

ABSTRACT
Cancer-associated fibroblasts (CAFs) are essential for tumor microenvironment remodeling and correlate with tumor progression. However, interactions between CAFs and tumor cells and immune cells in triple-negative breast cancer (TNBC) are still poorly explored. Here, we investigate the role of CAFs in TNBC and potential novel mediators of their functions. The clustering of classic markers was applied to estimate the relative abundance of CAFs in TNBC cohorts. Primary fibroblasts were isolated from normal and tumor samples. The RNA and culture medium of fibroblasts were subjected to RNA sequencing and mass spectrometry to explore the upregulated signatures in CAFs. Microdissection and single-cell RNA sequencing datasets were used to examine the expression profiles. CAFs were associated with hallmark signalings and immune components in TNBC. Clustering based on CAF markers in the literature revealed different CAF infiltration groups in TNBC: low, medium and high. Most of the cancer hallmark signaling pathways were enriched in the high CAF infiltration group. Furthermore, RNA sequencing and mass spectrometry identified biglycan (BGN), a soluble secreted protein, as upregulated in CAFs compared to normal cancer-adjacent fibroblasts (NAFs). The expression of biglycan was negatively correlated with CD8 + T cells. Biglycan indicated poor prognostic outcomes and might be correlated with the immunosuppressive tumor microenvironment (TME). In conclusion, CAFs play an essential role in tumor progression and the TME. We identified an extracellular protein, biglycan, as a prognostic marker and potential therapeutic target in TNBC.
KEYWORDS: Triple-negative breast cancer, cancer-associated fibroblast, biglycan, immune microenvironment, extracellular matrix
Introduction
Breast cancer is the most common malignant tumor in women worldwide.1 Triple-negative breast cancer (TNBC) is a special molecular subtype that is characteristic of negative hormone receptors (estrogen and progesterone receptors) and human epidermal growth factor receptor 2 (HER2). Neither endocrine therapy nor routine targeted therapy is effective for TNBC. There are limited therapeutic selections for TNBC and some patients are still burdened by low efficacy of treatment response and high risk of recurrence or metastasis.2 Hence, it is crucial to explore the mechanism underlying TNBC development and progression, which may lay a foundation for its diagnosis and treatment.
The crucial role of the tumor microenvironment (TME), which serves as the soil for seeds (cancer cells), has been proven in many studies.3–5 Cells in the TME mainly include stromal cells and immune cells. Recently, increasing evidence has highlighted that appropriate stromal cells are crucial for the development of tumors.6–8 Among them, cancer-associated fibroblasts (CAFs) represent the main fraction, and accumulating evidence has indicated their role in cancer proliferation, progression and invasion.9,10 It is well acknowledged that CAFs interact with malignant cells and orchestrate the metastasis of breast cancer.11–13 Although various clinical trials targeting CAFs have been performed in recent years, such as targeting surface markers, reducing CAF infiltration and normalizing CAF functions, most of them are still ongoing.10 Previous studies have identified many CAF markers, but few of them have moved into clinical practice.14 This may be due to the internal heterogeneity of CAFs. The CAFs seemed to originate from diverse cell types, such as fibrocytes, stellate cells, endothelial cells and mesenchymal stem cells.14 It is well accepted that most activated fibroblasts are derived from fibroblasts of adjacent normal tissues and induced by oxidative stress or specific cytokines and chemokines from cancer cells.15 Hence, distinct subclusters have been identified by previous studies. CAFs in human breast cancer can be divided into CAF-S1 to CAF-S4 according to specific signatures, such as CD29, FAP, FSP1, α-SMA, CAV1 and DPP4.16 Furthermore, the CAF-S1 cluster contributes to a poor response to immunotherapy.17 Immunotherapy has become a novel strategy for breast cancer treatment in recent years.18 Therefore, focusing on the function and mechanism of fibroblasts in the tumor microenvironment may provide a strategy for breast cancer treatment, especially immunotherapy.
In this study, we explored the relative infiltration level of fibroblasts in triple-negative breast cancer and the correlation between CAFs and immune components in the TME. We further identified an upregulated secreted protein, biglycan, in CAFs compared with normal cancer-adjacent fibroblasts (NAFs) using RNA sequencing and mass spectrometry methods. Biglycan is encoded by the BGN gene and is mainly expressed in the stromal part of tumors. Biglycan is a protein that belongs to the small leucine-rich proteoglycan (SLRP) family. We found that the expression level of BGN is correlated with the extracellular matrix, lymphangiogenesis, epithelial-mesenchymal transition, angiogenesis and TGF-β signaling. Single-cell sequencing results show that BGN is mainly expressed in stromal fibroblasts. Moreover, BGN is highly expressed in CAFs of TNBC compared with other cell subpopulations. The role of BGN in the TME and its mechanism underlying how to affect the microenvironment remain unknown.
Establishing the relevance of the role of fibroblasts and biglycan in human triple-negative breast cancer using publicly available datasets and clinical samples raises the probability that targeting biglycan may yield clinical utility.
Methods
Datasets and tissue specimens
The Cancer Genome Atlas (TCGA) dataset was obtained using the TCGAbiolinks package in R software.19,20 The transcripts per million (TPM) value was estimated at the transcript level. Patients who were diagnosed with breast cancer histologically and available for transcriptomic data were included. To further distinguish triple-negative breast cancer samples from the breast cancer cohort (BRCA), patients with immunohistochemically negative estrogen receptor (ER), progesterone receptor (PR) and human epidermal growth factor receptor 2 (HER2) were included. A total of 116 patients were enrolled in the TCGA-TNBC cohort. Overall survival (OS) was assessed using vital status and days from diagnosis to death or the last follow-up date. Only patients with active follow-up information were included in survival analysis.
The processed data of the Molecular Taxonomy of Breast Cancer International Consortium (METABRIC) dataset were downloaded from cBioPortal (http://www.cbioportal.org/) according to the website’s guidance.21 No identification information of participants was involved during download and analysis. Patients diagnosed with breast cancer and available for active follow-up information were included. Triple-negative breast cancer samples were also enrolled in the METABRIC-TNBC cohort as described above.
For other TNBC datasets (GSE25066, GSE103091, GSE21653 and GSE88715), the expression matrices were downloaded from the Gene Expression Omnibus (GEO) (https://www.ncbi.nlm.nih.gov/geo/).22–25 The probes were mapped using the corresponding annotation platforms. The expression values were further normalized by the limma package if necessary.26 The secreted genes were predicted as previously reported.27 Single-cell sequencing (scRNA-seq) datasets were acquired and analyzed according to the guidance of previous studies. The cell types were annotated using the SingleR package if necessary.28,29 GSE114727, GSE136206, GSE138536 were used to observe the expression of CAF markers in single-cell level.30–32 The BGN expression was also analyzed in cohorts (Bassez A, et al.; GSE118389; Wu SZ, et al.).33–35 These datasets are shown in Table S1. No identification information of participants was involved during download and analysis.
Tissues for immunohistochemistry and primary cell isolation were obtained at Sun Yat-sen University Cancer Center (SYSUCC) between January 2010 and June 2021. The clinical breast cancer specimens were collected with permission from the Institutional Review Board of the SYSUCC and informed consent was obtained from participants. The study was conducted according to the principles stated in the Declaration of Helsinki.
