

Abstract
A series of acyclic nucleoside phosphonates (ANPs) was designed as inhibitors of bacterial adenylate cyclases (ACs), where adenine was replaced with 2-amino-4-arylthiazoles. The target compounds were prepared using the Halogen Dance reaction. Final ACs inhibitors were evaluated in cell-based assays (prodrugs) and cell-free assays (phosphono diphosphates). Novel ANPs were potent inhibitors of adenylate cyclase toxin (ACT) from Bordetella pertussis and edema factor (EF) from Bacillus anthracis, with substantial selectivity over mammalian enzymes AC1, AC2, and AC5. Six of the new ANPs were more potent or equipotent ACT inhibitors (IC50 = 9–18 nM), and one of them was more potent EF inhibitor (IC50 = 12 nM), compared to adefovir diphosphate (PMEApp) with IC50 = 18 nM for ACT and IC50 = 36 nM for EF. Thus, these compounds represent the most potent ACT/EF inhibitors based on ANPs reported to date. The potency of the phosphorodiamidates to inhibit ACT activity in J774A.1 macrophage cells was somewhat weaker, where the most potent derivative had IC50 = 490 nM compared to IC50 = 150 nM of the analogous adefovir phosphorodiamidate. The results suggest that more efficient type of phosphonate prodrugs would be desirable to increase concentrations of the ANP-based active species in the cells in order to proceed with the development of ANPs as potential antitoxin therapeutics.
Keywords: Acyclic nucleoside phosphonates, Adenylate cyclase, Bordetella pertussis, Inhibitors, Prodrugs
Graphical Abstract
Novel acyclic nucleoside phosphonates (ANPs) derived from substituted 2-aminothiazole represent the most potent inhibitors (in the form of phosphono diphosphates in enzymatic assays) of bacterial adenylate cyclases (adenylate cyclase toxin and edema factor) based on ANPs. The target ANPs were synthesized using Halogen Dance reaction.
Introduction
Adenylate cyclases (ACs) form a large and diverse family of enzymes, which are broadly expressed in different kingdoms of life.[1] ACs catalyze an intramolecular conversion of ATP to cyclic adenosine 3′,5′-monophosphate (cAMP), a universal second messenger involved in passing on signals, that reach the cellular membrane, to intracellular targets.[2] cAMP is involved in a wide range of key physiological processes, from virulence mechanisms in various bacteria to regulation of metabolism and gene transcription in mammals.[3,4] The cytosolic levels of cAMP are strictly regulated mostly by its synthesis by diverse ACs and by cAMP degradation by phosphodiesterases (PDEs).[1,2] Substantial alteration in the intracellular cAMP concentration has a profound effect on essential cellular processes and can be exploited to facilitate the invasion and survival of various pathogens in the host, thus contributing to pathogenesis of infectious diseases.[5-7] It has been speculated, that inhibition of microbial AC-based toxins could be exploited as potential prophylactic or post-exposure treatment strategy to combat infectious diseases.[7-9]
It has been reported that adefovir diphosphate (PMEApp, Fig. 1), the active metabolite of prodrug bis(POM)PMEA (Fig. 1), was active against both adenylate cyclase toxine (ACT) from Bordetella pertussis[10] and adenylate cyclase-based edema factor (EF) from Bacillus anthracis.[11] Furthermore PMEApp and its mimics were shown to inhibit ACs derived from rat brain.[12]
Figure 1.
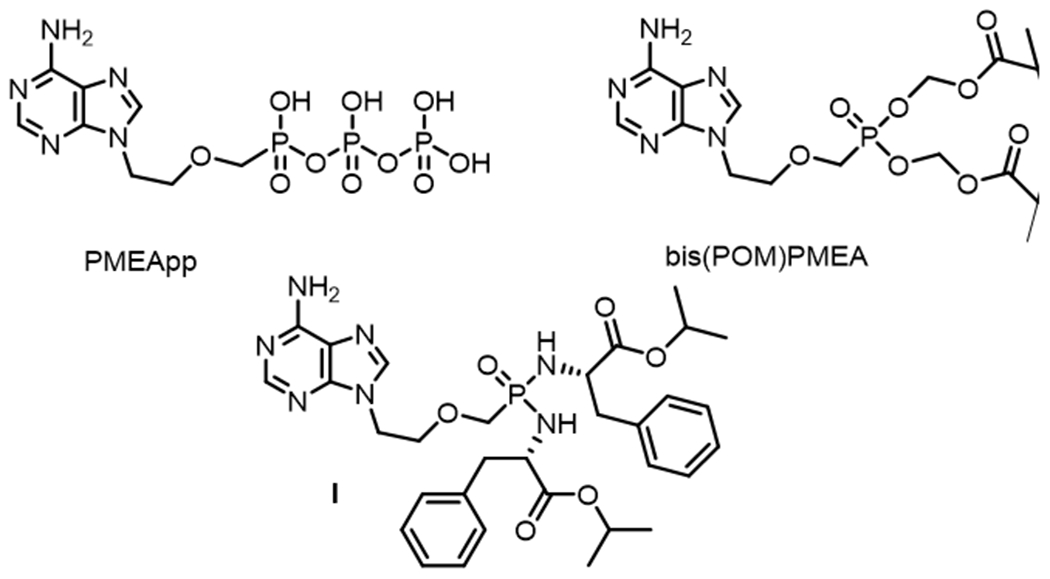
Structures of metabolically active adefovir disphosphate (PMEApp) and PMEA prodrugs.
Subsequently, an extensive structure-activity relationship study was performed with structurally modified adefovir (PMEA) analogues as inhibitors of bacterial adenylate cyclase toxins: the SAR study covered adefovir derivatives modified either at the nucleobase[13,14] or at the aliphatic moiety.[15,16] Some of the compounds also exhibited an ability to selectively modulate mammalian ACs (especially AC1), with an anticipated potential for development of non-opioid alternatives for the treatment of inflammatory and neuropathic pain.[14]
It has been recently reported,[17] that the adenine moiety in adefovir could be replaced by another heterocycle that is able to mimic the adenine nucleobase, namely 2-aminothiazole, leading to compounds II (Fig. 2). The design of such compounds was based on compound MB05032 (Fig. 2),[18-21] a potent and selective inhibitor of fructose 1,6-bisphosphatase (FBPase). It was shown that 2-aminothiazoles II bearing an aryl substituent in C5 position and a phosphonomethoxyethyl (PME) moiety in C4 position were potent inhibitors of bacterial AC toxins both as phosphorodiamidate prodrugs in J744A.1 murine macrophages or as phosphono diphosphates (nucleoside triphosphate analogues) in a cell-free assay.[17]
Figure 2.
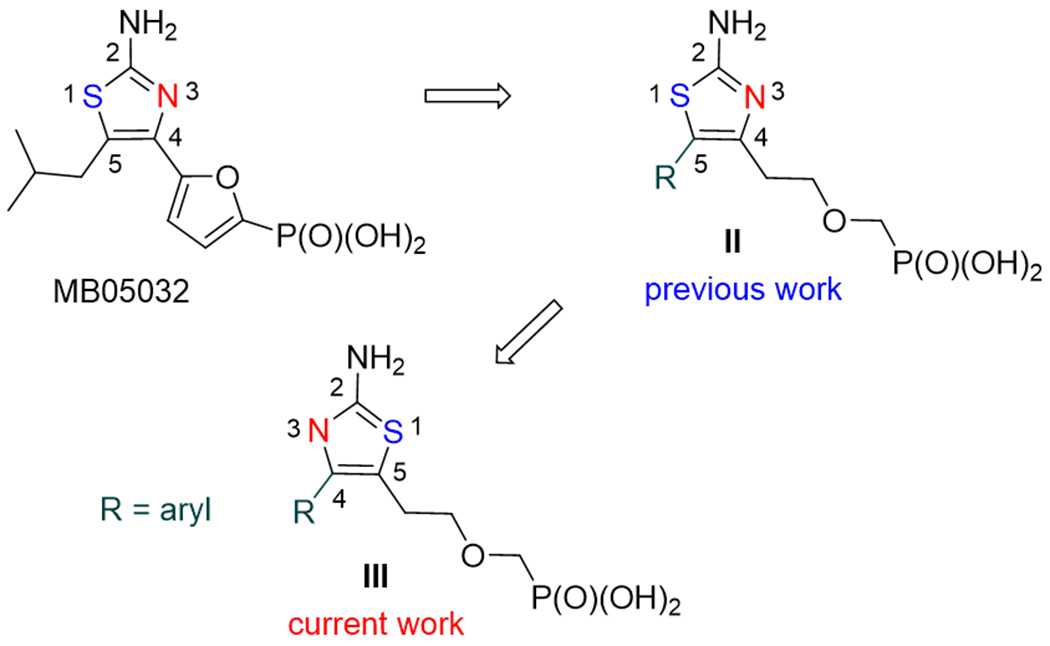
Structures of MB 05032, recently reported 5-aryl-4-PME-2-aminothiazoles II, and their 4-aryl-5-PME-2-aminothiazole regioisomers III prepared in this work.
The goal of the current study was to synthesize and evaluate a series of regioisomers derived from the previously reported 5-aryl-4-PME-2-aminothiazoles II, i.e. 4-aryl-5-PME-2-aminothiazoles III (Fig. 2).
In order to get an access to target compounds III (Fig. 2), synthetic methodology was developed and optimized starting from commercially available Boc-protected 2-aminothiazole. The final compounds were prepared in the form of their phosphorodiamidate prodrugs[22-24] for cell-based assays and as their phosphono diphosphates for enzymatic assays. The cell-permeable phosphorodiamidate prodrugs bearing non-toxic l-phenylalanine isopropyl ester moieties were selected based on the previous observation,[25] that PMEA prodrug I (Fig. 1) was significantly less cytotoxic and showed better plasma stability profiles compared to bis(POM)PMEA (Fig. 1).
Results and Discussion
Synthesis
Starting Boc-protected 2-aminothiazole 1[26] was converted into the key intermediate, 2-amino-5-bromothiazole derivative 2 (Scheme 1), in a 74% yield using N-bromosuccinimide (NBS). 5-Bromothiazole derivative 2 was then subjected to the Halogen Dance (HD) reaction,[27,28] using LDA and iodine as an electrophile, but desired 4-bromo-5-iodothiazole 3 was obtained only in a 37% yield. The next step was the transformation of 5-iodothiazole 3 into 5-vinylthiazole 4 using Suzuki-Miyaura cross-coupling and this step required an optimization of the reaction conditions: the reaction conversion was low with 1 equiv. of vinyltrifluoroborate, while large excess (5 equiv.) of the reagent led to the increased formation of undesired 4,5-divinyl-2-aminothiazole product (MW: 252.3, observed by UPLC-MS); eventually, the use of 3 equiv. of vinyltrifluoroborate afforded desired 5-vinylthiazole 4 in a 65% yield (Scheme 1). Nevertheless, the low yield (37%) of the HD reaction, overall high number of synthetic steps, and the necessity to optimize the subsequent transformation of 5-vinyl group into the 2-hydroxyethyl moiety compelled us to rethink the whole strategy at this point.
Scheme 1.
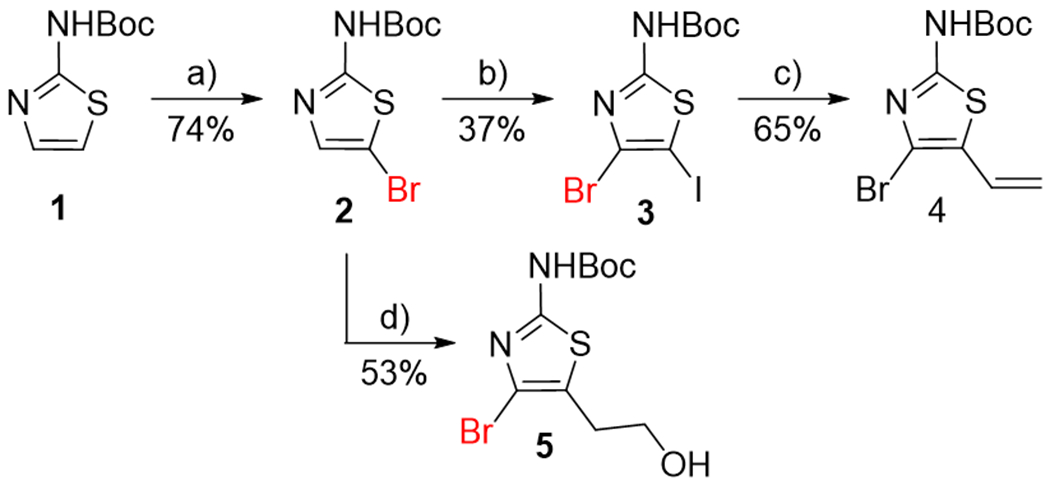
Synthesis of 2-(thiazol-5-yl)ethan-1-ol derivative 5. Reagents and conditions: a) NBS, THF, RT; b) LDA, THF, 0 °C, then I2, THF, RT; c) potassium vinyltrifuoroborate, iPr2NEt, Pd(dppf)Cl2·CH2Cl2, EtOH, 100 °C; d) LDA, THF, 0 °C, then oxirane, THF, 0–10 °C, then H2O.
We decided to introduce the 2-hydroxyethyl moiety directly using the HD reaction with oxirane as an electrophile. The HD reaction of 5-bromothiazole 2 with LDA followed by treatment with oxirane afforded the desired product as a lithium salt, which after hydrolysis gave compound 5 (Scheme 1) in a 53% yield. However, we decided to directly exploit the lithium salt of compound 5 for the subsequent in situ alkylation of the 2-hydroxyethyl moiety.
A sequential in situ treatment of 5-bromothiazole 2 with LDA, oxirane, and subsequently with TfOCH2P(O)(OiPr)2 afforded the desired phosphonate intermediate 6 (Scheme 2) in an overall 62% yield. It should be mentioned, that this satisfactory yield was obtained after a thorough optimization process and that several crucial factors should be taken into consideration for this one pot synthesis. First, each reaction step demanded a specific temperature: optimal temperature for the HD reaction was −20 °C, for the oxirane opening +10 °C, and for the final alkylation −40 °C. The second key factor was the presence of 3 equiv. of each of the reagents, i.e. LDA, oxirane, and the alkylating agent. The use of 1 or 2 equiv. of LDA dramatically decreased the HD reaction conversion, which is in accordance with the literature.[29] Similarly, significantly lower yields of compound 6 were obtained when only 1–2 equiv. of oxirane or of the alkylating agent were employed. Thirdly, the reaction time was crucial too: the reaction was ideally stopped 2 h after the addition of the alkylating agent despite the incomplete conversion (UPLC-MS), as after 3 h, the amount of bis-alkylated by-product 7 started to increase substantially (UPLC-MS).
Scheme 2.
