

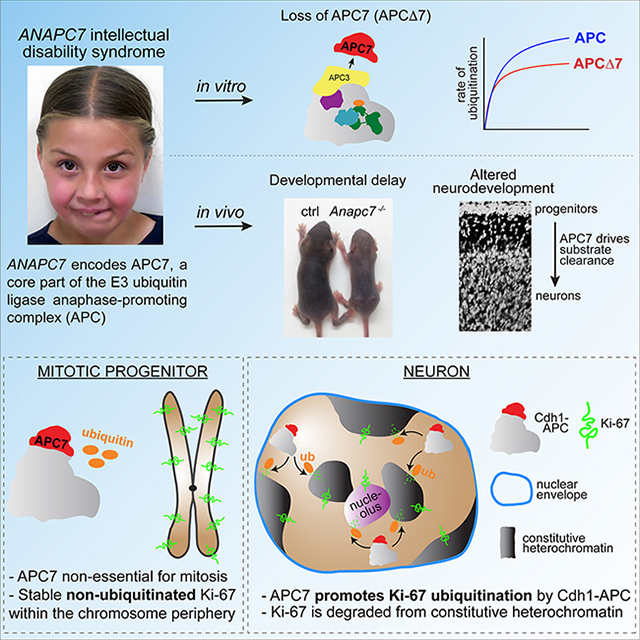
SUMMARY
Neurodevelopmental cognitive disorders provide insights into mechanisms of human brain development. Here, we report an intellectual disability syndrome caused by loss of APC7, a core component of the E3 ubiquitin ligase anaphase promoting complex (APC). In mechanistic studies, we uncover a critical role for APC7 during recruitment and ubiquitination of APC substrates. In proteomics analyses of the brain from mice harboring the patient-specific APC7 mutation, we identify the chromatin-associated protein Ki-67 as an APC7-dependent substrate of the APC in neurons. Conditional knockout of the APC coactivator protein Cdh1, but not Cdc20, leads to accumulation of Ki-67 protein in neurons in vivo, suggesting that APC7 is required for the function of Cdh1-APC in the brain. Deregulated neuronal Ki-67 upon APC7 loss localizes predominantly to constitutive heterochromatin. Together, our findings define an essential function for APC7 and Cdh1-APC in neuronal heterochromatin regulation, with implications for understanding human brain development and disease.
In brief
Ferguson et al. report that APC7 is required for the development of the mammalian brain, when APC7 promotes ubiquitination and clearance of the chromatin-associated factor Ki-67 from constitutive heterochromatin after neuronal progenitors exit mitosis.
Graphical Abstract
INTRODUCTION
Protein degradation is fundamental to nearly every aspect of the development and physiology of eukaryotic cells. Control of protein turnover via ubiquitination and proteasomal degradation is mediated by the sequential activity of E1 (ubiquitin-activating), E2 (ubiquitin-conjugating), and E3 (ubiquitin ligase) enzymes. Among E3 enzymes, RING-domain ubiquitin ligases represent the major class, with >600 genes in humans (Deshaies and Joazeiro, 2009). Despite the fundamental importance of E3s to cellular function, our understanding of their mechanisms of catalysis and substrate selection, biological roles and contribution to human disease remains limited.
The 1.2 MDa anaphase promoting complex (APC, no relation to adenomatous polyposis coli) is a ~20 subunit complex that represents a prototypical member of the class of RING E3 ubiquitin ligases (Deshaies and Joazeiro, 2009). The structure of the APC is conventionally viewed as an assembly configured from platform and TPR lobe subcomplexes (Figure 1A) (Chang et al., 2015; Watson et al., 2018). The platform (also known as the catalytic core) includes the catalytic cullin-RING components APC2 and APC11 and scaffolding subunits, whereas the TPR lobe (also known as the arc lamp) proteins APC3, APC6, APC7 and APC8 are enriched in tetratricopeptide repeat (TPR) motifs that mediate protein-protein interaction (Zeytuni and Zarivach, 2012). Although most APC subunits are conserved throughout eukaryotes, APC7 is unique for its origin in metazoans (Pál et al., 2007). Previous structural studies of wild-type apo-APC showed that the TPR subunits homodimerize (Chang et al., 2014; Zhang et al., 2013), and therefore the TPR lobe constitutes a sizeable fraction of the total mass of the APC. Although the roles of catalytic components of the APC have been characterized, the mechanisms by which non-catalytic subunits of the TPR lobe regulate APC function remain poorly defined.
Figure 1: APC7 is not required for the conformation of the APC or its assembly. See also Figure S1.
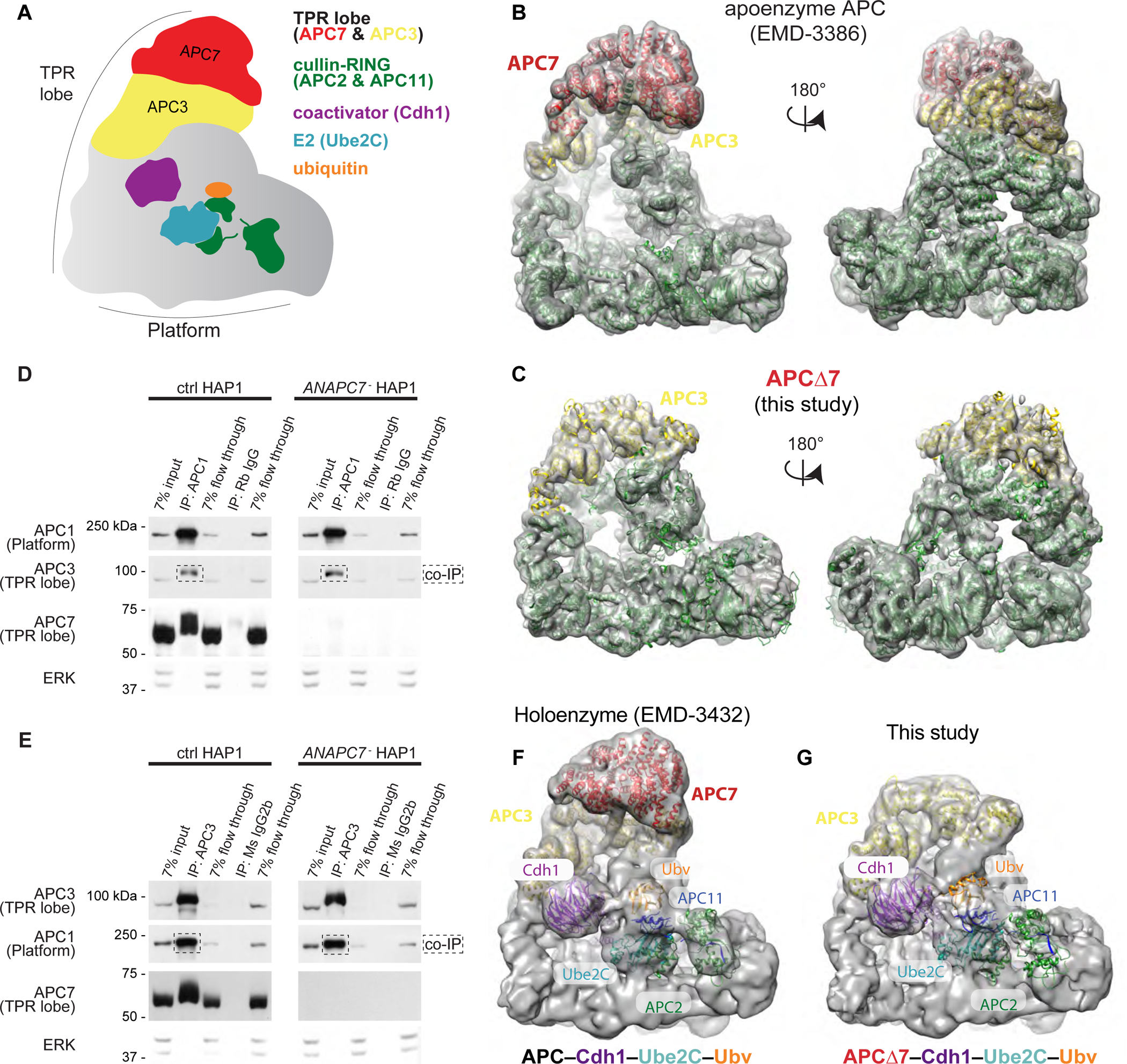
A. Schematic of the APC.
B. Published model of wild-type human APC (EMD-3386) showing the APC7 dimer (Chang et al., 2015).
C. Empirically determined Cryo-EM structure of APCΔ7. APC subunits from published coordinates (PDB 4L9U) were rigid-body docked using Chimera (Pettersen et al., 2004).
D. IP of APC1 from HAP1 cells followed by immunoblot (IB) for APC subunits. Flow through fractions reflect the remaining protein after antibody binding.
E. IP of APC3 (TPR lobe) from HAP1 cells followed by IB for APC subunits.
F. Published Cryo-EM structure of the APC holoenzyme (APC–Cdh1–Ube2C–substrate–Ubv) trapped by chemical crosslinking (Brown et al., 2016).
G. Cryo-EM structure of APCΔ7–Cdh1–Ube2C–substrate–Ubv with Ube2C poised for ubiquitin transfer. Published coordinates (PDB 4L9U) were rigid-body docked using Chimera.
Ubiquitination of target proteins of the APC proceeds in multiple stages (Brown et al., 2016; Watson et al., 2018). Peptide degrons such as the D-box and KEN box motifs are recognized by one of the homologous coactivators Cdc20 or Cdh1 (no relation to cadherin 1) (Peters, 2006). Coactivator binding to the APC activates the complex, exposing the binding site for the E2 enzyme Ube2C (Brown et al., 2015; Chang et al., 2014), which transfers ubiquitin onto substrates. Following Ube2C-mediated substrate ubiquitination, the APC employs the E2 Ube2S to extend K11-linked ubiquitin chains that drive proteasomal substrate degradation (Brown et al., 2016; Garnett et al., 2009; Williamson et al., 2009; Wu et al., 2010).
Although initially identified for its essential role in mitosis (Craney and Rape, 2013; Sivakumar and Gorbsky, 2015), the APC also controls differentiation and maintenance of cell identity through multiple mechanisms (Eguren et al., 2011; Huang and Bonni, 2016; Kimata, 2019; Oh et al., 2020). In post-mitotic neurons, the APC drives major steps in differentiation, including axon patterning (Konishi et al., 2004; Lasorella et al., 2006), dendrite morphogenesis (Kim et al., 2009), synapse differentiation (Yang et al., 2009) and synaptic plasticity (Huang et al., 2015). However, the mechanistic substrates that underlie APC-mediated developmental processes are incompletely defined. Furthermore, despite its central role in fundamental aspects of cell and neuronal biology, the contribution of APC subunit disruption to human disease is largely unknown.
In this study, we employ structural, biochemical, and genetic approaches to uncover critical functions of the TPR lobe subunit APC7. We find that APC7 is essential for stable recruitment and ubiquitination of substrates. We identify an inherited intellectual disability syndrome caused by loss of APC7, demonstrating the essential function for APC7 in human brain development. In proteomics analyses of the brain from mice harboring the patient-specific APC7 mutation, we find that the chromatin-associated protein Ki-67 represents an APC7-dependent substrate of the APC in post-mitotic neurons. These findings unveil a surprising role for APC7 and ubiquitin signaling in chromatin regulation during human brain development, with important implications for our understanding of neurodevelopmental disorders of cognition.
RESULTS
APC7 loss has minimal effect on the assembly and structure of the APC
Proteins such as APC7 that contain multiple tandem TPR motifs often promote protein-protein interactions (Zeytuni and Zarivach, 2012). Therefore, we began by ascertaining the structural consequences of loss of APC7 on the conformation of the APC. Human APC lacking APC7 (APCΔ7) was purified in the absence of cofactors (apo-APC) and subjected to cryo-electron microscopy (cryo-EM) to yield a 7.4 Å structure (see Table S1 for imaging metrics and parameters). Compared with wild-type apo-APC (Chang et al., 2015; Zhang et al., 2016), the structure of apo-APCΔ7 was remarkably similar, aside from loss of the density attributable to the APC7 dimer atop the TPR lobe (Figure 1B, 1C, S1A–D). In a haploid human HAP1 cell line subjected to CRISPR-induced null mutation of the ANAPC7 gene that encodes APC7 (referred to as ANAPC7− in figures), co-immunoprecipitation (IP) analysis of endogenous APC showed that loss of APC7 had little or no effect on the association of the TPR lobe and platform sub-complexes, whether the APC was isolated using antibodies to the TPR lobe (APC3) or the platform (APC1) (Figure 1D, 1E).
APC7 makes extensive contacts with underlying APC3, which interacts with the Isoleucine-Arginine (IR) tail of the coactivators Cdh1 and Cdc20 (Vodermaier et al., 2003). To explore the structural consequences of APC7 loss on the conformation of the holoenzyme (i.e. with cofactors), APCΔ7–Cdh1–Ube2C was trapped to mimic substrate ubiquitination (Brown et al., 2015, 2016) (Figure 1F, S1A–D). Surprisingly, our empirically solved cryo-EM map of APCΔ7–Cdh1–Ube2C failed to reveal defects in the recruitment or positioning of Cdh1 or the E2 enzyme Ube2C, despite the obvious absence of APC7. Direct comparisons with our published holoenzyme show similar overall topology with only minor changes in occupancy of elements of the catalytic core, consistent with their dynamic nature and corresponding limitations of the resolution of our density map (Brown et al., 2015, 2016) (Figure 1G). These data indicate that APC7 is not required for association with the key regulatory protein Cdh1 or the E2 enzyme Ube2C, leading us to conclude that the specialized function of APC7 lies downstream of catalytic assembly.
Loss of APC7 causes defects in APC-induced substrate ubiquitination
To explore the effects of loss of APC7 on the enzymatic function of the APC, we isolated the APC from control and APC7 mutant HAP1 cells via anti-APC3 IP. We supplied purified ubiquitin, Cdh1, E2 enzymes, E1 (Ube1), and fluorescent substrates to complete the reaction (Figure S2A). Loss of APC7 impaired ubiquitination of the APC substrates securin and cyclin B1 N-terminal domain (CycB-NTD) under diverse reaction conditions, including over time with Ube2C (Figure S2B), with both E2 enzymes Ube2C and Ube2S (Figure S2C), and with increasing Ube2C or the coactivator Cdh1 (Figure S2D, S2E). These results suggest that loss of APC7 in cells compromises the E3 ubiquitin ligase activity of the APC.
To dissect the function of APC7 at different stages in the ubiquitination cycle, we characterized the ubiquitin ligase activity of APCΔ7 in vitro using recombinant proteins, which permits precise control over reaction conditions (Jarvis et al., 2016). During APC–Cdh1–Ube2C-mediated polyubiquitination of securin, loss of APC7 reduced the appearance of highly ubiquitinated products over time (Figure 2A). In the presence of saturating amounts of securin and Ube2C, loss of APC7 reduced both Vmax and Km of ubiquitination in response to escalating Cdh1 (Figure 2B, 2C).
Figure 2: APC7 controls substrate recruitment and processive ubiquitination by Ube2C. See also Figure S2.
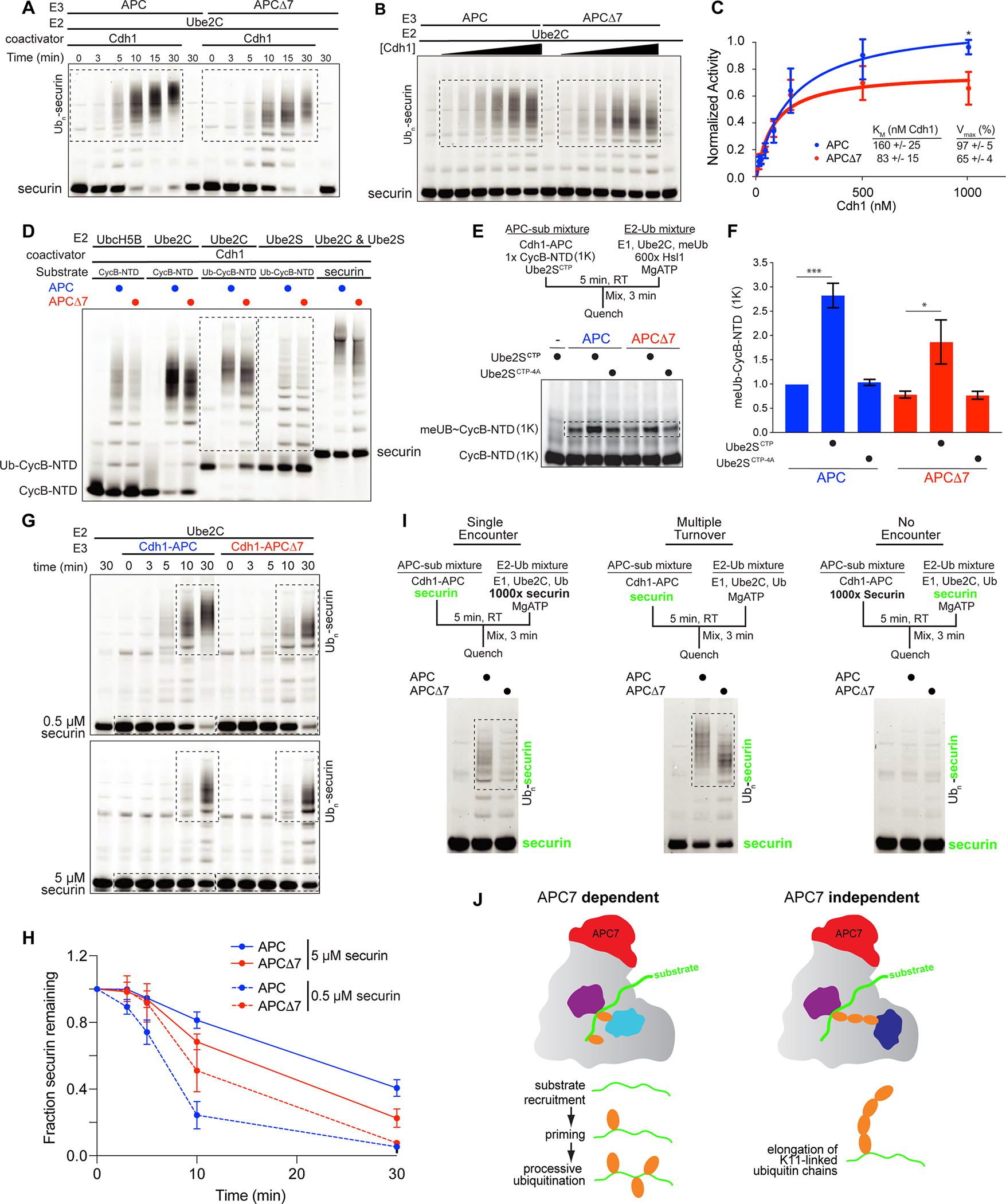
A. Time course of ubiquitination of securin by recombinant APC and APCΔ7. Boxed areas show reaction products.
B. Kinetic evaluation of APCΔ7-dependent ubiquitination in response to escalating Cdh1.
C. Kinetic analysis of substrate ubiquitination plotted using the Michaelis-Menten equation. Error bars SEM, N = 5. (*p = 0.02 by two-way ANOVA with Bonferroni test).
D. In vitro ubiquitination assays using recombinant proteins. Boxed areas show products from reactions using the E2 enzymes Ube2C and Ube2S.
E. The C-terminal peptide of Ube2S (Ube2SCTP) activates the APC to prime substrates during the initiation of ubiquitination. Shown are single encounter experiments with CycB-NTD (1K), a CycB-NTD variant with a single lysine for ubiquitin transfer. Use of methylated ubiquitin (meUb) ensures reactions terminate after priming. The E2-Ub sub mixture contained excess unlabeled substrate (Hsl1). Ube2SCTP−4A served as a negative control.
F. Quantitation of ubiquitinated CycB-NTD (1K). Error bars SEM, N = 3 (p-value by one-way ANOVA with Tukey test).
G. Securin ubiquitination at low (0.5 μM) and high (5 μM) substrate concentration.
H. Quantitation of unmodified securin over time. Error bars SEM, N = 3.
I. Ube2C-mediated processive ubiquitination in single and multiple encounter assays. Fluorescent securin detected during gel imaging is shown in green.
J. APC7 is required for the function of the E2 Ube2C at multiple stages. During ubiquitin chain elongation by Ube2S, APC adopts a distinct catalytic architecture that does not require APC7.