Tumor microenvironment estimation
The Estimate the Proportion of Immune and Cancer cells (EPIC), xCell, and Microenvironment Cell Populations-counter (MCP-counter) algorithms were applied to calculate the cancer-associated fibroblast scores in datasets.36–38 To analyze the correlation among fibroblasts and immune cells, fractions of 22 immune cells were estimated using the Cell-type Identification By Estimating Relative Subsets Of RNA Transcripts (CIBERSORT) algorithm.39–41 The estimation of stromal and immune cells in malignant tumor tissues using expression data (ESTIMATE) was applied to calculate the overall stromal and immune scores in cancer.42
Molecular markers and CAF clustering
Representative immune-related genes (IRGs) and CAF signatures were obtained from previous studies.3,43,44 The IFN-γ signature (IFNG1, STAT1, CXCL10, IDO1, CXCL9), myeloid lineage (CD14, CD163, ARG1, CD68, OLR1, NOS2, CD33), inhibitory immune ligands/receptors (HAVCR2, CTLA4, LAG3, PDCD1, CD274), immune modulators (ENTPD1, NT5E) and activating immune receptors (TNFRSF4, TNFRSF9, CD40, CD80, CD27, ICOS) were analyzed as previously reported.45 The edgeR and limma packages were used to calculate the fold change of genes between groups.26,46
Several classic CAF markers were adopted from previous studies to estimate the relative abundance of fibroblasts in cancer samples, including PDGFRA, PDGFRB, ACTA2, THY1, PDPN, COL1A1 and FAP.47–50 Clustering was performed in individual datasets, and the samples were further classified into high-, medium-, and low-infiltration groups using the ComplexHeatmap package.51 To further confirm the clustering results, the principal components analysis (PCA) method was applied as previously desribed.52 Inflammatory and myofibroblastic CAF features were also included to assess the internal characteristics. Comparisons of biological markers among different CAF infiltration groups are shown by the ggheatmap and ComplexHeatmap packages.
Functional analysis
The GSVA package was used for gene set variation analysis (GSVA).53 The GSVA results were compared between the high- and low-CAF infiltration groups and are displayed. Gene set enrichment analysis (GSEA) was used to explore the biological functions of BGN and performed using GSEA 4.1.0.54 Hallmark and gene ontology gene sets were obtained from the MSigDB Collections (http://www.gsea-msigdb.org/gsea). The TGF-β response signatures (TBRS) of T cells (T-TBRS), fibroblasts (F-TBRS), endothelial cells (End-TBRS) and macrophages (Ma-TBRS) were calculated and compared between different CAF infiltration groups as previously reported.55
Primary cell culture, RNA sequencing and mass spectrometry
Primary fibroblasts were isolated from immunohistochemically confirmed breast cancer samples obtained from surgery. For normal cancer-adjacent fibroblasts (NAFs), tissues >2 cm away from cancer were used. Primary fibroblasts were isolated as described in previous studies.56 Briefly, tissues were digested with hyaluronidase (Sigma), collagenase type I (Sigma) and collagenase type III (Worthinton) at 37°C with agitation for 3–4 hours. The digested tissues were then incubated without shaking for 10 min, and the supernatant enriched with stromal cells was moved to new tubes and centrifuged at 250 g for 10 min. The supernatant was discarded, and the pellet was harvested and cultured in DMEM with 10% FBS. Fibroblasts were passaged within 5 population doublings after isolation. The RNA of the primary cells was extracted using TRIzol reagent (Invitrogen). More than 2 μg of high-quality RNA with a 260/280 absorbance ratio of 1.8–2.2 per sample was used for subsequent experiments. The rRNA-depleted RNA was prepared by the Ribo-off™ rRNA depletion kit (Vazyme, N406-01) and further applied to synthesize the first cDNA using Total RNA-seq Library Prep Kit (Vazyme, NR603). After library construction, fragments were further enriched by PCR amplification (NEST Biotechnology) and screened before sequencing. The Illumina NovaSeq 6000 sequencing platform was used for sequencing and raw reads were generated following the recommended protocol from the vendor. The transcripts per million (TPM) were calculated at the transcript level based on counts data.
The culture medium of primary cells was collected and centrifuged at 500 g for 10 min at 4°C and 10,000 g for 30 min at 4°C to remove cellular debris. The supernatant was further filtered using a 0.22 μm filter. Briefly, the protein was extracted, digested by trypsin and further applied for label-free mass spectrometry. The results were further searched in Maxquant software and quantified using LFQ method.
Immunohistochemistry
The samples were fixed with paraformaldehyde and embedded in paraffin. The samples were then sectioned into 3 μm slides. Antigen retrieval was performed using a pressure cooker with citrate buffer (CWBIO) for 10–15 min. Goat serum (ZSGB-BIO) was used to block nonspecific binding to the samples. The samples were further incubated with specific anti-biglycan (1:150, Proteintech) or anti-CD8 (ready to use, ZSGB-BIO) antibodies at 4°C overnight and a secondary antibody (ZSGB-BIO) at room temperature for 1 hour. The expression of protein was detected using DAB (Dako) following the manufacturers’ instructions. The density of infiltrating CD8 + T cells was estimated using HALO software (Indica Labs).
Western blot
For cell lines, cells were lysed using RIPA buffer (Beyotime) at 4°C directly. Phenylmethanesulfonyl fluoride (PMSF, Beyotime) was added to reduce protein degradation during extraction. Proteins were separated in SDS-polyacrylamide gels and transferred to PVDF membranes, and nonfat milk was used to block the nonspecific binding sites on the membrane. The membranes were incubated with primary antibodies against biglycan (1:1000, Proteintech) and GAPDH (1:3000, SAB) at 4°C overnight. The secondary antibody (1:5000, Bioss) was applied on the following day, and the reaction was detected using enhanced chemiluminescence solution (ECL, Affinity).
Statistical methods
The best cutoff values for specific markers in each cohort were determined using the survminer package. The survival package was used for Kaplan-Meier overall survival analysis, and the log-rank test was applied for comparison. The hazard ratio (HR) was calculated via univariate Cox regression. Immune signatures were divided into two groups according to the median value and calculated by Cox regression in. Student’s t-test or Wilcoxon rank-sum test were used for comparison of normally and non-normally distributed variables in unpaired groups, respectively. The paired Student’s t-test was performed for paired samples. Chi-square test and Fisher’s exact test were applied for comparison of clinical features. The Spearman method was applied for correlation analysis. All P values were two-tailed. Statistical analysis was performed using R software (Version 3.5.3, https://www.r-project.org).
Results
CAF marker-based classifier identified three clusters with different CAF infiltration in TNBC cohorts
The relative abundance of fibroblasts in TNBC was estimated by expression profile clustering using classic CAF markers reported in previous studies, including ACTA2, FAP, PDGFRA, PDGFRB, PDPN, THY1 and COL1A1. The expression level of the above markers in CAFs of breast cancer was examined using single-cell sequencing datasets (Figure S1). Samples from the TCGA-TNBC, METABRIC-TNBC, GSE25066-TNBC, GSE21653 and GSE103091 cohorts were analyzed. The clustering results of TCGA-TNBC, METABRIC-TNBC and GSE25066-TNBC are shown in Figure 1a, S2a and S2c, and patients were divided into different CAF infiltration groups, namely, low, medium and high infiltration. The distributions of clinical or biological features, such as the ESTIMATE stromal score, immune score, mutation type, age at diagnosis, TNM stage, subtypes, amongst others, were displayed under the heatmap. PCA plots were employed to verify the clustering efficacy in the cohorts (Figure 1b, S2b, S2d). To further examine whether the clustering groups reflect the relative abundance of CAFs in the TME, we compared the CAF scores reported in previous studies and demonstrated an ascending tendency of MCP-counter and xCell scores in the low, medium and high CAF infiltration groups of representative cohorts (Figure 2a, 2d, 2g). Moreover, the expression of iCAF and myCAF markers was also compared. The results showed that lots of CAF markers were upregulated in the high and medium infiltration groups (Figure 2b, 2e, 2h). The results indicated that samples in these groups showed abundant infiltrating CAFs. The CAF infiltration clusters of all TNBC cohorts in this study are shown in Figure S3a. The overall survival did not show a significant difference among the CAF infiltration groups (Figure S3b-f).
Figure 1.