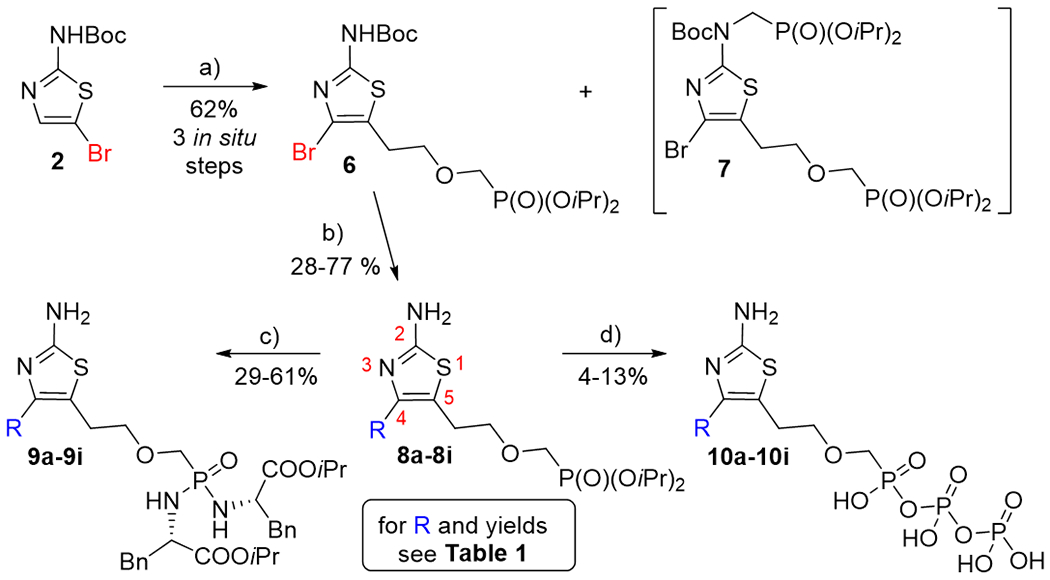
Synthesis of ANPs 8, the corresponding phosphorodiamidate prodrugs 9, and phosphono disphosphates 10. Reagents and conditions: a) LDA, THF, −20 °C, then oxirane, THF, 10 °C, then TfOCH2P(O)(OiPr)2, −40 °C; b) XPhos Pd G2, R-B(OH)2, K3PO4, THF/H2O (4:1), 40 °C, then TFA, RT, 5 min; c) TMSBr, pyridine, RT, then (L)-NH2CH(Bn)COOiPr·HCl, Aldrithiol™, PPh3, Et3N, pyridine, 70 °C; d) (i) TMSBr, pyridine, RT, (ii) 2M aq. TEAB and TBAH, (iii) CDI, (Bu3N)2P2O7, DMSO.
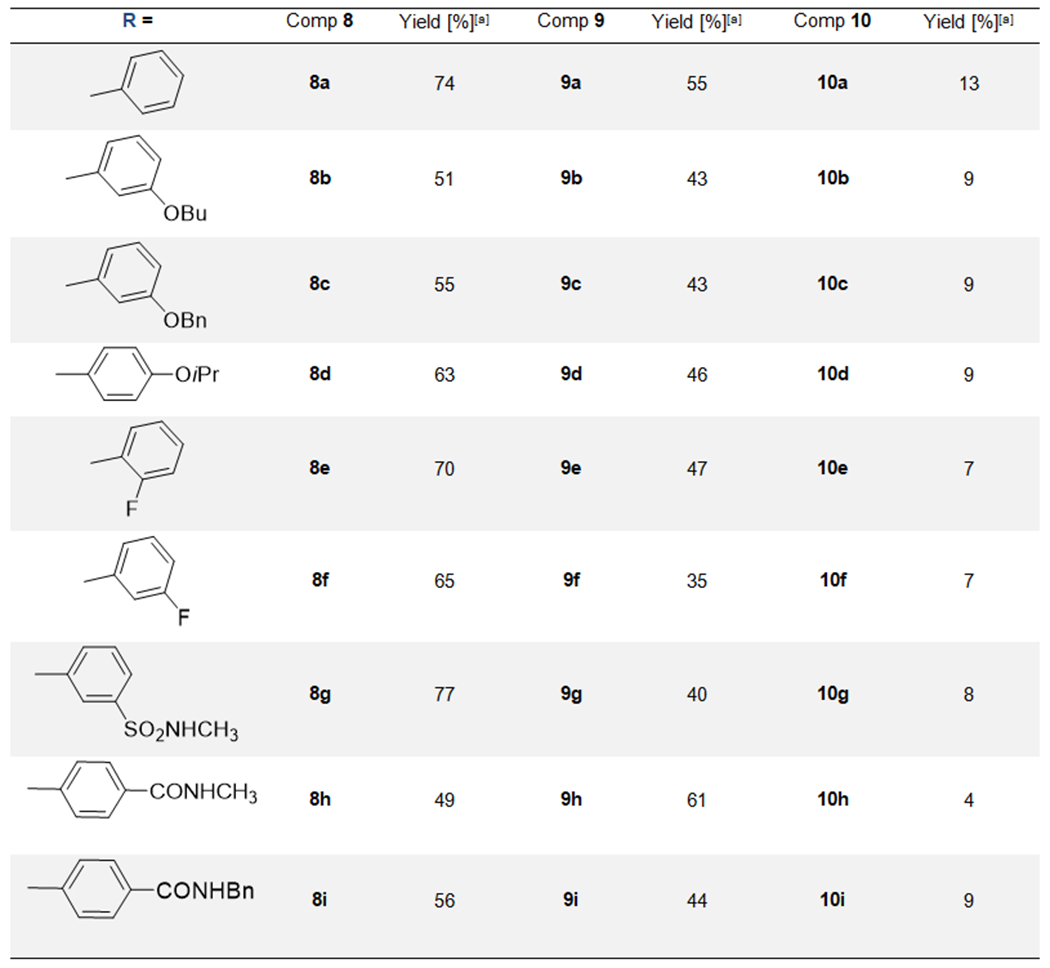
Key 4-bromothiazole intermediate 6 was used in the Suzuki-Miyaura coupling for the synthesis of a series of 4-aryl substituted 2-aminothiazole-based ANPs 8a–8i (Scheme 2) in 28–77% yields (Table 1). Phosphonate diesters 8a–8i were subsequently converted into final phosphorodiamidate prodrugs 9a–9i (29–61%) by their transformation into the corresponding silyl esters first, followed by the treatment with L-phenylalanine isopropyl ester under the standard reaction conditions (Aldrithiol-2, pyridine, Et3N, 70 °C) previously reported by our group.[30] Finally, compounds 8a–8i were used for the preparation of corresponding phosphono diphosphates 10a–10i (4–13%), the active triphosphate analogues (Table 1): transformation of 8a–8i into the corresponding silyl esters, followed by the treatment with carbonyldiimidazole and tributylammonium pyrophosphate.[31] The low yields of compounds 10a–10i were primarily caused by the low conversion, by relative instability of the phosphono diphosphates, and by their laborious purification process.
Table 1:
Yields of Suzuki coupling products 8a–8i, phosphorodiamidate prodrugs 9a–9i, and phosphono diphosphates 10a–10i (Scheme 2)
|
Isolated yields.
It should be noted, that the assignment of the regiochemistry of compounds 8a–8i was based on 2D NMR spectroscopy measurements, namely H,C-HMBC experiments, which correlate hydrogen and carbon signals usually via two or three covalent bonds. The chemical shifts of carbon atoms in positions C4 and C5 of the thiazole ring can be easily distinguished because C5 (neighboring the sulfur atom) is found in the region of 115–120 ppm while C4 (next to the nitrogen) can be found close to 145 ppm. The aromatic hydrogen atoms in ortho position to the thiazole ring have a clear three-bond correlation with carbon C4 in the HMBC spectra, while the aliphatic CH2 hydrogen atoms next to the thiazole ring have both the two-bond correlation to C5 and three-bond correlation to C4. The hydrogen atoms of the second next CH2 group have only three-bond correlations with carbon C5. The structure of the thiazole derivatives prepared earlier[17] was also unequivocally determined by HMBC experiments.
Inhibition of ACT in cell-based assays
The general putative cleavage of phosphorodiamidates[32] releasing the parent ANPs is similar to the metabolic activation of phosphoramidates (ProTides),[33] which is well-described.[34] The released phosphonates need to be further phosphorylated to the active species, phosphono diphosphates (nucleoside triphosphate analogues), as it was previously shown,[17] that parent ANPs are not active in the enzymatic assays. Thus, all prepared phosphorodiamidates 9a–9i were tested for their ability to inhibit B. pertussis adenylate cyclase toxin (ACT) activity in J774A.1 macrophage cells (Table 2). Murine J774A.1 cells were incubated with various concentrations of tested compounds and subsequently exposed to ACT. The cells were lysed and the amount of cAMP was determined. Two of the prepared compounds (9b and 9f) exhibited a single digit μM activity. The best ACT inhibitor from the series proved to be compound 9d with IC50 = 490 nM, which was only about three times less potent than phosphorodiamidate prodrug of adefovir I (IC50 = 147 nM, Table 2).[14] In general, the novel 4-aryl-5-PME-2-aminothiazole analogues 9a–9i exhibited somewhat lower potency to inhibit ACT in the cell-based assay compared to previously published regioisomeric 5-aryl-4-PME-2-aminothiazoles (Table 2, values in red color),[17] with the exception of compound 9d, (4-isopropoxyphenyl)thiazole derivative (IC50 = 490 nM), which is slightly (1.4 fold) more potent compared to its corresponding regioisomer II (IC50 = 680 nM). On the other hand, the most potent inhibitor from the previous series was 5-[3-(benzyloxy)phenyl]thiazole derivative (IC50 = 260 nM, Table 2),[17] while the 4-[3-(benzyloxy)phenyl]thiazole regioisomer 9c from the current study was inactive (IC50 >10 μM, Table 2). Thus, no clear correlation as for the ACT inhibition in the cell-based assay can be made between the two regioisomeric series of compounds.
Table 2.
ACT inhibition (J774A.1 cells), cytotoxic effects (J774A.1 cells), and mammalian AC1, AC2 and AC5 inhibition (HEK293 cells) of phosphorodiamidate prodrugs 9a–9i. The ACT inhibition of prodrugs 9a–9i is compared with the potency of their previously reported[17] regioisomers II (Fig. 2).
| Comp | ACT IC50 [μM][a] | ACT for II[17] IC50 [μM][a] | Viability [%][b] | % FSK (5 μM) Stimulated
Activity[d] |
||||
|---|---|---|---|---|---|---|---|---|
| AC1 | AC2 | AC5 | ||||||
| I [c] | 0.15 ± 0.04 | -- | 93 | 64 ± 9 | 200 ± 45 | 99 ± 8 | ||
|
| ||||||||
| 9a | >10 | 1.82 ± 0.58 | 112 | 61 ± 12 | 99 ± 26 | 79 ± 16 | ||
| 9b | 8.72 ± 1.33 | 1.62 ± 0.36 | 106 | 90 ± 15 | 137 ± 30 | 81 ± 15 | ||
| 9c | >10 | 0.26 ± 0.05 | 103 | 88 ± 14 | 126 ± 33 | 91 ± 9 | ||
| 9d | 0.49 ± 0.10 | 0.68 ± 0.08 | 103 | 71 ± 18 | 150 ± 28 | 87 ± 10 | ||
| 9e | >10 | ND | 101 | 74 ± 13 | 98 ± 16 | 81 ± 17 | ||
| 9f | 5.19 ± 0.76 | 0.45 ± 0.17 | 110 | 79 ± 22 | 139 ± 60 | 73 ± 7 | ||
| 9g | >10 | 4.30 ± 0.29 | 101 | 86 ± 41 | 115 ± 32 | 81 ± 9 | ||
| 9h | >10 | 0.66 ± 0.18 | 101 | 119 ± 15 | 112 ± 28 | 84 ± 5 | ||
| 9i | 13.1 ± 0.80 | ND | 108 | 102 ± 21 | 115 ± 33 | 81 ± 12 | ||
|
| ||||||||
| −FSK | ND | ND | ND | 0 ± 0 | 0 ± 0 | 0 ± 0 | ||
| +FSK | ND | ND | ND | 100 ± 0 | 100 ± 0 | 100 ± 0 | ||
| ST034307 | ND | ND | ND | 0 ± 0 | ND | ND | ||
| SKF83566 | ND | ND | ND | ND | 15 ± 12 | ND | ||
Data represent the mean ± SD of at least three independent experiments; IC50 = concentration of a compound causing a 50% decrease in ACT-induced cAMP accumulation.
Data represent the percentage of cell viability at a fixed prodrug concentration (10 μM) versus untreated control.
Previously published data for compound I, ref. [14].
Data represent the mean ± SD of at least three independent experiments at a fixed prodrug concentration (30 μM).
FSK; forskolin. ND: not determined.
Inhibition of mammalian ACs
All prepared phosphorodiamidates 9a–9i were tested against human transmembrane AC isoforms at a single concentration of 30 μM (Table 2). Human AC1, AC2, and AC5, representing the three major mAC subfamilies, were overexpressed in HEK293 cells, which exhibit drastically reduced forskolin (FSK)-stimulated cAMP formation in the absence of recombinant AC.[35] Most of the compounds failed to markedly inhibit any of the mACs tested, thus revealing promising selectivity for the bacterial ACs. From the whole series, compound 9a was the most potent AC1 inhibitor (61% of remaining AC activity) with a good to moderate selectivity over AC2 (99%) and AC5 (79%). Treatment of AC2 with 9d resulted in a moderate increase in activity, reaching 150% activity compared to FSK-stimulated control, a phenomenon that has previously been noted with some AC inhibitors.[14,36] Only two compounds inhibited AC5 activity more than 20%: 9a and 9f inhibited this isoform to 79% and 73% activity, respectively. Previously described isoform selective inhibitors of AC1 and AC2 were also included as controls and performed as expected:[36,37] the AC1-specific ST034307 reduced activity by over 99%, and SKF83566, an AC2-selective inhibitor, resulted in 85% inhibition of AC2.
