

Abstract
Brain derived neurotrophic factor (BDNF) is a neuropeptide that plays numerous important roles in synaptic development and plasticity. While its importance in fundamental physiology is well-established, studies of BDNF often produce conflicting and unclear results, and the scope of existing research makes the prospect of setting future directions difficult. In this review, we examine the importance of spatial and temporal factors on BDNF activity, particularly in processes such as synaptogenesis, Hebbian plasticity, homeostatic plasticity, and the treatment of psychiatric disorders. Understanding the fundamental physiology of when, where, and how BDNF acts and new approaches to control BDNF signaling in time and space can contribute to improved therapeutics and patient outcomes.
Overview of the pleotropic functions of BDNF-TrkB signaling
In 1982, a novel neurotrophic factor was found to support the survival and growth of chick embryo dorsal root ganglia (Barde et al., 1978). It was thus purified and became designated as brain derived neurotrophic factor (BDNF). BDNF was the second member of the neurotrophin family to be identified; other members of this family also include neurotrophin 3, neurotrophin 4, and nerve growth factor (Cohen and Levi-Montalcini, 1956; Ip et al., 1992; Maisonpierre et al., 1990).
Since its discovery, a vast number of studies have reported on numerous aspects of BDNF signaling in the central nervous system, as well as BDNF’s role in neuronal development, synaptic plasticity and the pathology and treatment of psychiatric disorders (Autry and Monteggia, 2012; Kowiański et al., 2018; Lu, 2003; Park and Poo, 2013). In the last two decades, BDNF has emerged as a key factor that underlies the efficacy of neuropsychiatric treatments in addition to being a critical factor for neuronal development and synaptic plasticity.
Yet, here we are faced with an embarrassment of riches. On one hand, there is strong evidence for the importance of BDNF in nervous system function and pathology that are at the core of what the field studies and hopes to achieve as potential treatments in central nervous system disorders. On the other hand, the multi-functional aspects of BDNF signaling combined with the large landscape of findings linked to BDNF makes formulating specific hypotheses or directions for future research rather daunting. Therefore, it is understandable that BDNF research, despite its fully substantiated importance, may trigger a level of frustration in the field.
In this review, we aim to present an overview of the pleiotropic functions of BDNF in multiple forms of synaptic plasticity, with emphasis on the findings that suggest timing and location of BDNF action as a critical component of its downstream effectors. While BDNF is a single factor, it can trigger multiple and often contradictory functional consequences based on its mechanism of expression, site of release, and site of action. Overall, we posit that future studies focusing on BDNF should account for its specific loci and temporal properties when pursuing its putative plastic and therapeutic effects.
From transcription to downstream signaling pathways
BDNF expression is regulated at multiple levels with several redundant mechanisms at the transcriptional level. The human BDNF gene has nine promoters that generate distinct mRNAs with different noncoding exons but encode the same protein. The human BDNF gene contains 11 exons (I-IX, Vh, VIIIh), two more than the rat and mouse BDNF genes, suggesting increased complexity in humans. BDNF transcript expression appears to have cell-specific and activity-dependent properties, and different transcripts can have unique effects on molecular and behavioral expression (Kokaia et al., 1994; Maynard et al., 2015). For instance, disruption of the BDNF promoter IV in the cortex leads to fewer inhibitory synapse numbers and reduced inhibitory presynaptic markers. These mice do not have the hyperphagia and obesity characteristic of BDNF knockout mice, suggesting a promoter-specific effect (Hong et al., 2008). BDNF mRNAs are polyadenylated at either of the two alternative sites, leading to 2 mRNA populations – one with short 3’ untranslated region (UTR) and those with a long 3’ UTR (Timmusk et al., 1993). Using a mouse mutant in which long BDNF 3’ UTR is disrupted, it was found that few BDNF mRNAs were present in dendrites, while total amounts of BDNF mRNA and protein remained the same. These mutant mice also had thinner and less dense hippocampal spines, as well as reduced long-term potentiation. (An et al., 2008).
BDNF protein is synthesized in cell bodies of neurons and glia, and it is found across the brain but is expressed in the highest amounts in the hippocampus and cortex (Poo, 2001). In human neurons, the BDNF transcript is translated into pre-pro-BDNF in the neuronal cell body, which is cleaved into the precursor pro-BDNF. Some controversy in the field exists about whether pro-BDNF is released. Some groups propose that only the mature form of BDNF is secreted (Matsumoto et al., 2008), while others have found that proBDNF is secreted by neurons to preferentially bind to the p75 receptor and facilitate long term depression (LTD) and pro-apoptotic pathways (Yang et al., 2009). As more groups study this phenomenon, it appears that much of the evidence suggests that proBDNF is both converted intracellularly and secreted (and subsequently converted) extracellularly, a process that helps modulate the differential roles of pro vs mBDNF’s role in synaptic processes such as long term potentiation (LTP) (Pang et al., 2004).
If cleaved intracellularly, BDNF can be packaged into dense core vesicles that are transported to axon terminals and dendritic compartments (Dieni et al., 2012; Egan et al., 2003). Following transport, BDNF can be released constitutively presumably by a Ca2+-dependent mechanism, its most effective form of secretion is activity-dependent via patterned electrical stimulation (Balkowiec and Katz, 2002). A recent study found that presynaptic N-methyl-D-aspartate receptors (NMDARs) at hippocampal synapses may contribute to BDNF release via increasing presynaptic calcium (Lituma et al., 2021). BDNF’s release is inhibited by synaptotagmin-IV, and work with an exogenously expressed BDNF reporter has shown its release is driven by distinct soluble NSF-attachment protein receptors (SNARE) complexes comprised of synaptobrevin-2, Synaptosomal-Associated Protein (SNAP25), and SNAP47 (Dean et al., 2009; Shimojo et al., 2015) (Figure 2).
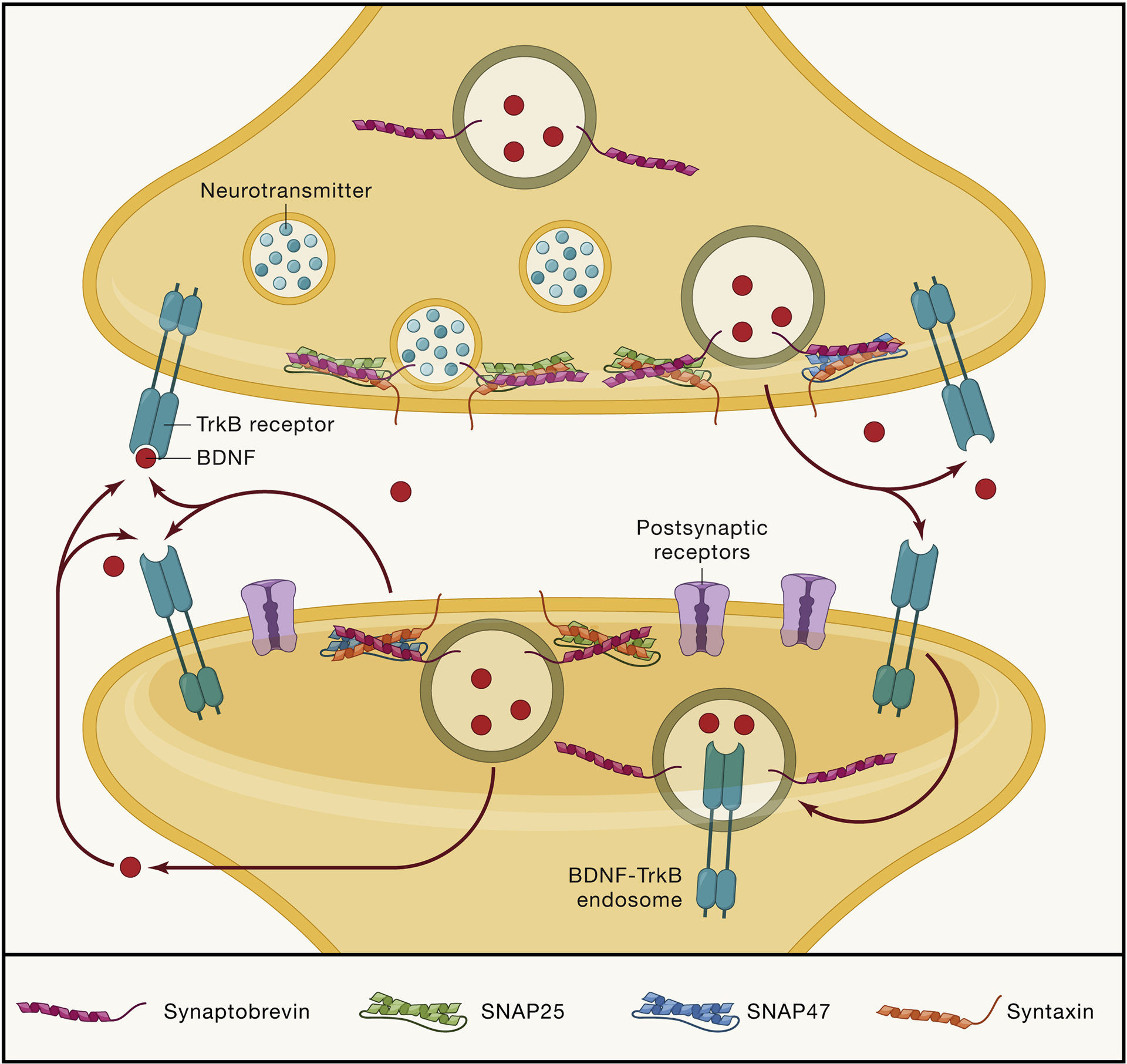
Figure 2: Schematic demonstrating the pre- and postsynaptic actions of BDNF-TrkB activity.
The release and action of BDNF is precisely regulated and has specific effects depending on the location of its binding to TrkB receptors. After BDNF is cleaved into its mature form, it can be trafficked to both presynaptic boutons and postsynaptic dendritic sites, where it is stored in dense core vesicles. Upon arrival to the synapse, BDNF is released with the help with SNARE proteins such as SNAP25, SNAP47, and synaptobrevin-2 (Syb2). Upon release into the extracellular space, BDNF then acts locally on cis- and trans-synaptic TrkB receptors. Activation of receptors at different locations differentially affects processes such as synaptogenesis to synaptic plasticity. Once BDNF binds to its high affinity TrkB receptor, it can activate downstream TrkB signaling pathways, as well as be endocytosed into an BDNF-TrkB containing endosome to be trafficked for further intracellular signaling or recycling of receptors.
BDNF, like all neurotrophins, is positively charged at physiological pH and readily binds to the cell membrane and the extracellular matrix, which limits its diffusional capabilities once secreted (Park and Poo, 2013). BDNF binds with high affinity to the Tropomyosin receptor kinase B (TrkB) receptor, which activates signaling pathways that can modulate a number of synaptic processes. TrkB is located on glial cells, as well as neuronal cell bodies, presynaptic terminals, and sites of postsynaptic specializations on dendrites (Frisen et al., 1993). The expression of TrkB on plasma membranes is dynamic and can be enhanced by excitatory synaptic activity, particularly high frequency stimulation (Du et al., 2000). TrkB receptors can also be internalized via clathrin-mediated endocytosis into intracellular vesicles where it can be transported to various regions of the neurons to affect regulatory processes (Meyer-Franke et al., 1998).
Activation of TrkB by BDNF triggers several downstream pathways important in neuronal function and survival. TrkB activation occurs via trans-phosphorylation of its tyrosine residues, leading to activation of src homolog domain 2 (SH2) and phosphorylation of the PLC-γ (Phospholipase C), PI3K-Akt (Phosphoinositide 3-kinase/ protein kinase B) activation, and mitogen-activated protein kinase—extracellular signal-regulated kinases (MAPK-ERK) pathways (Figure 1). PI3K/Akt activation promotes neuronal survival, and MAPK signaling supports synaptic plasticity and neuronal function. PLC-γ pathway activation leads to generation of IP3 and subsequent release of calcium from internal calcium stores that can activate calcium-dependent protein kinases to influence processes such as synaptic plasticity (Minichiello, 2009).
