

Summary
Nicotinamide riboside supplements (NRS) have been touted as a nutraceutical that promotes cardiometabolic and musculoskeletal health by enhancing nicotinamide adenine dinucleotide (NAD+) biosynthesis, mitochondrial function, and/or the activities of NAD-dependent sirtuin deacetylase enzymes. This investigation examined the impact of NRS on whole body energy homeostasis, skeletal muscle mitochondrial function, and corresponding shifts in the acetyl-lysine proteome, in the context of diet-induced obesity using C57BL/6NJ mice. The study also included a genetically modified mouse model that imposes greater demand on sirtuin flux and associated NAD+ consumption, specifically within muscle tissues. In general, whole body glucose control was marginally improved by NRS when administered at the midpoint of a chronic high-fat diet, but not when given as a preventative therapy upon initiation of the diet. Contrary to anticipated outcomes, the study produced little evidence that NRS increases tissue NAD+ levels, augments mitochondrial function, and/or mitigates diet-induced hyperacetylation of the skeletal muscle proteome.
Subject areas: Physiology, Proteomics, Nutrition
Graphical abstract
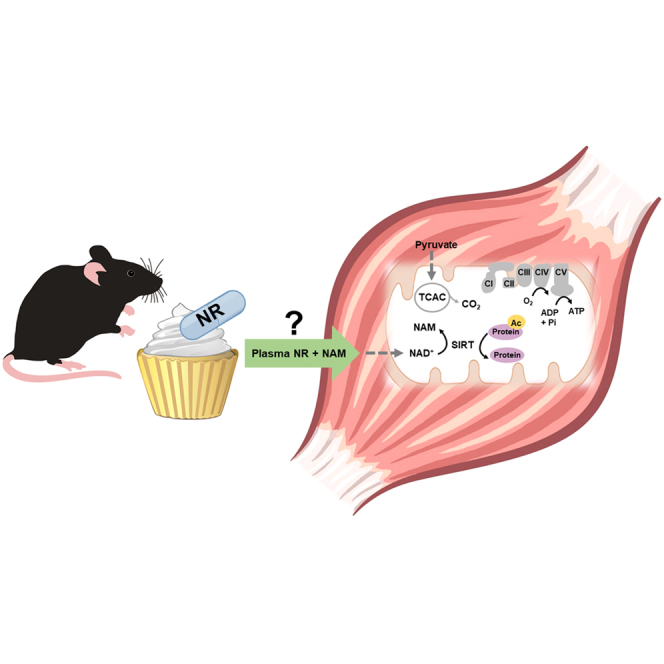
Highlights
-
•
Dietary NR supplementation (NRS) raises plasma and muscle NAM levels in obese mice
-
•
NRS given post obesity slightly improved glucose control and mitochondrial function
-
•
NRS did not oppose obesity-induced remodeling of the muscle acetyl-proteome
-
•
NRS during weight gain did not protect glucose control and mitochondrial function
Physiology; Proteomics; Nutrition
Introduction
Modern technology is increasingly recognized as a double-edged sword in the escalating battle against obesity and its attendant co-morbidities. On one side, the alarming upswing in obesity rates during the past fifty years has paralleled the growth of the processed food industry and the spread of the infamous Westernized diet. Indeed, decades of alarming epidemiological data offer convincing evidence that the massive proliferation of highly processed, inexpensive, nutrient poor, energy dense, and irresistibly palatable foods has contributed to an obesity pandemic that now affects nearly every industrialized society (Askari et al., 2020). Moreover, the worldwide surge in obesity has led to a corresponding rise in the prevalence of chronic metabolic disorders such as type 2 diabetes, heart disease, and non-alcoholic steatohepatitis. The pathophysiological consequences of these diseases accelerate and/or exacerbate age-related deterioration of multiple organ systems, including the skeletal muscle—a principal determinant of whole body glucose homeostasis, exercise tolerance, mobility, and lifelong functional independence.
On the contrary, advances in biomedical technologies and resulting scientific breakthroughs are paving exciting inroads toward a new era of precision medicine, including nutraceutical therapies that offer promise to thwart obesity-related disease progression and age-related deterioration of muscle performance and physical stamina. Prominent among the nutritional supplements that have gained attention and traction in the fields of energy metabolism, musculoskeletal health and anti-aging research is nicotinamide riboside (NR), a naturally occurring B3 vitamin and a precursor for nicotinamide adenine dinucleotide (NAD+) biosynthesis (Bieganowski and Brenner, 2004). NAD+ and its phosphorylated form, NADP+, hold prominent positions in energy metabolism by serving as cofactors for a broad range of dehydrogenase enzymes involved in glycolysis, the tricarboxylic acid (TCA) cycle, oxidative phosphorylation, and antioxidant defense (Ernster and Schatz, 1981; Stein and Imai, 2012). NAD+ also participates in processes that regulate DNA repair and functions as an obligatory substrate for a family of sirtuin deacylase enzymes that play important roles in metabolism, mitochondrial biogenesis, and mitochondrial quality control (Belenky et al., 2007). Notably, under basal conditions, sirtuin activity is estimated to account for over one-third of cellular NAD+ consumption (Liu et al., 2018). Moreover, sirtuin-associated pressure on NAD+ homeostasis presumably rises during aging, chronic high-fat feeding, and obesity, due to increased protein acetylation in these settings (Hirschey et al., 2011; Davies et al., 2016).
Interest in the therapeutic potential of NR first emerged after a series of studies—employing model systems ranging from yeast to mice—showed that targeted deletions of genes that regulate NAD+ homeostasis have a profound impact on energy metabolism, healthspan, and lifespan (Belenky et al., 2007). These observations sparked a flurry of preclinical studies that, in aggregate, support the idea that nutritional supplementation with NR or other NAD+ precursors can combat age- and/or obesity-related perturbations in liver metabolism, whole body glucose control, mitochondrial respiratory capacity, skeletal muscle function, and exercise endurance (Yoshino et al., 2018). Remarkably, in two different mouse models with genetically engineered deficiencies in skeletal muscle NAD+ biosynthesis (Nampt-Mlc1f Cre mice (Frederick et al., 2016) and Nampt-HSA Cre mice (Basse et al., 2021)), the accompanying skeletal muscle degeneration was reversed by NR supplementation (NRS). Importantly, however, multiple investigations have found that circulating NR is primarily cleared and metabolized by the liver, which in turn exports nicotinamide (NAM) and NMN as the major NAD precursors that are delivered to organs such as skeletal muscle and heart (Liu et al., 2018; Frederick et al., 2016). Additionally, the field has discovered that other factors, such as the gut microbiome, can influence the impact of NR and NAM supplements on systemic and tissue NAD+ levels (Shats et al., 2020).
Preclinical research aiming to delineate the mechanisms whereby NRS benefits metabolic health has focused heavily on the role of NAD+ as an essential substrate for sirtuin family members, SIRT1 and SIRT3, both of which act as deacetylase enzymes that remove acetyl groups on lysine residues of target proteins (Zhang et al., 2016; Gariani et al., 2016; Brown et al., 2014; Cerutti et al., 2014). SIRT1 acts in the nucleus and cytoplasm, whereas SIRT3 resides mainly in the mitochondrial matrix (Blander and Guarente, 2004). Both have been tightly linked to mitochondrial quality and performance—SIRT1 through its role in transcriptional regulation of mitochondrial biogenesis and SIRT3 via its direct action to deacetylate and regulate a broad network of oxidative enzymes. Nonetheless, strong evidence that NRS can actually promote skeletal muscle mitochondrial biogenesis and/or respiratory function in animals that do not harbor a targeted genetic lesion in NAD+ metabolism remains sparse. For this reason, the current investigation sought to examine the impact of NRS on whole body energy homeostasis, skeletal muscle metabolism, and mitochondrial function in the context of diet-induced obesity (DIO). Additionally, because the benefits of NRS have been linked to its role in supporting the activities of sirtuin enzymes, this study included a genetically modified mouse model that imposes greater demand on sirtuin flux and associated NAD+ consumption due to muscle-specific ablation of carnitine acetyltransferase (CrAT). Loss of this enzyme, which buffers the mitochondrial pool of acetyl CoA, leads to more extreme diet-induced acetylation of mitochondrial proteins, increased sirtuin flux, and more severe muscle insulin resistance (Williams et al., 2020; Davies et al., 2016).
Herein, we report that whole body glucose control was modestly improved by NRS when administered as an intervention at the midpoint of a 30-week high-fat (HF) diet, but not when given as a preventative therapy upon initiation of chronic HF feeding. Moreover, contrary to the anticipated outcomes, the study produced little evidence that NRS enhances mitochondrial function and/or mitigates diet-induced hyperacetylation of the skeletal muscle proteome, regardless of the genetic background.
Results
NRS alters the plasma and muscle NAD metabolome in DIO mice
The prevailing view suggests that the liver metabolizes orally administered NR and exports nicotinamide (NAM) to the general circulation. Upon clearance by the muscle, NAM is presumably converted to NAD+, which then participates in metabolic regulation and the activation of nuclear and mitochondrial sirtuins (Trammell et al., 2016a; Liu et al., 2018; Canto et al., 2012; Khan et al., 2014) (Figure 1A). The present study sought to validate this model by examining the impact of NRS on muscle NAD metabolites, mitochondrial performance, and changes in the lysine acetylproteome. To induce metabolic dysfunction and promote lysine acetylation (KAc), mice were fed a 45% high-fat (HF) diet for 15 weeks prior to NRS, which was then provided as a diet admixture for an additional 18 weeks in the context of HF feeding. To impose further stress on the sirtuin system, the study included a cohort of mice with muscle-specific deficiency of CrAT, a genetic manipulation that exacerbates diet-induced lysine acetylation of mitochondrial proteins (Davies et al., 2016; Williams et al., 2020). CrAT knockout (KO) mice and floxed controls (FC) were divided into two cohorts that continued feeding on the HF diet, either with or without the addition of NR at a dose of 350 mg NR/kg/day (Figure 1B). Prior to assessing the NAD metabolome in mice treated with NR, the LC/MS-based assays were first validated using purified mitochondria isolated from skeletal muscles of mice fed standard chow (Figures S1J–S1L). Results showed a near perfect correlation between mitochondrial protein input and concentrations of NAD, NAM, and NMN. Next, NRS-induced changes in the NAD metabolome were measured in plasma and muscle lysates (Figures 1 and S1). Plasma NR levels, which were lower in HF CrAT KO mice compared with controls, increased ∼20% in mice receiving NRS (Figure 1C). Moreover, NRS led to a robust 2-fold increase in plasma NAM (Figure 1D). Regardless of genotype, NRS did not alter muscle NAD+ levels (Figure 1E) or gene expression of NAD-processing enzymes (Figures S1M–S1O). NRS increased muscle NAM content in the FC group but not the KO mice (Figure 1G), and a similar trend was evident in the muscle NMN pool (Figure 1H). Plasma NMN and muscle NR concentrations were below the detectable range and therefore are not reported. Measurement of liver NAD metabolites in FC mice revealed an elevation in NR and NADH levels and a smaller trend toward an increase in NMN (Figures S1D–S1I). These results support the notion that NR exerts tissue-dependent effects on the NAD+ metabolome (Trammell et al., 2016a; Liu et al., 2018).
Figure 1.

NR supplementation alters the plasma and muscle NAD metabolome in DIO CrAT-deficient and control mice
(A) Prevailing view whereby oral NR administration impacts skeletal muscle metabolism.
(B) Intervention cohort experimental design in CrATfl/fl (floxed control, FC) and CrATMCK (knock out, KO) mice.
(C) Plasma NR.
(D) Plasma NAM.
(E) Skeletal muscle NAD.
(F) Skeletal muscle NADH.
(G) Skeletal muscle NAM.
(H) Skeletal muscle NMN. Data are represented as mean ± SEM.
(C–H) N = 5–8 per group. Data were analyzed by two-way ANOVA. ∗ represents a main effect of treatment, # represents a main effect of genotype, and ★ represents an interaction between treatment and genotype. ∗P≤0.05. N represents biological replicates.