Direct inhibition of ACT and EF activity
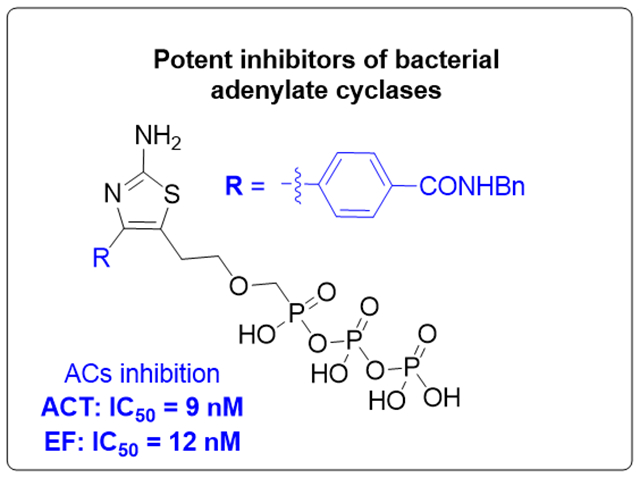
In order to observe direct enzymatic effect of the studied compounds (without the influence of the cell-penetration and prodrug biotransformation), triphosphate analogues 10a–10i were tested in a cell-free assay, along with PMEApp as a reference compound, for their inhibitory activity against two commercially available bacterial adenylate cyclases, adenylate cyclase toxin (ACT from B. pertussis) expressed recombinantly in E. coli, and edema factor (EF) from Bacillus anthracis. Compounds 10a–10i (Table 3) proved to be very potent (mostly submicromolar) inhibitors of both ACT and EF, and (with the exception of 10h) more potent compared to the previously reported regioisomers II (Fig. 2).[17] In general, ACT seems to be more susceptible to inhibition by 2-aminothiazole-based ANPpp 10a–10i than EF, which is also in agreement with previously reported regioisomeric analogues II.[17] Six of the compounds (10b, 10c, 10d, 10f, 10g, and 10i) are more potent or equipotent ACT inhibitors (IC50 = 9.0–18.4 nM) compared to PMEApp (IC50 = 17.8 nM) and, thus, they represent the most potent ANP-based inhibitors of bacterial ACs known up to date. From the whole series, derivative 10i is the most potent ACT (IC50 = 9 nM) and EF (IC50 = 12 nM) inhibitor, twice and three times more potent against ACT and EF, respectively, than PMEApp (Table 3).
Table 3.
Direct inhibition of bacterial ACs (ACT from B. pertussis and EF from B. anthracis) by prepared ANPpp 10a–10i and their comparison with the previously reported[17] regioisomers II (Fig. 2), where available.
| Compound | IC50
[nM][a] |
Regioisomers
IIb - IC50 [nM][a] |
||
|---|---|---|---|---|
| ACT | EF | ACT | EF | |
| 10a | 25.2 ± 5.7 | 192.7 ± 16.4 | -- | -- |
| 10b | 18.4 ± 6.6 | 174.9 ± 35.7 | -- | -- |
| 10c | 16.7 ± 0.4 | 67.3 ± 5.4 | 192 ± 13 | 235 ± 37 |
| 10d | 17.1 ± 7.1 | 263.8 ± 44 | 185 ± 31 | 428 ± 11 |
| 10e | 311.9 ± 7.3 | 7,992 ± 1,222 | -- | -- |
| 10f | 12 ± 0.2 | 120.9 ± 37.3 | 313 ± 55 | 1,335 ± 375 |
| 10g | 15.4 ± 4.5 | 5,792 ± 685 | -- | -- |
| 10h | 115.7 ± 22.4 | 162.3 ± 54.3 | 37.4 ± 7.6 | 704 ± 50 |
| 10i | 8.96 ± 0.07 | 11.6 ± 1.7 | -- | -- |
|
| ||||
| PMEApp | 17.8 ± 6.9 | 36.0 ± 2.1 | -- | -- |
Data represent the mean ± SD of at least three independent experiments.
Data taken from ref. [17].
Conclusion
Nine novel acyclic nucleoside phosphonates (ANPs) containing the 4-aryl-2-aminothiazole moiety as a replacement for the nucleobase (adenine) were designed and prepared. The compounds were prepared as phosphonate diesters (8a–8i), in the form of phosphorodiamidate prodrugs (9a–9i), and as phosphono diphosphate analogues (10a–10i). Several prodrugs (namely 9b, 9d, and 9f) were found to be potent (single digit μM or submicromolar) inhibitors of adenylate cyclase toxin (ACT) from Bordetella pertussis in the macrophage cell-based assay, they were non-cytotoxic, and did not markedly inhibit any of the tested mammalian ACs (mACs). The observed selectivity for ACT over mACs qualifies the 2-aminothiazole-based ANPs for potential therapeutic applications at concentrations that effectively neutralize bacterial toxins without deleterious side-effects through interference with the host ACs. The most potent compound of the series, 2-amino-4-(4-isopropoxyphenyl)thiazole derivative 9d, inhibits ACT with IC50 = 490 nM, while it only slightly (by 29%) decreased AC1 activity and moderately (by 50%) increased AC2 activity. On the other hand, compound 9a proved to be the most efficacious (61% of remaining AC activity) AC1 inhibitor, with some observed selectivity over AC2 and AC5 (with 99% and 79% remaining AC activity, respectively). In general, the phosphono diphosphate analogues 10a–10i proved to be very potent inhibitors of bacterial adenylate cyclase toxins, where ACT from B. pertussis was more sensitive than EF from B. anthracis. Phosphono diphosphate 10i was the most potent inhibitor of both ACT (IC50 = 9 nM) and EF (IC50 = 11.6 nM) in the series and represents the most potent ANP-based inhibitor of bacterial adenylate cyclases (ACT and EF) known to date. The noticeable discrepancy between the potency of studied analogues in cell-based (prodrugs) vs. enzymatic (phosphono diphosphates) assays could be explained by somewhat varying ability of the studied compounds to cross cell membranes and/or to undergo the biotransformation of the phosphorodiamidates into the enzymatically active triphosphate analogues. At the moment, development of more efficient prodrug strategy is in progress in order to evaluate the most potent analogues in the mouse models of pertussis and/or anthrax.
Experimental Section
Chemistry – general methods
Unless otherwise stated, solvents were evaporated at 40 °C/2 kPa and the compounds were dried over P2O5 at 2 kPa. The microwave-assisted reactions were carried out in CEM Discover (Explorer) microwave apparatus. Chemicals and reagents were obtained from commercial sources (Sigma–Aldrich, Fluorochem Ltd.). Solvents were dried by standard procedures; pyridine was stored over molecular sieves (4 Å); tetrahydrofuran (THF) was freshly distilled from LiAlH4 pellets under Ar. TLC was performed on plates of Kieselgel 60 F254 (Merck). Preparative TLC was performed on plates of UNIPLATE, SILICA GEL GF, 2000 μm, 20 × 20 cm (Analtech). Preparative HPLC purifications were performed on columns packed with 10 μm C18 reversed phase resin (Phenomenex Gemini 10 μm 21 × 250 mm) on Waters Delta 600 chromatography system in ca. 200 mg batches of crude mixtures using gradient MeOH/H2O as eluent. Flash chromatography on normal phase and on reversed phase was performed on Reveleris Flash Chromatography System. The deionization was performed on column Redisep®Rf GOLD C18 Teledyne ISCO. NMR spectra were recorded on Bruker Avance III 500 (1H at 500 MHz, 13C at 125.8 MHz and 31P at 202.4 MHz) and/or Bruker Avance III 400 (1H at 401 MHz, 13C at 100.8 MHz) spectrometers with TMS as internal standard or referenced to the residual solvent signal. Mass spectra were measured on UPLC-MS (Waters SQD-2) and HR-MS were taken on a LTQ Orbitrap XL spectrometer using electrospray ionization (ESI). The purity of target compounds was determined by HPLC (H2O-CH3CN, linear gradient) and was higher than 95%.
Method A. General procedure for Suzuki-Miyaura cross-coupling reactions of 2-amino-4-bromo-thiazole derivatives
A solution of 6 (0.50 g, 1.0 mmol) in THF (2 mL) under Ar was added to a mixture of precatalyst Xphos Pd G2 (31 mg, 0.04 mmol) and corresponding boronic acid (1.5 mmol) in degassed aqueous THF (21 mL, THF:H2O, 4:1) under Ar, followed by a degassed solution of K3PO4 (640 mg, 3.0 mmol) in H2O (2 mL). The resulting mixture was stirred at 50 °C overnight and then cooled down. Aqueous NH4Cl solution (50%, 10 mL) was added and the mixture was extracted with EtOAc (3 × 25 mL). Combined organic layers were dried (Na2SO4), volatiles were removed in vacuo and the residue was dissolved in conc. TFA (5 mL). After 5 min at RT, the volatiles were removed in vacuo and the residue was codistilled with water (3 × 10 mL). The crude product was adsorbed onto the C18 silica gel and purified by reverse phase flash chromatography (0–100% H2O in CH3CN with 0.1 % HCOOH) to give the desired product.
Method B. General procedure for synthesis of bis-(L-phenylalanine ethyl ester) prodrugs of phosphonates[30]
TMSBr (1 mL) was added to the corresponding phosphonate diester 8 (1.0 mmol) dissolved in dry pyridine (10 mL) and the reaction mixture was stirred at 20 °C overnight. Volatiles were removed in vacuo, while the moisture sensitive intermediate was permanently kept under Ar. Solid isopropyl ester L-phenylalanine hydrochloride (0.97 g, 4.0 mmol) was added to the intermediate, followed by dry pyridine (8 mL) and dry Et3N (2 mL) under Ar. The mixture was preheated to 70 °C and freshly prepared solution of Aldrithiol-2 (1.37 g, 6.2 mmol) and triphenylphosphine (1.64 g, 6.2 mmol) in pyridine (10 mL) was added. The resulting mixture was stirred at 70 °C for 72 h. Reaction mixture was evaporated in vacuo and the residue was purified by column chromatography (0–100% MeOH in CHCl3) followed by C18 reversed phase column chromatography (0–100% CH3CN in water). The UV absorbing fractions were collected and evaporated in vacuo. The product was dissolved in dioxane (2 mL) and lyophilized to give a white amorphous solid.
Method C. General procedure for synthesis of phosphono diphosphates[31]
TMSBr (1 mL) was added to the corresponding phosphonate diester 8 (1.0 mmol) dissolved in dry pyridine (10 mL) under Ar. The reaction mixture was stirred at 20 °C overnight and evaporated. Aqueous TEAB solution (2 M, 2 mL), 1.0 M aqueous tetrabutylammonium hydroxide solution (2.5 mL, 2.5 mmol), and dioxane (5 mL) were added and volatiles were evaporated. The residue was dissolved in water (10 mL) and applied on C18 column (0–100% CH3CN in water). Collected fractions with product were evaporated, the residue was codistilled with dioxane (3 × 5 mL) and dissolved in DMSO (10 mL). Carbonyldiimidazole (1.62 g, 10.0 mmol) was added and the mixture was stirred at RT for 2 h. MeOH (0.40 mL, 10.0 mmol) was added and the mixture was stirred at RT for another 30 min. Tributylammonium pyrophosphate (5.47 g, 10.0 mmol) was added and the mixture was stirred at RT overnight. Reaction mixture was diluted with water (5 mL) and applied on the RPC18 column (0–100% CH3CN in water). UV absorbing fractions with the product were evaporated and purified on POROS 50 HQ column (TEAB 0.05–1 M) and applied on Dowex 50 in Na+ cycle. The product was lyophilized to give a white amorphous solid.
Preparation of fresh LDA solution
2.7 M BuLi in hexane (0.48 mL, 1.3 mmol) was added dropwise to a solution of N,N-diisopropylamine (0.2 mL, 1.4 mmol) in THF (3.5 mL) cooled to −78 °C under Ar. The resulting solution was directly used in the next reaction step.
Compounds synthesis and characterization
5-Bromo-2-(tert-butoxycarbonylamino)thiazole (2).
A suspension of 2-(tert-butoxycarbonylamino)thiazole (6.3 g, 31.5 mmol) and N-bromosuccinimide (6.2 g, 34.6 mmol) in THF (100 mL) was stirred at room temperature for 12 h. The reaction mixture was filtered and purified by flash chromatography (linear gradient from hexane to 10% MeOH/EtOAc) to give 2 (6.5 g, 74%) as a white solid. ESI-MS, m/z (%): 301 [M+Na+] (100). 1H NMR (DMSO-d6): δ 11.73 (brs, 1H, NH); 7.43 (s, 1H, CH-4); 1.48 (s, 9H, CH3). 13C NMR (DMSO-d6): δ 160.16 (C-2); 152.97 (OCON); 139.00 (C-4); 100.59 (C-5); 81.72 (C-CH3); 27.84 (CH3). HR-MS (ESI+): m/z [M + Na]+ calculated for: C8H11O2N2BrNaS, 300.9617, found: 300.9620.
4-Bromo-5-iodo-2-(tert-butoxycarbonylamino)thiazole (3).
A solution of 2 (1.0 g, 3.6 mmol) in dry THF (4 mL) was added dropwise to a freshly prepared THF solution of LDA (11.9 mmol) under Ar at 0 °C. The resulting solution was stirred at 0 °C for further 15 min. The reaction mixture was allowed to warm up to RT and a solution of iodine (3.0 g, 11.9 mmol) in THF (5 mL) was added dropwise. The reaction mixture was stirred at RT for 1 h and then diluted with EtOAc (150 mL) and with 50 % sat. NH4Cl/H2O solution (40 mL). The organic layer was washed with water (100 mL) and brine (100 mL), and dried (Na2SO4). The solvent was evaporated and the crude product was purified by flash chromatography (10% EtOAc/cyclohexane) to give 3 (0.5 g, 37%) as a white solid. ESI-MS, m/z (%): 404.9 [M+H+] (60). 1H NMR (CDCl3): δ 9.06 (brs, 1H, NH); 1.56 (s, 9H, CH3). 13C NMR (CDCl3): δ 164.62 (C-2); 151.80 (OCON); 129.86 (C-4); 83.88 (C-CH3); 65.07 (C-5); 28.28 (CH3). HR-MS (ESI+): m/z [M + Na]+ calculated for: C8H10O2N2BrINaS, 426.8583, found: 426.8585.
4-Bromo-5-vinyl-2-(tert-butoxycarbonylamino)thiazole (4).
N,N-Diisopropylethylamine (0.04 mL, 0.25 mmol) was added to the mixture of 3 (0.10 g, 0.25 mmol), Pd(dppf)Cl2 · CH2Cl2 (0.02 g, 10 mol%) and potassium vinyltrifluoroborate (0.10 g, 0.74 mmol) in degassed 96% aq. EtOH (5 mL) under Ar. The reaction mixture was heated to 100 °C for 4 h and then allowed to cool down to RT. Volatiles were removed in vacuo, the residue was dissolved in Et2O (10 mL) and washed with 20% aq. NH4Cl solution (2 × 10 mL) and brine (10 mL). Organic layer was dried (Na2SO4) and evaporated in vacuo. The residue was purified by preparative TLC (10% cyclohexane/EtOAc) to give 4 (75 mg, 65%) as a white solid. ESI-MS, m/z (%): 327.0 [M+Na+] (100). 1H NMR (DMSO-d6): δ 11.88 (brs, 1H, NH); 6.65 (dd, J = 17.4, J = 11.0 Hz, 1H, H-1′); 5.49 (d, J = 17.3 Hz, 1H, H-2′a); 5.28 (d, J = 11.3 Hz, 1H, H-2′b); 1.48 (s, 9H, CH3). 13C NMR (DMSO-d6): δ 164.32 (C-2); 152.65 (OCON); 126.53 (C-1′); 124.24 (C-5); 121.14 (C-4); 115.87 (C-2′) 82.12 (C-CH3); 27.80 (CH3). HR-MS (ESI+): m/z [M + Na]+ calculated for: C10H13O2N2BrNaS, 326.9773, found: 326.9776.
