

Abstract
Alternative splicing is an essential mechanism of gene regulation, giving rise to remarkable protein diversity in higher eukaryotes. Epithelial-mesenchymal transition (EMT) is a developmental process that plays an essential role in metazoan embryogenesis. Recent studies have revealed that alternative splicing serves as a fundamental layer of regulation that govern cells to undergo EMT. In this review, we summarize recent findings on the functional impact of alternative splicing in EMT and EMT-associated activities. We then discuss the regulatory mechanisms that control alternative splicing changes during EMT.
Keywords: Epithelial-Mesenchymal Transition, Alternative Splicing, RNA Binding Proteins, Tumor Metastasis
INTRODUCTION
Epithelial-mesenchymal transition (EMT) is a conserved developmental process critical for metazoan embryogenesis[Thiery, 2003; Thiery et al., 2009]. First described in a chick primitive streak formation model, EMT is a process where epithelial cells acquire mesenchymal characteristics such as increased motility and invasive capacity [Nieto, 2013]. During embryogenesis, EMT is an essential step for epithelial cell migration to various future organ locations[Kalluri, and Weinberg, 2009]. At the secondary sites, these cells undergo mesenchymal-epithelial transition (MET), the reverse process of EMT, to differentiate and establish future organs[Acloque et al., 2009]. EMT and MET are two fundamental processes that provide cells with the flexibility to switch cell states according to developmental needs, and they are tightly regulated to ensure proper tissue and organ formation.
EMT is activated primarily by extensive crosstalk between extracellular stimuli, EMT-inducing signaling pathways and transcription factors[Nieto et al., 2016; Polyak, and Weinberg, 2009]. During EMT, epithelial cells lose their characteristic apical-basal polarity and intercellular junctions, detaching cells from the basement membrane. Their cobblestone-like morphology transitions towards a spindle-like, elongated morphology while acquiring increased migration and invasion capacity[Pérez-Pomares, and Muñoz-Chápuli, 2002; Thiery, and Sleeman, 2006; Acloque et al., 2009]. Mesenchymal cells can disintegrate the extracellular matrix and invade surrounding tissues, and acquire elevated resistance against apoptosis[Nieto, 2013; Lamouille et al., 2014; Radisky et al., 2005; Morel et al., 2012; Brabletz et al., 2018]. During development, cycles of EMT and MET are especially important for gastrulation, neural crest cell emigration, and proper formation of the embryonic heart [Acloque et al., 2009; Kim et al., 2017; Nakajima et al., 2000]. In adult tissues, normal EMT is mainly associated with wound healing and tissue regeneration, whereas pathological dysregulation of EMT can lead to organ fibrosis, carcinogenesis and metastasis[Kalluri, and Weinberg, 2009; Dongre, and Weinberg, 2019]. Classification of EMT into three categories is widely used: Type I EMT refers to EMT during embryonic development, Type II EMT is involved in myofibroblast production and tissue fibrosis, and Type III EMT is related to cancer[Kalluri, and Weinberg, 2009]. In this review, we will focus on Type III cancer-related EMT and one of its regulators, alternative splicing, which is especially important for cellular plasticity.
EMT plays a critical role in various key aspects of tumorigenesis [Brabletz et al., 2018; Tsai, and Yang, 2013; Pastushenko, and Blanpain, 2019]. In tumors, the EMT process elevates chemoresistance and promotes resistance to apoptosis[Chaffer et al., 2016], increasing likelihood of recurrence, which poses significant obstacles to treatment. EMT also increases the migration and invasion capacity of carcinogenic epithelial cells to form the invasive front, placing EMT as one of the major contributors to metastasis. It affects various aspects of the metastatic cascade including invasion, migration, intravasation/extravasation, dissemination and angiogenesis [Yeung, and Yang, 2017; Chaffer, and Weinberg, 2011]. Although the importance of EMT during metastasis is widely acknowledged, its direct contribution has been a topic of intense debate. Recent studies have proposed a theory that EMT is not required for metastasis in breast and pancreatic cancer models[Fischer et al., 2015; Zheng et al., 2015]. However, limitations in experimental design still cannot rule out the determining role of EMT during the metastatic cascade[Ye et al., 2017; Aiello et al., 2017; Nieto, 2017].
As tumors are not homogeneous, the ability of cells to shift towards a more mesenchymal state, where it can resist radiation and apoptosis, but also transition back towards a rapidly dividing epithelial state is critical to establishing a metastatic tumor[Chaffer et al., 2007]. Stable mesenchymal cells have been shown to be ineffective metastatic progenitors[Chao et al., 2010; Lecharpentier et al., 2011]. Circulating tumor cells (CTCs) displaying both epithelial and mesenchymal characteristics are found in the bloodstream, and more mesenchymal CTCs are found in patients who responded poorly to chemotherapy[Nieto, 2013; Aktas et al., 2009; Kasimir-Bauer et al., 2012; Yu et al., 2013; Aceto et al., 2014]. These observations suggest that the ability to undergo EMT and MET in cancer cells provides maximum plasticity for effective dissemination, metastatic colonization, and therapeutic resistance.
EMT is a complex biological process orchestrated by multiple layers of regulation. Extensive research in the EMT field has focused on transcription factors such as TWIST1, SNAI1/2, and ZEB1/2, signaling pathways such as TGF-β, Wnt/β-catenin and Notch, as well as cytokines and miRNA regulation[Lamouille et al., 2014; Peinado et al., 2007]. In addition to these well-studied master regulators of EMT, recent research has also highlighted other mechanisms of gene regulation, such as epigenetics, post-transcriptional RNA processing and post-translational modifications, that are essential for fine-tuning EMT[Pradella et al., 2017; Lee, and Kong, 2016]. Alternative splicing is one of the key post-transcriptional RNA processes that regulates crucial activities involving EMT such as signaling, invasion and migration[Biamonti et al., 2012; Baralle, and Giudice, 2017; Pradella et al., 2017; Aiello, and Kang, 2019]. This review will focus on the essential role of alternative splicing and how it is regulated during EMT.
ALTERNATIVE SPLICING AND EMT
Alternative splicing is a critical component of gene regulation and represents a major contributor to mRNA complexity and proteome diversity, emerging as one of the key regulators of tissue identity and cell state. Pre-mRNA is processed into mature mRNA by splicing out introns and properly joining together exons. This essential step is carried out by the spliceosome, which is a protein complex composed of small nuclear ribonucleoproteins (snRNPs) along with other protein and RNA components[Wahl et al., 2009; Shi, 2017]. It is estimated that up to 95% of human genes undergo alternative splicing, where specific exons are skipped or included to produce different mRNA products[Wang et al., 2008; Pan et al., 2008]. Common modes of alternative splicing include cassette exon (also called exon skipping), mutually exclusive exons, using alternative 3’ splice sites or 5’ splice sites, and intron retention (Fig 1A) [Wang et al., 2008; Liu, and Cheng, 2013]. Less common modes of alternative splicing such as circular RNA splicing also exist and can produce unique transcripts with functional significance (Fig 1A)[Jeck, and Sharpless, 2014]. Exon skipping is the most prevalent mode of alternative splicing, accounting for about 40% of alternative splicing events[Keren et al., 2010].
Fig 1. Alternative splicing is a fundamental regulator of EMT.
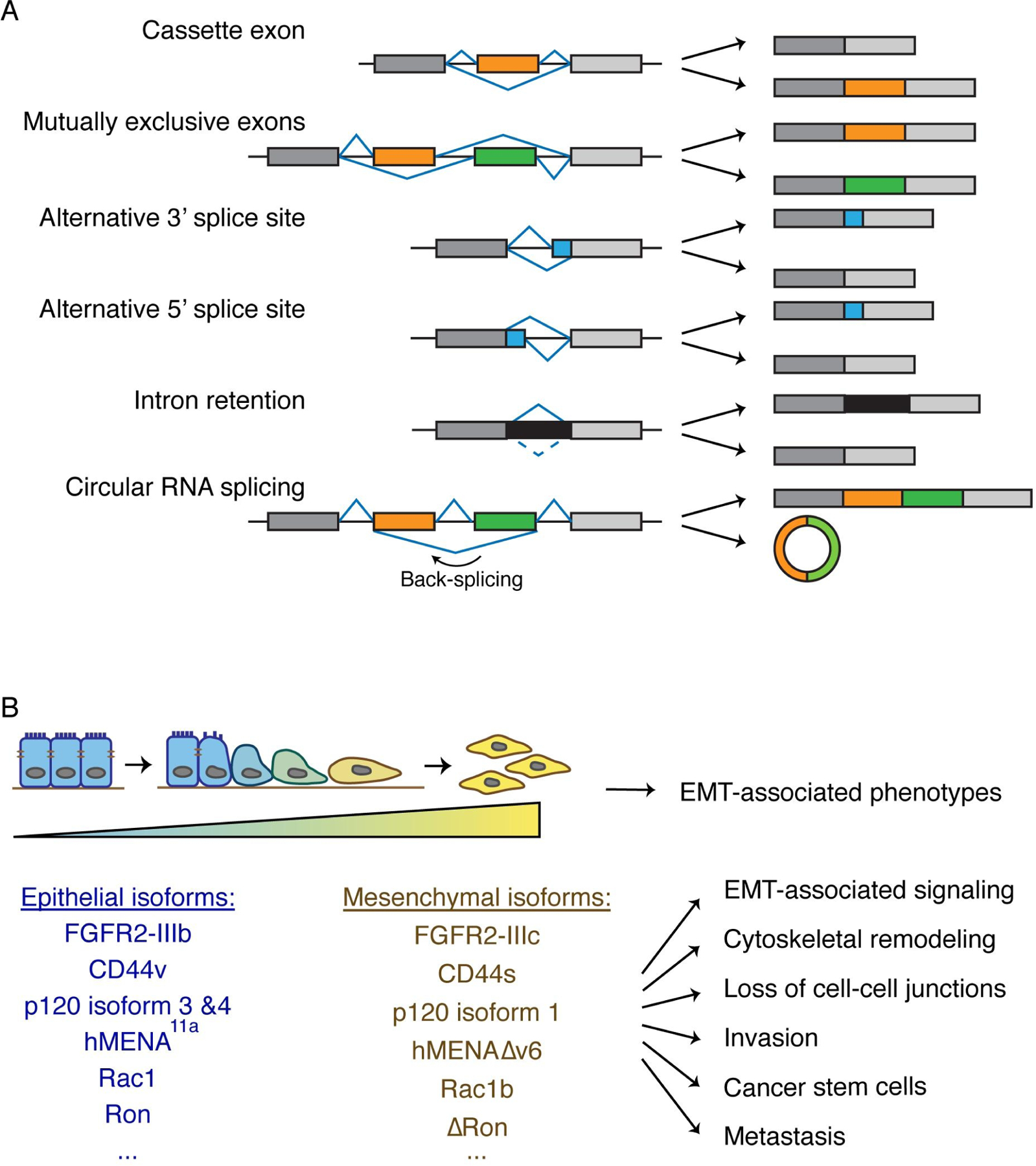
(A) Schematic depiction of the common modes of alternative splicing. Exons are depicted as rectangles and introns as lines.
