMED: 24363935] [DOI] [PMC free article] [PubMed] [Google Scholar]
Oster 2012
- Oster HS, Neumann D, Hoffman M, Mittelman M. Erythropoietin: the swinging pendulum. Leukemia Research 2012;36(8):939‐44. [DOI] [PubMed] [Google Scholar]
Parker 2012
- Parker JA, Barroso F, Stanworth SJ, Spiby H, Hopewell S, Doree CJ, et al. Gaps in the evidence for prevention and treatment of maternal anaemia: a review of systematic reviews. BMC Pregnancy and Childbirth 2012;12:56. [PUBMED: 22727258] [DOI] [PMC free article] [PubMed] [Google Scholar]
Pena‐Rosas 2012
- Pena‐Rosas JP, De‐Regil LM, Dowswell T, Viteri FE. Daily oral iron supplementation during pregnancy. Cochrane Database of Systematic Reviews 2012, Issue 12. [DOI: 10.1002/14651858.CD004736.pub4] [DOI] [PMC free article] [PubMed] [Google Scholar]
Portenoy 2006
- Portenoy RK. Fatigue measurement. Fatigue measurement. In: Max MB, Lynn J, editors. Interactive textbook of symptom research. Bethesda (MD): National Institutes of Health. Available at: http://painconsortium.nih.gov/symptomresearch/chapter_9/sec6/crps6pg1.htm. Accessed August 2013.
Reveiz 2011
- Reveiz L, Gyte GML, Cuervo LG, Casasbuenas A. Treatments for iron‐deficiency anaemia in pregnancy. Cochrane Database of Systematic Reviews 2011, Issue 10. [DOI: 10.1002/14651858.CD003094.pub3] [DOI] [PubMed] [Google Scholar]
RevMan 2014 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.3. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Richter 1995
- Richter C, Huch A, Huch R. Erythropoiesis in the postpartum period. Journal of Perinatal Medicine 1995;23(1‐2):51‐9. [DOI] [PubMed] [Google Scholar]
Rohde 2014
- Rohde JM, Dimcheff DE, Blumberg N, Saint S, Langa KM, Kuhn L, et al. Health care‐associated infection after red blood cell transfusion: a systematic review and meta‐analysis. JAMA 2014;311(13):1317‐26. [PUBMED: 24691607] [DOI] [PMC free article] [PubMed] [Google Scholar]
Salpeter 2014
- Salpeter SR, Buckley JS, Chatterjee S. Impact of more restrictive blood transfusion strategies on clinical outcomes: a meta‐analysis and systematic review. American Journal of Medicine 2014;127(2):124.e3‐131.e3. [PUBMED: 24331453] [DOI] [PubMed] [Google Scholar]
Schmitz 1999
- Schmitz N, Kruse J, Heckrath C, Alberti L, Tress W. Diagnosing mental disorders in primary care: the General Health Questionnaire (GHQ) and the Symptom Check List (SCL‐90‐R) as screening instruments. Social Psychiatry and Psychiatric Epidemiology 1999;34(7):360‐6. [DOI] [PubMed] [Google Scholar]
Shander 2010
- Shander A, Hofmann A, Ozawa S, Theusinger OM, Gombotz H, Spahn DR. Activity‐based costs of blood transfusions in surgical patients at four hospitals. Transfusion 2010;50(4):753‐65. [DOI] [PubMed] [Google Scholar]
SHOT Report 2011
- SHOT Steering Group. Annual SHOT report 2011. http://www.shotuk.org/wp‐content/uploads/2012/07/SHOT‐ANNUAL‐REPORT_FinalWebVersionBookmarked_2012_06_22.pdf (accessed September 2013) 2011.