NRS improves insulin tolerance with little effect on other markers of glucose homeostasis
Metabolic phenotyping tests performed 12 weeks after HF feeding but before the start of NRS (noted as Post HF/Pre NR) showed that the control and treatment cohorts were similar prior to the intervention (Figures S2A–S2D). However, analysis of the insulin tolerance test (ITT) by three-way ANOVA revealed a significant interaction between time and NRS treatment (Figure S2D). Consistent with previous reports (Muoio et al., 2012; Davies et al., 2016; Williams et al., 2020), KO mice fed an HF diet exhibited more severe intolerance to an oral glucose challenge than their FC counterparts (Figure S2A). Subsequent examination of body weight, food consumption, energy expenditure, respiratory exchange ratio (RER), and glucose homeostasis revealed little to no effect of NRS in both genotypes (Figures 2, S3, and S4). Likewise, the intervention did not improve oral glucose tolerance (OGTT) (Figure 2C); although data analysis by three-way ANOVA revealed a main effect of NRS on insulin during the OGTT (Figure 2D). When all groups were analyzed together, a two-way ANOVA detected main effects of NRS on fasting insulin (Figure 2E), baseline blood glucose prior to the ITT (Figure S2E), and ITT area above the curve (AAC) (Figures 2F and 2G). Analysis of the ITT results by three-way ANOVA revealed a main effect of NRS on blood glucose during the ITT (Figure 2F), but without an NRS (treatment) x time interaction. Additionally, regression analysis of the entire cohort revealed that insulin tolerance AAC correlated positively (R = 0.50, R2 = 0.246, p = 0.01) with plasma NAM (Figure 2H). By contrast, plasma and/or muscle NAM did not correlate with fasting insulin. Taken together, these results provide solid evidence that NRS altered whole body glucose homeostasis while also hinting toward modest treatment effects on insulin sensitivity.
Figure 2.

Insulin tolerance is improved in NR-treated mice whereas other markers of glucose homeostasis and energy balance remain largely unaffected
(A) Growth curve.
(B) Respiratory Exchange Ratio (RER).
(C) Oral glucose tolerance.
(D) Insulin during the oral glucose tolerance test.
(E) 5h fasting insulin.
(F) Insulin tolerance.
(G) ITT Area Above the Curve (AAC).
(H) ITT-AAC vs. Plasma NAM. Data are represented as mean ± SEM.
(A–H) N = 5–8 per group. Data in (A), (C), and (D) were analyzed by three-way ANOVA (treatment x genotype x time). In (A), § represents a time × treatment interaction, and Δ represents a time x genotype × treatment interaction. Data in (F) were analyzed by two-tailed Student's t-test and three-way ANOVA (treatment x genotype x time). In (F), ∗ represents a significant difference between HF- and HF + NR-fed mice by two-tailed Student's t-test, and ‡ represents a main effect of treatment by three-way ANOVA. Data in (B), (E), and (G) were analyzed by two-way ANOVA. In (E) and (G), ∗ represents a main effect of treatment. ∗P≤0.05. N represents biological replicates.
See also Figures S2–S4.
NRS led to modest improvements in respiratory function of mitochondria from FC but not KO muscles
Functional assessments of mitochondria from muscle of CrAT-deficient KO compared with FC mice have been described previously (Muoio et al., 2012; Seiler et al., 2015; Williams et al., 2020). Here, the impact of NRS on respiratory function was evaluated using high-resolution respirometry to measure maximal rates of oxygen consumption in isolated muscle mitochondria fueled by malate plus octanoyl-carnitine (OcM), palmitoyl-carnitine (PcM), or glutamate (GM) ± succinate, all performed in the absence and presence of 1mM ADP to stimulate ATP synthesis (Figure 3). In mitochondria from FC muscles, NRS produced modest increases in respiration fueled by octanoyl-carnitine (a medium chain fatty acid substrate) and glutamate (Figures 3A and 3C). By contrast, we found no evidence of improved respiratory capacity in mitochondria from KO mice exposed to NRS. Likewise, a pyruvate sensitivity assay performed by measuring rates of oxygen consumption during a substrate titration in the presence of malate and 1mM ADP (Figure 3D) revealed a subtle enhancement of Vmax in mitochondria from FC mice receiving NRS as compared against controls; whereas the same assay proved unremarkable in KO mitochondria. The slight NRS-induced increase in pyruvate-supported respiration in FC mitochondria was not accompanied by changes in muscle PDHE1a phosphorylation (Figures 3D andS5, Table S1) or expression levels of proteins involved in oxidative phosphorylation (Figure S6). A multivariate pairwise correlation analysis revealed positive associations (R = 0.43–0.50, R2 = 0.19–0.25) between mitochondrial respiratory capacity and plasma NAM, the latter of which correlated positively (R = 0.57, R2 = 0.32, p<0.001) with muscle NAM (Figure 3E). Interestingly, this analysis also identified a negative relationship between mitochondrial respiration and body weight (R = −0.32 to −0.50), as well as OcM-supported respiration and ITTAUC (ITTa, R= −0.47 and R2 = 0.22).
Figure 3.

NR slightly increases muscle mitochondrial respiratory function in HF-fed control but not CrAT-deficient mice
(A–C) Maximal, ADP-supported mitochondrial oxygen consumption (JO2) was assayed in isolated skeletal muscle mitochondria fueled by (A) octanoyl-carnitine/malate/ADP/cytochrome c, (B) palmitoyl-carnitine/malate/ADP, or (C) glutamate/malate/ADP/succinate.
(D) Pyruvate titration conducted in the presence of ADP and malate.
(E) Correlation matrix. Red lines indicate the line of best fit through the data points and the blue shaded region represents 95% confidence intervals. Data are represented as mean ± SEM.
(A–E) N = 5–8 per group. (A–D) Data were analyzed by two-tailed Student's t-test. ∗ represents a significant difference between HF- and HF + NR-fed mice. ∗P≤0.05. N represents biological replicates.
See also Figure S5.
NRS decreased expression of genes involved in the mitochondrial unfolded protein response (mtUPR)
Considering previous reports showing that NRS affects the mitochondrial unfolded protein response in liver (mtUPR) (Gariani et al., 2016; Khan et al., 2014; Zhang et al., 2016), we questioned if HF feeding and/or NRS might alter the mtUPR program in skeletal muscle. Interestingly, RT-qPCR analysis of tibialis anterior (TA) muscles from FC mice fed an HF as compared with a standard chow (SC) diet revealed elevated mRNA expression levels of mtUPR genes Lonp1 and Hspd1 (Figure 4A). Conversely, subsequent analysis of TA from both FC and KO mice fed HF ± NRS showed a dampening effect of NR on gene expression of mtUPR targets ClpP and Lonp1, and a slight increase in Hspe1 (Figures 4B–4F). In sum, NRS decreased gene expression of ClpP and Lonp1 regardless of genotype, but the specificity of this effect remains unclear.
Figure 4.

NR decreased gene expression for several markers of the mitochondrial unfolded protein response (mtUPR)
mRNA was extracted from tibialis anterior (TA) muscles and qPCR was performed for all experiments.
(A) Gene expression for markers of the mtUPR in FC standard chow (SC) and HF-fed mice. Gene expression of (B) Chop, (C) ClpP, (D) Lonp1, (E) Hspd1, and (F) Hspe1 from FC and KO mice fed either an HF or HF + NR diet. Data are represented as mean ± SEM. (A–F) N = 5 per group. Data in (A) were analyzed by two-tailed Student's t-test and data in (B–F) were analyzed by two-way ANOVA. In (A), ∗ represents a significant difference between SC and HF mice analyzed by two-tailed Student's t-test; and in (C), (D), and (F), ∗ represents a main effect of treatment analyzed by two-way ANOVA. ∗P≤0.05. N represents biological replicates.
NRS did not diminish lysine acetylation on a proteome-wide scale
We next sought to investigate whether NRS-mediated improvements in insulin tolerance, respiratory function, and/or mtUPR gene expression corresponded with sirtuin activation and a global shift in KAc. Because the impact of CrAT deficiency on the mitochondrial acetylome has been reported previously (Davies et al., 2016; Williams et al., 2020), this study focused on surveying the effects of NRS in FC mice, which in general, were more responsive to the treatment. Using quantitative nLC-MS/MS-based acetylproteomics and the workflow in Figure 5A, we quantified 744 acetyl-peptides (from 1,240 identified at 1% FDR) in TA tissue, with 275 mapping to 79 mitochondrial proteins (Calvo et al., 2016; Deutsch et al., 2017). Contrary to our original hypothesis, NRS treatment in FC mice tended to raise rather than diminish the acetyl-lysine landscape (Figures 5B and S6). However, this slight shift in the muscle acetyl-proteome did not reach statistical significance. While unexpected, these findings are similar to a prior study demonstrating that NAM treatment in HF-fed mice increased lysine acetylation in liver, as assessed by western blot analysis of whole tissue lysates (Mitchell et al., 2018). Moreover, upon further analyses of the skeletal muscle proteome and phosphoproteome (Figures S6B, S6C, and Table S1), we found no evidence of an NRS effect on acetylated Sod2 or expression of proteins related to energy expenditure, fuel preference, mitochondrial OxPHOS, mtUPR, or NAD-consuming enzymes (Table S1).
Figure 5.

The muscle mitochondrial acetylproteome is largely unaffected by NR treatment
(A) Proteomics workflow. Quadriceps muscle tissue was obtained from HF- and HF + NR-fed FC mice. Proteins were extracted, enzymatically digested, labeled with unique tandem mass tag (TMT) 10plex reagents, and pooled. After retaining a small portion of the input fraction, acetyl-peptides were enriched via immunoprecipitation, and the flow-through was subsequently used for phosphoenrichment via IMAC. After analyzing all fractions by nanoflow liquid chromatography-tandem mass spectrometry (nLC-MS/MS), the quantitative data was analyzed using Proteome Discoverer 2.4 (PD 2.4) and in-house Python code.
(B) Volcano plot of acetyl-peptide relative occupancy (protein-normalized acetylation) versus −log (p value), comparing FC mice with and without NR supplementation. Open and closed dots refer to non-mitochondrial and mitochondrial peptides, respectively, with size inversely correlated with quantitative FDR. N = 5 per group. N represents biological replicates.
NRS is ineffective when administered as a preventive therapy at the onset of a high-fat diet
Because modest improvements in whole body glucose homeostasis and mitochondrial respiration were observed in DIO mice that received NRS as a post-obesity intervention, we next sought to determine if the supplement might be more effective as a preventive therapy. To this end, 12-week-old male wild-type C57BL/6NJ (WT) mice were randomly assigned to receive an HF diet ± NRS and measures of energy balance and glucose homeostasis were monitored longitudinally for 18 weeks (Figure 6A). Metabolic assessments were identical between groups at the start of the treatment (Figures S7A–S7C), and surprisingly, remained largely similar through 18 weeks of HF feeding ± NRS (Figures 6B, 6C, 6E, and 6F). After 10 weeks of HF feeding, metabolic responses to an acute fast (18 h) and refeeding (6 h) challenge were monitored by gas exchange using a CLAMS (Figures 6D and S7D–S7O), and compared against age-matched wild-type C57BL6/NJ (WT) mice fed an SC diet. As expected, diurnal variations in oxygen consumption (VO2) and RER were greatly reduced by HF feeding (Figures S7F and S7L). NRS resulted in a subtle but significant increase in energy expenditure (EE), which was detectable during the fed but not the 18 h fasted or 6 h refed states (Figures 6D, S7G, S7H, S7M, and S7N). This NRS-associated increase in energy expenditure at 10 weeks was subsequently diminished when measured again at 16 weeks (Figure 6D). When total energy expenditure and body weight at 16 weeks were analyzed by ANCOVA, a significant group effect was detected. Thus, at that time point, energy expenditure was different between the groups after accounting for body weight (Figure S7N). NRS led to modest increases in both oxygen consumption (VO2) and carbon dioxide production (VCO2); thus, RER was unchanged between groups (Figures S7D–S7F and S7J–S7L).
Figure 6.

NR modestly increases energy expenditure wild-type C57BL6/NJ mice without impacting glucose tolerance when NR is administered at the onset of a high-fat diet
(A) Experimental design for the prevention cohort.
(B) Growth curve.
(C) Body composition at 8 and 18 weeks post HF or HF + NR diet.
(D) Energy expenditure in the fed state monitored over 24h at 10 and 16 weeks post HF or HF + NR diet.
(E) Oral glucose tolerance.
(F) 5h fasting insulin at 15 weeks post HF or HF + NR diet. Data are represented as mean ± SEM.
(A–F) N = 10 per group. Data were analyzed by two-tailed student's t-test. ∗ represents a significant difference between HF- and HF + NR-fed mice. ∗P≤0.05. N represents biological replicates.
See also Figure S7.
