

Abstract
Elevated expression of EZH2, the enzymatic subunit of polycomb repressive complex 2 (PRC2), often occurs in cancer. EZH2 expression results in the silencing of genes that suppress tumor formation and metastasis through trimethylation of histone H3 at lysine 27 (H3K27me3) at said gene promoters. However, inhibitors of EZH2 enzymatic activity have not shown the expected efficacy against cancer in clinical trials, suggesting a need for other strategies to address EZH2 overexpression. Here we show that SUMOylation, a post-translational modification characterized by covalent attachment of small ubiquitin-like modifier (SUMO) proteins to a lysine (Lys) residue on target proteins, enhances EZH2 transcription. Either knockdown of the SUMO-activating enzyme SAE2 or pharmacological inhibition of SUMOylation resulted in decreased levels of EZH2 mRNA and protein as well as reduced H3K27me3 levels. SUMOylation regulated EZH2 expression by enhancing binding of the E2F1 transcriptional activator to the EZH2 promoter. Inhibition of SUMOylation not only resulted in reduced EZH2 mRNA and protein levels but also increased expression of genes silenced by EZH2, such as E-cadherin which suppresses epithelial-mesenchymal transition and metastasis. In more than 6,500 patient tumor samples across different cancer types, expression of UBA2 and EZH2 were positively correlated. Taken together, our findings suggest that inhibition of SUMOylation may serve as a potential strategy to address EZH2 overexpression and improve current cancer therapeutic approaches.
Keywords: SUMOylation, EZH2, E-cadherin, E2F1, EMT
Introduction
Enhancer of zeste homolog 2 (EZH2) is the enzymatic component of polycomb repressive complex 2 (PRC2), which catalyzes trimethylation of histone H3 on lysine 27 (i.e. H3K27me3), a mark of transcriptional silencing (1,2). PRC2-mediated epigenetic silencing plays an important role in stem cell differentiation, maintenance of stem cell fate, early embryonic development, X chromosome inactivation, and imprinting. Aberrations in PRC2, including EZH2 mutations and overexpression, have been linked to the development of cancer and promotion of cancer metastasis through epigenetic silencing of tumor suppressor genes and suppressors of metastasis, such as E-cadherin (3–5), and promotion of epithelial-mesenchymal transition (EMT) (6,7). Several studies have reported that EZH2 is essential for colorectal and breast cancer cell growth and EZH2 expression is associated with poor outcome in colorectal and breast cancer (8–11). Thus, development of EZH2 inhibitors for application in cancer treatment has been actively pursued. EZH2 inhibitors appear to be effective in lymphomas carrying EZH2 activating mutations; however, EZH2 inhibitors have shown only limited efficacy in clinical trials in solid tumors like colorectal and breast cancers which present higher level of EZH2 (12). Gene expression analysis of public database of patient samples has shown that both colorectal and breast cancers consistently overexpress EZH2 (Supplementary Fig. S1A, B) and EZH2 overexpression correlates to poor survival (Supplementary Fig. S2). An improved understanding of mechanisms of EZH2 overexpression in cancers could lead to new strategies for treating cancer.
One potential mechanism by which EZH2 may be regulated is SUMOylation, a post-translational modification characterized by covalent attachment of small ubiquitin-like modifier (SUMO) proteins to a lysine (Lys) residue on target proteins (13). SUMOylation requires several steps and is catalyzed by enzymes generally known as E1, E2, and E3. SUMOylation is first activated by E1 (a heterodimer of SAE1 and SAE2, also known as UBA2), then conjugated with E2 (also known as Ubc9 or UBE2I); finally, a SUMO protein is ligated to the sidechain of a Lys residue, catalyzed by one of approximately ten E3 ligases (14–16). SUMO modification adds a new docking site to target proteins, and thus enables new protein–protein interactions through the SUMO-interacting motif during signaling events (17,18). SUMOylation enzymes are expressed at higher levels in cancer cells than in normal cells; their elevated expression is required for tumor progression, cancer metastasis, and cancer stem cell maintenance and self-renewal, and is usually associated with poor survival (13,19,20).
In this study, we show that knockdown of the SUMO E1 catalytic subunit (SAE2, also known as UBA2) expression or treatment with a SUMO E1 small molecule inhibitor greatly decreased EZH2 mRNA and protein levels as well as trimethylation at histone H3 Lys27 (H3K27me3) in colorectal and breast cancer cells. We identified that SUMOylation of E2F1 enhanced E2F1 occupancy on the EZH2 promoter and enhanced EZH2 transcription. Analysis of gene expression datasets of more than 6,500 patient samples across different cancer types, including breast, colorectal cancer, stomach cancer, prostate cancer, melanoma, hepatic carcinoma, lung adenocarcinoma, acute lymphoid leukemia (ALL) and acute myeloid leukemia (AML), showed significantly positive correlation between UBA2 and EZH2 mRNA levels. In addition, inhibition of SUMOylation also increased the expression of genes silenced by EZH2, such as E-cadherin, which suppresses epithelial-mesenchymal transition and metastasis. Our data suggest that inhibiting the SUMO E1 enzyme is a novel therapeutic strategy to block EZH2 overexpression-mediated cancer progression, and that inhibition of SUMOylation would enhance current cancer therapeutic approaches.
Material and Methods
Cell culture and lentiviral transduction.
Colorectal cancer cell lines HCT116 and HT29, and the breast cancer cell line MDA-MB-231 (obtained from ATCC), were grown in Dulbecco’s modified eagle medium (DMEM; 10013CV, Corning) supplemented with 10% heat inactivated fetal calf serum (Omega Scientific, Inc.), 2 mM L-glutamine, and 1% antibiotic-antimycotic (Life Technology). The breast cancer cell line HCC1937 (a gift from Dr. Shiuan Chen) was cultured in mammary epithelial basal medium (MEBM; CC-3151, Lonza) supplemented with a Mammary Epithelial Cell Growth Medium SingleQuots Kit containing supplements and growth factors (CC-4136, Lonza). Mycoplasma was routinely tested using MycoAlertTM Mycoplasma Detection Kit (LT07-318, Lonza). A GIPZ shRNA-eGFP vector with a short hairpin RNA (shRNA) sequence targeting the 3’-UTR of SAE2 was purchased from GE Dharmacon (CloneId: V2LHS 68112). A GIPZ non-silencing lentiviral shRNA vector was used as control (Ctrl-shRNA). Both vectors contain enhanced green fluorescent protein (eGFP) and a puromycin resistance marker. Tet-on pTRIPZ vectors containing inducible SAE2 shRNAs were purchased from GE Dharmacon (V2THS_254939 and V2THS_68114). For lentiviral generation, the envelope plasmid pCMV-VSVG and the packaging plasmid pCMV-dR8.2-dvpr were obtained from Addgene (8454 and 8455, provided by Dr Bob Weinberg). 293T producer cells were transfected with the lentiviral expression vector and packaging DNAs by DNA transfection reagent (Lipofectamine LTX; Invitrogen). The supernatant containing lentiviral particles was collected 24–48 h after transfection.For HT29 and MDA-MB-231 cell lines, lentiviral particles containing SAE2 shRNA (shSAE2) or non-silencing shRNA (shCtrl) were added to the media and puromycin was added at 5 μg/ml after 48 h transduction. Single colonies with strong eGFP expression were picked for cell expansion. Stable lines with constitutively expressed SAE2 shRNA (HT29 shSAE2 and MDA-MB-231 shSAE2) were established and knockdown of SAE2 was confirmed by western blot. Control stable lines (HT29 shCtrl and MDA-MB-231 shCtrl) were also established. For patient-derived primary cultured cancer cells, lentiviral particles expressing shSAE2 or shCtrl were added and cells were harvested after 72 h for later use. HCT116 and HCC1937 cells were stably transfected with the tetracycline (Tet) suppressor (TR) expression plasmid pcDNA6/TR before transduction with lentivirus containing Tet-On shRNA targeting SAE2 mRNA (Tet-On shSAE2). Stably transfected cells were selected using 5 μg/ml blasticidin. For doxycycline (Dox) induced SAE2 knockdown, 5 μg/ml Dox was added to cells for 3–5 days to induce knockdown.