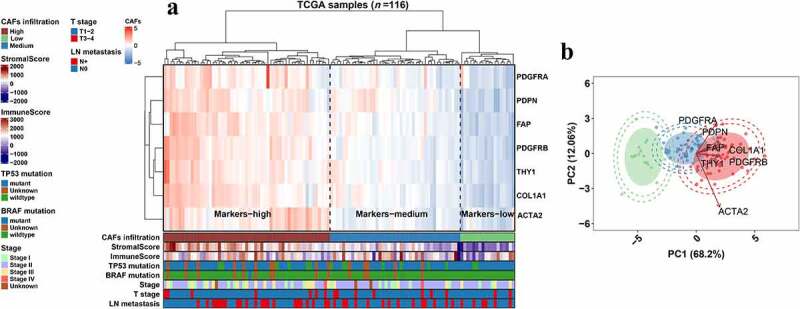
Clustering of classic CAF markers in the TCGA-TNBC cohort. a, Heatmap showing the expression level of CAF markers and clustering of CAF infiltration in the TCGA-TNBC (N = 116) cohort. Bars under the heatmap display the distributions of clinical features and ESTIMATE scores. b, Scatter plots showing the PCA result of the TCGA-TNBC cohort based on the classic CAF markers (x and y axes). External circle, 98% confidence interval; Internal circle, 95% confidence interval; Eclipse, 80%. Arrows, the tendency of marker profiling; low, medium and high infiltration levels are shown as green, blue and red, respectively.
Figure 2.
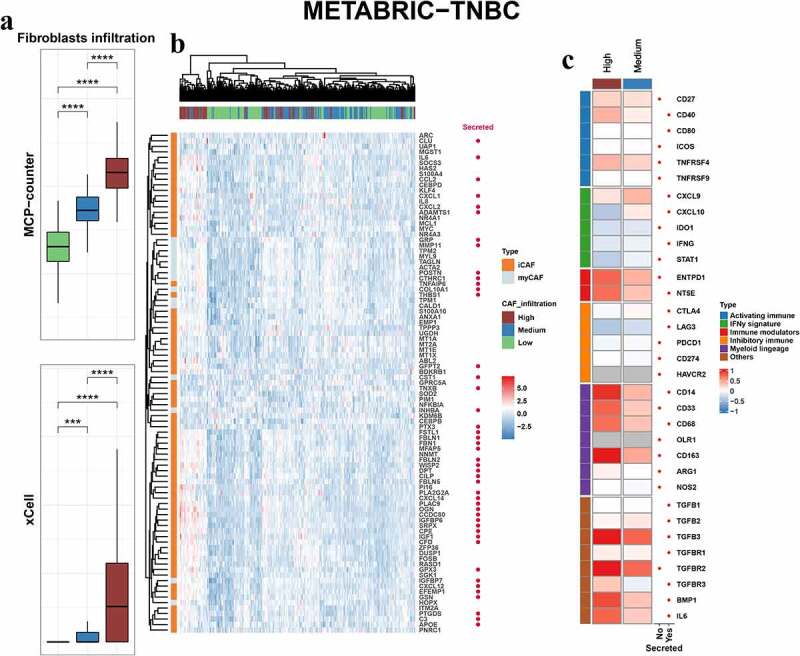
Fibroblastic and immune features in CAF infiltration groups. Box plots comparing the fibroblastic scores from MCP-counter (upper) and xCell (lower) in different infiltration groups of the METABRIC-TNBC (a), TCGA-TNBC (d) and GSE25066-TNBC (g) cohorts. P values from Student’s t-test. Heatmap showing the expression profile of myCAF and iCAF features in different infiltration groups of the METABRIC-TNBC (b), TCGA-TNBC (e) and GSE25066-TNBC (h) cohorts. Heatmap showing the fold change of the immune features in high and medium groups compared with the low CAF infiltration group (high vs. low, medium vs. low) in the METABRIC-TNBC (c), TCGA-TNBC (f) and GSE25066-TNBC (i) cohorts. P values are reported as: ns, nonsignificant; *, P < .05; **, P < .01; ***, P < .001; ****, P < .0001.
Figure 2.

Continued.
Figure 2.

Continued.
Correlation between CAFs infiltration and immune features
Previous studies have indicated the crucial role of CAFs in immune modulation of the microenvironment. Here, we sought to examine the correlation between CAFs and the immune landscape. First, we calculated the fold change of genes between different infiltration groups (high vs. low, medium vs. low) in METABRIC-TNBC, TCGA-TNBC and GSE25066-TNBC cohorts and the immune-related genes were shown (Figure 2c, 2f, 2i). The results indicated that some of them were upregulated in the high and medium infiltration groups of different cohorts, such as CD14, TGFB3, ENTPD1, NT5E and BMP1. Other upregulated markers were not consistent in different cohorts, such as LAG3, CD33, CD68, CXCL9, and IL6. Second, the GSVA scores of hallmark signaling pathways in cancer were compared between the high- and low-infiltration groups (Figure 3a-c). Some of the immune-related signaling pathways were significantly enriched in the high infiltration group, such as TGF-β, IL2-STAT5, complement, TNF-α-NF-κB and IL6-JAK-STAT3 signaling. Finally, as CAFs are one of the producers of TGF-β, we compared the expression levels of the TGF-β response signatures of fibroblasts (F-TBRS), T cells (T-TBRS), macrophages (Ma-TBRS) and endothelial cells (End-TBRS). Most signatures showed an ascending tendency from the low- to high-infiltration groups (Figure S4).
Figure 3.
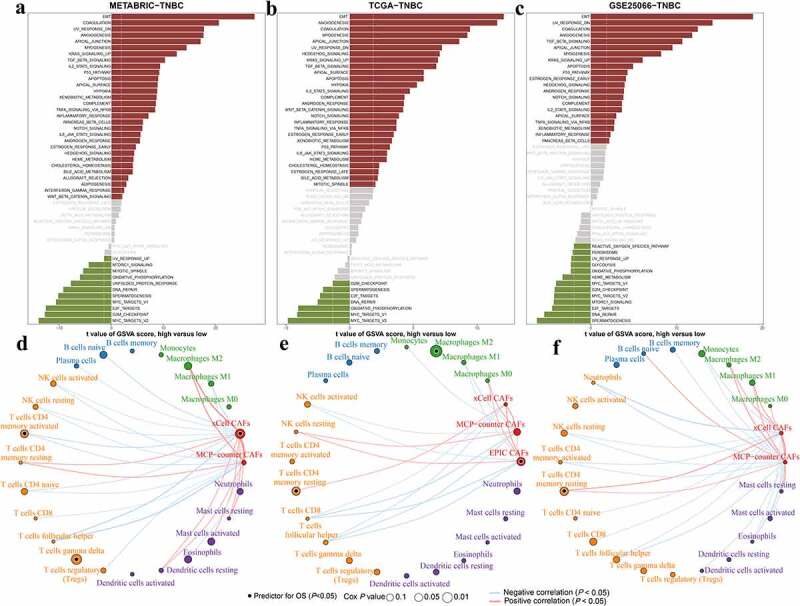
Correlation between CAFs and hallmark signaling and immune cells. a-c, Bar plots showing the GSVA scores of cancer hallmark signaling in the high infiltration group compared with the low CAF infiltration group. Significantly upregulated and downregulated signaling pathways are shown in red and green, respectively. Nonsignificant signalings are shown as gray. d-f, Network showing the correlation among CAFs and CIBERSORTx-derived immune cells. Significantly positive and negative correlations are shown as red and blue lines, respectively. The color and size of the nodes indicate the type of cells and P values from Cox regression. Prognostic signatures for overall survival are marked with dark dots in the nodes.
Based on the results above and previous studies, CAFs might exert their function by affecting tumors and immune cells. We evaluated the correlation between previously reported CAF scores and CIBERSORTx immune cells (Figure 3d-f). The CAF scores estimated by xCell, EPIC and MCP-counter were analyzed. CAFs were negatively correlated with cytotoxic cells, including CD8 + T cells and activated NK cells. In the METABRIC-TNBC cohort, MCP-counter-CAF and xCell-CAF were negatively correlated with activated NK cell, respectively. But only MCP-counter-CAF was negatively correlated with CD8 + T cell. In the TCGA-TNBC cohort, EPIC-CAF, MCP-counter-CAF and xCell-CAF were all negatively correlated with CD8 + T cell. But only EPIC-CAF and MCP-counter-CAF were negatively correlated with activated NK cell. In the GSE25066-TNBC cohort, only a negative correlation between xCell-CAF and CD8 + T cell was observed. Moreover, CAFs might exhibit significantly positive correlations with M2 macrophages and this result was only observed in METABRIC-TNBC and GSE25066-TNBC cohorts. Furthermore, xCell-CAF and EPIC-CAF were risk factors for overall survival in METABRIC-TNBC and TCGA-TNBC cohorts, respectively.