Comprehensive phenotyping of muscle mitochondria is unremarkable after 20 weeks of NRS
During this investigation, our laboratory developed a new platform for comprehensive analysis of mitochondrial function under physiologically relevant energetic demands (Fisher-Wellman et al., 2018) (Figure 7). A key component of the platform is its application of a modified version of the creatine kinase (CK) energetic clamp technique, which permits precise control of the extramitochondrial ATP:ADP ratio and the free energy of ATP hydrolysis (ΔGATP) by supplying excess CK and known concentrations of phosphocreatine (PCr), creatine (Cr), and adenylates (Messer et al., 2004; Glancy et al., 2008). Using the O2K respirometry system in combination with high-sensitivity spectrofluorometry, absolute rates of oxygen consumption (JO2) are measured in parallel with assessments of mitochondrial membrane potential (ΔΨm), NAD(P)H/NAD+ redox state, and H2O2 emissions. Recent applications of the platform showed that chronic HF feeding disrupts muscle mitochondrial redox balance in a manner that promotes H2O2 emission (Williams et al., 2020). In light of these results, and considering the effects of NRS observed during the intervention arm of the current study, we sought to examine the impact of NRS on mitochondrial bioenergetics at a much deeper level than afforded by more conventional approaches (Figure 3). To this end, freshly isolated muscle mitochondria from WT mice fed an HF diet ± NRS were analyzed using the CK clamp technique in the presence of pyruvate/malate (Pyr/M), palmitoyl-carnitine/malate (Pc/M), and glutamate/malate (G/M). In these experiments, respiratory conductance—the slope of the JO2 response (Figure 7A), ΔΨm (Figure 7B), NAD(P)H/NAD+ redox state (Figure 7C), respiratory efficiency—the relationship between JO2 and ΔΨm (Figure 7D), and H2O2 emissions (Figure 7E) were unaffected by NRS. Thus, in contrast to the crude respiratory phenotypes observed during the NRS intervention (Figure 3), we found no evidence that the supplement enhanced mitochondrial bioenergetics when provided as a preventative treatment and followed by extensive assessment of respiratory fluxes studied under more physiologically relevant energetic settings.
Figure 7.

NR does not alter muscle mitochondrial respiratory kinetics or H2O2 emissions in high-fat fed C57BL6/NJ mice under physiologically relevant energetic conditions
Mitochondria were isolated from skeletal muscles of wild-type C57BL6/NJ mice fed an HF or HF + NR diet.
(A–C) Relationship between (A) JO2, (B) ΔΨ, and (C) NAD(P)H/NAD(P)+ redox state versus Gibb's Energy of ATP hydrolysis (ΔGATP) measured in mitochondria fueled by pyruvate + malate (Pyr/M), palmitoyl-carnitine + malate (Pc/M), or glutamate + malate (G/M).
(D) Mitochondrial respiratory efficiency represented as JO2 plotted against ΔΨ.
(E) H2O2 emissions (JH2O2). Data are represented as mean ± SEM.
(A–E) N = 5 per group. Data were analyzed by two-tailed Student's t-test. ∗ represents a significant difference between HF- and HF + NR-fed mice. ∗P≤ 0.05. N represents biological replicates.
Discussion
To our knowledge, this is the first preclinical study to integrate comprehensive metabolic and mitochondrial phenotyping along with mass spectrometry-based proteomics to examine the effects of oral NRS on skeletal muscle metabolism and molecular reprogramming in the context of chronic HF feeding. Profiling of the plasma and muscle NAD+ metabolome in the intervention cohort revealed a positive correlation between plasma NAM and insulin responsiveness; and higher plasma and muscle levels of NAM were associated with marginal improvements in mitochondrial function. However, in the intervention arm of the study, we found no evidence that the modest NR-associated improvements in key endpoint measurements (including the muscle NAD metabolome, glucose homeostasis, and mitochondrial function) were related to a proteome-wide depression in lysine acetylation and/or increased abundance of proteins that regulate fuel preference and/or energy metabolism. Additionally, although NRS conferred marginal benefits on whole body and muscle metabolism when administered as an intervention at the midpoint of a 33-week HF diet, we were unable to reproduce the salutary outcomes during a second study in which the supplement was provided at the initiation of 22-week HF diet.
At first glance, the results described here appear to conflict with previous studies reporting more impressive NRS-induced changes in tissue NAD+ levels and/or energy homeostasis (Canto et al., 2012; Gariani et al., 2016; Crisol et al., 2020; Ryu et al., 2016). However, upon close examination, earlier studies reporting positive outcomes in response to NRS involved animal models in which tissue NAD+ levels were substantially diminished due to advanced age and/or a targeted genetic lesion that results in severe muscle dysfunction. For example, NR treatment was found to rejuvenate skeletal muscle satellite cell (MuSC) in mice that were approximately two years of age, and also prevented MuSC senescence in the mdx (C57BL/10ScSn-Dmdmdx/J) mouse model of muscular dystrophy (Zhang et al., 2016). Likewise, when applied to a genetic model of mitochondrial myopathy due to whole body deletion of a gene involved in mtDNA repair, NRS was found to delay both early- and late-stage disease progression by inducing mitochondrial biogenesis and mtUPR in skeletal muscle, which mitigated abnormalities in mitochondrial ultrastructure and mtDNA (Khan et al., 2014). NRS was also shown to rescue respiratory function assessed in muscle mitochondria derived from 7-month-old mice lacking NAMPT (Frederick et al., 2016). In the present study, mice with muscle-specific KO of CrAT were used as a model that imposes additional demand on the mitochondrial pool of NAD+. CrAT deficiency disrupts acetyl CoA balance, leading to hyperacetylation of mitochondrial proteins and increased substrate for NAD-dependent sirtuins (Davies et al., 2016; Williams et al., 2020). We therefore hypothesized that like other models that challenge NAD+ homeostasis, the KO mice might gain extra benefit from NRS. Notably, NRS raised circulating NAM levels by 2-fold in both the FC and KO groups, which were positively correlated with both muscle NAM and whole-body ITT-AAC. Although these results hint toward improvements in insulin sensitivity, the interpretation is complicated by a main effect of treatment on fasting blood glucose levels measured prior to the administration of insulin. Additionally, contrary to our predictions, NRS-mediated enhancement of mitochondrial function was modest in the FC mice and completely absent in KO mice. However, a multivariate pairwise correlation analysis did reveal a positive association between plasma NAM and mitochondrial oxygen consumption, as well as a negative correlation between respiratory capacity and body weight.
It is important to underscore that previous work concluding that NR enhances oxidative metabolism and protects against DIO in ∼20-week-old C57BL/6J mice did not perform functional assessments of skeletal muscle metabolism and/or mitochondrial performance (Canto et al., 2012). Also noteworthy is that many previous studies were performed using C57BL/6J mice, which harbor a loss-of-function mutation in the NAD nucleotide transhydrogenase (NNT) gene (Zhang et al., 2016; Canto et al., 2012; Trammell et al., 2016b; Gariani et al., 2016; Frederick et al., 2015). Because NNT resides on the mitochondrial inner membrane and uses energy from the proton motive force to produce NADPH while regenerating NAD+, this genetic background might be more susceptible to physiological insults that stress NAD homeostasis. Moreover, whereas some investigations provide evidence that NRS can augment whole body glucose control in diverse models of obesity and aging, a growing number of animal studies have reported negative outcomes (Elhassan et al., 2019; Shi et al., 2019; Crisol et al., 2020). Finally, several recent clinical trials performed in cohorts of middle- and older-aged humans have found little or no benefit of NRS on muscle function and/or mitochondrial bioenergetics, despite positive effects of the supplement on the muscle NAD metabolome (Elhassan et al., 2019; Dollerup et al., 2018, 2020).
In summary, the emergence of precision nutrition and nutrigenetics has helped to boost a global dietary supplements market now valued at over 125 billion USD. The economic future, scientific legitimacy, and clinical potential of this industry will depend heavily on the development of nutraceuticals with proven efficacy in combatting age-related metabolic disorders in humans. Credible evidence to justify major investments in large-scale human trials typically stems from rigorous and reproducible preclinical studies that firmly establish biological plausibility. To this point, the current study sought to corroborate and advance the science that underlies expanding interests in NR and the growing view that this supplement deserves serious attention from clinical/translational researchers. The findings reported here underscore a need for additional preclinical studies to further evaluate and validate the therapeutic potential of NRS in animal models of disease.
Limitations of the study
The current investigation examined the impact of NRS on skeletal muscle metabolism in middle-aged male mice subjected to chronic high-fat feeding. We did not perform extensive analysis of the liver and we did not study the effect of the supplement on other organ systems such as brown adipose tissue (BAT)—a prominent thermogenic tissue implicated in the NRS-mediated increase in energy expenditure (Crisol et al., 2018; Nascimento et al., 2021; Yamaguchi et al., 2019). Additionally, insulin sensitivity assessments were based on GTTs and ITTs rather than the hyperinsulinemic-euglycemic clamp technique, which is recognized as the gold standard measure of muscle insulin action. The mass spectrometry-based assays of protein acetylation and phosphorylation were intended to capture proteome-wide snapshots of these PTMs, but might not provide sufficient sensitivity to detect modest NRS-mediated changes in a small subset of specific proteins. The finding that NRS did not increase tissue NAD+ concentrations conflicts with some previous reports (Canto et al., 2012; Gariani et al., 2016; Crisol et al., 2020; Ryu et al., 2016) and implies that NRS might be most beneficial in pathophysiological settings that severely disrupt systemic NAD+ homeostasis. In addition, because NAD+ is constantly in flux, NAD metabolites including NAAD, meNAM, me-2-py, and me-4-py can collectively provide an improved picture of NAD+ utilization and were not measured in this study.
Another important limitation to consider is that the current study did not evaluate NR stability in the diet, which is an issue of growing concern in this field but rarely reported. A thorough summary of literature reporting on mode of administration, dosing, and stability of NR is provided in Table S2. The two viable options for long-term dosing studies in rodents include a diet admixture or administration via the drinking water. Herein, a diet admixture was used after consulting with the manufacturer of the supplement regarding compound stability. The major concern for long-term stability is exposure to high temperatures (above normal room temperatures), and therefore, application of heat when drying the pellets is not advised. For this reason, our pellets were dried overnight at room temperature under a hood, as recommended. Additionally, the NRS food pellets were refreshed daily and replaced weekly. However, without direct measures, we cannot be certain that some degradation of NR to NAM did not occur. We further recognize that we cannot definitively describe the process by which NR is converted to NAM—this might occur to some small degree prior to ingestion, or it might occur in the plasma and/or as a result of metabolism of NR by the liver. Nonetheless, the study sought to test the working model presented in Figure 1, which reflects the prevailing view that orally administered NR is converted into NAM and eventually NAD, and thereby participates in metabolic regulation via activation of nuclear and mitochondrial sirtuins.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| PDHE1a (S232) | Calbiochem | Millipore Cat# AP1063; RRID:AB_10616070 |
| PDHE1a (S293) | Calbiochem | Millipore Cat# AP1062; RRID:AB_10616069 |
| PDHE1a (S300) | Calbiochem | Cat #1064; RRID:AB_2043466 |
| Total PDHE1a | Abcam | Cat #ab110330; RRID:AB_10858459 |
| Total Rodent OxPhos WB antibody cocktail | Abcam (MitoSciences) | Cat #ab110413; RRID:AB_2629281 |
| Chemicals, peptides, and recombinant proteins | ||
| MOPS Free Acid | Millipore Sigma | Cat# M1254; CAS# 1132-61-2 |
| MES Potassium Salt | Millipore Sigma | Cat# M0895; CAS# 39946-25-3 |