Reverse transcription (RT)-PCR and real-time quantitative PCR (qPCR).
Total cellular RNA was extracted using an RNeasy Mini Kit (Qiagen). Total RNA (2 μg) was reverse-transcribed using an Omniscript RT kit (Qiagen) and oligo dT primer. Real-time qPCR was performed using the SYBR-Green Master Mix (Applied Biosystems) in an ABI3000 instrument (ABI).
Microarray and GSEA methods.
Two cell lines, HT29-shCtrl and HT29-shSAE2, were used for microarray. Global mRNA expression profiling was performed by the City of Hope Integrative Genomics Core Facility using the Affymetrix Genechip HG-U133_Plus_2. Gene set enrichment analysis (GSEA) was performed with gene sets downloaded from the Broad Institute’s MSigDB website (www.broad.mit.edu/gsea/) and data analysis was performed using web-based software. Permutation of gene set data was used to determine statistically significant enrichment of the gene sets using the signal-to-noise ratio of shCtrl versus shSAE2. A false discovery rate (FDR)-adjusted p-value less than 0.05 and FDR-adjusted q-value less than 0.25 were used as cutoffs to determine significance.
Immunoprecipitation (IP) and western blotting.
Cells were lysed in RIPA buffer with protease inhibitor cocktail (cOmplete, EDTA-free, Roche), phosphatase inhibitor cocktail (PhosSTOP, Roche), and 20 mM N-Ethylmaleimide (NEM, Sigma). After removal of cell debris by centrifugation, 1 μg of antibody and 50 μl Protein G agarose dynabeads (Invitrogen) were added to 500 μg of protein extraction and incubated overnight at 4°C. Beads were then washed three times and boiled with 2X SDS loading buffer for western blotting. Cells or tumor tissues were lysed in Laemmli sample buffer (5% SDS, 25% glycerol, 150 mM Tris-HCl pH6.8, 0.01% bromophenol blue). After protein concentration was measured using BCA protein assay, 0.7 M β-mercaptoethanol was added and protein samples were boiled for 10 min. Protein samples were separated by SDS-PAGE, and protein was transferred onto a polyvinylidene fluoride membrane (Immobilon-P membrane, Millipore). Specific antibodies to EZH2 (1:1,000, #5246, Cell Signaling Tech), SAE2 (1:1,000, ab58451, abcam), Ubc9 (1:1,000, #4918, Cell Signaling Tech), H3K27me3 (1:1,000, #5246, Cell Signaling Tech), Histone H3 (1:1,000, #14269, Cell Signaling Tech), SUMO1 (1:500, #4930, Cell Signaling Tech), E2F1 (1:1,000, #3742, Cell Signaling Tech), E-cadherin (1:1,000, #3195, Cell Signaling Tech), HA-Tag (1:1,000, #3724, Cell Signaling Tech), and GAPDH (1:1,000, sc-20357, Santa Cruz) were detected using the appropriate secondary antibodies (Licor) and visualized using an Odyssey detection system (Licor).
DNA transient transfection and Small interfering RNA (siRNA) transfection.
Transient transfection of plasmid DNA was performed using Lipofectamine 2000 (Life Technology) and cell lysates were collected after 48-hour transfection. siRNA transfection was performed using Lipofectamine RNAiMax (Life Technology) following the manufacturer’s instruction. Three days after transfection, cells were harvested for analysis. siRNAs against SAE2, UBC9 and a scramble control were obtained from GE Dharmacon SMARTpool.
Reporter assays.
The EZH2 promoter region (−1,063 to +12 bp) was amplified and subcloned into the pGL3 luciferase basic vector (Promega). The CDH1 promoter (−178 to +92) luciferase reporter plasmid proE-cad178-Luc was a gift from Kumiko UiTei (Addgene plasmid # 42081) (21). pCMVHA E2F1 was a gift from Kristian Helin (Addgene plasmid # 24225) (22). pcDNA3-E2F4 was a gift from Robert Weinberg (Addgene plasmid # 10914) (23). pCMV-Neo-Bam E2F6 was a gift from Jacqueline Lees (Addgene plasmid # 37965) (24). pCMVHA hEZH2 was a gift from Kristian Helin (Addgene plasmid # 24230) (25). HCT116 cells were transfected with the EZH2 promoter luciferase reporter plasmid, pTK-Renilla normalization plasmid (Promega), and either empty vector (Ctrl) or E1F1-, E2F4-, E2F6-, SAE2-, UBC9- or SENP1-encoding plasmids, then incubated for 48 h. HCT116 cells were transfected with proE-Cad178-Luc reporter plasmid with either control siRNA (sc-37007, Santa Cruz) or UBA2 siRNA (L-005248-01-0005, GE Dharmacon) in the presence of pCMVHA-hEZH2 or pCMV empty vector transfection. The Dual-Luciferase Reporter Assay System (Promega) was used to quantify luminescence from transfected cells, and normalized results were analyzed using a two-tailed Student t-test.
Chromatin immunoprecipitation (ChIP) assay.
To detect the binding occupancy of E2F1 on the human EZH2 promoter, ChIP analysis was conducted. 2×107 HCT116 cells were incubated in culture medium containing 1% formaldehyde for 10 min at room temperature, after which the cross-linking reaction was quenched with addition of glycine to a final concentration of 0.125 M. Cells were washed with PBS and harvested, followed by sonication to produce chromatin of primarily mononucleosome size. Fragmented chromatin was then incubated with E2F1 antibody at 4°C overnight. Protein–DNA complexes were recovered using protein G agarose beads, washed, and eluted with elution buffer. Crosslinks were reversed at 65°C in 0.25 M NaCl overnight, then the DNA was digested with proteinase K for 2 h at 50°C. The immunoprecipitated DNAs were subsequently isolated and used for PCR.
Colorectal cancer xenograft mice experiments.
All experiments using mice were approved by the Beckman Research Institute Animal Care and Use Committee (IACUC #10026) and complied with all relevant federal guidelines and institutional policies. HCT116 cells expressing Dox-inducible shSAE2 were injected subcutaneously into 6- to 8-week-old male Nu/J mice (3×106 cells per mouse). After 5 days, mice were given either 5% sucrose water (-Dox group), or 5% sucrose water containing 2 mg/ml Dox (+Dox group). Tumor volume was measured with calipers until tumor diameter reached 1.80 cm. Then mice were euthanized, and tumor tissues were harvested for analysis. Stable cell lines HT29-shCtrl and HT29-shSAE2 were injected with Matrigel subcutaneously into the flanks of 6- to 8-week-old female NSG mice (5,000 cells per mouse). Mice were monitored for 2 months to observe tumor formation and growth. Mice were euthanized and tumor tissues were harvested for analysis.
PDX primary culture.
A PDX model was generated by subcutaneously implanting human colorectal tumor tissue into 6-to 8- week-old female NSG mice. Human colorectal cancer tissue was obtained with patient consent, as approved by the Research Ethics Board at City of Hope (IRB13389). Primary culture was conducted as previously described (20). Tumor tissue for use in xenografts was washed in PBS, minced, and incubated with collagenase (235 U/ml) and hyaluronidase (850 U/ml) (Sigma-Aldrich) for 90–120 min at 37°C. DMEM with 10% FBS was added to stop enzymatic digestion. Digested tissues were serially filtered through a 70-μm then a 40-μm cell strainer. Cells were centrifuged (300×g, 10 min), then resuspended with 1X ice cold red blood cell lysis buffer (Santa Cruz Tech) and incubated for 2 min to lyse red blood cells. Cells were pelleted again and washed with 1x PBS, the cells were used for downstream applications. For further purification to remove stromal cells and fibroblasts, magnetic sorting was carried out using an EpCAM positive selection kit (StemCell Tech).
Survival data and gene expression analysis from public datasets.