(B) Alternative splicing changes during EMT give rise to various EMT-associated phenotypes. Blue to yellow gradient represents the transition from the epithelial state to mesenchymal state. Alternatively spliced isoforms during EMT are depicted and they contribute to many aspects of EMT-associated phenotypes such as EMT-associated signaling, cytoskeletal remodeling, loss of cell-cell junctions, invasion, cancer stem cells, and metastasis.
Alternative splicing produces functionally diverse transcripts. By including or excluding exons encoding functional domains, alternative splicing can alter enzymatic activity, change binding affinity for other proteins and form diverse protein complexes[Baralle, and Giudice, 2017; Romero et al., 2006]. Similarly, alternative splicing can change subcellular localization and protein solubility[Kelemen et al., 2013]. Many alternative exons are divisible by three, maintaining the open reading frame with insertion or deletion of a segment of peptides. However, alternative exons that are not divisible by three can alter the open reading frame to produce completely different proteins from the same gene locus[Xing, and Lee, 2005; Magen, and Ast, 2005]. They can also trigger non-sense mediated decay (NMD) through the generation of a premature stop codon, resulting in reduction of mRNA expression. Alternative splicing of terminal exons may produce mRNA transcripts with distinct 3’UTRs, thus affecting gene expression by including different miRNA binding sites[Sandberg et al., 2008]. Some RNA transcripts generated through alternative splicing are non-protein coding but have regulatory roles, such as long noncoding RNAs (lncRNA) and circular RNAs (circRNA)[Marchese et al., 2017; Kung et al., 2013; Kristensen et al., 2019; Barrett, and Salzman, 2016].
Well-coordinated regulation of alternative splicing ensures normal cellular function, while aberrant regulation of alternative splicing is increasingly recognized as a driver of disease. Mis-regulation of alternative splicing is prominent in many aspects of the hallmarks of cancer[Liu, and Cheng, 2013], one of which is EMT. As a matter of fact, alternative splicing of FGFR2 during EMT was considered as one of the first generation of EMT markers[Orr-Urtreger et al., 1993; Savagner et al., 1994]. In recent years, various splicing events have been shown to be critical during EMT. Here we will summarize the functional importance of alternative splicing that impact the EMT process. We will then discuss the mechanisms of alternative splicing regulation during EMT.
FUNCTIONAL IMPORTANCE OF ALTERNATIVE SPLICING CHANGES DURING EMT
The EMT process is tightly regulated by orchestrating signaling cascades, resulting in drastic changes in cellular morphology and how cells interact with the surrounding extracellular matrix. Alternative splicing events during EMT play an indispensable role, as splicing changes are found in genes involved in EMT-associated signaling, cytoskeletal remodeling, invasiveness, tumor initiating capacity, and metastasis (Fig 1B). Most of these alternatively spliced genes do not change significantly at gene expression levels, indicating that these genes are more functionally relevant to EMT at the post-transcriptional regulation level. These results demonstrate that alternative splicing regulation is an important process of gene regulation distinct from gene expression.
EMT-associated signaling
Several genes encoding surface receptors and kinases undergo alternative splicing during EMT. The signaling function of EMT-associated splice isoforms is altered through protein structure changes, accelerating EMT. Early research found the fibroblast growth factor receptor 2 (FGFR2) to be alternatively spliced during EMT. FGFR2 has 2 mutually exclusive exons IIIb and IIIc. In epithelial cells, FGFR2-IIIb is the predominant isoform, and in mesenchymal cells, FGFR2-IIIc is majorly expressed, changing the structure of an immunoglobin-like domain to alter binding affinity to FGF (Fig 2A)[Orr-Urtreger et al., 1993; Savagner et al., 1994; Yeh et al., 2003; Zhang et al., 2006]. This splicing event, along with the splicing of another FGF receptor, FGFR1, are further observed in breast cancer and clear cell renal carcinoma[Warzecha et al., 2009; Zhao et al., 2013; Hopkins et al., 2017]. Functionally, FGFR splice isoforms differ in ligand binding preferences. The mesenchymal isoforms have stronger affinity for FGF, enhancing FGFR downstream signaling to increase cancer cell invasiveness[Shirakihara et al., 2011; Hopkins et al., 2017; Zhang et al., 2006].
Fig 2. Critical alternative splicing events during EMT.
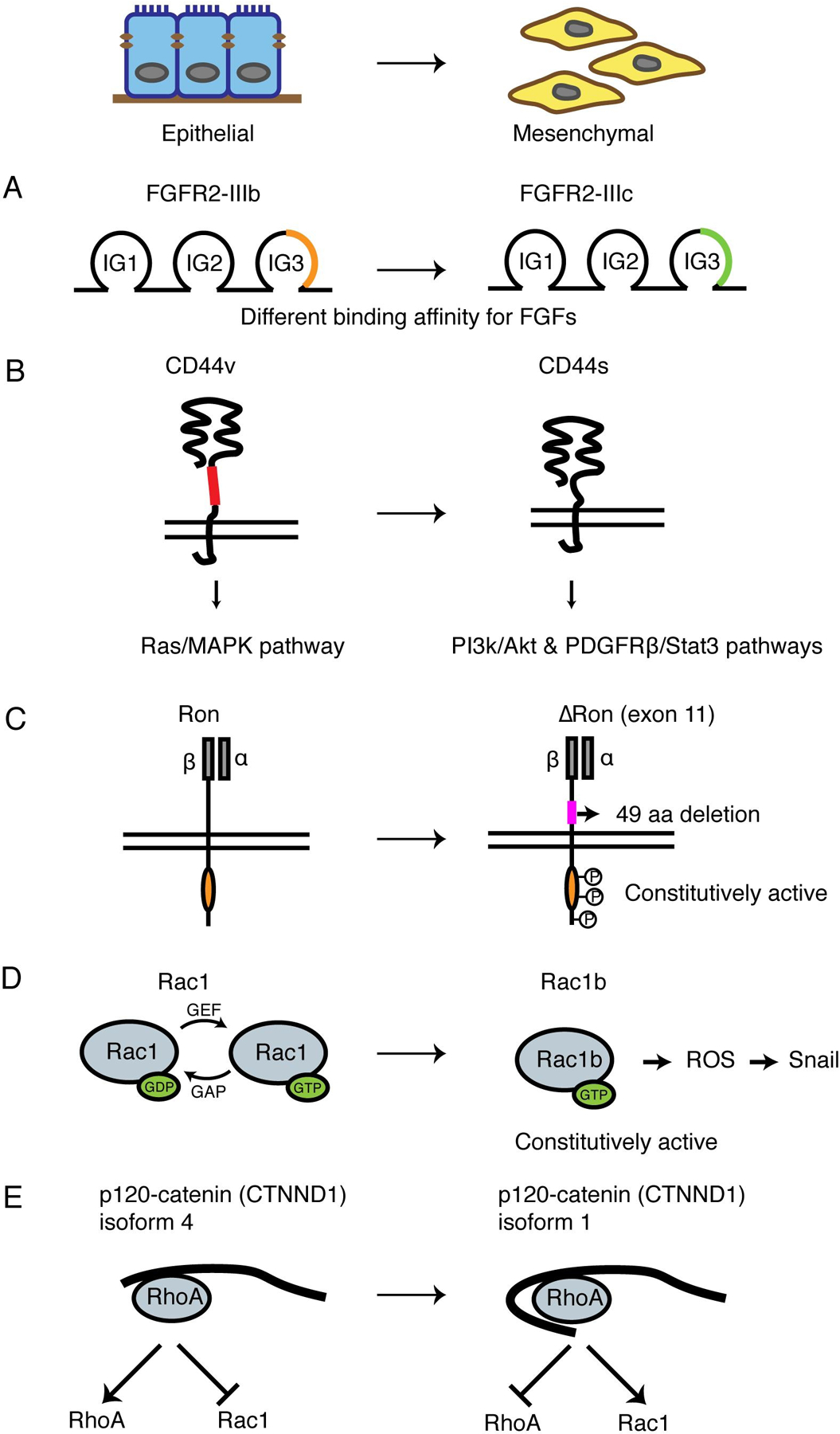
(A) Schematic of FGFR2 alternative splicing during EMT. Isoform switching from the epithelial FGFR2-IIIb to the mesenchymal FGFR2-IIIc changes the structure of an immuoglobin-like domain to enhance the binding affinity to FGF.
(B) Schematic of CD44 alternative splicing during EMT. Epithelial isoform CD44v has a longer extracellular stem region than mesenchymal isoform CD44s. The two isoforms activate Ras/MAPK pathway and PI3K/Akt, PDGFRβ/Stat3 pathways, respectively.
(C) Schematic of Ron alternative splicing during EMT. The mesenchymal isoform ∆Ron lacks a 49-amino acid region in the extracellular domain of the β-subunit and is constitutively active.
(D) Schematic of Rac1 alternative splicing during EMT. The mesenchymal Rac1b isoform is in the GTP-bound active conformation, generating ROS and induces SNAI1 expression.
(E) Schematic of p120-catenin (CTNND1) splicing during EMT. Mesenchymal isoform 1 is the longest isoform that contains a coiled-coil domain and a regulatory domain, inhibiting RhoA activity and promoting Rac1 activity. The epithelial isoform 4 lacks both domains and promotes RhoA activity.