| Bovine Serum Albumin (Fatty Acid Free) | Millipore Sigma | Cat# A3803; CAS# 9048-46-6 |
| EDTA | Millipore Sigma | Cat# E0270; CAS# 65501-24-8 |
| Trypsin from Porcine Pancreas (Trypsin) | Millipore Sigma | Cat# T4799; CAS# 9001-51-8 |
| Potassium Chloride | Millipore Sigma | Cat# P5405; CAS# 7447-40-7 |
| Magnesium Chloride Hexahydrate | Millipore Sigma | Cat# M2670; CAS# 7791-18-6 |
| EGTA | Millipore Sigma | Cat# E4378; CAS# 67-42-5 |
| Potassium Dihydrogen Phosphate | Millipore Sigma | Cat# P9791; CAS# 7778-77-0 |
| Creatine Monohydrate | Millipore Sigma | Cat# C3630; CAS# 6020-87-7 |
| Tris Salt of Phosphocreatine | Millipore Sigma | Cat# P1937; CAS# 108321-17-1 |
| Tris Salt of ATP | Millipore Sigma | Cat# A9062; CAS# 102047-34-7 |
| Hexokinase | Millipore Sigma | Cat# H4502; CAS# 9002-07-7 |
| Palmitoyl-L-carnitine | Millipore Sigma | Cat# P1645; CAS# 18877-64-0 |
| Octanoyl-L-carnitine | Millipore Sigma | Cat# 50892; CAS# 25243-95-2 |
| Malic Acid (Malate) | Millipore Sigma | Cat# M1000; CAS# 97-67-6 |
| Glutamic Acid (Glutamate) | Millipore Sigma | Cat# G1501; CAS# 6382-01-0 |
| Succinic Acid (Succinate) | Millipore Sigma | Cat# S3674; CAS# 110-15-6 |
| Adenosine Diphosphate (ADP) | Millipore Sigma | Cat# A5285; CAS# 72696-48-1 |
| Potassium Pyruvate | Combi-Blocks | Cat# QA-1116; CAS# 4151-33-1 |
| Creatine Kinase from Rabbit Muscle | Roche | Cat# 10127566001 |
| Amplex Ultra Red Reagent (AUR) | ThermoFisher | Cat# A36006 |
| Tetramethylrhodamine Methyl Ester (TMRM) | ThermoFisher | Cat# T668 |
| Auranofin | Millipore Sigma | Cat# A6733 |
| Potassium Cyanide | Millipore Sigma | Cat# 60178; CAS# 151-50-8 |
| Peroxidase from Horseradish (HRP) | Millipore Sigma | Cat# P8375; CAS# 9003-99-0 |
| Superoxide Dismutase (SOD) | Millipore Sigma | Cat# S9697; CAS# 9054-89-1 |
| Alamethicin | Enzo Life Sciences | Cat# BML-A150-0025 |
| CDNB | Millipore Sigma | Cat# 237329; CAS# 97-00-7 |
| Cytochrome c | Millipore Sigma | Cat#C2506 |
| Methanol | Millipore Sigma | Cat# 439193; CAS# 67-56-1 |
| Protease Inhibitor Cocktail | Millipore Sigma | Cat# P8340 |
| Phosphatase Inhibitor Cocktail 2 | Millipore Sigma | Cat# P5726 |
| Phosphatase Inhibitor Cocktail 3 | Millipore Sigma | Cat# P0044 |
| Nicotinamide | Millipore Sigma | Cat# N3376; CAS# 98-92-0 |
| Pierce Reversible Protein Stain Kit for Nitrocellulose Membranes (Memcode) | ThermoFisher Scientific | Cat# 24580 |
| 4-15% Criterion TGX Stain-Free Protein Gel, 18well | Biorad | Cat# 5678084 |
| 10X Tris Glycine SDS Running Buffer | Biorad | Cat# 1610732 |
| 10X Tris Buffered Saline | Biorad | Cat# 1706435 |
| Fish Gelatin | Millipore Sigma | Cat# G7765 |
| Casein | Millipore Sigma | Cat# C0626 |
| Sodium Nitrate | Millipore Sigma | Cat# S8032 |
| Roche cOmplete ULTRA EDTA-free Protease Inhibitor Mini Tablet | Millipore Sigma | Cat# 05892791001 |
| Roche 1x PhosSTOP Phosphatase Inhibitor Cocktail Tablets | Millipore Sigma | Cat# 04906837001 |
| Lysyl Endopeptidase, Mass Spectrometry Grade | Wako Chemicals | Cat# 125-05061 |
| Sequencing Grade Modified Trypsin | Promega | Cat# V5113 |
| tC18 SEP-PAK Solid Phase Extraction Columns (50 mg) | Waters | Cat# WAT054960 |
| tC18 SEP-PAK Solid Phase Extraction Columns (100 mg) | Waters | Cat# WAT036820 |
| Triethylammonium bicarbonate (TEAB) | ThermoFisher | Cat# 90114 |
| TMT10plex™Isobaric Label Reagent Set (0.8 mg per tag) | ThermoFisher | Cat# 90110 |
| MyTaq Red 2x Mix | Bioline | Cat# BIO-25044 |
| TRIzol Reagent | ThermoFisher | Cat# 15596026 |
| Chloroform | Millipore Sigma | Cat# C2432; CAS# 67-66-3 |
| Humulin R U-100 | Lilly USA | |
| εNAD | Millipore Sigma | Cat# N2630; CAS# ; 38806-38-1 |
| Nicotinamide riboside chloride | ChromaDex | Part # ASB-00014315-050 |
| Critical commercial assays | ||
| Pierce Quantitative Colorimetric Peptide Assay | ThermoFisher | Cat# 23275 |
| PTMScan Acetyl-Lysine Motif [Ac-K] Kit | Cell Signaling Technology | Cat# 13416 |
| Pierce High pH Reversed-Phase Peptide Fractionation Kit | ThermoFisher | Cat# 84868 |
| Pierce BCA Protein Assay | ThermoFisher | Cat# 23225 |
| RNeasy Mini Kit | Qiagen | Cat# 74106 |
| ALPCO STELLUXChemi Rodent ELISA Kit | ALPCO | Cat# 80-INSMR-CH01 |
| iSCRIPT cDNA Synthesis Kit | Biorad | Cat# 1708840 |
| TaqMan Gene Expression Assay on Demand for Chop | ThermoFisher | Assay ID Mm01135937_g1 |
| TaqMan Gene Expression Assay on Demand for Hspe1 | ThermoFisher | Assay ID Mm00434083_m1 |
| TaqMan Gene Expression Assay on Demand for Clpp | ThermoFisher | Assay ID Mm00489940_m1 |
| TaqMan Gene Expression Assay on Demand for Lonp1 | ThermoFisher | Assay ID Mm_01236887_m1 |
| Primetime qPCR Primer Assay for Hspd1 | IDT | Assay ID Mm.PT.58.13557954 |
| TaqMan Gene Expression Assay on Demand for GAPDH | ThermoFisher | Assay ID Mm99999915_g1 |
| TaqMan Gene Expression Assay on Demand for Rpl19 | ThermoFisher | Assay ID mM02601633-gd |
| Primetime qPCR Primer Assay for Rplp0 | IDT | Assay ID Mm.PT.58.43894205 |
| Deposited data | ||
| Proteomics Raw Data Files | This Publication | PRIDE Accession: PXD027904 |
| Experimental models: Organisms/strains | ||
| CrAT floxed mice | Dr. Randall Mynatt (Pennington Biomedical Research Center) | Crattm1.1Pbrc/Crattm1.1Pbrc [MGI:5427430] |
| MCK-Cre mice | The Jackson Laboratory | Tg(Ckmm-cre)5Khn/0 [MGI:2182095] |
| C57BL/6NJ mice | The Jackson Laboratory | Stock #005304 |
| Oligonucleotides | ||
| qPCR Primer Assay for CypA - Forward | IDT | Custom synthesized ; F: 5’-TATTCCAGG ATTCATGTGCCAGGG-3’ |
| qPCR Primer Assay for CypA - Reverse | IDT | Custom synthesized ; R: 5’-ATGCCAGG ACCTGTATGCTTTAGG-3’ |
| qPCR Primer Assay for CypA - Probe | IDT | Custom synthesized ; P: 5’ -/5HEX/TACA CGCCATAATGGCACTGGC/-3’ |
| Software and algorithms | ||
| Proteome Discoverer 2.4 | ThermoFisher | N/A |
| Web-based ΔGATP calculator | Fisher-Wellman et al., 2018 | https://dmpio.github.io/bioenergetic-calculators/ck_clamp/ |
| Omin | This publication | https://github.com/dmpio/omin; https://doi.org/10.5281/zenodo.5750799 |
| Proteomic Statistical Analysis | This publication | https://github.com/dmpio/nr_hdf_wt_acetyl_phospho_tmt10plex; https://doi.org/10.5281/zenodo.5750793 |
| CalR | Mina et al., 2018 | https://calrapp.org |
| Other | ||
| Oxygraph-2k | Oroboros Instruments | Cat# O2k-Core |
| QuantaMaster Spectrofluorometer | Horiba Scientific | Cat# QM-400 |
| Spectromax M2E Spectrophotometer | Molecular Devices | Part#: M2E |
| Thermo Fisher Scientific Q Exactive Plus Orbitrap Mass Spectrometer | ThermoScientific | Cat#: 0726030 |
| Thermo Fisher Scientific nanoEASY nLC | ThermoScientific | Cat #: LC140 |
| Waters Xevo TQ-S triple quadrupole mass spectrometer coupled to a Waters Acquity UPLC system | Waters | Part#: Xevo TQ-S |
| Bio-Rad Turboblot Transfer System | Biorad | Cat# 1704150EDU |
| TissueLyser II | Qiagen | Cat# 85300 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Deborah Muoio (muoio@duke.edu).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Mouse models
All animal studies were approved by the Duke University Institutional Animal Care and Use Committee and conducted in Association for Assessment and Accreditation of Laboratory Animal Care-accredited facilities. Mice were housed in a light (12h light/12h dark) and temperature (22°C) controlled room and had ad libitum access to food and water throughout all experiments unless noted otherwise. Male mice on a C57BL/6NJ background were used for all experiments. Two separate research designs (intervention and prevention) were implemented and are summarized in Figures 1B and 6A. For the intervention cohort, mice with a skeletal muscle and heart-specific deletion of CrAT (CrATM-/-, noted herein as knock outs or KO) and control animals (CrATfl/fl, noted herein as floxed controls or FC) were generated as previously described (Muoio et al., 2012). Mice were fed a standard chow diet (Lab Diet PicoLab Rodent Diet 20 5053) until approximately 12 weeks of age. At 12 weeks of age, mice were fed a high fat (HF) diet with additional sucrose (45% kcal as fat and 25% kcal as sucrose, Research Diets D03021303i) for 15 weeks. After 15 weeks, mice were assigned to one of two experimental groups: HF or HF+NR and fed handmade food pellets containing either HF diet + vehicle (water) or HF diet + NR (350 mg/kg/day) for 14 weeks prepared as previously described (Gariani et al., 2016). Briefly, group housed mice were weighed after 14 weeks of HF diet feeding; and body weight plus estimated daily food intake were used to calculate the amount (in grams) of NR chloride (MW: 290.7 g/mol) to add to the diet to achieve a dose that approximates 350 mg NR/kg body weight/day. Calculations were adjusted throughout the study to account for mouse body weight gain over time. On the day of diet preparation, NR chloride was weighed and dissolved in a known volume of double distilled (dd) water at room temperature. Frozen diet was powdered via food processor, weighed, and mixed with NR chloride dissolved in dd water (HF+NR) or dd water without NR chloride (HF). Food was pressed into small pellets by hand; and pellets were dried under laminar flow hood overnight at room temperature and stored in airtight bags at -20°C until use. To minimize potential NR degradation, fresh food pellets were added to the cages every 24-48 hours and provided in an amount intended to minimize excess. To assure that even small amounts of the food did not remain in the cages for extended periods, all food was emptied from the cages at least once per week. For the prevention cohort, male C57BL/6NJ mice were purchased from Jackson Laboratories. Mice were fed a standard chow diet until approximately 13 weeks of age and divided into two experimental groups: HF or HF+NR and fed handmade pellets containing either HF diet + vehicle (water) or HF diet + NR (350mg/kg/day) for 21-22 weeks.
Method details
Chemicals and reagents
All chemicals were purchased from Sigma-Aldrich unless otherwise stated. Creatine kinase from rabbit muscle was purchased from Roche Life Science Publishing. Tetramethylrhodamine methyl ester (TMRM) and Amplex Ultra Red were purchased from Thermo Fisher Scientific. Potassium pyruvate was purchased from Combi-Blocks. NR chloride was a gift from ChromaDex, Inc.
Determination of the NAD+ metabolome in plasma, muscle, and liver
Assays were first optimized and validated during a pilot study that measured NAD metabolites in isolated muscle mitochondria resuspended at a concentration of 10 μg/μL. Increasing ug quantities were aliquoted into methanol washed tubes and samples were centrifuged at 9,000 xg for 5 min at 4°C. Supernatant containing mitochondrial isolation buffer was carefully removed and mitochondrial pellets were snap frozen on dry ice. Mitochondria were homogenized on ice in 100 ul 100% methanol using a chilled pestle. This pilot study produced a near perfect correlation between skeletal muscle mitochondrial protein input and NAD, NAM, and NMN (Figures S2G–S2I). For the NRS experiments, muscle, liver, and plasma samples were obtained from 2h fasted mice and freeze clamped in liquid nitrogen and stored a -80°C until analysis. Fifty microliters (ul) of plasma were aliquoted into a methanol washed tube. Quadriceps muscles and livers were homogenized in 100% methanol at a concentration of 100 mg/ml. One hundred microliters (ul) of the muscle homogenate was transferred to a methanol washed tube. 50 ul of water was added to the sample, vortexed, and 100 ul of the sample was transferred to a separate tube. 10 ul of 100 uM internal standard ε-NAD (Sigma, MO, USA) was added to 100 ul of the tissue sample and 10 ul of 10 uM ε-NAD to 50 ul of plasma. The samples were precipitated with methanol, the supernatants were dried under N2, reconstituted in 200 ul of water, and analyzed on a Waters Xevo TQ-S triple quadrupole mass spectrometer coupled to a Waters Acquity UPLC system. The analytical column (Waters Acquity UPLC HSS T3 Column, 1.8 μm, 2.1 × 100 mm) was used at 30°C, 10 ul of the sample was injected onto the column, and eluted at a flow rate of 0.3 ml/min. The gradient began with 100% eluent A (5 mM ammonium acetate in water) and was then programmed as follows: 1.0 to 4.0 min – gradient to 15% eluent B (acetonitrile); 4.0 to 4.1 min - gradient to 50% eluent B; 4.1 to 5.1 min - hold at 50% eluent B, return to 100% A and re-equilibrate the column at initial conditions for 2 minutes. Mass transitions of m/z 664 → 428 for NAD, 666 → 514 for NADH, 335 → 123 for NMN, 255 → 123 for NR, 123 → 80 for NAM, and 688 → 566 for ε-NAD were monitored in a positive ion electrospray ionization mode. The analyte concentrations were determined using calibrators constructed from authentic compounds (Sigma, MO, USA) and dialyzed Fetal Bovine Serum. Importantly, the absolute values for NAD metabolites measured in the current study align with those reported previously (Dollerup et al., 2020; Remie et al., 2020; Gariani et al., 2016; Trammell et al., 2016a, 2016b; Costford et al., 2018; Catz et al., 2005).