Survival data and gene expression data from colorectal cancer (GSE17537) and breast cancer (GSE12276) were downloaded from the PrognoScan website (26). UBA2, EZH2 and CDH1 expressions were extracted from databases available at cBioPortal for Cancer Genomics (27,28). The correlation between UBA2 and EZH2 mRNA expression from selected data was plotted and analyzed.
Statistical analyses.
No animals or samples were excluded from analysis. For all experiments, P values were derived using a two-tailed Student’s t-test or ANOVA. Estimated variation is indicated as SD in each figure. For all graphs, * p < 0.05, ** p < 0.01 and *** p < 0.001.
Results
Inhibition of SUMOylation decreases EZH2 expression
We established stable cell lines that can be induced to express shRNA targeting the catalytic subunit of the SUMO E1 enzyme, SAE2, in the colorectal cancer cell line HCT116 and the breast cancer cell line HCC1937 (3). To rule out non-specific shRNA effects, we used two independent shRNAs targeting SAE2. Here, we show that EZH2 mRNA levels decreased in HCT116 and HCC1937 cells after Dox-induced SAE2 knockdown (Fig. 1A). To confirm the broad effect of SAE2 knockdown on EZH2 expression, we established additional colorectal cancer (HT29) and breast cancer (MB-MDA-231) cell lines stably expressing an SAE2-targeting shRNA (shSAE2) or non-silencing shRNA (shCtrl). HCT116 and HT29 represents different types of colorectal cancers, microsatellite instable (MSI, HCT116) and stable (MSS, HT29) as well KRas mutant (HCT116) and wildtype (HT29) colorectal cancers. Breast cancer cell lines MDA-MB-231 and HCC1937 are both the triple-negative breast cancer subtype (TNBC), which disproportionally affects patients with high unmet medical needs. We observed a reduction of EZH2 mRNA in the stable shSAE2 cell lines compared to the matched control cell lines (Fig. 1B). We used western blotting to show that stable knockdown (shRNA) of SAE2 in HCT116 and HT29 cells reduced both EZH2 and H3K27me3 levels (Fig. 1C). Transient (siRNA) knockdown of SAE2 or UBC9 also reduced EZH2 and H3K27me3 level (Fig. 1D). Similarly, inhibition of the enzymatic activity of SUMO E1 by the small molecule inhibitor ML-792 (29) reduced EZH2 protein in a dose-dependent manner (Fig. 1E), indicating that SAE2-mediated EZH2 expression is dependent on SUMO E1 enzymatic activity.
Figure 1. Inhibition of SUMOylation decreases EZH2 mRNA and protein levels.

(A) EZH2 mRNA levels were determined using real-time qPCR in HCT116 cells (left panel) and HCC1937 cells (right panel). Doxycycline (Dox, 5μg/ml) was added to induce SAE2 knockdown (+Dox) in HCT116 cells and HCC1937 cells for 3 days. Knockdown for 3 days was necessary to decrease enzyme level due to the slow turnover rate of the SUMO E1. (B) EZH2 mRNA levels decreased in HT29 and MB-MDA-231 cell lines by stable expression of shSAE2 in comparison to the matched control cell line (shCtrl). (C) Western blot shows SAE2, EZH2, UBC9, EZH2, H3K27me3 (tri-methylation at residue lysine 27 of histone H3) levels and SUMO-2,3 modifications in HCT116 and HT29 stable cell lines with SAE2 knockdown by shRNA. GAPDH and Histone H3 are used as loading controls. (D) Transient knockdown of SAE2 or UBC9 by siRNA transfection reduced EZH2 and H3K27me3 levels and SUMO-2,3 modifications in HCT116 and HT29 cells as determined by western blot. (E) Western blot for EZH2 and global SUMOylation (SUMO1 and SUMO2,3) levels in HCT116 cells treated with 0, 0.2, or 1.0 μM ML-792 SUMO E1 inhibitor for 24 h. Estimated variation is indicated as SD, p values were derived using a two-tailed Student’s t-test. * p < 0.05, ** p < 0.01.
To confirm that EZH2 expression is regulated by SUMOylation in vivo, we examined tumor tissue from cell line xenografts and primary PDX cells. For nude mice xenografted with HCT116 cells expressing Dox-inducible shSAE2, tumor weight was significantly reduced following Dox-induced SAE2 knockdown in comparison to uninduced control mice (Fig. 2A). Similarly, for NSG mice xenografted with HT29 cells stably expressing shSAE2, tumors were smaller than for shCtrl control mice (Fig. 2B). We used western blot of the xenograft tumor tissues to show that knockdown of SAE2 significantly reduced EZH2 level in both HCT116 (Fig. 2D) and HT29 (Fig. 2E) xenografts. We observed similar effects in primary colorectal tumor cells from PDX tissues. We transduced cells from two PDX tissues, PDX356531 and PDX354313, with SAE2-targeting shRNA. Knockdown of SAE2 significantly decreased PDX cancer cell viability (Fig. 2C). EZH2 protein level was reduced upon SAE2 knockdown in these primary cancer cells (Fig. 2F), indicating that the regulation of EZH2 expression by SAE2 is not restricted to cell lines. It has been reported that EZH2 is essential for colorectal and breast cancer cell proliferation (8,10). We conducted EZH2 knockdown by siRNA transfection in colorectal cancer cell lines HCT116, HT29 and breast cancer cell lines HCC1937, MDA-MB-231. EZH2 knockdown reduced cancer cell growth in all 4 cell lines (Supplementary Fig S3A&B). We also found that restoring EZH2 level in SAE2 knockdown cells partially compensated cell viability loss due to SAE2 knockdown (Supplementary Fig. S3C), suggesting inhibition of EZH2 expression by SAE2 knockdown contributes to cell growth inhibition.
Figure 2. Knockdown of SAE2 in colorectal cancer cell line xenograft mouse model and PDX primary colorectal cancer cells suppresses expression of EZH2.
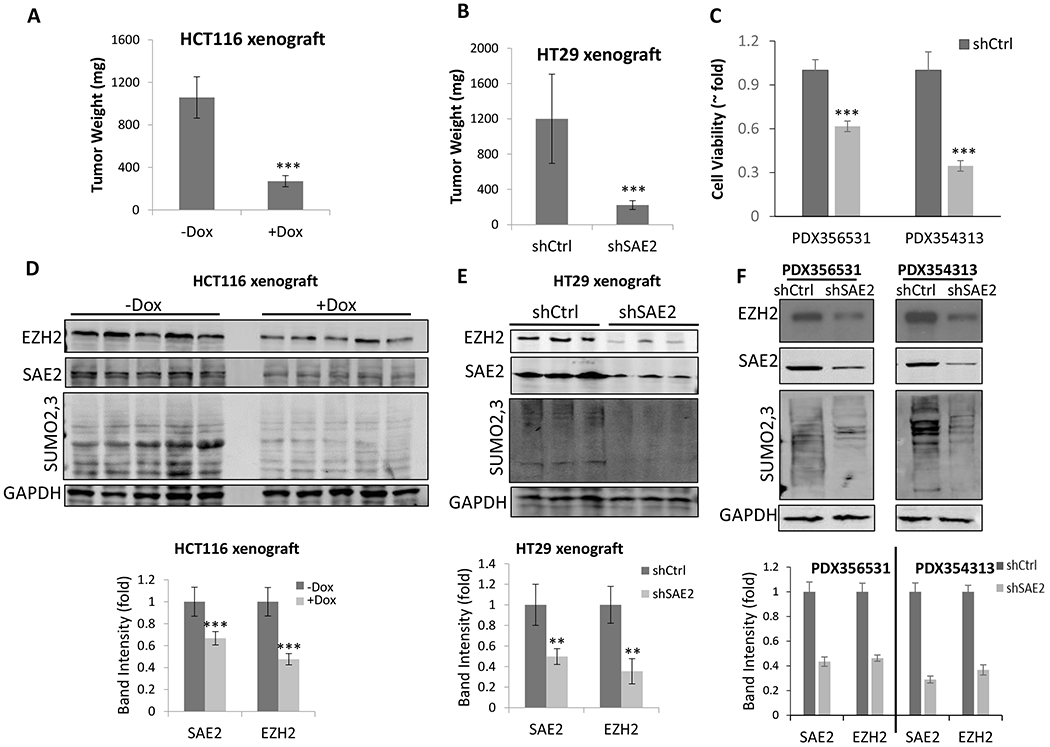
(A) Nude mice were xenografted with HCT116 cells transduced with Dox-inducible SAE2 shRNA by subcutaneous injection. Recipient animals were treated with (+Dox) or without (-Dox) Dox and tumor weight was measured after 16 days. (B) NSG mice were xenografted with HT29 cells transduced with SAE2-targeting shRNA lentivirus (shSAE2) or control shRNA (shCtrl) and tumor weight was measured after 35 days. (C) Two different colorectal PDX tissues, PDX356531 and PDX354313, were cultured and transduced with lentivirus containing SAE2-targeting shRNA (shSAE2) or control shRNA (shCtrl). Cell viability was determined after 72 h. SAE2 and EZH2 levels and SUMO2,3 modifications in tumor tissue from HCT116 xenografts (D) or HT29 xenografts (E) were determined using western blot (top) and quantified using Image J (bottom). (F) PDX356531 and PDX354313 cells with SAE2-targeting shRNA lentivirus (shSAE2) or control shRNA (shCtrl) were harvested after 72 h, then SAE2 and EZH2 levels and SUMO-2,3 modifications were determined using western blot (top) and quantified (bottom). GAPDH was used as a loading control. Estimated variation is indicated as SD, p values were derived using a two-tailed Student’s t-test. * p < 0.05, ** p < 0.01, *** p < 0.001.