We next determined the effect of APC7 loss on the function of different E2 enzymes. To assess substrate ubiquitination by Ube2C, we monitored modification of the APC substrates CycB-NTD and securin. To assess ubiquitin chain elongation by Ube2S, we generated a fusion protein in which the ubiquitin coding sequence was added upstream of CycB-NTD (Ub-CycB-NTD) (Jarvis et al., 2016). Ubiquitination by APCΔ7 was impaired to a much greater degree with Ube2C than with Ube2S or UbcH5B/Ube2D, a nonspecific E2 (Figure 2D), indicating that APC7 participates in Ube2C-mediated ubiquitination. We recently reported that a peptide consisting of the C-terminal 18 amino acids of Ube2S (Ube2SCTP) independently functions to allosterically promote conformational activation of the APC via a mechanism that is distinct from Cdh1 binding (Martinez-Chacin et al., 2020). Ube2SCTP therefore serves as an independent method of stimulating the APC during substrate ubiquitination. To determine whether Ube2SCTP might rescue the effects of loss of APC7 in Ube2C-dependent substrate priming (initial ubiquitin transfer), we employed single encounter experiments. Priming by APCΔ7 was slightly reduced in the absence of Ube2SCTP (Figure 2E, 2F). Although Ube2SCTP enhanced the ability of wild-type APC to induce substrate priming by 3-fold, priming by APCΔ7 was only induced 2-fold by Ube2SCTP (Figure 2E, 2F). The negative control peptide Ube2SCTP−4A with alanine substitutions at 4 critical amino acids had little or no effect on substrate priming (Figure 2F) (Martinez-Chacin et al., 2020). Together, these observations suggest that loss of APC7 diminishes the ability of Cdh1 or Ube2SCTP to induce the ubiquitin ligase activity of the APC.
To determine how effects of loss of APC7 vary with substrate abundance, we assessed ubiquitination in vitro at high and low concentrations of securin. In APC ubiquitin ligase assays employing low concentrations (0.5 μM) of securin, loss of APC7 impaired ubiquitination to a greater degree than with high (5 μM) securin (Figure 2G, 2H). To directly test the ability of the APC to stably recruit substrates during priming and processive ubiquitination, we performed single encounter assays in which Cdh1-APC was preincubated with low abundance fluorescent securin before adding a mixture containing Ube2C, ubiquitin and excess unlabeled securin (Figure 2I, left panel). Because Cdh1-APC encounters unlabeled substrate after dissociating from the fluorescent substrate, reaction products reflect ubiquitination from a single binding event. Loss of APC7 reduced APC-induced substrate ubiquitination in both single encounter and multiple turnover experiments (Figure 2I, middle panel), indicating a requirement for APC7 in stable retention of substrates during processive ubiquitination. Taken together, our results show that loss of APC7 leads to global substrate-dependent catalytic defects affecting substrate recruitment, APC activation and Ube2C-mediated ubiquitination (see model in Figure 2J).
ANAPC7 mutation causes an intellectual disability syndrome
Having established that APC7 is critical for APC-dependent substrate ubiquitination, we next asked whether APC7 plays an important role in human development. Fortuitously, we discovered patients with syndromic intellectual disability due to loss of APC7. Chromosomal microarray analysis of 11 young people of Old Order Amish heritage who displayed developmental delay and intellectual disability revealed a deletion within the ANAPC7 locus. Thermal asymmetric interlaced PCR refined the boundaries of an 8054 bp deletion that included exons 4–6 of the ANAPC7 gene (Figure 3A, S3A, S3B). The p.Ile171GlufsX14 allele (ANAPC7 c.511–2480_919+3276del, NC_000012.12(NM_016238.2):c.511–2480_919+3276del) results in a frameshift in exon 7 and loss of the APC7 protein (Figure 3B). All affected patients were homozygous for the mutant ANAPC7 allele, whereas heterozygous siblings and parents were unaffected clinically. A pedigree spanning 10 generations connected all probands (Figure S3C), consistent with a founder effect in this population. Skeletal and/or craniofacial deformities were seen in a subset of patients (Figure 3C, Table S2) and body and head size were low, though microcephaly was found in only one patient (Figure S3D). Together with intellectual disability, acquisition of early motor milestones was also delayed (Figure S3E).
Figure 3: APC7 is required for human and mouse brain development. See also Figures S3, S4 and S5.
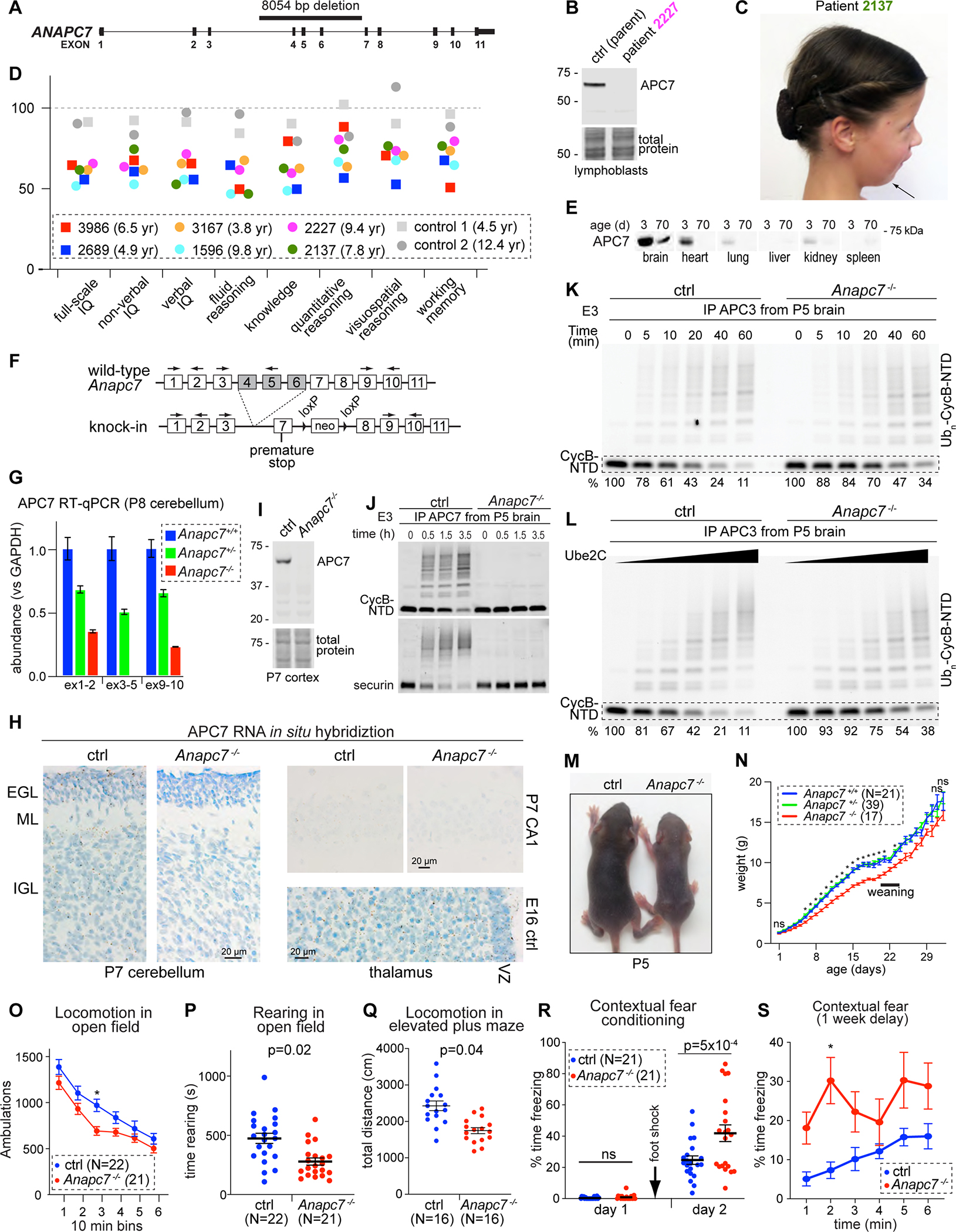
A. 8054 bp deletion in the human ANAPC7 gene.
B. APC7 IB in human lymphoblastoid cells.
C. Patient 2137 with the ANAPC7 intellectual disability syndrome exhibits Pierre-Robin sequence.
D. Stanford-Binet testing of IQ and subdomains. Age at testing is shown.
E. APC7 IB in different mouse tissues.
F. Recapitulation of the patient mutation in mice.
G. RT-qPCR for APC7 in mouse cerebellum. The location of primers is shown in 3F. Error bars SEM, N = 4.
H. APC7 in situ hybridization. Brown dots represent mRNA molecules. (EGL, external granule layer; ML, molecular layer; IGL, internal granule layer; VZ, ventricular zone).
I. APC7 IB.
J. In vitro ubiquitination assay of fluorescent APC substrates. The source of E3 was IP of APC7 from P5 brain.
K. Ubiquitination of CycB-NTD by the APC isolated by APC3 IP from mouse brain at P5.
L. Ubiquitination of CycB-NTD in response to increasing Ube2C. E3 (APC) was isolated by anti-APC3 IP from the brain at P5.
M. Littermates at P5.
N. Growth of mutant mice. Error bars SEM (*p < 0.001, mutant versus each of Anapc7+/− and Anapc7+/+, by mixed-effects ANOVA analysis and Tukey test).
O. Total ambulations during open field test. Error bars SEM (*p = 0.01 by repeated measure ANOVA and Bonferroni test).
P. Time rearing in the open field test. Error bars SEM (p-value by Kolmogorov-Smirnov test).
Q. Total ambulatory distance in the elevated plus maze. Error bars SEM (p-value by Kolmogorov-Smirnov test).
R. Assessment of contextual fear memory. Foot shock was applied on day 1 after habituation to the context environment. Freezing (conditioned response) was assessed on day 2 upon reintroduction to the context environment. Error bars SEM (p-value by one-way ANOVA with Tukey test).
S. Long-term fear conditioning was assessed by reintroducing mice to the context environment after a one-week delay. Error bars SEM (*p = 0.01 by repeated measure ANOVA and Bonferroni test).
To characterize cognitive function in patients with the ANAPC7 intellectual disability syndrome, we administered the Stanford-Binet IQ test (Roid, 2003). Full-scale IQ (FSIQ) ranged from 52 to 65 in 6 tested patients, compared to 91 and 92 in two control siblings (Figure 3D). Impairment was similar for verbal and non-verbal IQ, and extended across all tested indices including fluid reasoning, knowledge, quantitative reasoning, visuospatial processing, and working memory. Male and female patients were affected to similar degrees. Deficiencies in all major facets of intelligence in patients with the ANAPC7 syndrome suggests a fundamental function of APC7 in brain development and function.
APC7-mediated ubiquitination is required for mouse brain development
After establishing that APC7 is required for human neurodevelopment, we explored the biochemical and cellular functions of APC7 in the developing mammalian brain. In wild-type mice, APC7 was more abundant in the brain relative to other tissues (Figure 3E). APC7 and other APC subunits including the APC coactivator proteins Cdh1 and Cdc20 declined during mouse brain development (Figure S4A–C). In fractionated lysates of the mouse cerebral cortex and cerebellum, APC7 and APC3 were enriched in the nucleus relative to other APC subunits (Figure S4B, S4C). When combined with purified ubiquitin, Cdh1, E1, E2 (Ube2C), and the substrate CycB-NTD (Jarvis et al., 2016), immunoprecipitated APC from the embryonic brain showed higher activity than APC isolated from the brain of adult mice (Figure S4D, S4E). These findings suggest APC7 operates primarily in the nucleus during brain development.
To characterize the function of APC7 in neural development, we recapitulated the patient-specific APC7 mutation in knock-in mice, generated to harbor a mutant Anapc7 allele with deletion of exons 4–6 (Anapc7−/− in figures) (Figure 3F, S5A, S5B). RT-qPCR analyses in the brain and primary neuronal cultures showed reduction of APC7 mRNA in proportion to the number of copies of the mutant allele (Figure 3G, S5C), suggesting the mutant transcript is degraded via nonsense-mediated decay (Laf and Basu, 2019). Using in situ RNA hybridization in P7 mouse cerebellum, we detected the APC7 mRNA in neuronal progenitors within the external granule layer (EGL) and differentiated neurons of the internal granule layer (IGL) (Figure 3H). In contrast, minimal residual transcript was detected in mice homozygous for the patient-specific APC7 mutation. APC7 expression also persisted beyond the proliferative ventricular zone in the developing thalamus on embryonic day 16 (E16) and was detected in CA1 of the hippocampus of wild-type mice on postnatal day 7 (P7) (Figure 3H). In agreement, single-cell RNA sequencing of human brain showed that APC7 is expressed in both neuronal progenitors and mature neurons in the cortex and the cerebellum (Song et al., 2021). In Anapc7−/− mice, APC7 protein was undetectable in the cerebral cortex (Figure 3I), and IP of APC7 from the brain of control mice supported ubiquitin ligase activity in vitro, whereas no activity was observed following IP of APC7 from the mutant brain (Figure 3J). These results establish that APC7 is expressed throughout neuronal development, when APC7 associates with enzymatically active APC, and that the patient mutation in ANAPC7 leads to loss of APC7 protein.
To explore the biochemical basis of the ANAPC7 syndrome, we first determined whether APC7 is required for the formation of the APC in the brain. Co-IP of endogenous APC from lysates of P5 mouse brain showed little or no effect of APC7 loss on the assembly of the APC (Figure S5D). To determine how loss of APC7 in the brain impacts catalysis in ubiquitin ligase assays, we immunoprecipitated the APC from the mouse brain using antibodies to APC3 and added additional components as purified proteins. Loss of APC7 impaired APC-induced ubiquitination of CycB-NTD over time or upon addition of increasing amounts of Ube2C (Figure 3K, 3L). At the completion of these reactions, the amount of substrate that remained was threefold higher upon loss of APC7, confirming that APC activity is diminished in the mutant brain.
The most conspicuous phenotype in juvenile APC7 mutant mice was growth delay, with the maximum difference in body weight around weaning (Figure 3M, 3N). However, loss of APC7 had no major effect on embryogenesis, birth weight or adult size (Figure 3N, S5E, S5F). Surprisingly, histologic analysis of the cerebral cortex, hippocampus and cerebellum showed that loss of APC7 did not alter the anatomy of these regions (Figure S5G), suggesting that APC7 is not required for histogenesis of the brain.
We next characterized the effects of Anapc7 mutation on behavior in adult mice. Mutant mice showed reduced voluntary locomotion, impaired swimming and difficulty maneuvering on a pole (Figure 3O–Q, S5H–K), phenotypes suggestive of sensorimotor dysfunction. To test memory, we employed Pavlovian paradigms that pair a conditioned stimulus with an aversive unconditioned stimulus, leading to a freezing behavior upon re-exposure to the conditioned stimulus after learning (Khuchua et al., 2003; Wozniak et al., 2007). While the APC7 mutant mice showed minimal freezing under baseline conditions, exaggerated freezing was seen upon re-exposure to the context environment (but not the auditory cue), an effect that persisted after a one-week delay (Figure 3R, 3S, S5L). Mutant mice showed no deficits in memory retrieval during the Morris Water maze test of spatial memory (Figure S5K). These results implicate APC7 in the regulation of short and long-term memory retrieval of the contextual fear response. We conclude that a mouse model of the patient-specific ANAPC7 mutation observed in humans exhibits phenotypes consistent with defects in nervous system function.
APC7 loss has little or no effect on mitosis
Because the APC is a central regulator of mitosis, we characterized the role of APC7 in mitosis. Human TERT RPE-1 cells are widely used in studies of mitosis (Bodnar et al., 1998). We used a CRISPR-induced knockout approach to determine the effect of APC7 depletion on mitosis in RPE-1 cells. We tested 4 guide RNAs (gRNAs) targeting APC7 in cells expressing doxycycline-inducible Cas9 (TetOn-Cas9). APC7 was depleted upon doxycycline treatment in cells expressing gRNAs 2 and 4 (Figure 4A). Loss of APC7 failed to prolong the period between nuclear envelope breakdown (NEBD) and anaphase (length of mitosis) or the frequency of lagging chromosomes (Figure 4B–4D).
Figure 4: APC7 loss has little or no effect on mitosis. See also Figure S6.
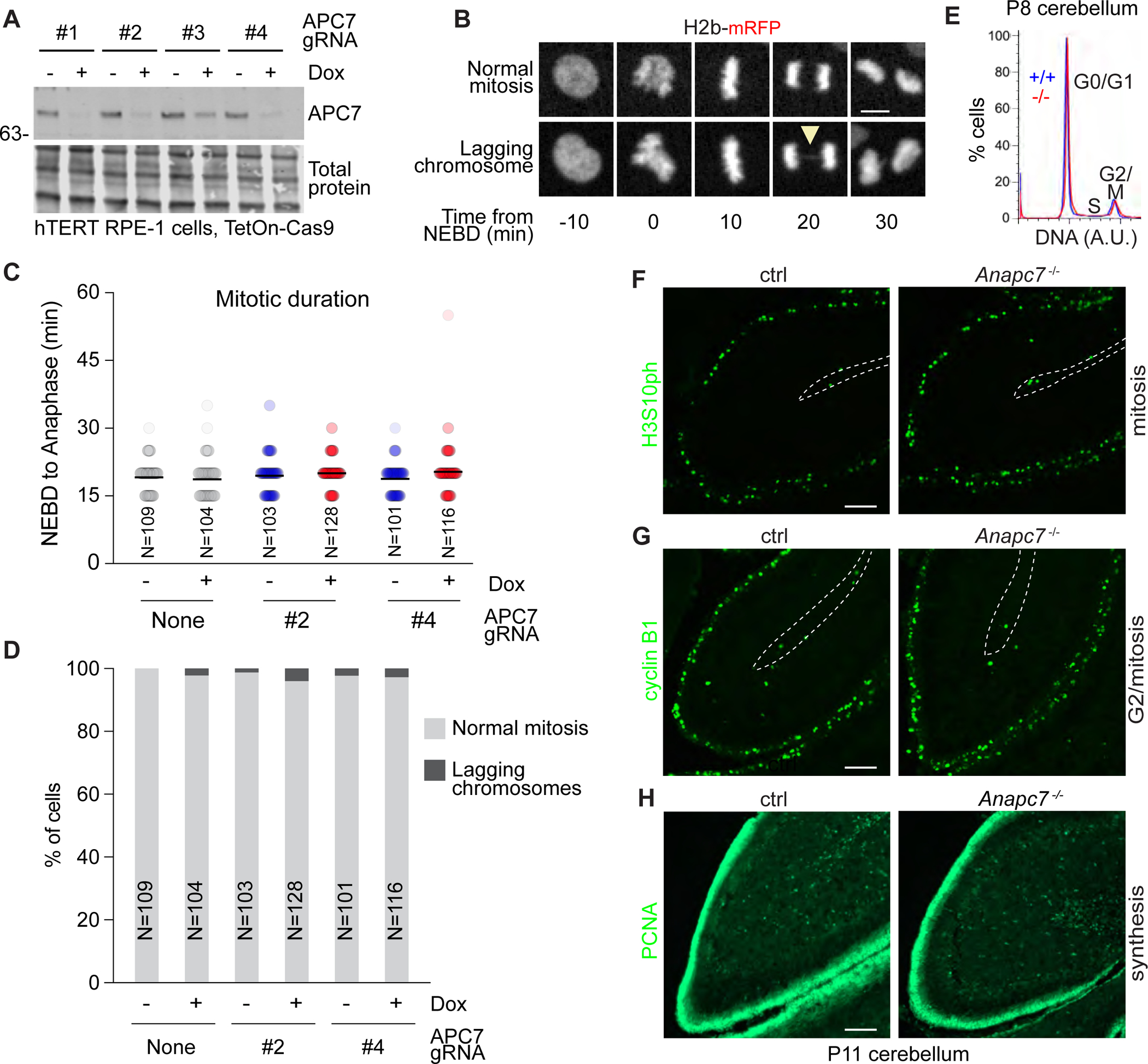
A. APC7 IB in hTERT RPE-1 cells expressing Dox-inducible Cas9 and transfected with 4 CRISPR guide RNAs targeting APC7.
B. Example images of hTERT RPE-1 cells expressing H2b-mRFP. Arrowhead indicates a lagging chromosome.
C. Quantitation of the length of mitosis (NEBD to anaphase). The number of cells analyzed is shown.
D. Quantitation of lagging chromosomes. The number of cells analyzed is shown.
E. Flow-cytometry analysis of DNA content in dissociated P8 cerebellum stained with propidium iodide.
F. H3S10ph immunofluorescence (IF).
G. Cyclin B1 IF.
H. Proliferating Cell Nuclear Antigen (PCNA) IF.
We next assessed the effect of APC7 loss on mitosis in the mouse brain. In dissociated P8 cerebellum stained with the DNA dye propidium iodide and subjected to flow cytometric quantitation of DNA content, loss of APC7 had little or no effect on the number of tetraploid cells in G2/mitosis (Figure 4E). Similarly, immunohistochemical (IHC) analysis of the developing mouse cerebellum failed to show an effect of APC7 loss on the number or distribution of cells in mitosis (H3S10ph), G2/mitosis (cyclin B1) or S phase (PCNA) (Figure 4F–H). Similar findings were seen in other brain regions (Figure S6A–E). Finally, migration of post-mitotic neurons, which is coupled to cell cycle exit (Godin and Nguyen, 2014), was not altered upon loss of APC7 in the developing cerebellar and cerebral cortex (Figure S6F, S6G). These data show that APC7 is not essential for mitosis.