Measurements of glucose metabolism and energy balance
Mice were singly housed and fasted on Alpha-dri bedding with access to water for 2h for all insulin tolerance tests and 5h for all oral glucose tolerance tests (OGTTs). Due to their high metabolic rate, mice are significantly stressed when subjected to an overnight fast. Thus, a 5-6 h fast is standard practice prior to an OGTT (McGuinness et al., 2009). By contrast, ITTs were performed after a 2 h fast to further minimize metabolic stress and prevent severe hypoglycemia upon exogenous insulin administration. At the start of the test, mice were weighed and a baseline blood sample was obtained from the tail for the determination of fasting blood glucose (Bayer Contour Blood Glucose Monitoring System) and plasma insulin. For all OGTTs, mice were gavaged with a 1.5 g/kg body weight glucose solution (45% glucose diluted in tap water). Subsequent blood glucose samples were obtained via the tail at 0, 15, 30, 60, 90, and 120min post-gavage. Blood from the tail was collected at 0, 15, 30, and 60min post-gavage using heparinized capillary tubes (Sarstedt Microvette CB 300 LH) and plasma was isolated for the determination of circulating insulin. Plasma insulin was determined using the ALPCO STELLUX Chemi Rodent ELISA kit (ALPCO) per manufacturer's instructions. For all ITTs, mice were injected with insulin at a dose of 1.25U/kg body weight for all post-HF pre-NR ITTs and 1.5U/kg body weight for all post-HF or HF+NR ITTs. OGTT AUC and ITT AAC were calculated using the trapezoid method and differences in fasting glucose were considered. Body composition was determined using a Bruker NMR Body Composition – LF90 Instrument. Indirect calorimetry was determined using an 8-chamber CLAMS system Columbus Instruments, Columbus, OH, USA). Raw data were uploaded into CalR for data visualization and analysis (https://calrapp.org)(Mina et al., 2018)).
Mitochondrial isolation from skeletal muscle
Mice were fasted for 2 h with access to water to ensure that all animals were in a similar postprandial state at the time of harvest. Skeletal muscle (gastrocnemius and quadriceps) was excised and immediately placed in ice-cold Buffer A (phosphate buffered saline (PBS) supplemented with 10 mM EDTA (pH 7.4)). Mitochondria were isolated using differential centrifugation (Frezza et al., 2007). The buffers for all isolations are as follows: Buffer B – 100 mM KCl, 50 mM MOPS, 1 mM EGTA, 5 mM MgSO4 (pH 7.1); and Buffer C – Buffer B supplemented with 2 g/L fatty-acid free bovine serum albumin (BSA). Tissues were minced in 1.5 ml Buffer A, transferred to a 50-mL conical, and incubated with 10 ml of Buffer A supplemented with 0.05% trypsin on ice for 5min with vortexing every 30sec. The skeletal muscle suspension was centrifuged at 200 xg for 5min at 4°C and the supernatant with trypsin was discarded. The tissue pellet was suspended in 10 ml Buffer C and homogenized with an ice-chilled Teflon pestle and borosilicate glass vessel and centrifuged at 800 xg for 10 min at 4°C. The supernatant was filtered through two layers of gauze and centrifuged at 9,000 xg for 10 min at 4°C. Mitochondrial pellets were suspended in 1.4 ml Buffer C using an ice-chilled Teflon pestle and centrifuged at 9,000 xg for 3 min at 4°C. Pellets were suspended in 1ml Buffer B and centrifuged at 9,000 xg for 3 min at 4°C. Buffer B was aspirated from each tube and mitochondrial pellets were suspended in 80-100 μL Buffer B. Protein content was determined using the Pierce BCA protein assay and mitochondria were resuspended at a final concentration of 10 mg/mL for all functional assays.
Maximal ADP-supported mitochondrial respiration
High-resolution respirometry was performed using the OROBOROS Oxygraph-2k (O2K, Oroboros Instruments). Measurements of oxygen consumption were conducted at 37°C in 2 ml Buffer Z supplemented with creatine monohydrate (20 mM), glucose (5 mM), 1 mM EGTA, and 1U/ml hexokinase (HK). The following substrate combinations were used: Palmitoyl-carnitine/Malate (Pc/M; 20 uM/2 mM), Octanoyl-carnitine/Malate (Oc/M; 200 uM/2 mM), and Glutamate/Malate/Succinate (G/M/S; 10/2/10 mM). The ADP concentration for all titrations was 1 mM. Cytochrome c (10 μM) was added at the end of the Oc/M titration to test mitochondrial membrane integrity. The pyruvate titration was performed in the presence of 2 mM Malate and 1mM ADP.
Mitochondrial respiratory control
Mice were fasted for 2h with access to water and mitochondria were isolated from skeletal muscle as described above. Steady-state oxygen consumption rates (JO2) ranging from state 2 (i.e. resting or non-phosphorylating) to ∼95% of maximal state 3 were sequentially determined using a modified version of the creatine kinase (CK) energetic clamp (Glancy et al., 2008, 2013; Messer et al., 2004) in the presence of excess CK and known amounts of creatine (Cr), phosphocreatine (PCr), and ATP. The CK reaction is used to couple the conversion of ATP and ADP to PCr and Cr. The buffer for all assays was Buffer Z supplemented with Cr (5 mM), PCr (1.5 mM), CK (20 U/mL), 2.5 mg/ml fatty-acid free BSA, and 1 mM EGTA (pH 7.2). At the start of each assay, isolated mitochondria (0.025 mg/ml) were added to the assay buffer, followed by the addition of respiratory substrates and ATP (5mM). Next, sequential additions of PCr were added to the chamber to achieve the following final concentrations: 3, 6, 9, 12, 15 mM. The purpose of the sequential additions is to alter energy demand or the ATP:ADP and gradually slow JO2 and bring it back down to baseline levels. Under the buffer conditions described above, the free energy of ATP hydrolysis (ΔGATP) after each PCr addition was calculated using the following equation:
where ΔG′°ATP is the standard apparent transformed Gibbs energy (under a specified pH, ionic strength, free magnesium and pressure), R is the gas constant (8.3145 J/kmol) and T is temperature in kelvin (310.15). After each PCr addition, the calculation of both ΔGATP and K′CK were adjusted for changes in buffer strength and free magnesium according to (Golding et al., 1995; Teague et al., 1996) and calculation of ΔGATP at each titration point was performed using a recently developed online tool (https://dmpio.github.io/bioenergetic-calculators/).
Respiratory sensitivity, or the ability of mitochondria to respond to a given energy demand (ΔGATP), was calculated by plotting the JO2 against the corresponding ΔGATP. The slope of the linear portion (a linear forceflow relationship) represents the conductance or elasticity of the entire respiratory system under specified substrate conditions. The following substrate conditions were tested: Pyruvate/Malate – (Pyr/M; 5/2.5 mM), Palmitoyl-carnitine/Malate – (Pc/M; 20 μM/2.5 mM), and Glutamate/Malate – (G/M; 10/2.5 mM). Additional information regarding the underlying rationale for each substrate combination is described in detail in (Fisher-Wellman et al., 2018).
Mitochondrial membrane potential (ΔΨ) and NAD(P)H/NAD(P)+ redox
Fluorescent measurements of mitochondrial membrane potential (ΔΨ) and NAD(P)H/NAD(P)+ redox were determined simultaneously using a QuantaMaster Spectrofluorometer (QM-400; Horiba Scientific). All assays were conducted at 37°C in a 0.2 mL reaction buffer. The buffer for all assays was Buffer Z supplemented with creatine (Cr; 5 mM), phosphocreatine (PCr; 1.5 mM), creatine kinase (CK; 20 U/ml) and TMRM (0.2 μM). At the start of each assay, isolated mitochondria (0.1 mg/ml) were added to the assay buffer, followed by the addition of respiratory substrates (G/M, Pyr/M, Pc/M, Pyr/Pc/M/Carn), ATP (5mM), and then sequential additions of PCr to obtain the following final concentrations: 3, 6, 9, 12, 15 mM. After the final PCr addition, cyanide (4 mM) was added to induce a state of 100% reduction within the NAD(P)H/NAD(P)+ couple and alamethicin (12.5 μg/mL) was added to permeabilize the mitochondria. NAD(P)H/NAD(P)+ during the experiment was expressed as a percent reduced according to the formula: % Reduction = (F-F0%)/(F100%-F0%), where the 0% reduction state is represented as the fluorescent signal (Ex/Em, 340/450nm) recorded in the presence of alamethicin. Mitochondrial membrane potential (ΔΨ) was determined using TMRM by taking the fluorescence ratio of the following excitation/emission parameters [Ex/Em, (572/590nm)/(551/590nm)] as previously described (Scaduto and Grotyohann, 1999). Data were converted from the 572/551nm ratio to millivolts (mV) following a KCl standard curve in the presence of valinomycin (Krumschnabel et al., 2014).
Mitochondrial H2O2 emission (JH2O2)
Rates of mitochondrial H2O2 emissions (JH2O2) were determined fluorometrically (QuantaMaster Spectrofluorometer; Ex:Em 565:600nm) using the Amplex Ultra Red (AUR)/horseradish peroxidase (HRP) detection system. All assays were conducted at 37°C in a 0.2 mL reaction buffer (Buffer Z supplemented with creatine (Cr; 5 mM), phosphocreatine (PCr; 1.5 mM), creatine kinase (CK; 20 U/mL), AUR (10 μM), HRP (1 U/ml) and superoxide dismutase (20 U/ml). At the start of each assay, isolated mitochondria (0.1mg/ml) were added to the assay buffer, followed by respiratory substrates (G/M, Pyr/M, Pc/M), auranofin (AF; 0.1uM), ATP (5 mM), and then sequential additions of PCr to obtain the following final concentrations: 6 and 15mM. After the final PCr addition, 1-chloro-2,4-dinitrobenzene (CDNB) was added to approximate maximal mitochondrial JH2O2.
Western blot analysis
Tissues were freeze clamped, powdered under liquid nitrogen, and homogenized in Cell Lytic-M or 2x SDS buffer supplemented with protease inhibitor cocktail, phosphatase inhibitor cocktails 2 and 3, and 10 mM nicotinamide using an ice-cold Potter-Elvehjem tissue grinder. Samples were centrifuged for 20 min at 13,000 xg for 20min at 4°C, the supernatant was transferred into a clean tube, and samples were stored at -80°C. Protein content was determined via the Pierce BCA protein assay. Samples were diluted in Cell Lytic-M and mixed with an appropriate volume of 5x loading buffer. Fifty micrograms of protein per well were loaded onto a 4-15% Criterion TGX Stain Free SDS-PAGE gel (Bio-Rad), transferred to a nitrocellulose membrane via the Bio-Rad TurboBlot Transfer system, and total protein was visualized using the Pierce Reversible Stain MemCodeKit– a rapid and sensitive alternative to Ponceau S stain for protein detection on nitrocellulose membranes after transfer from polyacrylamide gels (ThermoFisher, https://www.thermofisher.com/order/catalog/product/24580) or via stain-free gels. Membranes were blocked in fish gelatin or 5% BSA (for acetyl-lysine blots) with tris buffered saline (TBS) for 1h at room temperature and incubated with primary antibodies overnight at 4°C. Following primary incubation, membranes were washed in TBS+0.05% Tween, incubated with appropriate secondary antibody (LICOR Biosciences) diluted in fish gelatin or 5% BSA in TBS, washed, and imaged using the Odyssey Imager (LICOR Biosciences). Band intensities were determined using Image Studio version 3.1 (LICOR Biosciences) and ImageJ where appropriate. The following primary antibodies and dilutions were used: PDHE1a (S232) (Calbiochem #1063, 1:1000), PDHE1a (S293) (Calbiochem #AP1062, 1:1000), PDHE1a (S300) (Calbiochem #1064, 1:1000), Total PDHE1a (Abcam #ab110330, 1:1000), OxPhos cocktail (Abcam #ab110413, 1:1000).