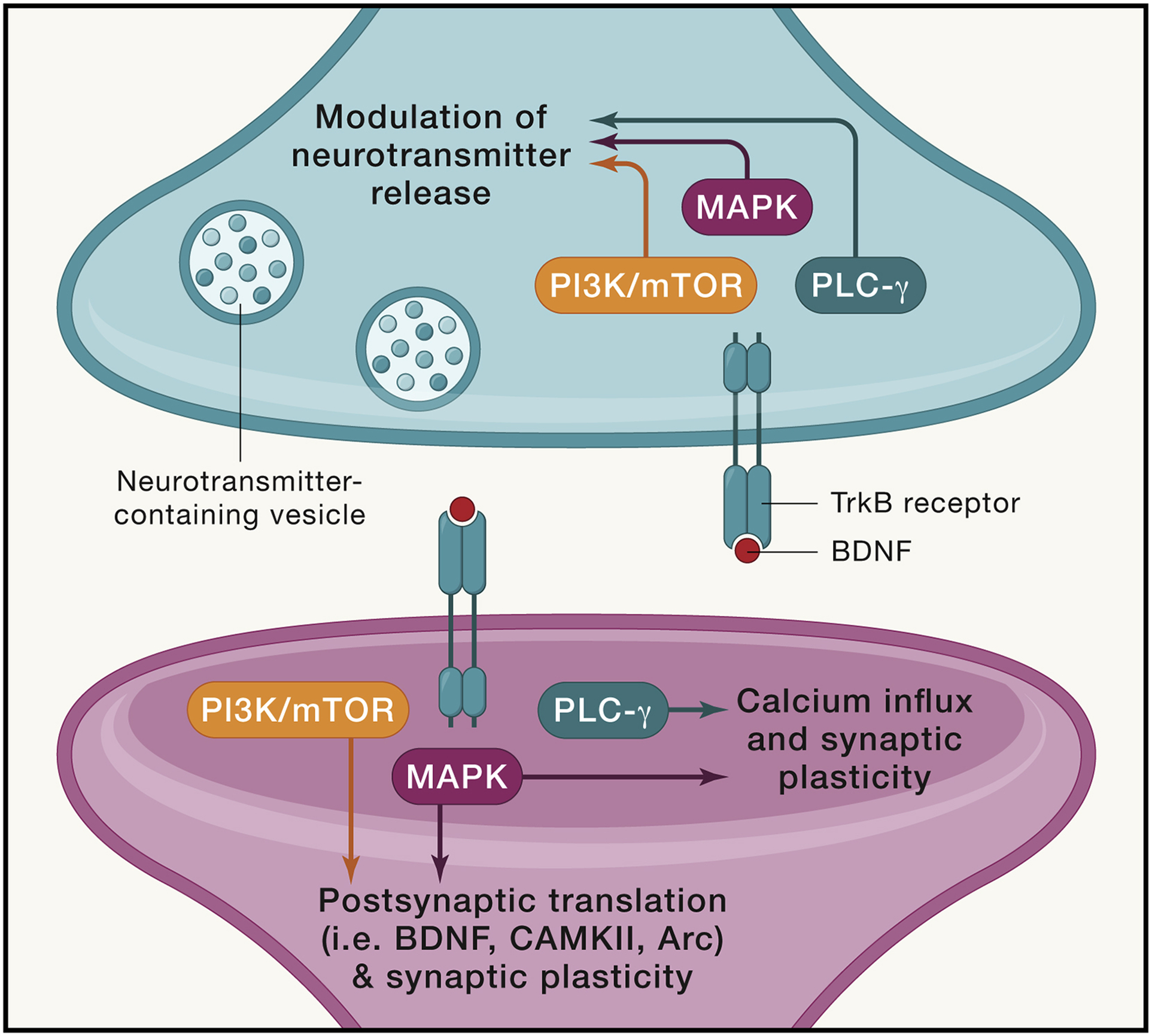
Figure 1: BDNF-TrkB receptor binding leads to activation of distinct downstream signaling pathways.
The binding of BDNF to its high affinity TrkB receptor induces the activation of distinct signaling pathways. The TrkB receptor has several phosphorylation sites, which can activate several unique pathways at both the pre- and post-synaptic specializations. The PI3K/mTOR and MAPK pathways can activate protein translation (including BDNF, CAMKII, and Arc) at dendritic boutons. The PLC-gamma and MAPK pathways can impact calcium influx, which in turn lead to activation of CAMKII and trigger and subsequent synaptic plasticity (i.e. synaptic strengthening via increased AMPAR trafficking). At presynaptic terminals, activation of TrkB leads to changes in neurotransmitter release (i.e. increased release at hippocampal neurons). All three downstream pathways have been implicated in this effect. The action of BDNF at synapses is specific to its binding of TrkB receptors, though the location at and circumstances under which this binding occurs can lead to widely different effects on synaptic activity.
Synaptogenesis
Multiple studies have found that synapse formation is modulated by BDNF-TrkB signaling (Luikart et al., 2005; Park and Poo, 2013; Vicario-Abejón et al., 2002). In addition to its cell-specific regulation, the mode of BDNF delivery (i.e. exogenous application vs endogenous expression) can affect findings. BDNF can regulate synapse formation in three main ways: increasing the arborization of axons and dendrites, inducing axonal and dendritic bouton formation, and stabilizing existing synapses. Furthermore, BDNF affects synaptogenesis in a temporally and spatially relevant manner.
Cell type specificity and spatial range have emerged as important determinants of BDNF action. In visual cortical slice cultures, BDNF application increased the length and complexity of basal dendrites in layer 4, while other layers did not respond, suggesting a laminar specificity of BDNF signaling in cortical brain regions (McAllister et al., 1995). BDNF also has a limited diffusional capacity, leading to effects located locally of its release or application. Chronic application of BDNF on hippocampal slice cultures increased the length of apical dendrites without affecting basal dendritic properties, and the effect on apical dendrites have been shown to be blocked by inhibiting the MEK pathway (Alonso et al., 2004). Notably, the method of BDNF application matters for its effect on synaptogenesis. Acute application of BDNF (and acute activation of TrkB) augmented the size of the dendritic spine head, whereas gradually eliciting BDNF-TrkB activation caused spine length elongation and increased filopodia protrusions (Ji et al., 2010). While the ability to examine the kinetics of TrkB activation in vivo remains limited, one may surmise that gradual activation may occur from constitutive BDNF secretion, or release from a distant source, while more acute activation may occur at local receptors due to intense neuronal firing. Studies of endogenous BDNF have also supported the local distribution of its effects. BDNF overexpressing “donor” neurons in ferret cortical slice cultures could elicit increased dendritic growth and branching at “recipient” neurons within ~4.5 um of the site of BDNF secretion (Horch and Katz, 2002). Studies of both exogenous application and endogenous release support the localized effects of BDNF.
Improved genetic tools allow more precise characterization, and indeed the usage of three different Cre-expressing transgenic mice revealed unique temporal and spatial configurations of TrkB activity in formation of pre- vs postsynaptic specializations. During the critical development period, deletion of TrkB at presynaptic sites impaired presynaptic terminal formation, deletion of TrkB in postsynaptic sites impaired formation postsynaptic specializations, and deletion of both impaired formation on both sides. After the developmental period had completed and synapses had been formed, however, TrkB deletion did not impact synaptic formation (Luikart et al., 2005).
The effects of BDNF on synaptogenesis involve localized binding to TrkB receptors. Together, the spatio-temporal precision of these mechanisms allows BDNF to specifically modulate synaptogenesis in the “critical period” of development and adulthood.
Hebbian Plasticity
Hebbian plasticity is the most widely studied form of activity-dependent adaptation of synaptic strength, which includes long term potentiation and depression (LTP and LTD), as well as presynaptic facilitation and depression (a temporary change in the probability of neurotransmitter release in response to an action potential, following a particular stimulus pattern). Hallmark features of Hebbian plasticity include associative, rapidly induced, and input-specific changes. It is a positive feedback process – for instance, upon LTP induction, the synaptic connection becomes stronger via an increased response to the same stimuli (Malenka and Bear, 2004) (Figure 3). Hebbian plasticity is thought to form the mechanistic basis for learning and memory, and BDNF has been shown to be involved in presynaptic and postsynaptic mechanisms underlying plasticity.
Figure 3: Principles of Hebbian vs Homeostatic Forms of Plasticity.
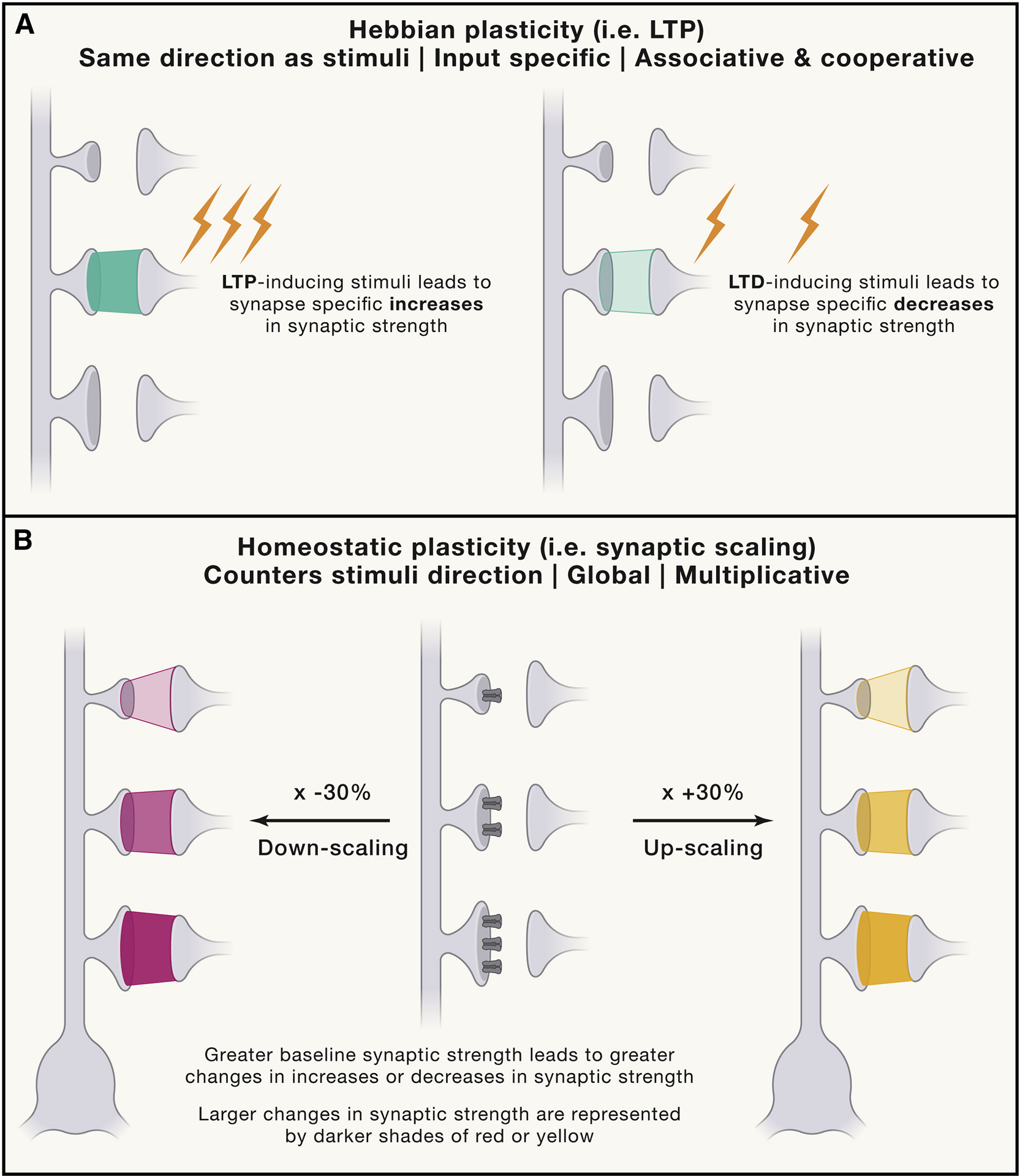
Hebbian plasticity, including LTP, is the most classically studied form of plasticity (A). It occurs in a positive feedback loop, whereby an axon repeatedly and persistently fires to induce an increase in the baseline efficiency of neurotransmission. It is input specific, associative and occurs on a rapid timescale of seconds to minutes. Homeostatic plasticity, on the other hand, counters the direction of stimuli over the course of hours to days to return a neuron to its homeostatic set point (B). This change is global and multiplicative, such that synapses that have a higher efficacy of transmission at baseline will experience a greater change in homeostatic plasticity. This is represented in the figure by synapses with a larger number of receptors having a higher baseline level of transmission. These more efficacious synapses experience a larger change in synaptic scaling upon chronic increases or decreases in activity, as shown by a darker shade of color (red for downscaling, yellow for upscaling) at the synapse.
Acute modulation of neurotransmitter release
Lohof et al. was one of the first to show that BDNF acutely augmented synaptic transmission at the frog neuromuscular junction, which was followed by a series of studies in mammalian central synapses (Lohof et al., 1993). Since then, as further elucidated in the following paragraphs, studies have further reported the effects of BDNF on excitatory versus inhibitory release, spontaneous versus evoked release, as well as downstream TrkB signaling pathways involved in these processes.