CD44 is a transmembrane glycoprotein that takes part in multiple signaling cascades [Ponta et al., 2003]. The CD44 gene undergoes extensive alternative splicing, changing the length of the extracellular stem region. Human CD44 encodes nine variable exons flanked by a total of nine constitutive exons. Alternative splicing generates two major families of CD44 splice isoforms. The CD44 variant isoforms (CD44v) are a collection of isoforms that contain at least one of the variable exons, whereas the CD44 standard isoform (CD44s) is devoid of all variable exons. CD44v and CD44s both contain the same ligand binding domain for hyaluronan (HA), but CD44s shows higher affinity[Aruffo et al., 1990; Culty et al., 1990; Miyake, 1990]. Aside from binding canonical ligands, CD44v contains additional binding sites. For example, CD44v3 contains heparin-binding domains that bind to HB-EGF[Bennett et al., 1995]. Different isoforms of CD44 are known to activate distinct pathways important for respective cell states. CD44v6, one of the epithelial dominant isoforms of CD44, helps sustain a Ras/MAPK feedback loop that promotes cell proliferation[Matter et al., 2002; Cheng, 2006]. The mesenchymal dominant isoform, CD44s, sustains PI3K/Akt signaling and activates PDGFRβ/Stat3 pathway to enhance cancer stem cell (CSC) characteristics (Fig 2B)[Zhang et al., 2019a; Liu, and Cheng, 2017].
Ron is a receptor tyrosine kinase that contains important docking sites for many downstream signaling adaptors. When phosphorylated, Ron promotes cell growth, controls cell motility and supports invasion of extracellular matrices[Camp et al., 2005]. The variable exon 11 of Ron encodes 49 amino acids in the extracellular domain of the β-subunit (Fig 2C). Skipping of exon 11 produces the ∆Ron isoform that is constitutively active even in the absence of its ligand MSP [Ghigna et al., 2005]. ∆Ron is upregulated in tumors and during EMT. Overexpression of ∆Ron promotes cell motility and induces a mesenchymal morphology[Ghigna et al., 2005; Zhou et al., 2003].
Rac1 is a member of the Rho GTPase family important for regulation of actin cytoskeleton organization. Rac1 contains a variable exon 3b, inclusion of which leads to a 19-amino acid insertion in the GDP/GTP binding region[Singh et al., 2004; Schnelzer et al., 2000]. This mesenchymal isoform, Rac1b, exists predominantly in the GTP-bound active conformation and stimulates the NF-κB pathway, promoting cell cycle progression and enhancing anti-apoptotic response. Aberrant splicing of Rac1b stimulates mitochondrial production of ROS, which promotes EMT through inducing SNAI1 expression (Fig 2D)[Gonçalves et al., 2009; Radisky et al., 2005; Matos, and Jordan, 2005]. These examples demonstrate that alternative splicing during EMT produces isoforms that sustain EMT signaling and subsequently causes morphological and functional changes associated with mesenchymal cells.
Cytoskeleton remodeling, cell junctions and invasiveness
During EMT, cells undergo extensive cytoskeletal remodeling to disrupt cell-cell junctions, change cellular morphology, and enhance invadopodia formation. Multiple splicing events have been shown to directly affect cell-cell junctions and the invasive front.
p120-catenin (CTNND1), a crucial regulator of adherens junctions, is alternatively spliced during EMT. The full-length isoform 1, containing a coiled-coil domain and a regulatory domain, is overexpressed in mesenchymal cells and tumors, promoting invasiveness through inhibition of RhoA GTPase activity. In contrast, p120 isoforms 3 and 4 which lack the coiled-coil and regulatory domains, are dominantly expressed in epithelial cell state, blocking invasiveness by increasing binding to and stabilization of E-cadherin at adherens junctions (Fig 2E)[Ishiyama et al., 2010; Yanagisawa et al., 2008; Slorach et al., 2011; Faux et al., 2021]. TJP1 (ZO-1), a junction protein located at the cytoplasmic membrane surface of tight junctions, is also regulated by alternative splicing in a similar manner. By excluding its variable exon 20, TJP1 loses its activity at tight junctions but enhances actin stress fiber assembly, increasing migration[Kim et al., 2019].
Alternative splicing also regulates genes involved in actin regulation and invadopodia formation. MENA (ENAH) is an actin regulatory protein that antagonizes actin capping proteins[Gertler et al., 1996; Barzik et al., 2005; Applewhite et al., 2007]. hMENA∆v6 is expressed in invasive cancer cells while the epithelial isoform, hMENA11a, is expressed in premalignant cells and facilitates F-actin organization. The two isoforms have antagonistic functions. The hMENA∆v6 mesenchymal isoform shows increased invasive ability while the hMENA11a epithelial isoform reduces migration and invasion by decreasing the amount of filopodia[Modugno et al., 2012]. Other examples of alternative splicing involved in cytoskeletal rearrangement include Exo70-M that regulates actin branching, CD44s isoform that is required for formation of invadopodia, and Rac1b isoform that serves as an important regulator of the cytoskeleton[Lu et al., 2013; Zhao et al., 2016; Ishii et al., 2014]. In summary, alternative splicing of both cytoskeletal modulators and junction proteins are crucial to the regulation of cell-cell and cell-matrix interactions during EMT.
Tumor initiating capacity
Cancer stem cells (CSCs), also called tumor initiating cells, are recognized as cells that are highly resistant to treatment and have strong tumor initiating capacity. CD44 is a widely used CSC marker in various types of cancers. Previous work has illustrated the causal role of CD44 isoform switching in EMT and metastasis[ Brown et al., 2011; Mima et al., 2012; Park et al., 2016; Preca et al., 2015; Roca et al., 2013; Tripathi et al., 2016]. Both CD44s and CD44v are shown to promote tumor progression in a tissue-specific manner, playing distinct roles. Mesenchymal-dominant CD44s promotes triple-negative breast cancer cell survival and metastasis[Brown et al., 2011; Xu et al., 2014; Zhang et al., 2019a; Zhao et al., 2016; Ouhtit et al., 2007; Hiraga et al., 2013; Mima et al., 2012] whereas epithelial-dominant CD44v stimulates tumor cell proliferation and promotes gastric cancer progression[Cheng, 2006; Todaro et al., 2014; Zeilstra et al., 2014; Lau et al., 2014; Yoshikawa et al., 2013]. Switching isoform expression between CD44s and CD44v determines breast CSC and non-CSC cell states[Zhang et al., 2019a]. CD44s can activate the PDGFRβ/Stat3 pathway and ZEB1 transcription factor to promote mammosphere-formation ability[Zhang et al., 2019a; Wellner et al., 2009; Preca et al., 2015]. This is also found in ovarian cancer models where CD44s expression increases aldehyde dehydrogenase activity, tumor spheroid formation, and chemoresistance[Bhattacharya et al., 2018]. CD44s is more potent at initiating tumor formation and promoting breast tumor recurrence than CD44v in mice, and CD44s is positively correlated with a mammary stem cell signature in TCGA[Brown et al., 2011; Zhang et al., 2019a].
Other alternative splicing events involved in CSC characteristics include the splicing of the surface molecule α6 integrin. Within a CD44high/CD24low CSC population, the epithelial-like cells express α6A, while the mesenchymal-like cells express α6B, and the two isoforms differ in their cytoplasmic domains. The α6B splice isoform promotes CSC characteristics and enhances tumor initiation potential through sustaining VEGF signaling[Goel et al., 2014]. It will be interesting to investigate whether there are additional alternative splicing events that are crucial to maintaining the CD44high/CD24low CSC population and its tumor initiation capacity.
Noncoding RNAs produced from alternative splicing
Apart from producing protein-coding RNA variants, alternative splicing also regulates noncoding RNAs. This includes long noncoding RNAs (lncRNAs) and circular RNAs (circRNAs) that are functionally important for EMT regulation, such as MALAT1, PNUTS, and CiRS-7[Gugnoni, and Ciarrocchi, 2019; Conn et al., 2015; Grelet et al., 2017; Dhamija, and Diederichs, 2016]. As an example, alternative splicing of the PNUTS gene generates a PNUTS mRNA and a lncRNA-PNUTS by using different splice acceptor sites of exon 12. The lncRNA-PNUTS is upregulated during EMT and correlates with ZEB1/ZEB2 mesenchymal marker expression. Functionally, the lncRNA-PNUTS antagonizes the epithelial regulator miR-205, serving as a miRNA sponge to promote EMT. When overexpressed, lncRNA-PNUTS promotes tumor initiation and metastasis[Grelet et al., 2017].
circRNAs are produced from a particular type of alternative splicing, where the splice donor of one exon is ligated to the splice acceptor site of an upstream exon (Fig 1A) [Jeck, and Sharpless, 2014; Conn et al., 2015]. RNA sequencing analysis has identified hundreds of circRNAs whose expression is increased during EMT. Many of them are produced from genes involved in EMT including SMARCA5, POLE2, SHPRH and DOCK1. These results suggest that circRNA may play important roles in regulating EMT, an area warranting further investigation[Conn et al., 2015; Pillman et al., 2018].
In recent years, RNA-seq has been extensively involved in discovering global alternative splicing changes in processes including EMT. Numerous splicing events have been identified, and their encoding genes are commonly involved in actin cytoskeleton regulation, cell-cell junctions, migration and wound healing [Shapiro et al., 2011; Vanharanta et al., 2014; Li et al., 2018; Pillman et al., 2018; Ahuja et al., 2020]. While at this stage, only a handful of these splicing changes have been connected to functional properties, it will be important to investigate these many other alternative splicing events on their biological roles in EMT in the future.
CELL STATE-SPECIFIC REGULATION OF ALTERNATIVE SPLICING DURING EMT
In order to generate cell-state specific isoforms during EMT, alternative splicing needs to be tightly controlled by cellular machinery. The 5’ and 3’ splice sites located at intron-exon boundaries are essential for recognition by the spliceosome. Variations in the splice site sequences determine their strength. Weaker splice sites are less often recognized by the spliceosome, resulting in exon skipping, whereas stronger splice sites are more likely to be recognized by the spliceosome, leading to exon inclusion in the mature mRNA product. Regulation of the recognition of splice sites, especially the weaker ones, are modulated by cis-acting elements contained within the intron and exon sequences. These cis-acting elements include exonic splicing enhancers (ESEs), intronic splicing enhancers (ISE), exonic splicing silencers (ESSs), and intronic splicing silencers (ISSs) (Fig 3) [Fu, and Jr, 2014]. Other cis-acting elements, including secondary and tertiary RNA structures, are also determinants of alternative splicing [Huang et al., 2017]. These sequence and structural elements can be recognized and bound by trans-acting elements, which are mainly RNA binding proteins (RBPs). Through binding to the cis-acting enhancer or silencer sequences, these RBPs either enhance or reduce recruitment of the spliceosome and thus promote exon inclusion or skipping, respectively (Fig. 3). Importantly, RBPs are dynamically regulated during EMT to control splicing events for enhanced oncogenic signaling, cytoskeletal remodeling, and increased cell death resistance[Hopkins et al., 2017; Reinke et al., 2012; Yang, and Carstens, 2017; Valacca et al., 2010; Venables et al., 2013; Shirakihara et al., 2011; Horiguchi et al., 2012; Gordon et al., 2019; Pillman et al., 2018].