Gene expression
RNA was isolated from approximately 25 mg tibilas anterior (TA) muscles using a Trizol/chloroform extraction method paired with RNeasy Mini spin columns (Qiagen) with on column DNase treatment. RNA concentration was determined using a NanoDrop and RNA quality for samples used in Figures 4A and 4B was determined via denaturing gel. cDNA was synthesized using the iScript cDNA Synthesis Kit (Bio-Rad). Quantitative PCR (qPCR) was performed using Taqman Master Mix or IDT Primetime Master Mix and the following assays: Ddit3 (Chop): Mm01135937_g1 (Taqman), Hspe1: Mm00434083_m1 (Taqman), Clpp: Mm00489940_m1 (Taqman), Lonp1: Mm_01236887_m1 (Taqman), GAPDH: Mm99999915_g1 (Taqman), Hspd1: Mm.PT.58.13557954 (IDT), Rpl19: mM02601633-gd (Taqman), Rplp0: Mm.PT.58.43894205 (IDT), and CypA (IDT, F: 5’-TATTCCAGGATTCATGTGCCAGGGG-3’, R: 5’-ATGCCAGGACCTGTATGCTTTAGG-3’, P: 5’-/5HEX/TACACGCCATAATGGCACTGGCGGCAGGT/3IAbkFQ/-3’). Each target gene was paired (duplexed) with one of three reference genes: Rpl19, GAPDH, and Rplp0. These reference genes were chosen based on their use in similar real-time PCR experiments in studies of the mtUPR (Mohrin et al., 2015; Ryu et al., 2016; Xu et al., 2015). The duplexed primer/probe sets were validated prior to all qPCR experiments using serial dilution of a common sample to establish that 1) the duplexed primer/probe sets were compatible and 2) the Ct values were linear across a range of cDNA concentrations which included the 20ng per well used in subsequent assays. Samples used for the serial dilution represented a pooled sample of cDNA from FC HF and FC HF+NR mice. Data from the primer/prove validation studies showed each standard curve obtained from the multiplexed reactions was linear and roughly parallel within the concentration range tested, with an acceptable efficiency (approximately 100% or a slope of -3.32) indicating that each target/reference gene could be duplexed. The efficiency of each reaction was calculated using the following equation: . mRNA quantities in Figures 4 and S1D were determined by comparing the reported Ct value of each target/reference gene to a standard curve performed in the same RT-PCR run. Data were normalized to CypA (Figure S1D) or the geometric mean of mRNA quantities of the reference genes (Figures 4A–4F).
Tissue lysis, digestion, and TMT labeling for proteomics
Approximately 15 mg of powdered quadriceps muscle were resuspended in ice-cold Urea Lysis Buffer (8M urea in 50 mM Tris, pH 8.0, 40 mM NaCl, 2 mM MgCl2 supplemented with 10 mM nicotinamide, 10 μM trichostatin A, and 1x Roche cOmplete ULTRA EDTA-free protease inhibitor mini tablet) and samples were disrupted with a TissueLyzer (Qiagen) for 30 sec at 30Hz. Samples were removed, frozen in liquid nitrogen, subjected to three freeze-thaw cycles, and further disrupted by sonication with a probe sonicator (three 5 sec bursts, power setting of 3). Samples were centrifuged at 10,000 xg for 10 min at 4°C, protein content was determined via the Pierce BCA protein assay, 500 μg of protein was transferred to a clean tube, and volumes were normalized with Urea Lysis Buffer (2.5 mg/mL). Samples were reduced with 5 mM DTT at 32°C for 30 min, cooled to room temperature, alkylated with 15 mM iodoacetamide for 30min in the dark and unreacted iodoacetamide was quenched by the addition of DTT to 15 mM. Samples were digested with LysC (Wako Chemicals; 1:100 w:w; 5 μg enzyme per 500 μg protein) for 4 h at 32°C; the urea diluted to 1.5 M with 50 mM Tris (pH 8.0), 5 mM CaCl2 and digested with trypsin (50:1 w/w, protein:enzyme) overnight at 32°C. Samples were acidified to 0.5% v/v trifluoroacetic acid (TFA) and centrifuged at 10,000 xg for 10 min at room temperature to pellet any undigested material. The supernatant containing soluble peptides was desalted on a 50 mg tC18 SEP-PAK Solid Phase Extraction (SPE) column (Waters) and peptides were eluted once with 500 μL of 25% acetonitrile/0.1% and twice with 500 μL 50% acetonitrile/0.1% TFA. The eluate was frozen and dried overnight in a speed vac. All samples were suspended in 100 μL of 200 mM triethylammonium bicarbonate (TEAB, ThermoFisher Scientific), mixed with a unqiue 10-plex Tandem Mass Tag (TMT, 0.8 mg in 50 μL 100% acetonitrile, ThermoFisher Scientific), and vortexed for 4 h at room temperature. Samples were quenched with 0.8 μL 50% hydroxylamine, vortexed for an additional 15 min at room temperature, combined, frozen, and dried overnight in a speed vac. Samples were re-suspended in 1 mL 0.5% TFA and desalted on a 100 mg tC18 SEP-PAK SPE column (Waters) as described above. The eluate was vortexed and ∼5% was transferred to a separate tube for quantification of unmodified peptides (“input” material) and assessment of TMT labeling efficiency, and the larger portion (95%) was retained for PTM enrichment. Both portions were frozen on dry ice, dried overnight in a speed vac, and stored at -80°C.
PTM enrichment for proteomics
Acetylpeptide enrichment was performed first, using the Cell Signaling PTMScan Acetyl-lysine Motif Kit (#13416). The larger peptide aliquot (95%) was re-suspended in 1.4 mL IAP buffer and centrifuged at 10,000 xg for 5 min at 4°C to remove any insoluble material. The supernatant was transferred to a tube containing PBS-washed antibody beads and incubated on a rotator overnight at 4°C. Samples were centrifuged at 2,000 xg for 30 sec to pellet the beads and beads were washed twice with IAP buffer and three times with chilled milli-Q filtered water. The flow-through and wash fractions were saved for phosphopeptide analysis (described below). After the last wash step, all remaining liquid was carefully removed, and bound peptides were eluted twice with 100 μL of 0.15% TFA. The eluate was acidified to 0.5% TFA and desalted on a 50 mg tC18 SEP-PAK SPE column (Waters) as previously described, the eluate frozen, dried overnight in a speed vac, Acetylpeptide samples were re-suspended in 12 μL 0.1% formic acid, and stored at -80°C. The flow-through and wash fractions from the acetyl IP described above were combined, acidified, and desalted using a 100 mg tC18 SEP-PAK SPE column. The elutate was dried in a speed vac and subjected to phosphopeptide enrichment via immobilized metal affinity chromatography (IMAC) using Ni-NTA Magnetic Agarose Beads. Briefly, the beads were washed three times with water, incubated in 40 mM EDTA, pH 8.0 for 30 minutes while shaking, and subsequently washed with water three times. The beads were then incubated with 100 mM FeCl3 for 30 minutes while shaking, and were washed four times with 80% acetonitrile/0.15% TFA. Samples were re-suspended in 1 mL 80% acetonitrile/0.15% TFA, added to the beads, and incubated for 30 minutes at room temperature while shaking. Samples were subsequently washed three times with 1 mL 80% acetonitrile/0.15% TFA and eluted for 1 minute by vortexing in 100 μL of 50% acetonitrile, 0.7% NH4OH. Eluted phosphopeptides were acidified immediately with 50 μL 4% formic acid, frozen and dried in a speed vac, re-suspended in 22.5 μL 0.1% FA and stored at -80°C.
Processing of the input fraction for proteomics
The input material was processed using the Pierce High pH Reversed-Phase Peptide Fractionation Kit (ThermoFisher Scientific #84868). Dried input material was dissolved in 300 μL of 0.1% TFA. A 150 μL aliquot was removed, diluted 1:1 with 150 μL 0.1% TFA, and loaded onto a spin column while the remaining 150 μL was stored at -80°C. The eluate was collected for a total of eight fractions per TMT kit, based on the manufacturer’s instructions. All fractions were frozen and dried overnight in a speed vac. Dried samples were resuspended in 10 μL 0.1% formic acid and peptides were quantified using the Pierce Quantitative Colormetric Peptide Assay (ThermoFisher Scientific #23275). Samples were suspended to a maximum final concentration of 0.2 μg/μL with 0.1% formic acid and stored at -80°C.
nLC-MS/MS for proteomics
All samples were subjected to nanoLC-MS/MS analysis using an EASY-nLC UHPLC system (Thermo) system coupled to a Q Exactive Plus Hybrid Quadrupole-Orbitrap mass spectrometer (ThermoFisher Scientific) via a nanoelectrospray ionization source Phosphopeptide samples were analyzed in triplicate injections of 6 μL, while acetylpeptide (6 μL) and input (8 μL, 1 ug maximum) fractions were each analyzed with a single injection. After trapping on an Acclaim PepMap 100 C18 trapping column (3 μm particle size, 75 μm × 20 mM, Thermo) at a variable flow rate dictated by max pressure of 500 Bar, each sample underwent analytical separation on an Acclaim PepMap RSLC C18 analytical column (2 μm particle size, 75 μm × 500 mM column, Thermo) over a 105 min gradient (flow rate of 300 nL/min) of 5 to 40% solvent B (90% ACN/0.1% FA), with a column temperature of 55°C. MS1 (precursor ions) was performed with default settings of 70,000 resolution, an AGC target of 3x106 ions, and a maximum injection time (IT) of 60 ms. MS2 spectra (product ions) were collected by data-dependent acquisition (DDA) of the top 10 (loop count) most abundant precursor ions with a charge greater than 1 per MS1 scan with dynamic exclusion enabled for a window of 30 sec. Precursor ions were filtered with a 0.7 m/z isolation window and fragmented with a normalized collision energy (NCE) of 30. MS2 scans were performed at 35,000 resolution with an AGC target of 1x105 ions and a maximum IT of 60 ms.
Proteomics data analysis
Data were searched against the UniProt mouse complete proteome database of reviewed (Swiss-Prot) and unreviewed (TrEMBL) proteins, which consisted of 55,398 sequences on the date of download (4/27/2020). Data analysis was performed using Proteome Discoverer 2.4, searching with both Sequest HT and MS Amanda 2.0 with the following default parameters: oxidation (15.995 Da on M) as a variable modification and carbamidomethyl (57.021 Da on C) and TMT10plex (229.163 Da on peptide N-term and K) as fixed modifications, and 2 missed cleavages (full trypsin specificity). TMT labeling efficiency was assessed as a quality control measure by searching for N-terminal TMT as a variable modification—confirming labeling efficiency at ∼93%. Phosphopeptide runs added phosphorylation (79.96633 Da on S,T, Y) as a variable modification. Acetylpeptide runs included acetylation (42.011 Da on K) as a variable modification and changed TMT to a variable modification on K (remaining fixed on peptide N-term). Data were searched with a 10 ppm precursor mass and 0.02 Da product ion tolerance. The maximum number of missed cleavages was set to a default value of 2 (but changed to 4 for acetyl runs) and enzyme specificity was trypsin (full). Considering each data type (acetyl, phospho, input) separately, peptide spectral matches (PSMs) from each search algorithm were filtered to a 1% false discovery rate (FDR) using Percolator (Kall et al., 2007) and PTM site localization probabilities were determined using ptmRS (Taus et al., 2011). PSMs were grouped to unique peptides while maintaining a 1% FDR for peptides and a 90% localization threshold for PTMs. Peptides from phospho, acetyl, and input fractions were grouped to proteins using the rules of strict parsimony and proteins were filtered to 1% FDR using the Protein FDR Validator. Reporter ion intensities for all PSMs with co-isolation interference below 50% (percentage of the ion current in the isolation window) and an average S/N >5 for reporter ions were summed together at the peptide and protein level, but quantification for each data type (acetyl, input) were kept separate. Peptides shared between protein groups were excluded from protein quantitation calculations.
Proteomics statistical analysis
Protein and peptide isoforms tabs from the PDv2.4 results were exported as tab delimited .txt files and analyzed with an in-house Python module (Omin) based on a workflow previously described previously (Fisher-Wellman et al., 2019) with some modifications. Omin contains algorithms that normalize TMT-based quantitative proteomics data generated from Proteome Discoverer Software (Thermo Fischer), based on previously developed logic (Wenger et al., 2011; Phanstiel et al., 2011). The system uses keywords to link PTM-enriched fractions to their respective inputs and automatically normalize PTM and protein relative abundance for any differences in total peptide signal on each TMT channel in the input fraction. The source code can be found at https://github.com/dmpio/omin and the acetyl-proteomics data analysis for this paper can be found at https://github.com/dmpio/nr_hdf_wt_acetyl_phospho_tmt10plex. Briefly, peptide group reporter intensities for each peptide group in the input material were summed together for each TMT channel, each channel’s sum was divided by the average of all channels' sums, resulting in channel-specific loading control normalization factors to correct for any deviation from equal protein/peptide input into the ten-sample comparison. Reporter intensities for peptide isoforms from the phosphopeptide and acetylpeptide runs and proteins from the input fraction runs were divided by the loading control normalization factors for each TMT channel, respectively. All loading control-normalized quantifications were converted to log2 space. Protein-level quantification was performed exclusively on Master Proteins—the most statistically significant protein representing a group of parsimonious proteins containing common peptides identified at 1% FDR. Phosphopeptide and acetylpeptide measurements were calculated alone (abundance) and with normalization to any change in the corresponding Master Protein (relative occupancy), calculated by subtracting Log2 Master Protein values from PTM-containing peptide quantitation values on a sample-specific basis.