APC7 mediates ubiquitination and degradation of Ki-67 in the brain
Having identified an essential role for APC7 in the development of the mammalian brain, we sought to identify APC7-dependent APC substrates that contribute to the pathogenesis of the ANAPC7 syndrome. Because APC7 increases the kinetics of ubiquitin ligase activity, we reasoned that loss of APC7 should impair degradation of select protein substrates that require maximal APC activity. We therefore deployed TMT-based quantitative mass spectrometry (Navarrete-Perea et al., 2018) to measure protein abundance in the brain of APC7 mutant and control littermate mice. We chose the cerebellum because it contains a highly abundant and relatively homogenous population of granule neurons, which facilitates biochemical analyses (Huang and Bonni, 2016; Kim et al., 2009; Yamada et al., 2013; Yang et al., 2010). The levels of APC7 protein were profoundly reduced in the cerebellum of APC7 mutant mice (Figure 5A, 5B), validating the TMT proteomics method. We found 24 proteins that were elevated in the cerebellum of APC7 mutant mice, many of which function in chromosome architecture and integrity (Table S3). Among these, we observed elevation of the chromosome passenger complex subunits Aurora B and INCENP but not the canonical APC substrates cyclin B1 and cyclin A2 in the mutant brain (Figure 5B) (Geley et al., 2001; Yu et al., 1996). Strikingly, the chromatin-associated protein Ki-67 was the most highly and consistently elevated protein in the APC7 mutant cerebellum (Figure 5A, 5B). Immunoblot (IB) analyses showed elevation of Ki-67 throughout cerebellar development in APC7 mutant mice, whereas the level of the Ki-67 mRNA was unchanged (Figure 5C, 5D). Importantly, Ki-67 was elevated in lymphoblasts from a patient with the ANAPC7 syndrome (Figure 5E). The APC, in purified recombinant form, supplemented with Ube2C and Cdh1 induced ubiquitination of Ki-67 which was immunoprecipitated from the cerebellum of wild-type P7 mice (Figure 5F, 5G). The polyubiquitinating E2 enzyme Ube2S triggered elongation of ubiquitin chains from pre-existing Ki-67-linked ubiquitin (Figure 5H). Finally, APCΔ7 was deficient in ubiquitinating full-length Ki-67 that was immunoprecipitated from mouse cerebellum (Figure 5I, 5J). To determine whether the APC might recognize Ki-67 through degrons characteristically found within substrates of the APC (Davey and Morgan, 2016), we generated fluorescently labeled purified proteins corresponding to amino acids 1–300 and 765–1000 of human Ki-67 and used these as substrates in in vitro enzyme assays. Both fragments were robustly ubiquitinated by the APC, and ubiquitination was dependent upon the KEN box motifs located at residues 254, 933 and 939 (Figure 5K, 5L). These results demonstrate that Ki-67 is a substrate of the APC, and that Ki-67 ubiquitination requires APC7.
Figure 5: Identification of Ki-67 as an APC7-dependent substrate of the APC in neurons. See also Figure S7.
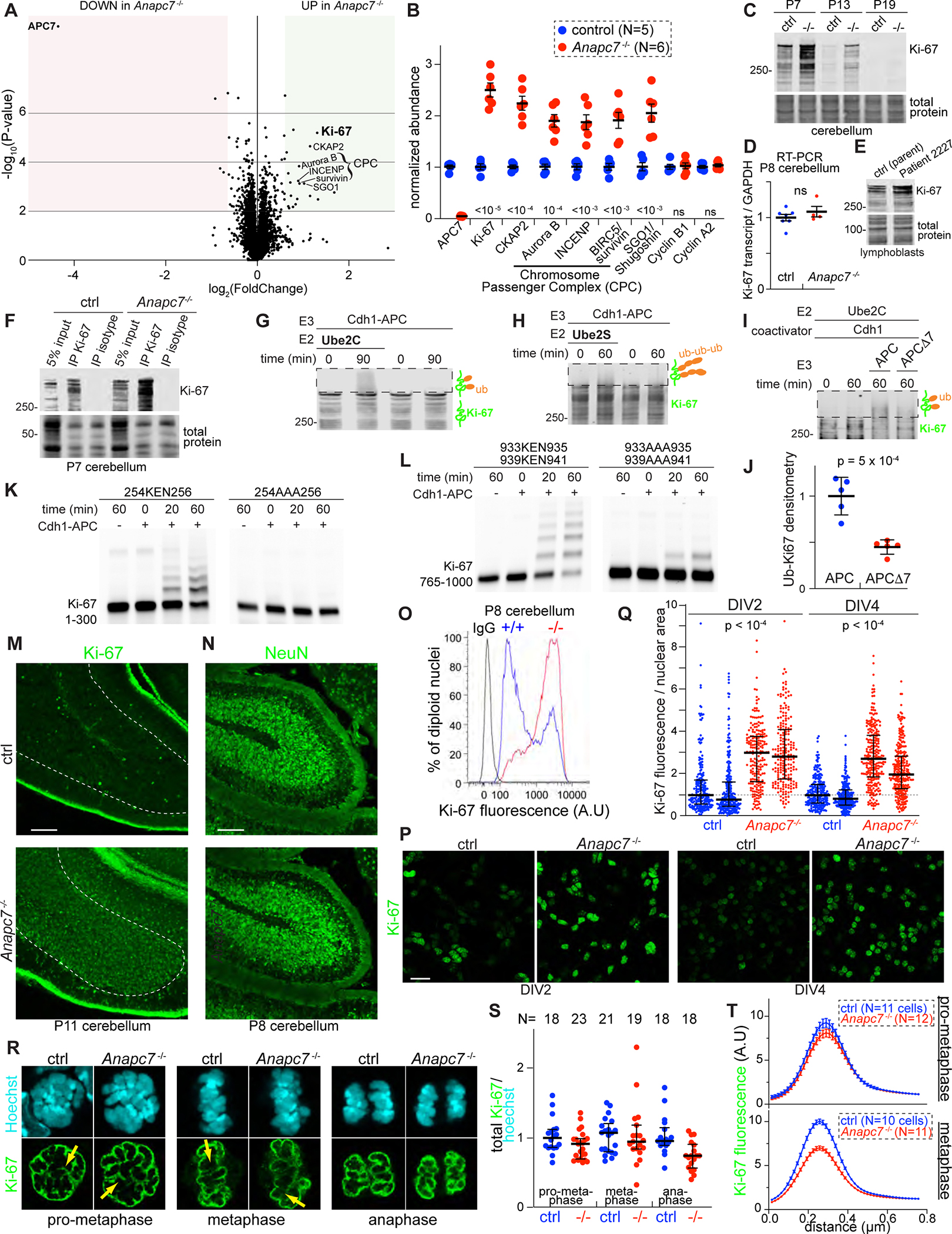
A. Volcano plot of protein abundance in P8 cerebellum as assessed by Tandem Mass Tag (TMT) proteomics (N=5 control, N=6 APC7 mutant). Shaded regions indicate a 1.5-fold change in abundance with p-value < 0.01 by two-tailed unpaired t-test.
B. Normalized TMT ratios for relevant proteins (p-values by two-tailed unpaired t-test).
C. Ki-67 IB in mouse cerebellum.
D. Ki-67 RT-qPCR in P8 cerebellum (N=7 control, N=4 APC7 mutant). Not significant by two-tailed unpaired t-test.
E. Ki-67 IB in human lymphoblasts.
F. IP of Ki-67 from P8 cerebellum followed by Ki-67 IB.
G. In vitro ubiquitination of Ki-67 by recombinant APC. Ki-67 was isolated by IP from wild-type P7 cerebellum. Boxed areas represent reaction products detected by Ki-67 IB.
H. In vitro polyubiquitination of Ki-67 by recombinant Cdh1-APC and Ube2S. Ki-67 was isolated by IP from wild-type P7 cerebellum.
I. In vitro ubiquitination assay using recombinant APC and APCΔ7. Ki-67 was isolated by IP from wild-type P7 cerebellum.
J. Densitometry-based measurement of reaction products following ubiquitination of immunoprecipitated Ki-67 by APC and APCΔ7. Errors bars SEM (N = 5, p-value by two-tailed unpaired t-test).
K. In vitro ubiquitination of amino acids 1–300 of human Ki-67 by recombinant APC. KEN to AAA substitution occurred at the indicated residues.
L. In vitro ubiquitination of amino acids 765–1000 of human Ki-67 by recombinant APC. KEN to AAA substitutions occurred at the indicated residues
M. Ki-67 IF in P11 cerebellum.
N. NeuN IF in P11 cerebellum.
O. Flow cytometry of diploid nuclei isolated from P8 cerebellum labeled with anti-Ki-67 or isotype antibody.
P. Confocal IF of Ki-67 in primary mouse cerebellar granule neuron cultures. The day in vitro (DIV) is indicated.
Q. Quantitation of Ki-67 in individual neuronal nuclei normalized to nuclear area. Two biological replicates are shown for each genotype and DIV. Error bars interquartile range (p-values by one-way ANOVA with Tukey test).
R. Confocal optical sections of anti-Ki-67 IF in neuronal precursors during mitosis. Yellow arrows indicate regions sampled in Figure 5T.
S. Quantitation of Ki-67 IF during mitosis. Ki-67 intensity was normalized to Hoechst. The number of cells analyzed is shown. Error bars SEM.
T. Quantitation of the intensity of the chromosome periphery. For each cell, 3–5 segments were analyzed. Errors bars SEM.
Next, we identified the cells in the cerebellum in which Ki-67 abundance was increased by loss of APC7. Remarkably, IHC analysis of the cerebellum in mutant mice revealed that ectopic Ki-67 was present in granule neurons expressing the mature neuron marker NeuN within the IGL (Figure 5M, 5N). In contrast, Ki-67 was restricted to proliferating granule neuron precursors in the outer EGL of control mice (Figure S7A). Ki-67 was also present outside of progenitor zones in other developing regions of the APC7 mutant brain (Figure S7B–F). Flow cytometry-based detection of anti-Ki-67 immunolabeling confirmed the elevation of Ki-67 in diploid nuclei from the cerebellum of APC7 mutant mice (Figure 5O, S7G). Finally, to assess Ki-67 abundance in neurons over time, we employed primary cultures of cerebellar granule cells, which rapidly differentiate ex vivo (Bilimoria and Bonni, 2008). Using confocal immunofluorescence (IF) imaging, we observed a shift toward higher levels of Ki-67 in nuclei of mutant neurons on day in vitro 2 (DIV2) and after maturation on DIV4 (Figure 5P, 5Q). IB analysis confirmed Ki-67 elevation in mutant primary granule neurons (Figure S7H, S7I) with no significant effect on the Ki-67 mRNA (Figure S7J). Finally, we confirmed the link between APC7 and Ki-67 using a transgenic Ki-67-RFP reporter line (Basak et al., 2014) using anti-RFP IP and confocal imaging of primary granule neurons from Anapc7−/−,Ki-67-RFP mice (Figure S7K–M). These data establish a biochemical pathway by which ubiquitination regulates the abundance of Ki-67 in post-mitotic neurons.
During mitosis, Ki-67 constitutes a major component of the surfactant-like chromosome periphery compartment required for chromosome integrity and heterochromatin organization (Booth and Earnshaw, 2017; Booth et al., 2016; Cuylen et al., 2016; Sobecki et al., 2016). However, in IF analyses, loss of APC7 had no effect on total or chromosome-associated levels of Ki-67 in primary granule neuron precursors during pro-metaphase, metaphase and anaphase (Figure 5R–T). Thus, APC7 selectively regulates APC-induced ubiquitination and degradation of Ki-67 in postmitotic neurons.
APC7 regulates the E3 ubiquitin ligase Cdh1-APC but not Cdc20-APC in the brain
The homologous APC coactivator proteins Cdh1 and Cdc20 play critical yet functionally distinct roles in brain development (Huang and Bonni, 2016). We therefore generated conditional knockout mice in which Cdh1 or Cdc20 was disrupted in the granule cell lineage using Math1-promoter driven Cre (García-Higuera et al., 2008; Manchado et al., 2010; Matei et al., 2005). The hypoplastic cerebellum of conditional Cdc20 knockout mice displayed a malformed pattern of vertical foliation with Purkinje neuron heterotopias in the white matter, consistent with abnormal proliferation (Figure 6A, 6B). In contrast, mutation of Cdh1, like the patient-specific APC7 mutation, did not alter cerebellar morphology (Figure 6A). Recombinant Cdh1 and Cdc20 stimulated APC-dependent ubiquitination of full-length Ki-67 immunoprecipitated from wild-type mouse cerebellum (Figure 6C). However, Cdh1-APC, but not Cdc20-APC, robustly ubiquitinated the KEN-box containing fragments of the Ki-67 N-terminus (Figure 6D). Accordingly, accumulation of Ki-67 was only observed in the brain of Cdh1 conditional knockout mice but not conditional Cdc20 knockout mice (Figure 6E, 6F). Finally, IF analyses showed accumulation of Ki-67 in DIV2 and DIV4 primary granule neurons from conditional Cdh1 knockout mice (Figure 6G, 6H). Taken together, these results suggest that APC7 and Cdh1 act in concert to drive APC-induced ubiquitination and degradation of Ki-67 in neurons.
Figure 6: APC7 operates in the context of Cdh1-APC to drive Ki-67 degradation in neurons.
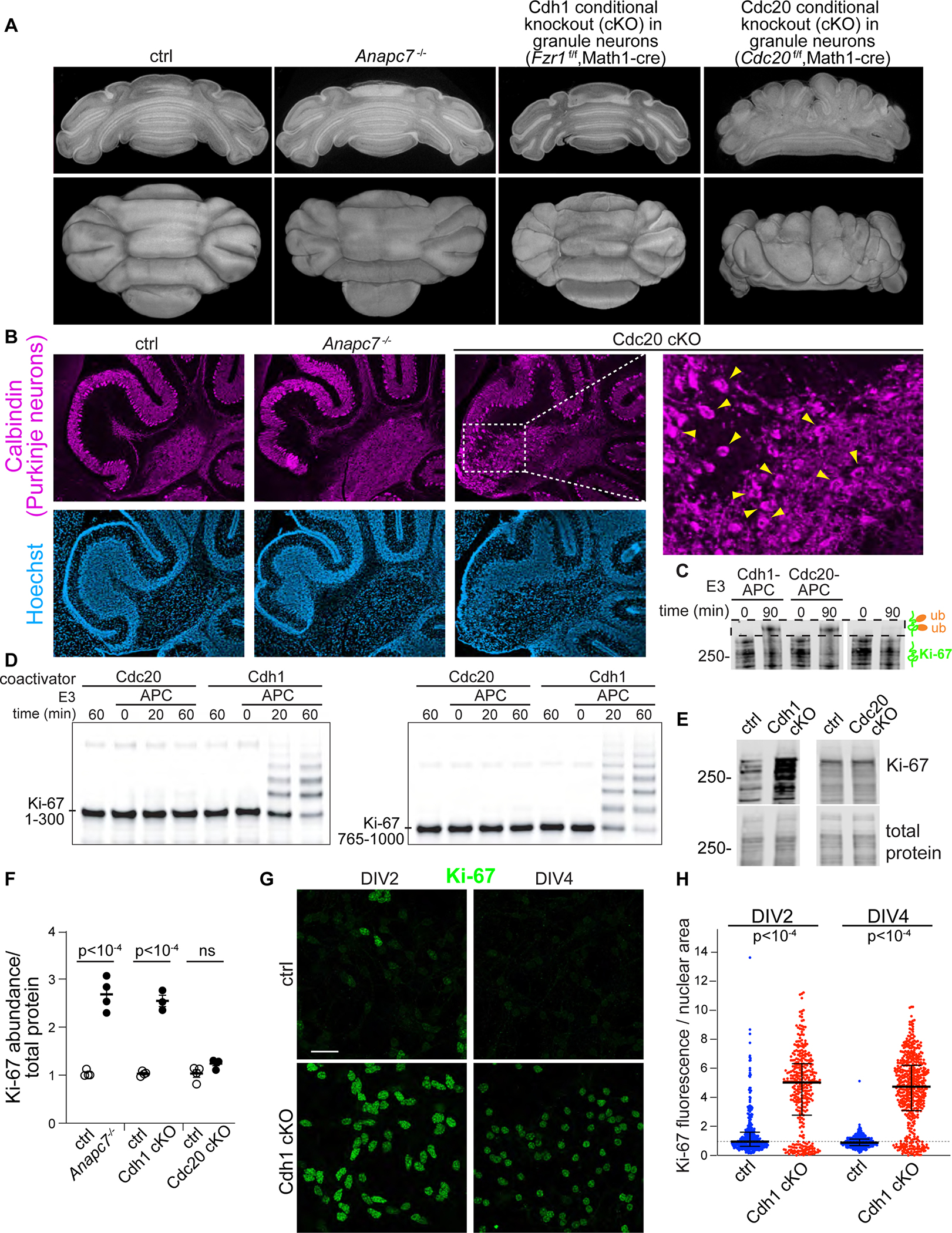
A. Electron tomography of P8 cerebellum.
B. Calbindin IF in sagittal sections of P12 cerebellum. An expanded region from the Cdc20f/f,Math1-cre is shown (PCL, Purkinje cell layer).
C. In vitro ubiquitination of full-length Ki-67 by recombinant Cdh1-APC and Cdc20-APC. Ki-67 was immunoprecipitated from wild-type P7 cerebellum and detected by Ki-67 IB.
D. In vitro ubiquitination of human Ki-67 amino acids 1–300 (left) and 765–1000 (right) by Cdh1-APC and Cdc20-APC.
E. Ki-67 IB in P12 cerebellum.
F. Densitometry quantitation of Ki-67 IB. Mutants were normalized and compared to littermates. Error bars SEM (N = 4, p-value by two-tailed unpaired t-test).
G. IF of Ki-67 in primary cerebellar granule neuron cultures.
H. Quantitation of Ki-67 IF normalized to nuclear area in cultured neurons on the indicated DIV. Error bars interquartile range (p-values by Kruskal-Wallis test for non-parametric data followed by Dunn test).
APC7 regulates Ki-67 turnover in constitutive heterochromatin
The finding that APC7 is selectively required for APC-induced turnover of the protein Ki-67 in post-mitotic neurons led us to ascertain whether ubiquitin signaling might operate in a previously unidentified context within nuclei of neurons. Ki-67 immunoreactivity was localized to chromocenters labeled by the DNA dye Hoechst (bisbenzimide) in neurons throughout the brain including in the cerebral cortex (Figure 7A, 7B). Chromocenters contain densely compacted DNA within constitutive heterochromatin (Ostromyshenskii et al., 2018). In primary granule neurons, loss of APC7 or Cdh1 led to extensive colocalization of Ki-67 with constitutive heterochromatin, identified using Hoechst, but not with facultative heterochromatin, identified using the histone modification H3K27me3 (Figure 7C–E). To assess the role of APC7 in constitutive heterochromatin, we performed RT-qPCR for sequences that are enriched in constitutive heterochromatin. Loss of APC7 reduced transcription of pericentromeric major satellite DNA, but did not change the expression of LINE-1 and IAP sequences derived from endogenous retroviruses (Figure 7F). These results suggest that APC7 regulates constitutive heterochromatin.
Figure 7: The APC regulates Ki-67 protein turnover in heterochromatin in neurons.