Bromo-5-(ethanol-2-yl)-2-(tert-butoxycarbonylamino)thiazole (5).
A solution of 2 (0.50 g, 1.8 mmol) in dry THF (10 mL) was added dropwise to a freshly prepared THF solution of LDA (5.9 mmol) under Ar at 0 °C. The resulting solution was stirred at 0 °C for further 15 min and 3.0 M THF solution of oxirane (1.97 mL, 5.9 mmol) was added dropwise. The temperature was kept at 0 °C for further 30 min and then the reaction mixture was allowed to warm up to 10 °C and stirred for additional 2 h. The resulting slurry was diluted with water (10 mL) and volatiles were evaporated in vacuo. The residue was purified by flash chromatography (0.5–3% MeOH/toluene, checked at λmax = 270 nm) to give 5 (0.31 g, 53%) as a white solid. ESI-MS, m/z (%): 345.0 [M+Na+] (100). 1H NMR (DMSO-d6): δ 11.55 (brs, 1H, NH); 4.93 (m, 1H, OH); 3.56 (m, 2H, O-CH2); 2.77 (t, J (CH2,CH2) = 6.3 Hz, 2H, 5-CH2); 1.48 (s, 9H, CH3). 13C NMR (DMSO-d6): δ 158.55 (C-2); 152.70 (OCON); 122.99 (C-5); 118.70 (C-4); 81.53 (C-CH3); 60.25 (O-CH2); 30.57 (5-CH2); 27.84 (CH3). HR-MS (ESI+): m/z [M + Na]+ calculated for: C10H15O3N2BrNaS, 344.9879, found: 344.9882.
4-Bromo-2-(tert-butoxycarbonylamino)-5-{2-[(diisopropoxyphosphoryl)methoxy]ethyl}-thiazole (6).
A solution of 2 (1.0 g, 3.6 mmol) in dry THF (20 mL) was added dropwise to a freshly prepared THF solution of LDA (11.9 mmol) under AR at −20 °C and after 15 min, THF solution of oxirane (3 M, 4 mL, 11.9 mmol) was added dropwise at −20 °C. The reaction mixture was allowed to warm up to 10 °C (1 h), when the brown solution changed into a brownish slurry. The reaction mixture was cooled down to −40 °C and a solution of diisopropylphosphonomethyl triflate (3.9 g, 11.9 mmol) in THF (30 mL) was added dropwise. The reaction mixture was stirred at −40 °C for 2 h. Then, 50 % sat. NH4Cl/H2O solution (40 mL) was added and the mixture was washed with EtOAc (150 mL). The organic layer was washed by water (100 mL) and brine (100 mL) and dried (Na2SO4). The solvent was evaporated and the crude product was purified by flash chromatography (linear gradient from cyclohexane to 10% MeOH/EtOAc) to give 6 (1.15 g, 64%) as a white solid. ESI-MS, m/z (%): 523.0 [M+Na+] (100). 1H NMR (DMSO-d6): δ 11.58 (s, 1H, NH); 4.70–4.48 (m, 2H, CH-iPr); 3.76 (d, J(CH2,P) = 8.3 Hz, 2H, P-CH2); 3.68 (t, J(CH2,CH2) = 5.9 Hz, 2H, O-CH2); 2.88 (t, J(CH2,CH2) = 6.1 Hz, 2H, 5-CH2); 1.47 (s, 9H, CH3); 1.24–1.19 (m, 12H, 2 x CH3). 13C NMR (DMSO-d6): δ 158.75 (C-2); 152.70 (OCON); 122.44 (C-5); 119.12 (C-4); 81.58 (C-CH3); 71.41 (d, J = 11.8 Hz, OCH2); 70.24 (d, J = 6.4 Hz, CH-iPr); 64.81 (d, J = 164.1 Hz, P-CH2); 27.83 (C-CH3); 27.36 (C-1′); 23.82, 23.79, 23.71, 23.67 (4 x CH3). HR-MS (ESI+): m/z [M + Na]+ calculated for: C17H30O6N2BrNaPS, 523.0638, found: 523.0643.
2-Amino-5-{2-[(diisopropoxyphosphoryl)methoxy]ethyl}-4-phenylthiazole (8a).
Treatment of 6 (0.40 g, 0.8 mmol) by Method A afforded 8a (0.24 g, 74%) as a yellowish oil. ESI-MS, m/z (%): 399 [M+H+] (100). 1H NMR (DMSO-d6): δ 7.58–7.46 (m, 2H, H-2′); 7.35–7.40 (m, 2H, H-3′); 7.32–7.23 (m, 1H, H-4′); 6.84 (bs, 2H, NH2); 4.57 (m, 2H, CH-iPr); 3.74 (d, J(CH2, P) = 8.1 Hz, 2H, P-CH2); 3.69 (t, J(CH2,CH2) = 6.5 Hz, 2H, O-CH2); 2.92 (t, J(CH2,CH2) = 6.5 Hz, 2H, 5-CH2); 1.22 and 1.21 2 x (d, J(CH3,CH) = 7.7 Hz, 6H, CH3). 13C NMR (DMSO-d6): δ 165.46 (C-2); 146.23 (C-4); 135.40 (C-1′); 128.23 and 128.07 (C-2′, 3′); 127.02 (C-4′); 116.82 (C-5); 72.98 d, J(C-O-C-P) = 11.5 Hz, (O-CH2); 71.15 (d, J(C-O-P) = 6.4 Hz, CH-iPr); 64.81 (d, J(C-P) = 163.9 Hz, CH2-P); 27.16 (5-CH2); 23.84, 23.82, 23.80 and 23.71 (CH3-iPr). HR-MS (ESI+): m/z [M + H]+ calculated for: C18H28N2O4PS, 399.1502, found: 399.1502.
2-Amino-5-{2-[(diisopropoxyphosphoryl)methoxy]ethyl}-4-(3-butoxyphenyl)thiazole (8b).
Treatment of 6 (0.40 g, 0.8 mmol) by Method A afforded 8b (0.19 g, 51%) as a yellowish oil. ESI-MS, m/z (%): 471 [M+H+] (100).1H NMR (DMSO-d6): δ 7.24–7.29 (m, 1H, H-5’); 7.12–7.00 (m, 2H, H-2′, 6′); 6.84–6.86 (m, 3H, C-4′, NH2); 4.51–4.63 (m, 2H, CH-iPr); 3.97 (t, J(CH2,CH2) = 6.5 Hz, 2H, 3′-O-CH2); 3.74 (d, J(CH2,P) = 8.1 Hz, 2H, P-CH2); 3.69 (t, J(CH2,CH2) = 6.5 Hz, 2H, O-CH2); 2.94 (t, J(CH2,CH2) = 6.5 Hz, 2H, 5-CH2); 1.79–1.63 (m, 2H, -O-CH2-CH2); 1.58–1.35 (m, 2H, CH2-CH3); 1.23 (d, J(CH3,CH) = 8.0 Hz, 6H, CH3); 1.21 (d, J(CH3,CH) = 8.0 Hz, 6H, CH3); 0.94 (t, J(CH3,CH2) = 7.4 Hz, 3H, CH2CH3). 13C NMR (DMSO-d6): δ 165.38 (C-2); 158.48 (C-3′); 146.15 (C-4); 136.78 (C-1′); 128.97 (C-5′); 120.42 (C-6′); 117.00 (C-5); 114.01 (C-2′); 113.48 (C-4′); 72.96 d, J(C-O-C-P) = 11.3 Hz, (O-CH2); 71.16 (d, J(C-O-P) = 6.3 Hz, CH-iPr); 67.04 ( 3′-O-CH2); 64.85 d, J(C-P) = 164.1 Hz, (CH2-P); 30.80 (3″-O-CH2-CH2); 27.19 (5-CH2); 23.84, 23.81, 23.71 and 23.66 (CH3-iPr); 18.77 (CH2-CH3); 13.72 (CH2-CH3). HR-MS (ESI+): m/z [M + H]+ calculated for: C22H36N2O5PS, 471.2077, found: 471.2075.
2-Amino-5-{2-[(diisopropoxyphosphoryl)methoxy]ethyl}-4-[3-(benzyloxy)phenyl]thiazole (8c).
Treatment of 6 (0.40 g, 0.8 mmol) by Method A afforded 8c (0.18 g, 55%) as a colourless oil. ESI-MS, m/z (%):505 [M+H+] (100). 1H NMR (DMSO-d6): δ 7.45 (m, 2H, H-2″); 7.38 (m, 2H, H-3″); 7.07–7.33 (m, 5H, H-2′, 4′, 5′, 6′, 4″); 6.84 (bs, 2H, NH2); 5.13 (s, 2H, 1″-CH2); 4.63–4.51 (m, 2H, CH-iPr); 3.74 (d, J(CH2,P) = 8.1 Hz, 2H, P-CH2); 3.67 (t, J(CH2,CH2) = 6.4 Hz, 2H, O-CH2); 2.90 (t, J(CH2,CH2) = 6.5 Hz, 2H, 5-CH2); 1.23 (d, J(CH3,CH) = 8.3 Hz, 6H, CH3); 1.21 (d, J(CH3,CH) = 8.3 Hz, 6H, CH3). 13C NMR (DMSO-d6): δ 165.45 (C-2); 158.12 (C-3′); 146.03 (C-4); 137.19 (C-1″); 136.80 (C-1′); 129.89 (C-5′); 128.46 (C-3″); 127.79 (C-4″); 127.62 (C-2″); 120.89 (C-6′); 117.23 (C-5); 114.43 (C-2′); 113.84 (C-4′); 72.98 d, J(C-O-C-P) = 11.5 Hz, (O-CH2); 70.24 (d, J(C-O-P) = 6.3 Hz, CH-iPr); 69.12 (3′-O-CH2); 64.88 d, J(C-P) = 164.0 Hz, (CH2-P); 27.19 (5-CH2); 23.86, 23.83, 23.73 and 23.68 (CH3-iPr). HR-MS (ESI+): m/z [M + H]+ calculated for: C25H34N2O5PS, 505.1921, found: 505.1919.
2-Amino-5-{2-[(diisopropoxyphosphoryl)methoxy]ethyl}-4-(4-isopropoxyphenyl)thiazole (8d).
Treatment of 6 (0.50 g, 1.0 mmol) by Method A afforded 8d (0.29 g, 63%) as a yellowish oil. ESI-MS, m/z (%): 457 [M+H+] (100). 1H NMR (DMSO-d6): δ 7.48–7.37 (m, 2H, H-2′); 7.02–6.76 (m, 2H, H-3′); 4.62–4.58 (m, 3H, 4′-O-CH, CH-iPr); 3.75 (d, J(CH2,P) = 8.1 Hz, 2H, P-CH2); 3.69 (t, J(CH2,CH2) = 6.4 Hz, 2H, O-CH2); 2.89 (t, J(CH2,CH2) = 6.4 Hz, 2H, 5-CH2); 1.26 (d, 6H, J(CH3,CH) = 6.0 Hz, 4′-O-CH-CH3); 1.23 (m, 6H, CH3); 1.21 (m, 6H, CH3). 13C NMR (DMSO-d6): δ 166.07 (C-2); 156.99 (C-4′); 146.26 (C-4); 129.73 (C-2′, C-6′); 127.91 (C-1′); 115.47 (C-5); 115.17 (C-3′, 5′); 72.59 d, J(C-O-C-P) = 11.6 Hz, (O-CH2); 70.15 (d, J(C-O-P) = 6.4 Hz, CH-iPr); 69.16 (4′-O-CH); 64.80 d, J(C-P) = 164.0 Hz, (CH2-P); 26.96 (5-CH2); 23.84, 23.80, 23.71 and 23.66 (CH3-iPr); 21.82 (4″-O-CH-CH3). HR-MS (ESI+): m/z [M + H]+ calculated for: C21H34N2O5PS, 457.1921, found: 457.1919.
2-Amino-5-{2-[(diisopropoxyphosphoryl)methoxy]ethyl}-4-(2-fluorophenyl)thiazole (8e).
Treatment of 6 (0.50 g, 1.0 mmol) by Method A afforded 8e (0.29 g, 70%) as a brownish oil. ESI-MS, m/z (%): 417 [M+H+] (100). 1H NMR (DMSO-d6): δ 7.35–7.47 (m, 2H, H-4″, 6″); 7.19–7.28 (m, 2H, H-3″, 5″); 6.86 (bs, 2H, NH2); 4.45–4.65 (m, 2H, CH-iPr); 3.70 (d, J(P,CH2) = 8.2 Hz, 2H, P-CH2); 3.60 (t, J(CH2,CH2) = 6.6 Hz, 2H, O-CH2); 2.71 (t, J(CH2,CH2) = 6.4 Hz, 2H, 5-CH2); 1.22 (d, J(CH3,CH) = 6.2 Hz, 6H, CH3); 1.20 (d, J(CH3,CH) = 6.2 Hz, 6H, CH3). 13C NMR (DMSO-d6): δ 166.00 (C-2); 159.08 (d, J(2″-F) = 245.8 Hz, C-2″); 140.61 (C-4); 131.80 (d, J(6″-F) = 3.4 Hz, C-6″); 129.65 (d, J(4″-F) = 8.2 Hz, C-4″); 124.15 (d, J(5″-F) = 3.3 Hz, C-5″); 123.22 (d, J(1″-F) = 15.0 Hz, C-1″); 118.75 (C-5); 115.72 (d, J(3″-F) = 22.4 Hz, C-3″); 72.67 d, J(C-O-C-P) = 11.5 Hz, (O-CH2); 70.15 J(C-O-P) = 6.4 Hz, (CH-iPr); 64.75 d, J(C-P) = 164.1 Hz, (CH2-P); 27.14 (5-CH2); 23.84, 23.80, 23.70 and 23.65 (CH3-iPr). HR-MS (ESI+): m/z [M + H]+ calculated for: C18H27N2O4FPS, 417.1408, found: 417.1408.
2-Amino-5-{2-[(diisopropoxyphosphoryl)methoxy]ethyl}-4-[3-(fluoro)phenyl]thiazole (8f).