BDNF appears to increase excitatory evoked release, but it has less consistent effects on spontaneous release. Exogenous BDNF has been repeatedly found to increase evoked response frequency and amplitude in both primary neuron cultures and brain slice preparations (Kang and Schuman, 1995; Levine et al., 1995; Li et al., 1998a). These effects are dependent on TrkB activation, as overexpression of a dominant negative form of this receptor blocked BDNF’s effect on presynaptic release (Li et al., 1998b). While BDNF-TrkB signaling increases evoked release, its effect on spontaneous neurotransmission is less clear. In the presence of tetrodotoxin (TTX), a potent voltage-gated sodium channel blocker, BDNF application increased the frequency of miniature excitatory postsynaptic currents (mEPSCs) (Li et al., 1998a; Tyler and Pozzo-Miller, 2001), while others have found that BDNF has no effect on spontaneous release (Figurov et al., 1996; Patterson et al., 1996).
BDNF’s effect on inhibitory neurotransmission has been studied less extensively; however, BDNF appears to largely decrease inhibitory neurotransmission. BDNF reduces paired-pulse depression of the inhibitory postsynaptic currents (IPSC), suggesting a presynaptic locus of action (Frerking et al., 1998; Wardle et al., 2003). Endocannabinoids have been suggested to be released as retrograde messengers in response to postsynaptic TrkB activation in the cortex, to decrease release probability of GABAergic transmission. This suggests that BDNF-TrkB may decrease inhibitory transmission via endocannabinoid signaling, and thus affect short term plasticity mechanisms (Lemtiri-Chlieh and Levine, 2010). However, other studies using hippocampal cultures found that application of BDNF over minutes to days did not affect inhibitory neurotransmitter release (Shinoda et al., 2014). In sum, it appears that BDNF increases excitatory evoked release, may modulate excitatory spontaneous release, and overall attenuates inhibitory neurotransmission.
Several mechanisms have been proposed that may mediate BDNF’s effect on spontaneous and evoked release. Importantly, the synaptic location of TrkB activation may be significant for these actions (Figure 2). Earlier work suggested that BDNF acts postsynaptically to acutely modulate neurotransmission (Levine et al., 1995). Moreover, a study using targeted genetic manipulation of the CA3-CA1 hippocampal circuitry reported that presynaptic, but not postsynaptic TrkB, receptors underlie BDNF’s effect on evoked neurotransmitter release (Lin et al., 2018).
The TrkB receptor activates the MAPK and PLC-γ pathways, both of which have been implicated in BDNF’s ability to affect presynaptic neurotransmitter release. BDNF-TrkB binding that activates MAPK signaling has been found to regulate neurotransmitter release. According to this hypothesis, the MAPK pathway phosphorylates synapsin, a synaptic vesicle associated protein that in turn modifies synaptic vesicle pool dynamics (Cheng et al., 2017). BDNF increases synapsin I phosphorylation and glutamate release within minutes, and concomitant inhibition of the MAPK pathway suppresses this response (Jovanovic et al., 2000). These results support the notion that BDNF may act through the MAPK pathway to regulate synaptic vesicle pool dynamics.
The PLC-γ signaling pathway has also been implicated in BDNF’s effect on neurotransmission, notably through modulating intracellular calcium levels. Calcium levels play an important role in neurotransmission; for instance, it triggers evoked release and modulates spontaneous neurotransmission (Südhof, 2012). Several studies have shown that BDNF increases calcium levels at the cellular and synaptic level (Amaral and Pozzo-Miller, 2007a; Lang et al., 2007). Whether BDNF-induced calcium release occurs at the pre- or postsynaptic sites, however, remains to be clearly elucidated. One study demonstrated that the increases in calcium co-localized with postsynaptic markers (Lang et al., 2007), while another found that BDNF-induced calcium changes may be presynaptic in origin as well (Cheng et al., 2017). A potential source of calcium influx may be from transient receptor potential cation (TRPC) channels, which are activated by the PLC- γ pathway. Pharmacological and genetic approaches have found that blocking TRPC activity attenuates BDNF-mediated calcium influx, suggesting a significant contribution of this channel in BDNF’s action (Amaral and Pozzo-Miller, 2007a, 2007b; Li et al., 1999). More studies are needed, but it may be that presynaptic and postsynaptic calcium changes are not mutually exclusive, and they are involved in distinct pathways to affect BDNF-induced changes in neurotransmission.
Notably, the location of BDNF- TrkB signaling at different brain regions can lead to different effects on calcium signaling. For instance, in contrast to observations in hippocampal synapses, BDNF slows calcium channel activation and inhibits exocytosis and endocytosis in the calyx of Held synapses of the brain stem (Baydyuk et al., 2015). Overall, these studies bolster the premise that when examining the effects of BDNF on calcium signaling, it is important to consider the brain region being studied and how BDNF effect in a particular region may impact a larger synaptic network.
Long term potentiation
Long term potentiation (LTP) is a form of synaptic plasticity whereby brief strong stimulation or temporal pairing of pre-and postsynaptic stimulation potentiates, or increases, the subsequent response to the baseline level of stimulation (Figure 3). This process has been strongly implicated to form the basis for learning and memory (Morris, 2003). LTP is broken down into three main phases: induction, maintenance, and expression. Induction involves the application of high frequency stimuli or a “pairing protocol” to trigger LTP, maintenance involves persistent biochemical changes at the synapse, and expression is characterized by long lasting cellular changes induced by biochemical changes (Malenka and Bear, 2004).
Early studies found that genetic deletion of BDNF led to impairments in induction and potentiation magnitude of LTP, and infusion of recombinant BDNF rescued this deficit (Korte et al., 1995; Patterson et al., 1996). And indeed, further study of BDNF has found that BDNF-TrkB signaling plays both permissive and instructive roles in different phases of LTP (Lin et al., 2018), and these effects are affected by factors including stage of synapse development and TrkB receptor location.
Role of BDNF-TrkB activation kinetics and synaptic developmental stage
The effects of BDNF-TrkB signaling may depend on the developmental age and the kinetics of BDNF application. Hippocampal slices at postnatal day 8–9 did not have significant differences in LTP with exogenous BDNF. However, BDNF promoted LTP in rat hippocampal slices at postnatal day 12–13, suggesting that BDNF’s ability to affect synaptic potentiation may increase with synaptic maturity (Figurov et al., 1996). Furthermore, exogenous BDNF application may differently affect potentiation depending on the method of delivery. Perfusion of exogenous BDNF at a slow rate did not show potentiation, while increasing the rate of BDNF application led to synaptic potentiation (Kang et al., 1996). Perhaps these results help explain why the results of exogenous BDNF application are often conflicting and varying. Furthermore, as it is demonstrated by recent work, BDNF acts locally and specifically on TrkB receptors (Lin et al., 2018), so it may be that manipulation of endogenous BDNF may be able to discern effects with a greater spatial specificity compared to broadly applying BDNF on neural tissue.
Studies that manipulate endogenous BDNF have found that the location of BDNF’s release and TrkB activation matters in long term potentiation. BDNF and TrkB receptors in the CA3-CA1 pathway were genetically deleted to discern the role of pre- versus postsynaptic signaling. It was found that postsynaptic TrkB receptors are involved in LTP induction, while both pre- and postsynaptic TrkB receptors are involved in LTP maintenance. Both induction and maintenance of LTP were activated by BDNF release from presynaptic CA3 neurons (Lin et al., 2018). In earlier work, a similar deficit could be rescued by infusion of BDNF into presynaptic CA3 neurons, but not at postsynaptic neurons (Zakharenko et al., 2003). Furthermore, BDNF can act in an autocrine signaling system within a single spine. Postsynaptic BDNF release, induced by stimulation, activates TrkB receptors on the same postsynaptic spine which was essential for structural and functional LTP (Harward et al., 2016). These findings suggest that both pre- and postsynaptic BDNF are critical for LTP expression, and the spatial location of TrkB at synapses is important in affecting LTP (Figure 2).
A number of TrkB downstream pathways have been implicated in the effects of BDNF-TrkB activation on LTP. A mutation at the PLC-γ phosphorylation docking site of the TrkB receptor showed that the PLC-γ pathway is involved in early and late LTP. The PLC-γ pathway forms IP3, which binds to its receptors on the sarcoplasmic reticulum to increase intracellular calcium levels and facilitate increased α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) expression and cellular potentiation (Gruart et al., 2007). TrkB has also been shown to bind and activate Fyn (a Src-family tyrosine kinase that phosphorylates NMDARs), which increases the open probability of NMDAR channels and increase the calcium influx and likelihood of synaptic potentiation (Hildebrand et al., 2016). Based on these proposed mechanisms, it appears that it is the specific activation of TrkB, and subsequent phosphorylation of downstream proteins, that mediate BDNF’s effect on LTP.
Learning and memory
BDNF has been shown to be increased after learning tasks such as contextual fear conditioning (Hall et al., 2000). This suggests that BDNF is endogenously modulated by learning tasks and may contribute to the neurobiology of learning and memory. Genetic approaches using heterozygous BDNF KO mice showed impairments in learning, such as in fear conditioning (Liu et al., 2004).
BDNF’s role in learning and memory has been debated over the years, without conclusive evidence. It may be that BDNF is modulated in different brain regions to modulate different types of learning. For instance, BDNF in the hippocampus may mediate learning in novel object recognition tasks (Hall et al., 2000; Minichiello et al., 1999). Moreover, a study utilizing the CA3-CA1 pathway found that TrkB receptor activation at both the pre- and postsynaptic sites were critical for performance in the novel recognition memory task (Lin et al., 2018).
Human studies have further corroborated the importance of BDNF in memory performance. A common single nucleotide polymorphism called Val66Met polymorphism, with a Met to Val substitution at codon 66 that impairs BDNF release from neurons. This polymorphism is very frequent, with up to 30% Met carriers in a European sample, and Egan et al. demonstrated that this polymorphism has been associated with performance in memory tasks (Egan et al., 2003). However, this study also has several caveats – the number of met/met groups is limited, and there were no genotype effects within the patient group. These findings suggest that in animal studies, BDNF is involved in learning and memory, though extrapolating these results to human studies must come with careful consideration.
Homeostatic plasticity
While Hebbian plasticity is critical for processes such as information storage, its positive feedback loop of reinforcing the direction of synaptic changes may lead to runaway excitation or inhibition. Homeostatic plasticity maintains activity around a set point, such that an error signal is generated to regulate activity back towards a target set point. For instance, upon chronic blockade of cortical culture activity, the amplitude of mEPSCs were increased. Furthermore, blocking GABAergic signaling initially increased network activity, but after 48 hours the activity returned to baseline values. These homeostatic changes were found to scale multiplicatively, and they were thus deemed “synaptic scaling” (Turrigiano et al., 1998) (Figure 3).
Extensive evidence suggests that BDNF signaling is critical for homeostatic forms of plasticity, in addition to the its roles in classical Hebbian plasticity (Desai et al., 1999; Rutherford et al., 1997). If this is the case, how can the same signaling pathway be involved in these two separate categories of synaptic plasticity that are expected to counter one another? Several lines of thought have arisen to address this apparent conundrum, largely targeted towards the location and duration of BDNF signaling involved in specific categories of synaptic plasticity. Another clue may arise from recent studies that suggest a permissive and modulatory role for BDNF signaling in adjusting the specific amplitude of Hebbian plasticity (Lin et al., 2018), in contrast to BDNF signaling as instructive and essential for certain forms of homeostatic plasticity (Rutherford et al., 1998).
BDNF - TrkB signaling has been shown to be a key player in homeostatic plasticity in numerous studies. In cortical cultures treated with TTX over days, exogenous BDNF application prevented synaptic upscaling, while blocking endogenous TrkB receptors mimicked the effects of activity blockade. This suggests that when activity rises and BDNF production increases, the network properties favor inhibition; whereas when activity falls and BDNF production is reduced, the network properties favor excitation (Rutherford et al., 1998). The effects of BDNF in synaptic scaling has been replicated across other studies as well (Desai et al., 1999). Interestingly, a recent study found that inhibiting spontaneous inhibitory neurotransmission drives increases in BDNF transcription, in a manner independent of excitatory transmission. This increase in BDNF feeds back to downregulate excitatory synaptic strength (Horvath et al., 2021).
Similar to its involvement in other processes, BDNF may have differential effects depending on its spatial location and timescale of application. Chronic BDNF-TrkB activity was found to mediate down-scaling of AMPAR expression in nucleus accumbens (NAc) primary culture (Reimers et al., 2014). This contrasts with its chronic effects in mediating synaptic upscaling in cortical neurons (Rutherford et al., 1998), suggesting a regional specificity of effects on homeostatic plasticity. Furthermore, the effect of BDNF on AMPA responses may depend on the postsynaptic cell type and synaptic location (Jakawich et al., 2010; Rutherford et al., 1998). The timescale of BDNF release also affects its effect on plasticity. Acute treatment of BDNF in the NAc increased AMPAR subunit expression, while its chronic activity mediates down-scaling (Reimers et al., 2014). This suggests a bidirectional effect on plasticity depending on the timescale of application, such that rapid increases in BDNF mediate synaptic upscaling, while chronic elevations lead to the opposite effect in synaptic downscaling (Figure 3). BDNF is an important mediator of homeostatic plasticity, and thus it is critical to consider the effects of its timescale and spatial location on activity.