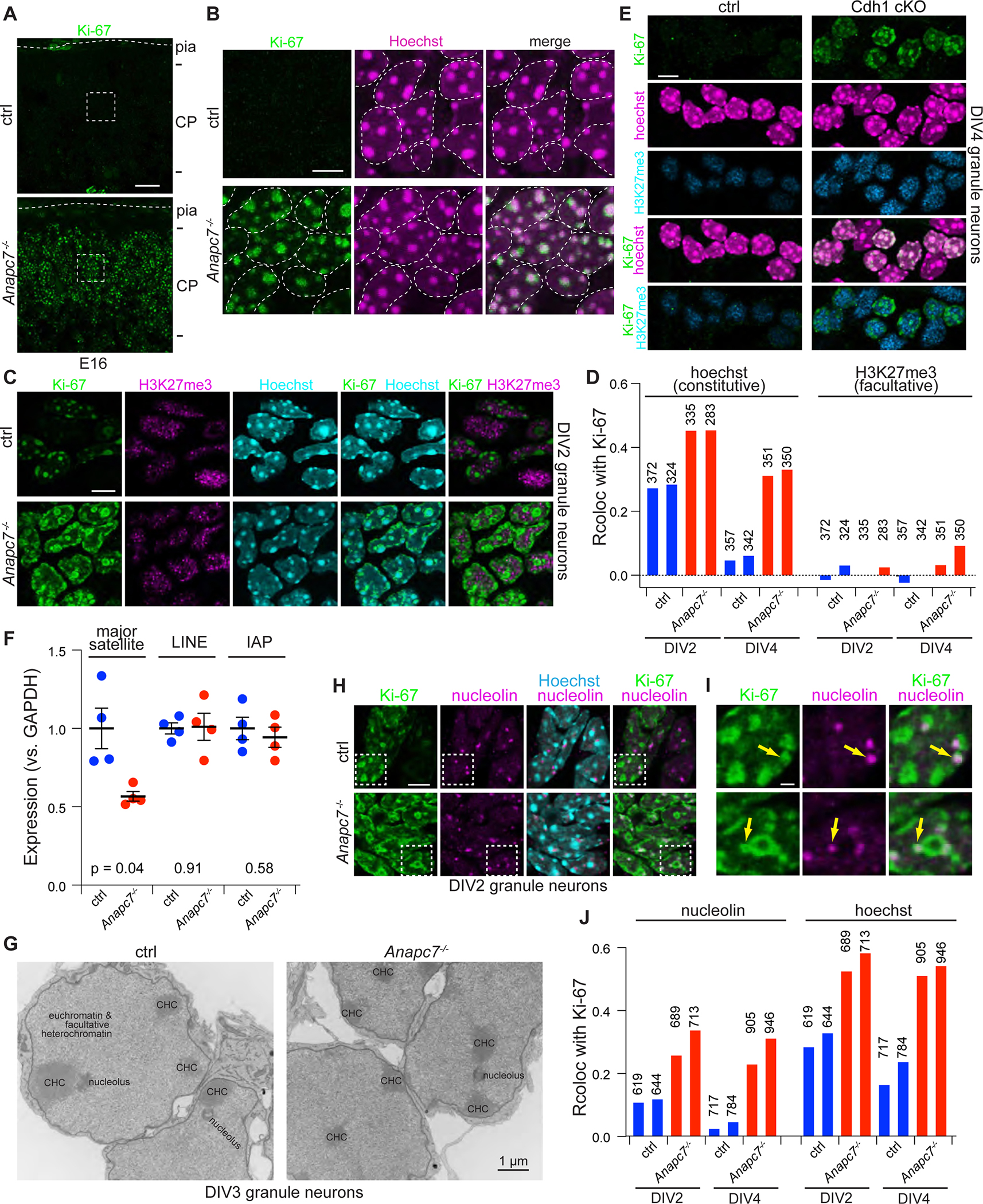
A. Ki-67 IF in the cortical plate (CP) at E16.
B. Expanded area from part A. Outlines delimit nuclei.
C. IF of Ki-67 and H3K27me3 in primary cerebellar granule neurons on DIV2.
D. Quantitation of colocalization between Ki-67 and Hoechst versus Ki-67 and H3K27me3. The number of neurons analyzed is shown.
E. Ki-67, H3K27me3 and Hoechst in DIV4 cultured granule neurons from Cdh1 cKO mice.
F. RT-qPCR analysis of repeat expression in P11 cerebellum. Error bars SEM (p-values by two-tailed Welch’s t-test)
G. Transmission EM of cultured neurons at DIV3. Constitutive heterochromatin appears electron dense and resides within lamin-associated and nucleolar-associated domains.
H. Fluorescence imaging of Ki-67, nucleoli and Hoechst in cultured neurons.
I. Magnified areas from 7H, arrows demonstrate colocalization of Ki-67 and nucleolin.
J. Quantitation of colocalization between Ki-67 and nucleolin, versus Ki-67 and Hoechst in cultured neurons. The number of cells analyzed is shown.
During interphase, Ki-67 localizes to nucleoli in non-neuronal cells (Ian R. Kill, 1996). Therefore, we examined the nucleolus in APC7 mutant mice. In transmission EM analyses, the stereotyped spatial relationship between nucleoli and the adjacent constitutive heterochromatin was preserved in granule neurons from mice with APC7 mutation (Figure 7G). Notably, quantitative IHC analyses demonstrated that a smaller fraction of Ki-67 was localized in nucleoli than in constitutive heterochromatin in neurons from APC7 mutant mice (Figure 7H–J). Collectively, our findings reveal that APC7 operates in the context of the E3 ubiquitin ligase Cdh1-APC to drive ubiquitination and degradation of Ki-67 primarily in neuronal heterochromatin in the brain.
DISCUSSION
In this study, we defined an enzymatic function for the major E3 ubiquitin ligase anaphase-promoting complex (APC) in regulating heterochromatin in the developing mammalian brain. Although the APC retains its structure in the absence of APC7, this core subunit is essential for recruiting and ubiquitinating substrates. We found that the chromatin-associated protein Ki-67 represents the major APC7-dependent substrate in post-mitotic neurons. Conditional knockout of the coactivator Cdh1 also led to Ki-67 accumulation, indicating that APC7 operates in the context of Cdh1-APC in the brain. Finally, deregulated Ki-67 in mutant mice accumulates within constitutive heterochromatin of post-mitotic neurons, a finding which carries implications for our understanding of human brain development and neurodevelopmental disorders of cognition.
Identification of patients with the ANAPC7 syndrome establishes that the major E3 ubiquitin ligase the APC plays a fundamental role in human brain development. Although studies of the APC in model systems have identified several critical functions in neurons (Huang and Bonni, 2016; Yang et al., 2010), no mutations in any of the large number of core APC subunits have been previously reported in genetic brain disorders. Loss of APC7 diminishes but does not abolish the E3 ubiquitin ligase activity of the APC, and submaximal APC activity does not significantly disrupt cell proliferation in the brain. Together with the observation that APC7 is expressed in post-mitotic neurons, these observations suggest that APC7 is critical for neuronal differentiation in the brain.
Our discovery that the chromatin-associated mitotic protein Ki-67 is a major APC7-dependent substrate in post-mitotic neurons unveils a previously unrecognized role for the APC in chromatin regulation. We found that as progenitors exit mitosis, the localization of Ki-67 shifts from the chromosome periphery to become concentrated within constitutive heterochromatin in post-mitotic neurons. Because Ki-67 is rich in intrinsically disordered repeat domains (Cuylen et al., 2016; Sobecki et al., 2016), Ki-67 accumulation in mutant neurons may influence condensation of constitutive heterochromatin, which forms through the process of phase separation (Larson et al., 2017; Strom et al., 2017). Imaging analyses in APC7 mutant neurons showed enrichment of Ki-67 on the surface of chromocenters relative to their core, potentially a consequence of a perturbed phase boundary between heterochromatin and euchromatin. Our observation that APC7 loss also affects the chromosomal passenger complex (CPC), a phase separated complex that regulates histone phosphorylation, could point to a broader role for APC7 in regulating epigenetic states via biomolecular condensates (Trivedi and Stukenberg, 2020). Because APC7 is required for processive ubiquitination, bulky substrates such as Ki-67 and the CPC that are prone to polymerization may be differentially perturbed in the ANAPC7 syndrome.
As cells exit mitosis, Ki-67 promotes chromosome clustering prior to reformation of the nuclear envelope (Cuylen-Haering et al., 2020). Our data suggest that Ki-67 accumulates along the inner surface of nuclei in APC7 mutant neurons, potentially altering the integrity of nuclear membrane and lamin-associated domains. Elucidating how deregulated Ki-67 alters heterochromatin, nuclear architecture and ultimately gene expression in APC7 mutant neurons in future studies should further advance our understanding of mechanisms of genome regulation in the brain.
Mutation of proteins that regulate chromatin often cause neurodevelopmental disorders of cognition (Avagliano et al., 2020). Among these proteins, chromatin remodelers including the CHD (chromodomain helicase DNA-binding) family and the regulators of genome architecture CTCF and Cohesin have direct roles in chromatin biology (Avagliano et al., 2020; Goodman and Bonni, 2019). The function of chromatin regulatory proteins is often coordinated, and therefore it will be essential to determine how the APC/Ki-67 ubiquitin signaling link influences other chromatin factors.
In conclusion, we have identified an intellectual disability syndrome that triggered the discovery of a connection between APC7-dependent ubiquitin signaling and heterochromatin regulation in the brain. Further studies of the pathogenesis of the ANAPC7 syndrome should yield clues for treating neurodevelopmental disorders of cognition.
Limitations of the Study
Although our study provides insight into the genetic, biochemical and cellular regulation of brain development, gaps in our understanding remain. We do not fully understand the biological consequences of deregulated Ki-67 in isolation on the function of constitutive heterochromatin. Furthermore, the challenges associated with purifying large proteins limit our ability to comprehensively explore the recognition of Ki-67 by the APC, and other domains of Ki-67 might directly bind to the APC via APC7 or another subunit. Although many of the putative APC7-dependent substrates identified in TMT proteomics are implicated in chromosome architecture and integrity, how these substrates contribute to phenotypes in the brain upon APC7 loss remains to be explored. Finally, the physiological processes that are perturbed by APC7 loss and how these aberrations contribute to dysfunction of neural circuits during memory formation and retrieval remain to be defined.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact:
Further information and requests for resources and reagents should be directed to the Lead Contact, Azad Bonni (bonni@wustl.edu).
Materials availability:
All mouse lines, cell lines, purified protein or any other reagent generated or used in the course of this study will be made freely available to interested researchers.
Data and code availability:
The TMT proteomics dataset “APC7 KO vs controls” has been deposited at ProteomeXchange via the PRIDE database and is publicly available as of the date of publication. Cryo-EM structure data have been deposited to the EM Data Bank and are publicly available as of the date of publication. Accession numbers for these datasets are listed in the key resources table. Original/source data have been deposited on Mendeley Data and are publicly available as of the date of publication. DOIs are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-APC1 | Bethyl | Cat# A301–653A, RRID:AB_1210875 |
| Mouse monoclonal (AF3.1) anti-CDC27/APC3 (immunoprecipitation) | Thermo Fisher Scientific | Cat# MA1–24749, RRID:AB_2075283 |
| Rabbit polyclonal anti-CDC27/APC3 (immunoblotting) | Bethyl | Cat# A301–184A, RRID:AB_890565 |
| Rabbit polyclonal anti-APC4 | Bethyl | Cat# A301–176A, RRID:AB_2227071 |
| Rabbit polyclonal anti-APC5 | Bethyl | A301–026A, RRID:AB_2227119 |
| Rabbit polyclonal anti-CDC16/APC6 | Bethyl | Cat# A310–389A, RRID:AB_890557 |
| Rabbit polyclonal anti-APC7 N-term (Immunoprecipitation) | Bethyl | A302–550A, RRID:AB_1998915 |
| Rabbit polyclonal anti-APC7 C-term (Immunoblotting) |
Bethyl | Cat# A302–551A, RRID:AB_1998911 |
| Rabbit polyclonal anti-APC8 | Bethyl | Cat# A301–182A |
| Rabbit polyclonal anti-APC16 | Santa Cruz Biotechnology | Cat# sc-135452, RRID:AB_10988755 |
| Rabbit polyclonal anti-CDC20 | Bethyl | Cat# A301–180A, RRID:AB_890559 |
| Mouse monoclonal (DH01) anti-FZR1/CDH1 | Thermo Fisher Scientific | Cat# MA5–11496, RRID:AB_10979615 |
| Rabbit polyclonal anti-p44/42 MAPK (Erk1/2) Antibody #9102 | Cell Signaling Technology | Cat# 9102, RRID:AB_330744 |
| Mouse monoclonal anti-14–3-3 beta (A-6) | Santa Cruz Biotechnology | Cat# sc-25276, RRID:AB_626617 |
| Rabbit polyclonal anti-PARP | Cell Signaling Technology | Cat# 9542, RRID:AB_2160739 |
| Mouse monoclonal anti-BrdU (G3G4) | DSHB | G3G4(AntiBrdUrd), RRID:AB_1157913 |
| Mouse monoclonal anti-PCNA (PC10, 3F81) | Thermo Fisher Scientific | Cat# 13–9910-82, RRID:AB_466977 |
| Mouse monoclonal anti-NeuN | Millipore | Cat# MAB377, RRID:AB_2298772 |
| Mouse monoclonal anti-cyclin B1 (V152) | Abcam | Cat# ab72, RRID:AB_305751 |
| Rat monoclonal anti-Ki-67 (SolA15) | Thermo Fisher Scientific | Cat# 14–5698-82, RRID:AB_10854564 |
| Rabbit polyclonal anti-Calbindin (D-28K) | SWANT | Cat# CB38, RRID:AB_2721225 |
| Rabbit anti-nucleolin | Bethyl | Cat# IHC-00083, RRID:AB_2267070 |
| Normal rabbit IgG | Millipore | Cat# NI01–100UG, RRID:AB_10681285 |
| Mouse IgG2b, kappa monoclonal (MPC-11) | Abcam | Cat# NI01–100UG, RRID: not found |
| Rat IgG2a kappa light chain | Thermo Fisher Scientific | Cat# 14–4321-82, RRID:AB_470105 |
| Goat anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary, Alexa Fluor 568 | Thermo Fisher Scientific | Cat# A-11031, RRID:AB_144696 |
| Goat anti-Rat IgG (H+L) Cross-Adsorbed Secondary, Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A-11006, RRID:AB_2534074 |
| Donkey anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary, Alexa Fluor 568 | Thermo Fisher Scientific | Cat# A10042, RRID:AB_2534017 |
| Donkey Anti-Mouse IgG IRDye 680 Conjugated antibody | LI-COR Biosciences | Cat# 926–32222, RRID:AB_621844 |
| IRDye 800CW Donkey anti-Rabbit IgG antibody | LI-COR Biosciences | Cat# 925–32213, RRID:AB_2715510 |
| IRDye 800CW Goat anti-Rat IgG antibody | LI-COR Biosciences | Cat# 926–32219, RRID:AB_1850025 |
| RFP-Trap Agarose | Chromotek | rta-10 |
| Bacterial and Virus Strains | ||
| Baculovirus | (Weissmann et al., 2016) | N/A |
| Lentivirus; H2b-mRFP1.3 | (Meitinger et al., 2020) | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Ube1 (E1) | (Jarvis et al., 2016) | N/A |
| CDH1 protein | (Jarvis et al., 2016) | N/A |
| CDC20 protein | (Jarvis et al., 2016) | N/A |
| Ube2C (E2) | (Jarvis et al., 2016) | N/A |
| Ube2S (E2) | (Jarvis et al., 2016) | N/A |
| UBCH5 (E2) | (Dou et al., 2012) | N/A |
| APC apoenzyme | (Jarvis et al., 2016) | N/A |
| APCΔ7 apoenzyme | (Jarvis et al., 2016) | N/A |
| APC holoenzyme | (Brown et al., 2016) | N/A |
| APCΔ7 holoenzyme | This paper | N/A |
| UBE2SCTP and UBE2SCTP-4A | (Martinez-Chacin et al., 2020) | N/A |
| Fluorescent cyclin B1-NTD (CycB-NTD) | (Jarvis et al., 2016) | N/A |
| Fluorescent Ub-CycB-NTD | (Jarvis et al., 2016) | N/A |
| Fluorescent securin | (Jarvis et al., 2016) | N/A |
| Methyl-ubiquitin | (Jarvis et al., 2016) | N/A |
| ubiquitin | (Jarvis et al., 2016) | N/A |
| Benzonase | Millipore | E1014 |
| Hoechst 33258 solution | Millipore Sigma | Cat# 94403 |
| TMT11 reagent | Thermo-Fisher Scientific | A37727 |
| Pierce Trypsin Protease, MS Grade | Thermo-Fisher Scientific | Cat#90058 |
| Lys-C, Mass Spectrometry Grade | Wako Chemicals | Barcode#4987481427648 |
| Pierce Phosphatase Inhibitor Mini Tablets | Thermo Fisher Scientific | A32957 |
| Pierce Protease Inhibitor Mini Tables | Thermo Fisher Scientific | A32953 |
| Protease inhibitor cocktail | Sigma | P8340 |
| phosphatase inhibitor cocktail #3 | Sigma | P0044 |
| PMSF | Sigma | PMSF-RO |
| HyClone Fetal Bovine Serum | Cytiva | SH30071.03IH25–40 |
| 5-Bromo-2′-deoxyuridine (BrdU) | Sigma | B5002 |
| Polybrene | EMD Millipore | TR-1003-G |
| Affymetrix CytoScan HD Assay | Thermo Fisher Scientific | Cat# 901835 |
| Pierce BCA Protein Assay | Thermo Fisher Scientific | Cat# 23223 |
| Bradford regent | Bio-Rad | Cat# 5000006 |
| Revert total protein stain | LI-COR | Cat# 926–11011 |
| iScript cDNA synthesis kit | Bio-Rad | Cat# 1708890 |
| Deposited Data | ||
| TMT proteomics data PXD016683 | ProteomeXchange | (http://www.ebi.ac.uk/pride), PXD016683 |
| Cryo-EM structure of apo-APC7 | EM Data Bank | EMDB-25026 |
| Cryo-EM structure of holo-APC7 | EM Data Bank | EMDB-25027 |
| Original/source data | Mendeley Data | DOI: 10.17632/hrk4ztwrpn.1 |
| Experimental Models: Cell Lines | ||
| Lymphoblasts from patient 2227 | Coriell | Coriell Cat# GM25252, RRID:CVCL_LN92 |
| Lymphoblasts from mother of patient 2227 | Coriell | Coriell Cat# GM25253, RRID:CVCL_LN93 |
| HAP1 ANAPC7 null | Horizon Discovery | RRID:CVCL_SC47 |
| HAP1 wild type | Horizon Discovery | |
| 129S5/SvEvTac EDJ22 ES cells | Washington University Mouse Genetics Core | http://escore.im.wustl.edu/SubMenu_celllines/wtEScell_linesEDJ22.html |
| High Five (BTI-Tn-5B1–4) cells | Thermo Fisher Scientific | BTI-TN-5B1–4 |
| RPE-1 CEP192-mNeonGreen TetOn-Cas9 | (Meitinger et al., 2020) | ODCL0191 |
| RPE-1 CEP192-mNeonGreen TetOn-Cas9 ANAPC7 gRNA #1 | This study | ODCL0271 |
| RPE-1 CEP192-mNeonGreen TetOn-Cas9 ANAPC7 gRNA #2 | This study | ODCL0272 |
| RPE-1 CEP192-mNeonGreen TetOn-Cas9 ANAPC7 gRNA #3 | This study | ODCL0273 |
| RPE-1 CEP192-mNeonGreen TetOn-Cas9 ANAPC7 gRNA #4 | This study | ODCL0274 |
| RPE-1 CEP192-mNeonGreen TetOn-Cas9 pEF1alpha-H2B-mRFP | This study | ODCL0275 |
| RPE-1 CEP192-mNeonGreen TetOn-Cas9 ANAPC7 gRNA #2 pEF1alpha-H2B-mRFP | This study | ODCL0276 |
| RPE-1 CEP192-mNeonGreen TetOn-Cas9 ANAPC7 gRNA #2 pEF1alpha-H2B-mRFP | This study | ODCL0277 |
| Experimental Models: Organisms/Strains | ||
| Anapc7 mutant on hybrid C57BL/6 x 129 | This paper | N/A |
| Anapc7 mutant on Jackson C57BL/6 | This paper | N/A |
| CDC20f/f mice on C57BL/6 | (Manchado et al., 2010) | N/A |
| CDH1f/f mice on C57BL/6 | (García-Higuera et al., 2008) | N/A |
| Math1/Atoh1-cre mice on C57BL/6 | The Jackson Laboratory | Cat# 011104 |
| Wild-type Jackson C57BL/6 | The Jackson Laboratory | Cat# 000664 |
| Sox2-cre mice on C57BL/6 | The Jackson Laboratory | Cat# 008454 |
| Ki67-RFP reporter on C57BL/6 | The Jackson Laboratory | Cat# 029802 |
| Wild-type outbred mice on CD1 | Charles River | Cat# 022 |
| Oligonucleotides | ||
| See Table S4 | ||
| Recombinant DNA | ||
| Anapc7 knock-in targeting vector with deletion of exons 4–6, floxed neo cassette and G>A substitution | (Adams et al., 2005) | BAC ID# bMQ-440B18 |
| Amino acids 1–300 and 765–100 of human Ki-67 cloned into pGEX | ||
| Software and Algorithms | ||
| ImageJ | (Schneider et al., 2012) | https://imagej.nih.gov/ij/ |
| UCSF Chimera | (Pettersen et al., 2004) | https://www.cgl.ucsf.edu/chimera/ |
| Image Studio | LI-COR | N/A |
| ImageLab | Bio-rad | N/A |
| Chromosome Analysis Suite | Affymetrix | N/A |
| Proteomics software pipeline | SEQUEST | http://fields.scripps.edu/yates/wp/?page_id=17 |
| Gautomatch | MRC Laboratory of Molecular Biology | https://www2.mrc-lmb.cam.ac.uk/research/locally-developed-software/zhang-software/ |
| RELION-3.0 | (Zivanov et al., 2018) | https://www3.mrc-lmb.cam.ac.uk/relion/index.php/Main_Page |
| QuantStudio 6 real-time PCR system | Bio-Rad | N/A |
| Dragonfly | Object Research Systems | https://www.theobjects.com/dragonfly/index.html |
| SerialEM | (Mastronarde, 2003) | http://bio3d.colorado.edu/SerialEM/ |
| SEQUEST-based in-house platform | Thermo-Fisher Scientific (Eng et al., 1994; Huttlin et al., 2010) |
N/A |
| Other | ||
| QUANTIFOIL® R2.1, 100 Holey Carbon Films, Grids: Cu 200 mesh | Quantifoil Micro Tools GmbH | https://www.quantifoil.com |
| Custom In situ hybridization probe for mouse Anapc7 | ACDbio | https://acdbio.com |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice:
Animals were cared for in accordance with NIH guidelines. All Experimental methods were approved by Washington University Institutional Committee on the Use and Care of Animals under the protocol number 20180303. Animals were housed in a 12:12 light:dark cycle. We employed trio breeding and only used unaffected animals as parents. Pups were genotyped at P5 by tail biopsy and weaned into sex-matched cages at P21 to be bred at 7–8 weeks. Animals were allocated to experimental versus control groups based on genotype. Sex matched-littermate controls were used in all experiments, and the age at which animals were used is reported in figure legends.