BGN is upregulated and mainly expressed in stromal CAFs
Here, we sought to investigate effective biomarkers derived from CAFs and their corresponding prognostic value. Normal cancer-adjacent fibroblasts and cancer-associated fibroblasts were isolated from tissue samples after surgical resection. RNA sequencing and label-free mass spectrometry were further applied to explore the transcriptomic expression profile and differentially expressed genes (DEGs) in the NAF and CAF groups (Figure 4a). The top 40 expressed DEGs in CAFs are shown in Figure 4b. The label-free mass spectrometry identified the top 40 secreted proteins (Figure 4c). BGN, an extracellular protein, was upregulated in CAFs compared to NAFs. Previous studies indicated the roles of BGN in therapy resistance and immune activity.57–60 Overexpression of biglycan induced resistance to 5-FU and rapamycin in cancer cells via NF-κB signaling pathway. Besides, most mechanistic studies mainly focused on the interaction between biglycan and toll-like receptors (TLR2/4).61 However, the mechanism underlying how biglycan affects tumor microenvironment in other ways is still poorly understood despite its abundance in TME. The TPM values of BGN from RNA sequencing were compared (Figure 4d). We further compared the BGN level in the TCGA-TNBC cohort and observed a significantly higher level of BGN in tumor tissues (Figure 4e). We further verified our results at the protein level by Western blotting and observed a significantly higher level of biglycan protein in CAFs of patient 1, 2 and 3 (Figure 4f).
Figure 4.

BGN is upregulated in cancer-associated fibroblasts. a, Illustration showing the procedure of RNA sequencing and mass spectrometry analysis for tissue-derived fibroblasts and culture medium, respectively. Representative image of primary fibroblasts is shown. b, Heatmap showing the top 40 expressed genes upregulated in CAFs. c, Heatmap showing the top 40 secreted proteins identified by label-free mass spectrometry in culture medium derived from CAFs and the corresponding NAFs. d. Box plot comparing the RNA level of biglycan in NAFs and CAFs. P value from paired Student’s t-test. e, Box plot comparing BGN expression in normal and tumor tissues in the TCGA-TNBC cohort. P value from Student’s t-test. f, Western blot showing the expression of biglycan protein in fibroblasts isolated from normal and cancer tissues of TNBC. g, Violin plot comparing the BGN expression level in the epithelial and stromal components derived from microdissection (GSE88715). P value from paired Wilcoxon rank-sum test.
To further confirm the expression profile of BGN in different tumor components, we analyzed its expression pattern using microdissection and single-cell sequencing datasets. Microdissection analysis (GSE88715) indicated a higher level of BGN expression in the stromal part of the tumor (Figure 4g). Moreover, BGN expression showed an ascending tendency from the low to high CAF infiltration group described above (Figure S5a). The expression level of BGN was also positively correlated with classic CAF markers in each cohort (Figure S5b).
To further confirm its expression feature in TNBC, we sought to analyze the BGN expression level in TNBC scRNA-seq datasets. BGN was mainly expressed in the overall fibroblasts of TNBC cohorts (Bassez A, et al.; GSE118389; Figure 5a, 5b). In another dataset with more detailed cell clusters, BGN was mainly expressed in CAFs and perivascular-like (PVL) subpopulations (Wu SZ, et al.; Figure 5c-e). The BGN expression level was significantly correlated with the scores of fibroblasts and endothelial cells estimated by MCP-counter and xCell in these cohorts, respectively (Figure 6a-c-). This result provided additional evidence for the derivation of biglycan. These results further indicated that BGN might be a CAF-specific biomarker in TNBC and exert its function in the extracellular matrix.
Figure 5.

BGN expression in single-cell RNA sequencing datasets of TNBC. a-c, Violin plots showing the expression level of BGN in cells of scRNA-seq datasets (Bassez A ; GSE118389; Wu SZ). d, Scatter plot showing the cell clusters in c (Wu SZ). e, Scatter plot showing the distribution of cells expressing a high level of BGN in c.
Figure 6.

The correlation of BGN and CAF scores and pathway enrichment. a-c, Scatter plots showing the correlation between BGN and CAFs and endothelial cells. P and r values from Spearman correlation analyses. d-f, Enrichment scores of the representative enriched signaling pathways in the high-BGN group compared with the low-BGN group. Enrichment Score from the Gene Ontology enrichment.
Biglycan correlates biological features and clinical outcomes
The BGN gene encodes biglycan, an extracellular soluble protein that might exert its function via intercellular contact. Here, we sought to explore its role in tumor development and its prognostic value in clinical practice. GO signaling enrichment was performed using BGN-correlated genes, and the representative results are shown in Figure 6d-f. Representative signaling pathways involved in cancer progression and TME modulation were significantly enriched in METABRIC-TNBC, TCGA-TNBC, GSE25066-TNBC cohorts (FDR<0.05). The high BGN level indicates the extension of new blood vessels from the existing vessels (SPROUTING_ANGIOGENESIS), lymph vessel formation (LYMPHANGIOGENESIS), extracellular matrix remodeling (COLLAGEN_CONTAINING_ECM) and promotion of tumor cell metastasis (POSITIVE_REGULATION_OF_EMT). Furthermore, BGN was also correlated with the formation of an immunosuppressive microenvironment (TGF_BETA_RECEPTOR_SIGNALING). These results indicated that biglycan might be associated with tumor angiogenesis, TME remodeling and tumor metastasis in TNBC. Hence it might serve as a protumor and immunosuppressive factor in these cohorts. Therefore, we further analyzed the association between BGN and the immune signatures. First, the correlation between BGN and immune-related genes was analyzed (Figure 7a). BGN was positively correlated with some immune-inhibitory molecules, including TGFB1, VEGFA, VEGFB and CD276, especially in METABRIC-TNBC and GSE21653 cohorts. However, we did not observe significant association with classic checkpoint molecules except for CTLA4 and CD274 (PD-L1) in GSE103091 and BTLA in GSE21653. These results imply that BGN might affect immune cell infiltration or modulate immune activity. Hence, we calculated the correlation between BGN expression and ESTIMATE scores (Figure 7b). The BGN was significantly positively correlated with the stromal scores in each cohort. We observed significant correlations between BGN and the immune scores in METABRIC-TNBC (negative, P < .05), GSE21653 (negative, P < .05) and GSE103091 (negative, P < .1). Finally, we examined the correlation between BGN and immune components. BGN was significantly correlated with both CD8 + T cells (negative, P < .05) and activated NK cells (negative, P < .05) in the METABRIC-TNBC and TCGA-TNBC cohorts, respectively. Moreover, BGN was correlated with M0 macrophages in all cohorts (positive, P < .05) except for GSE21653 and M2 macrophages in METABRIC-TNBC, GSE25066-TNBC and GSE103091 (positive, P < .05) (Figure 7c).
Figure 7.

The correlation between BGN and immune features. a, Heatmap showing the correlation between BGN and immune-related genes. b, Bubble plot showing the correlation between BGN and ESTIMATE stromal and immune scores. c, Bubble plot showing the correlation between BGN and CIBERSORT-derived immune cell scores. Positive and negative correlations are shown as red and blue, respectively. Correlation coefficients and P values from the Spearman method.
