

Abstract
The translocation t(8;9)(p22;p24) results in the production of a chimeric PCM1‐JAK2 fusion protein leading to the constitutive activation of the Janus Kinase 2 that renders this disease potentially sensitive to ruxolitinib. Here, we report an interesting case of PCM1‐JAK2 myeloproliferative neoplasm evolving in myeloid sarcoma and B precursor ALL.
Keywords: acute B‐lymphoblastic leukemia, case report, myeloid sarcoma, PCM1‐JAK2 myeloproliferative neoplasm, ruxolitinib
The identification of a specific molecular marker offered the possibility to monitor the depth of remission after transplant and to start an early preemptive therapy with ruxolitinib as soon as any evidence of disease reappraisal was documented.

1. BACKGROUND
The translocation t(8;9)(p22;p24) results in the production of a chimeric PCM1‐JAK2 fusion protein leading to the constitutive activation of the Janus Kinase 2. 1 It represents the driver mutation of a specific, extremely rare entity, recognized as provisional by the WHO (World Health Organization) classification: “Myeloid/Lymphoid neoplasm with eosinophilia and PCM1‐JAK2 gene rearrangement”. 1 , 2
The hematological trait shares common features with myeloproliferative neoplasm and myelodysplastic/myeloproliferative neoplasm. 3 , 4 Hyperactivation of the JAK‐STAT pathway renders this disease potentially sensitive to ruxolitinib; however, there are conflicting results about the duration of response and whether consolidation with allogeneic hematopoietic stem cell transplantation (alloHSCT) is indicated when a response is achieved. 5
Here, we report an interesting case of PCM1‐JAK2 myeloproliferative neoplasm evolving in myeloid sarcoma and B precursor acute lymphoblastic leukemia (BCP‐ALL), which required composite clinical management as well as an advanced molecular diagnostic approach in the follow‐up.
2. THE CASE
In November 2017, a 53‐year‐old woman was diagnosed with Myelofibrosis documented by histopathologic analysis showing a marrow hypercellularity, myeloid and erythroid hyperplasia, MF‐2. BCR‐ABL1 gene rearrangement and JAK2 mutations were negative. She was initially managed with a watch‐and‐wait approach. One year later, in March 2019, due to a rapidly worsening coxalgia, she underwent a PET/CT (Positron Emission Tomography/Computed Tomography) scan showing a neoformation infiltrating the right ileo‐psoas muscle (Figure 1A). The ileo‐psoas biopsy showed a myeloid infiltrate with medium‐sized blasts with dispersed nuclear chromatin with one or two nucleoli; blasts were positive for MPO, CD33 e CD45 resulted in a diagnosis of myeloid sarcoma (Figure 2A,B). A bone marrow reassessment showed the presence of a blastic infiltration with dual phenotype: the major infiltrate consisting of 60% lymphoid blasts (CD10, CD19, CD34, CD45, CD52, TdT, CD38 positive but negative for intracytoplasmic immunoglobulins); the minor one consisting of myeloid blasts (Myeloperoxidase, CD33, CD45 positive) equal to 25–30%. Background hematopoietic cell composition was compatible with the underlying myeloproliferative disorder (Figure 3). Peripheral blood test showed normal values of hemoglobin (Hb 11.6 g/dl), leukocytes (6.64 × 109/L), and platelets/424 × 109/L), without evidence of blast. Cytogenetic analysis on the bone marrow specimen revealed the presence of the translocation(t(8;9)(p22;p24)) leading to the presence of PCM1‐JAK2 fusion protein, as demonstrated by FISH analysis and by PCR (Polymerase chain reaction) and Nested PCR (Figure 4). Molecular biology for NPM1, FLT3‐ ITD, and point mutations, IDH1, IDH2, JAK2V617F , CALR tested using PCR resulted was negative. Targeted next‐generation sequencing (NGS) performed using a capture‐based method (Sophia Myeloid Solution, Sophia Genetics SA, Saint Sulpice, Switzerland selecting 30 gene regions associated with myelodysplastic syndrome, myeloproliferative neoplasms, and leukemia) was negative. Based on genetic results, we revised the initial diagnosis as PCM1‐JAK2 neoplasm with a synchronous evolution into an isolated bone myeloid sarcoma and a BCP‐ALL. In order to tackle myeloid and lymphoid diseases, the patient started induction with standard chemotherapy according to FLAI scheme (Fludarabine 30 mg/m2 for 5 days, Cytarabine 2 g/m2 for 5 days, and Idarubicine 10 mg/m2 for 3 days), without combination with tyrosine kinase or JAK inhibitors. A bone marrow evaluation after induction revealed no evidence of residual AML. She was then consolidated with a second cycle of FLAI followed by a cycle with high‐dose methotrexate and cytarabine, also for CNS prophylaxis. Finally, radiotherapy consolidation (30 Gy administered in 15 fractions) on the ileo‐psoas myeloid sarcoma was performed. Restaging marrow showed a complete hematological response, but FISH studies revealed the persistence of the t(8;9) translocation in 23% of bone marrow (BM) cells. MRD analysis of immunoglobulin rearrangement showed a positive though not quantifiable signal; PET imaging showed complete remission of myeloid sarcoma (Figure 1B). Taken together, these results pointed toward an effective eradication of both myeloid sarcoma and BCP‐ALL, yet with a persisting residual myeloproliferative background.