For the Anapc7 knock-in mutant mouse strain, we characterized mutant mice derived from two independent ES cell lines on the 129S5/SvEvTac EDJ22 background (42 and 83). Chimeric founder males were crossed with female Jackson C57BL/6 mice expressing Sox2-cre for removal of the floxed neomycin resistance cassette introduced by the targeting vector. The Anapc7 mutation was henceforth maintained on this hybrid C57BL/6 × 129 mixed background through cousin matings and is currently at F6. These animals served as the source for primary granule neuronal cultures and all imaging experiments. They were also used for body weight measurements, in which we followed a cohort of consecutively born animals from multiple cages. The Anapc7 mutant allele was also backcrossed onto the Jackson C57BL/6 background, and after 5 generations the percent conversion by SNP typing found to be >93% (Dartmouse). For behavior studies and TMT proteomics, we used Anapc7−/− mutants and their littermate controls which had been backcrossed between 7 and 9 generations onto the Jackson C57BL/6 background. For experiments with APC7 mutant mice, Anapc7+/− mice served as controls when available, but Anapc7+/+ were also used if heterozygotes unavailable.
The ANAPC7 syndrome affects males and females, therefore we carefully considered the effects of sex on the phenotype of each mutant strain included in this study. In proteomics and behavioral assays, approximately equal numbers of male and female animals were included, and the data for males and females was interspersed and overall concordant, both in the case of control as well as mutant mice. Loss of APC7 had no effect on the body weight of adult male or female mice. For imaging experiments that included a biological replicate, we included both sexes whenever possible and verified every phenotype in both sexes. In sum, we looked for but failed to detect any relationship between sex and the severity of the molecular, cellular and behavioral phenotypes associated with loss of APC7.
For crosses involving mice harboring floxed Fzr1/Cdh1, floxed Cdc20, and Math1-cre, all mice were maintained on the congenic Jackson C57BL/6 background, having being back crossed for more than 5 generations. To generate experimental animals, mice homozygous for each floxed allele were crossed with mice heterozygous for the floxed allele and positive for Math1-cre. We brought in the Math1-cre from both the paternal and maternal sides to assess the possibility of germline inactivation of cre. Mice which were homozygous for the floxed allele and positive for Math1-cre were used as experimental animals, while sex-matched littermates that were floxed heterozygotes and positive for Math1-cre were used preferentially as controls if available, although other genotypes were allowed if necessary.
Ki67-RFP mice expressing RFP-tagged Ki-67 from the endogenous Mki67 locus were purchased from Jackson Labs. We validated this line using anti-RFP immunoprecipitation. To generate APC7 mutant mice that expressed the Ki67-RFP fusion protein, we bred C57BL/6 mice harboring Ki67-RFP for two generations with N9 Anapc7+/− mutant mice on the C57BL/6 background.
Wild-type CD1 outbred mice were used for brain fractionation and organ lysates.
Human subjects:
Patients with the ANAPC7 syndrome were identified and characterized genetically and clinically at the Clinic for Special Children (https://clinicforspecialchildren.org), which specifically serves children and adults who suffer from genetic and other complex medical disorders. According to their agreement with the clinic, these patients and their families have consented to being included in all research studies that arise from the identification of disease alleles.
Characterizing the developmental of patients with the ANAPC7 syndrome is a major focus of this study. Clinical data, including age and sex, can be found in Table S1.
Cell lines:
Lymphoblastoid cell lines were purchased from Coriell. These cell lines were established from peripheral lymphocytes of female patient 2227 (GM25252) and her mother (GM25253). Cells were immortalized by transduction of Epstein-Barr Virus. Loss of APC7 was confirmed in immunoblots, validating the patient mutation. Cells were maintained in suspension in 5% CO2 at 37 degrees in RPMI with 12% calf serum plus antibiotic. Cells were split 1:3 every 2–3 days.
The mostly haploid HAP1 cell line was originally derived from human male chronic myelogenous leukemia cells. HAP1 cells were subjected to CRISPR-mediated mutagenesis by Horizon Discovery to generate a null allele in ANAPC7, which was confirmed via immunoblot and immunoprecipitation. Cells were then maintained according to the manufacturer’s protocol at 37 degrees in 5% CO2 IMDM with 12% calf serum plus antibiotic and split 1:10 every 2–3 days.
RPE-1 (hTERT-RPE-1) cells expressing doxycycline-inducible Cas9 and in situ-tagged Cep192-mNeonGreen were previously described (Meitinger et al., 2020). Cells were maintained in DMEM/F-12 media supplemented with 10% fetal bovine serum plus antibiotics and were grown at 37 degrees in 5% CO2. Cells were split 1:10 every 3–4 days.
METHOD DETAILS
Array Comparative Genome Hybridization
Chromosomal microarray analysis (CMA) was performed with the Affymetrix CytoScan HD Assay (Thermo Fisher Scientific) following the manufacturers protocol. Briefly, 250 ng of genomic DNA was digested with the restriction enzyme NspI and then ligated to proprietary adaptors. Samples were subsequently amplified by polymerase chain reaction using a primer complimentary to the adaptor sequence and the resulting products were fragmented and labelled prior to a 16–18 hour hybridization with the array. Arrays were washed and stained using the Affymetrix F450 Fluidics Station and then scanned using the Affymetrix 7G GeneChip Scanner. All CMA data were analyzed and visualized with the Affymetrix Chromosome Analysis Suite (ChAS). Copy number variants were then exported to a local FileMaker database for further analysis.
Thermal Interlaced Asymmetric PCR
To identify the deletion breakpoints, we used high efficiency TAIL-PCR to amplify and sequence DNA from the breakpoint region as determined by the chromosomal microarray (Liu and Chen, 2007). Using nested gene-specific primers with longer arbitrarily degenerate (LAD) primers, we first isolated the flanking genomic sequences near the putative CMA-based breakpoint. We followed the thermal cycling conditions for hiTAIL-PCR, but we modified the LAD primers as follows: the four fixed and two degenerate bases at the 3’ end of the primers were replaced with the 6 base pair recognition sequences for a variety of restriction enzymes. For example, the LAD primer for XbaI restriction sites was 5’-ACGATGGACTCCAGAGCGGCCGCVNBNNNTCTAGA-3’. This modification prioritized amplification products for sequencing based on the known restriction enzyme map of the region. Products of unusual or unexpected sizes as assessed by agarose gel electrophoresis were sequenced since they likely represented products that spanned the deletion breakpoint.
Intelligence testing
All participants were assessed using the established measure of intellectual ability, the Stanford Binet Tests of Intelligence (Roid, 2003). Differences in age were accounted for by normalizing to standard scores at each age.
Generation of knock-in mutation in Anapc7
A mouse 129 BAC (bacterial artificial chromosome) containing the mouse Anapc7 genomic locus was used to generate the Anapc7 knock-out/knock-in targeting vector (BAC ID# bMQ-440B18) (Adams et al., 2005). The mouse Anapc7 gene is composed of 11 exons spanning a genomic region of 22.47kb and is represented by a 2.751 kb transcript encoding for a 565 AA protein. The desired G>A mutation (chr5: 122,438,172) was introduced into the BAC using galK based recombineering (Warming et al., 2005). This mutation was later confirmed in the BAC by sequencing. Genomic fragments containing the G>A mutation and the targeting vector homology arms were generated from the engineered BAC using gap repair (Lee et al., 2001) into a plasmid vector containing a diphtheria toxin cassette for negative selection in 129S5/SvEvTac EDJ22 ES cells. Anapc7 exons 4 – 6 (chr5: 122,432,741 – 122,435,661) were not included in the targeting vector in order to generate the knock-out allele in vivo. A loxP-neo-loxP expression cassette was inserted within a non-conserved region of the intron between exons 7 and 8 via recombineering (Lee et al., 2001) to provide a means for positive selection in ES cells. The targeting vector was linearized for ES cell electroporation with AscI upstream of the long homology arm. PCR analysis using an external primer 3’ of the short arm and an internal primer within the neomycin cassette and southern blot analysis confirmed homologous recombination and copy number in ES cells. The G>A mutation was also confirmed by sequencing. ES cells were injected into blastocysts to generate chimeras and assess germline transmission. For excision of the neomycin resistance cassette, founder male mice were crossed with female C57BL/6 mice expressing Sox2-cre. Neo excision was confirmed by PCR genotyping.
Additional mouse lines
The Fzr1/Cdh1 (García-Higuera et al., 2008) and Cdc20 mutant mice (Manchado et al., 2010) were previously described. The Math1-cre (Atoh1) was acquired from Jackson Labs.
Imaging developing embryos
Pregnant females were anesthetized with isoflurane prior to dissection. After removing the placenta and membranes, embryos were immersion fixed overnight in 4% paraformaldehyde (PFA). Imaging was carried out on an Olympus SZX16 equipped with a digital camera.
Body weight of mutant mice
A cohort of consecutive animals on the mixed C57BL/6 × 129SvJ background were weighed nearly every day through the age of 5 weeks and weekly thereafter. Because litters were asynchronous, we could collect daily data on most but not all days. Therefore, the ages for which data are missing differs from animal to animal but in a random fashion that should not bias statistical analysis with ANOVA.
One-hour locomotor activity
To evaluate general activity levels and possible alterations in emotionality, mice were evaluated over a 1-h period in transparent (47.6 × 25.4 × 20.6 cm high) polystyrene enclosures. Each cage was surrounded by a frame containing a 4 × 8 matrix of photocell pairs, the output of which was fed to an on-line computer (Hamilton-Kinder, LLC, Poway, CA). The system software (Hamilton-Kinder, LLC) was used to define a 33 × 11 cm central zone and a peripheral or surrounding zone that was 5.5 cm wide with the sides of the cage being the outermost boundary. Variables that were analyzed included the total number of ambulations and rearing on hindlimbs, as well as the number of entries, the time spent, and the distance traveled in the center area as well as the distance traveled in the periphery surrounding the center.
Sensorimotor battery
Walking initiation, ledge, platform, pole, and inclined and inverted screen tests were performed as previously described (Wozniak et al., 2004). Time in each task was manually recorded. The average for two trials was used for analyses. Test duration was 60 s, except for the pole test, which was extended to 120 s. For walking initiation, time for an animal to leave a 21 × 21 cm square on a flat surface was recorded. For ledge and platform tests, the time the animal was able to balance on an acrylic ledge (0.75 cm wide and 30 cm high), and on a wooden platform (1.0 cm thick, 3.0 cm in diameter and elevated 47 cm) was recorded, respectively. The pole test was used to evaluate fine motor coordination by quantifying time to turn 180° and climb down a vertical pole. The screen tests assessed a combination of coordination and strength by quantifying time to climb up or hang onto a mesh wire grid measuring 16 squares per 10 cm, elevated 47 cm and inclined (60° or 90°) or inverted.
Morris water maze
The Morris water maze (MWM) was conducted as previously described (Wozniak et al., 2004). Briefly, cued, place and probe trials were conducted in a galvanized steel pool, measuring 120 cm in diameter, and filled with opaque water. The PVC escape platform measured 11.5 cm in diameter. A digital video camera connected to a PC computer and the computer software program ANY-maze (Stoelting Co., Wood Dale, IL) tracked the swimming pathway of the mouse to the escape platform and quantified path length, latency to find escape platform, and swimming speeds. On two consecutive days, animals received four cued trials, separated by 1 hr, during which a red tennis ball atop a rod was attached to the escape platform and served as a visual cue. To prevent spatial learning, the escape platform was moved to a different quadrant location for each trial. The mouse was released from the quadrant opposite to the platform location and allowed 60 s to locate the platform. Once the mouse found the platform, it was allowed to remain there for 10 s before being returned to its home cage. Three days following visible platform testing, the cue was removed from the platform and it was submerged 1cm under the water for the hidden platform tests. Animals received two blocks of two consecutive trials on five consecutive days, with an inter-trial interval between 30–90 s and approximately 2 hr separating trial blocks. The escape platform remained in the same quadrant location for all trials and distal cues were placed on the walls of the room to support spatial learning. The mouse was released from a different location for each trial on each day. The mouse was allowed 60 s to find the escape platform and allowed to sit on it for 10 s before being returned to its home cage. Cued and hidden platform trials were combined into blocks of two or four trials for analyses, respectively. Following completion of hidden platform trials on the 5th day of training, the escape platform was removed from the pool and one 60 s probe trial was conducted to assess memory retention for the location of the platform.
Elevated plus maze
EPM was conducted as previously described (Wozniak et al., 2013). Briefly, in a dimly lit room, animals were placed on a black acrylic surface measuring 5 × 5 cm and elevated 63 cm above the floor equipped with photo beam pairs. Four arms (35 cm long, 5cm wide; two open and two with 15 cm high walls) extended from a central area. The MotorMonitor software (Kinder Scientific, LLC, Poway, CA) quantified beam breaks as duration, distance traveled, entries, and time at rest in each zone (closed arms, open arms and center area) during a 5 min session.
Conditioned Fear
A previously described protocol (Wozniak et al., 2007) was used to train and test mice using two clear-plastic conditioning chambers (26 × 18 × 18 cm) (Med-Associates, St. Albans, VT) which were easily distinguished by different olfactory, visual, and tactile cues present in each chamber. On day 1, each mouse was placed into the conditioning chamber for 5 min and freezing behavior was quantified during a 2 min baseline period. Freezing (no movement except that associated with respiration) was quantified using FreezeFrame image analysis software (Actimetrics, Evanston, IL) which allows for simultaneous visualization of behavior while adjusting for a “freezing threshold” during 0.75 s intervals. After baseline measurements, a conditioned stimulus (CS) consisting of an 80 dB tone (white noise) was presented for 20 sec followed by an unconditioned stimulus (US) consisting of a 1 s, 1.0 mA continuous foot shock. This tone-shock (T/S) pairing was repeated each minute over the next 2 min, and freezing was quantified after each of the three tone-shock pairings. Twenty-four hours after training, each mouse was placed back into the original conditioning chamber to test for fear conditioning to the contextual cues in the chamber. This involved quantifying freezing over an 8 min period without the tone or shock being present. Twenty-four hours later, the mice were evaluated on the auditory cue component of the conditioned fear procedure, which included placing each mouse into the other chamber containing distinctly different cues. Freezing was quantified during a 2 min “altered context” baseline period as well as over a subsequent 8 min period during which the auditory cue (CS) was presented. Shock sensitivity was evaluated following completion of the conditioned fear test as previously described (Khuchua et al., 2003).
Following a one-week delay, mice were returned to the conditioning chamber (context, CS), and freezing (conditioned response) was assessed over a 6 min period in the absence of a shock or tone. On the subsequent day, the mice were evaluated on the auditory cue component of the conditioned fear procedure as described above.
Generation of lysates from viscera
Total lysates from organs of wild-type P3 and P70 mice were generated by dicing tissue and then homogenizing by Dounce A then B in RIPA buffer containing 1 mM DTT, protease inhibitor cocktail (Sigma P8340), phosphatase inhibitor cocktail #3 (Sigma P0044), and 0.2 mM PMSF. Lysates were clarified by centrifugation at > 20,000 × g.
Protein quantitation in lysates
We used the Bradford reagent to quantify protein throughout this study. Each sample was assayed in triplicate and the median value taken. Lysis buffer served as the blank.
Fractionation of mouse brain
Dissected whole brain, cortex or cerebellum were diced and then subjected to 10 strokes with Dounce A in cold hypotonic buffer (10 mM HEPES pH 7.9, 1.5 mM MgCl2 and 10 mM KCl) containing 1 mM DTT, protease inhibitor cocktail (Sigma P8340), phosphatase inhibitor cocktail #3 (Sigma P0044), and 0.2 mM PMSF. Samples were rotated at 4 degrees for 15 min before 20 more strokes with Dounce A. Each sample was then split in two. For total fractions, NaCl and triton X-100 were then added to final concentrations of 250 mM and 0.1%, respectively, before vortexing to ensure nuclear lysis. Samples rotated for 10 more min at 4 degrees before centrifugation at >20000 × g for 10 min to remove insoluble chromatin. The other portion of the total lysate was subjected to centrifugation at 800 × g for 5 min. The cytoplasmic fraction was the supernatant after this low-speed spin. The nucleus-containing pellet that resulted from low-speed centrifugation was then washed 2 times in hypotonic buffer before lysis in 250 mM NaCl plus 0.1% triton X-100 and 10% glycerol. After vortexing, insoluble chromatin was removed by centrifugation at >20000 × g for 10 min. This supernatant represented the nucleoplasmic lysate fraction.
Immunoblotting
For immunoblots we used Invitrogen NuPage Bis-Tris 4–12% gels and LDS sample buffer followed by transfer onto Nitrocellulose membranes via tank immersion. To quantitate loading, prior to blocking membranes were incubated with the ReVert total protein stain (LI-COR) and imaged on a LI-COR Odyssey CLx. This stain was then removed using the manufacturer’s method. Blocking and antibody-incubation steps were performed in Tris-buffered saline with 0.1% Tween-20 and 2% BSA. Infrared fluorescent secondary antibodies were detected by the LI-COR Odyssey CLx. Blots were imaged at 300 dpi and we did not perform any further processing of raw images after the acquisition step. Optical densitometry and relative protein abundance were measured using the LI-COR Image Studio software. Exposure time was chosen to avoid pixel saturation.
For Ki-67 immunoblot in brain, we isolated nuclei with hypotonic buffer as described above. The pellet was then resuspended in hypotonic buffer with 0.1% Triton X-100. Endonuclease digestion with benzonase was performed for 1 hour at 4 degrees while rotating. Nuclei were lysed by addition of NaCl to 250 mM, vortex and 15 additional min of rotation at 4 degrees. The resulting solution was opaque but uniform and could be accurately pipetted without the need for centrifugation, and therefore represented a total nuclear lysate that included chromatin.
For lysis of patient lymphoblasts, cells were spun down and the pellet washed in PBS prior to lysis in hypotonic buffer with 0.1% triton, DTT and protease and phosphatase inhibitors. DNA was digested using benzonase for 1 hour at 4 degrees prior to adding NaCl to a final concentration of 300 mM and vortexing. Lysis of primary mouse cerebellar granule neurons for the purpose of Ki-67 immunoblotting was performed using a similar approach after using a cell scraper to remove neurons from plates.
Immunoprecipitation
For immunoprecipitation of APC7, APC1 and APC3 from brain, we generated total cell lysates (cytoplasmic plus nuclear without chromatin) via two-stage lysis as described above (hypotonic buffer then followed by 250 mM NaCl and Triton X-100 and centrifugation to pellet chromatin). During the immunoprecipitation, antibodies were present at 1–2 μg / mg of lysate. After antibody binding for 4 hours, 15 μL of washed protein G agarose beads (GE Healthcare) were added per immunoprecipitation. Bead binding proceeded for 1 hour before washing beads 3 times in lysis buffer with triton and NaCl at a final concentration equivalent to lysates. For downstream immunoblotting, LDS sample buffer was added and samples were heated to 70 degrees for 10 min. A similar strategy was used for isolating the APC from HAP1 cells. Cells were trypsinized and the cell pellet was washed with PBS prior to two-stage cytoplasmic then nuclear lysis as above with centrifugation at >20,000 × g to pellet insoluble material prior to immunoprecipitation.
For Ki-67 immunoprecipitation, nuclei were isolated and treated with benzonase as above prior to nuclear lysis with 250 mM NaCl and a spin to remove the insoluble chromatin. The resulting supernatant was used as input for immunoprecipitation of Ki-67 as described above using protein G beads. Immunoprecipitation of Ki67-RFP was carried out in a similar fashion but capture was via anti-RFP trap resin. In both cases beads were washed in lysis buffer before adding LDS sample buffer and heating to 70 degrees for 10 min and immunoblotting.