Fig 3. Regulation of alternative splicing by splicing enhancers and silencers and RNA binding proteins.
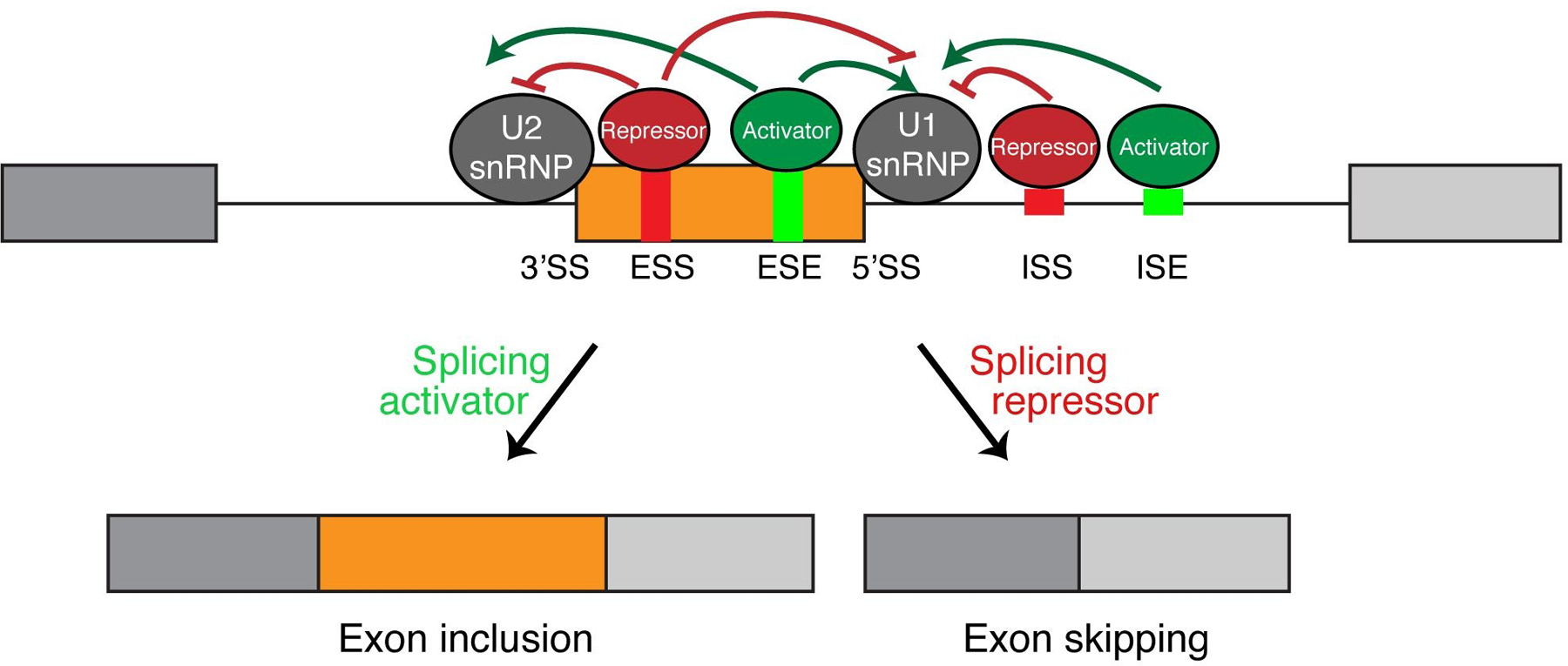
Exonic splicing enhancers and silencers (ESE, ESS) and intronic splicing enhancers and silencers (ISE, ISS) are shown in the alternative exon and downstream intron flanking the alternative exon. These short sequences are bound by RNA binding proteins which either function as a splicing repressor or activator. Binding of these RBPs directly act upon the U1 and U2 snRNP, two core components of the spliceosome, to dictate splicing outcome.
Regulation of epithelial splicing patterns
The epithelial splicing regulatory proteins 1/2 (ESRP1/2) are the first RBPs identified to specifically regulate epithelial alternative splicing[Warzecha et al., 2009]. The ESRPs are by far the most well characterized epithelial-type specific master regulators of alternative splicing and epithelial cell state. Loss of the ESRPs is an essential event during EMT while re-expression of the ESRPs in the mesenchymal state can partially restore an epithelial splicing program along with epithelial marker expression. As EMT occurs during early stages of embryogenesis, ESRP1 knockout mice was neonatal lethal due to cleft lip and cleft palate[Bebee et al., 2015; Lee et al., 2020]. Similar palate development defects were also observed in ESRP1/2 knockout zebrafish, demonstrating the conserved function of ESRPs in vertebrate development[Carroll et al., 2020]. ESRP1 and ESRP2 double knockout embryos fail to form lung and salivary glands, indicating that the ESRPs are essential for branching morphogenesis. Moreover, these double knockout mice exhibit epidermal hypoplasia and hair follicle developmental defects[Bebee et al., 2015]. These phenotypes are consistent with the phenotype of FGFR2-IIIb knockout mouse, which causes developmental defects in the lung, thymus, salivary gland and kidney[Moerlooze et al., 2000]. ESRP1 knockout also demonstrates tissue-specific defects in mice. ESRP1 knockout disrupts cochlear development, resulting in hearing loss, while germline knockout of ESRP1 in female mice caused infertility. Both phenotypes are caused by aberrant splicing of FGFR2 after ESRP1 loss [Rohacek et al., 2017; Yu et al., 2021]. These results indicate that ESRPs play an indispensable role in embryogenesis through regulating downstream alternative splicing events such as FGFR2 during developmental EMT.
ESRP1 binds a consensus GU-rich motif generally located in the proximal introns of a variable exon[Warzecha et al., 2010]. It regulates a large cohort of splicing events found to be critical during EMT, such as FGFR2, CTNND1, CD44, ENAH, Exo70, α6 integrin, and ARHGEF11[Warzecha et al., 2009; Brown et al., 2011; Modugno et al., 2012; Lu et al., 2013; Goel et al., 2014; Lee et al., 2018; Faux et al., 2021]. When ESRP1 is depleted, epithelial cells undergo accelerated transition to the mesenchymal state in response to EMT stimuli[Brown et al., 2011]. The ESRP1 promoter contains E-boxes, which recruits the SNAI1 transcription factor, resulting in transcriptional suppression of ESRP1 during EMT, further affecting a large cohort of downstream splicing events[Reinke et al., 2012].
Other RBPs regulating epithelial cell state have been identified. Loss of RBM47 during EMT enhances breast cancer metastasis[Vanharanta et al., 2014]. This effect is mediated through both regulation of alternative splicing and mRNA stability [Vanharanta et al., 2014; Yang et al., 2016]. RBM47 depletion during EMT increases TJP1-α-production to increase invasion[Kim et al., 2019]. AKAP8 is another recently discovered epithelial RBP that suppresses breast cancer metastasis by antagonizing mesenchymal splicing of CD44 and CLSTN1[Hu et al., 2020]. Specific RNA secondary structures can also act as important regulators of alternative splicing. One example is the RNA G-quadruplex, which contains two or more planar structures formed by Hoogsteen hydrogen bonding of four guanines. Recent work has shown that the CD44 intron contains RNA G-quadruplexes, which facilitate the recruitment of hnRNPF. hnRNPF promotes exon inclusion in G-quadruplex-containing introns, therefore inhibiting EMT by regulating inclusion of important EMT-related alternative splicing events such as CD44[Huang et al., 2017]. hnRNPF regulation of splicing can be perturbed by disrupting the G-quadruplex structure[Zhang et al., 2019b]. While most of these epithelial RBPs are studied in the context of cancer, it will be interesting to investigate whether they play an equally important role in developmental EMT.
Regulation of mesenchymal splicing patterns
hnRNPM, RBFOX1/2 and QKI are examples of RBPs that play an indispensable role in regulating mesenchymal splicing patterns. hnRNPM, a member of the hnRNP family, is often associated with splicing suppression[Han et al., 2010]. It was found to be a key regulator of mesenchymal splicing[Xu et al., 2014]. hnRNPM inhibits CD44 variable exon inclusion and promotes the production of CD44s mesenchymal isoform. By acting on CD44 alternative splicing, hnRNPM potentiates TGF-b signaling, accelerates EMT, and promotes breast cancer lung metastasis[Xu et al., 2014]. In addition to CD44, hnRNPM also promotes splice isoform switching of a large set of genes, including previously identified EMT-related splicing events EXOC7 and TCF7L2 [Harvey et al., 2018; Lu et al., 2013; Weise et al., 2010].
RBFOX factors alter splicing direction by binding up or downstream of the alternative exon. Binding upstream tends to repress splicing and vice versa[Yeo et al., 2009; Underwood et al., 2005; Zhang et al., 2008]. RBFOX2 expression is induced by TGF-β in breast cancer cell lines, which promotes the mesenchymal splicing of TAK1 exon 12 to sustain multiple signaling pathways including JNK, p38/MAPK and NF-κB[Tripathi et al., 2019]. On a global scale, depleting RBFOX2 in mesenchymal cells can induce phenotypes similar to an epithelial state[Shapiro et al., 2011], whereas its overexpression promotes several mesenchymal-specific splicing patterns [Braeutigam et al., 2014]. The STAR family RBP Quaking (QKI), along with RBFOX1, a homolog of RBFOX2, are identified as critical regulators that induce a mesenchymal cell state in breast cancer cells [Li et al., 2018; Pillman et al., 2018]. QKI promotes a broad spectrum of EMT-associated alternative splicing events. Similar to the binding position dependency of RBFOX factors, QKI binding upstream of an alternative exon often promotes skipping, while binding downstream promotes exon inclusion[Pillman et al., 2018]. It will be interesting to investigate the function of these splicing events in EMT in more detail.