Quantification and statistical analysis
Data are presented as means ± SEM. Statistical analyses were preformed using GraphPad Prism 9.0 (GraphPad Software, San Diego, CA) using two-tailed Student's t-tests, two-way and three-way ANOVAs, and repeated measures ANOVA. The goal of our study was to evaluate the effects of NRS in two different genetic backgrounds. We hypothesized a priori that the Crat KO mouse line would be particularly responsive to NRS, attributable to their previously established susceptibility to diet-induced hyper-acetylation of mitochondrial proteins. Accordingly, assessment of mitochondrial bioenergetics was a primary outcome measure of the study. Because the study was designed to compare the effect of NRS within each genotype, muscle mitochondria were harvested from two mice per day and assayed in parallel for data shown in Figure 3 (e.g. FC HF and FC HF+NR or KO HF and KO HF+NR). Given the foregoing design and execution of these experiments, a two-tailed t-test was viewed as the most appropriate statistical test and no direct comparisons can be drawn between genotypes in affected outcomes. Multivariate pairwise correlation analysis was generated using JMP 15.1 (SAS Institute, Cary, NC). Figures were generated using GraphPad Prism 9.0. Statistical tests and symbols are defined in each relevant figure legend. For data analyzed via two-tailed Student’s t-test, ∗ represents a significant difference; and for data analyzed via two-way ANOVAs, ∗ represents a main effect of treatment, # represents a main effect of genotype, and ★ represents an interaction between treatment and genotype. Lastly, for data analyzed via three-way ANOVAs (treatment x genotype x time), ‡ represents a main effect of treatment, # represents a main effect of genotype, ε represents a time x genotype interaction, and § represents a time x treatment interaction. The level of significance for all experiments was set at P ≤ 0.05.
Acknowledgments
The authors would like to thank the members of the Muoio laboratory for intellectual discussions regarding these data and Tara Narowski for her technical assistance with the preventative NRS cohort. We also thank Dr. Elizabeth Hauser, Blair Chesnut, and Colette Blach for code review and consultation on best practices for data provenance. We would also like to thank the DMPI Metabolomics Core Lab (supported by Diabetes and Endocrine Research Center grant P30 DK124723) for the metabolomics data and ChromaDex Inc. for generously supplying the NR Chloride as a gift. This work was supported by National Institutes of Health, (F32 DK105922 (ASW), R01 DK089312 (DMM), and the American Heart Association (18CDA34110216) (PAG)). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author contributions
A.S.W., P.A.G., and D.M.M. contributed to the conception and design of experiments. A.S.W., T.R.K., D.S., Y.P., O.I., and P.A.G. contributed to data acquisition. A.S.W., T.R.K., Y.P., O.I., J.A.D., and P.A.G. analyzed data. A.S.W., P.A.G., and D.M.M. interpreted data. A.S.W. and D.M.M drafted the manuscript. All authors helped to revise the manuscript and provided critical intellectual content. All authors approved the manuscript for publication.
Declaration of interest
The authors received NR Chloride as a gift from ChromaDex Inc.
Inclusion and diversity
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science.
Published: January 21, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.103635.
Supplemental information
Supplementary Excel file containing proteomics data, displayed on the following four spreadsheet tabs: Tab 1) Key. Includes descriptions of information included in columns of subsequent tabs. Tab 2) Acetylpeptides summary.Acetylpeptide Relative Abundance is normalized for total peptide amount on each TMT channel in the “input” fraction. Acetylpeptide Relative Occupancy is normalized for any biological changes in the “Master Protein” to which a given acetylated peptide maps. Tab 3) Phosphopeptides summary.Phosphopeptide Relative Abundance is normalized for total peptide amount on each TMT channel in the “input” fraction. Phosphopeptide Relative Occupancy is normalized for any biological changes in the “Master Protein” to which a given phosphorylated peptide maps. Tab 4) Proteins summary.Summary of relative quantitation statistics for all “Master” proteins.Protein Relative Abundance is normalized for total peptide amount on each TMT channel. Related to Figure 5.
Data and code availability
-
•
Proteomics raw data have been deposited at the Proteome Xchange Consortium (Deutsch et al., 2017) and are publicly available as of the date of publication. The accession number is listed in the key resources table. Proteomics data reported in this paper will be shared by the lead contact upon request.
-
•
All original code for Omin (proteomics data analysis) has been deposited at GitHub and is publicly available at https://github.com/dmpio/omin and the acetyl-proteomics data analysis for this paper is publicly available at https://github.com/dmpio/nr_hdf_wt_acetyl_phospho_tmt10plex as of the date of publication.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Askari M., Heshmati J., Shahinfar H., Tripathi N., Daneshzad E. Ultra-processed food and the risk of overweight and obesity: a systematic review and meta-analysis of observational studies. Int. J. Obes. (Lond.) 2020;44:2080–2091. doi: 10.1038/s41366-020-00650-z. [DOI] [PubMed] [Google Scholar]
- Basse A.L., Agerholm M., Farup J., Dalbram E., Nielsen J., Ortenblad N., Altintas A., Ehrlich A.M., Krag T., Bruzzone S., et al. Nampt controls skeletal muscle development by maintaining Ca(2+) homeostasis and mitochondrial integrity. Mol. Metab. 2021;53:101271. doi: 10.1016/j.molmet.2021.101271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belenky P., Bogan K.L., Brenner C. NAD+ metabolism in health and disease. Trends Biochem. Sci. 2007;32:12–19. doi: 10.1016/j.tibs.2006.11.006. [DOI] [PubMed] [Google Scholar]
- Bieganowski P., Brenner C. Discoveries of nicotinamide riboside as a nutrient and conserved NRK genes establish a Preiss-Handler independent route to NAD+ in fungi and humans. Cell. 2004;117:495–502. doi: 10.1016/s0092-8674(04)00416-7. [DOI] [PubMed] [Google Scholar]
- Blander G., Guarente L. The Sir2 family of protein deacetylases. Annu. Rev. Biochem. 2004;73:417–435. doi: 10.1146/annurev.biochem.73.011303.073651. [DOI] [PubMed] [Google Scholar]
- Brown K.D., Maqsood S., Huang J.Y., Pan Y., Harkcom W., Li W., Sauve A., Verdin E., Jaffrey S.R. Activation of SIRT3 by the NAD(+) precursor nicotinamide riboside protects from noise-induced hearing loss. Cell Metab. 2014;20:1059–1068. doi: 10.1016/j.cmet.2014.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo S.E., Clauser K.R., Mootha V.K. MitoCarta2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 2016;44:D1251–D1257. doi: 10.1093/nar/gkv1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canto C., Houtkooper R.H., Pirinen E., Youn D.Y., Oosterveer M.H., Cen Y., Fernandez-Marcos P.J., Yamamoto H., Andreux P.A., Cettour-Rose P., et al. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 2012;15:838–847. doi: 10.1016/j.cmet.2012.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catz P., Shinn W., Kapetanovic I.M., Kim H., Kim M., Jacobson E.L., Jacobson M.K., Green C.E. Simultaneous determination of myristyl nicotinate, nicotinic acid, and nicotinamide in rabbit plasma by liquid chromatography-tandem mass spectrometry using methyl ethyl ketone as a deproteinization solvent. J. Chromatogr.B Analyt Technol. Biomed. Life Sci. 2005;829:123–135. doi: 10.1016/j.jchromb.2005.10.003. [DOI] [PubMed] [Google Scholar]
- Cerutti R., Pirinen E., Lamperti C., Marchet S., Sauve A.A., Li W., Leoni V., Schon E.A., Dantzer F., Auwerx J., et al. NAD(+)-dependent activation of Sirt1 corrects the phenotype in a mouse model of mitochondrial disease. Cell Metab. 2014;19:1042–1049. doi: 10.1016/j.cmet.2014.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costford S.R., Brouwers B., Hopf M.E., Sparks L.M., Dispagna M., Gomes A.P., Cornnell H.H., Petucci C., Phelan P., Xie H., et al. Skeletal muscle overexpression of nicotinamide phosphoribosyl transferase in mice coupled with voluntary exercise augments exercise endurance. Mol. Metab. 2018;7:1–11. doi: 10.1016/j.molmet.2017.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crisol B.M., Veiga C.B., Braga R.R., Lenhare L., Baptista I.L., Gaspar R.C., Munoz V.R., Cordeiro A.V., da Silva A.S.R., Cintra D.E., et al. NAD(+) precursor increases aerobic performance in mice. Eur. J. Nutr. 2020;59:2427–2437. doi: 10.1007/s00394-019-02089-z. [DOI] [PubMed] [Google Scholar]
- Crisol B.M., Veiga C.B., Lenhare L., Braga R.R., Silva V.R.R., da Silva A.S.R., Cintra D.E., Moura L.P., Pauli J.R., Ropelle E.R. Nicotinamide riboside induces a thermogenic response in lean mice. Life Sci. 2018;211:1–7. doi: 10.1016/j.lfs.2018.09.015. [DOI] [PubMed] [Google Scholar]
- Davies M.N., Kjalarsdottir L., Thompson J.W., Dubois L.G., Stevens R.D., Ilkayeva O.R., Brosnan M.J., Rolph T.P., Grimsrud P.A., Muoio D.M. The acetyl group buffering action of carnitine acetyltransferase offsets macronutrient-induced lysine acetylation of mitochondrial proteins. Cell Rep. 2016;14:243–254. doi: 10.1016/j.celrep.2015.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutsch E.W., Csordas A., Sun Z., Jarnuczak A., Perez-Riverol Y., Ternent T., Campbell D.S., Bernal-Llinares M., Okuda S., Kawano S., et al. The ProteomeXchange consortium in 2017: supporting the cultural change in proteomics public data deposition. Nucleic Acids Res. 2017;4:D1100–D1106. doi: 10.1093/nar/gkw936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dollerup O.L., Christensen B., Svart M., Schmidt M.S., Sulek K., Ringgaard S., Stodkilde-Jorgensen H., Moller N., Brenner C., Treebak J.T., Jessen N. A randomized placebo-controlled clinical trial of nicotinamide riboside in obese men: safety, insulin-sensitivity, and lipid-mobilizing effects. Am. J. Clin.Nutr. 2018;108:343–353. doi: 10.1093/ajcn/nqy132. [DOI] [PubMed] [Google Scholar]
- Dollerup O.L., Chubanava S., Agerholm M., Sondergard S.D., Altintas A., Moller A.B., Hoyer K.F., Ringgaard S., Stodkilde-Jorgensen H., Lavery G.G., et al. Nicotinamide riboside does not alter mitochondrial respiration, content or morphology in skeletal muscle from obese and insulin-resistant men. J. Physiol. 2020;598:731–754. doi: 10.1113/JP278752. [DOI] [PubMed] [Google Scholar]
- Elhassan Y.S., Kluckova K., Fletcher R.S., Schmidt M.S., Garten A., Doig C.L., Cartwright D.M., Oakey L., Burley C.V., Jenkinson N., et al. Nicotinamide riboside augments the aged human skeletal muscle NAD(+) metabolome and induces transcriptomic and anti-inflammatory signatures. Cell Rep. 2019;28:1717–1728.e6. doi: 10.1016/j.celrep.2019.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernster L., Schatz G. Mitochondria: a historical review. J.Cell Biol. 1981;91:227s–255s. doi: 10.1083/jcb.91.3.227s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher-Wellman K.H., Davidson M.T., Narowski T.M., Lin C.T., Koves T.R., Muoio D.M. Mitochondrial diagnostics: a multiplexed assay platform for comprehensive assessment of mitochondrial energy fluxes. Cell Rep. 2018;24:3593–3606.e10. doi: 10.1016/j.celrep.2018.08.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher-Wellman K.H., Draper J.A., Davidson M.T., Williams A.S., Narowski T.M., Slentz D.H., Ilkayeva O.R., Stevens R.D., Wagner G.R., Najjar R., et al. Respiratory phenomics across multiple models of protein hyperacylation in cardiac mitochondria reveals a marginal impact on bioenergetics. Cell Rep. 2019;26:1557–1572.e8. doi: 10.1016/j.celrep.2019.01.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederick D.W., Davis J.G., Davila A., Jr., Agarwal B., Michan S., Puchowicz M.A., Nakamaru-Ogiso E., Baur J.A. Increasing NAD synthesis in muscle via nicotinamide phosphoribosyltransferase is not sufficient to promote oxidative metabolism. J. Biol. Chem. 2015;290:1546–1558. doi: 10.1074/jbc.M114.579565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederick D.W., Loro E., Liu L., Davila A., Jr., Chellappa K., Silverman I.M., Quinn W.J., 3rd, Gosai S.J., Tichy E.D., Davis J.G., et al. Loss of NAD homeostasis leads to progressive and reversible degeneration of skeletal muscle. Cell Metab. 2016;24:269–282. doi: 10.1016/j.cmet.2016.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frezza C., Cipolat S., Scorrano L. Organelle isolation: functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nat. Protoc. 2007;2:287–295. doi: 10.1038/nprot.2006.478. [DOI] [PubMed] [Google Scholar]