FIGURE 1.
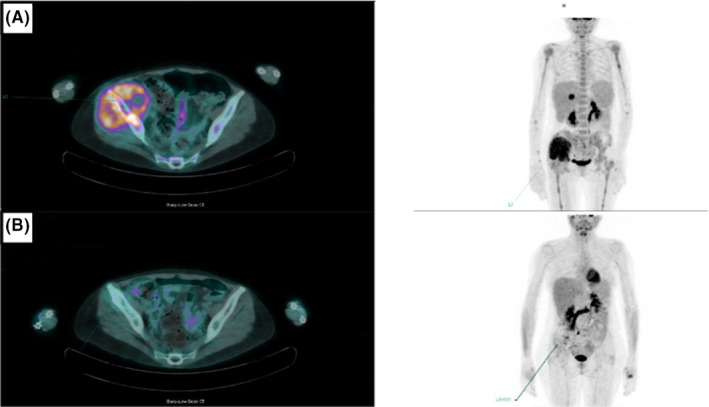
(A, B) PET/CT scan showing ileo‐psoas and iliac crest involvement before (Panel A) and after (Panel B) treatment
FIGURE 2.
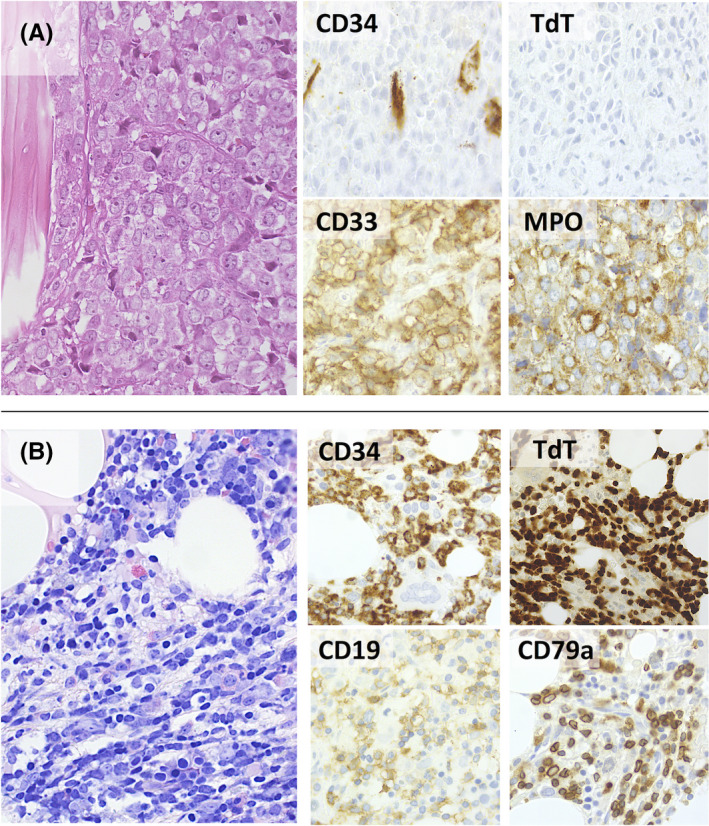
(A, B) Myeloid sarcoma presented as a large osteolytic mass with necrobiotic artifacts, consisting of sheets of large myeloblasts with finely dispersed chromatin and large nucleoli. The cells were negative for CD34 and TdT and positive for the myeloid‐associated markers CD33 and MPO (A). Staging bone marrow biopsy disclosed an interstitial and diffuse infiltrate of smaller blastoid cells with fine chromatin and scant cytoplasm. These cells expressed CD34, TdT, CD19, and CD79a, consistent with B‐cell lymphoblastic lymphoma/leukemia. (H&E, Giemsa and peroxidase stain; original magnification: 40× and 63×) (B)
FIGURE 3.