SUMOylation enhances EZH2 promoter transcriptional activity through E2F1
Because SUMO E1 regulates EZH2 mRNA expression, we investigated whether SUMOylation regulates the transcriptional activity of the EZH2 promoter. We overexpressed SAE2, UBC9, or SUMO1 in HCT116 cells, then used a luciferase reporter to measure EZH2 promoter activity. EZH2 promotor activity was significantly increased by overexpression of SAE2, UBC9, or SUMO1 (Fig. 3A). Consistent with this, EZH2 mRNA level was also significantly increased by SAE2, UBC9, or SUMO1 overexpression (Fig. 3B). Conversely, EZH2 promoter activity was significantly reduced when SAE2 was knocked down (Fig. 3C). This reduced activity was partially compensated by overexpression of UBC9 or SUMO1 (Fig. 3C). Taken together, these data indicate that SUMOylation activity regulates the EZH2 promoter.
Figure 3. SUMOylation enhances EZH2 promoter transcriptional activity and mRNA level through E2F1.

(A) EZH2 promoter transcriptional activity was enhanced by SAE2, UBC9, and SUMO1 overexpression. HCT116 cells were transfected with empty vector (Ctrl) or SAE2, UBC9, or SUMO1 expression plasmids, together with EZH2 promoter luciferase reporter and Renilla plasmids. Dual-luciferase activity was measured after 48 h. (B) EZH2 mRNA level was enhanced by SAE2, UBC9, and SUMO1 overexpression. HCT116 cells were transfected with empty vector (Ctrl) or SAE2, UBC9, or SUMO1 expression plasmids. mRNA was extracted after 48 h for real-time qPCR. (C) Knockdown of SAE2 decreased EZH2 promoter activity and overexpression of UBC9 or SUMO1 showed partial rescue effects. HCT116 cells without (−Dox) or with (+Dox) SAE2 knockdown were transfected with empty vector (Ctrl), or plasmids expressing UBC9, or SUMO1, then EZH2 promoter activity was measured using dual-luciferase assay. (D) EZH2 promoter transcriptional activity was significantly enhanced by E2F1 expression, but not by E2F4 or E2F6 expression. HCT116 cells were transfected with empty vector (Ctrl) or E2F1, E2F4, or E2F6 expression plasmids at two doses, 100 ng/well (+) and 200 ng/well (++), together with EZH2 promoter luciferase reporter and Renilla plasmids. Dual-luciferase activity was measured after 48 h. (E) The EZH2 promoter transcriptional activity of E2F1 was enhanced by co-expression of SAE2 or SUMO1 but suppressed by SENP1 expression in HCT116 cells. (F) EZH2 mRNA expression was induced by E2F1 overexpression and the induction was enhanced by co-transfection of SAE2 or SUMO1 but decreased by SENP1 in HCT116 cells. (G) E2F1 partially restores EZH2 mRNA level in HCT116 cells with SAE2 knockdown. HCT116 cells without (-Dox) or with (+Dox) SAE2 knockdown were transfected with empty vector (Ctrl) or HA-tagged E2F1 plasmid (E2F1), then EZH2 mRNA level was measured by real-time qPCR. Estimated variation is indicated as SD, p values were derived using a two-tailed Student’s t-test or ANOVA. ns, not significant, * p < 0.05, ** p < 0.01, *** p < 0.001.
Next, we investigated how SUMOylation regulates EZH2 expression. Previous studies have reported EZH2 as downstream as pRB-E2F1 pathway (25,30,31). To verify the transcription factors involved in regulating EZH2 expression in a SUMOylation-dependent manner, we focused on the transcription factors E2F1, E2F4, and E2F6, which were shown to bind to the EZH2 promoter in genome-wide ChIP-seq studies using UCSC Genome Browser (genome.ucsc.edu) (32,33). We transfected plasmids expressing E2F1, E2F4, or E2F6 into HCT116 cells, then performed EZH2 promoter luciferase reporter assay. EZH2 promoter activity was significantly increased by E2F1 overexpression in a dose-dependent manner, but was not affected by E2F4 or E2F6 overexpression (Fig. 3D). This is consistent with previous studies showing that E2F1 is a transcription factor for EZH2 expression (25,30,31). E2F1-mediated activation of the EZH2 promoter was further enhanced by co-expression of SAE2 or SUMO1 but suppressed by co-expression of SENP1, a de-SUMOylation enzyme (Fig. 3E). Consistent with this, EZH2 mRNA expression was significantly increased by E2F1 overexpression and the increase was enhanced by co-expression of SAE2 or SUMO1, but decreased by co-expression of SENP1 (Fig. 3F). Overexpression of E2F1 in SAE2 knockdown cells showed partially restoring EZH2 mRNA level (Fig. 3G), indicating SUMOylation is critical for the transcriptional activity of E2F1. Together, these data indicate that E2F1 is a key transcriptional activator of EZH2 and that SUMOylation enhances the activity of E2F1.
We further investigated how SUMOylation regulates E2F1 transcriptional activity. We showed that E2F1 protein level did not change upon SAE2 knockdown in HCT116 cells (Fig. 4A), suggesting that SUMOylation does not regulate E2F1 expression. We used IP to show that SUMO modification of E2F1 was reduced in SAE2 knockdown cells (Fig. 4A). E2F1 is known to be modified by SUMO1 at Lys-226 (34). To confirm Lys-226 as the SUMOylation site, we used site-directed mutagenesis to replace Lys-226 with arginine (Arg), which cannot be SUMOylated, to create a mutant K266R expression plasmid. We conducted IP in cell lysates from HCT116 cells co-expressing E2F1 wild-type (WT) or K266R mutant with UBC9 and SUMO1. E2F1 WT protein was modified by SUMO1, but E2F1 K266R mutant was not, validating K266 as the only SUMOylation site on E2F1 (Fig. 4B). Using the EZH2 promoter luciferase reporter assay in HCT116 cells, we showed that the transcriptional activity of the E2F1 WT protein was significantly reduced by SAE2 knockdown (Fig. 4C) and significantly increased by SUMO1 overexpression (Fig. 4D). In contrast, the transcriptional activity of the E2F1 K266R mutant was not affected by SAE2 knockdown (Fig. 4C) or SUMO1 co-expression (Fig. 4D). We then used ChIP to evaluate the binding of endogenous E2F1 to the EZH2 promoter in HCT116 cells in the presence or absence of SAE2 knockdown. Four pairs of primers were used to cover ~ 1kb promoter region. SAE2 knockdown significantly reduced E2F1 occupancy throughout the EZH2 promoter region (Fig. 4F). We used ChIP assay in HCT116 cells overexpressing E2F1 WT or K266R mutant protein to show significantly reduced E2F1 occupancy on the EZH2 promoter for the K266R mutant compared to WT E2F1 (Fig. 4G). Consistent with this, GSEA analysis shows that E2F1-target genes were suppressed in shSAE2 knockdown cells compared to shCtrl cells (Fig. 4H). Taken together, our studies demonstrate that SUMOylation enhances EZH2 promoter transcriptional activity by modifying E2F1 to regulate E2F1 binding to the EZH2 promoter.