Combinatorial regulation of alternative splicing
Multiple studies regarding alternative splicing regulation have created a blueprint for constructing networks consisting of alternative splicing events co-regulated by a set of RBPs. It is increasingly recognized that a single splicing event is co-regulated by multiple RBPs in different cell states. Furthermore, a single RBP can regulate a cohort of downstream events that are interconnected with other RBP regulation networks. For instance, although the ESRPs are strong epithelial regulators, other RBPs have shown the ability to regulate splicing events in both cell states. RBM47 regulates mostly epithelial splicing programs, but can antagonize some ESRP-regulated events[Yang et al., 2016]. RBFOX2 regulates a majority of mesenchymal splicing events, but can enhance some epithelial splicing patterns[Braeutigam et al., 2014; Venables et al., 2013; Lapuk et al., 2010; Dittmar et al., 2012]. Thus, it is important to understand how RBPs are tightly regulated in a tissue and cell state-specific manner, and how the expression of epithelial-specific and mesenchymal-specific RBPs act antagonistically on the same splicing event to determine splicing outcome.
An early example of co-regulation of the same event by multiple RBPs during EMT was demonstrated by the event of Ron splicing. SRSF1 promotes production of mesenchymal ∆Ron, while hnRNP A1 inhibits its production[Bonomi et al., 2013]. The ratio of ESRP1 and RBFOX2 expression dictates the outcome of hMENA exon 11a splicing in breast cancer[Ahuja et al., 2020]. In CD44 alternative splicing, inclusion of variable exons is promoted by ESRP1, hnRNPF, and AKAP8 in the epithelial state, while skipping of variable exons is enhanced by hnRNPM (Fig 4A) [Brown et al., 2011; Huang et al., 2017; Hu et al., 2020; Xu et al., 2014]. In fact, ESRP1 and hnRNPM compete for binding at the same GU-rich motifs, co-regulating a large cohort of splicing events antagonistically during EMT[Harvey et al., 2018].
Fig 4. Combinatorial regulation of EMT-associated alternative splicing by RNA binding proteins.
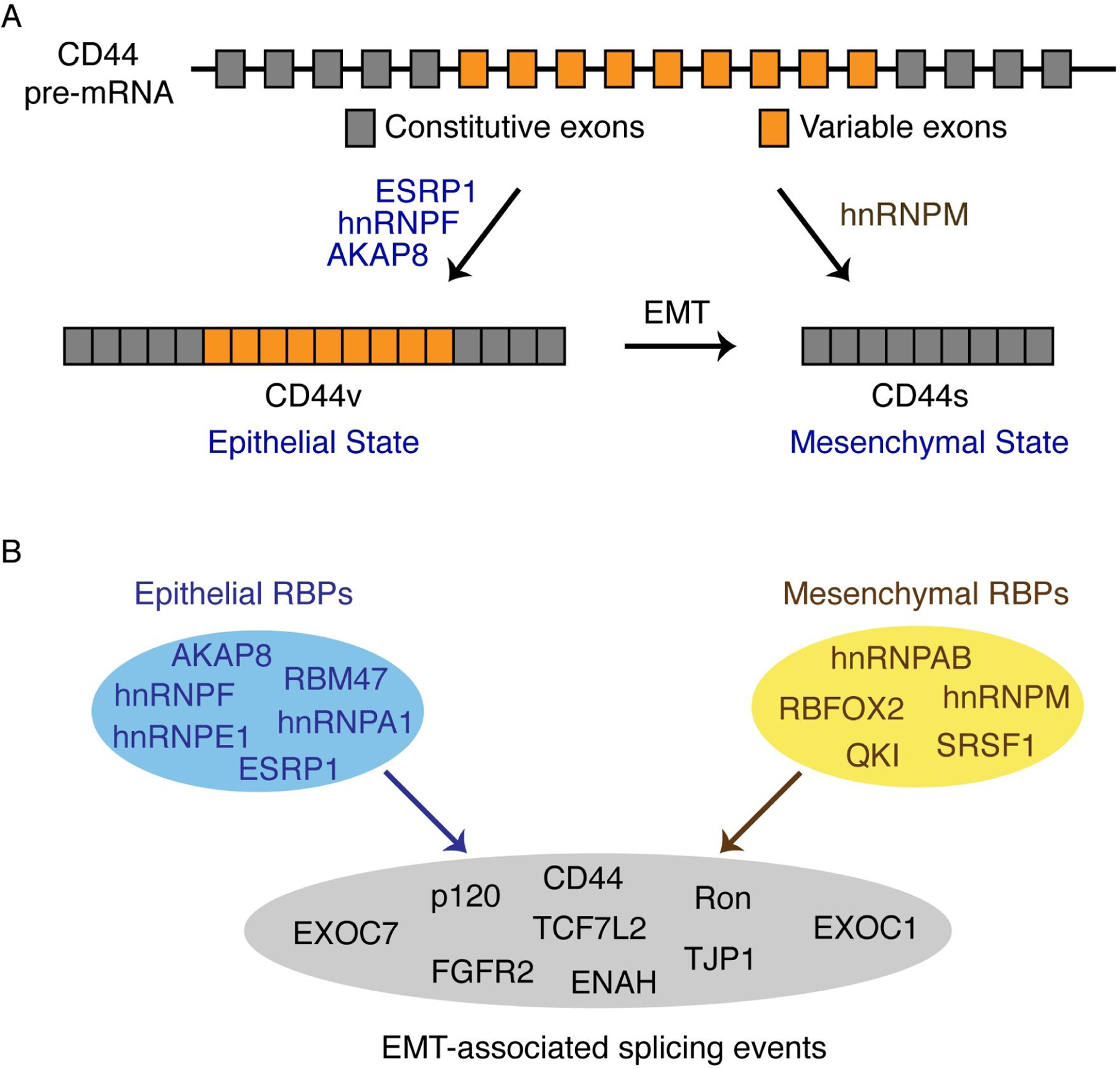
(A) CD44 splice isoform switching during EMT is co-regulated by several RBPs. ESRP1, hnRNPF, and AKAP8 promotes the epithelial CD44v splicing pattern, while hnRNPM promotes the mesenchymal CD44s splicing pattern.
(B) Epithelial and mesenchymal RBPs share overlapping alternative splicing targets. Cooperative or competitive regulation of RBPs on EMT-associated splicing events determine splicing outcome, which further affects EMT cell states.
Recently, through analysis of breast cancer specimens in BRCA-TCGA datasets, a combinatorial network of splicing regulation by RBPs was identified as a common mechanism in controlling EMT-associated alternative splicing events[Qiu et al., 2020]. In support of previous experimental findings, this study demonstrates that RBPs have strongly overlapping regulation networks, and that cooperative or competitive modes of RBP regulation on alternative splicing are essential for splicing outcome during EMT (Fig 4B).
CONCLUSIONS AND PERSPECTIVES
EMT is a critical step during normal embryogenesis and wound healing, but dysregulation of EMT is common in many types of cancer and affects invasion, migration, survival and metastasis. EMT is also critical for phenotypic heterogeneity in tumors as cells can transition between epithelial and mesenchymal states to obtain a balance between proliferation and survival. Alternative splicing, as one of the major contributors to proteome diversity, provides a mode of versatile gene regulation during EMT. Dysregulation of alternative splicing leads to disruption of epithelial cell state, increasing metastasis and enhancing survival. EMT-associated alternative splicing are mostly found in genes involved in signaling, cytoskeletal remodeling, invasiveness, cancer stem cells, and metastasis. RBPs are critical trans-acting regulators of alternative splicing that specifically regulates epithelial or mesenchymal splicing patterns. Balanced regulation of RBPs determines the outcome of global alternative splicing through an intricate interaction network between RBPs and alternative splicing events. Thus, the regulation of RBPs serves as an essential mechanism to control EMT and metastasis in cancer.
As EMT-related alternative splicing is an intricately regulated process that contributes to the heterogenous nature of solid tumors, its regulators and resulting isoforms have become targets of interest for precision medicine. Although research in this area is still in its early stages, therapeutic agents against pathogenic isoforms in diseases such as spinal muscular atrophy have been tested and approved for patient care. An antisense oligo against SMN2 exon 7 became the first FDA-approved drug against a pathogenic isoform in spinal muscular atrophy (SMA)[Hua et al., 2010]. Targeting the regulators and products of alternative splicing holds enormous promise for developing novel therapeutics.
Although hundreds of splicing changes have been identified during EMT, much is still unknown concerning the functional role underlying the switched expression of splice isoforms. A key future direction is to understand the function and regulation of EMT-associated alternative splicing isoforms. It would also be interesting to understand how alternative splicing cooperates with other post-transcriptional regulatory methods, such as RNA modifications and epigenetic regulation to ensure rapid and flexible gene regulation during EMT.
Acknowledgement and Funding Sources:
We thank members in the Cheng lab for insightful inputs. CC acknowledges funding from NIH (R35GM131876) and Cancer Prevention and Research Institute of Texas (RR160009) to support this work.
Footnotes
Conflict of Interest Statement:
The authors declare no conflict of interest.