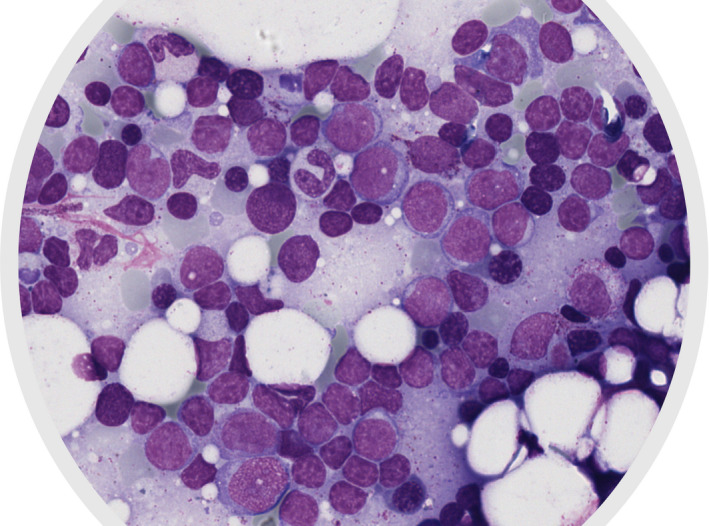
Diagnostic bone marrow smear showing several lymphoblasts with a high nuclear‐cytoplasmatic ratio and variably condensed chromatin
FIGURE 4.
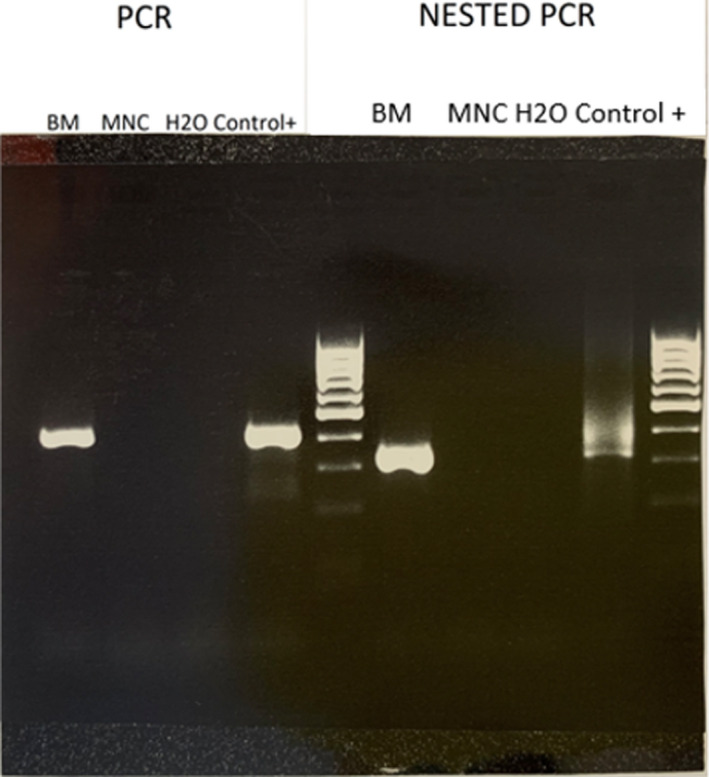
Gel electrophoresis of DNA fragments generated by polymerase chain reaction (PCR) on the left, and Nested PCR on the right. The first column of the left panel shows the strong presence of PCM1‐JAK2 fragment from bone marrow‐derived sample, in concordance with positive control column; this finding is further confirmed by analogous results from Nested PCR (right panel)
AlloHSCT was performed in September 2019, following myeloablative conditioning based on Busulphan 12.8 mg/kg and Fludarabine 160 mg/m2, considering the aggressive disease course of this entity 6 and the availability of a fully matched unrelated donor. Pre‐transplant in vivo T‐cell depletion with anti‐thymocyte immunoglobulins (ATG, 5 mg/kg, Sanofi‐Genzyme) followed by cyclosporine and short‐course methotrexate post‐transplant was used as GvHD (graft vs. host disease) prophylaxis.
To address the challenge of appropriate molecular monitoring after alloHSCT, we generated two different qPCR‐based MRD assays exploiting not only the above described, patient‐specific IgH recombination probe to monitor BP‐ALL 7 but also a PCM1‐JAK2 fusion gene‐specific probe to detect residual myeloproliferative disease.