Treatment of 6 (0.50 g, 1.0 mmol) by Method A afforded 8f (0.27 g, 65%) as a brownish oil. ESI-MS, m/z (%): 417 [M+H+] (80). 1H NMR (DMSO-d6): δ 7.39–7.47 (m, 2H, H-5′, 6′); 7.29–7.36 (m, 1H, H-2′); 7.09–7.17 (m, 1H, H-4′); 6.89 (bs, 2H, NH2); 4.57 (sept, J(CH, CH3) = 6.2 Hz, 2H, CH-iPr); 3.75 (d, J(P,CH2) = 8.2 Hz, 2H, P-CH2); 3.70 (t, J(CH2,CH2) = 6.4 Hz, 2H, O-CH2); 2.95 (t, J(CH2,CH2) = 6.4 Hz, 2H, 5-CH2); 1.22 (d, J(CH3,CH) = 6.2 Hz, 6H, CH3); 1.20 (d, J(CH3,CH) = 6.2 Hz, 6H, CH3). 13C NMR (DMSO-d6): δ 166.02 (C-2); 162.46 (d, J(2′,F) = 242.7 Hz, C-2′); 145.35 (d, J(4,F) = 2.4 Hz, C-4); 138.25 (d, J(1′,F) = 8.2 Hz, C-1′); 130.48 (d, J(5′,F) = 8.5 Hz, C-5′); 124.69 (d, J(6′,F) = 2.5 Hz, C-6′); 118.66 (C-5); 115.19 (d, J(2′,F) = 22.1 Hz, C-2′); 114.23 (d, J(4′,F) = 20.9 Hz, C-4′); 73.35 d, J(C-O-C-P) = 11.6 Hz, (O-CH2); 70.63 J(C-O-P) = 6.3 Hz, (CH-iPr); 65.32 d, J(C-P) = 164.0 Hz, (CH2-P); 27.59 (5-CH2); 24.29, 24.25, 24.15 and 24.11 (CH3-iPr). HR-MS (ESI+): m/z [M + H]+ calculated for: C18H27N2O4FPS, 417.1408, found: 417.1408.
2-Amino-5-{2-[(diisopropoxyphosphoryl)methoxy]ethyl}-4-[3-(N-methylsulfamoyl)phenyl]thiazole (8g).
Treatment of 6 (0.50 g, 1.0 mmol) by Method A afforded 8g (0.38 g, 77%) as a brownish oil. ESI-MS, m/z (%): 492 [M+H+] (100). 1H NMR (DMSO-d6): δ 9.78 (bs, 1H, SO2NH); 7.46–6.93 (m, 6H, H-2′, 4′ 5′, 6′, NH2); 4.57 (sept., J(CH,CH3) = 6.2 Hz, 2H, CH-iPr); 3.75 (d, J(P,CH2) = 8.2 Hz, 2H, P-CH2); 3.69 (d, J(CH2,CH2) = 6.4 Hz, 2H, O-CH2); 2.97 (s, 3H, CH3NH); 2.92 (t, J(CH2,CH2) = 6.4 Hz, 2H, 5-CH2); 1.23 (d, J(CH3,CH) = 6.2 Hz, 6H, CH3); 1.21 (d, J(CH3,CH) = 6.2 Hz, 6H, CH3). 13C NMR (DMSO-d6): δ 165.82 (C-2); 144.68 (C-4); 138.34 and 138.39 (C-3′, C-1′); 129.30 (C-5′); 123.83 (C-6′); 119.76 (C-2′); 118.60 (C-4′); 117.30 (C-5); 72.76 d, J(C-O-C-P) = 11.5 Hz, (O-CH2); 70.21 (d, J(C-O-P) = 6.4 Hz, CH-iPr); 64.79 d, J(C-P) = 164.1 Hz, (CH2-P); 39.31 (NH-CH3); 27.06 (5-CH2); 23.85, 23.82, 23.72 and 23.68 (CH3-iPr). HR-MS (ESI+): m/z [M + H]+ calculated for: C19H31N3O6PS2, 492.1386, found: 492.1386.
2-Amino-5-{2-[(diisopropoxyphosphoryl)methoxy]ethyl}-4-[4-(methylcarbamoyl)phenyl]thiazole (8h).
Treatment of 6 (0.50 g, 1.0 mmol) by Method A afforded 8h (0.22 g, 49%) as a brownish oil. ESI-MS, m/z (%): 456 [M+H+] (50); 478 [M+Na+]. 1H NMR (DMSO-d6): δ 8.43 (q, 1H, J(NH,CH3) = 4.2 Hz, NHCH3); 7.80–7.85 (m, 2H, H-2′, 6′); 7.58–7.63 (m, 2H, H-3′, 5′); 6.89 (bs, 2H, NH2); 4.57 (sept., J(CH,CH3) = 6.2 Hz, 2H, CH-iPr); 3.74 (d, J(P,CH2) = 8.1 Hz, 2H, P-CH2); 3.70 (t, J(CH2,CH2) = 6.4 Hz, 2H, O-CH2); 2.95 (t, J(CH2,CH2) = 6.4 Hz, 2H, 5-CH2); 2.79 (d, J(CH3,NH) = 4.5 Hz, 3H, NH-CH3); 1.22 (d, J(CH3,CH) = 6.2 Hz, 6H, CH3); 1.20 (d, J(CH3,CH) = 6.2 Hz, 6H, CH3). 13C NMR (DMSO-d6): δ 166.34 (CON); 165.58 (C-2); 145.40 (C-4); 137.87 (C-4′); 132.84 (C-1′); 127.95 (C-2′); 126.93 (C-3′); 118.25 (C-5); 72.88 d, J(C-O-C-P) = 11.7 Hz, (O-CH2); 70.17 J(C-O-P) = 6.4 Hz, (CH-iPr); 64.82 d, J(C-P) = 164.0 Hz, (CH2-P); 27.19 (5-CH2); 26.26 (NH-CH3); 23.85, 23.81, 23.71 and 23.67 (CH3-iPr). HR-MS (ESI+): m/z [M + H]+ calculated for: C20H31N3O5PS, 456.1717, found: 456.1717.
2-Amino-5-{2-[(diisopropoxyphosphoryl)methoxy]ethyl}-4-[4-(benzylcarbamoyl)phenyl]thiazole (8i).
Treatment of 6 (0.50 g, 1.0 mmol) by Method A afforded 8i (0.30 g, 56%) as a brownish oil. ESI-MS, m/z (%): 554 [M+Na+] (100). 1H NMR (DMSO-d6): δ 9.05 (t, J(NH,CH2) = 6.1 Hz, CO-NH); 7.87–7.94 (m, 2H, H-3′); 7.60–7.66 (m, 2H, H-2′); 7.29–7.36 (m, 4H, H-2″, 3″); 7.21–7.28 (m, 1H, H-4″); 6.88 (bs, 2H, NH2); 4.57 (sept., J(CH,CH3) = 6.2 Hz, 2H, CH-iPr); 4.49 (d, J(CH2,NH) = 6.0 Hz, 2H, NH-CH2); 3.74 (d, J(P,CH2) = 8.2 Hz, 2H, P-CH2); 3.70 (t, J(CH2,CH2) = 6.4 Hz, 2H, O-CH2); 2.96 (t, J(CH2,CH2) = 6.4 Hz, 2H, 5-CH2); ); 1.22 (d, J(CH3,CH) = 6.2 Hz, 6H, CH3); 1.20 (d, J(CH3,CH) = 6.2 Hz, 6H, CH3). 13C NMR (DMSO-d6): δ 165.92 (CO-NH); 165.58 (C-2); 145.48 (C-4); 139.75 (C-1″); 138.15 (C-1′); 132.63 (C-4′); 128.27 (C-3″); 127.98 (C-2′); 127.19 (C-3′); 127.15 (C-2″); 126.72 (C-4″); 118.35 (C-5); 72.88 d, J(C-O-C-P) = 11.4 Hz, (O-CH2); 70.17 J(C-O-P) = 6.3 Hz, (CH-iPr); 64.82 d, J(C-P) = 163.9 Hz, (CH2-P); 27.19 (5-CH2); 23.85, 23.81, 23.71 and 23.67 (CH3-iPr). HR-MS (ESI+): m/z [M + H]+ calculated for: C26H34N3O5PSNa, 554.1849, found: 554.1847.
Bis(l-phenylalanine isopropyl ester) prodrug of 2-amino-5-[2-(phosphorylmethoxy)ethyl]-4-(phenyl)thiazole (9a).
Treatment of 8a (0.14 g, 0.35 mmol) by Method B afforded 9a (0.13 g, 55%) as a colourless foam. ESI-MS, m/z (%): 693 [M+H+] (100). 1H NMR (DMSO-d6): δ 7.50 (m, 2H, H-2″); 7.37 (m, 2H, H-3″); 7.11–7.31 (m, 11H, H-2′, 3′, 4′, 4″); 6.85 (bs, 2H, NH2); 4.81 and 4.77 2 × (sept., J(CH,CH3) = 6.3 Hz, 1H, CH-iPr); 4.42 and 4.09 2 × (t, J(NH,CH) = J(NH,P) = 11.3 Hz, 1H, NH); 3.84–3.97 (m, 2H, NH-CH); 3.50 (t, J(CH2,CH2) = 7.1 Hz, 2H, O-CH2); 3.20–3.30 (m, 2H, P-CH2); 2.87 (t, J = 7.1 Hz, 2H, 5-CH2); 2.74–2.91 (m, 4H, CH2Ph); 1.15, 1.11, 1.06 and 1.01, 4 × (d, J(CH3,CH) = 6.3 Hz, 3H, CH3). 13C NMR (DMSO-d6): δ 172.50 (d J(C-C-N-P) = 2.9, COO); 172.34 (d J(C-C-N-P) = 5.1, COO); 165.59 (C-2); 146.44 (C-4); 137.19 and 137.34 (C-1′); 135.64 (C-1″); 129.71 (C-2′); 128.38 (C-3′); 128.31, 128.28 (C-2″, 3″); 127.25 (C-4″); 126.71 and 126.66 (C-4′); 116.54 (C-5); 73.04 d, J(C-O-C-P) = 11.0 Hz, (O-CH2); 68.17 and 68.04 (CH-iPr); 67.75 d, J(C-P) = 134.9 Hz, (CH2-P); 54.18 and 54.14 (NH-CH); 39.90 (CH2Ph); 27.36 (5-CH2); 21.72, 21.66, 21.60 and 21.54 (CH3-iPr); HR-MS (ESI+): m/z [M + H]+ calculated for: C36H46N4O6PS, 693.2870, found: 693.2879.
Bis(l-phenylalanine isopropyl ester) prodrug of 2-amino-5-[2-(phosphorylmethoxy)ethyl]-4-(3-butoxyphenyl)thiazole (9b).
Treatment of 8b (0.17 g, 0.35 mmol) by Method B afforded 9b (0.12 g, 43%) as a colourless solid. ESI-MS, m/z (%): 765 [M+H+] (100). 1H NMR (DMSO-d6): 7.11–7.27 (m, 11H, H-2′, 3′, 4′, 5″); 7.02–7.06 (m, 2H, H-2″, 6″); 6.84–6.86 (m, 3H, C-4″, NH2); 4.81 and 4.77 2 × (sept., J(CH,CH3) = 6.3 Hz, 1H, CH-iPr); 4.41 and 4.06 2 × (t, J(NH,CH) = J(NH,P) = 11.3 Hz, 1H, NH); 3.95 (t, 2H, J(CH2,CH2) = 6.5 Hz, 3″-O-CH2); 3.83–3.97 (m, 2H, NH-CH); 3.49 (t, J(CH2,CH2) = 7.2 Hz, 2H, O-CH2); 3.19–3.28 (m, 2H, P-CH2); 2.73–2.90 (m, 6H, 5-CH2, CH2Ph); 1.68 (m, 2H, 3″-O-CH2-CH2); 1.42 (m, 2H, CH2-CH3); 1.15, 1.11, 1.06 and 1.01, 4 × (d, J(CH3,CH) = 6.3 Hz, 3H, CH3). 13C NMR (DMSO-d6): δ 172.49 (d J(C-C-N-P) = 3.0, COO); 172.33 (d J(C-C-N-P) = 5.1, COO); 165.54 (C-2); 158.72 (C-3″); 146.24 (C-4); 137.34 and 137.20 (C-1′); 136.97 (C-1″); 129.72 (C-2′); 129.38 (C-5″); 128.30 and 128.27 (C-3′); 126.70 and 126.65 (C-4′); 120.54 (C-6″); 116.73 (C-5); 114.18 (C-2″); 113.67 (C-4″); 72.98 d, J(C-O-C-P) = 10.7 Hz, (O-CH2); 68.17 and 68.04 (CH-iPr); 67.71 d, J(C-P) = 134.7 Hz, (CH2-P); 67.25 ( 3″-O-CH2); 54.18 and 54.14 (NH-CH); 39.90 (CH2Ph); 31.02 (3″-O-CH2-CH2); 27.40 (5-CH2); 21.72, 21.66, 21.60 and 21.54 (CH3-iPr); 19.01 (CH2-CH3); 13.97 (CH2-CH3). HR-MS (ESI+): m/z [M + H]+ calculated for: C40H54N4O7PS, 765.3445, found: 765.3435.
Bis(l-phenylalanine isopropyl ester) prodrug of 2-amino-5-[2-(phosphorylmethoxy)ethyl]-4-[3-(benzyloxy)phenyl]thiazole (9c).
Treatment of 8c (0.18 g, 0.35 mmol) by Method B afforded 9c (0.12 g, 43%) as an amorphous yellowish solid. ESI-MS, m/z (%): 799 [M+H+] (100). 1H NMR (DMSO-d6): δ 7.45 (m, 2H, H-2‴); 7.38 (m, 2H, H-3‴); 7.07–7.33 (m, 14H, H-2′, 3′, 4′, 2″, 5″, 6″, 4‴); 6.85 (bs, 2H, NH2); 5.11 (s, 2H, 1‴-CH2); 4.80 and 4.76 2 × (sept., J(CH,CH3) = 6.3 Hz, 1H, CH-iPr); 4.41 and 4.07 2 × (t, J(NH,CH) = J(NH,P) = 11.3 Hz, 1H, NH); 3.83–3.97 (m, 2H, NH-CH); 3.48 (t, J(CH2,CH2) = 7.1 Hz, 2H, O-CH2); 3.20–3.28 (m, 2H, P-CH2); 2.73–2.90 (m, 6H, 5-CH2, CH2Ph); 1.15, 1.10, 1.06 and 1.01, 4 × (d, J(CH3,CH) = 6.3 Hz, 3H, CH3). 13C NMR (DMSO-d6): δ 172.49 (d J(C-C-N-P) = 3.0, COO); 172.33 (d J(C-C-N-P) = 5.1, COO); 165.56 (C-2); 158.36 (C-3″); 146.15 (C-4); 137.34 and 137.33 (C-1′, 1‴); 137.19 (C-1′); 137.03 (C-1″); 129.71 (C-2′); 129.42 (C-5″); 128.66 (C-3‴) 128.30 and 128.27 (C-3′); 128.01 (C-4‴); 127.81 (C-2‴); 126.70 and 126.65 (C-4′); 120.96 (C-6″); 116.88 (C-5); 114.60 (C-2″); 113.96 (C-4″); 72.98 d, J(C-O-C-P) = 10.7 Hz, (O-CH2); 69.30 (1‴-CH2); 68.16 and 68.03 (CH-iPr); 67.76 d, J(C-P) = 134.9 Hz, (CH2-P); 54.17 and 54.13 (NH-CH); 39.90 (CH2Ph); 27.36 (5-CH2); 21.71, 21.65, 21.59 and 21.53 (CH3-iPr). HR-MS (ESI+): m/z [M + H]+ calculated for: C43H52N4O7PS, 799.3289, found: 799.3277.