Figure 4. SUMOylation of E2F1 at K226 controls E2F1 binding to the EZH2 promoter.

(A) E2F1 expression was not affected by SAE2 knockdown. Western blot of E2F1 and SUMO1 in HCT116 cells with (+Dox) or without (−Dox) SAE2 knockdown and HT29 cells with (shSAE2) or without (shCtrl) SAE2 knockdown. E2F1 SUMO1 modification was reduced by SAE2 knockdown. (B) E2F1 is SUMOylated at lysine 266. HCT116 cells were transfected with HA-tagged E2F1 WT or K226R mutant expression plasmid together with a UBC9-expression plasmid. The expression of WT and K226 mutant E2F1 was detected by western blots with an anti-HA-tag antibody in input (left panel). Cell lysates were immunoprecipitated with an anti-HA-tag antibody followed by western blot with an anti-HA-tag antibody (middle panel) or an anti-SUMO1 antibody (right panel). (C) K226R mutation significantly decreased E2F1 transcriptional activation of the EZH2 promoter and the transcriptional activity of E2F1-K226R mutant was not affected by SAE2 knockdown. HCT116 cells containing Dox-inducible SAE2 shRNA were pretreated with (+Dox) or without (−Dox) Dox for 24 h. Cells were transfected with E2F1 WT or K226R mutant, together with EZH2 promoter luciferase reporter and Renilla plasmids. After transfection for 6 h, cells were treated with or without Dox for another 48 h and dual-luciferase assay was performed. (D) SUMO1 increased transcriptional activity on EZH2 promoter when co-expressed with WT E2F1 but showed no effect when co-expressed with E2F1 K266R mutant. HCT116 cells were transfected with E2F1 WT or K226R mutant along with a SUMO1-expressing plasmid (SUMO1) or empty vector (EV). All wells were transfected with the same amount of EZH2 promoter luciferase reporter and Renilla plasmids. Dual-luciferase assay was performed after 48 h. (E) Schematic of the genomic region surrounding the transcriptional start site (TSS) of the human EZH2 gene and the E2F1 binding region on the promoter. The annealing positions of primers used for ChIP experiments (labeled as P1, P2, P3, and P4) are indicated at the bottom of the figure. (F) SAE2 knockdown decreased the occupancy of E2F1 on the EZH2 promoter as measured by ChIP assay. ChIP was performed using an anti-E2F1 antibody in HCT116 cells without (−Dox) or with (+Dox) SAE2 knockdown. The occupancy was normalized to DNA input and calculated relative to IgG control. (G) E2F1 SUMOylation site mutation significantly decreased the occupancy of E2F1 on the EZH2 promoter. HCT116 cells were transfected with HA-tagged E2F1 WT or K226R expression plasmids, followed by ChIP assay using an anti-HA antibody. (H) GSEA analysis shows that “REN_BOUND_BY_E2F” gene set was enriched in HT29 control group (shCtrl) compared to the SAE2 knockdown group (shSAE2). Estimated variation is indicated as SD, p values were derived using a two-tailed Student’s t-test or ANOVA. ns, not significant, * p < 0.05, ** p < 0.01, *** p < 0.001.
SUMOylation regulates genes involved in EMT
We investigated the effect of SUMOylation on EZH2 downstream targets. E-cadherin (CDH1) is a well-established EZH2-regulated gene. We analyzed public patient cohort and found reduced expression of E-cadherin occurs in breast and colorectal cancers (Supplementary Fig. S4A–D). Loss of E-cadherin is thought to promote EMT and cancer metastasis (35). We used a CDH1 promoter luciferase reporter assay in HCT116 cells to show that transient knockdown of SAE2 caused a significant increase in CDH1 promoter activity compared to control; this effect was reversed by overexpression of EZH2 (Fig. 5A). Similarly, HCT116 cells with Dox-induced shSAE2 showed a significant increase (approximately 4.5 fold) in CDH1 mRNA expression (Fig. 5B) and a significant increase in E-cadherin protein level shown by western blot (Fig. 5C) compared to shCtrl cells; these effects were also reversed by EZH2 overexpression. SAE2-mediated induction of CDH1 promoter activity and mRNA expression can be suppressed by exogenous EZH2 expression, supporting a role for SUMOylation in regulating the CDH1 promoter through EZH2. Consistent with this, E-cadherin protein level increased upon Dox-induced SAE2 knockdown in HCT116 cells (Fig. 5D). SAE2 knockdown also induced ZO-1 upregulation, which is known to limit cell invasion (36) (Fig. 5D). In contrast, proteins promoting EMT (Snail and Slug) (37) were suppressed by SAE2 knockdown (Fig. 5D). Consistently, GSEA analyses also show that SAE2 knockdown suppressed gene pathways known to promote metastasis (Fig. 5E).
Figure 5. SUMOylation represses an EZH2 target gene CDH1.

(A) Knockdown of SAE2 increased CDH1 gene promoter activity and overexpression of EZH2 reversed the effect. HCT116 cells were transfected with control siRNA (siCtrl) or SAE2-targeting siRNA (siSAE2) together with an empty vector (Ctrl) or EZH2-expression plasmid (EZH2). CDH1 promoter reporter plasmid and Renilla plasmid were also transfected and CDH1 promoter activity was measured using dual-luciferase assay after 72 h. (B) Stable knockdown of SAE2 increased CDH1 mRNA level and overexpression of EZH2 reversed the effect. (C) Knockdown of SAE2 increased and overexpression of EZH2 suppressed E-cadherin protein level. HCT116 cells without (−Dox) or with (+Dox) SAE2 knockdown were transfected with empty vector (Ctrl) or EZH2-expression plasmid (EZH2). CDH1 coding protein E-cadherin was detected using western blot. Relative protein band intensity was quantified using Image J, normalized to GAPDH and labeled below each blotting band. SAE2 level was blotted to confirm Dox-induced SAE2 knockdown and HA-tag was blotted to confirm the expression of transfected HA-EZH2. GAPDH was used as loading control. (D) Knockdown of SAE2 increased E-cadherin and ZO-1 but decreased N-cadherin, Snail, and Slug. Protein was extracted from HCT116 cells without (−Dox) or with (+Dox) SAE2 knockdown and SAE2, EZH2, E-cadherin, ZO-1, Snail, and Slug were detected using western blot. Relative protein band intensity was quantified using Image J, normalized to GAPDH, and labeled below each blotting band. (E) GSEA revealed that “Alonso_Metastasis_UP” (top) and “Bidus_Metastasis_UP” (middle) gene sets were enriched in the HT29 control group (shCtrl) in comparison to the SAE2 knockdown group (shSAE2), whereas “Bidus_Metastasis_DN” gene sets (bottom) were enriched in the SAE2 knockdown group compared to the control group. Estimated variation is indicated as SD, p values were derived using a two-tailed Student’s t-test. * p < 0.05, ** p < 0.01, *** p < 0.001.