Homeostatic Plasticity as a Target for Psychiatric Treatment
Homeostatic plasticity may be involved in psychiatric disease treatment mechanisms as well as be an important target for future therapeutics. Both ketamine and lithium, which are approved treatments for mood disorders, appear to involve synaptic scaling in their targeting mechanism. Ketamine, an antidepressant that can elicit a response within hours of administration, is proposed to mechanistically act via inhibiting NMDA receptors involved in spontaneous transmission. This blockade activates a cascade of homeostatic responses including inhibition of the calcium-calmodulin dependent kinase, eEF2 kinase, which rapidly triggers BDNF protein synthesis and induces synaptic potentiation in the hippocampus (Autry et al., 2011). Consistent with this proposed mechanism, eEF2 kinase KO mice do not show an antidepressant-like response to ketamine nor exhibit a ketamine-induced potentiation (Nosyreva et al., 2013). A recent study found that while both ketamine-mediated deactivation of eEF2K and subsequent protein translation and retinoic acid (RA) dependent regulation of dendritic protein translation elicit homeostatic synaptic up-scaling, the two pathways operate largely independent of each other. Nevertheless, as the two pathways converge on homeostatic scaling as a synaptic end point, they both lead to rapid antidepressant-like responses (Suzuki et al., 2021). These findings suggest a causal link between signaling pathways that target synaptic up-scaling and rapid antidepressant action.
Importantly, ketamine’s impact on BDNF signaling and rapid antidepressant effects are not mimicked by another NMDAR antagonist, the closely related compound memantine, which is FDA-approved in the treatment of Alzheimer’s Disease. This disparity was found to be due to memantine’s limited efficacy in blocking NMDA receptors near resting membrane potentials, and thus it does not trigger key intracellular signaling pathways such as inhibition of eEF2 or BDNF protein expression (Gideons et al., 2014). Some studies have found that ketamine-induced potentiation is distinct from and does not affect NMDA-dependent LTP (Crawford et al., 2017), while other studies have found that ketamine may demonstrate metaplastic effects in blocking LTP (Kang et al., 2020). This interaction between ketamine and other forms of plasticity such as LTP remain unclear, and further studies are warranted. Further adding to the complexity of BDNF’s role in ketamine action, there appears to be an age- dependency of ketamine’s synaptic and behavioral effects. In juvenile animals, ketamine did not produce an antidepressant behavioral response, nor did it trigger potentiation in juvenile hippocampal slices. This suggests that developmentally mature hippocampal synapses are required to mediate ketamine’s physiological and behavioral effects (Nosyreva et al., 2014).
Lithium is an effective treatment for bipolar disorder, though its mechanisms are not fully understood. Lithium’s mechanism of action has been found to involve BDNF and synaptic down-scaling in its antimanic effects, though unlike in ketamine, it induced an increase in BDNF which is stable and long lasting. In conditional BDNF KO mice, the hyperactivity was not altered in lithium treated mice, suggesting a requirement or BDNF in the anti-manic effects of lithium. Furthermore, lithium causes a downward scaling of AMPAR amplitudes via post-synaptic mechanisms. This effect was found to be mediated by BDNF-TrkB signaling, which causes dynamin-mediated endocytosis of AMPAR subunits (Gideons et al., 2017). These results suggest that BDNF-TrkB signaling is involved in the antimanic, synaptic downscaling effects of lithium.
Ketamine and lithium appear to involve BDNF signaling to enact opposing effects on synaptic scaling; however, this may be due to the different localization (e.g. axon vs dendrite) and time course of BDNF release induced by the two drugs. Ketamine-induced increases in BDNF protein is acute and brief, while lithium is used as a chronic treatment and leads to a more sustained BDNF rise (Figure 4). Studies of both ketamine and lithium provide evidence that synaptic scaling plays a role in the treatment response, and these findings suggest that other neuropsychiatric treatments may also involve forms of homeostatic plasticity. Future approaches to expand upon these findings may include targeting the signaling pathways involved in homeostatic plasticity, such as eEF2K (Autry et al., 2011; Nosyreva et al., 2013) or retinoic acid signaling (Suzuki et al., 2021; Wang et al., 2011).
Figure 4: Importance of the acute versus chronic nature of BDNF activity on plasticity.
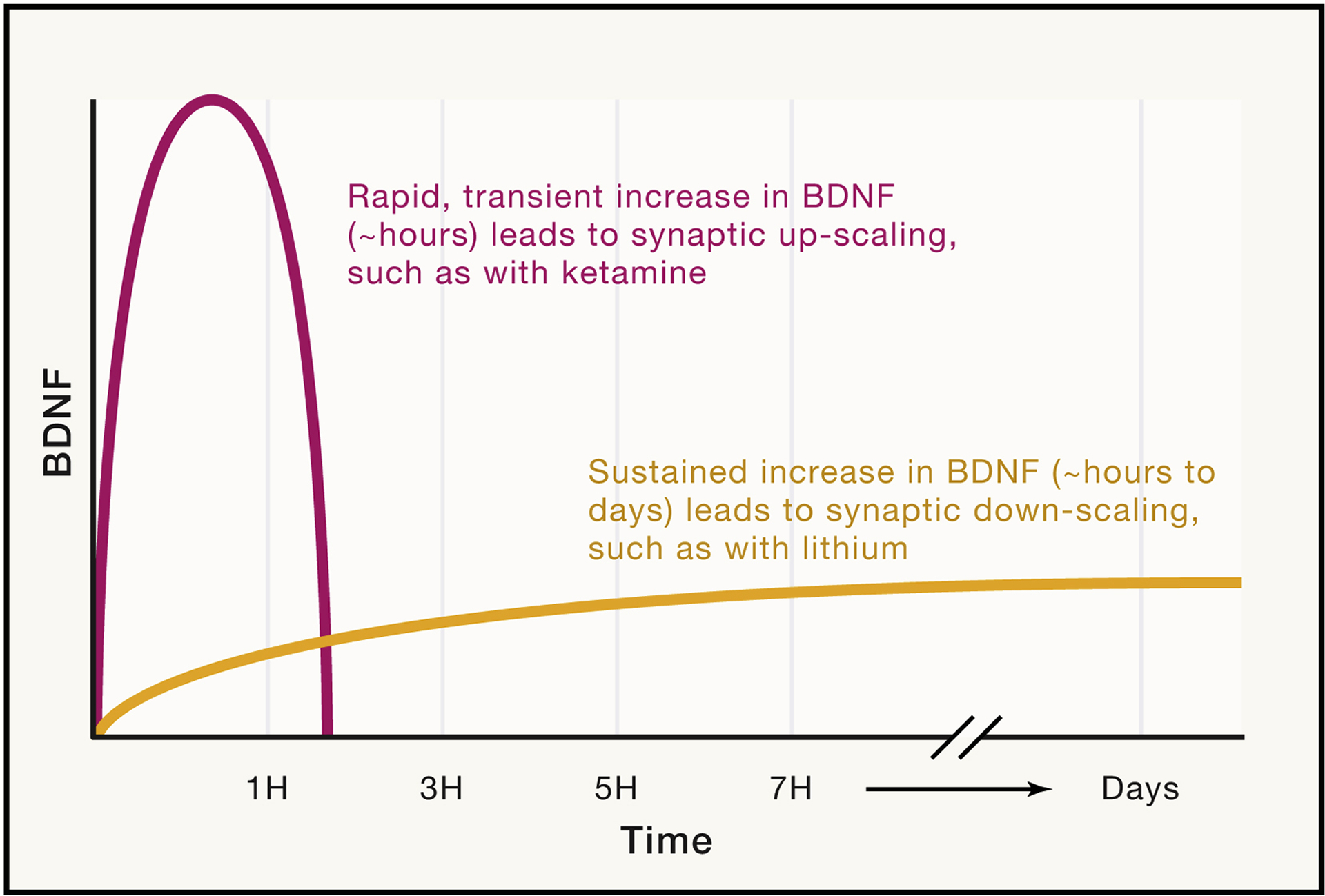
BDNF plays an important role in synaptic scaling, which involves the modulation of synaptic strength to counter chronic changes in activity, as a mechanism to maintain the relative strength of synaptic connections. Ketamine, a rapid antidepressant that inhibits NMDA receptors, induces a rapid increase in BDNF that leads to synaptic upscaling. On the other hand, lithium is a mood stabilizer that is an effective treatment for bipolar disorder. Lithium has been found to induce chronic, lasting increases in BDNF, and furthermore, has been shown to induce synaptic downscaling. Though both drugs increase BDNF levels, differences in the time course of this increase can elicit directly opposing effects. Furthermore, different types of synapses respond differentially to BDNF (i.e. inhibitory versus excitatory synapses). Taking into account the direction of change, the timescale, and the type of synaptic formations can hugely impact the effect of BDNF at synapses and ultimately behavior.
Psychiatric diseases are difficult to model due to the polygenetic nature of these disorders, and even so, baseline changes in homeostatic plasticity as a result of the disease process may be distinct from those involved in the treatment response. Certainly, homeostatic plasticity remains a field with many open questions, and it is important to consider any maladaptive consequences of manipulating these processes. By further elucidating the connection between psychiatric disease and homeostatic plasticity, these forms of non-Hebbian plasticities may emerge as important targets for future therapeutics (Kavalali and Monteggia, 2020).
Psychiatric Disease and Treatment
The biological underpinnings of many psychiatric illnesses remain unclear. Multiple genetic and environmental factors contribute, though understanding these factors to a sufficient degree to create a robust preclinical model remains elusive. Thus, caution should be taken in extrapolating effects from preclinical model studies onto human disease and treatment. Furthermore, within a single diagnosis, human patients can exhibit a heterogenous combination of symptoms. Despite how little we understand about mechanisms underlying psychiatric conditions, we have some therapeutics that demonstrate efficacy in managing symptoms. Furthermore, some of these treatments are used across different diagnoses – for instance, antidepressants are first line treatments for post- traumatic stress disorder (PTSD) as well as obsessive-compulsive disorder (OCD) (Vaswani et al., 2003).
Different psychiatric illnesses implicate varying and diverse mechanisms in their development. However, we often utilize overlapping pharmacologic agents such as antidepressants to treat these different diseases, which suggests that these therapeutics likely do not enact their function by reversing disease pathophysiology. Perhaps, current psychiatric drug treatments manage symptoms, but do not cure diseases. The mechanism of some of these pharmacologic agents, particularly antidepressants and mood stabilizers, is thought to involve BDNF-TrkB signaling (Figure 5). A better understanding of how BDNF is involved in various psychiatric treatments can help improve our understanding of how and when to target this pathway to improve disease outcomes. In the following sections, we will discuss how BDNF may or may not be involved in the mechanisms underlying various psychiatric therapeutics.
Figure 5: Effects of psychiatric drugs on BDNF activity in different brain regions.