Bis(l-phenylalanine isopropyl ester) prodrug of 2-amino-5-[[2-(phosphorylmethoxy)ethyl]-4-(4-isopropoxyphenyl)thiazole (9d).
Treatment of 8d (0.16 g, 0.35 mmol) by Method B afforded 9d (0.12 g, 46%) as an amorphous white solid. ESI-MS, m/z (%): 751 [M+H+] (100). 1H NMR (DMSO-d6): δ 7.41 (m, 2H, H-2″); 7.11–7.26 (m, 10H, H-2′, 3′, 4′); 6.89 (m, 2H, H-3″); 6.80 (bs, 2H, NH2); 4.81 and 4.77 2 × (sept., J(CH,CH3) = 6.3 Hz, 1H, CH-iPr); 4.58 (sept., 2H, J(CH3,CH) = 6.0 Hz, 4″-O-CH); 4.43 and 4.09 2 × (t, J(NH,CH) = J(NH,P) = 11.3 Hz, 1H, NH); 3.84–3.97 (m, 2H, NH-CH); 3.49 (m, 2H, O-CH2); 3.21–3.30 (m, 2H, P-CH2); 2.75–2.91 (m, 6H, 5-CH2, CH2Ph); 1.26 (d, 6H, J(CH3,CH) = 6.0 Hz, 4″-O-CH-CH3); 1.15, 1.11, 1.06 and 1.01, 4 × (d, J(CH3,CH) = 6.3 Hz, 3H, CH3). 13C NMR (DMSO-d6): δ 172.49 (d J(C-C-N-P) = 3.0, COO); 172.32 (d J(C-C-N-P) = 5.1, COO); 165.33 (C-2); 156.66 (C-4″); 146.26 (C-4); 137.34 and 137.18 (C-1′); 129.71 (C-2′); 129.62 (C-2″); 128.30 and 128.27 (C-3′); 127.91 (C-1″); 126.69 and 126.65 (C-4′); 115.25 (C-3″); 114.98 (C-5); 73.10 d, J(C-O-C-P) = 10.9 Hz, (O-CH2); 69.24 (4″-O-CH); 68.16 and 68.03 (CH-iPr); 67.76 d, J(C-P) = 134.9 Hz, (CH2-P); 54.17 and 54.15 (NH-CH); 39.90 (CH2Ph); 27.39 (5-CH2); 22.08 (4″-O-CH-CH3); 21.71, 21.59, 21.65 and 21.53 (CH3-iPr). HR-MS (ESI+): m/z [M + H]+ calculated for: C39H52N4O7PS, 751.3289, found: 751.3280.
Bis(l-phenylalanine isopropyl ester) prodrug of 2-amino-5-[2-(phosphorylmethoxy)ethyl]-4-(2-fluorophenyl)thiazole (9e).
Treatment of 8e (0.15 g, 0.35 mmol) by Method B afforded 9e (0.12 g, 47%) as an amorphous white solid. ESI-MS, m/z (%): 711 [M+H+] (100). 1H NMR (DMSO-d6): δ 7.37–7.44 (m, 2H, H-4″, 6″); 7.10–7.26 (m, 12H, H-2′, 3′, 4′, 3″, 5″); 6.87 (bs, 2H, NH2); 4.80 and 4.76 2 × (sept., J(CH,CH3) = 6.3 Hz, 1H, CH-iPr); 4.39 and 4.06 2x (t, J(NH,CH) = J(NH,P) = 11.3 Hz, 1H, NH); 3.82–3.95 (m, 2H, NH-CH); 3.40 (t, J(CH2,CH2) = 7.2 Hz, 2H, O-CH2); 3.16–3.24 (m, 2H, P-CH2); 2.73–2.90 (m, 4H, CH2Ph); 2.63 (t, J(CH2,CH2) = 7.1 Hz, 2H, 5-CH2); 1.15, 1.10, 1.06 and 1.01, 4 × (d, J(CH3,CH) = 6.3 Hz, 3H, CH3). 13C NMR (DMSO-d6): δ 172.47 (d J(C-C-N-P) = 3.1, COO); 172.34 (d J(C-C-N-P) = 5.2, COO); 166.13 (C-2); 159.25 (d, J(2″,F) = 245.6 Hz, C-2″); 140.78 (C-4); 137.33 and 137.20 (C-1′); 131.98 (d, J(6″,F) = 3.2 Hz, C-6″); 129.91 (d, J(4″,F) = 8.2 Hz, C-4″); 129.71 (C-2′); 128.29 and 128.25 (C-3′); 126.69 and 126.65 (C-4′); 124.45 (d, J(5″,F) = 3.2 Hz, C-5″); 123.40 (d, J(1″,F) = 15.1 Hz, C-1″); 118.47 (C-5); 115.96 (d, J(3″,F) = 22.4 Hz, C-3″); 72.76 d, J(C-O-C-P) = 11.2 Hz, (O-CH2); 68.16 and 68.02 (CH-iPr); 67.66 d, J(C-P) = 135.1 Hz, (CH2-P); 54.17 and 54.12 (NH-CH); 39.90 (CH2Ph); 27.28 (d, J = 3.1 Hz, 5-CH2); 21.71, 21.65, 21.59 and 21.53 (CH3-iPr). HR-MS (ESI+): m/z [M + H]+ calculated for: C36H45N4O6FPS, 711.2776, found: 711.2768.
Bis(l-phenylalanine isopropyl ester) prodrug of 2-amino-5-[2-(phosphorylmethoxy)ethyl]-4-(3-fluorophenyl)thiazole (9f).
Treatment of 8f (0.15 g, 0.35 mmol) by Method B afforded 9f (0.09 g, 35%) as an amorphous solid. ESI-MS, m/z (%): 733 [M+Na+] (100). 1H NMR (DMSO-d6): δ 7.41 (m, 1H, H-5″); 7.36 (dt, J(6″,5″) = 7.8 Hz, J(6″,2″) = 1.3 Hz, 1H, H-6″); 7.29 (ddd, J(2″,F) = 10.6 Hz, J(2″,4″) = 2.7 Hz, J(2″,6″) = 1.5 Hz, 1H, H-2″); 7.10–7.26 (m, 11H, H-2′, 3′, 4′, 4″); 6.91 (bs, 2H, NH2); 4.80 and 4.77 2 × (sept., J(CH,CH3) = 6.3 Hz, 1H, CH-iPr); 4.41 and 4.08 2x (t, J(NH,CH) = J(NH,P) = 11.3 Hz, 1H, NH); 3.84–3.97 (m, 2H, NH-CH); 3.51 (t, J(CH2,CH2) = 7.0 Hz, 2H, O-CH2); 3.22–3.30 (m, 2H, P-CH2); 2.74–2.90 (m, 6H, CH2Ph, 5-CH2); 1.15, 1.10, 1.06 and 1.01, 4 × (d, J(CH3,CH) = 6.3 Hz, 3H, CH3). 13C NMR (DMSO-d6): δ 172.49 (d J(C-C-N-P) = 2.9, COO); 172.33 (d J(C-C-N-P) = 5.1, COO); 165.71 (C-2); 162.24 (d, J(3″,F) = 242.7 Hz, C-3″); 144.95 (d, J(4,F) = 2.5 Hz, C-4); 137.95 (d, J(1″,F) = 8.0 Hz, C-1″); 137.33 and 137.19 (C-1′); 130.34 (d, J(5″-F) = 8.5 Hz, C-5″); 129.71 and 129.70 (C-2′); 128.30 and 128.26 (C-3′); 126.69 and 126.65 (C-4′); 124.36 (d, J(6″,F) = 2.6 Hz, C-6″); 117.90 (C-5); 114.91 (d, J(2″,F) = 22.1 Hz, C-2″); 114.03 (d, J(4″,F) = 20.9 Hz, C-4″); 72.86 d, J(C-O-C-P) = 10.8 Hz, (O-CH2); 68.16 and 68.03 (CH-iPr); 67.76 d, J(C-P) = 134.6 Hz, (CH2-P); 54.17 and 54.13 (NH-CH); 39.90 (CH2Ph); 27.33 (5-CH2); 21.71, 21.65, 21.59 and 21.53 (CH3-iPr). HR-MS (ESI+): m/z [M + H]+ calculated for: C36H45N4O6FPS, 711.2776, found: 711.2767.
Bis(l-phenylalanine isopropyl ester) prodrug of 2-amino-5-[2-(phosphorylmethoxy)ethyl]-4-[3-(N-methylsulfamoyl)phenyl]thiazole (9g).
Treatment of 8g (0.17 g, 0.35 mmol) by Method B afforded 9g (0.11 g, 40%) as a yellowish amorphous solid. ESI-MS, m/z (%): 808 [M+Na+] (100). 1H NMR (DMSO-d6): δ 9.85 (bs, 1H, SO2NH); 7.43 (t, J(2″,4″) = J(2″, 6″) = 2.0 Hz, 2H, H-2″); 7.32 (t, J(5″,4″) = J(5″,6″) = 7.9 Hz, 2H, H-5″); 7.10–7.26 (m, 12H, H-2′, 3′, 4′, 4″,6″); 6.88 (bs, 2H, NH2); 4.80 and 4.77 2 × (sept., J(CH,CH3) = 6.3 Hz, 1H, CH-iPr); 4.45 (t, J(NH,CH) = J(NH,P) = 11.4 Hz, 1H, NH); 4.12 (dd, J(NH,CH) = J(NH,P) = 12.2 Hz, 1H, NH); 3.83–3.99 (m, 2H, NH-CH); 3.51 (t, J(CH2,CH2) = 7.1 Hz, 2H, O-CH2); 3.21–3.32 (m, 2H, P-CH2); 2.74–2.91 (m, 6H, 5-CH2, CH2Ph); 1.15, 1.10, 1.06 and 1.01, 4 × (d, J(CH3,CH) = 6.3 Hz, 3H, CH3). 13C NMR (DMSO-d6): δ 172.49 (d J(C-C-N-P) = 3.0, COO); 172.31 (d J(C-C-N-P) = 5.0, COO); 165.64 (C-2); 145.98 (C-4); 138.58 (C-1″) and 131.61 (C-3″); 137.33 and 137.16 (C-1′); 129.72 and 129.70 (C-2′); 129.26 (C-5″); 128.31 and 128.27 (C-3′); 126.71 and 126.66 (C-4′); 123.83 (C-6″); 119.68 (C-2″); 118.46 (C-4″); 117.05 (C-5); 72.97 d, J(C-O-C-P) = 10.6 Hz, (O-CH2); 68.18 and 68.05 (CH-iPr); 67.72 d, J(C-P) = 134.9 Hz, (CH2-P); 54.14 (NH-CH); 39.90 (CH2Ph); 39.30 (NH-CH3); 27.38 (5-CH2); 21.71, 21.65, 21.59 and 21.53 (CH3-iPr). HR-MS (ESI+): m/z [M + H]+ calculated for: C37H49N5O8PS2, 786.2755, found: 786.2743.
Bis(l-phenylalanine isopropyl ester) prodrug of 2-amino-5-[2-(phosphorylmethoxy)ethyl]-4-[4-(methylcarbamoyl)phenyl]thiazole (9h).
Treatment of 8h (0.16 g, 0.35 mmol) by Method B afforded 9h (0.16 g, 61%) as an amorphous white solid. ESI-MS, m/z (%): 772 [M+Na+] (100). 1H NMR (DMSO-d6): δ 8.45 (q, J(NH,CH3) = 4.5 Hz, 1H, CONH); 7.85 (m, 2H, H-3″); 7.59 (m, 2H, H-2″); 7.10–7.26 (m, 10H, H-2′, 3′, 4′); 6.90 (bs, 2H, NH2); 4.81 and 4.77 2 × (sept., J(CH,CH3) = 6.3 Hz, 1H, CH-iPr); 4.42 (t, J(NH,CH) = J(NH,P) = 11.3 Hz, 1H, NH); 4.09 (dd, J(NH,CH) = 10.6 Hz, J(NH,P) = 12.0 Hz, 1H, NH); 3.83–3.97 (m, 2H, NH-CH); 3.51 (t, J(CH2,CH2) = 7.1 Hz, 2H, O-CH2); 3.21–3.30 (m, 2H, P-CH2); 2.74–2.91 (m, 6H, 5-CH2, CH2Ph); 2.78 (d, J(CH3-NH) = 4.6 Hz, 3H, NH-CH3); 1.15, 1.10, 1.06 and 1.01, 4 × (d, J(CH3,CH) = 6.3 Hz, 3H, CH3). 13C NMR (DMSO-d6): δ 172.50 (d J(C-C-N-P) = 2.8, COO); 172.34 (d J(C-C-N-P) = 5.1, COO); 166.54 (CON); 165.70 (C-2); 145.56 (C-4); 138.10 (C-1″) 137.33 and 137.19 (C-1′); 133.05 (C-4″); 129.71 (C-2′); 128.30 and 128.27 (C-3′); 128.10 (C-2″); 127.24 (C-3″); 126.70 and 126.65 (C-4′); 117.95 (C-5); 72.88 d, J(C-O-C-P) = 10.8 Hz, (O-CH2); 68.17 and 68.04 (CH-iPr); 67.75 d, J(C-P) = 134.9 Hz, (CH2-P); 54.17 and 54.12 (NH-CH); 39.90 (CH2Ph); 27.39 (5-CH2); 26.50 (NH-CH3); 21.71, 21.65, 21.59 and 21.53 (CH3-iPr). HR-MS (ESI+): m/z [M + H]+ calculated for: C38H49N5O7PS, 750.3085, found: 750.3092.
Bis(l-phenylalanine isopropyl ester) prodrug of 2-amino-5-[2-(phosphorylmethoxy)ethyl]-4-[4-(benzylcarbamoyl)phenyl]thiazole (9i).