SUMOylation of EZH2 does not alter PRC2 enzymatic function
PRC2 has been reported to be SUMO-modified (38). In order to determine if there is an enzymatic functional role for direct SUMOylation of EZH2, we carried out in vitro SUMOylation on purified PRC2 complex to identify intrinsic SUMOylation sites. PRC2 subunits EZH2, SUZ12 and EED can be modified by SUMO (Fig. 6A&B). However, SUMO-modified PRC2 didn’t increase trimethylation of H3K27 and removal of SUMOylation by co-incubation with SENP1 didn’t affect H3K27me3 level in in vitro Histone methyl-transferase assay (Fig. 6B). Liquid chromatography-tandem mass spectrometry (LC–MS/MS) analysis of PRC2 identified K307 of EZH2 as a SUMO1 modification site and K419 and K421 as SUMO2 modification sites. To examine the effect of SUMOylation of EZH2, we made a SUMO-defective mutant of EZH2 by replacing the 3 lysine with arginine and also replaced a predicted SUMOylation site K20 (4KR). Transfection of the EZH2 4KR mutant in cells followed by immunoprecipitation and western blotting showed that the 4KR mutant could not completely abolish SUMOylation but showed significantly less SUMOylation compared to WT (Fig. 6C). However, SUMOylation sites mutation doesn’t affect EZH2 protein stability (Fig. 6D). In CDH1 promoter luciferase assay, both WT and 4KR EZH2 showed similar dose-dependent suppression of the CDH1 promoter (Fig. 6E). Taken together, these data suggest that SUMOylation regulates EZH2 function mainly through its expression and not through direct SUMO modification of EZH2.
Figure 6. EZH2 SUMOylation doesn’t affect its histone methyltranferase activity, protein stability or its suppression of CDH1 promoter.

(A) A representative blot of in vitro SUMOylation of purified PRC2 (Creative-Biomart) by incubating with recombinant SUMO E1 (SAE1/SAE2), SUMO E2 enzyme (UBC9), and SUMO1 or SUMO2, and without or with RanBP2 (an E3) at 30ºC for 3 h or 16 h. EZH2 was detected by western blot. (B) A representative blot of in vitro histone methytransferase assay with PRC2 after PRC2 in vitro SUMOylation. SUMOylated PRC2 from in vitro SUMOylation was incubated with or without SENP1 for 30 min, and then histone H3 and S-adenosyl methionine were added for 60 min. Then, western blots were performed to detect H3K27me3 level. (C) Top, schematic diagram indicating the three detected and one predicted SUMOylation sites of EZH2—K20, K307, K419 and K421. 293T cells were transfected with plasmids expressing HA-tagged WT or mutant EZH2 with 4 SUMOylation sites mutated to arginine (4KR) along with an UBC9-expressing vector. Then, the expression of both WT and mutant EZH2 were detected by western blots with an anti-HA antibody. Cell lysates were immunoprecipitated with an anti-HA antibody under denaturing condition followed by immunoblotting with an anti-HA antibody and SUMO1 antibody. (D) SUMOylation did not affect EZH2 protein stability. Representative western blot of EZH2 in HCT116 cells transfected with HA-tagged EZH2 WT or 4KR mutant expression plasmid for 2 days, followed by treatment with 100 μg ml−1 CHX for indicated time. GAPDH was used as a loading control. (E) SUMOylation doesn’t affect EZH2 function in suppressing CDH1 promoter. HCT116 cells were transfected with empty vector (Ctrl), EZH2 WT or 4KR mutant together with CDH1 promoter reporter plasmid and renilla plasmid. EZH2 WT or 4KR plasmids were transfect with different doses (100 ng/well “+” and 200 ng/well “++”). CDH1 promoter activity was measured 48 h post transfection and normalized to renilla. Estimated variation is indicated as SD, p values were derived using a two-tailed Student’s t-test or ANOVA. ns, not significant, * p < 0.05, ** p < 0.01, *** p < 0.001.
Positive correlation between UBA2 and EZH2 expression in patient tumor tissues and the association of UBA2 expression with therapeutic outcome
To validate our findings, we analyzed published gene expression datasets of primary patient samples. We identified statistically positive correlations between UBA2 and EZH2 expression in datasets containing more than 6,500 patient samples across different cancer types including breast, colorectal cancer, stomach cancer, prostate cancer, melanoma, hepatic carcinoma, lung adenocarcinoma, ALL and AML (Supplementary Table S1). This analysis supports our finding that SUMOylation regulates the expression of EZH2.
Further analysis suggests that the correlation of UBA2 and EZH2 expression is also associated with treatment resistance and patient survival. Breast cancers contain various subtypes that are treated by different modalities. We analyzed dataset GSE9893 which came from a cohort of 132 patients (ER+ or PR+ or both positive) who received adjuvant tamoxifen therapy (39). We also analyzed dataset GSE7378 which came from a cohort of 54 node-negative ER+ breast cancer patients treated with tamoxifen or chemotherapy (40). High UBA2 (SAE2) level was associated with poor outcomes in both cohorts (Fig. 7A&C). The correlation of UBA2 expression with EZH2 expression exists in these cohorts (Fig. 7B&D). Such correlation is not limited to these breast cancers, but also exists in colorectal cancers (Fig. 7E&F) and other breast cancer cohorts (Supplementary Fig. S5A&B). Dataset GSE76360 is a cohort of 50 HER2+ breast cancer patients with target therapy of trastuzumab (herceptin) treatment and with clinical response information. Non-responders (NOR) showed statistically significant higher UBA2 level than complete and partial responders (Rec) (Supplementary Fig. S5C). The data indicates that high UBA2 expression contributed to therapy resistance and poor survival.
Figure 7. UBA2 level is associated with treatment outcome and correlates with EZH2 expression in patient specimens.

(A, B) Analysis for cohorts of 132 ER+ and/or PR+ breast cancer patients (GSE9893) treated with adjuvant tamoxifen therapy. (C, D) Analysis of cohort of 54 node-negative ER+ breast cancer patients (GSE7378) treated with tamoxifen or chemotherapy. The expression of SAE2 (UBA2) correlates to poor survival for both cohorts (A and C). Patients with high SAE2 (UBA2) (UBA2high group) showed higher EZH2 levels than patients with low SAE2 (UBA2) (UBA2low group) in both cohorts (B and D). A colon cancer cohort (GSE17537) also showed similar correlation (E and F). Kaplan-Meier plots were generated using online server Prognoscan. Solid lines represent two patient groups— high SAE2 level group (red) and low SAE2 level group (blue). Dotted lines indicate the 95% confidence intervals for each group. P values were derived using a two-tailed Student’s t-test. * p < 0.05, *** p < 0.001. (G) Schematic showing the mechanism of SUMOylation of E2F1 leads to increased EZH2 transcription, resulting in reduced E-cadherin levels.
Discussion
Our study uncovered a mechanism in which SUMOylation regulates EZH2 expression and H3K27me3, as well as EZH2-H3K27me3-regulated genes (Fig. 7G). Our data indicate that the SUMOylation-dependent regulation of EZH2 occurs through SUMO modification of the transcription factor E2F1. Inhibition of SUMOylation reduced E2F1 binding to the EZH2 promoter, thereby reducing EZH2 expression. E2F transcriptional activators promote cell cycle progression and are negatively regulated by binding to the tumor suppressor Rb protein (41). Our findings reveal that SUMO1 regulation of E2F1 binding to the EZH2 promoter is an important post-translational mechanism for controlling E2F1 function. A previous study suggested that EZH2 expression could be upregulated by Myc through downregulation of miRNA-101 (5). Our group also found that inhibition of SUMOylation reduces c-Myc mRNA and protein levels (3,42). However, our genome-wide miRNA expression analysis showed that SAE2 knockdown did not affect miR-101 expression, suggesting that SAE2 does not regulate EZH2 expression through c-Myc/miR-101 in the cell lines used (Supplementary Fig. S6). The connection between SUMOylation activity and EZH2 expression is supported by our genome-wide gene expression analysis of patient specimens, which showed that greater SUMO activation enzyme expression correlates with greater EZH2 expression (Table S1 and Fig. 7).