REFERENCES
- Aceto N, Bardia A, Miyamoto DT, Donaldson MC, Wittner BS, Spencer JA, et al. : Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 2014;158:1110–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acloque H, Adams MS, Fishwick K, Bronner-Fraser M, Nieto MA: Epithelial-mesenchymal transitions: the importance of changing cell state in development and disease. J Clin Invest 2009;119:1438–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahuja N, Ashok C, Natua S, Pant D, Cherian A, Pandkar MR, et al. : Hypoxia-induced TGF-β-RBFOX2-ESRP1 axis regulates human MENA alternative splicing and promotes EMT in breast cancer. NAR Cancer 2020;2:zcaa021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aiello NM, Brabletz T, Kang Y, Nieto MA, Weinberg RA, Stanger BZ: Upholding a role for EMT in pancreatic cancer metastasis. Nature; London 2017;547:E7–E8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aiello NM, Kang Y: Context-dependent EMT programs in cancer metastasis. J Exp Med 2019;216:1016–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aktas B, Tewes M, Fehm T, Hauch S, Kimmig R, Kasimir-Bauer S: Stem cell and epithelial-mesenchymal transition markers are frequently overexpressed in circulating tumor cells of metastatic breast cancer patients. Breast Cancer Research 2009;11:R46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Applewhite DA, Barzik M, Kojima S-I, Svitkina TM, Gertler FB, Borisy GG: Ena/VASP proteins have an anti-capping independent function in filopodia formation. Mol Biol Cell 2007;18:2579–2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aruffo A, Stamenkovic I, Melnick M, Underhill CB, Seed B: CD44 is the principal cell surface receptor for hyaluronate. Cell 1990;61:1303–1313. [DOI] [PubMed] [Google Scholar]
- Baralle FE, Giudice J: Alternative splicing as a regulator of development and tissue identity. Nature Reviews Molecular Cell Biology; London 2017;18:437–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett SP, Salzman J: Circular RNAs: analysis, expression and potential functions. Development 2016;143:1838–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barzik M, Kotova TI, Higgs HN, Hazelwood L, Hanein D, Gertler FB, et al. : Ena/VASP proteins enhance actin polymerization in the presence of barbed end capping proteins. J Biol Chem 2005;280:28653–28662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bebee TW, Park JW, Sheridan KI, Warzecha CC, Cieply BW, Rohacek AM, et al. : The splicing regulators Esrp1 and Esrp2 direct an epithelial splicing program essential for mammalian development. eLife 2015;4. DOI: 10.7554/eLife.08954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett KL, Jackson DG, Simon JC, Tanczos E, Peach R, Modrell B, et al. : CD44 isoforms containing exon V3 are responsible for the presentation of heparin-binding growth factor. Journal of Cell Biology 1995;128:687–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya R, Mitra T, Ray Chaudhuri S, Roy SS: Mesenchymal splice isoform of CD44 (CD44s) promotes EMT/invasion and imparts stem-like properties to ovarian cancer cells. Journal of Cellular Biochemistry 2018;119:3373–3383. [DOI] [PubMed] [Google Scholar]
- Biamonti G, Bonomi S, Gallo S, Ghigna C: Making alternative splicing decisions during epithelial-to-mesenchymal transition (EMT). Cell Mol Life Sci 2012;69:2515–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonomi S, di Matteo A, Buratti E, Cabianca DS, Baralle FE, Ghigna C, et al. : HnRNP A1 controls a splicing regulatory circuit promoting mesenchymal-to-epithelial transition. Nucleic Acids Res 2013;41:8665–8679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brabletz T, Kalluri R, Nieto MA, Weinberg RA: EMT in cancer. Nature Reviews Cancer 2018;18:128–134. [DOI] [PubMed] [Google Scholar]
- Braeutigam C, Rago L, Rolke A, Waldmeier L, Christofori G, Winter J: The RNA-binding protein Rbfox2: an essential regulator of EMT-driven alternative splicing and a mediator of cellular invasion. Oncogene 2014;33:1082–1092. [DOI] [PubMed] [Google Scholar]
- Brown RL, Reinke LM, Damerow MS, Perez D, Chodosh LA, Yang J, et al. : CD44 splice isoform switching in human and mouse epithelium is essential for epithelial-mesenchymal transition and breast cancer progression. Journal of Clinical Investigation 2011;121:1064–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camp ER, Liu W, Fan F, Yang A, Somcio R, Ellis LM: RON, a tyrosine kinase receptor involved in tumor progression and metastasis. Ann Surg Oncol 2005;12:273–281. [DOI] [PubMed] [Google Scholar]
- Carroll SH, Macias Trevino C, Li EB, Kawasaki K, Myers N, Hallett SA, et al. : An Irf6-Esrp1/2 regulatory axis controls midface morphogenesis in vertebrates. Development 2020;147. DOI: 10.1242/dev.194498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaffer CL, San Juan BP, Lim E, Weinberg RA: EMT, cell plasticity and metastasis. Cancer Metastasis Rev 2016;35:645–654. [DOI] [PubMed] [Google Scholar]
- Chaffer CL, Thompson EW, Williams ED: Mesenchymal to Epithelial Transition in Development and Disease. Cells Tissues Organs 2007;185:7–19. [DOI] [PubMed] [Google Scholar]
- Chaffer CL, Weinberg RA: A Perspective on Cancer Cell Metastasis. Science 2011;331:1559–1564. [DOI] [PubMed] [Google Scholar]
- Chao YL, Shepard CR, Wells A: BRerseearachst carcinoma cells re-express E-cadherin during mesenchymal to epithelial reverting transition 2010;18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng C: A positive feedback loop couples Ras activation and CD44 alternative splicing. Genes & Development 2006;20:1715–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn SJ, Pillman KA, Toubia J, Conn VM, Salmanidis M, Phillips CA, et al. : The RNA Binding Protein Quaking Regulates Formation of circRNAs. Cell 2015;160:1125–1134. [DOI] [PubMed] [Google Scholar]
- Culty M, Miyake K, Kincade PW, Sikorski E, Butcher EC, Underhill C, et al. : The hyaluronate receptor is a member of the CD44 (H-CAM) family of cell surface glycoproteins. J Cell Biol 1990;111:2765–2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhamija S, Diederichs S: From junk to master regulators of invasion: lncRNA functions in migration, EMT and metastasis. Int J Cancer 2016;139:269–280. [DOI] [PubMed] [Google Scholar]
- Dittmar KA, Jiang P, Park JW, Amirikian K, Wan J, Shen S, et al. : Genome-wide determination of a broad ESRP-regulated posttranscriptional network by high-throughput sequencing. Mol Cell Biol 2012;32:1468–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dongre A, Weinberg RA: New insights into the mechanisms of epithelial–mesenchymal transition and implications for cancer. Nature Reviews Molecular Cell Biology 2019;20:69–84. [DOI] [PubMed] [Google Scholar]
- Faux MC, King LE, Kane SR, Love C, Sieber OM, Burgess AW: APC regulation of ESRP1 and p120-catenin isoforms in colorectal cancer cells. Mol Biol Cell 2021;32:120–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer KR, Durrans A, Lee S, Sheng J, Li F, Wong STC, et al. : Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015;527:472–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu X-D, Jr MA: Context-dependent control of alternative splicing by RNA-binding proteins. Nature Reviews Genetics 2014;15:689–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gertler FB, Niebuhr K, Reinhard M, Wehland J, Soriano P: Mena, a relative of VASP and Drosophila Enabled, is implicated in the control of microfilament dynamics. Cell 1996;87:227–239. [DOI] [PubMed] [Google Scholar]
- Ghigna C, Giordano S, Shen H, Benvenuto F, Castiglioni F, Comoglio PM, et al. : Cell Motility Is Controlled by SF2/ASF through Alternative Splicing of the Ron Protooncogene. Molecular Cell 2005;20:881–890. [DOI] [PubMed] [Google Scholar]
- Goel HL, Gritsko T, Pursell B, Chang C, Shultz LD, Greiner DL, et al. : Regulated Splicing of the α6 Integrin Cytoplasmic Domain Determines the Fate of Breast Cancer Stem Cells. Cell Rep 2014;7:747–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonçalves V, Matos P, Jordan P: Antagonistic SR proteins regulate alternative splicing of tumor-related Rac1b downstream of the PI3-kinase and Wnt pathways. Human Molecular Genetics 2009;18:3696–3707. [DOI] [PubMed] [Google Scholar]
- Gordon MA, Babbs B, Cochrane DR, Bitler BG, Richer JK: The long non-coding RNA MALAT1 promotes ovarian cancer progression by regulating RBFOX2-mediated alternative splicing. Molecular Carcinogenesis 2019;58:196–205. [DOI] [PubMed] [Google Scholar]
- Grelet S, Link LA, Howley B, Obellianne C, Palanisamy V, Gangaraju VK, et al. : A regulated PNUTS mRNA to lncRNA splice switch mediates EMT and tumour progression. Nature Cell Biology 2017;19:1105–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gugnoni M, Ciarrocchi A: Long Noncoding RNA and Epithelial Mesenchymal Transition in Cancer. Int J Mol Sci 2019;20. DOI: 10.3390/ijms20081924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han SP, Tang YH, Smith R: Functional diversity of the hnRNPs: past, present and perspectives. Biochem J 2010;430:379–392. [DOI] [PubMed] [Google Scholar]
- Harvey SE, Xu Y, Lin X, Gao XD, Qiu Y, Ahn J, et al. : Coregulation of alternative splicing by hnRNPM and ESRP1 during EMT. RNA 2018;24:1326–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiraga T, Ito S, Nakamura H: Cancer stem-like cell marker CD44 promotes bone metastases by enhancing tumorigenicity, cell motility, and hyaluronan production. Cancer Res 2013;73:4112–4122. [DOI] [PubMed] [Google Scholar]
- Hopkins A, Coatham ML, Berry FB: FOXC1 Regulates FGFR1 Isoform Switching to Promote Invasion Following TGFβ-Induced EMT. Molecular Cancer Research 2017;15:1341–1353. [DOI] [PubMed] [Google Scholar]
- Horiguchi K, Sakamoto K, Koinuma D, Semba K, Inoue A, Inoue S, et al. : TGF-β drives epithelial-mesenchymal transition through δEF1-mediated downregulation of ESRP. Oncogene 2012;31:3190–3201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Harvey SE, Zheng R, Lyu J, Grzeskowiak CL, Powell E, et al. : The RNA-binding protein AKAP8 suppresses tumor metastasis by antagonizing EMT-associated alternative splicing. Nat Commun 2020;11:486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua Y, Sahashi K, Hung G, Rigo F, Passini MA, Bennett CF, et al. : Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev 2010;24:1634–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Zhang J, Harvey SE, Hu X, Cheng C: RNA G-quadruplex secondary structure promotes alternative splicing via the RNA-binding protein hnRNPF. Genes Dev 2017;31:2296–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii H, Saitoh M, Sakamoto K, Kondo T, Katoh R, Tanaka S, et al. : Epithelial splicing regulatory proteins 1 (ESRP1) and 2 (ESRP2) suppress cancer cell motility via different mechanisms. J Biol Chem 2014;289:27386–27399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiyama N, Lee S-H, Liu S, Li G-Y, Smith MJ, Reichardt LF, et al. : Dynamic and static interactions between p120 catenin and E-cadherin regulate the stability of cell-cell adhesion. Cell 2010;141:117–128. [DOI] [PubMed] [Google Scholar]