At day +60 after alloHSCT, a bone marrow evaluation revealed the persistence of the PCM1‐JAK2 fusion transcript, while no residual BP‐ALL MRD signal was detected. Therefore, we decided to start ruxolitinib treatment at the dosage of 10 mg/BID. By day +90 after alloHSCT, we documented a successful negativization of the PCM1‐JAK2 MRD chimeric signal. Due to peripheral edema, ruxolitinib was decreased at day+120; this dose reduction was followed by a reappraisal of a weak MRD signal on bone marrow and peripheral blood. For this reason, a full ruxolitinib dosage was promptly reintroduced, and MRD negativity in peripheral blood was restored. The patient remains on ruxolitinib treatment, without significant side effects or infections at day +260 (Figure 5).
FIGURE 5.
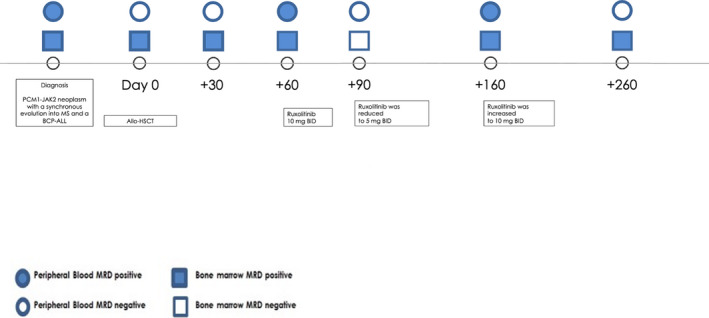
Timeline of the PCM1‐JAK2 fusion gene‐specific probe evaluated on peripheral blood and on bone marrow. AlloHSCT, allogeneic hematopoietic stem cell transplantation, MS, myeloid sarcoma; BCP‐ALL, B‐cell precursor acute lymphoblastic leukemia
3. DISCUSSION AND CONCLUSIONS
We report an unusual case of myeloproliferative neoplasm with the PCM1‐JAK2 fusion t(8;9)(p22;p24), evolving in blast crisis of both lymphoid and myeloid lineages. To the best of our knowledge, this is the first case reporting the synchronous evolution in acute myeloid and acute lymphoblastic leukemia of a myeloproliferative neoplasm with PCM1‐JAK2 fusion. 8 , 9 The unique scenario of our case bestowed significant challenges in both diagnosis and treatment.
Identifying the PCM1‐JAK2 fusion gene‐specific probe is essential for a proper diagnostic workup 3 that should guide personalized and risk‐adapted therapy decisions, such as using ruxolitinib as targeted therapy and the early allocation of suitable patients to alloHSCT. In this patient, a more accurate molecular initial diagnosis could have prevented the rapid myeloid and lymphoid blastic transformation and perhaps the intensive cycles of chemotherapy, but most likely not the transplant choice. Moreover, this case report highlights the challenges in the clinical management of neoplasm involving different cell lineages. We speculated that a pediatric‐like protocol for ALL or a conventional regimen was likely not sufficiently active to tackle this disease. Accordingly, fludarabine containing high‐dose protocol was chosen and proved partially effective. The management of the underlying chronic neoplasm was another challenge since it could have led to an early recurrence of the disease. For this reason, as soon as complete remission was obtained, the patient underwent alloHSCT, and ruxolitinib therapy was started as post‐transplant maintenance.
Ruxolitinib has been reported to be active against PCM1‐JAK2 fusion genes neoplasms, and its use as bridging therapy before alloHSCT has been described. 5 , 6 , 10 In our case, identifying a specific molecular marker offered the possibility to monitor the depth of remission after transplant and start an early preemptive therapy with ruxolitinib as soon as any evidence of disease reappraisal was documented. However, it is worthy to mention that the use of ruxolitinib in the post‐transplant setting was not supported by evidence. In this patient, we decided to start ruxolitinib mainly taking into consideration the preliminary evidences in favor of a possible ability in suppressing the malignant clone 5 , 6 , 10 and balancing the risk of recurrence that we considered significantly higher compared with potential risks of adverse effects. The reappraisal of a weak MRD signal after ruxolitinib dose reduction and the negativity with full ruxolitinib dosage suggests that ruxolitinib was responsible for the successful therapeutic effect in the post‐transplant in our patient, although the additive effect of the withdrawal of immunosuppression cannot formally be ruled out. In addition, it remains to be addressed whether this treatment should be continued indefinitely or if it could be discontinued.