Our study, combined with the literature, reveals that SUMOylation regulates EMT through modifications of multiple proteins. PRC2, which is regulated by SUMOylation as described here, regulates E-cadherin expression, the loss of which is characteristic of EMT (43). Consistent with reduced EZH2 expression through inhibition of SUMOylation, we showed that E-cadherin mRNA and protein levels were increased (Fig. 5). Another regulator of E-cadherin expression is ZEB2, a transcription factor regulated by SUMOylation (44). ZEB2 binds to an E-box at its regulatory gene promoters and represses the expression of its target genes, which include E-cadherin. Interestingly, PRC2 acts as an E3 ligase for ZEB2 SUMOylation (44). Since SUMOylation of ZEB2 is essential for its gene repression function (44), knockdown of SAE2 is expected to reduce SUMOylation of ZEB2, thereby removing the suppression of ZEB2 on E-cadherin transcription. PRC2 is recruited to the E-cadherin promoter by the transcription factor Snail (45). Snail protein stability is thought to be regulated by SUMOylation (46). Consistent with this, our data shows that inhibition of SUMOylation reduced both Snail and Slug protein levels (Fig. 6D). In addition to repressing epithelial genes, Snail activates genes that contribute to the mesenchymal phenotype (47). Taken together, this suggests that inhibiting SUMOylation inhibits multiple EMT-related mechanisms.
Our study along with the literature highlights how SUMOylation modulates the expression of a single gene by synergistic and feed forward mechanisms. In the regulation of E-cadherin expression, multiple transcription factors, E2F1, Snail and ZEB2, are substrates of SUMOylation. SUMOylation regulates the different transcriptional factors in different ways – enhancing transcriptional activation (E2F1), enhancing transcriptional repression (ZEB2), or controlling protein stability (Snail). However, the effect of SUMOylation of these different transcription factors on E-cadherin expression is the same. As discussed in the previous paragraph, SUMOylation of E2F1 increases the expression of EZH2 that leads to repression of E-cadherin expression. SUMOylation of Snail increases its protein stability, whereby enhancing its repression of E-cadherin (46). For ZEB2, SUMOylation directly enhances its transcriptional repression on E-cadherin. Therefore, despite the different mechanisms of how SUMOylation regulates these different transcriptional factors, the outcome of SUMOylation inhibition on E-cadherin is the same through these different mechanisms. Additionally, like one of our recent findings (3), the regulation of PRC2 and E-cadherin by SUMOylation likely involves feed-forward loops. Besides its histone methylation function, PRC2 also has SUMO E3 ligase function that stimulates ZEB2 SUMOylation. Thus, reduced EZH2 expression by inhibiting SUMOylation could lead to reduced SUMO E3 ligase function that would further inhibit SUMOylation of ZEB2 and other PRC2 targets, further reducing the ZEB2 repression on E-cadherin. These findings represent an example of SUMOylation as a critical node of multiple mechanisms involved in regulating a critical gene.
Our finding suggests that targeting the SUMO E1 enzyme in EZH2 overexpression tumors could improve current targeted and other therapeutic approaches. In breast cancers, for which targeted therapies for specific subtypes are used, high UBA2 expression correlates to high EZH2 expression and treatment resistance to tamoxifen, trastuzumab, and chemotherapy (Fig. 7, Supplementary Fig. S5). High UBA2 expression also correlates to high EZH2 expression and poor survival for colorectal and other cancers (Fig. 7, Supplementary Fig. S5). Therefore, inhibition of SUMOylation could improve outcome of the existing therapies. Beside tumor-intrinsic roles, SUMOylation also has roles in anti-tumor immunity. Inhibition of SUMOylation in dendritic cells activates type I interferon expression and thus is expected to promote T-cell priming (48,49). Additionally, SUMOylation is required for regulatory T cell development and their immune suppressive function (50). Therefore, inhibition of SUMOylation is expected to enhance anti-tumor immune response. At present, a first-in-class SUMOylation inhibitor has been developed by Takeda Pharmaceuticals and has entered clinical trials. Further studies of pharmacological inhibition of SUMOylation will provide new insights into new strategies to inhibit tumor progression and immune evasion to advance cancer therapy.
Supplementary Material
Significance.
Findings provide important biological insights into the mechanism of EZH2 overexpression in cancers and suggest that inhibiting SUMOylation may improve current cancer therapeutic approaches.
Acknowledgements
We thank the City of Hope Core facilities for excellent technical support. Research reported in this publication included work performed in the Analytical Cytometry, Light Microscopy, Integrative Genomics, and Bioinformatics Cores and the Animal Tumor Models Program supported by the National Cancer Institute of the National Institutes of Health under grant number P30CA033572. The study described here was supported by grants from NIH (R01GM086171, R01CA212119, R01CA216987) and California Institute of Regenerative Medicine (DISC2-10107). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We thank Professor Jing Yang (University of California, San Diego) for invaluable comments on the manuscript. We also thank Dr. Yu Zhou and Dr. Ebrahim Zandi in University of Southern California for mass spectrometry analysis and Dr. Sarah Wilkinson for editing and proof-reading of the manuscript.
Footnotes
Disclosure
M.G.F. reports grants and speaking fees from Amgen, speaking fees from Guardant, grants from Novartis and BMS, consulting fees from Bayer and Pfizer outside the submitted work. Y.C. reports equity ownership and consulting fees from Oncovalent Therapeutics, Inc. outside the submitted work.
References
- 1.Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature 2011;469(7330):343–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, et al. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 2002;298(5595):1039–43. [DOI] [PubMed] [Google Scholar]
- 3.Li YJ, Du L, Aldana-Masangkay G, Wang X, Urak R, Forman SJ, et al. Regulation of miR-34b/c-targeted gene expression program by SUMOylation. Nucleic acids research 2018;46(14):7108–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cao Q, Yu J, Dhanasekaran SM, Kim JH, Mani RS, Tomlins SA, et al. Repression of E-cadherin by the polycomb group protein EZH2 in cancer. Oncogene 2008;27(58):7274–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zheng M, Jiang YP, Chen W, Li KD, Liu X, Gao SY, et al. Snail and Slug collaborate on EMT and tumor metastasis through miR-101-mediated EZH2 axis in oral tongue squamous cell carcinoma. Oncotarget 2015;6(9):6797–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG, et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 2002;419(6907):624–9. [DOI] [PubMed] [Google Scholar]
- 7.Tiwari N, Tiwari VK, Waldmeier L, Balwierz PJ, Arnold P, Pachkov M, et al. Sox4 is a master regulator of epithelial-mesenchymal transition by controlling Ezh2 expression and epigenetic reprogramming. Cancer cell 2013;23(6):768–83. [DOI] [PubMed] [Google Scholar]
- 8.Fussbroich B, Wagener N, Macher-Goeppinger S, Benner A, Falth M, Sultmann H, et al. EZH2 depletion blocks the proliferation of colon cancer cells. PloS one 2011;6(7):e21651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen Z, Yang P, Li W, He F, Wei J, Zhang T, et al. Expression of EZH2 is associated with poor outcome in colorectal cancer. Oncology letters 2018;15(3):2953–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gonzalez ME, Li X, Toy K, DuPrie M, Ventura AC, Banerjee M, et al. Downregulation of EZH2 decreases growth of estrogen receptor-negative invasive breast carcinoma and requires BRCA1. Oncogene 2009;28(6):843–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kleer CG, Cao Q, Varambally S, Shen R, Ota I, Tomlins SA, et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proceedings of the National Academy of Sciences of the United States of America 2003;100(20):11606–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ribich S, Harvey D, Copeland RA. Drug Discovery and Chemical Biology of Cancer Epigenetics. Cell chemical biology 2017;24(9):1120–47. [DOI] [PubMed] [Google Scholar]