- Jeck WR, Sharpless NE: Detecting and characterizing circular RNAs. Nat Biotechnol 2014;32:453–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri R, Weinberg RA: The basics of epithelial-mesenchymal transition. J Clin Invest 2009;119:1420–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasimir-Bauer S, Hoffmann O, Wallwiener D, Kimmig R, Fehm T: Expression of stem cell and epithelial-mesenchymal transition markers in primary breast cancer patients with circulating tumor cells. Breast Cancer Research 2012;14:R15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelemen O, Convertini P, Zhang Z, Wen Y, Shen M, Falaleeva M, et al. : Function of alternative splicing. Gene 2013;514:1–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren H, Lev-Maor G, Ast G: Alternative splicing and evolution: diversification, exon definition and function. Nature Reviews Genetics 2010;11:345–355. [DOI] [PubMed] [Google Scholar]
- Kim DH, Xing T, Yang Z, Dudek R, Lu Q, Chen Y-H: Epithelial Mesenchymal Transition in Embryonic Development, Tissue Repair and Cancer: A Comprehensive Overview. J Clin Med 2017;7. DOI: 10.3390/jcm7010001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y-E, Won M, Lee S-G, Park C, Song C-H, Kim KK: RBM47-regulated alternative splicing of TJP1 promotes actin stress fiber assembly during epithelial-to-mesenchymal transition. Oncogene 2019;38:6521–6536. [DOI] [PubMed] [Google Scholar]
- Kristensen LS, Andersen MS, Stagsted LVW, Ebbesen KK, Hansen TB, Kjems J: The biogenesis, biology and characterization of circular RNAs. Nature Reviews Genetics 2019;20:675–691. [DOI] [PubMed] [Google Scholar]
- Kung JTY, Colognori D, Lee JT: Long noncoding RNAs: past, present, and future. Genetics 2013;193:651–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamouille S, Xu J, Derynck R: Molecular mechanisms of epithelial–mesenchymal transition. Nature Reviews Molecular Cell Biology 2014;15:178–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapuk A, Marr H, Jakkula L, Pedro H, Bhattacharya S, Purdom E, et al. : Exon-Level Microarray Analyses Identify Alternative Splicing Programs in Breast Cancer. Molecular Cancer Research 2010;8:961–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau WM, Teng E, Chong HS, Lopez KAP, Tay AYL, Salto-Tellez M, et al. : CD44v8–10 is a cancer-specific marker for gastric cancer stem cells. Cancer Res 2014;74:2630–2641. [DOI] [PubMed] [Google Scholar]
- Lecharpentier A, Vielh P, Perez-Moreno P, Planchard D, Soria JC, Farace F: Detection of circulating tumour cells with a hybrid (epithelial/mesenchymal) phenotype in patients with metastatic non-small cell lung cancer. Br J Cancer 2011;105:1338–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J-Y, Kong G: Roles and epigenetic regulation of epithelial–mesenchymal transition and its transcription factors in cancer initiation and progression. Cell Mol Life Sci 2016;73:4643–4660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Cieply B, Yang Y, Peart N, Glaser C, Chan P, et al. : Esrp1-Regulated Splicing of Arhgef11 Isoforms Is Required for Epithelial Tight Junction Integrity. Cell Reports 2018;25:2417–2430.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Sears MJ, Zhang Z, Li H, Salhab I, Krebs P, et al. : Cleft lip and cleft palate in Esrp1 knockout mice is associated with alterations in epithelial-mesenchymal crosstalk. Development 2020;147. DOI: 10.1242/dev.187369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Choi PS, Chaffer CL, Labella K, Hwang JH, Giacomelli AO, et al. : An alternative splicing switch in FLNB promotes the mesenchymal cell state in human breast cancer. eLife 2018;7. DOI: 10.7554/eLife.37184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Cheng C: Alternative RNA splicing and cancer. Wiley Interdisciplinary Reviews: RNA 2013;4:547–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Cheng C: Akt Signaling Is Sustained by a CD44 Splice Isoform–Mediated Positive Feedback Loop. Cancer Research 2017;77:3791–3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Liu J, Liu S, Zeng J, Ding D, Carstens RP, et al. : Exo70 Isoform Switching upon Epithelial-Mesenchymal Transition Mediates Cancer Cell Invasion. Dev Cell 2013;27:560–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magen A, Ast G: The importance of being divisible by three in alternative splicing. Nucleic Acids Res 2005;33:5574–5582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchese FP, Raimondi I, Huarte M: The multidimensional mechanisms of long noncoding RNA function. Genome Biology 2017;18:206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matos P, Jordan P: Expression of Rac1b stimulates NF-kappaB-mediated cell survival and G1/S progression. Exp Cell Res 2005;305:292–299. [DOI] [PubMed] [Google Scholar]
- Matter N, Herrlich P, König H: Signal-dependent regulation of splicing via phosphorylation of Sam68. Nature 2002;420:691–695. [DOI] [PubMed] [Google Scholar]
- Mima K, Okabe H, Ishimoto T, Hayashi H, Nakagawa S, Kuroki H, et al. : CD44s regulates the TGF-β-mediated mesenchymal phenotype and is associated with poor prognosis in patients with hepatocellular carcinoma. Cancer Res 2012;72:3414–3423. [DOI] [PubMed] [Google Scholar]
- Miyake K: Hyaluronate can function as a cell adhesion molecule and CD44 participates in hyaluronate recognition. Journal of Experimental Medicine 1990;172:69–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modugno FD, Iapicca P, Boudreau A, Mottolese M, Terrenato I, Perracchio L, et al. : Splicing program of human MENA produces a previously undescribed isoform associated with invasive, mesenchymal-like breast tumors. PNAS 2012;109:19280–19285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moerlooze LD, Spencer-Dene B, Revest J, Hajihosseini M, Rosewell I, Dickson C: An important role for the IIIb isoform of fibroblast growth factor receptor 2 (FGFR2) in mesenchymal-epithelial signalling during mouse organogenesis. Development 2000;127:483–492. [DOI] [PubMed] [Google Scholar]
- Morel A-P, Hinkal GW, Thomas C, Fauvet F, Courtois-Cox S, Wierinckx A, et al. : EMT inducers catalyze malignant transformation of mammary epithelial cells and drive tumorigenesis towards claudin-low tumors in transgenic mice. PLoS Genet 2012;8:e1002723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima Y, Yamagishi T, Hokari S, Nakamura H: Mechanisms involved in valvuloseptal endocardial cushion formation in early cardiogenesis: Roles of transforming growth factor (TGF)-β and bone morphogenetic protein (BMP). The Anatomical Record 2000;258:119–127. [DOI] [PubMed] [Google Scholar]
- Nieto MA: Epithelial Plasticity: A Common Theme in Embryonic and Cancer Cells. Science 2013;342. DOI: 10.1126/science.1234850 [DOI] [PubMed] [Google Scholar]
- Nieto MA: Context-specific roles of EMT programmes in cancer cell dissemination. Nature Cell Biology 2017;19:416–418. [DOI] [PubMed] [Google Scholar]
- Nieto MA, Huang RY-J, Jackson RA, Thiery JP: EMT: 2016. Cell 2016;166:21–45. [DOI] [PubMed] [Google Scholar]
- Orr-Urtreger A, Bedford MT, Burakova T, Arman E, Zimmer Y, Yayon A, et al. : Developmental localization of the splicing alternatives of fibroblast growth factor receptor-2 (FGFR2). Dev Biol 1993;158:475–486. [DOI] [PubMed] [Google Scholar]
- Ouhtit A, Abd Elmageed ZY, Abdraboh ME, Lioe TF, Raj MHG: In vivo evidence for the role of CD44s in promoting breast cancer metastasis to the liver. Am J Pathol 2007;171:2033–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ: Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nature Genetics 2008;40:1413–1415. [DOI] [PubMed] [Google Scholar]
- Park NR, Cha JH, Jang JW, Bae SH, Jang B, Kim J-H, et al. : Synergistic effects of CD44 and TGF-β1 through AKT/GSK-3β/β-catenin signaling during epithelial-mesenchymal transition in liver cancer cells. Biochem Biophys Res Commun 2016;477:568–574. [DOI] [PubMed] [Google Scholar]
- Pastushenko I, Blanpain C: EMT Transition States during Tumor Progression and Metastasis. Trends in Cell Biology 2019;29:212–226. [DOI] [PubMed] [Google Scholar]
- Peinado H, Olmeda D, Cano A: Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nature Reviews Cancer 2007;7:415–428. [DOI] [PubMed] [Google Scholar]
- Pérez-Pomares JM, Muñoz-Chápuli R: Epithelial-mesenchymal transitions: a mesodermal cell strategy for evolutive innovation in Metazoans. Anat Rec 2002;268:343–351. [DOI] [PubMed] [Google Scholar]
- Pillman KA, Phillips CA, Roslan S, Toubia J, Dredge BK, Bert AG, et al. : miR-200/375 control epithelial plasticity-associated alternative splicing by repressing the RNA-binding protein Quaking. EMBO J 2018;37. DOI: 10.15252/embj.201899016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polyak K, Weinberg RA: Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer 2009;9:265–273. [DOI] [PubMed] [Google Scholar]
- Ponta H, Sherman L, Herrlich PA: CD44: From adhesion molecules to signalling regulators. Nature Reviews Molecular Cell Biology 2003;4:33–45. [DOI] [PubMed] [Google Scholar]
- Pradella D, Naro C, Sette C, Ghigna C: EMT and stemness: flexible processes tuned by alternative splicing in development and cancer progression. Molecular Cancer 2017;16. DOI: 10.1186/s12943-016-0579-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preca B-T, Bajdak K, Mock K, Sundararajan V, Pfannstiel J, Maurer J, et al. : A self-enforcing CD44s/ZEB1 feedback loop maintains EMT and stemness properties in cancer cells. International Journal of Cancer 2015;137:2566–2577. [DOI] [PubMed] [Google Scholar]
- Qiu Y, Lyu J, Dunlap M, Harvey SE, Cheng C: A combinatorially regulated RNA splicing signature predicts breast cancer EMT states and patient survival. RNA 2020; DOI: 10.1261/rna.074187.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radisky DC, Levy DD, Littlepage LE, Liu H, Nelson CM, Fata JE, et al. : Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature 2005;436:123–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinke LM, Xu Y, Cheng C: Snail Represses the Splicing Regulator Epithelial Splicing Regulatory Protein 1 to Promote Epithelial-Mesenchymal Transition. J Biol Chem 2012;287:36435–36442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roca H, Hernandez J, Weidner S, McEachin RC, Fuller D, Sud S, et al. : Transcription factors OVOL1 and OVOL2 induce the mesenchymal to epithelial transition in human cancer. PLoS ONE 2013;8:e76773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohacek AM, Bebee TW, Tilton RK, Radens CM, McDermott-Roe C, Peart N, et al. : ESRP1 Mutations Cause Hearing Loss due to Defects in Alternative Splicing that Disrupt Cochlear Development. Dev Cell 2017;43:318–331.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero PR, Zaidi S, Fang YY, Uversky VN, Radivojac P, Oldfield CJ, et al. : Alternative splicing in concert with protein intrinsic disorder enables increased functional diversity in multicellular organisms. Proc Natl Acad Sci USA 2006;103:8390–8395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandberg R, Neilson JR, Sarma A, Sharp PA, Burge CB: Proliferating Cells Express mRNAs with Shortened 3’ Untranslated Regions and Fewer MicroRNA Target Sites. Science 2008;320:1643–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savagner P, Vallés AM, Jouanneau J, Yamada KM, Thiery JP: Alternative splicing in fibroblast growth factor receptor 2 is associated with induced epithelial-mesenchymal transition in rat bladder carcinoma cells. Molecular Biology of the Cell 1994;5:851–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnelzer A, Prechtel D, Knaus U, Dehne K, Gerhard M, Graeff H, et al. : Rac1 in human breast cancer: overexpression, mutation analysis, and characterization of a new isoform, Rac1b. Oncogene 2000;19:3013–3020. [DOI] [PubMed] [Google Scholar]