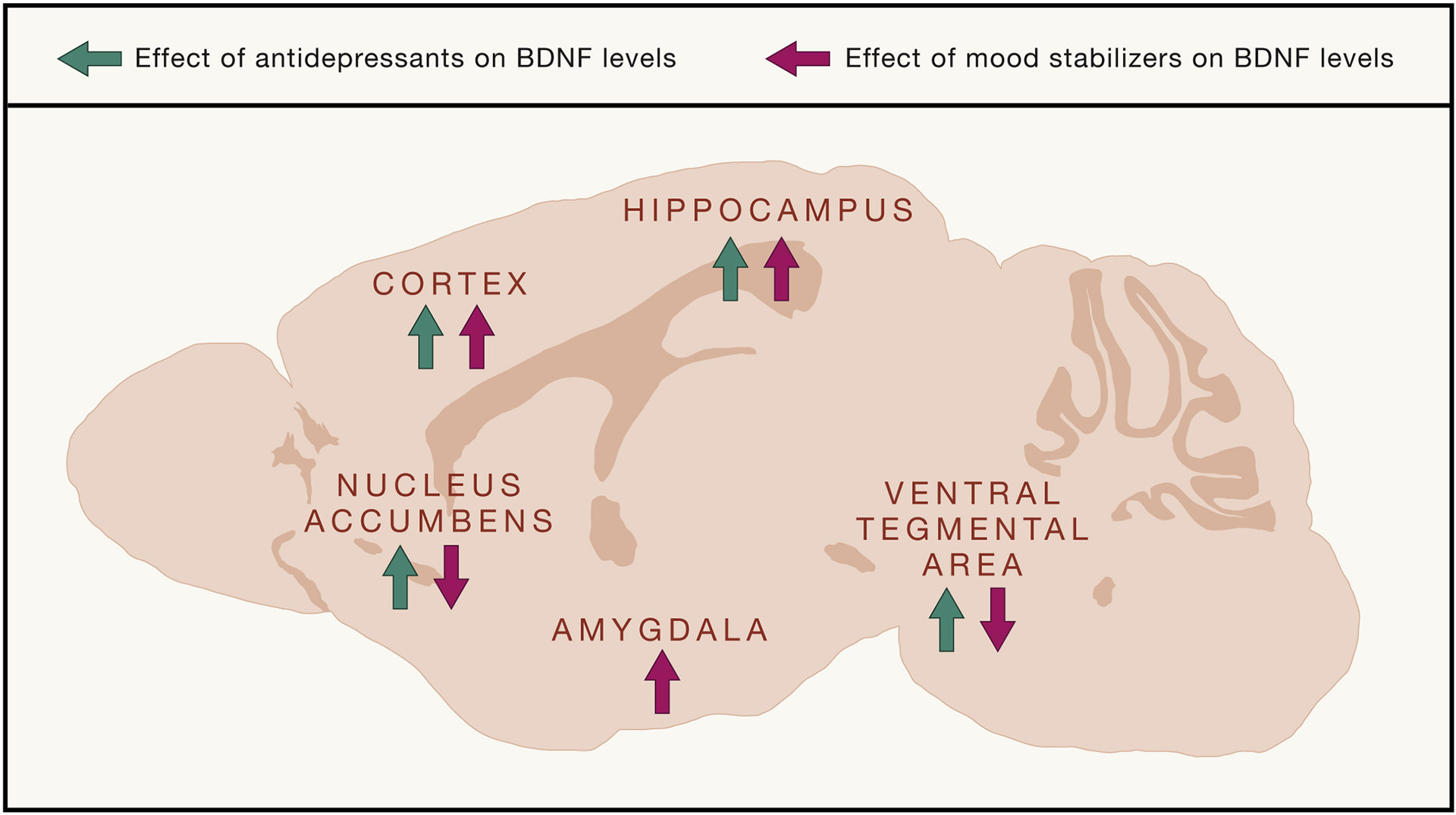
BDNF plays a critical role in the mechanism underlying psychiatric treatment, notably with antidepressants and mood stabilizers. Antidepressants, including SSRIs and ketamine, have been found to increase BDNF in the cortex and hippocampus, while decreasing protein levels in the nucleus accumbens and ventral tegmental area. Mood stabilizers, including lithium, have been found to increase BDNF levels in many brain regions including the cortex, hippocampus, ventral tegmental area, amygdala, and nucleus accumbens. The effect of psychiatric drugs on BDNF levels depends on the brain region studied, demonstrating a circuit level involvement of BDNF and psychiatric treatments.
The Antidepressant Response
Major depressive disorder (MDD) is a heterogeneous condition diagnosed by the DSM-5 Criteria, which includes symptoms such as loss of pleasure, fatigue, and sleep disturbances (American Psychological Association (APA), 2013). Pharmacologic agents targeting the monoaminergic system, such as serotonin-selective reuptake inhibitors (SSRIs), have become the mainstays for the treatment of MDD for several decades, and until recently, there were no new breakthroughs in expanding the repertoire of psychiatric pharmacology mechanisms. In the 2000s, however, a low intravenous ketamine treatment, an NMDA antagonist, was shown to have rapid antidepressant effects in patients with depression (Berman et al., 2000) or treatment-resistant depression (Zarate et al., 2006) which was a breakthrough as it introduced an entirely new mechanism of action. In 2019, the FDA approved a ketamine-derived nasal spray for treatment-resistant depression. Despite the apparently distinct mechanism between ketamine and more traditional antidepressants, as well as the differences in the time course of antidepressant action, BDNF has arisen as a factor common to the mechanism of both therapeutics (Kavalali and Monteggia, 2012; Monteggia et al., 2013). In the case of rapidly acting antidepressants such as ketamine and scopolamine, both their acute and sustained effects require BDNF signaling – in particular, BDNF driven transcriptional regulation (Kim et al., 2021). These results implicate BDNF as a potential biological target of convergence for a general antidepressant mechanism. Although the results are complex, the further study of BDNF may help improve future therapeutics.
Conventional antidepressants targeting the monoaminergic system increase BDNF and TrkB expression in a time course consistent with the antidepressant responses in cortical and subcortical structures such as the hippocampus (Nibuya et al., 1995; Rantamäki et al., 2007). In mice with conditional BDNF KO expression in forebrain regions or TrkB dominant negative genotypes, depressive phenotypes were not demonstrated. However, they had attenuated responses to antidepressants (Monteggia et al., 2004; Saarelainen et al., 2003). Furthermore, direct infusion of BDNF into the midbrain produces an antidepressant-like response (Siuciak et al., 1996). These results suggest that BDNF-TrkB signaling may not necessarily be involved in the depression pathophysiology; however, it may be a critical mediator of the antidepressant response.
Mechanisms surrounding TrkB involvement in the antidepressant response
Consistent with the notion that BDNF has regionally specific effects, BDNF was found to have different roles even within the hippocampus in the antidepressant response. BDNF infusion and deletion within the dentate gyrus induced and attenuated an antidepressant response, respectively. (Adachi et al., 2017; Shirayama et al., 2002). In a recent study, postsynaptic CA1 TrkB receptors were found to be critical for ketamine-induced synaptic potentiation and rapid antidepressant effects (Lin et al., 2021). These results demonstrate the importance of regional selectivity, even within the hippocampus, of BDNF-TrkB signaling in antidepressant action.
While neurogenesis in the hippocampus has been proposed to play a role in the antidepressant effect, the evidence surrounding this is not without controversy. Chronic antidepressant treatment, such as fluoxetine, increases the number of bromodeoxyuridine (BrdU)-labeled cells, a marker of DNA synthesis, suggesting that antidepressants may increase neuronal proliferation in the dentate gyrus (Malberg et al., 2000). Genetic and radiologic disruption of neurogenesis blocked the behavioral effects of antidepressants (Santarelli et al., 2003). However, this approach used x-irradiation to ablate cell proliferation, which has many non-specific side effects. Furthermore, a role for neurogenesis in antidepressant action has also been mired in some uncertainty as the behavioral effects of serotonin selective reuptake inhibitors can occur independent of neurogenesis in some mouse strains (Holick et al., 2008). The rapid timescale of antidepressant action with ketamine suggests neurogenesis does not appear to be strictly required for initiating antidepressant effects. However, TrkB-dependent neuronal differentiation has been reported to mediate the sustained antidepressant action of ketamine (Ma et al., 2017).
A recent study reported that both typical and fast-acting antidepressants bind directly to the TrkB receptor to facilitate its activation by BDNF (Casarotto et al., 2021). This was an unexpected finding given that the prior convention of understanding was that antidepressants increase BDNF expression, which subsequently activated TrkB receptors. Further work is required to replicate this study as well as understand how such diverse agents all bind and activate the TrkB receptor. Selective TrkB agonists have been put forward as potential therapeutics (Jang et al., 2010; Zhang et al., 2015), however the specificity of the TrkB agonists to activate downstream signaling has been called into question. If spatial and temporal factors are critical in BDNF’s effect, it is reasonable to imagine that TrkB activation would necessitate regional and contextual relevance as well.
BDNF plays an important role in classical as well as rapidly acting antidepressant mechanisms. This suggests that an antidepressant response is not necessarily a reversal of the pathophysiology of disease processes, but more of a “masking” effect on maladaptive thought or behavior patterns. As further explored in the topics below, antidepressants can treat a number of psychiatric illnesses that are thought to have vastly different underlying mechanisms and distinctly different symptom presentations, further supporting this separation between disease pathophysiology and treatment mechanism.
Treatment of Bipolar Disorder
The diagnosis of bipolar disorder hinges on a past or current history of a manic episode, or a hypomanic episode with a major depressive episode, as defined by the DSM-5 (i.e. inflated self-esteem, decreased need for sleep, pressured speech, etc.). Mood stabilizers are the pharmacological mainstays of treatment for acute mania, with lithium being the gold standard (McIntyre et al., 2020). Administration of lithium in rodent models has been found to involve BDNF in its antimanic effects. Chronic administration of lithium increased BDNF protein levels, particularly in the hippocampus and frontal cortex (Fukumoto et al., 2001). Additionally, lithium has been found to mediate synaptic downscaling via sustained endocytosis of AMPAR subunits in hippocampal cultures, an effect dependent on BDNF-TrkB signaling (Gideons et al., 2017). Valproic acid is another mood stabilizer, and its repeated administration increased BDNF expression in the hippocampus and cortex (Fukumoto et al., 2001). Both lithium and valproate were found to activate promoter IV of BDNF (Yasuda et al., 2009).
Overall, it appears that mood stabilizers may increase BDNF activity in brain regions such as the hippocampus, cortex, and VTA-PFC projections (Figure 5). Furthermore, lithium in particular may involve synaptic downscaling of AMPAR subunits to mediate its antimanic effects.
Post-Traumatic Stress Disorder
Post-traumatic stress disorder (PTSD) may develop after exposure to traumatic event(s), which lead to characteristic symptoms such as hypervigilance, avoidance, and mood/cognitive alterations. The first line treatment of PTSD is SSRIs due to its efficacy and low side effect profile (American Psychological Association (APA), 2013). The mechanism of PTSD remains unclear but is thought to involve fear learning and extinction. BDNF-TrkB signaling has been implicated in fear learning and extinction, particularly in the prefrontal cortex, hippocampus, and amygdala (Chhatwal et al., 2006; Heldt et al., 2007).
While there are distinct and diverse genetic and contextual contributions to an individual’s susceptibility to PTSD, the first line of treatment is the same as for major depression. This fact further highlights that we can treat some psychiatric conditions, despite knowing very little about the underlying mechanism, effectively with drugs approved for other psychiatric symptoms.
Schizophrenia
Schizophrenia is a psychiatric disorder involving chronic or recurrent psychosis, characterized by positive symptoms (delusions, hallucinations, disorganized speech) and negative symptoms (anhedonia, depressed mood). Antipsychotics treat positive symptoms by targeting the dopaminergic system, and they are the mainstays of treatment.
BDNF has been found to play a role in improving the survival and function of dopaminergic neurons, which are implicated in the symptomatic manifestations of schizophrenia (Hyman et al., 1991). Thus, it is plausible that schizophrenia and its treatment may involve BDNF signaling as a component of their mechanism. Animal models of drug-induced psychosis (i.e. ibotenic acid, phencyclidine) have found decreased BDNF levels (Snigdha et al., 2011); however, these changes could be causative or compensatory. Furthermore, there is a widely heterogenous collection of genetic and environmental factors contributing to the development of schizophrenia that makes it difficult to model in preclinical studies accurately (Lewis and Levitt, 2002).
The role of BDNF in antipsychotics is even less clear. Studies have found that while some antipsychotics reduce BDNF and TrkB expression in preclinical models (Parikh et al., 2004), others have no effect (Parikh et al., 2004), and still others increase it (Xu et al., 2002). BDNF-TrkB signaling has an unclear connection to the treatment of schizophrenia with antipsychotics, with more work necessary to clarify a potential link.
Obsessive Compulsive Disorder
Obsessive-compulsive disorder (OCD) is characterized by recurrent and intrusive thoughts that cause significant distress, for which the patient may employ repetitive, compensatory behaviors or mental acts to alleviate stress (i.e. washing their hands repetitively after touching a public toilet seat). Its first line treatment is SSRIs, which are strongly suggested to involve the BDNF-TrkB signaling pathway (Björkholm and Monteggia, 2016).
While OCD is difficult to model preclinically, there have been some genes and circuits that have been suggested to contribute to its pathogenesis. For instance, knocking out the SLITRK5 gene, whose encoded protein has neurite-modulating properties (Aruga et al., 2003), led to OCD-like behaviors in mice (i.e. pathologic over-grooming and increased anxiety-like behaviors that were normalized with SSRIs) (Shmelkov et al., 2010). SLITRK5 has been found to be involved in BDNF-TrkB signaling, as it acts as a TrkB co-receptor during BDNF-induced activity (Song et al., 2015). The cortico-striatal thalamo-cortical loop has been found to be a major site of synaptic dysfunction in patients with OCD (Ting and Feng, 2008), and BDNF may be involved in this dysfunction (Jing et al., 2017). Yet, the link between BDNF and OCD pathogenesis remains murky, and correlations between genetic variations in BDNF and OCD symptoms have been inconsistent (Alonso et al., 2008; Mössner et al., 2005).
More so than pathogenesis, BDNF may be involved in the treatment response of patients with OCD. Small clinical trials have suggested that BDNF may play a role in the treatment of OCD, and that BDNF genotyping could be useful for treatment personalization (Laje et al., 2012; Real et al., 2009)(Laje et al., 2012; Linkovski et al., 2019). Further supporting this idea, another study found that some genetic variants in BDNF were correlated with a better response to antidepressants in OCD (Real et al., 2009). These findings suggest that BDNF may have a clinically relevant effect in improving OCD disease symptomology as it relates to pharmacologic treatment, and that better understanding the natural variation in human alleles may assist in improving treatment approaches.