- 13.Hay RT. SUMO: a history of modification. Molecular cell 2005;18(1):1–12. [DOI] [PubMed] [Google Scholar]
- 14.Johnson ES. Protein modification by SUMO. Annual review of biochemistry 2004;73:355–82. [DOI] [PubMed] [Google Scholar]
- 15.Desterro JM, Rodriguez MS, Kemp GD, Hay RT. Identification of the enzyme required for activation of the small ubiquitin-like protein SUMO-1. The Journal of biological chemistry 1999;274(15):10618–24. [DOI] [PubMed] [Google Scholar]
- 16.Johnson ES, Schwienhorst I, Dohmen RJ, Blobel G. The ubiquitin-like protein Smt3p is activated for conjugation to other proteins by an Aos1p/Uba2p heterodimer. The EMBO journal 1997;16(18):5509–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Song J, Durrin LK, Wilkinson TA, Krontiris TG, Chen Y. Identification of a SUMO-binding motif that recognizes SUMO-modified proteins. Proceedings of the National Academy of Sciences of the United States of America 2004;101(40):14373–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Song J, Zhang Z, Hu W, Chen Y. Small ubiquitin-like modifier (SUMO) recognition of a SUMO binding motif: a reversal of the bound orientation. The Journal of biological chemistry 2005;280(48):40122–9. [DOI] [PubMed] [Google Scholar]
- 19.Kim JH, Choi HJ, Kim B, Kim MH, Lee JM, Kim IS, et al. Roles of sumoylation of a reptin chromatin-remodelling complex in cancer metastasis. Nature cell biology 2006;8(6):631–9. [DOI] [PubMed] [Google Scholar]
- 20.Du L, Li YJ, Fakih M, Wiatrek RL, Duldulao M, Chen Z, et al. Role of SUMO activating enzyme in cancer stem cell maintenance and self-renewal. Nature communications 2016;7:12326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mazda M, Nishi K, Naito Y, Ui-Tei K. E-cadherin is transcriptionally activated via suppression of ZEB1 transcriptional repressor by small RNA-mediated gene silencing. PloS one 2011;6(12):e28688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lukas J, Petersen BO, Holm K, Bartek J, Helin K. Deregulated expression of E2F family members induces S-phase entry and overcomes p16INK4A-mediated growth suppression. Molecular and cellular biology 1996;16(3):1047–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sardet C, Vidal M, Cobrinik D, Geng Y, Onufryk C, Chen A, et al. E2F-4 and E2F-5, two members of the E2F family, are expressed in the early phases of the cell cycle. Proceedings of the National Academy of Sciences of the United States of America 1995;92(6):2403–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Trimarchi JM, Fairchild B, Wen J, Lees JA. The E2F6 transcription factor is a component of the mammalian Bmi1-containing polycomb complex. Proceedings of the National Academy of Sciences of the United States of America 2001;98(4):1519–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bracken AP, Pasini D, Capra M, Prosperini E, Colli E, Helin K. EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. The EMBO journal 2003;22(20):5323–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mizuno H, Kitada K, Nakai K, Sarai A. PrognoScan: a new database for meta-analysis of the prognostic value of genes. BMC medical genomics 2009;2:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer discovery 2012;2(5):401–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science signaling 2013;6(269):pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dongre A, Rashidian M, Reinhardt F, Bagnato A, Keckesova Z, Ploegh HL, et al. Epithelial-to-Mesenchymal Transition Contributes to Immunosuppression in Breast Carcinomas. Cancer Res 2017;77(15):3982–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee SR, Roh YG, Kim SK, Lee JS, Seol SY, Lee HH, et al. Activation of EZH2 and SUZ12 Regulated by E2F1 Predicts the Disease Progression and Aggressive Characteristics of Bladder Cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 2015;21(23):5391–403. [DOI] [PubMed] [Google Scholar]
- 31.Beguelin W, Rivas MA, Calvo Fernandez MT, Teater M, Purwada A, Redmond D, et al. EZH2 enables germinal centre formation through epigenetic silencing of CDKN1A and an Rb-E2F1 feedback loop. Nature communications 2017;8(1):877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, et al. The human genome browser at UCSC. Genome research 2002;12(6):996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rosenbloom KR, Sloan CA, Malladi VS, Dreszer TR, Learned K, Kirkup VM, et al. ENCODE data in the UCSC Genome Browser: year 5 update. Nucleic acids research 2013;41(Database issue):D56–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang L, Lin C, Liu W, Zhang J, Ohgi KA, Grinstein JD, et al. ncRNA- and Pc2 methylation-dependent gene relocation between nuclear structures mediates gene activation programs. Cell 2011;147(4):773–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tong ZT, Cai MY, Wang XG, Kong LL, Mai SJ, Liu YH, et al. EZH2 supports nasopharyngeal carcinoma cell aggressiveness by forming a co-repressor complex with HDAC1/HDAC2 and Snail to inhibit E-cadherin. Oncogene 2012;31(5):583–94. [DOI] [PubMed] [Google Scholar]
- 36.Huang RY, Guilford P, Thiery JP. Early events in cell adhesion and polarity during epithelial-mesenchymal transition. J Cell Sci 2012;125(Pt 19):4417–22. [DOI] [PubMed] [Google Scholar]
- 37.Zhang Y, Weinberg RA. Epithelial-to-mesenchymal transition in cancer: complexity and opportunities. Front Med 2018;12(4):361–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Riising EM, Boggio R, Chiocca S, Helin K, Pasini D. The polycomb repressive complex 2 is a potential target of SUMO modifications. PloS one 2008;3(7):e2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chanrion M, Negre V, Fontaine H, Salvetat N, Bibeau F, Mac Grogan G, et al. A gene expression signature that can predict the recurrence of tamoxifen-treated primary breast cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 2008;14(6):1744–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou Y, Yau C, Gray JW, Chew K, Dairkee SH, Moore DH, et al. Enhanced NF kappa B and AP-1 transcriptional activity associated with antiestrogen resistant breast cancer. BMC Cancer 2007;7:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shan B, Chang CY, Jones D, Lee WH. The transcription factor E2F-1 mediates the autoregulation of RB gene expression. Molecular and cellular biology 1994;14(1):299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li YJ, Du L, Wang J, Vega R, Lee TD, Miao Y, et al. Allosteric Inhibition of Ubiquitin-like Modifications by a Class of Inhibitor of SUMO-Activating Enzyme. Cell Chem Biol 2019;26(2):278–88 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fujii S, Ito K, Ito Y, Ochiai A. Enhancer of zeste homologue 2 (EZH2) down-regulates RUNX3 by increasing histone H3 methylation. The Journal of biological chemistry 2008;283(25):17324–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Long J, Zuo D, Park M. Pc2-mediated sumoylation of Smad-interacting protein 1 attenuates transcriptional repression of E-cadherin. The Journal of biological chemistry 2005;280(42):35477–89. [DOI] [PubMed] [Google Scholar]
- 45.Herranz N, Pasini D, Diaz VM, Franci C, Gutierrez A, Dave N, et al. Polycomb complex 2 is required for E-cadherin repression by the Snail1 transcription factor. Molecular and cellular biology 2008;28(15):4772–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jang D, Kwon H, Choi M, Lee J, Pak Y. Sumoylation of Flotillin-1 promotes EMT in metastatic prostate cancer by suppressing Snail degradation. Oncogene 2019;38(17):3248–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barrallo-Gimeno A, Nieto MA. The Snail genes as inducers of cell movement and survival: implications in development and cancer. Development 2005;132(14):3151–61. [DOI] [PubMed] [Google Scholar]
- 48.Crowl JT, Stetson DB. SUMO2 and SUMO3 redundantly prevent a noncanonical type I interferon response. Proceedings of the National Academy of Sciences of the United States of America 2018;115(26):6798–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Portilho DM, Fernandez J, Ringeard M, Machado AK, Boulay A, Mayer M, et al. Endogenous TRIM5alpha Function Is Regulated by SUMOylation and Nuclear Sequestration for Efficient Innate Sensing in Dendritic Cells. Cell reports 2016;14(2):355–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ding X, Wang A, Ma X, Demarque M, Jin W, Xin H, et al. Protein SUMOylation Is Required for Regulatory T Cell Expansion and Function. Cell reports 2016;16(4):1055–66. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.