Since BGN plays an essential role in the biological process of cancer, we further sought to examine its clinical relevance. First, we observed a significantly lower level of BGN in TNBC than in non-TNBC samples of the TCGA and METABRIC cohorts (Figure S6a). BGN expression was significantly upregulated in tumors compared with paired normal tissues in the TCGA-TNBC cohort (Figure S6b). Second, the Cox regression model was conducted according to the best cutoff value for overall survival and indicated that BGN was a poor prognostic predictor in the METABRIC-TNBC, GSE103091 and GSE21653 cohorts (Figure S6c). Besides, FAP predicted poor prognosis in TCGA-TNBC and GSE21653 cohorts. PDGFRB and COL1A1 predicted poor outcomes in TCGA-TNBC and GSE103091 cohorts. However, PDGFRA was predictive of favorable outcomes in cohorts except for TCGA-TNBC and PDPN predicted favorable prognosis in METABRIC-TNBC and GSE25066-TNBC cohorts. Kaplan-Meier analysis demonstrated that a higher BGN expression level predicted worse overall survival outcomes in each cohort (Figure 8a). Finally, we also validated the prognostic value and immunosuppressive function of biglycan in the SYSUCC cohort. Patients in the high-biglycan group showed a lower level of total infiltrating CD8 + T cells (Figure 8b, 8c). Survival analyses demonstrated that biglycan might serve as a poor prognostic marker in TNBC (Figure 8d, Table S2). The univariate and multivariate Cox regression model for the SYSUCC cohort indicated that biglycan was an independent risk factor for overall survival (Table S3).
Figure 8.

Survival analysis of BGN in datasets and the SYSUCC cohort. a. Kaplan-Meier plots comparing the overall survival between the high-BGN and low-BGN groups according to the best cutoff value. P values from log-rank tests. High-BGN and low-BGN groups are shown as red and blue. b, Representative IHC staining images of stromal biglycan and CD8 in TNBC of SYSUCC (N = 100). Scale bar, 100 μm. c, Boxplot comparing the density of CD8 + T cells in high-biglycan and low-biglycan groups. P value from Student’s t-test. d, Kaplan-Meier plot comparing the overall survival between high-biglycan and low-biglycan groups. P value from log-rank tests. High-biglycan and low-biglycan groups are shown as red and blue.
Discussion
Recent studies have indicated the crucial roles of cancer-associated fibroblasts in breast cancer.15 Many of them focus on the heterogeneity and corresponding biological features of different CAF clusters in cancer.16,17,62 However, the detailed mechanism underlying how CAFs affect the TME has been a prominent topic in recent years.
Here, we have included the expression profile of the tumor bulk and single-cell RNA sequencing to explore the characteristics of fibroblasts in triple-negative breast cancer. We evaluated the relative abundance of fibroblasts in TNBC and further identified an extracellular secreted protein, biglycan, as a biomarker for CAFs and a predictor for poor prognosis in TNBC.
CAFs have been reported to be a tumor-promoting component of the stroma in most cancers.10 In previous studies, fibroblasts exerted their function by producing excreted factors, remodeling the extracellular matrix, influencing cancer cell metabolism and direct cell-cell interactions. For example, CAF-derived PLGF and BDNF promote NR4A1 expression in triple-negative breast cancer cells and enhance invasiveness.11 In addition, CCL2/7 are produced by lung-resident fibroblasts to modify cancer cell metabolism by activating cholesterol synthesis in TNBC cells in the lung and promoting angiogenesis.63 Furthermore, CAFs serve as leading cells for cancer cells during cell migration.64 Fibroblasts pave the way for subsequent malignant cells and lead to tumor invasion by direct cell-to-cell contact. Here, we sought to explore the effect of the relative abundance of CAFs in TNBC. The CAF infiltration level affects tumor cells and the TME with different CAF, immune and tumor cell expression profiles. Although some immune markers show an ascending tendency from low to high CAF infiltration levels (CD14, NT5E and ENTPD1, etc.), some other markers do not. This is consistent with a previous study which indicates that fibroblast-enriched samples might be clustered into fibrotic (F) and immune-enriched/fibrotic (IE/F) groups with distinct features.65 Furthermore, the simple relative abundance of CAF infiltration did not necessarily seem to be a prognostic predictor. This is consistent with previous studies showing that CAFs are highly heterogeneous and different subpopulations display various features. Tumor-infiltrating fibroblasts are commonly divided into inflammatory CAFs (iCAFs) with lower α-SMA and higher cytokine production, and myofibroblastic CAFs (myCAFs) with higher levels of α-SMA.34 Recently, a novel cluster of antigen-presenting CAFs (apCAFs) was identified, and CAFs were confirmed to directly participate in immune reactions.44 The reason why high infiltration group exhibited a higher level of BGN might be partially due to a larger amount of overall CAFs. But it is not capable of predicting the dominant role of BGN-expression CAFs. These results indicate that the heterogeneity of CAFs is more important than simply cell abundance. A single-cell sequencing cohort of TNBC with a large population and active follow-up data will help to clarify the role of the amounts of overall CAFs and specific dominant subclusters in predicting clinical outcomes in future studies.
Furthermore, internal heterogeneity in the cellular population poses a new challenge in modern medicine. In recent years, single-cell sequencing technology has helped to explore single-cell expression profiles and identify internal heterogeneous clusters of cells. Infiltrating CAF-S1 is enriched in TNBC, and further clustering by single-cell sequencing reveals 8 subclusters with distinct features, in which Clusters 0/3 are linked to immunotherapy resistance in cancer.17 Together, these results imply that specific CAF subpopulations might truly matter instead of simply the number of infiltrating fibroblasts in TNBC. Hence, it is more important to explore the expression profile of CAFs.
As CAF-derived proteins represent the most common way for intercellular crosstalk and might serve as biomarkers for cancer, we used RNA sequencing and mass spectrometry technology, and identified the coding gene for biglycan, BGN, to be mainly expressed in CAFs. The biglycan protein belongs to the SLRP family and plays a role in the extracellular matrix (ECM).66 Interestingly, although BGN was found to be upregulated in CAFs compared to NAFs, it also seemed to be expressed in endothelial cells. A previous study reported that BGN was approximately 100-fold more abundant in tumor endothelial cells than in corresponding normal endothelial cells.67 In another study, the results from scRNA-seq indicated that BGN is also expressed in perivascular-like subclusters except for CAFs.34 However, this is not contradictory to our findings, as endothelial cells and pericytes are commonly recognized as crucial origins for CAFs and transform into CAFs under certain circumstances.14 Furthermore, the number of fibroblasts was far greater than that of endothelial cells in scRNA-seq datasets, which indicates that CAFs are the major origin of biglycan proteins. BGN expression correlates with poor overall survival, cancer biological processes and microenvironmental components. Biglycan is a nonfibrillar component in the ECM, suggesting that extracellular biglycan might not only constitute the ECM but also exert other functions. BGN encodes a secreted protein that could bind to receptors on immune cells and it might also potentially interact with tumor cells, pericytes or endothelial cells.60 The Gene Ontology enrichment in this study implies the potential role of BGN in tumor vasculature and our findings are consistent with previous studies that CAFs promoted tumor angiogenesis mainly by releasing secreted factors, such as SDF1, MMPs and VEGFs.68,69 Biglycan is a vital component in bone and might promote the proliferation of osteosarcoma cells through an LPR6/β-catenin/IGFR-IR axis.70 Other studies indicate that biglycan might enhance drug resistance in cancer cells.58,59 These studies imply that biglycan promotes cancer progression by cell crosstalk. In previous studies, soluble biglycan was found to interact with CD14 and Toll-like receptor 2/4 (TLR2/4), leading to the activation of downstream NF-κB and ERK1/2 signaling.57,60 TLR2/4 are membrane receptors for various immune cells and modulate the immune response in the TME.71 Previous studies have confirmed the role of TLR4 in the polarization of tumor-associated macrophages and the conversion of CAFs, tumor-associated dendritic cells and myeloid-derived suppressor cells.60,61,72 On the basis of previous studies, we might hypothesize that biglycan exerts its function as a secreted ligand and modulates the downstream signalings by interacting with corresponding receptors on other cells. These results suggest that biglycan might play a crucial role in immune modulation and tumor angiogenesis.
We also realize the limitations of our study. First, the relative abundance of fibroblasts was evaluated by a clustering method using classic CAF markers with higher specificity instead of all well-known fibroblast markers. Comprehensive clustering method and single-cell sequencing data were applied to enhance the specificity, but the similarity of expression profiles in CAFs and vascular cells might still confound our analysis. Besides, the results are only comparable within each cohort. Second, sampling bias might occur because the fraction of the stromal part varies in different samples due to the internal heterogeneity of the tumor bulk. Bulk sequencing might not truly reflect the overall landscape of tumors. Third, the mechanical and chemical effects during isolation of primary cells from tissues and in vitro cells culture might change the phenotype of fibroblasts, leading to detection bias in the experiments. Finally, the mechanism underlying how biglycan affects the TME was not explored in this study. Further investigations about how biglycan affects tumor cells and immune components, such as CD8 + T cells and macrophages, will advance our understanding of the roles of CAFs in TNBC.