TrkB agonists as therapeutics
Given the link between BDNF-TrkB signaling in the involvement of neuropsychiatric treatments, it is not surprising that there has been interest in therapeutics that directly target this pathway. However, since BDNF is a peptide, it does not cross the blood brain barrier making it difficult as a therapeutic. There have been attempts with the infusion of the peptide into the brain, though an indiscriminate amount of BDNF infusion leads to undesired side effects (Lähteinen et al., 2003).
Several groups have developed small molecule TrkB agonists to treat neuropsychiatric diseases that involve BDNF in their mechanism (Massa et al., 2010; Schmid et al., 2012), including the well-studied 7,8-dihydroxyflavone (Liu et al., 2016). However, a recent study found that none of the reported TrkB agonists actually work in their purported function, raising doubt on the efficacy and specificity of these compounds to activate TrkB downstream pathways (Boltaev et al., 2017; Lowe, 2017). While small molecule TrkB agonists may indeed have effects in animal studies, these could be due to mechanisms independent of direct TrkB activation. Furthermore, the ability of TrkB agonists, similar to exogenous BDNF, is expected to be limited in mimicking endogenous BDNF action, due to the precise spatial and temporal aspects of BDNF signaling outlined here.
Conclusion
In this article, we aimed to discuss the multifaceted effects of BDNF-TrkB signaling in its regulation of structural as well as Hebbian and homeostatic forms of synaptic plasticity. Akin to signaling pathways directed by neurotransmitters such as glutamate and GABA, BDNF elicits diverse instructive as well as permissive effects on synaptic signaling, in some cases leading to seemingly contradictory forms of regulation. However, emerging studies from our group as well as others suggest that BDNF’s effects on plasticity depend on the precise subcellular location of signaling as well as temporal profile of BDNF signals. Indeed, the importance of its specificity of release and site of binding provides an explanation of why broad application of exogenous BDNF can provide conflicting results. In contrast, studies that manipulate endogenous BDNF lead to more converging findings. Therefore, development of approaches for spatially and temporally specific manipulation of BDNF will likely be more fruitful in further understanding BDNF’s actions. Furthermore, BDNF acts specifically and locally on pre- and postsynaptic TrkB receptors to activate distinct downstream signaling pathways that are involved in synaptogenesis, short- and long-term plasticity.
While the role of BDNF in disease pathophysiology remains unclear, BDNF appears to play a critical role in the mechanism underlying psychiatric therapeutics, in particular antidepressants and mood stabilizers. Given the emerging consensus on the key role of BDNF in treatments of psychiatric disorders, future basic as well as translational studies – in particular potential BDNF-TrkB therapeutics – will need to take into account the precise spatial and temporal relevance of BDNF actions to achieve the desired treatment outcomes.
Table 1.
Impact of different psychiatric treatments on BDNF at varying timescales and brain region location
| BDNF effect/treatment timescale | Brain region | Antidepressant treatment | ADT effect on BDNF activity | Reference(s) |
|---|---|---|---|---|
| Chronic (days) | CA3 | ECS, sertraline, desipramine, BDNF infusion | increase | Nibuya et al., 1995; Shirayama et al., 2002 |
| Chronic (days) | CA1 | BDNF infusion, desipramine and citalopram | none | Adachi et al., 2008; Shirayama et al., 2002 |
| Chronic tx (days) | CA1 | ECS, sertraline, desipramine | increase | Nibuya et al., 1995 |
| Chronic BDNF (days) | DG | fluoxetine and imipramine, BDNF infusion, desipramine and citalopram | increase | Adachi et al., 2008; Nibuya et al., 1995; Santarelli et al., 2003; Shirayama et al., 2002 |
| Chronic tx (days) | frontal cortex | ECS | increase | Nibuya et al., 1995 |
| BDNF forebrain deletion | forebrain | desipramine | increase | Monteggia et al., 2004 |
| Chronic (days) | anterior cingulate cortex | imipramine, fluoxetine, citalopram | increase | Rantamäki et al., 2007; Saarelainen et al., 2003 |
| Chronic | prefrontal cortex | imipramine, fluoxetine, ketamine | increase | Saarelainen et al., 2003 |
| Chronic (days) | VTA-NAc | fluoxetine, imipramine | decrease | Berton et al., 2006 |
| Chronic (days) | visual cortex | fluoxetine | increase | Maya Vetencourt et al., 2008 |
| Timescale of treatment | Brain region | Mood stabilizer | Drug effects on BDNF | Reference |
| Chronic tx | cortical neurons | lithium, valproic acid | increase BDNF and promoter i.v.-driven BDNF | Yasuda et al., 2009 |
| Chronic tx | hippocampus | lithium | increase | Gideons et al., 2017 |
| Timescale of treatment | Brain region | Antipsychotic | Drug effects on BDNF | Reference |
| Chronic | cortex | lurasidone (SGA) | increase | Fumagalli et al., 2012 |
| Single injection | DG and PFC | clozapine (SGA) and haloperidol (FGA) | decrease | Lipska et al., 2001 |
| Chronic (days) | hippocampus | haloperidol (FGA) | no change | Shirayama et al., 2002 |
| Chronic | hippocampus | lurasidone (SGA) | increases | Fumagalli et al., 2012 |
| Chronic | hippocampus | quetiapine (SGA) | increase | Xu et al., 2002 |
ECS, electroconvulsive seizures; tx, treatment; DG, dentate gyrus; i.v., intravenous; SGA, second-generation antipsychotics; and FGA, first-generation antipsychotics.
Acknowledgements
We would like to thank current and former members of Kavalali and Monteggia laboratories for numerous invaluable discussions. We acknowledge that the field of BDNF is vast, and we apologize that we were unable to cite many important papers due to space limitations. Figures 1, 2, and 5 were created with Biorender.com. This work was supported by National Institute of Mental Health grants, MH081060 and MH070727 (LMM), and MH66198 (ETK).
Declaration of interests
The authors declare no competing interests. L.M.M. has received funding from Astellas Pharmaceuticals on an unrelated project and is on the scientific advisory board of Gilgamesh and Rodin Therapeutics.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adachi M, Autry AE, Mahgoub M, Suzuki K, and Monteggia LM (2017). TrkB Signaling in Dorsal Raphe Nucleus is Essential for Antidepressant Efficacy and Normal Aggression Behavior. Neuropsychopharmacology 42, 886–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso M, Medina JH, and Pozzo-Miller L (2004). ERK1/2 Activation Is Necessary for BDNF to Increase Dendritic Spine Density in Hippocampal CA1 Pyramidal Neurons. Learning and Memory 11, 172–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso P, Gratacòs M, Menchón JM, Saiz-Ruiz J, Segalàs C, Baca-García E, Labad J, Fernández-Piqueras J, Real E, Vaquero C, et al. (2008). Extensive Genotyping of the BDNF and NTRK2 Genes Define Protective Haplotypes Against Obsessive-Compulsive Disorder. Biological Psychiatry 62, 619–628. [DOI] [PubMed] [Google Scholar]
- Amaral MD, and Pozzo-Miller L (2007a). BDNF induces calcium elevations associated with IBDNF, a nonselective cationic current mediated by TRPC channels. Journal of Neurophysiology 98, 2476–2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaral MD, and Pozzo-Miller L (2007b). TRPC3 channels are necessary for brain-derived neurotrophic factor to activate a nonselective cationic current and to induce dendritic spine formation. Journal of Neuroscience 27, 5179–5189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Psychological Association (APA) (2013). Diagnostic and Statistical Manual of Mental Disorders: Depressive Disorders.
- An JJ, Gharami K, Liao GY, Woo NH, Lau AG, Vanevski F, Torre ER, Jones KR, Feng Y, Lu B, et al. (2008). Distinct Role of Long 3′ UTR BDNF mRNA in Spine Morphology and Synaptic Plasticity in Hippocampal Neurons. Cell 134, 175–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aruga J, Yokota N, and Mikoshiba K (2003). Human SLITRK family genes: genomic organization and expression profiling in normal brain and brain tumor tissue. Gene 315, 87–94. [DOI] [PubMed] [Google Scholar]
- Autry AE, and Monteggia LM (2012). Brain-Derived Neurotrophic Factor and Neuropsychiatric Disorders. Pharmacological Reviews 64, 238–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Autry AE, Adachi M, Nosyreva E, Na ES, Los MF, Cheng P, Kavalali ET, and Monteggia LM (2011). NMDA Receptor Blockade at Rest Triggers Rapid Behavioural Antidepressant Responses. Nature 475, 91–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balkowiec A, and Katz DM (2002). Cellular mechanisms regulating activity-dependent release of native brain-derived neurotrophic factor from hippocampal neurons. The Journal of Neuroscience : The Official Journal of the Society for Neuroscience 22, 10399–10407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barde YA, Lindsay RM, Monard D, and Thoenen H (1978). New factor released by cultured glioma cells supporting survival and growth of sensory neurones. Nature 274, 818. [DOI] [PubMed] [Google Scholar]
- Baydyuk M, Wu X-S, He L, and Wu L-G (2015). Brain-derived neurotrophic factor inhibits calcium channel activation, exocytosis, and endocytosis at a central nerve terminal. The Journal of Neuroscience : The Official Journal of the Society for Neuroscience 35, 4676–4682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS, and Krystal JH (2000). Antidepressant effects of ketamine in depressed patients. Biological Psychiatry 47, 351–354. [DOI] [PubMed] [Google Scholar]
- Björkholm C, and Monteggia LM (2016). BDNF - A key transducer of antidepressant effects. Neuropharmacology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boltaev U, Meyer Y, Tolibzoda F, Jacques T, Gassaway M, Xu Q, Wagner F, Zhang Y-L, Palmer M, Holson E, et al. (2017). Multiplex quantitative assays indicate a need for reevaluating reported small-molecule TrkB agonists. Science Signaling 10. [DOI] [PubMed] [Google Scholar]
- Casarotto PC, Girych M, Fred SM, Kovaleva V, Moliner R, Enkavi G, Biojone C, Cannarozzo C, Sahu MP, Kaurinkoski K, et al. (2021). Antidepressant drugs act by directly binding to TRKB neurotrophin receptors. Cell 184, 1299–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Q, Song SH, and Augustine GJ (2017). Calcium-dependent and synapsin-dependent pathways for the presynaptic actions of BDNF. Frontiers in Cellular Neuroscience 11, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chhatwal JP, Stanek-Rattiner L, Davis M, and Ressler KJ (2006). Amygdala BDNF signaling is required for consolidation but not encoding of extinction. Nature Neuroscience 9, 870–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S, and Levi-Montalcini R (1956). A nerve growth-stimulating factor isolated from snake venom. Proceedings of the National Academy of Sciences 42, 571–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford DC, Ramirez DMO, Trauterman B, Monteggia LM, and Kavalali ET (2017). Selective molecular impairment of spontaneous neurotransmission modulates synaptic efficacy. Nature Communications 8, 14436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean C, Liu H, Dunning FM, Chang PY, Jackson MB, and Chapman ER (2009). Synaptotagmin-IV modulates synaptic function and long-term potentiation by regulating BDNF release. Nature Neuroscience 12, 767–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai NS, Rutherford LC, and Turrigiano GG (1999). BDNF regulates the intrinsic excitability of cortical neurons. Learning and Memory 6, 284–291. [PMC free article] [PubMed] [Google Scholar]