Ubiquitination assays with immunoprecipitated APC
For assessment of APC-mediated ubiquitination in P5 brain, total lysates without chromatin were prepared as above. Immunoprecipitation was also carried out as above with antibodies to APC3 or APC7, however after binding to protein G beads and washing in lysis buffer, beads were then washed with hypotonic buffer 2 times to remove detergent from the bead supernatant, which inhibits enzyme activity. Beads were resuspended in a master mix that contained Ube2C, Cdh1, and fluorescent substrate and allowed to come to 37 degrees before adding a second mix with ubiquitin and the E1 Ube1 to start the reaction. The final concentration of ubiquitin was 100 μM, Ube1 100 nM, Ube2C 500 nM (ranging from 0.06 to 1 M for “dose response” experiments), Cdh1 500 nM and fluorescent substrate 250 nM (Jarvis et al., 2016). Reactions were performed in a 37 degree incubator shaking at 200 RPM to keep beads suspended. Reactions were stopped by addition of sample buffer and boiling followed by running out the supernatant on Tris-glycine SDS-PAGE gels. For experiments in which the abundance of a single component was adjusted (e.g. Cdh1 or Ube2C), reactions proceeded for 45 min. Fluorescent cyclin-B1 NTD and securin were detected on a Bio-Rad Chemi-Doc with 300 dpi resolution. There was no further processing of raw images after the acquisition step. Quantitation of substrate modification was carried out using the LI-COR Image Studio software. All pixels were within the dynamic range because the exposure settings were chosen to avoid signal saturation.
For in vitro ubiquitination of full-length Ki-67, we began by performing Ki-67 immunoprecipitation from benzonase-treated nuclear extracts from wild-type P7 cerebellum as described above. Purified recombinant human APC or APCΔ7 was generated as described above (Jarvis et al., 2016) and used as the source of E3 enzyme at a final concentration of 15 nM. Ubiquitin, E1, E2, Cdh1 and Cdc20 were supplied at concentrations listed above (Jarvis et al., 2016). A phosphomimetic version of the APC that can function with both coactivators Cdc20 and Cdh1 was used (Qiao et al., 2016). Reactions were carried out in a 37 degree incubator shaking at 200 RPM to keep beads suspended. At the end of the reaction, beads were washed twice in hypotonic buffer to remove soluble reaction components before adding LDS sample buffer and placing at 70 degree for 10 min. For assessing polyubiquitination of Ki-67 by Ube2S, lysis and immunoprecipitation were performed as before with the exception that all steps were carried out in the presence of the deubiquitinase inhibitor iodoacetamide at 0.1 M. In all cases, Ki-67 was detected by anti-Ki-67 immunoblot.
Purification of APCΔ7 for cryo-electron microscopy
Recombinant APC for cryo-electron microscopy (Cryo-EM) was expressed as described previously (Brown et al., 2016; Weissmann et al., 2016). Briefly, APC and Cdh1 were expressed independently in Baculovirus-infected High Five insect cells (Thermo Fisher Scientific). Following sonication, the cleared lysate was treated to a Strep-affinity chromatography selection for a twin-strep tag at the N-terminus of APC4. For apo-APCΔ7, Strep elution fractions containing protein were further purified by Size-exclusion chromatography using Superose 6 10/300 column (GE Healthcare). APCΔ7-Cdh1 with chemically stabilize proxy or the UBE2C–ubiquitin–substrate transition state, referred to as Flag-UBE2C~Ubv~Substrate, was prepared as described previously (Brown et al., 2016). Cells expressing APC were mixed with cells expressing Cdh1 for colysis by sonication. Following sonication, cleared lysate was applied to a Strep-affinity chromatography selection for a twin-strep tag at the N-terminus of APC4. Strep eluant was incubated with Flag-UBE2C~Ubv~Substrate, TEV protease, and HRV14 3C-protease for 1 hour. This mixture was further purified through FLAG affinity chromatography and eluted with antigenic peptides. 125 μg of purified APCΔ7-Cdh1 with FLAG-UBE2C~Ubv~Substrate was loaded onto a 10%–40% glycerol gradient containing 50 mM HEPES pH 8.0, 200 mM NaCl, 2 mM MgCl2. For particle fixation by GraFix (Kastner et al., 2008), the gradient also contained 0.025% and 0.1% glutaraldehyde in the lighter and denser glycerol solution, respectively, creating an additional glutaraldehyde gradient from top to bottom (0.025–0.1%). Centrifugation was performed at 34,000 RPM in a SW55TI rotor (Beckman) for 15 hr at 4°C and fractions containing APC were subjected to a buffer exchange procedure using Zeba spin columns (Pierce) to remove the glycerol prior to EM grid preparation. For both complexes, APC particles were allowed to adsorb on a thin film of carbon for 5 min, transferred onto a cryo-EM grid (Quantifoil R2/1, Jena) and then plunged into liquid ethane under controlled environmental conditions of 4 °C and 100% humidity in a vitrification device (Vitrobot Mark IV, FEI Company, Eindhoven).
Cryo-EM imaging and data processing
Micrographs were recorded on a K2 direct detector (operated in electron counting mode) under liquid-nitrogen conditions with a Titan Krios electron microscope (Thermo Fisher Scientific) equipped with a GIF quantum energy filter (20 eV) (Gatan). Automated data collection was carried out using SerialEM (Mastronarde, 2005). Movies were recorded at a nominal magnification of 105,000x that corresponds to 1.34 A ° /pixel at the specimen level. A total exposure of ~60 e−/A2 was distributed across 50 frames, with a defocus range of −1.2 μm to −3.6 μm. Movies were motion-corrected using MotionCor2 (Zheng et al., 2017) with dose weighting. Contrast transfer function was estimated using CTFFIND (Rohou and Grigorieff, 2015). Particles were picked with Gautomatch (K. Zhang, MRC Laboratory of Molecular Biology). Reference free two-dimensional classification was performed in RELION-3.0 (Zivanov et al., 2018) and resulting 2D classes were manually examined and selected for further processing. Selected particles underwent further 3D classification, refinement, post-processing and particle polishing. The resolution was estimated by applying a soft mask around the protein density and using the gold standard Fourier shell correction (FSC) = 0.143 criterion, as implemented in the RELION post-processing routine. Similar procedure was followed for both APCΔ7 and APCΔ7 with Flag-UBE2C~Ubv~Substrate, and details are listed in Table S2. UCSF Chimera (Pettersen et al., 2004) was used for rigid-body docking and figure making.
In vitro ubiquitination assays with recombinant APC
Purified proteins were generated as previously described (Dou et al., 2012; Jarvis et al., 2016). A phosphomimetic version of the APC that can function with both coactivators Cdc20 and Cdh1 was used (Qiao et al., 2016). The Ubiquitination assay reactions were prepared by mixing ubiquitination components in assay buffer (20 mM Hepes pH 8, 200 mM NaCl) on ice, warming them to room temperature, and adding ubiquitin to initiate the reactions. Reactions were quenched using 6x SDS loading buffer at designated time points and separated by SDS-PAGE. Unmodified substrates and modified products were visualized by scanning gels using the Amersham Typhoon 5 to detect fluorescently labeled substrates. Substrates and products were measured and quantified using either Bio-rad ImageLab or ImageQuant software.
Qualitative assays probing APC activity with different E2s were performed with 20 nM APC or APCΔ7, 1 μM E1, 1 μM Cdh1, 0.5 μM substrate, 2 μM Ube2C and/or Ube2S, or 8 μM UbcH5B, and 125 μM Ub. Substrates were fluorescent CycB-NTD, Ub-CycB-NTD and securin. Reactions were quenched at 10 min.
Ubiquitination assays monitoring the rate of substrate depletion in the presence of Ube2C at high and low substrate concentrations were performed with 20 nM APC or APCΔ7, 2 μM Ube2C, 1 μM E1, 125 μM Ub. Bands corresponding to unmodified substrate were quantitated and normalized to the reaction at time zero.
Kinetic ubiquitination assays monitoring the dose-dependent effects of Cdh1 were carried out using 20 nM APC, with 1 μM E1, 2 μM Ube2C, 0.5 μM substrate, 125 μM Ub, and a range of 0.05 to 5 μM Cdh1. Reaction kinetics were plotted and fit using Michaelis-Menten Kinetics and GraphPad Prism.
Single encounter ubiquitination assays
Single encounter experiments were used to examine ubiquitination of substrate during a single substrate binding event on APC. Ubiquitination assays using methyl-ubiquitin and fluorescein labeled substrate with a single lysine were performed to examine Ube2C-dependent substrate priming. For processive multiubiquitination, fluorescent securin was used as the substrate with wild-type ubiquitin. This was monitored by using a fluorescein-labeled substrate and excess unlabeled Hsl1, such that Cdh1-APC dissociated from labeled substrate is rapidly sequestered with unlabeled substrate. Reaction components were prepared in two independent mixes. The first mix, APC-substrate, consisted of APC, Cdh1, fluorescent CycB-NTD(1K) or securin, and Ube2SCTP or Ube2SCTP−4A, when indicated. This was incubated on ice for 30 min. The second mix, E2-Ub, consisted of E1, E2, MgCl2, ATP, and 600-fold excess of unlabeled Hsl1 or 1000-fold excess unlabeled Securin. Ubiquitin or methylated-ubiquitin was added to the second mix 5 min before completion of the 30 min incubation of APC-substrate mixture. Both mixes were allowed to reach room temperature for 5 min. Immediately, both reactions were co-mixed and quenched at 3 min. Final concentrations of all components after initiating reactions were: 100 nM E1, 200 nM APC, 0.5 μM Cdh1, 80 nM cycB-NTD(1K) or 160 nM securin, 48 μM Hsl1 or 160 μM securin, 10 μM Ube2SCTP or Ube2SCTP−4A, and 132 μM wild-type Ub or 320 μM meUb. Bands corresponding to primed substrate and unmodified substrate were quantitated. Values were plotted as a fraction of primed substrate to unmodified substrate over time.
Cloning and purification of Ki-67 fragments
Truncated Fragments of the human Ki-67 protein corresponding to amino acids 1–300 and 765–1000 were subcloned into pGEX vectors to generate GST-tagged proteins after PCR amplification from the full-length Ki-67 cDNA (a gift from Daniel Fisher). DNA fragments containing the KEN to AAA mutations were ordered as synthetic DNA molecules from Genewiz, and we utilized convenient restriction sites in the Ki-67 cDNA to substitute these fragments harboring AAA substitutions. Ki-67 fragments were then expressed as a fusion protein with GST and His6-tag at its N- and C-terminus, respectively. Protein fragments were purified by a glutathione and nickel affinity chromatography. After TEV-cleavage to remove the GST from Ki-67, the fragments were fluorescently labeled by a sortase reaction (Martinez-Chacin et al., 2020).
TMT proteomics
We used Anapc7−/− mutant mice and sex-matched littermates of both sexes on the C57BL/6 background. P8 cerebellum was flash frozen in liquid nitrogen and processed using the streamlined tandem mass tag (TMT) labelling protocol (Navarrete-Perea et al., 2018). Samples were lysed in 8 M urea in 200 mM EPPS pH 8.5 with protease (Pierce A32953) and phosphatase (Pierce A32957) inhibitors, and passed through a 21-guage needle 10x. Samples were reduced with 5 mM TCEP, alkylated with 10 mM iodoacetamide, and quenched with 5 mM DTT, followed by methanol/chloroform precipitation of 150 μg protein. Pellets were reconstituted in 200 mM EPPS pH 8.5, digested overnight with LysC (Wako 129–02541) at 1:100 while shaking at room temperature, followed by digestion with trypsin (Pierce 90305) at 1:100 while shaking at 37 degrees. Anhydrous acetonitrile (Honeywell AS017–0100) was added to ~30%, followed by labelling with TMT11 (Thermo Fisher A37727) reagent. 1% of each labeled sample was combined and analyzed unfractionated to ensure labeling efficiency was >97% and that the samples are mixed at a 1:1 (total amount) ratio across all conditions. After mixing, labelled peptide samples were de-salted using a 200 mg Sep-Pak cartridge (Waters WAT054925), followed by drying in a rotary evaporator. Samples were then reconstituted in 5% ACN 10 mM ammonium bicarbonate for basic reverse phase fractionation on an Agilent 300extend-C18 column (3.5 μm, 4.6 × 250 mm) using an Agilent Infinity 1260 HPLC. Peptides were subjected to a 75 min linear gradient from 13% to 42% of Buffer B (10 mM ammonium bicarbonate, 90% ACN, pH 8) at a flow rate of 0.6 mL/min, resulting in a total of 96 fractions which were consolidated into 24 by combining (in a chessboard pattern) four alternating wells down columns of the 96-well plate. Assuming adjacent fractions contain overlapping peaks, only 12 non-adjacent samples were analyzed by the mass spectrometer. The pooling scheme has been illustrated previously (Navarrete-Perea et al., 2018; Paulo et al., 2016a). Each eluted fraction was desalted via StageTip for SPS-MS3 analysis.
Mass spectra were collected on Orbitrap Lumos mass spectrometer (Thermo Fisher Scientific) coupled to a Proxeon EASY-nLC 1200 LC pump (Thermo Fisher Scientific). Peptides were separated on a 35 cm column (i.d. 100 μm, Accucore, 2.6 μm, 150 Å) packed in-house using a 90 min gradient (from 5% - 30% acetonitrile with 0.1% formic acid) at 500 nL/min. Each analysis used an SPS-MS3-based TMT method (McAlister et al., 2014; Ting et al., 2011), which has been shown to reduce ion interference compared to MS2-based quantification (Paulo et al., 2016b). MS1 data were collected using the Orbitrap (120,000 resolution; maximum injection time 50 ms; AGC 4e5, 400–1400 m/z). Determined charge states between 2 and 5 were required for sequencing and a 90 s dynamic exclusion window was used. MS2 scans consisted of collision-induced dissociation (CID), quadrupole ion trap analysis, automatic gain control (AGC) 2E4, NCE (normalized collision energy) 34, q-value 0.25, maximum injection time 35 ms, and isolation window of 0.7 Da using a Top10 method. Through Thermo Fisher Scientific’s instrument application-programming interface (iAPI) for Tribrid mass spectrometers, an on-line real-time search (RTS) algorithm (using the UniProt Mouse database, December 2018) was used to trigger MS3 scans for quantification (Erickson et al., 2019; Schweppe et al., 2020). MS3 scans were collected in the Orbitrap at a resolution of 50,000, NCE of 45% (11-plex), maximum injection time of 100 ms, and AGC of 1.5e5. Data for the 12 fractions used a close-out of 2, which limited up to two MS3 measurements per protein per fraction, and all were combined and filtered to achieve a <1% FDR.
Mass spectra were processed using a SEQUEST-based software pipeline (Eng et al., 1994; Huttlin et al., 2010). Data were searched against the UniProt Mouse database (December 2018), using a 20-ppm precursor ion tolerance for total protein-level analysis and 0.9 Da product ion tolerance. TMT tags on lysine residues and peptide N-termini (+229.163 Da) and carbamidomethylation of cysteine residues (+57.021 Da) were set as static modifications, while oxidation of methionine residues (+15.995 Da) was set as a variable modification. Peptide-spectrum matches (PSMs) were identified, quantified, and filtered to a 1% peptide false discovery rate (FDR) and then collapsed further to a final protein-level FDR of 1%. Proteins were quantified by summing reporter ion counts across all matching PSMs. Briefly, a 0.003 Da (3 millidalton) window around the theoretical m/z of each reporter ion was scanned and the maximum intensity nearest the theoretical m/z was used. Reporter ion intensities were adjusted to correct for the isotopic impurities of the different TMT reagents according to manufacturer specifications and adjusted to normalize ratios across labelling channels. Lastly, for each protein, signal-to-noise (S:N) measurements of the peptides were summed and then normalized to 100.
Cerebellar granule neuronal culture
Cultures of mouse primary cerebellar granule neurons were generated as previously described (Bilimoria and Bonni, 2008). For imaging experiments, primary granule neurons were plated onto acid treated, poly-L ornithine coated optical cover glass in 24 well dishes with 750–900 thousand cells per well. For RT-PCR and immunoblotting, neurons were plated onto PLO coated plastic 3.5 cm dishes with 4.5 million cells per well. High plating density delayed differentiation, facilitating the study of the early post-mitotic period of neuronal differentiation.
Quantitative reverse transcriptase PCR (RT-qPCR)
Total RNA was isolated using Trizol and RNeasy (Qiagen) columns. For assessing the abundance of the APC7 and Ki-67 transcripts, we generated cDNA using the Bio-Rad iScript with polyA selection. A no RT control was used for each amplicon to ensure there was not spurious amplification of contaminating DNA. Each amplicon was designed to be 100–250 nucleotides and to span an intron >1 kb. We used the SYBR green reagent and the QuantStudio 6 machine (Applied Biosystems).
For assessment of expression of repetitive sequences that are enriched in constitutive heterochromatin (e.g. major satellite, LINE and IAP), we began by isolating RNA as described above. We then treated RNA with DNase I (NEB) according to the manufacturer’s protocol. We generated cDNA using Bio-Rad iScript without polyA selection and included the essential no RT control in our experiment. The abundance of each transcript was normalized to GAPDH we subtracted the signal observed under no RT conditions from the amount of transcript detected in the RT condition to compensate for the small amount of DNA remaining after DNase treatment.
Histology
Brains were immersion fixed in 4% PFA overnight at 4 degrees before processing for routine histology with H&E staining. Brightfield imaging was performed on Olympus BX-51 equipped with Olympus SC180 camera.
In situ hybridization
Custom RNAScope probes to detect the APC7 mRNA were designed by and purchased from ACDbio. Staining was carried out on a Ventana system with antigen retrieval and brightfield imaging on an Olympus BX-53 with Olympus SC180 camera.
Immunofluorescence in tissue
Mice were anesthetized with isoflurane (>P10) or decapitated (<P10) before brain dissection and immersion fixation of the whole brain in 4% paraformaldehyde overnight at 4 degrees. Brain tissue was then placed in 30% sucrose in PBS until it sank, rocked in equal parts OCT & 30% sucrose until homogeneous, and placed in pure OCT overnight. Tissue was flash frozen using liquid nitrogen and then sectioned at 20 μm in a Leica cryostat. Triton X-100 was used for permeabilization and goat serum for blocking. Primary antibody binding occurred overnight at 4 degrees and secondary binding took 2 hours at room temperature. Hoechst was applied during secondary antibody incubation at 1:3000. Antigen retrieval was required for labeling with anti-Ki67, PCNA, and BrdU. This was accomplished by placing slides in boiling sodium citrate buffer (0.1 M, pH 6.8 with 0.1% Tween-20) for 10 min prior to blocking.
BrdU injection
BrdU was introduced into pups or pregnant females by intraperitoneal injection with 200 mg / kg BrdU. We detected labeled cells using anti-BrdU immunofluorescence in tissue prepared at the ages indicated in figure legends.
Immunocytochemistry
Cultured primary cerebellar granule neurons were fixed in 4% paraformaldehyde for 15 min at room temperature for 15 min before washing in PBS, permeabilization in 0.3% triton X-100 and blocking in 3% goat serum. Primary antibodies were incubated for 2 hours at room temperature and secondary antibodies for 45 min at room temperature. Hoechst was applied during secondary antibody incubation at 1:3000.
Epifluorescence imaging
Zeiss Axioimager was used to image tissue in Figures 5M, S5A, S5C, S5D, S5E, S5F, S5G and throughout Figure S6). The Apotome device was employed while imaging anti-Calbindin IF (Figure 6B) to generate 1 μm Z-stacks over 5 μm, which were subsequently flattened in ImageJ by making maximum intensity Z-projections. Post-processing entailed adjusting brightness and contrast and was performed in Photoshop.
Confocal imaging
We used the Zeiss 880 LSM2 equipped with Airyscan hexagonal detector array for near super-resolution confocal microscopy of cultured neurons (Figures 5, 6 and 7), E16 cortical plate (Figure 7A, B) and cerebellum (Figure S5B). Stacks of 10 images with 0.8 μm Z-steps were acquired using the 63x objective without digital zoom followed by Airyscan post-processing with the default settings. For displayed images, Z-stacks were flattened in ImageJ by making maximum intensity Z-projections. Each image contained an average of between ~30 and 80 cells. For quantitative image analysis, raw images were analyzed. We attempted to blind ourselves to genotype during imaging, but the Ki-67 phenotypes is so obvious that cultures could essentially be genotyped via anti-Ki67 staining. Therefore, to ensure there was no bias in the choice of individual fields for imaging, each field was chosen based exclusively upon the Hoechst channel with the microscopist agnostic to the signal in other channels.
For imaging of mitotic cells, the 63x objective was combined with 7x digital zoom to acquire 0.16 μm optical sections prior to Airyscan post-processing with default settings. Post-processing was performed in Photoshop only for images which were displayed, whereas raw images were used for quantitation.
Image analysis – total fluorescence
Raw images of primary cerebellar granule neurons were acquired as above and processed in ImageJ (Schneider et al., 2012) in a completely blinded fashion. Z projections were made through a sum function rather than maximum intensity. The Hoechst channel was used to identify regions of interest (cell nuclei) by converting into a binary image and applying the watershed tool. We set bounds on nuclear circularity and size to exclude inappropriately partitioned nuclei or nuclei from non-granule neurons. This caught most errors in selecting nuclei, however, the few remaining abnormally partitioned nuclei were manually excluded from analysis. The Hoechst binary image was used to calculate the area of each nucleus. The total intensity of Ki-67 staining of each nucleus was measured and would be later divided by its area. Background fluorescence was defined as the single nucleus with the lowest level of area-normalized Ki-67 intensity in either genotype at any stage of culture, and this value was subtracted from the values for all the other nuclei for the purpose of display.