Notably, we observed that biglycan was upregulated and secreted in cancer-associated fibroblasts compared with normal cancer-adjacent fibroblasts. The expression of BGN was significantly correlated with overall clinical survival and biological processes in TNBC, suggesting that biglycan might play a crucial role in cell-to-cell crosstalk between CAFs and cancer cells. Moreover, we confirmed the prognostic value of biglycan in TNBC using the cancer tissue microarray. Interestingly, BGN was not significantly correlated with classic checkpoint molecules in most cohorts. But we observed an evident positive correlation between BGN and TGF-β, a well-known immunosuppressive molecule, indicating that BGN might exert its effect on immune activity in a certain way. Besides, BGN might participate in tumor angiogenesis which is also a prominent target to synergize with immunotherapy.
In conclusion, we identified that CAF-derived biglycan is crucial for TNBC progression. The upregulation of biglycan and the mechanism underlying how biglycan exerts its function in the TME may serve as a promising diagnostic biomarker and may provide more promising strategies for cancer treatment.
Supplementary Material
Acknowledgments
We thank to all participants in public datasets, including GEO, TCGA research network, METABRIC, TISCH and SYSUCC. We also thank all the authors who contribute their valuable methods and data and make them public (Table S1). We thank the Jianming Zeng and Biotrainee as they helped us with bioinformatic analysis. We thank BioRender for providing technical help. Many thanks for the developers of the “survminer” and other packages in R software that helped us with our study (Alboukadel Kassambara, Marcin Kosinski and Przemyslaw Biecek, etc). We also thank all the members in our research team for their contribution in this study.
Funding Statement
This work was supported by the National Natural Science Foundation of China [82003066]; Guangzhou Basic and Applied Basic Research Project [202102020168]; Guangdong Province Basic and Applied Basic Research Fund Project [2020A1515110034]; Guangzhou Science and Technology Plan Project [202102020114].
Contributors
All authors participated in the study planning and analysis or laboratory experiments. X.X., S.Z. and W.W. designed the study. S.Z., Y.Z., Y.T., A.Y., J.L. and L.W. collected, processed and analyzed the datasets. L.Y., X.X. and J.X. verified the data. S.Z., L.W., A.Y., W.T. and W.X. performed the experiments. W.T. and L.W. collected the clinical information. Y.Z., Y.T. and J.L. performed the statistical analysis. J.L. and X.X. provided critical suggestions. All authors have read and approved the final version of the manuscript.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data sharing statement
All datasets involved in this study can be viewed in Gene Expression Omnibus (GEO), The Cancer Genome Atlas (TCGA) and cBioPortal (http://www.cbioportal.org/), or data availability part of the corresponding articles. The details of publicly available datasets are listed in Table S1. All the transcriptomic data could be obtained and analyzed according to the requirements and guidance of the providers.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Siegel RL, Miller KD, Fuchs HE, Jemal A . Cancer statistics, 2021. CA Cancer J Clin. 2021. Jan;71(1):7–16. [DOI] [PubMed] [Google Scholar]
- 2.Waks AG, Winer EP.. Breast cancer treatment: a review. JAMA. 2019;321(3):288–300. doi: 10.1001/jama.2018.19323. [DOI] [PubMed] [Google Scholar]
- 3.Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, T-h OY. The immune landscape of cancer. Immunity. 2018;48:4. doi: 10.1016/j.immuni.2017.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zheng S, Zou Y, Xie X, Liang JY, Yang A, Yu K, Wang J, Tang H, Xie X. Development and validation of a stromal immune phenotype classifier for predicting immune activity and prognosis in triple-negative breast cancer. Int J Cancer. 2020;147(2):542–553. doi: 10.1002/ijc.33009. [DOI] [PubMed] [Google Scholar]
- 5.Tamborero D, Rubio-Perez C, Muiños F, Sabarinathan R, Piulats JM, Muntasell A, Dienstmann R, Lopez-Bigas N, Gonzalez-Perez A. A pan-cancer landscape of interactions between solid tumors and infiltrating immune cell populations. Clin Cancer Res: Off Am J Cancer Res. 2018;24(15):3717–3728. doi: 10.1158/1078-0432.CCR-17-3509. [DOI] [PubMed] [Google Scholar]
- 6.Lambrechts D, Wauters E, Boeckx B, Aibar S, Nittner D, Burton O, Bassez A, Decaluwé H, Pircher A, Van den Eynde K, et al. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat Med. 2018;24(8):1277–1289. [DOI] [PubMed] [Google Scholar]
- 7.Auciello FR, Bulusu V, Oon C, Tait-Mulder J, Berry M, Bhattacharyya S, Tumanov S, Allen-Petersen BL, Link J, Kendsersky ND, et al. A stromal lysolipid-autotaxin signaling axis promotes pancreatic tumor progression. Cancer Discov. 2019;9(5):617–627. doi: 10.1158/2159-8290.CD-18-1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bertero T, Oldham WM, Grasset EM, Bourget I, Boulter E, Pisano S, Hofman P, Bellvert F, Meneguzzi G, Bulavin DV, et al. Tumor-stroma mechanics coordinate amino acid availability to sustain tumor growth and malignancy. Cell Metab. 2019;29(1):124–40 e10. doi: 10.1016/j.cmet.2018.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boyle ST, Poltavets V, Kular J, Pyne NT, Sandow JJ, Lewis AC, Murphy KJ, Kolesnikoff N, Moretti PAB, Tea MN, et al. ROCK-mediated selective activation of PERK signalling causes fibroblast reprogramming and tumour progression through a CRELD2-dependent mechanism. Nat Cell Biol. 2020;22(7):882–895. doi: 10.1038/s41556-020-0523-y. [DOI] [PubMed] [Google Scholar]
- 10.Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM, Fearon D, Greten FR, Hingorani SR, Hunter T, et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat Rev Cancer. 2020;20(3):174–186. doi: 10.1038/s41568-019-0238-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gagliano T, Shah K, Gargani S, Lao L, Alsaleem M, Chen J, Ntafis V, Huang P, Ditsiou A, Vella V, et al. PIK3Cdelta expression by fibroblasts promotes triple-negative breast cancer progression. J Clin Invest. 2020;130(6):3188–3204. doi: 10.1172/JCI128313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alcaraz LB, Mallavialle A, David T, Derocq D, Delolme F, Dieryckx C, Mollevi C, Boissière-Michot F, Simony-Lafontaine J, Du Manoir S, et al. A 9-kDa matricellular SPARC fragment released by cathepsin D exhibits pro-tumor activity in the triple-negative breast cancer microenvironment. Theranostics. 2021;11(13):6173–6192. doi: 10.7150/thno.58254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Al-Ansari MM, Hendrayani SF, Shehata AI, Aboussekhra A. p16(INK4A) represses the paracrine tumor-promoting effects of breast stromal fibroblasts. Oncogene. 2013;32(18):2356–2364. doi: 10.1038/onc.2012.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen X, Song E. Turning foes to friends: targeting cancer-associated fibroblasts. Nat Rev Drug Discov. 2019;18(2):99–115. doi: 10.1038/s41573-018-0004-1. [DOI] [PubMed] [Google Scholar]
- 15.Chen Y, McAndrews KM, Kalluri R. Clinical and therapeutic relevance of cancer-associated fibroblasts. Nat Rev Clin Oncol. 2021;18:792–804. doi: 10.1038/s41571-021-00546-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Costa A, Kieffer Y, Scholer-Dahirel A, Pelon F, Bourachot B, Cardon M, Sirven P, Magagna I, Fuhrmann L, Bernard C, et al. Fibroblast heterogeneity and immunosuppressive environment in human breast cancer. Cancer Cell. 2018;33(3):463–79 e10. doi: 10.1016/j.ccell.2018.01.011. [DOI] [PubMed] [Google Scholar]