- Dieni S, Matsumoto T, Dekkers M, Rauskolb S, Ionescu MS, Deogracias R, Gundelfinger ED, Kojima M, Nestel S, Frotscher M, et al. (2012). BDNF and its pro-peptide are stored in presynaptic dense core vesicles in brain neurons. Journal of Cell Biology 196, 775–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Feng L, Yang F, and Lu B (2000). Activity- and Ca2+-dependent modulation of surface expression of brain-derived neurotrophic factor receptors in hippocampal neurons. Journal of Cell Biology 150, 1423–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan MF, Kojima M, Callicott JH, Goldberg TE, Kolachana BS, Bertolino A, Zaitsev E, Gold B, Goldman D, Dean M, et al. (2003). The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell 112, 257–269. [DOI] [PubMed] [Google Scholar]
- Figurov A, Pozzo-Miller LD, Olafsson P, Wang T, and Lu B (1996). Regulation of synaptic responses to high-frequency stimulation and LTP by neurotrophins in the hippocampus. Nature 381, 706–709. [DOI] [PubMed] [Google Scholar]
- Frerking M, Malenka RC, and Nicoll RA (1998). Brain-derived neurotrophic factor (BDNF) modulates inhibitory, but not excitatory, transmission in the CA1 region of the hippocampus. Journal of Neurophysiology 80, 3383–3386. [DOI] [PubMed] [Google Scholar]
- Frisen J, Verge VM, Fried K, Risling M, Persson H, Trotter J, Hokfelt T, and Lindholm D (1993). Characterization of glial trkB receptors: differential response to injury in the central and peripheral nervous systems. Proceedings of the National Academy of Sciences 90, 4971–4975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukumoto T, Morinobu S, Okamoto Y, Kagaya A, and Yamawaki S (2001). Chronic lithium treatment increases the expression of brain-derived neurotrophic factor in the rat brain. Psychopharmacology 158, 100–106. [DOI] [PubMed] [Google Scholar]
- Gideons ES, Kavalali ET, and Monteggia LM (2014). Mechanisms underlying differential effectiveness of memantine and ketamine in rapid antidepressant responses. Proceedings of the National Academy of Sciences 111, 8649–8654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gideons ES, Lin P-Y, Mahgoub M, Kavalali ET, and Monteggia LM (2017). Chronic lithium treatment elicits its antimanic effects via BDNF-TrkB dependent synaptic downscaling. ELife 6, e25480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruart A, Sciarretta C, Valenzuela-Harrington M, Delgado-García JM, and Minichiello L (2007). Mutation at the TrkB PLCγ-docking site affects hippocampal LTP and associative learning in conscious mice. Learning and Memory 14, 54–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall J, Thomas KL, and Everitt BJ (2000). Rapid and selective induction of BDNF expression in the hippocampus during contextual learning. Nature Neuroscience 3, 533–535. [DOI] [PubMed] [Google Scholar]
- Harward SC, Hedrick NG, Hall CE, Parra-Bueno P, Milner TA, Pan E, Laviv T, Hempstead BL, Yasuda R, and McNamara JO (2016). Autocrine BDNF-TrkB signalling within a single dendritic spine. Nature 538, 99–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heldt S, Stanek L, Chhatwal J, and Ressler K (2007). Hippocampus-specific deletion of BDNF in adult mice impairs spatial memory and extinction of aversive memories. Molecular Psychiatry 12, 656–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrand ME, Xu J, Dedek A, Li Y, Sengar AS, Beggs S, Lombroso PJ, and Salter MW (2016). Potentiation of Synaptic GluN2B NMDAR Currents by Fyn Kinase Is Gated through BDNF-Mediated Disinhibition in Spinal Pain Processing. Cell Reports 17, 2753–2765. [DOI] [PubMed] [Google Scholar]
- Holick KA, Lee DC, Hen R, and Dulawa SC (2008). Behavioral effects of chronic fluoxetine in BALB/cJ mice do not require adult hippocampal neurogenesis or the serotonin 1A receptor. Neuropsychopharmacology 33, 406–417. [DOI] [PubMed] [Google Scholar]
- Hong EJ, McCord AE, and Greenberg ME (2008). A Biological Function for the Neuronal Activity-Dependent Component of Bdnf Transcription in the Development of Cortical Inhibition. Neuron 60, 610–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horch HW, and Katz LC (2002). BDNF release from single cells elicits local dendritic growth in nearby neurons. Nature Neuroscience 5, 1177–1184. [DOI] [PubMed] [Google Scholar]
- Horvath PM, Chanaday NL, Alten B, Kavalali ET, and Monteggia LM (2021). A subthreshold synaptic mechanism regulating BDNF expression and resting synaptic strength. Cell Reports 36, 109467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman C, Hofer M, Barde YA, Juhasz M, Yancopoulos GD, Squinto SP, and Lindsay RM (1991). BDNF is a neurotrophic factor for dopaminergic neurons of the substantia nigra. Nature 350, 230–232. [DOI] [PubMed] [Google Scholar]
- Ip NY, Ibanez CF, Nye SH, McClain J, Jones PF, Gies DR, Belluscio L, Le Beau MM, Espinosa R, Squinto SP, et al. (1992). Mammalian neurotrophin-4: Structure, chromosomal localization, tissue distribution, and receptor specificity. Proceedings of the National Academy of Sciences of the United States of America 89, 3060–3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakawich SK, Nasser HB, Strong MJ, McCartney AJ, Perez AS, Rakesh N, Carruthers CJL, and Sutton MA (2010). Local Presynaptic Activity Gates Homeostatic Changes in Presynaptic Function Driven by Dendritic BDNF Synthesis. Neuron 68, 1143–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang S-W, Liu X, Yepes M, Shepherd KR, Miller GW, Liu Y, Wilson WD, Xiao G, Blanchi B, Sun YE, et al. (2010). A selective TrkB agonist with potent neurotrophic activities by 7,8-dihydroxyflavone. Proceedings of the National Academy of Sciences 107, 2687–2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Y, Lu Y, Yang F, Shen W, Tang TT-T, Feng L, Duan S, and Lu B (2010). Acute and gradual increases in BDNF concentration elicit distinct signaling and functions in neurons. Nature Neuroscience 13, 302–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H, and Schuman E (1995). Long-lasting neurotrophin-induced enhancement of synaptic transmission in the adult hippocampus. Science 267, 1658–1662. [DOI] [PubMed] [Google Scholar]
- Kang H, Jia LZ, Suh KY, Tang L, and Schuman EM (1996). Determinants of BDNF-induced hippocampal synaptic plasticity: Role of the Trk B receptor and the kinetics of neurotrophin delivery. Learning Memory 3, 188–196. [DOI] [PubMed] [Google Scholar]
- Kang H, Park P, Han M, Tidball P, Georgiou J, Bortolotto ZA, Lodge D, Kaang B-K, and Collingridge GL (2020). (2S,6S)- and (2R,6R)-hydroxynorketamine inhibit the induction of NMDA receptor-dependent LTP at hippocampal CA1 synapses in mice. Brain and Neuroscience Advances 4, 2398212820957847–2398212820957847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavalali ET, and Monteggia LM (2012). Synaptic Mechanisms Underlying Rapid Antidepressant Action of Ketamine. American Journal of Psychiatry 169, 1150–1156. [DOI] [PubMed] [Google Scholar]
- Kavalali ET, and Monteggia LM (2020). Targeting Homeostatic Synaptic Plasticity for Treatment of Mood Disorders. Neuron 106, 715–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J-W, Autry AE, Na ES, Adachi M, Björkholm C, Kavalali ET, and Monteggia LM (2021). Sustained effects of rapidly acting antidepressants require BDNF-dependent MeCP2 phosphorylation. Nature Neuroscience. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokaia Z, Metsis M, Kokaia M, Bengzon J, Elmér E, Smith M-L, Timmusk T, Siesjö BK, Persson H, and Lindvall O (1994). Brain Insults in Rats Induce Increased Expression of the BDNF Gene through Differential Use of Multiple Promoters. European Journal of Neuroscience 6, 587–596. [DOI] [PubMed] [Google Scholar]
- Korte M, Carroll P, Wolf E, Brem G, Thoenen H, and Bonhoeffer T (1995). Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proceedings of the National Academy of Sciences of the United States of America 92, 8856–8860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowiański P, Lietzau G, Czuba E, Waśkow M, Steliga A, and Moryś J (2018). BDNF: A Key Factor with Multipotent Impact on Brain Signaling and Synaptic Plasticity. Cellular and Molecular Neurobiology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lähteinen S, Pitkänen A, Koponen E, Saarelainen T, and Castrén E (2003). Exacerbated status epilepticus and acute cell loss, but no changes in epileptogenesis, in mice with increased brain-derived neurotrophic factor signaling. Neuroscience 122. [DOI] [PubMed] [Google Scholar]
- Laje G, Lally N, Mathews D, Brutsche N, Chemerinski A, Akula N, Kelmendi B, Simen A, McMahon FJ, Sanacora G, et al. (2012). Brain-derived neurotrophic factor Val66Met polymorphism and antidepressant efficacy of ketamine in depressed patients. Biological Psychiatry 72, e27–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang SB, Stein V, Bonhoeffer T, and Lohmann C (2007). Endogenous brain-derived neurotrophic factor triggers fast calcium transients at synapses in developing dendrites. The Journal of Neuroscience : The Official Journal of the Society for Neuroscience 27, 1097–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemtiri-Chlieh F, and Levine ES (2010). BDNF evokes release of endogenous cannabinoids at layer 2/3 inhibitory synapses in the neocortex. Journal of Neurophysiology 104, 1923–1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine ES, Dreyfus CF, Black IB, and Plummer MR (1995). Brain-derived neurotrophic factor rapidly enhances synaptic transmission in hippocampal neurons via postsynaptic tyrosine kinase receptors. Proceedings of the National Academy of Sciences of the United States of America 92, 8074–8077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis DA, and Levitt P (2002). Schizophrenia as a Disorder of Neurodevelopment. Annual Review of Neuroscience 25, 409–432. [DOI] [PubMed] [Google Scholar]
- Li HS, Xu XZS, and Montell C (1999). Activation of a trpc3-dependent cation current through the neurotrophin bdnf. Neuron 24, 261–273. [DOI] [PubMed] [Google Scholar]
- Li YX, Zhang Y, Lester HA, Schuman EM, and Davidson N (1998a). Enhancement of neurotransmitter release induced by brain-derived neurotrophic factor in cultured hippocampal neurons. Journal of Neuroscience 18, 10231–10240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YX, Xu Y, Ju D, Lester HA, Davidson N, and Schuman EM (1998b). Expression of a dominant negative TrkB receptor, T1, reveals a requirement for presynaptic signaling in BDNF-induced synaptic potentiation in cultured hippocampal neurons. Proceedings of the National Academy of Sciences of the United States of America 95, 10884–10889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin P-Y, Kavalali ET, and Monteggia LM (2018). Genetic Dissection of Presynaptic and Postsynaptic BDNF-TrkB Signaling in Synaptic Efficacy of CA3-CA1 Synapses. Cell Reports 24, 1550–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin P-Y, Ma ZZ, Mahgoub M, Kavalali ET, and Monteggia LM (2021). A synaptic locus for TrkB signaling underlying ketamine rapid antidepressant action. Cell Reports 36, 109513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linkovski O, Wheaton MG, Zwerling J, Odgerel Z, Van Roessel P, Filippou-Frye M, Lombardi A, Wright B, Steinman SA, Simpson HB, et al. (2019). Exploring Brain-Derived Neurotrophic Factor Val66Met Polymorphism and Extinction Learning-Based Treatment Outcome in Obsessive-Compulsive Disorder: A Pilot Study. Journal of Clinical Psychopharmacology 39, 91–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lituma PJ, Kwon H-B, Alviña K, Luján R, and Castillo PE (2021). Presynaptic NMDA receptors facilitate short-term plasticity and BDNF release at hippocampal mossy fiber synapses. ELife 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Chan CB, and Ye K (2016). 7,8-dihydroxyflavone, a small molecular TrkB agonist, is useful for treating various BDNF-implicated human disorders. Translational Neurodegeneration 5, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu IYC, Lyons WE, Mamounas LA, and Thompson RF (2004). Brain-derived neurotrophic factor plays a critical role in contextual fear conditioning. Journal of Neuroscience 24, 7958–7963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohof AM, Ip NY, and Poo M (1993). Potentiation of developing neuromuscular synapses by the neurotrophins NT-3 and BDNF. Nature 363, 350–353. [DOI] [PubMed] [Google Scholar]
- Lowe D (2017). Those Compounds Aren’t What You Think They Are.