Mitotic cells were identified based upon chromosomal morphology and imaged as described above. Regions of interest (individual mitotic genomes) were manually outlined and the total fluorescence intensity of the Hoechst and Ki-67 channels was measured. Ki-67 fluorescence was then normalized to Hoechst. These values were then normalized to the average of control cells in pro-metaphase.
Image analysis – linear intensity
Cells at different stages of mitosis were identified based upon chromosomal morphology and imaged as described above before being analyzed in a blinded fashion. To assess the fraction of Ki-67 on the chromosome periphery, fluorescence intensity was measured in ImageJ along a line starting from the outside (0 μm) and bisecting the chromosome periphery perpendicularly. 3–6 lines were drawn per image. Fluorescence intensity values were then aligned based upon peak fluorescence.
Image analysis – colocalization
Triple labeled 63x images of primary granule neurons were acquired as described above. Maximum intensity Z-projections of the raw confocal micrographs were analyzed using the ImageJ Colocalization plug-in. We recorded the Rcoloc value of each fluorescence channel. Nuclei were then counted by generating a binary using the Hoechst channel as described above. We then calculated a weighted average for the Rcoloc value of each image by correcting for the fraction of the total cells that were present in a given image.
Generation of inducible ANAPC7 CRISPR-Cas9 lines
To generate inducible ANAPC7 knock-out RPE-1 lines, we used a similar strategy as described before (Meitinger et al., 2020). In brief, four gRNAs targeting ANAPC7 (see Table S4) originating from the Brunello gRNA library (Doench et al., 2016) were cloned into the lentiGuide-Puro vector (a gift from F. Zhang; addgene plasmid 52963). Lentiviral particles were generated by transfecting the constructs into HEK-293T cells using Lenti-X Packaging Single Shots (Takara Bio USA). Forty-eight hours after transfection, virus-containing culture supernatant was collected.
RPE-1 cells expressing Cep192-mNeonGreen (in situ-tagged) and doxcycline-inducible Cas9 (Meitinger et al., 2020) were infected with the gRNA lentiviruses in the presence of 8 μg/ml of polybrene (EMD Millipore) and selected with 8 μg/ml of puromycin. To induce Cas9 expression, cells were treated with 0.1 μg/ml of doxycycline for 24 hr. After washing doxycycline off, cells were incubated for 48hr. Cells were then collected and their genomic DNA was extracted and amplified by PCR using Q5 DNA polymerase (NEB) and specific oligo pairs for each gRNA (see Table S4). PCR products were analyzed by Sanger sequencing followed by TIDE analysis to score for the induction of indels upon Cas9 expression. This analysis revealed that gRNAs #2 and #4 were the most efficient and showed the least amount of indels in the absence of doxycycline. Cells were further transduced with lentivirus encoding for mRFP-Histone H2b and RFP-positive populations selected by FACS-sorting.
Live-cell imaging
RPE-1 cells were treated with doxycycline as described above and seeded into 96-well polystyrene plates (Greiner; cat #655090) 4 hr prior to imaging. Live-cell Imaging was performed on a CQ-1 spinning disk confocal system (Yokogawa) with a 20x (0.75 NA) objective and a 2560 × 2160 pixel sCMOS camera. Image acquisition was performed using CellVoyager software. The humidity-controlled imaging chamber was maintained at 37°C and 5% CO2. 5 fields per well were imaged and a 2×2 binning protocol was used. 4 × 3 μm z-sections in the 561 nm (20% laser power, 200 ms exposure) channel was captured in each field at 5-minute intervals for 14 hours. Imaging analysis was done using ImageJ.
Flow cytometry analysis of cell cycle
Dissociated cerebellum was prepared as for primary granule neurons cultures and fixed in 70% ethanol overnight. Lymphoblasts were washed and fixed overnight in ethanol. To ensure only DNA was detected, cells were treated with RNase A (NEB) overnight degrees at 4 degrees in the presence of propidium iodide at 100 μg/mL to stain DNA. Cells were flowed through an LSR Fortessa or BD Canto cytometer and excited by a 488 nm laser. Subsequent analysis was carried out using FlowJo software.
Flow cytometry analysis of Ki-67 labeling in nuclei
Nuclei were isolated from P8 cerebellum as described above. Nuclei were then fixed in 4% PFA, permeabilized in triton X-100, blocked in 2.5 % goat serum and incubated with anti-Ki67 or Rat IgG isotype control overnight at 4 degrees while rotating. Nuclei were then washed and incubated with secondary antibodies and DNA was stained with Hoechst. Nuclei were flowed through an LSR Fortessa cytometer with 405 nm and 488 nm lasers and analyzed with FlowJo software.
X-ray microscopy
Cerebellum was dissected from P9 mice after immersion fixation overnight at 4 degrees in 4% PFA, followed by processing and Lugol staining as previously described (Degenhardt et al., 2010). Images were acquired on a Zeiss Xradia Versa 520 XRM and processed using Dragonfly software.
Transmission electron microscopy
A fresh solution of 2.5% glutaraldehyde + 2% paraformaldehyde in 0.15 M cacodylate buffer pH 7.4 with 2 mM calcium chloride was warmed to 37 degrees and applied to primary cerebellar granule neuron cultures and incubated at 37 degrees for 5 min. Samples were fixed for one additional hour at room temperature before transfer to ice and overnight fixation at 4 degrees. Coverslips were then rinsed in 0.15 M cacodylate buffer 3 times for 10 min each, and then subjected to a secondary fixation for one hour in 1% osmium tetroxide/0.3% potassium ferrocyanide in cacodylate buffer on ice. Subsequently, samples were washed in ultrapure water 3 times for 10 min each and en bloc stained for 1 hour with 2% aqueous uranyl acetate. Post staining, samples were briefly washed in ultrapure water, dehydrated in a graded acetone series (50%, 70%, 90%, 100% x2) for 10 min in each step, infiltrated with microwave assistance (Pelco BioWave Pro, Redding, CA) into LX112 resin, and flat embedded in a polypropylene petri dish and cured in an oven at 60°C for 48 hours. Once the resin blocks were cured, coverslips were exposed with a razor blade and etched off with concentrated hydrofluoric acid. Small pieces of the resin containing cells were then cut out by saw and mounted onto blank resin stubs before 70 nm thick sections were cut in the cell culture growth plane. Sections were placed onto Formvar/Carbon Support Film Hexagonal 100 mesh Cu Grids (Electron Microscopy Sciences, Hatfield, PA). Each grid was then counter-stained with 2% aqueous uranyl acetate for 10 min and rinsed with ultrapure water three times, and then stained once more with Reynold’s lead citrate for 5 min and rinsed with ultrapure water three times. Stained grids were imaged on JEOL 1200 EX equipped with a digital camera.
QUANTIFICATION AND STATISTICAL ANALYSIS
We used GraphPad Prism 8 software to perform the statistical tests described in figure legends. For parametric data with two comparators, we used the two-tailed student’s t-test. For nonparametric data with two comparators, we used the Kolmogorov-Smirnov test. One-way ANOVA with Tukey’s post-hoc test was used for comparisons of parametric data between three or more groups. To compare genotypes over time, we used repeated measures ANOVA when data were complete followed by Bonferroni or Tukey’s post-hoc tests, as indicated in figure legends. A mixed-effects ANOVA model was used when data were incomplete, for example with daily body weight measurements. For details of the quantitative methods used in TMT proteomics and cryo-EM, please refer to the above sections.
Supplementary Material
Table S3 (Related to Figure 5): Tandem-mass tag (TMT) proteomics in control and Anapc7−/− cerebellum at P8.
Highlights.
ANAPC7 mutation causes a recessive intellectual disability syndrome
APC7 is required for efficient ubiquitination by the E3 ubiquitin ligase APC
APC7 promotes degradation of Ki-67 during post-mitotic neuronal differentiation
Ki-67 clearance from constitutive heterochromatin of neurons requires APC7 and Cdh1-APC
ACKNOWLEDGMENTS
We thank the Bonni laboratory for critical feedback. We also thank C. Yuede, D. Wozniak, S. Conyers, J. Gunn, S. Joh, R. Lewis, M. Wallace, H. Stark, D. Haselbach, D. Pizzo, L. Kaube, D. Bolhuis, F. Meitinger, K. Williams, L. Poskitt, M. Young and T. Morlet. This work was supported by the NIH grant NS051255 (AB); NIH K08HD099314 (CJF); NIH R35GM128855 and UCRF (NGB); NIH R37GM065930, NIH P30CA021765 and the Max Planck Society (BAS); NIH GM67945 (SPG); NIH T32GM008570 and NSF DGE-1650116 (TB); NIH R01GM074215 (AD); and NIH OD021629 (WUCCI).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests. Azad Bonni is an employee of Roche. Brenda Schulman is on the Scientific Advisory Board for BioTheryX and Interline Therapeutics and a shareholder of Interline Therapeutics, and a co-inventor of intellectual property licensed to Cinsano.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Adams DJ, Quail MA, Cox T, Van Der Weyden L, Gorick BD, Su Q, Chan WI, Davies R, Bonfield JK, Law F, et al. (2005). A genome-wide, end-sequenced 129Sv BAC library resource for targeting vector construction. Genomics 86, 753–758. [DOI] [PubMed] [Google Scholar]
- Avagliano L, Parenti I, Grazioli P, Di Fede E, Parodi C, Mariani M, Kaiser FJ, Selicorni A, Gervasini C, and Massa V (2020). Chromatinopathies: A focus on Cornelia de Lange syndrome. Clin. Genet. 97, 3–11. [DOI] [PubMed] [Google Scholar]
- Basak O, van de Born M, Korving J, Beumer J, van der Elst S, van Es JH, and Clevers H (2014). Mapping early fate determination in Lgr5+ crypt stem cells using a novel Ki67-RFP allele. EMBO J. 33, 2057–2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilimoria PM, and Bonni A (2008). Cultures of cerebellar granule neurons. Cold Spring Harb. Protoc. 3, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu C, Morin GB, Harley CB, Shay JW, Lichtsteiner S, and Wright WE (1998). Extension of Life-Span by Introduction of Telomerase into Normal Human Cells. 279. [DOI] [PubMed] [Google Scholar]
- Brown NG, VanderLinden R, Watson ER, Qiao R, Grace CRR, Yamaguchi M, Weissmann F, Frye JJ, Dube P, Ei Cho S, et al. (2015). RING E3 mechanism for ubiquitin ligation to a disordered substrate visualized for human anaphase-promoting complex. Proc. Natl. Acad. Sci. USA 112, 5272–5279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown NG, VanderLinden R, Watson ER, Weissmann F, Ordureau A, Wu KP, Zhang W, Yu S, Mercredi PY, Harrison JS, et al. (2016). Dual RING E3 architectures regulate multiubiquitination and ubiquitin chain elongation by APC/C. Cell 165, 1440–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang L, Zhang Z, Yang J, McLaughlin SH, and Barford D (2014). Molecular architecture and mechanism of the anaphase-promoting complex. Nature 17, 13–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang L, Zhang Z, Yang J, McLaughlin SH, and Barford D (2015). Atomic structure of the APC/C and its mechanism of protein ubiquitination. Nature 522, 450–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craney A, and Rape M (2013). Dynamic regulation of ubiquitin-dependent cell cycle control. Curr. Opin. Cell Biol. 25, 704–710. [DOI] [PubMed] [Google Scholar]
- Cuylen-Haering S, Petrovic M, Hernandez-Armendariz A, Schneider MWG, Samwer M, Blaukopf C, Holt LJ, and Gerlich DW (2020). Ki-67-regulated chromosome clustering excludes cytoplasm during nuclear assembly. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuylen S, Blaukopf C, Politi AZ, Muller-Reichert T, Neumann B, Poser I, Ellenberg J, Hyman AA, and Gerlich DW (2016). Ki-67 acts as a biological surfactant to disperse mitotic chromosomes. Nature 535, 308–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey NE, and Morgan DO (2016). Building a Regulatory Network with Short Linear Sequence Motifs: Lessons from the Degrons of the Anaphase-Promoting Complex. Mol. Cell 64, 12–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degenhardt K, Wright AC, Horng D, Padmanabhan A, and Epstein JA (2010). Rapid Three-Dimensional Phenotyping of Cardiovascular Development in Mouse Embryos by Micro-CT with Iodine Staining. Circ Cardiovasc Imaging 3, 314–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshaies RJ, and Joazeiro C a P. (2009). RING domain E3 ubiquitin ligases. Annu. Rev. Biochem. 78, 399–434. [DOI] [PubMed] [Google Scholar]
- Deutsch EW, Csordas A, Sun Z, Jarnuczak A, Perez-Riverol Y, Ternent T, Campbell DS, Bernal-Llinares M, Okuda S, Kawano S, et al. (2017). The ProteomeXchange consortium in 2017: Supporting the cultural change in proteomics public data deposition. Nucleic Acids Res. 45, D1100–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R, et al. (2016). Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 34, 184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou H, Buetow L, Sibbet GJ, Cameron K, and Huang DT (2012). BIRC7-E2 ubiquitin conjugate structure reveals the mechanism of ubiquitin transfer by a RING dimer. Nat. Struct. Mol. Biol. 19, 876–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eguren M, Manchado E, and Malumbres M (2011). Non-mitotic functions of the Anaphase-Promoting Complex. Semin. Cell Dev. Biol. 22, 572–578. [DOI] [PubMed] [Google Scholar]
- Eng JK, McCormack AL, and Yates JRI (1994). An approach to correlate MS/MS data to amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 5, 976–989. [DOI] [PubMed] [Google Scholar]
- Erickson BK, Mintseris J, Schweppe DK, Navarrete-Perea J, Erickson AR, Nusinow DP, Paulo JA, and Gygi SP (2019). Active Instrument Engagement Combined with a Real-Time Database Search for Improved Performance of Sample Multiplexing Workflows. J. Proteome Res. 18, 1299–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Higuera I, Manchado E, Dubus P, Cañamero M, Méndez J, Moreno S, and Malumbres M (2008). Genomic stability and tumour suppression by the APC/C cofactor Cdh1. Nat. Cell Biol. 10, 802–811. [DOI] [PubMed] [Google Scholar]
- Garnett MJ, Mansfeld J, Godwin C, Matsusaka T, Wu J, Russell P, Pines J, and Venkitaraman AR (2009). UBE2S elongates ubiquitin chains on APC/C substrates to promote mitotic exit. Nat. Cell Biol. 11, 1363–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geley S, Kramer E, Gieffers C, Gannon J, Peters JM, and Hunt T (2001). Anaphase-promoting complex/cyclosome-dependent proteolysis of human cyclin A starts at the beginning of mitosis and is not subject to the spindle assembly checkpoint. J. Cell Biol. 153, 137–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman JV, and Bonni A (2019). Regulation of neuronal connectivity in the mammalian brain by chromatin remodeling. Curr. Opin. Neurobiol. 59, 59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, and Bonni A (2016). A decade of the anaphase-promoting complex in the nervous system. Genes Dev. 30, 622–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Ikeuchi Y, Malumbres M, and Bonni A (2015). A Cdh1-APC/FMRP Ubiquitin Signaling Link Drives mGluR-Dependent Synaptic Plasticity in the Mammalian Brain. Neuron 86, 726–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttlin EL, Jedrychowski MP, Elias JE, Goswami T, Rad R, Beausoleil SA, Villén J, Haas W, Sowa ME, and Gygi SP (2010). A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143, 1174–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kill Ian R. (1996). Localisation of the Ki-67 antigen within the nucleolus. Evidence for a fibrillarin-deficient region of the dense fibrillar component. J. Cell Sci. 109, 1253–1263. [DOI] [PubMed] [Google Scholar]
- Jarvis M, Brown N, Schulman B, and Peters Ja. (2016). Measuring APC/C-Dependent Ubiquitylation In Vitro Marc. Methods Mol. Biol. 1342, 157–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastner B, Fischer N, Golas MM, Sander B, Dube P, Boehringer D, Hartmuth K, Deckert J, Hauer F, Wolf E, et al. (2008). GraFix: Sample preparation for single-particle electron cryomicroscopy. Nat. Methods 5, 53–55. [DOI] [PubMed] [Google Scholar]
- Khuchua Z, Wozniak DF, Bardgett ME, Yue Z, McDonald M, Boero J, Hartman RE, Sims H, and Strauss AW (2003). Deletion of the N-terminus of murine MAP2 by gene targeting disrupts hippocampal CA1 neuron architecture and alters contextual memory. Neuroscience 119, 101–111. [DOI] [PubMed] [Google Scholar]
- Kim AH, Puram SV, Bilimoria PM, Ikeuchi Y, Keough S, Wong M, Rowitch D, and Bonni A (2009). A Centrosomal Cdc20-APC Pathway Controls Dendrite Morphogenesis in Postmitotic Neurons. Cell 136, 322–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimata Y (2019). APC/C Ubiquitin Ligase: Coupling Cellular Differentiation to G1/G0 Phase in Multicellular Systems. Trends Cell Biol. 29, 591–603. [DOI] [PubMed] [Google Scholar]
- Konishi Y, Stegmuller J, Matsuda T, Bonni S, and Bonni A (2004). Cdh1-APC Controls Axonal Growth and Patterning in the Mammalian Brain. Science (80-. ). 303, 1026–1030. [DOI] [PubMed] [Google Scholar]
- Laf B, and Basu U (2019). Biology of RNA Surveillance in Development and Disease. Trends Cell Biol. 29, 428–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson AG, Elnatan D, Keenen MM, Trnka MJ, Johnston JB, Burlingame AL, Agard DA, Redding S, and Narlikar GJ (2017). Liquid droplet formation by HP1α suggests a role for phase separation in heterochromatin. Nature 547, 236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasorella A, Stegmüller J, Guardavaccaro D, Liu G, Carro MS, Rothschild G, de la Torre-Ubieta L, Pagano M, Bonni A, and Iavarone A (2006). Degradation of Id2 by the anaphase-promoting complex couples cell cycle exit and axonal growth. Nature 442, 471–474. [DOI] [PubMed] [Google Scholar]
- Lee EC, Yu D, Martinez De Velasco J, Tessarollo L, Swing DA, Court DL, Jenkins NA, and Copeland NG (2001). A highly efficient Escherichia coli-based chromosome engineering system adapted for recombinogenic targeting and subcloning of BAC DNA. Genomics 73, 56–65. [DOI] [PubMed] [Google Scholar]
- Liu YG, and Chen Y (2007). High-efficiency thermal asymmetric interlaced PCR for amplification of unknown flanking sequences. Biotechniques 43, 649–656. [DOI] [PubMed] [Google Scholar]
- Manchado E, Guillamot M, de Cárcer G, Eguren M, Trickey M, García-Higuera I, Moreno S, Yamano H, Cañamero M, and Malumbres M (2010). Targeting Mitotic Exit Leads to Tumor Regression In Vivo: Modulation by Cdk1, Mastl, and the PP2A/B55α,δ Phosphatase. Cancer Cell 18, 641–654. [DOI] [PubMed] [Google Scholar]
- Martinez-Chacin RC, Bodrug T, Bolhuis DL, Kedziora KM, Bonacci T, Ordureau A, Gibbs ME, Weissmann F, Qiao R, Grant GD, et al. (2020). Ubiquitin chain-elongating enzyme UBE2S activates the RING E3 ligase APC/C for substrate priming. Nat. Struct. Mol. Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastronarde DN (2005). Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36–51. [DOI] [PubMed] [Google Scholar]