- Shapiro IM, Cheng AW, Flytzanis NC, Balsamo M, Condeelis JS, Oktay MH, et al. : An EMT– Driven Alternative Splicing Program Occurs in Human Breast Cancer and Modulates Cellular Phenotype. PLOS Genetics 2011;7:e1002218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y: Mechanistic insights into precursor messenger RNA splicing by the spliceosome. Nature Reviews Molecular Cell Biology 2017;18:655–670. [DOI] [PubMed] [Google Scholar]
- Shirakihara T, Horiguchi K, Miyazawa K, Ehata S, Shibata T, Morita I, et al. : TGF-β regulates isoform switching of FGF receptors and epithelial–mesenchymal transition. EMBO J 2011;30:783–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A, Karnoub AE, Palmby TR, Lengyel E, Sondek J, Der CJ: Rac1b, a tumor associated, constitutively active Rac1 splice variant, promotes cellular transformation. Oncogene 2004;23:9369–9380. [DOI] [PubMed] [Google Scholar]
- Slorach EM, Chou J, Werb Z: Zeppo1 is a novel metastasis promoter that represses E-cadherin expression and regulates p120-catenin isoform expression and localization. Genes Dev 2011;25:471–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiery JP: Epithelial–mesenchymal transitions in development and pathologies. Current Opinion in Cell Biology 2003;15:740–746. [DOI] [PubMed] [Google Scholar]
- Thiery JP, Acloque H, Huang RYJ, Nieto MA: Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009;139:871–890. [DOI] [PubMed] [Google Scholar]
- Thiery JP, Sleeman JP: Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol 2006;7:131–142. [DOI] [PubMed] [Google Scholar]
- Todaro M, Gaggianesi M, Catalano V, Benfante A, Iovino F, Biffoni M, et al. : CD44v6 Is a Marker of Constitutive and Reprogrammed Cancer Stem Cells Driving Colon Cancer Metastasis. Cell Stem Cell 2014;14:342–356. [DOI] [PubMed] [Google Scholar]
- Tripathi V, Shin J-H, Stuelten CH, Zhang YE: TGF-β-induced alternative splicing of TAK1 promotes EMT and drug resistance. Oncogene 2019;38:3185–3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi V, Sixt KM, Gao S, Xu X, Huang J, Weigert R, et al. : Direct Regulation of Alternative Splicing by SMAD3 through PCBP1 Is Essential to the Tumor-Promoting Role of TGF-β. Molecular Cell 2016;64:549–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai JH, Yang J: Epithelial–mesenchymal plasticity in carcinoma metastasis. Genes Dev 2013;27:2192–2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Underwood JG, Boutz PL, Dougherty JD, Stoilov P, Black DL: Homologues of the Caenorhabditis elegans Fox-1 protein are neuronal splicing regulators in mammals. Mol Cell Biol 2005;25:10005–10016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valacca C, Bonomi S, Buratti E, Pedrotti S, Baralle FE, Sette C, et al. : Sam68 regulates EMT through alternative splicing–activated nonsense-mediated mRNA decay of the SF2/ASF proto-oncogene. The Journal of Cell Biology 2010;191:87–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanharanta S, Marney CB, Shu W, Valiente M, Zou Y, Mele A, et al. : Loss of the multifunctional RNA-binding protein RBM47 as a source of selectable metastatic traits in breast cancer. Elife 2014;3. DOI: 10.7554/eLife.02734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venables JP, Brosseau J-P, Gadea G, Klinck R, Prinos P, Beaulieu J-F, et al. : RBFOX2 Is an Important Regulator of Mesenchymal Tissue-Specific Splicing in both Normal and Cancer Tissues. Molecular and Cellular Biology 2013;33:396–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl MC, Will CL, Lührmann R: The Spliceosome: Design Principles of a Dynamic RNP Machine. Cell 2009;136:701–718. [DOI] [PubMed] [Google Scholar]
- Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, et al. : Alternative isoform regulation in human tissue transcriptomes. Nature; London 2008;456:470–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warzecha CC, Jiang P, Amirikian K, Dittmar KA, Lu H, Shen S, et al. : An ESRP-regulated splicing programme is abrogated during the epithelial–mesenchymal transition. The EMBO Journal 2010;29:3286–3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warzecha CC, Sato TK, Nabet B, Hogenesch JB, Carstens RP: ESRP1 and ESRP2 Are Epithelial Cell-Type-Specific Regulators of FGFR2 Splicing. Molecular Cell 2009;33:591–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weise A, Bruser K, Elfert S, Wallmen B, Wittel Y, Wöhrle S, et al. : Alternative splicing of Tcf7l2 transcripts generates protein variants with differential promoter-binding and transcriptional activation properties at Wnt/beta-catenin targets. Nucleic Acids Res 2010;38:1964–1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellner U, Schubert J, Burk UC, Schmalhofer O, Zhu F, Sonntag A, et al. : The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nature Cell Biology 2009;11:1487–1495. [DOI] [PubMed] [Google Scholar]
- Xing Y, Lee CJ: Protein modularity of alternatively spliced exons is associated with tissue-specific regulation of alternative splicing. PLoS Genet 2005;1:e34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Gao XD, Lee J-H, Huang H, Tan H, Ahn J, et al. : Cell type-restricted activity of hnRNPM promotes breast cancer metastasis via regulating alternative splicing. Genes Dev 2014;28:1191–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagisawa M, Huveldt D, Kreinest P, Lohse CM, Cheville JC, Parker AS, et al. : A p120 Catenin Isoform Switch Affects Rho Activity, Induces Tumor Cell Invasion, and Predicts Metastatic Disease. Journal of Biological Chemistry 2008;283:18344–18354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Carstens RP: Alternative splicing regulates distinct subcellular localization of Epithelial splicing regulatory protein 1 (Esrp1) isoforms. Scientific Reports 2017;7:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Park JW, Bebee TW, Warzecha CC, Guo Y, Shang X, et al. : Determination of a Comprehensive Alternative Splicing Regulatory Network and Combinatorial Regulation by Key Factors during the Epithelial-to-Mesenchymal Transition. Molecular and Cellular Biology 2016;36:1704–1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye X, Brabletz T, Kang Y, Longmore GD, Nieto MA, Stanger BZ, et al. : Upholding a role for EMT in breast cancer metastasis. Nature 2017;547:E1–E3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh BK, Igarashi M, Eliseenkova AV, Plotnikov AN, Sher I, Ron D, et al. : Structural basis by which alternative splicing confers specificity in fibroblast growth factor receptors. Proceedings of the National Academy of Sciences 2003;100:2266–2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo GW, Coufal NG, Liang TY, Peng GE, Fu X-D, Gage FH: An RNA code for the FOX2 splicing regulator revealed by mapping RNA-protein interactions in stem cells. Nat Struct Mol Biol 2009;16:130–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung KT, Yang J: Epithelial–mesenchymal transition in tumor metastasis. Mol Oncol 2017;11:28–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshikawa M, Tsuchihashi K, Ishimoto T, Yae T, Motohara T, Sugihara E, et al. : xCT inhibition depletes CD44v-expressing tumor cells that are resistant to EGFR-targeted therapy in head and neck squamous cell carcinoma. Cancer Res 2013;73:1855–1866. [DOI] [PubMed] [Google Scholar]
- Yu L, Zhang H, Guan X, Qin D, Zhou J, Wu X: Loss of ESRP1 blocks mouse oocyte development and leads to female infertility. Development 2021;148. DOI: 10.1242/dev.196931 [DOI] [PubMed] [Google Scholar]
- Yu M, Bardia A, Wittner BS, Stott SL, Smas ME, Ting DT, et al. : Circulating Breast Tumor Cells Exhibit Dynamic Changes in Epithelial and Mesenchymal Composition. Science 2013;339:580–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeilstra J, Joosten SPJ, van Andel H, Tolg C, Berns A, Snoek M, et al. : Stem cell CD44v isoforms promote intestinal cancer formation in Apc(min) mice downstream of Wnt signaling. Oncogene 2014;33:665–670. [DOI] [PubMed] [Google Scholar]
- Zhang C, Zhang Z, Castle J, Sun S, Johnson J, Krainer AR, et al. : Defining the regulatory network of the tissue-specific splicing factors Fox-1 and Fox-2. Genes Dev 2008;22:2550–2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Brown RL, Wei Y, Zhao P, Liu S, Liu X, et al. : CD44 splice isoform switching determines breast cancer stem cell state. Genes Dev 2019a;33:166–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Harvey SE, Cheng C: A high-throughput screen identifies small molecule modulators of alternative splicing by targeting RNA G-quadruplexes. Nucleic Acids Res 2019b;47:3667–3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Ibrahimi OA, Olsen SK, Umemori H, Mohammadi M, Ornitz DM: Receptor Specificity of the Fibroblast Growth Factor Family: THE COMPLETE MAMMALIAN FGF FAMILY. Journal of Biological Chemistry 2006;281:15694–15700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao P, Xu Y, Wei Y, Qiu Q, Chew T-L, Kang Y, et al. : The CD44s splice isoform is a central mediator for invadopodia activity. Journal of Cell Science 2016;129:1355–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q, Caballero OL, Davis ID, Jonasch E, Tamboli P, Yung WKA, et al. : Tumor-Specific Isoform Switch of the Fibroblast Growth Factor Receptor 2 Underlies the Mesenchymal and Malignant Phenotypes of Clear Cell Renal Cell Carcinomas. Clinical Cancer Research 2013;19:2460–2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H, et al. : Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015;527:525–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y-Q, He C, Chen Y-Q, Wang D, Wang M-H: Altered expression of the RON receptor tyrosine kinase in primary human colorectal adenocarcinomas: generation of different splicing RON variants and their oncogenic potential. Oncogene 2003;22:186–197. [DOI] [PubMed] [Google Scholar]