- Lu B (2003). BDNF and activity-dependent synaptic modulation. Learning & Memory (Cold Spring Harbor, N.Y.) 10, 86–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luikart BW, Nef S, Virmani T, Lush ME, Liu Y, Kavalali ET, and Parada LF (2005). TrkB has a cell-autonomous role in the establishment of hippocampal Schaffer collateral synapses. The Journal of Neuroscience : The Official Journal of the Society for Neuroscience 25, 3774–3786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maisonpierre PC, Belluscio L, Squinto S, Ip NY, Furth ME, Lindsay RM, and Yancopoulos GD (1990). Neurotrophin-3: A neurotrophic factor related to NGF and BDNF. Science 247, 1446–1451. [DOI] [PubMed] [Google Scholar]
- Malberg JE, Eisch AJ, Nestler EJ, and Duman RS (2000). Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. Journal of Neuroscience 20, 9104–9110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, and Bear MF (2004). LTP and LTD: An Embarrassment of Riches. Neuron 44, 5–21. [DOI] [PubMed] [Google Scholar]
- Massa SM, Yang T, Xie Y, Shi J, Bilgen M, Joyce JN, Nehama D, Rajadas J, and Longo FM (2010). Small molecule BDNF mimetics activate TrkB signaling and prevent neuronal degeneration in rodents. Journal of Clinical Investigation 120, 1774–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto T, Rauskolb S, Polack M, Klose J, Kolbeck R, Korte M, and Barde YA (2008). Biosynthesis and processing of endogenous BDNF: CNS neurons store and secrete BDNF, not pro-BDNF. Nature Neuroscience 11, 131–133. [DOI] [PubMed] [Google Scholar]
- Maynard KR, Sukumar M, Lu B, Tessarollo L, Schloesser RJ, and Martinowich K (2015). Promoter-specific production of BDNF regulates social behaviors in mice. Biological Psychiatry 41, 1943–1955. [Google Scholar]
- McAllister AK, Lo DC, and Katz LC (1995). Neurotrophins regulate dendritic growth in developing visual cortex. Neuron 15, 791–803. [DOI] [PubMed] [Google Scholar]
- McIntyre RS, Berk M, Brietzke E, Goldstein BI, López-Jaramillo C, Kessing LV, Malhi GS, Nierenberg AA, Rosenblat JD, Majeed A, et al. (2020). Bipolar disorders. The Lancet 396, 1841–1856. [DOI] [PubMed] [Google Scholar]
- Meyer-Franke A, Wilkinson GA, Kruttgen A, Hu M, Munro E, Hanson MG, Reichardt LF, and Barres BA (1998). Depolarization and cAMP elevation rapidly recruit TrkB to the plasma membrane of CNS neurons. Neuron 21, 681–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minichiello L (2009). TrkB signalling pathways in LTP and learning. Nature Reviews. Neuroscience 10, 850–860. [DOI] [PubMed] [Google Scholar]
- Minichiello L, Korte M, Wolfer D, Kühn R, Unsicker K, Cestari V, Rossi-Arnaud C, Lipp HP, Bonhoeffer T, and Klein R (1999). Essential role for TrkB receptors in hippocampus-mediated learning. Neuron 24, 401–414. [DOI] [PubMed] [Google Scholar]
- Monteggia LM, Barrot M, Powell CM, Berton O, Galanis V, Gemelli T, Meuth S, Nagy A, Greene RW, and Nestler EJ (2004). Essential role of brain-derived neurotrophic factor in adult hippocampal function. Proceedings of the National Academy of Sciences of the United States of America 101, 10827–10832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteggia LM, Gideons E, and Kavalali ET (2013). The role of eukaryotic elongation factor 2 kinase in rapid antidepressant action of ketamine. Biological Psychiatry 73, 1199–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris RGM (2003). Long-term potentiation and memory. Philosophical Transactions of the Royal Society of London. Series B: Biological Sciences 358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mössner R, Walitza S, Lesch KP, Geller F, Barth N, Remschmidt H, Hahn F, Herpertz-Dahlmann B, Fleischhaker C, Schulz E, et al. (2005). Brain-derived neurotrophic factor V66M polymorphism in childhood-onset obsessive-compulsive disorder [2]. International Journal of Neuropsychopharmacology 8, 133–136. [DOI] [PubMed] [Google Scholar]
- Nibuya M, Morinobu S, and Duman RS (1995). Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. Journal of Neuroscience 15, 7539–7547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nosyreva E, Szabla K, Autry AE, Ryazanov AG, Monteggia LM, and Kavalali ET (2013). Acute suppression of spontaneous neurotransmission drives synaptic potentiation. Journal of Neuroscience 33, 6990–7002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nosyreva E, Autry AE, Kavalali ET, and Monteggia LM (2014). Age dependence of the rapid antidepressant and synaptic effects of acute NMDA receptor blockade. Frontiers in Molecular Neuroscience 7, 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang PT, Teng HK, Zaitsev E, Woo NT, Sakata K, Zhen S, Teng KK, Yung WH, Hempstead BL, and Lu B (2004). Cleavage of proBDNF by tPA/plasmin is essential for long-term hippocampal plasticity. Science 306, 487–491. [DOI] [PubMed] [Google Scholar]
- Parikh V, Khan MM, and Mahadik SP (2004). Olanzapine counteracts reduction of brain-derived neurotrophic factor and TrkB receptors in rat hippocampus produced by haloperidol. Neuroscience Letters 356, 135–139. [DOI] [PubMed] [Google Scholar]
- Park H, and Poo M (2013). Neurotrophin regulation of neural circuit development and function. Nature Reviews Neuroscience 14, 7–23. [DOI] [PubMed] [Google Scholar]
- Patterson SL, Abel T, Deuel TAS, Martin KC, Rose JC, and Kandel ER (1996). Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron 16, 1137–1145. [DOI] [PubMed] [Google Scholar]
- Poo M (2001). Neurotrophins as synaptic modulators. Nature Reviews Neuroscience 2, 24–32. [DOI] [PubMed] [Google Scholar]
- Rantamäki T, Hendolin P, Kankaanpää A, Mijatovic J, Piepponen P, Domenici E, Chao MV, Männistö PT, and Castrén E (2007). Pharmacologically diverse antidepressants rapidly activate brain-derived neurotrophic factor receptor TrkB and induce phospholipase-Cγ signaling pathways in mouse brain. Neuropsychopharmacology 32, 2152–2162. [DOI] [PubMed] [Google Scholar]
- Real E, Gratacòs M, Soria V, Escaramís G, Alonso P, Segalàs C, Bayés M, de Cid R, Menchón JM, and Estivill X (2009). A Brain-Derived Neurotrophic Factor Haplotype Is Associated with Therapeutic Response in Obsessive-Compulsive Disorder. Biological Psychiatry 66, 674–680. [DOI] [PubMed] [Google Scholar]
- Reimers J, Loweth J, and Wolf M (2014). BDNF contributes to both rapid and homeostatic alterations in AMPA receptor surface expression in nucleus accumbens medium spiny neurons. The European Journal of Neuroscience 39, 1159–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford LC, DeWan A, Lauer HM, and Turrigiano GG (1997). Brain-derived neurotrophic factor mediates the activity-dependent regulation of inhibition in neocortical cultures. Journal of Neuroscience. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford LC, Nelson SB, and Turrigiano GG (1998). BDNF has opposite effects on the quantal amplitude of pyramidal neuron and interneuron excitatory synapses. Neuron 21, 521–530. [DOI] [PubMed] [Google Scholar]
- Saarelainen T, Hendolin P, Lucas G, Koponen E, Sairanen M, MacDonald E, Agerman K, Haapasalo A, Nawa H, Aloyz R, et al. (2003). Activation of the TrkB neurotrophin receptor is induced by antidepressant drugs and is required for antidepressant-induced behavioral effects. Journal of Neuroscience 23, 349–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santarelli L, Saxe M, Gross C, Surget A, Battaglia F, Dulawa S, Weisstaub N, Lee J, Duman R, Arancio O, et al. (2003). Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science 301, 805–809. [DOI] [PubMed] [Google Scholar]
- Schmid DA, Yang T, Ogier M, Adams I, Mirakhur Y, Wang Q, Massa SM, Longo FM, and Katz DM (2012). A TrkB Small Molecule Partial Agonist Rescues TrkB Phosphorylation Deficits and Improves Respiratory Function in a Mouse Model of Rett Syndrome. Journal of Neuroscience 32, 1803–1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimojo M, Courchet J, Pieraut S, Torabi-Rander N, Sando R, Polleux F, and Maximov A (2015). SNAREs Controlling Vesicular Release of BDNF and Development of Callosal Axons. Cell Reports 11, 1054–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinoda Y, Ahmed S, Ramachandran B, Bharat V, Brockelt D, Altas B, and Dean C (2014). BDNF enhances spontaneous and activity-dependent neurotransmitter release at excitatory terminals but not at inhibitory terminals in hippocampal neurons. Frontiers in Synaptic Neuroscience 6, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirayama Y, Chen AC-H, Nakagawa S, Russell DS, and Duman RS (2002). Brain-derived neurotrophic factor produces antidepressant effects in behavioral models of depression. The Journal of Neuroscience : The Official Journal of the Society for Neuroscience 22, 3251–3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shmelkov SV, Hormigo A, Jing D, Proenca CC, Bath KG, Milde T, Shmelkov E, Kushner JS, Baljevic M, Dincheva I, et al. (2010). Slitrk5 deficiency impairs corticostriatal circuitry and leads to obsessive-compulsive-like behaviors in mice. Nature Medicine 16, 598–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siuciak JA, Lewis DR, Wiegand SJ, and Lindsay RM (1996). Antidepressant-like effect of brain-derived neurotrophic factor (BDNF). Pharmacology Biochemistry and Behavior 56, 131–137. [DOI] [PubMed] [Google Scholar]
- Snigdha S, Neill JC, McLean SL, Shemar GK, Cruise L, Shahid M, and Henry B (2011). Phencyclidine (PCP)-induced disruption in cognitive performance is gender-specific and associated with a reduction in Brain-Derived Neurotrophic Factor (BDNF) in specific regions of the female rat brain. Journal of Molecular Neuroscience 43, 337–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song M, Giza J, Proenca CC, Jing D, Elliott M, Dincheva I, Shmelkov SV, Kim J, Schreiner R, Huang SH, et al. (2015). Slitrk5 Mediates BDNF-Dependent TrkB Receptor Trafficking and Signaling. Developmental Cell 33, 690–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhof TC (2012). Calcium control of neurotransmitter release. Cold Spring Harbor Perspectives in Biology 4, a011353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, Kim J-W, Nosyreva E, Kavalali ET, and Monteggia LM (2021). Convergence of distinct signaling pathways on synaptic scaling to trigger rapid antidepressant action. Cell Reports 37, 109918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmusk T, Palm K, Metsis M, Reintam T, Paalme V, Saarma M, and Persson H (1993). Multiple promoters direct tissue-specific expression of the rat BDNF gene. Neuron 10, 475–489. [DOI] [PubMed] [Google Scholar]
- Ting JT, and Feng G (2008). Glutamatergic Synaptic Dysfunction and Obsessive-Compulsive Disorder. Current Chemical Genomics 2008, 62–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG, Leslie KR, Desai NS, Rutherford LC, and Nelson SB (1998). Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature 391, 892–896. [DOI] [PubMed] [Google Scholar]
- Tyler WJ, and Pozzo-Miller LD (2001). BDNF enhances quantal neurotransmitter release and increases the number of docked vesicles at the active zones of hippocampal excitatory synapses. The Journal of Neuroscience : The Official Journal of the Society for Neuroscience 21, 4249–4258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaswani M, Linda FK, and Ramesh S (2003). Role of selective serotonin reuptake inhibitors in psychiatric disorders: a comprehensive review. Progress in Neuro-Psychopharmacology and Biological Psychiatry 27, 85–102. [DOI] [PubMed] [Google Scholar]
- Vicario-Abejón C, Owens D, McKay R, and Segal M (2002). Role of neurotrophins in central synapse formation and stabilization. Nature Reviews Neuroscience 3, 965–974. [DOI] [PubMed] [Google Scholar]
- Wang H-L, Zhang Z, Hintze M, and Chen L (2011). Decrease in Calcium Concentration Triggers Neuronal Retinoic Acid Synthesis during Homeostatic Synaptic Plasticity. Journal of Neuroscience 31, 17764–17771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wardle RA, Poo M, Sullivan J, Arevalo M, Rodríguez-Tébar A, Meier J, Grantyn R, Fukata M, Poo M, Hell SW, et al. (2003). Brain-derived neurotrophic factor modulation of GABAergic synapses by postsynaptic regulation of chloride transport. The Journal of Neuroscience : The Official Journal of the Society for Neuroscience 23, 8722–8732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Qing H, Lu W, Keegan D, Richardson JS, Chlan-Fourney J, and Li XM (2002). Quetiapine attenuates the immobilization stress-induced decrease of brain-derived neurotrophic factor expression in rat hippocampus. Neuroscience Letters 321, 65–68. [DOI] [PubMed] [Google Scholar]
- Yang J, Siao CJ, Nagappan G, Marinic T, Jing D, McGrath K, Chen ZY, Mark W, Tessarollo L, Lee FS, et al. (2009). Neuronal release of proBDNF. Nature Neuroscience 14, 51–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda S, Liang MH, Marinova Z, Yahyavi A, and Chuang DM (2009). The mood stabilizers lithium and valproate selectively activate the promoter IV of brain-derived neurotrophic factor in neurons. Molecular Psychiatry 14, 51–59. [DOI] [PubMed] [Google Scholar]
- Zakharenko SS, Patterson SL, Dragatsis I, Zeitlin SO, Siegelbaum SA, Kandel ER, and Morozov A (2003). Presynaptic BDNF Required for a Presynaptic but Not Postsynaptic Component of LTP at Hippocampal CA1-CA3 Synapses. Neuron 39, 975–990. [DOI] [PubMed] [Google Scholar]
- Zarate CA, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, Charney DS, and Manji HK (2006). A Randomized Trial of an N-methyl-D-aspartate Antagonist in Treatment-Resistant Major Depression. Archives of General Psychiatry 63, 856–864. [DOI] [PubMed] [Google Scholar]
- Zhang J, Wu J, Fujita Y, Yao W, Ren Q, Yang C, Li S, Shirayama Y, and Hashimoto K (2015). Antidepressant Effects of TrkB Ligands on Depression-Like Behavior and Dendritic Changes in Mice After Inflammation. International Journal of Neuropsychopharmacology 18, pyu077. [DOI] [PMC free article] [PubMed] [Google Scholar]