CONFLICTS OF INTERESTS
The authors declare no conflicts of interest.
AUTHOR CONTRIBUTION
GR, ML, and SI designed the study, followed the clinical course of the patient, collected data, drafted the manuscript, and gave the final approval before manuscript submission. DT, MCM, FL, and GB followed the clinical course of the patient and gave the final approval before manuscript submission. MT, OS, MP, and LB processed the samples, performed molecular and chimerism analyses, and gave the final approval before manuscript submission. FL contributed to the final writing of the paper and gave the final approval before manuscript submission. AR and CG supervised the study, revised the manuscript, and gave the final approval before manuscript submission.
ETHICAL APPROVAL
The study was approved by the local Institutional Review Board, and it was conducted in accordance with the Declaration of Helsinki.
CONSENT
Written informed consent was obtained from the patient to publish this report in accordance with the journal's patient consent policy.
ACKNOWLEDGEMENTS
None.
Rizzuto G, Leoncin M, Imbergamo S, et al. Sequential allogeneic transplantation and ruxolitinib maintenance for a synchronous PCM1‐JAK2 positive myeloid sarcoma and acute B‐lymphoblastic leukemia. Clin Case Rep. 2022;10:e05212. doi: 10.1002/ccr3.5212
Giuliana Rizzuto and Matteo Leoncin contributed equally as co‐first authors.
Funding information
None
DATA AVAILABILITY STATEMENT
Data available on request from the authors.
REFERENCES
- 1. Masselli E, Mecucci C, Gobbi G, et al. Implication of MAPK1/MAPK3 signalling pathway in t(8;9)(p22;24)/PCM1‐JAK2 myelodysplastic/myeloproliferative neoplasms. Br J Haematol. 2013;162(4):563‐566. [DOI] [PubMed] [Google Scholar]
- 2. Bain BJ, Ahmad S. Should myeloid and lymphoid neoplasms with PCM1‐JAK2 and other rearrangements of JAK2 be recognized as specific entities? Br J Haematol. 2014;166(6):809‐817. [DOI] [PubMed] [Google Scholar]
- 3. Patterer V, Schnittger S, Kern W, Haferlach T, Haferlach C. Hematologic malignancies with PCM1‐JAK2 gene fusion share characteristics with myeloid and lymphoid neoplasms with eosinophilia and abnormalities of PDGFRA, PDGFRB, and FGFR1. Ann Hematol. 2013;92(6):759‐769. [DOI] [PubMed] [Google Scholar]
- 4. Bain BJ, Horny H‐P, Arber DA, Tefferi A, Hasserjian RP. Myeloid/Lymphoid neoplasms with eosinophilia and gene rearrangement. In: Swerdlow SH, Campo E , Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. International Agency for Research on Cancer (IARC); 2016:78‐79. [Google Scholar]
- 5. Rumi E, Milosevic JD, Selleslag D, et al. Efficacy of ruxolitinib in myeloid neoplasms with PCM1‐JAK2 fusion gene. Ann Hematol. 2015;94(11):1927‐1928. [DOI] [PubMed] [Google Scholar]
- 6. Schwaab J, Knut M, Haferlach C, et al. Limited duration of complete remission on ruxolitinib in myeloid neoplasms with PCM1‐JAK2 and BCR‐JAK2 fusion genes. Ann Hematol. 2015;94(2):233‐238. [DOI] [PubMed] [Google Scholar]
- 7. Bassan R, Spinelli O, Oldani E, et al. Improved risk classification for risk‐specific therapy based on the molecular study of minimal residual disease (MRD) in adult acute lymphoblastic leukemia (ALL). Blood. 2009;113(18):4153‐4162. [DOI] [PubMed] [Google Scholar]
- 8. Lee JM, Lee J, Han E, et al. PCM1‐JAK2 fusion in a patient with acute myeloid leukemia. Ann Lab Med. 2018;38(5):492‐494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Song I, Lee DH, Lee JH, Jang S, Huh JR, Seo EJ. A t(8;9)(p22;p24)/PCM1‐JAK2 translocation in a patient with myeloproliferative neoplasm and myeloid sarcoma: first report in Korea. Ann Lab Med. 2016;36(1):79‐81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lierman E, Selleslag D, Smits S, Billiet J, Vandenberghe P. Ruxolitinib inhibits transforming JAK2 fusion proteins in vitro and induces complete cytogenetic remission in t(8;9)(p22;p24)/PCM1‐JAK2–positive chronic eosinophilic leukemia. Blood. 2012;120(7):1529‐1531. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data available on request from the authors.