- Matei V, Pauley S, Kaing S, Rowitch D, Beisel KW, Morris K, Feng F, Jones K, Lee J, and Fritzsch B (2005). Smaller inner ear sensory epithelia in Neurog1 null mice are related to earlier hair cell cycle exit. Dev. Dyn. 234, 633–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlister GC, Nusinow DP, Jedrychowski MP, Wühr M, Huttlin EL, Erickson BK, Rad R, Haas W, and Gygi SP (2014). MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal. Chem. 86, 7150–7158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meitinger F, Ohta M, Lee KY, Watanabe S, Davis RL, Anzola JV, Kabeche R, Jenkins DA, Shiau AK, Desai A, et al. (2020). TRIM37 controls cancer-specific vulnerability to PLK4 inhibition. Nature 585, 440–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarrete-Perea J, Yu Q, Gygi SP, and Paulo JA (2018). Streamlined Tandem Mass Tag (SL-TMT) Protocol: An Efficient Strategy for Quantitative (Phospho)proteome Profiling Using Tandem Mass Tag-Synchronous Precursor Selection-MS3. J. Proteome Res. 17, 2226–2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh E, Mark KG, Mocciaro A, Watson ER, Prabu JR, Cha DD, Kampmann M, Gamarra N, Zhou CY, and Rape M (2020). Gene expression and cell identity controlled by anaphase-promoting complex. Nature 579, 136–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostromyshenskii DI, Chernyaeva EN, Kuznetsova IS, and Podgornaya OI (2018). Mouse chromocenters DNA content: Sequencing and in silico analysis. BMC Genomics 19, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pál M, Nagy O, Ménesi D, Udvardy A, and Deák P (2007). Structurally related TPR subunits contribute differently to the function of the anaphase-promoting complex in Drosophila melanogaster. J. Cell Sci. 120, 3238–3248. [DOI] [PubMed] [Google Scholar]
- Paulo JA, O’Connell JD, Everley RA, O’Brien J, Gygi MA, and Gygi SP (2016a). Quantitative mass spectrometry-based multiplexing compares the abundance of 5000 S. cerevisiae proteins across 10 carbon sources. J. Proteomics 148, 85–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulo JA, O’Connell JD, and Gygi SP (2016b). A Triple Knockout (TKO) Proteomics Standard for Diagnosing Ion Interference in Isobaric Labeling Experiments. J Am Soc Mass Spectrom 27, 1620–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Riverol Y, Xu QW, Wang R, Uszkoreit J, Griss J, Sanchez A, Reisinger F, Csordas A, Ternent T, Del-Toro N, et al. (2016). PRIDE inspector toolsuite: Moving toward a universal visualization tool for proteomics data standard formats and quality assessment of proteomexchange datasets. Mol. Cell. Proteomics 15, 305–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Riverol Y, Csordas A, Bai J, Bernal-Llinares M, Hewapathirana S, Kundu DJ, Inuganti A, Griss J, Mayer G, Eisenacher M, et al. (2019). The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 47, D442–D450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters J-M (2006). The anaphase promoting complex/cyclosome: a machine designed to destroy. Nat. Rev. Mol. Cell Biol. 7, 644–656. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera - A visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Qiao R, Weissmanna F, Masaya Yamaguchib, Brownb Nicholas G., Ryan VanderLindenb c, Richard Imred, Jarvisa Marc A., Brunnerb Michael R., Davidsona Iain F., Litosa Gabriele, Haselbache David, f, Karl Mechtlera d, Starke Holger, f 2, Schulmanb Brenda A., c, g, 2, and Jan-Mic 2, and AResearch (2016). Mechanism of APC/C-CDC20 activation by mitotic phosphorylation. PNAS. [Google Scholar]
- Rohou A, and Grigorieff N (2015). CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roid G (2003). Stanford-Binet Intelligence Scales. (Itasca, IL: Riverside; ). [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweppe DK, Eng JK, Bailey D, Rad R, Yu Q, Navarrete-Perea J, Huttlin EL, Erickson BK, Paulo JA, and Gygi SP (2020). Full-featured, real-time database searching platform enables fast and accurate multiplexed quantitative proteomics. J. or Proteome Res. 19, 2026–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivakumar S, and Gorbsky GJ (2015). Spatiotemporal regulation of the anaphase-promoting complex in mitosis. Nat. Rev. Mol. Cell Biol. 16, 82–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skene PJ, Illingworth RS, Webb S, Kerr ARW, James KD, Turner DJ, Andrews R, and Bird AP (2010). Neuronal MeCP2 Is Expressed at Near Histone-Octamer Levels and Globally Alters the Chromatin State. Mol. Cell 37, 457–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobecki M, Mrouj K, Camasses A, Parisis N, Nicolas E, Llères D, Gerbe F, Prieto S, Krasinska L, David A, et al. (2016). The cell proliferation antigen Ki-67 organises heterochromatin. Elife 5, e13722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song L, Pan S, Zhang Z, Jia L, Chen W, and Zhao X (2021). STAB : a spatio-temporal cell atlas of the human brain. Nucleic Acids Res. 49, 1029–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strom AR, Emelyanov AV, Mir M, Fyodorov DV, Darzacq X, and Karpen GH (2017). Phase separation drives heterochromatin domain formation. Nature 547, 241–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting L, Rad R, Gygi SP, and Haas W (2011). MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nat. Methods 8, 937–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trivedi P, and Stukenberg PT (2020). A Condensed View of the Chromosome Passenger Complex. Trends Cell Biol. 30, 676–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vodermaier HC, Gieffers C, Maurer-Stroh S, Eisenhaber F, and Peters JM (2003). TPR subunits of the anaphase-promoting complex mediate binding to the activator protein CDH1. Curr. Biol. 13, 1459–1468. [DOI] [PubMed] [Google Scholar]
- Warming S, Costantino N, Court DL, Jenkins NA, and Copeland NG (2005). Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res. 33, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson ER, Brown NG, Peters JM, Stark H, and Schulman BA (2018). Posing the APC/C E3 Ubiquitin Ligase to Orchestrate Cell Division. Trends Cell Biol. 29, 117–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissmann F, Petzold G, VanderLinden R, Huis In ‘t Veld P.J., Brown NG, Lampert F, Westermann S, Stark H, Schulman BA, and Peters J-M (2016). biGBac enables rapid gene assembly for the expression of large multisubunit protein complexes. Proc. Natl. Acad. Sci. U. S. A. 1604935113-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson A, Wickliffe KE, Mellone BG, Song L, Karpen GH, and Rape M (2009). Identification of a physiological E2 module for the human anaphase-promoting complex. Proc. Natl. Acad. Sci. USA 106, 18213–18218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozniak DF, Hartman RE, Boyle MP, Vogt SK, Brooks AR, Tenkova T, Young C, Olney JW, and Muglia LJ (2004). Apoptotic neurodegeneration induced by ethanol in neonatal mice is associated with profound learning/memory deficits in juveniles followed by progressive functional recovery in adults. Neurobiol. Dis. 17, 403–414. [DOI] [PubMed] [Google Scholar]
- Wozniak DF, Xiao M, Xu L, Yamada KA, and Ornitz DM (2007). Impaired spatial learning and defective theta burst induced LTP in mice lacking fibroblast growth factor 14. Neurobiol. Dis. 26, 14–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozniak DF, Diggs-Andrews KA, Conyers S, Yuede CM, Dearborn JT, Brown JA, Tokuda K, Izumi Y, Zorumski CF, and Gutmann DH (2013). Motivational Disturbances and Effects of L-dopa Administration in Neurofibromatosis-1 Model Mice. PLoS One 8, e66024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T, Merbl Y, Huo Y, Gallop JL, Tzur A, and Kirschner MW (2010). UBE2S drives elongation of K11-linked ubiquitin chains by the anaphase-promoting complex. Proc. Natl. Acad. Sci. USA 107, 1355–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada T, Yang Y, and Bonni A (2013). Spatial organization of ubiquitin ligase pathways orchestrates neuronal connectivity. Trends Neurosci. 36, 218–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Kim AH, Yamada T, Wu B, Bilimoria PM, Ikeuchi Y, de la Iglesia N, Shen J, and Bonni A (2009). A Cdc20-APC ubiquitin signaling pathway regulates presynaptic differentiation. Science (80-. ). 326, 575–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Kim AH, and Bonni A (2010). The dynamic ubiquitin ligase duo: Cdh1-APC and Cdc20-APC regulate neuronal morphogenesis and connectivity. Curr. Opin. Neurobiol. 20, 92–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, King RW, Peters JM, and Kirschner MW (1996). Identification of a novel ubiquitin-conjugating enzyme involved in mitotic cyclin degradation. Curr. Biol. 6, 455–466. [DOI] [PubMed] [Google Scholar]
- Zeytuni N, and Zarivach R (2012). Structural and functional discussion of the tetra-trico-peptide repeat, a protein interaction module. Structure 20, 397–405. [DOI] [PubMed] [Google Scholar]
- Zhang S, Chang L, Alfieri C, Zhang Z, Yang J, Maslen S, Skehel M, and Barford D (2016). Molecular mechanism of APC/C activation by mitotic phosphorylation. Nature 533, 260–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Chang L, Yang J, Conin N, Kulkarni K, and Barford D (2013). The four canonical TPR subunits of human APC/C form related homo-dimeric structures and stack in parallel to form a TPR suprahelix. J. Mol. Biol. 425, 4236–4248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng SQ, Palovcak E, Armache JP, Verba KA, Cheng Y, and Agard DA (2017). MotionCor2: Anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivanov J, Nakane T, Forsberg BO, Kimanius D, Hagen WJH, Lindahl E, and Scheres SHW (2018). New tools for automated high-resolution cryo-EM structure determination in RELION-3. Elife 7, 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S3 (Related to Figure 5): Tandem-mass tag (TMT) proteomics in control and Anapc7−/− cerebellum at P8.
Data Availability Statement
The TMT proteomics dataset “APC7 KO vs controls” has been deposited at ProteomeXchange via the PRIDE database and is publicly available as of the date of publication. Cryo-EM structure data have been deposited to the EM Data Bank and are publicly available as of the date of publication. Accession numbers for these datasets are listed in the key resources table. Original/source data have been deposited on Mendeley Data and are publicly available as of the date of publication. DOIs are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-APC1 | Bethyl | Cat# A301–653A, RRID:AB_1210875 |
| Mouse monoclonal (AF3.1) anti-CDC27/APC3 (immunoprecipitation) | Thermo Fisher Scientific | Cat# MA1–24749, RRID:AB_2075283 |
| Rabbit polyclonal anti-CDC27/APC3 (immunoblotting) | Bethyl | Cat# A301–184A, RRID:AB_890565 |
| Rabbit polyclonal anti-APC4 | Bethyl | Cat# A301–176A, RRID:AB_2227071 |
| Rabbit polyclonal anti-APC5 | Bethyl | A301–026A, RRID:AB_2227119 |
| Rabbit polyclonal anti-CDC16/APC6 | Bethyl | Cat# A310–389A, RRID:AB_890557 |
| Rabbit polyclonal anti-APC7 N-term (Immunoprecipitation) | Bethyl | A302–550A, RRID:AB_1998915 |
| Rabbit polyclonal anti-APC7 C-term (Immunoblotting) |
Bethyl | Cat# A302–551A, RRID:AB_1998911 |
| Rabbit polyclonal anti-APC8 | Bethyl | Cat# A301–182A |
| Rabbit polyclonal anti-APC16 | Santa Cruz Biotechnology | Cat# sc-135452, RRID:AB_10988755 |
| Rabbit polyclonal anti-CDC20 | Bethyl | Cat# A301–180A, RRID:AB_890559 |
| Mouse monoclonal (DH01) anti-FZR1/CDH1 | Thermo Fisher Scientific | Cat# MA5–11496, RRID:AB_10979615 |
| Rabbit polyclonal anti-p44/42 MAPK (Erk1/2) Antibody #9102 | Cell Signaling Technology | Cat# 9102, RRID:AB_330744 |
| Mouse monoclonal anti-14–3-3 beta (A-6) | Santa Cruz Biotechnology | Cat# sc-25276, RRID:AB_626617 |
| Rabbit polyclonal anti-PARP | Cell Signaling Technology | Cat# 9542, RRID:AB_2160739 |
| Mouse monoclonal anti-BrdU (G3G4) | DSHB | G3G4(AntiBrdUrd), RRID:AB_1157913 |
| Mouse monoclonal anti-PCNA (PC10, 3F81) | Thermo Fisher Scientific | Cat# 13–9910-82, RRID:AB_466977 |
| Mouse monoclonal anti-NeuN | Millipore | Cat# MAB377, RRID:AB_2298772 |
| Mouse monoclonal anti-cyclin B1 (V152) | Abcam | Cat# ab72, RRID:AB_305751 |
| Rat monoclonal anti-Ki-67 (SolA15) | Thermo Fisher Scientific | Cat# 14–5698-82, RRID:AB_10854564 |
| Rabbit polyclonal anti-Calbindin (D-28K) | SWANT | Cat# CB38, RRID:AB_2721225 |
| Rabbit anti-nucleolin | Bethyl | Cat# IHC-00083, RRID:AB_2267070 |
| Normal rabbit IgG | Millipore | Cat# NI01–100UG, RRID:AB_10681285 |
| Mouse IgG2b, kappa monoclonal (MPC-11) | Abcam | Cat# NI01–100UG, RRID: not found |
| Rat IgG2a kappa light chain | Thermo Fisher Scientific | Cat# 14–4321-82, RRID:AB_470105 |
| Goat anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary, Alexa Fluor 568 | Thermo Fisher Scientific | Cat# A-11031, RRID:AB_144696 |
| Goat anti-Rat IgG (H+L) Cross-Adsorbed Secondary, Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A-11006, RRID:AB_2534074 |
| Donkey anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary, Alexa Fluor 568 | Thermo Fisher Scientific | Cat# A10042, RRID:AB_2534017 |
| Donkey Anti-Mouse IgG IRDye 680 Conjugated antibody | LI-COR Biosciences | Cat# 926–32222, RRID:AB_621844 |
| IRDye 800CW Donkey anti-Rabbit IgG antibody | LI-COR Biosciences | Cat# 925–32213, RRID:AB_2715510 |
| IRDye 800CW Goat anti-Rat IgG antibody | LI-COR Biosciences | Cat# 926–32219, RRID:AB_1850025 |
| RFP-Trap Agarose | Chromotek | rta-10 |
| Bacterial and Virus Strains | ||
| Baculovirus | (Weissmann et al., 2016) | N/A |
| Lentivirus; H2b-mRFP1.3 | (Meitinger et al., 2020) | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Ube1 (E1) | (Jarvis et al., 2016) | N/A |
| CDH1 protein | (Jarvis et al., 2016) | N/A |
| CDC20 protein | (Jarvis et al., 2016) | N/A |
| Ube2C (E2) | (Jarvis et al., 2016) | N/A |
| Ube2S (E2) | (Jarvis et al., 2016) | N/A |
| UBCH5 (E2) | (Dou et al., 2012) | N/A |
| APC apoenzyme | (Jarvis et al., 2016) | N/A |
| APCΔ7 apoenzyme | (Jarvis et al., 2016) | N/A |
| APC holoenzyme | (Brown et al., 2016) | N/A |
| APCΔ7 holoenzyme | This paper | N/A |
| UBE2SCTP and UBE2SCTP-4A | (Martinez-Chacin et al., 2020) | N/A |
| Fluorescent cyclin B1-NTD (CycB-NTD) | (Jarvis et al., 2016) | N/A |
| Fluorescent Ub-CycB-NTD | (Jarvis et al., 2016) | N/A |
| Fluorescent securin | (Jarvis et al., 2016) | N/A |
| Methyl-ubiquitin | (Jarvis et al., 2016) | N/A |
| ubiquitin | (Jarvis et al., 2016) | N/A |
| Benzonase | Millipore | E1014 |
| Hoechst 33258 solution | Millipore Sigma | Cat# 94403 |
| TMT11 reagent | Thermo-Fisher Scientific | A37727 |
| Pierce Trypsin Protease, MS Grade | Thermo-Fisher Scientific | Cat#90058 |
| Lys-C, Mass Spectrometry Grade | Wako Chemicals | Barcode#4987481427648 |
| Pierce Phosphatase Inhibitor Mini Tablets | Thermo Fisher Scientific | A32957 |
| Pierce Protease Inhibitor Mini Tables | Thermo Fisher Scientific | A32953 |
| Protease inhibitor cocktail | Sigma | P8340 |
| phosphatase inhibitor cocktail #3 | Sigma | P0044 |
| PMSF | Sigma | PMSF-RO |
| HyClone Fetal Bovine Serum | Cytiva | SH30071.03IH25–40 |
| 5-Bromo-2′-deoxyuridine (BrdU) | Sigma | B5002 |
| Polybrene | EMD Millipore | TR-1003-G |
| Affymetrix CytoScan HD Assay | Thermo Fisher Scientific | Cat# 901835 |
| Pierce BCA Protein Assay | Thermo Fisher Scientific | Cat# 23223 |
| Bradford regent | Bio-Rad | Cat# 5000006 |
| Revert total protein stain | LI-COR | Cat# 926–11011 |
| iScript cDNA synthesis kit | Bio-Rad | Cat# 1708890 |
| Deposited Data | ||
| TMT proteomics data PXD016683 | ProteomeXchange | (http://www.ebi.ac.uk/pride), PXD016683 |
| Cryo-EM structure of apo-APC7 | EM Data Bank | EMDB-25026 |
| Cryo-EM structure of holo-APC7 | EM Data Bank | EMDB-25027 |
| Original/source data | Mendeley Data | DOI: 10.17632/hrk4ztwrpn.1 |
| Experimental Models: Cell Lines | ||
| Lymphoblasts from patient 2227 | Coriell | Coriell Cat# GM25252, RRID:CVCL_LN92 |
| Lymphoblasts from mother of patient 2227 | Coriell | Coriell Cat# GM25253, RRID:CVCL_LN93 |
| HAP1 ANAPC7 null | Horizon Discovery | RRID:CVCL_SC47 |
| HAP1 wild type | Horizon Discovery | |
| 129S5/SvEvTac EDJ22 ES cells | Washington University Mouse Genetics Core | http://escore.im.wustl.edu/SubMenu_celllines/wtEScell_linesEDJ22.html |
| High Five (BTI-Tn-5B1–4) cells | Thermo Fisher Scientific | BTI-TN-5B1–4 |
| RPE-1 CEP192-mNeonGreen TetOn-Cas9 | (Meitinger et al., 2020) | ODCL0191 |
| RPE-1 CEP192-mNeonGreen TetOn-Cas9 ANAPC7 gRNA #1 | This study | ODCL0271 |
| RPE-1 CEP192-mNeonGreen TetOn-Cas9 ANAPC7 gRNA #2 | This study | ODCL0272 |
| RPE-1 CEP192-mNeonGreen TetOn-Cas9 ANAPC7 gRNA #3 | This study | ODCL0273 |
| RPE-1 CEP192-mNeonGreen TetOn-Cas9 ANAPC7 gRNA #4 | This study | ODCL0274 |
| RPE-1 CEP192-mNeonGreen TetOn-Cas9 pEF1alpha-H2B-mRFP | This study | ODCL0275 |
| RPE-1 CEP192-mNeonGreen TetOn-Cas9 ANAPC7 gRNA #2 pEF1alpha-H2B-mRFP | This study | ODCL0276 |
| RPE-1 CEP192-mNeonGreen TetOn-Cas9 ANAPC7 gRNA #2 pEF1alpha-H2B-mRFP | This study | ODCL0277 |
| Experimental Models: Organisms/Strains | ||
| Anapc7 mutant on hybrid C57BL/6 x 129 | This paper | N/A |
| Anapc7 mutant on Jackson C57BL/6 | This paper | N/A |
| CDC20f/f mice on C57BL/6 | (Manchado et al., 2010) | N/A |
| CDH1f/f mice on C57BL/6 | (García-Higuera et al., 2008) | N/A |
| Math1/Atoh1-cre mice on C57BL/6 | The Jackson Laboratory | Cat# 011104 |
| Wild-type Jackson C57BL/6 | The Jackson Laboratory | Cat# 000664 |
| Sox2-cre mice on C57BL/6 | The Jackson Laboratory | Cat# 008454 |
| Ki67-RFP reporter on C57BL/6 | The Jackson Laboratory | Cat# 029802 |
| Wild-type outbred mice on CD1 | Charles River | Cat# 022 |
| Oligonucleotides | ||
| See Table S4 | ||
| Recombinant DNA | ||
| Anapc7 knock-in targeting vector with deletion of exons 4–6, floxed neo cassette and G>A substitution | (Adams et al., 2005) | BAC ID# bMQ-440B18 |
| Amino acids 1–300 and 765–100 of human Ki-67 cloned into pGEX | ||
| Software and Algorithms | ||
| ImageJ | (Schneider et al., 2012) | https://imagej.nih.gov/ij/ |
| UCSF Chimera | (Pettersen et al., 2004) | https://www.cgl.ucsf.edu/chimera/ |
| Image Studio | LI-COR | N/A |
| ImageLab | Bio-rad | N/A |
| Chromosome Analysis Suite | Affymetrix | N/A |
| Proteomics software pipeline | SEQUEST | http://fields.scripps.edu/yates/wp/?page_id=17 |
| Gautomatch | MRC Laboratory of Molecular Biology | https://www2.mrc-lmb.cam.ac.uk/research/locally-developed-software/zhang-software/ |
| RELION-3.0 | (Zivanov et al., 2018) | https://www3.mrc-lmb.cam.ac.uk/relion/index.php/Main_Page |
| QuantStudio 6 real-time PCR system | Bio-Rad | N/A |
| Dragonfly | Object Research Systems | https://www.theobjects.com/dragonfly/index.html |
| SerialEM | (Mastronarde, 2003) | http://bio3d.colorado.edu/SerialEM/ |
| SEQUEST-based in-house platform | Thermo-Fisher Scientific (Eng et al., 1994; Huttlin et al., 2010) |
N/A |
| Other | ||
| QUANTIFOIL® R2.1, 100 Holey Carbon Films, Grids: Cu 200 mesh | Quantifoil Micro Tools GmbH | https://www.quantifoil.com |
| Custom In situ hybridization probe for mouse Anapc7 | ACDbio | https://acdbio.com |