- 17.Kieffer Y, Hocine HR, Gentric G, Pelon F, Bernard C, Bourachot B, Lameiras S, Albergante L, Bonneau C, Guyard A, et al. Single-cell analysis reveals fibroblast clusters linked to immunotherapy resistance in cancer. Cancer Discov. 2020;10(9):1330–1351. [DOI] [PubMed] [Google Scholar]
- 18.Zou Y, Zou X, Zheng S, Tang H, Zhang L, Liu P, Xie X. Efficacy and predictive factors of immune checkpoint inhibitors in metastatic breast cancer: a systematic review and meta-analysis. Ther Adv Med Oncol. 2020;12:1758835920940928. doi: 10.1177/1758835920940928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mounir M, Lucchetta M, Silva TC, Olsen C, Bontempi G, Chen X, Noushmehr H, Colaprico A, Papaleo E. New functionalities in the TCGAbiolinks package for the study and integration of cancer data from GDC and GTEx. PLoS Comput Biol. 2019;15(3):e1006701. doi: 10.1371/journal.pcbi.1006701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Silva TC, Colaprico A, Olsen C, D’Angelo F, Bontempi G, Ceccarelli M, Noushmehr H. Analyze cancer genomics and epigenomics data using Bioconductor packages. F1000Res. 2016;5:1542. doi: 10.12688/f1000research.8923.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486(7403):346–352. doi: 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sabatier R, Finetti P, Cervera N, Lambaudie E, Esterni B, Mamessier E, Tallet A, Chabannon C, Extra J-M, Jacquemier J, et al. A gene expression signature identifies two prognostic subgroups of basal breast cancer. Breast Cancer Res Treat. 2011;126(2):407–420. doi: 10.1007/s10549-010-0897-9. [DOI] [PubMed] [Google Scholar]
- 23.Hatzis C, Pusztai L, Valero V, Booser DJ, Esserman L, Lluch A, Vidaurre T, Holmes F, Souchon E, Wang H, et al. A genomic predictor of response and survival following taxane-anthracycline chemotherapy for invasive breast cancer. JAMA. 2011;305(18):1873–1881. doi: 10.1001/jama.2011.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jézéquel P, Loussouarn D, Guérin-Charbonnel C, Campion L, Vanier A, Gouraud W, Lasla H, Guette C, Valo I, Verrièle V, et al. Gene-expression molecular subtyping of triple-negative breast cancer tumours: importance of immune response. Breast Cancer Res: BCR. 2015;17:43. doi: 10.1186/s13058-015-0550-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gruosso T, Gigoux M, Manem VSK, Bertos N, Zuo D, Perlitch I, Saleh SMI, Zhao H, Souleimanova M, Johnson RM, et al. Spatially distinct tumor immune microenvironments stratify triple-negative breast cancers. J Clin Invest. 2019;129(4):1785–1800. doi: 10.1172/JCI96313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Freire PP, Fernandez GJ, de Moraes D, Cury SS, Dal Pai-Silva M, Dos Reis PP, Rogatto SR, Carvalho RF. The expression landscape of cachexia-inducing factors in human cancers. J Cachexia Sarcopenia Muscle. 2020;11(4):947–961. doi: 10.1002/jcsm.12565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hao Y, Hao S, Andersen-Nissen E, Mauck WM, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184:13. doi: 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aran D, Looney AP, Liu L, Wu E, Fong V, Hsu A, Chak S, Naikawadi RP, Wolters PJ, Abate AR, et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat Immunol. 2019;20(2):163–172. doi: 10.1038/s41590-018-0276-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Azizi E, Carr AJ, Plitas G, Cornish AE, Konopacki C, Prabhakaran S, Nainys J, Wu K, Kiseliovas V, Setty M, et al. Single-cell map of diverse immune phenotypes in the breast tumor microenvironment. Cell. 2018;174(5):1293–308 e36. doi: 10.1016/j.cell.2018.05.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hollern DP, Xu N, Thennavan A, Glodowski C, Garcia-Recio S, Mott KR, He X, Garay JP, Carey-Ewend K, Marron D, et al. B cells and t follicular helper cells mediate response to checkpoint inhibitors in high mutation burden mouse models of breast cancer. Cell. 2019;179:5. doi: 10.1016/j.cell.2019.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gulati GS, Sikandar SS, Wesche DJ, Manjunath A, Bharadwaj A, Berger MJ, Ilagan F, Kuo A, Hsieh RW, Cai S, et al. Single-cell transcriptional diversity is a hallmark of developmental potential. Science (New York, NY). 2020;367(6476):405–411. doi: 10.1126/science.aax0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bassez A, Vos H, Van Dyck L, Floris G, Arijs I, Desmedt C, Boeckx B, MV Bempt, Nevelsteen I, Lambein K, et al. A single-cell map of intratumoral changes during anti-PD1 treatment of patients with breast cancer. Nat Med. 2021;27(5):820–832. doi: 10.1038/s41591-021-01323-8. [DOI] [PubMed] [Google Scholar]
- 34.Wu SZ, Roden DL, Wang C, Holliday H, Harvey K, Cazet AS, KJ Murphy, Pereira B, Al‐Eryani G, Bartonicek N, et al. Stromal cell diversity associated with immune evasion in human triple-negative breast cancer. EMBO J. 2020;39(19):e104063. doi: 10.15252/embj.2019104063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Karaayvaz M, Cristea S, Gillespie SM, Patel AP, Mylvaganam R, Luo CC, Specht MC, Bernstein BE, Michor F, Ellisen LW, et al. Unravelling subclonal heterogeneity and aggressive disease states in TNBC through single-cell RNA-seq. Nat Commun. 2018;9(1):3588. doi: 10.1038/s41467-018-06052-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Racle J, de Jonge K, Baumgaertner P, Speiser DE, Gfeller D. Simultaneous enumeration of cancer and immune cell types from bulk tumor gene expression data. eLife. 2017. Nov 13;6:e26476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Becht E, Giraldo NA, Lacroix L, Buttard B, Elarouci N, Petitprez F, Selves J, Laurent-Puig P, Sautès-Fridman C, Fridman WH, et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 2016;17(1):218. doi: 10.1186/s13059-016-1070-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aran D, Hu Z, Butte AJ. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017;18(1):220. doi: 10.1186/s13059-017-1349-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, Hoang CD, Diehn M, Alizadeh AA. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12(5):453–457. doi: 10.1038/nmeth.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li T, Fu J, Zeng Z, Cohen D, Li J, Chen Q, Li B, Liu XS. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 2020;48(W1):W509–W14. doi: 10.1093/nar/gkaa407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Newman AM, Steen CB, Liu CL, Gentles AJ, Chaudhuri AA, Scherer F, Khodadoust MS, Esfahani MS, Luca BA, Steiner D, et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat Biotechnol. 2019;37(7):773–782. doi: 10.1038/s41587-019-0114-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yoshihara K, Shahmoradgoli M, Martinez E, Vegesna R, Kim H, Torres-Garcia W, Treviño V, Shen H, Laird PW, Levine DA, et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun. 2013;4:2612. doi: 10.1038/ncomms3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brooks JM, Menezes AN, Ibrahim M, Archer L, Lal N, Bagnall CJ, Von Zeidler SV, Valentine HR, Spruce RJ, Batis N, et al. Development and validation of a combined hypoxia and immune prognostic classifier for head and neck cancer. Clin Cancer Res. 2019;25(17):5315–5328. doi: 10.1158/1078-0432.CCR-18-3314. [DOI] [PubMed] [Google Scholar]