Treatment of 8i (0.19 g, 0.35 mmol) by Method B afforded 9i (0.13 g, 44%) as an amorphous white solid. ESI-MS, m/z (%): 848 [M+Na+] (100). 1H NMR (DMSO-d6): δ 9.07 (t, J(NH,CH2) = 6.0 Hz, CO-NH); 7.92 (m, 2H, H-3″); 7.61 (m, 2H, H-2″); 7.30–7.34 (m, 4H, H-2‴,3‴); 7.10–7.26 (m, 11H, H-2′, 3′, 4′, 4‴); 6.92 (bs, 2H, NH2); 4.81 and 4.76 2 × (sept., J(CH,CH3) = 6.3 Hz, 1H, CH-iPr); 4.49 (d, J(CH2,NH) = 6.0 Hz, 2H, NH-CH2); 4.41 and 4.09 2x (t, J(NH,CH) = J(NH,P) = 11.3 Hz, 1H, NH); 3.83–3.98 (m, 2H, NH-CH); 3.51 (t, J(CH2,CH2) = 7.2 Hz, 2H, O-CH2); 3.22–3.30 (m, 2H, P-CH2); 2.74–2.92 (m, 6H, CH2Ph, 5-CH2); 1.14, 1.10, 1.06 and 1.01, 4 × (d, J(CH3,CH) = 6.3 Hz, 3H, CH3). 13C NMR (DMSO-d6): δ 172.50 (d J(C-C-N-P) = 2.9, COO); 172.34 (d J(C-C-N-P) = 5.1, COO); 166.11 (CO-NH); 165.73 (C-2); 145.57 (C-4); 139.96 (C-1‴); 138.32 (C-1″); 137.32 and 137.18 (C-1′); 132.86 (C-4″); 129.71 and 129.70 (C-2′); 128.49 (C-3‴); 128.30 and 128.26 (C-3′); 128.14 (C-2″); 127.46 (C-3″); 127.40 (C-2‴); 126.94 (C-4‴); 126.69 and 126.65 (C-4′); 118.04 (C-5); 72.88 d, J(C-O-C-P) = 10.7 Hz, (O-CH2); 68.17 and 68.04 (CH-iPr); 67.75 d, J(C-P) = 134.9 Hz, (CH2-P); 54.17 and 54.12 (NH-CH); 42.80 (NH-CH2); 39.90 (CH2Ph); 27.39 (5-CH2); 21.71, 21.65, 21.59 and 21.52 (CH3-iPr). HR-MS (ESI+): m/z [M + H]+ calculated for: C44H53N5O7PS, 826.3398, found: 826.3386.
{[2-(2-Amino-4-phenylthiazol-5-yl)ethoxy]methyl}phosphonic acid diphosphate (10a).
Treatment of 8a (0.10 g, 0.25 mmol) by Method C afforded 10a (0.015 g, 13%) as an amorphous white solid. ESI-MS, m/z (%): 473 [M-H] (100). 1H NMR (D2O): δ 7.43–7.52 (m, 5H, H-2′, 3′, 4′); 3.78–3.81 (m, 4H, CH2-O, P-CH2); 3.02 (t, J(CH2,CH2) = 6.2 Hz, 2H, 5-CH2). 13C NMR (D2O): δ 169.00 (C-2); 144.91 (C-4); 134.17 (C-1′); 129.40 (C-2′) and 129.37 (C-3′); 128.97 (C-4′); 120.34 (C-5); 73.44 d, J(C-O-C-P) = 10.4 Hz, (O-CH2); 67.24 d, J(C-P) = 163.0 Hz, (CH2-P); 27.15 (5-CH2). 31P NMR (121.4 MHz, D2O): 9.38 d J(P-O-P) = 26.2 Hz; −8.38 d J(P-O-P) = 19.9 Hz; −21.92 dd J(P-O-P) = 26.4 and 19.7 Hz. HR-MS (ESI−): m/z [M − H] calculated for: C12H16N2O10P3S: 472.9744, found: 472.9742.
{[2-(2-Amino-4-(3-butoxyphenyl)thiazol-5-yl)ethoxy]methyl}phosphonic acid diphosphate (10b).
Treatment of 8b (0.10 g, 0.21 mmol) by Method C afforded 10b (11 mg, 9%) as an amorphous white solid. ESI-MS, m/z (%): 545 [M-H] (100). 1H NMR (D2O): δ 7.42 (t, 2H, J(5′,4′) = J(5′,6′) = 7.9 Hz, H-5′); 7.12 (m, 1H, H-6′); 7.01–7.06 (m, 2H, H-2′, 4′); 4.10 (t, J(CH2,CH2) = 8.7 Hz, 2H, CH3(CH2)2CH2-); 3.77–3.81 (m, 4H, P-CH2, O-CH2); 3.01 (t, J(CH2,CH2) = 6.1 Hz, 2H, 5-CH2); 1.75 (m, 2H, CH3CH2CH2-); 1.46 (m, 2H, CH3CH2-); 0.94 (t, J(CH2,CH2) = 7.4 Hz, 2H, CH3). 13C NMR (D2O): δ 169.03 (C-2); 158.90 (C-3′); 143.78 (C-4); 135.33 (C-1′); 130.68 (C-5′); 122.33 (C-6′); 120.52 (C-5); 115.58 and 115.60 (C-2′ and C-4′); 73.31 d, J(C-O-C-P) = 10.6 Hz, (O-CH2); 67.23 d, J(C-P) = 163.4 Hz, (CH2-P); 63.90 (CH3CH2CH2CH2-); 31.19 (CH3CH2CH2-); 27.21 (5-CH2); 19.30 (CH3CH2-); 13.78 (CH3). 31P NMR (D2O) 9.41 d J(P-O-P) = 26.5 Hz; −8.88 m; −22.04 m. HR-MS (ESI−): m/z [M − H] calculated for: C16H24N2O11P3S: 545.0319, found: 545.0318.
{[2-(2-Amino-4-(3-(benzyloxy)phenyl)thiazol-5-yl)ethoxy]methyl}phosphonic acid diphosphate (10c).
Treatment of 8c (0.10 g, 0.20 mmol) by Method C afforded 10c (12 mg, 9%) as an amorphous white solid. ESI-MS, m/z (%): 579 [M-H] (100). 1H NMR (D2O): δ 7.37–7.45 (m, 6H, H-5′, 2″, 3″, 4″); 7.06–7.09 (m, 2H, H-4′, 6′); 6.94 (m, 1H, H-2′); 5.11 (s, 2H, 1″-CH2); 3.76 (d, 2H, J(H-C-P) = 8.7 Hz, P-CH2 ); 3.63 (t, J(CH2,CH2) = 5.8 Hz, 2H, CH2-O); 2.80 (t, J(CH2,CH2) = 5.8 Hz, 2H, 5-CH2). 13C NMR (D2O): δ 169.31 (C-2); 158.42 (C-3′); 139.38 (C-4); 137.09 (C-1″); 132.94 (C-1′); 130.94 (C-5′); 129.56 (C-3″); 129.10 (C-4″); 128.58 (C-2″); 122.43 (C-6′); 119.81 (C-5); 116.68 (C-4′); 115.82 (C-2′); 72.70 d, J(C-O-C-P) = 11.6 Hz, (O-CH2); 70.92 (1″-CH2); 67.14 d, J(C-P) = 163.8 Hz, (CH2-P); 27.02 (5-CH2). 31P NMR (D2O): 9.34 d J(P-O-P) = 26.2 Hz; −9.67 d J(P-O-P) = 19.7 Hz; −22.28 dd J(P-O-P) = 22.6 and 19.4 Hz. HR-MS (ESI−): m/z [M − H] calculated for: C19H22N2O11P3S: 579.0163, found: 579.0161.
{[2-(2-Amino-4-(4-isopropoxyphenyl)thiazol-5-yl)ethoxy]methyl}phosphonic acid diphosphate (10d).
Treatment of 8d (0.107 g, 0.23 mmol) by Method C afforded 10d (13 mg, 9%) as an amorphous white solid. ESI-MS, m/z (%): 531 [M-H] (100). 1H NMR (D2O): δ 7.42 (m, 2H, H-2′); 7.05 (m, 2H, H-3′); 4.72 (sept, 1H, CH-iPr); 3.81 (d, J(C-H-P) = 8.5 Hz, 2H, P-CH2); 3.77 (t, J(CH2,CH2) = 6.1 Hz, 2H, O-CH2); 2.97 (t, J(CH2,CH2) = 6.1 Hz, 2H, 5-CH2); 1.34 (d, J(CH3,CH) = 6.1 Hz, 6H, CH3). 13C NMR (D2O): δ 168.84 (C-2); 157.37 (C-4′); 143.26 (C-4); 130.76 (C-2′); 126.68 (C-1′); 119.19 (C-5); 116.95 (C-3′); 73.27 d, J(C-O-C-P) = 10.5 Hz, (O-CH2); 72.31 (CH-iPr); 67.19 d, J(C,P) = 163.4 Hz, (CH2-P); 27.10 (5-CH2) 21.72 (CH3-iPr). 31P NMR (D2O): 9.41 d J(P-O-P) = 26.0 Hz; −8.3 d J(P-O-P) = 18.3 Hz; −21.82 m. HR-MS (ESI−): m/z [M − H] calculated for: C15H22N2O11P3S: 531.0163, found: 531.0160.
{[2-(2-Amino-4-(2-fluorophenyl)thiazol-5-yl)ethoxy]methyl}phosphonic acid diphosphate (10e).
Treatment of 8e (0.104 g, 0.25 mmol) by Method C afforded 10e (0.010 g, 7%) as amorphous solid. ESI-MS, m/z (%): 491 [M-H] (100). 1H NMR (D2O): δ 7.46 (m, 1H, H-4′); 7.43 (td, J(6′,5′) = J(6′,F) = 7.6 Hz, J(6′,4′) = 1.8 Hz, 1H, H-6′); 7.29 (td, J(5′,6′) = J(5′,4′) = 7.5 Hz, J(5′,3′) = 1.1 Hz, 1H, H-5′); 7.25 (ddd, J(3′,F) = 10.5 Hz, J(3′,4’) = 8.3 Hz, J(3′,5′) = 1.2 Hz, 1H, H-3′); 3.78 (d, J(H-C-P) = 8.5 Hz, 2H, P-CH2); 3.75 (t, J(CH2,CH2) = 6.3 Hz, 2H, O-CH2); 2.87 (t, J(CH2,CH2) = 6.3 Hz, 2H, 5-CH2). 13C NMR (D2O): δ 169.20 (C-2); 160.44 (d, J(2′,F) = 245.0 Hz, (C-2′); 140.45 (C-4); 132.38 (C-6′); 131.21 (d, J(4′,F) = 8.3 Hz, (C-4′); 125.12 (d, J(5′,F) = 3.4 Hz, (C-5′); 122.81 (C-5); 116.54 (d, J(3′,F) = 22.1 Hz, (C-3′); 73.31 d, J(C-O-C-P) = 10.2 Hz, (O-CH2); 67.36 d, J(C-P) = 162.6 Hz, (CH2-P); 27.13 (5-CH2). 31P NMR (D2O): 9.16 d J(P-O-P) = 25.8 Hz; −5.79 m; −21.18 m. HR-MS (ESI−): m/z [M − H] calculated for: C12H15N2O10FP3S: 490.9650, found: 490.9648.
{[2-(2-Amino-4-(3-fluorophenyl)thiazol-5-yl)ethoxy]methyl}phosphonic acid diphosphate (10f).
Treatment of 8f (91 mg, 0.22 mmol) by Method C afforded 10f (8 mg, 7%) as an amorphous white solid. ESI-MS, m/z (%): 491 [M-H] (100). 1H NMR (D2O): δ 7.47 (ddd, J(5′,4′) = 8.5 Hz, J(5′,6′) = 7.7 Hz, J(5′,F) = 6.1 Hz, 1H, H-5′); 7.32 (ddd, J(6′,5′) = 7.7 Hz, J(6′,2′) = 1.5 Hz, J(6′,4′) = 0.9 Hz, 1H, H-6′); 7.25 (ddd, J(2′,F) = 10.1 Hz, J(2′,4′) = 2.6 Hz, J(2′,6′) = 1.5 Hz, 1H, H-2′); 7.16 (m, 1H, H-4′); 3.78–3.81 (m, 4H, O-CH2, P-CH2 ); 3.03 (t, J(CH2,CH2) = 6.3 Hz, 2H, 5-CH2). 13C NMR (D2O): δ 168.88 (C-2); 163.08 (d, J(3′,F) = 243.7 Hz, (C-3′); 144.78 (C-4); 136.75 (d, J(1′,F) = 8.2 Hz, (C-1′); 130.93 (d, J(5′,F) = 8.7 Hz, (C-5′); 125.20 (d, J(6′,F) = 2.8 Hz, (C-6′); 121.24 (C-5); 115.98 (d, J(2′,F) = 22.4 Hz, (C-2′); 115.40 (d, J(4′,F) = 21.0 Hz, (C-4′); 73.37 d, J(C-O-C-P) = 10.3 Hz, (O-CH2); 67.24 d, J(C-P) = 162.9 Hz, (CH2-P); 27.10 (5-CH2). 31P NMR (D2O): 9.30 d J(P-O-P) = 26.1 Hz; −7.37 m; −21.62 m. HR-MS (ESI−): m/z [M − H] calculated for: C12H15N2O10FP3S: 490.9650, found: 490.9646.
{[2-(2-Amino-4-(3-(N-methylsulfamoyl)phenyl)thiazol-5-yl)ethoxy]methyl}phosphonic acid diphosphate (10g).
Treatment of 8g (0.10 g, 0.20 mmol) by Method C afforded 10g (10 mg, 8%) as an amorphous white solid. ESI-MS, m/z (%): 565 [M-H] (100). 1H NMR (D2O): δ 7.81 (m, 2H, H-3′); 7.62 (m, 2H, H-2′); 3.78–3.82 (m, 4H, O-CH2, P-CH2 ); 3.04 (t, J(CH2,CH2) = 6.2 Hz, 2H, 5-CH2); 2.94 (s, 3H, CH3). 13C NMR (D2O): δ 171.12 (CON); 169.06 (C-2); 144.74 (C-4); 138.00 (C-1′); 133.61 (C-4′); 129.59 (C-2′); 127.92 (C-3′); 121.81 (C-5); 73.41 d, J(C-O-C-P) = 10.4 Hz, (O-CH2); 67.21 d, J(C-P) = 163.0 Hz, (CH2-P); 27.23 (5-CH2); 27.09 (CH3). 31P NMR (D2O): 9.40 d J(P-O-P) = 26.3 Hz; −8.65 m; −21.99 m. HR-MS (ESI−): m/z [M − H] calculated for: C13H19N3O12P3S2: 565.9629, found: 565.9626.
{[2-(2-Amino-4-(4-(methylcarbamoyl)phenyl)thiazol-5-yl)ethoxy]methyl}phosphonic acid diphosphate (10h).
Treatment of 8h (0.100 g, 0.22 mmol) by Method C afforded 10h (5 mg, 4%) as an amorphous white solid. ESI-MS, m/z (%): 530 [M-H] (100). 1H NMR (D2O): δ 7.81 (m, 2H, H-3′); 7.62 (m, 2H, H-2′); 3.78–3.82 (m, 4H, O-CH2, P-CH2 ); 3.04 (t, J(CH2,CH2) = 6.2 Hz, 2H, 5-CH2); 2.94 (s, 3H, CH3). 13C NMR (D2O): δ 171.12 (CON); 169.06 (C-2); 144.74 (C-4); 138.00 (C-1′); 133.61 (C-4′); 129.59 (C-2′); 127.92 (C-3′); 121.81 (C-5); 73.41 d, J(C-O-C-P) = 10.4 Hz, (O-CH2); 67.21 d, J(C-P) = 163.0 Hz, (CH2-P); 27.23 (5-CH2); 27.09 (CH3). 31P NMR (D2O): 9.40 d J(P-O-P) = 26.3 Hz; −8.65 m; −21.99 m. HR-MS (ESI−): m/z [M − H] calculated for: C14H19N3O11P3S: 529.9959, found: 529.9955.