Smets 1995
- Smets EM, Garssen B, Bonke B, Haes JC. The Multidimensional Fatigue Inventory (MFI) psychometric qualities of an instrument to assess fatigue. Journal of Psychosomatic Research 1995;39(3):315‐25. [PUBMED: 7636775] [DOI] [PubMed] [Google Scholar]
Smith 2013
- Smith HM, Shirey RS, Thoman SK, Jackson JB. Prevalence of clinically significant red blood cell alloantibodies in pregnant women at a large tertiary‐care facility. Immunohematology 2013;29(4):127‐30. [PUBMED: 24689681] [PubMed] [Google Scholar]
Toblli 2008
- Toblli JE, Cao G, Olivieri L, Angerosa M. Comparative study of gastrointestinal tract and liver toxicity of ferrous sulfate, iron amino chelate and iron polymaltose complex in normal rats. Pharmacology 2008;82(2):127‐37. [PUBMED: 18607114] [DOI] [PubMed] [Google Scholar]
Toblli 2013
- Toblli JE, Cao G, Oliveri L, Angerosa M. Effects of iron polymaltose complex, ferrous fumarate and ferrous sulfate treatments in anemic pregnant rats, their fetuses and placentas. Inflammation & Allergy Drug Targets 2013;12(3):190‐8. [PUBMED: 23547731] [DOI] [PubMed] [Google Scholar]
Villanueva 2013
- Villanueva C, Colomo A, Bosch A, Concepcion M, Hernandez‐Gea V, Aracil C, et al. Transfusion strategies for acute upper gastrointestinal bleeding. New England Journal of Medicine 2013;368(1):11‐21. [DOI] [PubMed] [Google Scholar]
Ware 2000
- Ware JE Jr. SF‐36 health survey update. Spine 2000;25(24):3130‐9. [DOI] [PubMed] [Google Scholar]
Weiskopf 1998
- Weiskopf RB, Viele MK, Feiner J, Kelley S, Lieberman J, Noorani M, et al. Human cardiovascular and metabolic response to acute, severe isovolemic anemia. JAMA 1998;279(3):217‐21. [PUBMED: 9438742] [DOI] [PubMed] [Google Scholar]
WHO 1999
- World Health Organization. Reduction of Maternal Mortality. A jointWHO/UNFPA/UNICEF/World Bank statement. Geneva: WHO, 1999. [Google Scholar]
WHO 2001
- World Health Organization. Iron Deficiency Anaemia ‐ Assessment, Prevention, and Control WHO/NHD/01.3 2001. Geneva: WHO, 2001. [Google Scholar]
WHO 2002
- Rastogi T, Mathers C. Global burden of Iron Deficiency Anaemia in the year 2000. http://www.who.int/healthinfo/statistics/bod_irondeficiencyanaemia.pdf (accessed August 4 2013) 2002.
WHO 2012a
- World Health Organization. Trends in maternal mortality: 1990 to 2010. WHO, UNICEF, UNFPA and The World Bank estimates. http://www.unfpa.org/webdav/site/global/shared/documents/publications/2012/Trends_in_maternal_mortality_A4‐1.pdf (accessed September 2013) 2012.
WHO 2012b
- World Health Organization. The WHO Application of ICD‐10 to deaths during pregnancy, childbirth and the puerperium: ICD‐MM. http://apps.who.int/iris/bitstream/10665/70929/1/9789241548458_eng.pdf (accessed August 4 2013) 2012.
WHO ICD 2010
- World Health Organization. International Classification of Diseases (ICD‐10). http://www.who.int/classifications/icd/ICD10Volume2_en_2010.pdf (accessed August 4 2013).
Wiesen 1994
- Wiesen AR, Hospenthal DR, Byrd JC, Glass KL, Howard RS, Diehl LF. Equilibration of hemoglobin concentration after transfusion in medical inpatients not actively bleeding. Annals of Internal Medicine 1994;121(4):278‐30. [DOI] [PubMed] [Google Scholar]
Wysowski 2010
- Wysowski DK, Swartz L, Borders‐Hemphill BV, Goulding MR, Dormitzer C. Use of parenteral iron products and serious anaphylactic‐type reactions. American Journal of Hematology 2010;85(9):650‐4. [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Dodd 2004
- Dodd J, Dare MR, Middleton P. Treatment for women with postpartum iron deficiency anaemia. Cochrane Database of Systematic Reviews 2004, Issue 4. [DOI: 10.1002/14651858.CD004222.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]