- Gariani K., Menzies K.J., Ryu D., Wegner C.J., Wang X., Ropelle E.R., Moullan N., Zhang H., Perino A., Lemos V., et al. Eliciting the mitochondrial unfolded protein response by nicotinamide adenine dinucleotide repletion reverses fatty liver disease in mice. Hepatology. 2016;63:1190–1204. doi: 10.1002/hep.28245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glancy B., Barstow T., Willis W.T. Linear relation between time constant of oxygen uptake kinetics, total creatine, and mitochondrial content in vitro. Am. J. Physiol.Cell Physiol. 2008;294:C79–C87. doi: 10.1152/ajpcell.00138.2007. [DOI] [PubMed] [Google Scholar]
- Glancy B., Willis W.T., Chess D.J., Balaban R.S. Effect of calcium on the oxidative phosphorylation cascade in skeletal muscle mitochondria. Biochemistry. 2013;52:2793–2809. doi: 10.1021/bi3015983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golding E.M., Teague W.E., Jr., Dobson G.P. AΔΨ ustment of K0 to varying pH and pMg for the creatine kinase, adenylate kinase and ATP hydrolysis equilibria permitting quantitative bioenergetic assessment. J. Exp. Biol. 1995;198:1775–1782. doi: 10.1242/jeb.198.8.1775. [DOI] [PubMed] [Google Scholar]
- Hirschey M.D., Shimazu T., Jing E., Grueter C.A., Collins A.M., Aouizerat B., Stancakova A., Goetzman E., Lam M.M., Schwer B., et al. SIRT3 deficiency and mitochondrial protein hyperacetylation accelerate the development of the metabolic syndrome. Mol.Cell. 2011;44:177–190. doi: 10.1016/j.molcel.2011.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kall L., Canterbury J.D., Weston J., Noble W.S., MacCoss M.J. Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat. Methods. 2007;4:923–925. doi: 10.1038/nmeth1113. [DOI] [PubMed] [Google Scholar]
- Khan N.A., Auranen M., Paetau I., Pirinen E., Euro L., Forsstrom S., Pasila L., Velagapudi V., Carroll C.J., Auwerx J., Suomalainen A. Effective treatment of mitochondrial myopathy by nicotinamide riboside, a vitamin B3. EMBO Mol. Med. 2014;6:721–731. doi: 10.1002/emmm.201403943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krumschnabel G., Eigentler A., Fasching M., Gnaiger E. Use of safranin for the assessment of mitochondrial membrane potential by high-resolution respirometry and fluorometry. Methods Enzymol. 2014;542:163–181. doi: 10.1016/B978-0-12-416618-9.00009-1. [DOI] [PubMed] [Google Scholar]
- Liu L., Su X., Quinn W.J., 3rd, Hui S., Krukenberg K., Frederick D.W., Redpath P., Zhan L., Chellappa K., White E., et al. Quantitative analysis of NAD synthesis-breakdown fluxes. Cell Metab. 2018;27:1067–1080.e5. doi: 10.1016/j.cmet.2018.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuinness O.P., Ayala J.E., Laughlin M.R., Wasserman D.H. NIH experiment in centralized mouse phenotyping: the Vanderbilt experience and recommendations for evaluating glucose homeostasis in the mouse. Am. J. Physiol. Endocrinol.Metab. 2009;297:E849–E855. doi: 10.1152/ajpendo.90996.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messer J.I., Jackman M.R., Willis W.T. Pyruvate and citric acid cycle carbon requirements in isolated skeletal muscle mitochondria. Am. J. Physiol.Cell Physiol. 2004;286:C565–C572. doi: 10.1152/ajpcell.00146.2003. [DOI] [PubMed] [Google Scholar]
- Mina A.I., LeClair R.A., LeClair K.B., Cohen D.E., Lantier L., Banks A.S. CalR: a web-based analysis tool for indirect calorimetry experiments. Cell Metab. 2018 doi: 10.1016/j.cmet.2018.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell S.J., Bernier M., Aon M.A., Cortassa S., Kim E.Y., Fang E.F., Palacios H.H., Ali A., Navas-Enamorado I., Di Francesco A., et al. Nicotinamide improves aspects of healthspan, but not lifespan, in mice. Cell Metab. 2018;27:667–676.e4. doi: 10.1016/j.cmet.2018.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohrin M., Shin J., Liu Y., Brown K., Luo H., Xi Y., Haynes C.M., Chen D. Stem cell aging. A mitochondrial UPR-mediated metabolic checkpoint regulates hematopoietic stem cell aging. Science. 2015;347:1374–1377. doi: 10.1126/science.aaa2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muoio D.M., Noland R.C., Kovalik J.P., Seiler S.E., Davies M.N., DeBalsi K.L., Ilkayeva O.R., Stevens R.D., Kheterpal I., Zhang J., et al. Muscle-specific deletion of carnitine acetyltransferase compromises glucose tolerance and metabolic flexibility. Cell Metab. 2012;15:764–777. doi: 10.1016/j.cmet.2012.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nascimento E.B.M., Moonen M.P.B., Remie C.M.E., Gariani K., Jorgensen J.A., Schaart G., Hoeks J., Auwerx J., van Marken Lichtenbelt W.D., Schrauwen P. Nicotinamide riboside enhances in vitro beta-adrenergic Brown adipose tissue activity in humans. J. Clin.Endocrinol.Metab. 2021;106:1437–1447. doi: 10.1210/clinem/dgaa960. [DOI] [PubMed] [Google Scholar]
- Phanstiel D.H., Brumbaugh J., Wenger C.D., Tian S., Probasco M.D., Bailey D.J., Swaney D.L., Tervo M.A., Bolin J.M., Ruotti V., et al. Proteomic and phosphoproteomic comparison of human ES and iPS cells. Nat. Methods. 2011;8:821–827. doi: 10.1038/nmeth.1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remie C.M.E., Roumans K.H.M., Moonen M.P.B., Connell N.J., Havekes B., Mevenkamp J., Lindeboom L., de Wit V.H.W., van de Weijer T., Aarts S., et al. Nicotinamide riboside supplementation alters body composition and skeletal muscle acetylcarnitine concentrations in healthy obese humans. Am. J. Clin. Nutr. 2020 doi: 10.1093/ajcn/nqaa072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu D., Zhang H., Ropelle E.R., Sorrentino V., Mazala D.A., Mouchiroud L., Marshall P.L., Campbell M.D., Ali A.S., Knowels G.M., et al. NAD+ repletion improves muscle function in muscular dystrophy and counters global PARylation. Sci. Transl. Med. 2016;8:361ra139. doi: 10.1126/scitranslmed.aaf5504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaduto R.C., Jr., Grotyohann L.W. Measurement of mitochondrial membrane potential using fluorescent rhodamine derivatives. Biophys. J. 1999;76:469–477. doi: 10.1016/S0006-3495(99)77214-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiler S.E., Koves T.R., Gooding J.R., Wong K.E., Stevens R.D., Ilkayeva O.R., Wittmann A.H., DeBalsi K.L., Davies M.N., Lindeboom L., et al. Carnitine acetyltransferase mitigates metabolic inertia and muscle fatigue during exercise. Cell Metab. 2015;22:65–76. doi: 10.1016/j.cmet.2015.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shats I., Williams J.G., Liu J., Makarov M.V., Wu X., Lih F.B., Deterding L.J., Lim C., Xu X., Randall T.A., et al. Bacteria boost mammalian host NAD metabolism by engaging the deamidated biosynthesis pathway. Cell Metab. 2020;31:564–579.e7. doi: 10.1016/j.cmet.2020.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi W., Hegeman M.A., Doncheva A., Bekkenkamp-Grovenstein M., de Boer V.C.J., Keijer J. High dose of dietary nicotinamide riboside induces glucose intolerance and white adipose tissue dysfunction in mice fed a mildly obesogenic diet. Nutrients. 2019;11 doi: 10.3390/nu11102439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein L.R., Imai S. The dynamic regulation of NAD metabolism in mitochondria. Trends Endocrinol.Metab. 2012;23:420–428. doi: 10.1016/j.tem.2012.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taus T., Kocher T., Pichler P., Paschke C., Schmidt A., Henrich C., Mechtler K. Universal and confident phosphorylation site localization using phosphoRS. J. Proteome Res. 2011;10:5354–5362. doi: 10.1021/pr200611n. [DOI] [PubMed] [Google Scholar]
- Teague W.E., Golding E.M., Dobson G.P. AΔΨ ustment of K0 for the creatine kinase, adenylate kinase and ATP hydrolysis equilibria to varying temperature and ionic strength. J. Exp. Biol. 1996;199:509–512. doi: 10.1242/jeb.199.2.509. [DOI] [PubMed] [Google Scholar]
- Trammell S.A., Schmidt M.S., Weidemann B.J., Redpath P., Jaksch F., Dellinger R.W., Li Z., Abel E.D., Migaud M.E., Brenner C. Nicotinamide riboside is uniquely and orally bioavailable in mice and humans. Nat. Commun. 2016;7:12948. doi: 10.1038/ncomms12948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trammell S.A., Weidemann B.J., Chadda A., Yorek M.S., Holmes A., Coppey L.J., Obrosov A., Kardon R.H., Yorek M.A., Brenner C. Nicotinamide riboside opposes type 2 diabetes and neuropathy in mice. Sci. Rep. 2016;6:26933. doi: 10.1038/srep26933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenger C.D., Phanstiel D.H., Lee M.V., Bailey D.J., Coon J.J. COMPASS: a suite of pre- and post-search proteomics software tools for OMSSA. Proteomics. 2011;11:1064–1074. doi: 10.1002/pmic.201000616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams A.S., Koves T.R., Davidson M.T., Crown S.B., Fisher-Wellman K.H., Torres M.J., Draper J.A., Narowski T.M., Slentz D.H., Lantier L., et al. Disruption of acetyl-lysine turnover in muscle mitochondria promotes insulin resistance and redox stress without overt respiratory dysfunction. Cell Metab. 2020;31:131–147.e11. doi: 10.1016/j.cmet.2019.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W., Barrientos T., Mao L., Rockman H.A., Sauve A.A., Andrews N.C. Lethal cardiomyopathy in mice lacking transferrin receptor in the heart. Cell Rep. 2015;13:533–545. doi: 10.1016/j.celrep.2015.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi S., Franczyk M.P., Chondronikola M., Qi N., Gunawardana S.C., Stromsdorfer K.L., Porter L.C., Wozniak D.F., Sasaki Y., Rensing N., et al. Adipose tissue NAD(+) biosynthesis is required for regulating adaptive thermogenesis and whole-body energy homeostasis in mice. Proc. Natl. Acad. Sci. U S A. 2019;116:23822–23828. doi: 10.1073/pnas.1909917116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshino J., Baur J.A., Imai S.I. NAD(+) intermediates: the biology and therapeutic potential of NMN and NR. Cell Metab. 2018;27:513–528. doi: 10.1016/j.cmet.2017.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H., Ryu D., Wu Y., Gariani K., Wang X., Luan P., D'Amico D., Ropelle E.R., Lutolf M.P., Aebersold R., et al. NAD(+) repletion improves mitochondrial and stem cell function and enhances life span in mice. Science. 2016;352:1436–1443. doi: 10.1126/science.aaf2693. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Excel file containing proteomics data, displayed on the following four spreadsheet tabs: Tab 1) Key. Includes descriptions of information included in columns of subsequent tabs. Tab 2) Acetylpeptides summary.Acetylpeptide Relative Abundance is normalized for total peptide amount on each TMT channel in the “input” fraction. Acetylpeptide Relative Occupancy is normalized for any biological changes in the “Master Protein” to which a given acetylated peptide maps. Tab 3) Phosphopeptides summary.Phosphopeptide Relative Abundance is normalized for total peptide amount on each TMT channel in the “input” fraction. Phosphopeptide Relative Occupancy is normalized for any biological changes in the “Master Protein” to which a given phosphorylated peptide maps. Tab 4) Proteins summary.Summary of relative quantitation statistics for all “Master” proteins.Protein Relative Abundance is normalized for total peptide amount on each TMT channel. Related to Figure 5.
Data Availability Statement
-
•
Proteomics raw data have been deposited at the Proteome Xchange Consortium (Deutsch et al., 2017) and are publicly available as of the date of publication. The accession number is listed in the key resources table. Proteomics data reported in this paper will be shared by the lead contact upon request.
-
•
All original code for Omin (proteomics data analysis) has been deposited at GitHub and is publicly available at https://github.com/dmpio/omin and the acetyl-proteomics data analysis for this paper is publicly available at https://github.com/dmpio/nr_hdf_wt_acetyl_phospho_tmt10plex as of the date of publication.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.