{[2-(2-Amino-4-(4-(benzylcarbamoyl)phenyl)thiazol-5-yl)ethoxy]methyl}phosphonic acid diphosphate (10i).
Treatment of 8i (0.103 g, 0.19 mmol) by Method C afforded 10i (12 mg, 9%) as an amorphous white solid. ESI-MS, m/z (%): 606 [M-H] (100). 1H NMR (D2O): δ 7.83 (m, 2H, H-3′); 7.60 (m, 2H, H-2′); 7.44–7.39 (m, 4H, H-2″,3″); 7.35 (m, 1H, H-4″); 4.59 (s, 2H, 1″-CH2); 3.80–3.82 (m, 4H, O-CH2, P-CH2 ); 3.04 (t, J(CH2,CH2) = 6.3 Hz, 2H, 5-CH2). 13C NMR (D2O): δ 171.02 (CON); 168.90 (C-2); 144.97 (C-4); 138.70 (C-1″); 138.28 (C-1′); 133.33 (C-4′); 129.47 and 129.45 (C-2′ and C-3″); 128.08 (C-4″); 127.97 and 127.86 (C-2″ and C-3′); 121.84 (C-5); 73.40 d, J(C-O-C-P) = 10.3 Hz, (O-CH2); 67.30 d, J(C-P) = 162.7 Hz, (CH2-P); 44.10 (1″-CH2); 27.23 (5-CH2). 31P NMR (D2O): 9.30 d J(P-O-P) = 26.1 Hz; −6.85 bd J(P-O-P) = 18.6 Hz; −21.40 m. HR-MS (ESI−): m/z [M − H] calculated for: C20H23N3O11P3S: 606.0272, found: 606.0268.
Inhibition of ACT – cell based assay
J774A.1 cells were seeded in a 96-well plate at 5×104 cells per well, and left to attach overnight. Prior to the experiment, cells were washed with HBSS (135 mM NaCl, 5.9 mM KCl, 1.5 mM CaCl2, 1.2 mM MgCl2, 25 mM glucose, 10 mM HEPES [pH 7.4]) and pre-incubated with the tested compounds at concentrations of 0.001–30 μM for 5 h. After that, cells were exposed to ACT (2 nM) from B.pertussis (Enzo Life Sciences, Palo Alto, CA; SA=115 μmol/min/mg) for 30 min. Finally, the cAMP content was determined by using the CatchPointcAMP immunoassay kit (Molecular Devices, Wokingham, UK). Briefly, the reaction was terminated by the addition of lysis buffer to the treated cells (50 μL per well), the cellular content was extracted by shaking the plate at 250 rpm for 10 min. The plate was then centrifuged to remove cell debris, the supernatant was transferred to the immunoassay plate, and immunoassays were carried out according to the manufacturer’s instructions. Fluorescence signal was acquired using an Infinite M1000 plate reader (Tecan Systems Inc., San Jose, CA, USA).
Effect on the viability of J774A.1 cells
J774A.1 cells were plated onto white 96-well assay plates at 5×104 cells per well and allowed to attach overnight. Cells were then washed with HBSS and treated with 10 μM compounds for 5 h. Cell viability was then assessed with the Cell Titer-Glo Luminescent Cell Viability assay (Promega, Madison, WI, USA) according to the manufacturer’s instructions. Measurement of luminescence signal was performed by use of a GENios micro plate reader (Tecan Systems). Data are expressed as a percentage of the viability of control (untreated) cells.
Assays with mACs
Materials: Forskolin (FSK) and 3-isobutyl-1-methylxanthine (IBMX) were purchased from Sigma–Aldrich (St. Louis, MO, USA). OptiMEM, DMEM, and antibiotic-antimycotic were purchased from Life Technologies (Grand Island, NY, USA). FCI and BCS were purchased from Hyclone (Logan, UT, USA). Geneticin was purchased from InvivoGen (San Diego, CA, USA). Methods: HEK293 cells with CRISPR-Cas9 knockout of endogenous AC3 and AC6 (HEKΔ3/6) stably expressing recombinant AC1, AC2, and AC5 were grown, cryopreserved, and assayed with the prepared compounds at 30 μM concentration as described previously.[35] Briefly, frozen cells were rapidly thawed in a 37 °C water bath, added to 9 ml of 37 °C OptiMEM, pelleted at 500 xg, resuspended in fresh OptiMEM to achieve a cell density of 1.75 × 105 cells/ml (AC1 and AC5) or 7 × 105 (AC2), and 10 μL cells were added to each well. Cells were allowed to recover for 1 hour at 37 °C, 5% CO2 prior to the addition of 5 μL of 4X concentrated test compound or vehicle control in OptiMEM for 30 minutes at 37 °C, 5% CO2. Accumulation of cAMP was stimulated with 4X forskolin (5 μM final) or vehicle control in OptiMEM and plate was incubated for 1 hour at ambient temperature, after which cAMP accumulation was terminated by addition of HTRF reagents (Cisbio, Bedford, MA) as previously described.[35] After 1 hour at ambient temperature, HTRF signal was measured using a Synergy Neo2 (Biotek, Winooski, VT), and cAMP was quantified as previously described, according to manufacturer directions.[35] Data were normalized to cAMP formation in the absence of forskolin as 0% activity, and in the presence of 5 μM forskolin in the absence of inhibitor to 100%. Data represent the mean of N = 3 experiments, each performed with duplicate wells.
Inhibition of ACT – cell free assay
AC enzymatic activity was evaluated by determining the conversion of [3H] ATP to [3H]cAMP. Each assay mixture contained 3 μM BSA, 20 mM HEPES (pH 7.4), 10 mM MnCl2, 1 mM EDTA, 1 μM CaCl2, 0.1 mM cold ATP, 20 μCi [2,8-3H]ATP (ARC, St. Louis, MO, USA; specific activity 20 Ci/mmol), 1.2 μM calmodulin and tested compound at a concentration of 0 – 100 μM. Inhibition of AC activity was evaluated towards three different commercially available bacterial adenylate cyclases: ACT (Sigma), specific activity 65 μmol/min/mg, ACT (Enzo), specific activity 115 μmol/min/mg and finally, EF-A (LBL), specific activity 830 μmol/min/mg, with the final enzyme concentration of 1.1 nM , 0.67 nM and 0.12 nM, respectively. The incubation was carried out for 30 min at 30 °C in a reaction volume of 50 μL. A 2 μL aliquot of the assay mixture was spotted onto a polyethyleneimine-cellulose TLC plate, and developed in 4M LiCl:1 M acetic acid (1:4). After developing the TLC plate, the spots containing ATP and cAMP were quantified using Radio-TLC scanner RITA (RAYTEST, Germany) equipped with the evaluation software GINA STAR TLC. Inhibition rates were calculated from the percentage of [3H]ATP to [3H]cAMP conversion. Ki values were calculated using the Graphpad Prism 5 software (San Diego, CA, USA). All assays were performed in duplicate with three independent experiments, and results are presented as mean values ± SD.
Acknowledgements
This research was funded by the Institute of Organic Chemistry and Biochemistry (RVO 61388963), the Ministry of Education, Youth and Sports (MŠMT in Czech) in the program INTER-EXCELLENCE (project LTAUSA18086), and the National Institutes of Health (MH101673).
Footnotes
The authors declare no competing financial interest.
References
- [1].Gancedo JM, Biol. Rev 2013, 88, 645–668. [DOI] [PubMed] [Google Scholar]
- [2].McKnight GS, Curr. Opin. Cell Biol 1991, 3, 213–217. [DOI] [PubMed] [Google Scholar]
- [3].Hanoune J, Defer N, Annu. Rev. Pharmacol. Toxicol 2001, 41, 145–174. [DOI] [PubMed] [Google Scholar]
- [4].Sunahara RK, Dessauer CW, Gilman AG, Annu. Rev. Pharmacol. Toxicol 1996, 36, 461–480. [DOI] [PubMed] [Google Scholar]
- [5].Ahuja N, Kumar P, Bhatnagar R, Crit. Rev. Microbiol 2004, 30, 187–196. [DOI] [PubMed] [Google Scholar]
- [6].Khannpnavar B, Mehta V, Qi C, Korkhov V, Curr. Opin. Struct. Biol 2020, 63, 34–41. [DOI] [PubMed] [Google Scholar]
- [7].McDonough K, Rodriguez A, Nat. Rev. Microbiol 2012, 10, 27–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Seifert R, Dove S, Trends Microbiol 2012, 20, 343–351. [DOI] [PubMed] [Google Scholar]
- [9].Ivarsson ME, Leroux J-C, Castagner B, Angew. Chem. Int. Ed 2012, 51, 4024–4045. [DOI] [PubMed] [Google Scholar]
- [10].Guo Q, Shen Y, Lee Y-S, Gibbs CS, Mrksich M, Tang W-J, EMBO J 2005, 24, 3190–3201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Shen Y, Zhukovskaya NL, Zimmer MI, Soelaiman S, Bergson P, Wang C-R, Gibbs CS, Tang W-J, Proc. Natl. Acad. Sci. USA 2004, 101, 3242–3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Shoshani I, Laux WHG, Périgaud C, Gosselin G, Johnson RA, J. Biol. Chem 1999, 274, 34742–34744. [DOI] [PubMed] [Google Scholar]
- [13].Česnek M, Jansa P, Šmídková M, Mertlíková-Kaiserová H, Dračínský M, Brust TF, Pávek P, Tretnar F, Watts VJ, Janeba Z, ChemMedChem 2015, 10, 1351–1364. [DOI] [PubMed] [Google Scholar]
- [14].Česnek M, Skácel J, Jansa P, Dračínský M, Šmídková M, Mertlíková-Kaiserová H, Soto-Velasquez MP, Watts VJ, Janeba Z, ChemMedChem 2018, 13, 1779–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Břehová P, Šmídková M, Skácel J, Dračínský M, Mertlíková-Kaiserová H, Soto-Velasquez MP, Watts VJ, Janeba Z, ChemMedChem 2016, 11, 2534–2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Frydrych J, Skácel J, Šmídková M, Mertlíková-Kaiserová H, Dračínský M, Gnanasekaran R, Lepšík M, Soto-Velasquez M, Watts VJ, Janeba Z, ChemMedChem 2018, 13, 199–206. [DOI] [PubMed] [Google Scholar]
- [17].Břehová P, Chaloupecká E, Česnek M, Skácel J, Dračínský M, Tloušťová E, Mertlíková-Kaiserová H, Soto-Velasquez MP, Watts VJ, Janeba Z, European J Med. Chem 2021, 222, 113581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Erion MD, van Poelje PD, Dang Q, Kasibhatla SR, Potter SC, Reddy MR, Reddy KR, Jiang T, Lipscomb WN, PNAS 2005, 102, 7970–7975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Dang Q, Kasibhatla SR, Reddy KR, Jiang T, Reddy MR, Potter SC, Fujitaki JM, van Poelje PD, Huang J, Lipscomb WN, Erion MD, J. Am. Chem. Soc 2007, 129, 15491–15502. [DOI] [PubMed] [Google Scholar]
- [20].Dang Q, Kasibhatla SR, Jiang T, Fan K, Liu Y, Taplin F, Schulz W, Cashion DK, Reddy KR, van Poelje PD, Fujitaki JM, Potter SC, Erion MD, J. Med. Chem 2008, 51, 4331–4339. [DOI] [PubMed] [Google Scholar]
- [21].Dang Q, Liu Y, Cashion DK, Kasibhatla SR, Jiang T, Taplin F, Jacintho JD, Li H, Sun Z, Fan Y, Da Re J, Tian F, Li W, Gibson T, Lemus R, van Poelje PD, Potter SC, Erion MD, J. Med. Chem 2011, 54, 153–165. [DOI] [PubMed] [Google Scholar]
- [22].Pradere U, Garnier-Amblard EC, Coats SJ, Amblard F, Schinazi RF, Chem. Rev 2014, 114, 9154–9218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Wiemer AJ, Wiemer DF, Top. Curr. Chem 2015, 360, 115–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Heidel KM, Dowd CS, Future Med. Chem 2019, 11, 1625–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Šmídková M, Dvořáková A, Tloušťová E, Česnek M, Janeba Z, Mertlíková-Kaiserová H, Antimicrob. Agents Chemother 2014, 58, 664–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Guianvarc’h D, Fourrey J-L, Maurisse R, Sun J-S, Benhida R, Org. Lett 2002, 4, 4209–4212. [DOI] [PubMed] [Google Scholar]
- [27].Schnürch M, Spina M, Khan AF, Mihovilovic MD, Stanetty P, Chem. Soc. Rev 2007, 36, 1046–1057. [DOI] [PubMed] [Google Scholar]
- [28].Stanetty P, Schnürch M, Mereiter K, Mihovilovic MD, J. Org. Chem 2005, 70, 567–574. [DOI] [PubMed] [Google Scholar]
- [29].Schnürch M, Khan AF, Mihovilovic MD, Stanetty P, Eur. J. Org. Chem 2009, 19, 3228–3236. [Google Scholar]
- [30].Jansa P, Baszczyňski O, Dračínský M, Votruba I, Zídek Z, Bahador G, Stepan G, Cihlar T, Mackman R, Holý A, Janeba Z, Eur. J. Med. Chem 2011, 46, 3748–3754. [DOI] [PubMed] [Google Scholar]
- [31].Holý A, Rosenberg I, Collect. Czech. Chem. Commun 1987, 52, 2801–2809. [Google Scholar]
- [32].Slusarczyk M, Ferrari V, Serpi M, Gönczy B, Balzarini J, McGuigan C, ChemMedChem 2018, 13, 2305–2316. [DOI] [PubMed] [Google Scholar]
- [33].Slusarczyk M, Serpi M, Pertusati F, Antivir. Chem. Chemother 2018, 26, 1–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Procházková E, Navrátil R, Janeba Z, Roithová J, Baszczyňski O, Org. Biomol. Chem 2019, 17, 315–320. [DOI] [PubMed] [Google Scholar]
- [35].Soto-Velasquez M, Hayes MP, Alpsoy A, Dykhuizen EC, Watts VJ, Mol Pharmacol 2018, 94, 963–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Brust TF, Alongkronrusmee D, Soto-Velasquez M, Baldwin TA, Ye Z, Dai M, Dessauer CW, van Rijn RM, Watts VJ, Sci. Signaling 2017, 10, eaah5381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Conley JM, Brand CS, Bogard AS, Pratt EP, Xu R, Hockerman GH, Ostrom R, Dessauer CW, Watts VJ, J. Pharmacol. Exp. Ther 2013, 347, 276–287. [DOI] [PMC free article] [PubMed] [Google Scholar]