- 44.Elyada E, Bolisetty M, Laise P, Flynn WF, Courtois ET, Burkhart RA, Teinor JA, Belleau P, Biffi G, Lucito MS, et al. Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals antigen-presenting cancer-associated fibroblasts. Cancer Discov. 2019;9(8):1102–1123. doi: 10.1158/2159-8290.CD-19-0094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zheng S, Zou Y, Liang JY, Xiao W, Yang A, Meng T, Lu S, Luo Z, Xie X. Identification and validation of a combined hypoxia and immune index for triple-negative breast cancer. Mol Oncol. 2020;14(11):2814–2833. doi: 10.1002/1878-0261.12747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer. 2016;16(9):582–598. doi: 10.1038/nrc.2016.73. [DOI] [PubMed] [Google Scholar]
- 48.Kawase A, Ishii G, Nagai K, Ito T, Nagano T, Murata Y, Hishida T, Nishimura M, Yoshida J, Suzuki K, et al. Podoplanin expression by cancer associated fibroblasts predicts poor prognosis of lung adenocarcinoma. Int J Cancer. 2008;123(5):1053–1059. doi: 10.1002/ijc.23611. [DOI] [PubMed] [Google Scholar]
- 49.Kisselbach L, Merges M, Bossie A, Boyd A. CD90 Expression on human primary cells and elimination of contaminating fibroblasts from cell cultures. Cytotechnology. 2009;59(1):31–44. doi: 10.1007/s10616-009-9190-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Madsen DH, Leonard D, Masedunskas A, Moyer A, Jürgensen HJ, Peters DE, Amornphimoltham P, Selvaraj A, Yamada SS, Brenner DA, et al. M2-like macrophages are responsible for collagen degradation through a mannose receptor-mediated pathway. J Cell Biol. 2013;202(6):951–966. doi: 10.1083/jcb.201301081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gu Z, Eils R, Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics (Oxford, England). 2016;32(18):2847–2849. doi: 10.1093/bioinformatics/btw313. [DOI] [PubMed] [Google Scholar]
- 52.Liang J-Y, Wang D-S, Lin H-C, Chen -X-X, Yang H, Zheng Y, Li Y-H. A novel ferroptosis-related gene signature for overall survival prediction in patients with hepatocellular carcinoma. Int J Biol Sci. 2020;16(13):2430–2441. doi: 10.7150/ijbs.45050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hänzelmann S, Castelo R, Guinney J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013;14:7. doi: 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102(43):15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Calon A, Espinet E, Palomo-Ponce S, Tauriello DVF, Iglesias M, Céspedes MV, Sevillano M, Nadal C, Jung P, Zhang X-F, et al. Dependency of colorectal cancer on a TGF-β-driven program in stromal cells for metastasis initiation. Cancer Cell. 2012;22(5):571–584. doi: 10.1016/j.ccr.2012.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, Carey VJ, Richardson AL, Weinberg RA. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121(3):335–348. doi: 10.1016/j.cell.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 57.Schaefer L, Babelova A, Kiss E, Hausser H-J, Baliova M, Krzyzankova M, Marsche G, MF Young, Mihalik D, Gotte M, et al. The matrix component biglycan is proinflammatory and signals through Toll-like receptors 4 and 2 in macrophages. J Clin Invest. 2005;115(8):2223–2233. doi: 10.1172/JCI23755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu B, Xu T, Xu X, Cui Y, Xing X. Biglycan promotes the chemotherapy resistance of colon cancer by activating NF-κB signal transduction. Mol Cell Biochem. 2018;449(1–2):285–294. doi: 10.1007/s11010-018-3365-1. [DOI] [PubMed] [Google Scholar]
- 59.Fang D, Lai Z, Wang Y. Overexpression of Biglycan is associated with resistance to rapamycin in human WERI-Rb-1 retinoblastoma cells by inducing the activation of the phosphatidylinositol 3-Kinases (PI3K)/Akt/nuclear factor kappa B (NF-κB) signaling pathway. Med Sci Monit. 2019;25:6639–6648. doi: 10.12659/MSM.915075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Roedig H, Nastase MV, Frey H, Moreth K, Zeng-Brouwers J, Poluzzi C, LT Hsieh, Brandts C, Fulda S, Wygrecka M, et al. Biglycan is a new high-affinity ligand for CD14 in macrophages. Matrix Biol. 2019. Apr;77:4-22. [DOI] [PubMed] [Google Scholar]
- 61.Roedig H, Damiescu R, Zeng-Brouwers J, Kutija I, Trebicka J, Wygrecka M, Schaefer L. Danger matrix molecules orchestrate CD14/CD44 signaling in cancer development. Semin Cancer Biol. 2020;62:31–47. doi: 10.1016/j.semcancer.2019.07.026. [DOI] [PubMed] [Google Scholar]
- 62.Dominguez CX, Muller S, Keerthivasan S, Koeppen H, Hung J, Gierke S, Breart B, Foreman O, Bainbridge TW, Castiglioni A, et al. Single-cell RNA sequencing reveals stromal evolution into LRRC15 + myofibroblasts as a determinant of patient response to cancer immunotherapy. Cancer Discov. 2020;10(2):232–253. doi: 10.1158/2159-8290.CD-19-0644. [DOI] [PubMed] [Google Scholar]
- 63.Yang X, Lin Y, Shi Y, Li B, Liu W, Yin W, Dang Y, Chu Y, Fan J, He R, et al. FAP promotes immunosuppression by cancer-associated fibroblasts in the tumor microenvironment via STAT3-CCL2 signaling. Cancer Res. 2016;76(14):4124–4135. doi: 10.1158/0008-5472.CAN-15-2973. [DOI] [PubMed] [Google Scholar]
- 64.Gaggioli C, Hooper S, Hidalgo-Carcedo C, Grosse R, Marshall JF, Harrington K, Sahai E. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat Cell Biol. 2007;9(12):1392–1400. doi: 10.1038/ncb1658. [DOI] [PubMed] [Google Scholar]
- 65.Bagaev A, Kotlov N, Nomie K, Svekolkin V, Gafurov A, Isaeva O, Osokin N, Kozlov I, Frenkel F, Gancharova O, et al. Conserved pan-cancer microenvironment subtypes predict response to immunotherapy. Cancer Cell. 2021;39:6. doi: 10.1016/j.ccell.2021.04.014. [DOI] [PubMed] [Google Scholar]
- 66.Weber CK, Sommer G, Michl P, Fensterer H, Weimer M, Gansauge F, Leder G, Adler G, Gress TM. Biglycan is overexpressed in pancreatic cancer and induces G1-arrest in pancreatic cancer cell lines. Gastroenterology. 2001;121(3):657–667. doi: 10.1053/gast.2001.27222. [DOI] [PubMed] [Google Scholar]
- 67.Yamamoto K, Ohga N, Hida Y, Maishi N, Kawamoto T, Kitayama K, Akiyama K, Osawa T, Kondoh M, Matsuda K, et al. Biglycan is a specific marker and an autocrine angiogenic factor of tumour endothelial cells. Br J Cancer. 2012;106(6):1214–1223. doi: 10.1038/bjc.2012.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Armulik A, Genové G, Betsholtz C. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell. 2011;21(2):193–215. doi: 10.1016/j.devcel.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 69.De Palma M, Biziato D, Petrova TV. Microenvironmental regulation of tumour angiogenesis. Nat Rev Cancer. 2017;17(8):457–474. doi: 10.1038/nrc.2017.51. [DOI] [PubMed] [Google Scholar]
- 70.Aggelidakis J, Berdiaki A, Nikitovic D, Papoutsidakis A, Papachristou DJ, Tsatsakis AM, Tzanakakis GN. Biglycan regulates MG63 osteosarcoma cell growth through a LPR6/β-Catenin/IGFR-IR signaling axis. Front Oncol. 2018;8:470. doi: 10.3389/fonc.2018.00470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Korneev KV, Atretkhany KN, Drutskaya MS, Grivennikov SI, Kuprash DV, Nedospasov SA. TLR-signaling and proinflammatory cytokines as drivers of tumorigenesis. Cytokine. 2017;89:127–135. doi: 10.1016/j.cyto.2016.01.021. [DOI] [PubMed] [Google Scholar]
- 72.Mai CW, Kang YB, Pichika MR. Should a Toll-like receptor 4 (TLR-4) agonist or antagonist be designed to treat cancer? TLR-4: its expression and effects in the ten most common cancers. Onco Targets Ther. 2013;6:1573–1587. doi: 10.2147/OTT.S50838. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.