

Abstract
Clear cell renal cell carcinoma (ccRCC) is characterized by large intracellular lipid droplets (LDs) containing free and esterified cholesterol; however, the functional significance of cholesterol accumulation in ccRCC cells is unknown. We demonstrate that, surprisingly, genes encoding cholesterol biosynthetic enzymes are repressed in ccRCC, suggesting a dependency on exogenous cholesterol. Mendelian randomization analyses performed on 31,000 individuals indicate a causal link between elevated circulating high-density lipoprotein (HDL) cholesterol and ccRCC risk. Depriving ccRCC cells of either cholesterol or HDL compromises proliferation and survival in vitro and tumor growth in vivo; in contrast, elevated dietary cholesterol promotes tumor growth. Scavenger Receptor B1 (SCARB1) is uniquely required for cholesterol import, and inhibiting SCARB1 is sufficient to cause ccRCC cell cycle arrest, apoptosis, elevated intracellular reactive oxygen species levels and decreased PI3K/AKT signaling. Collectively, we reveal a cholesterol dependency in ccRCC and implicate SCARB1 as a novel therapeutic target for treating kidney cancer.
Keywords: SCARB1, HDL, ROS, PI3K/AKT, ccRCC
INTRODUCTION
Renal cell carcinoma (RCC) ranks among the 10 most common cancers in men and women, with approximatively 74,000 new cases of RCC and 15,000 deaths in the United States in 2020 (American Cancer Society). ccRCC represents 70–80% of all RCC subtypes, with over 90% displaying copy number deletion, inactivating mutation and/or epigenetic silencing of the von Hippel-Lindau (VHL) tumor suppressor gene (1). VHL encodes the recognition component of a protein complex that catalyzes the oxygen-dependent ubiquitination and subsequent proteasomal degradation of the hypoxia inducible factor (HIF) transcriptional regulators (2). Recently, ccRCC has been classified as a “metabolic disease” due to common alterations in cellular metabolic pathways associated with cancer initiation and progression (3). HIFs are critical drivers of ccRCC metabolic rewiring, partly by promoting the “Warburg effect” that provides cancer cells with ATP and glycolytic intermediates to fulfill the anabolic demands of proliferating cells (4,5). Moreover, recent genome-wide, metabolic gene set analyses reveal that ccRCC tumors downregulate enzymes in the pathways opposing glycolysis, such as gluconeogenesis, as well as pathways that consume potential nutrients, such as the urea cycle pathway relative to normal tissues. For example, the rate-limiting gluconeogenic enzyme fructose-1,6-bisphosphatase 1 (FBP1) is universally depleted in ccRCC tumors, and its re-expression in ccRCC cells confirms an unexpected tumor suppressor activity (6). Similarly, expression of the urea cycle enzyme arginase 2 (ARG2) is also repressed in ccRCC, and ectopic ARG2 expression in ccRCC cells results in toxic polyamine accumulation and depletion of the essential biosynthetic cofactor pyridoxal phosphate (7).
Extensive accumulation of intracellular lipid droplets (LDs) and glycogen, which confer the “clear cell” histological phenotype, is an additional metabolic hallmark of ccRCC (8). LDs are dynamic cytoplasmic organelles protected by a phospholipid monolayer surrounding a neutral lipid core, composed primarily of cholesterol esters (CEs) and triacylglycerols (TGs), and have been linked with many cellular functions (9). We previously reported that reduced expression of the LD coat protein Perilipin-2 (PLIN2), or its transcriptional regulator HIF2α, impairs LD formation and reduces ccRCC cell viability (10). Cholesterol and esterified cholesterol are the most prominent lipids stored in ccRCC LDs, accumulating to 8-fold and 35-fold higher levels, respectively, compared to normal kidney tissue (11). Currently, the functional importance of cholesterol accumulation and turnover for ccRCC tumor growth and progression remains obscure.
In this present study, we performed integrative studies of human ccRCC tumors and cell lines and demonstrate that ccRCC cells suppress de novo cholesterol biosynthesis, despite accumulating high levels of cholesterol and cholesterol esters. We demonstrate that ccRCC cells are, surprisingly, functional auxotrophs for exogenous cholesterol and induce apoptotic cell death in its absence. Furthermore, elevated dietary cholesterol promotes, whereas dietary cholesterol restriction inhibits, ccRCC xenograft tumor growth. The scavenger receptor B1 (SCARB1) transports cholesterol into ccRCC cells in the form of high-density lipoproteins (HDLs), circulating 5–17 nm diameter particles composed of proteins (primarily apolipoproteins apo A-I and apo A-II), phospholipids, cholesterol, cholesterol esters, triglycerides, and other neutral lipids. Previous genome-wide association studies (GWAS) identified a common single nucleotide polymorphism (SNP) within the SCARB1 locus linked to increased ccRCC risk (12,13). We demonstrate that SCARB1 is functionally required for HDL transport in ccRCC; mutating the SCARB1 gene, reducing its expression, or inhibiting SCARB1 activity compromises ccRCC cell viability and tumor growth. Importantly, Mendelian randomization analyses of GWAS data indicate a causal relationship between genetic alleles associated with elevated circulating HDL (and HDL cholesterol) levels and ccRCC risk. Finally, we demonstrate that inhibiting SCARB1 or limiting cholesterol availability increases ROS levels in ccRCC cells, revealing an important role for cholesterol in redox homeostasis. The dependence of ccRCC on exogenous cholesterol import suggests a metabolic vulnerability that can be explored as a novel therapeutic strategy for ccRCC.
RESULTS
ccRCC cells harbor deregulated cholesterol metabolism
To investigate cholesterol metabolism in ccRCC, we conducted gene set enrichment analysis (GSEA) of TCGA RNAseq data, and observed that gene sets associated with cholesterol metabolism and biosynthesis (Figure 1A) are significantly downregulated in ccRCC tumors (Figure 1B, C and S1A). The majority of these genes encode rate-limiting enzymes in the de novo cholesterol biosynthetic pathway, including HMG-coA reductase (HMGCR), lanosterol synthase (LSS) and squalene monooxygenase (SQLE). Integrative analysis indicated that mRNA transcripts encoding other enzymes in the mevalonate pathway are coordinately repressed in ccRCC tumor samples relative to normal kidney (Figure 1D). These genes were similarly underexpressed in 12 pairs of ccRCC human patient samples, relative to surrounding normal tissue (Figure 1E). A large-scale proteogenomic study (14) also confirmed that mevalonate pathway gene and protein expression is downregulated in ccRCC (Figure S1B, C). In human A498 and 786-O ccRCC cell lines, HMGCR, LSS, SQLE, delta-24-sterol reductase (DHCR24), and farnesyl-diphosphate farnesyltransferase 1 (FDFT1) levels were reduced relative to the non-transformed HK-2 tubular epithelial cell line derived from normal kidney (Figure 1F).
Figure 1. ccRCC cells harbor deregulated cholesterol metabolism.
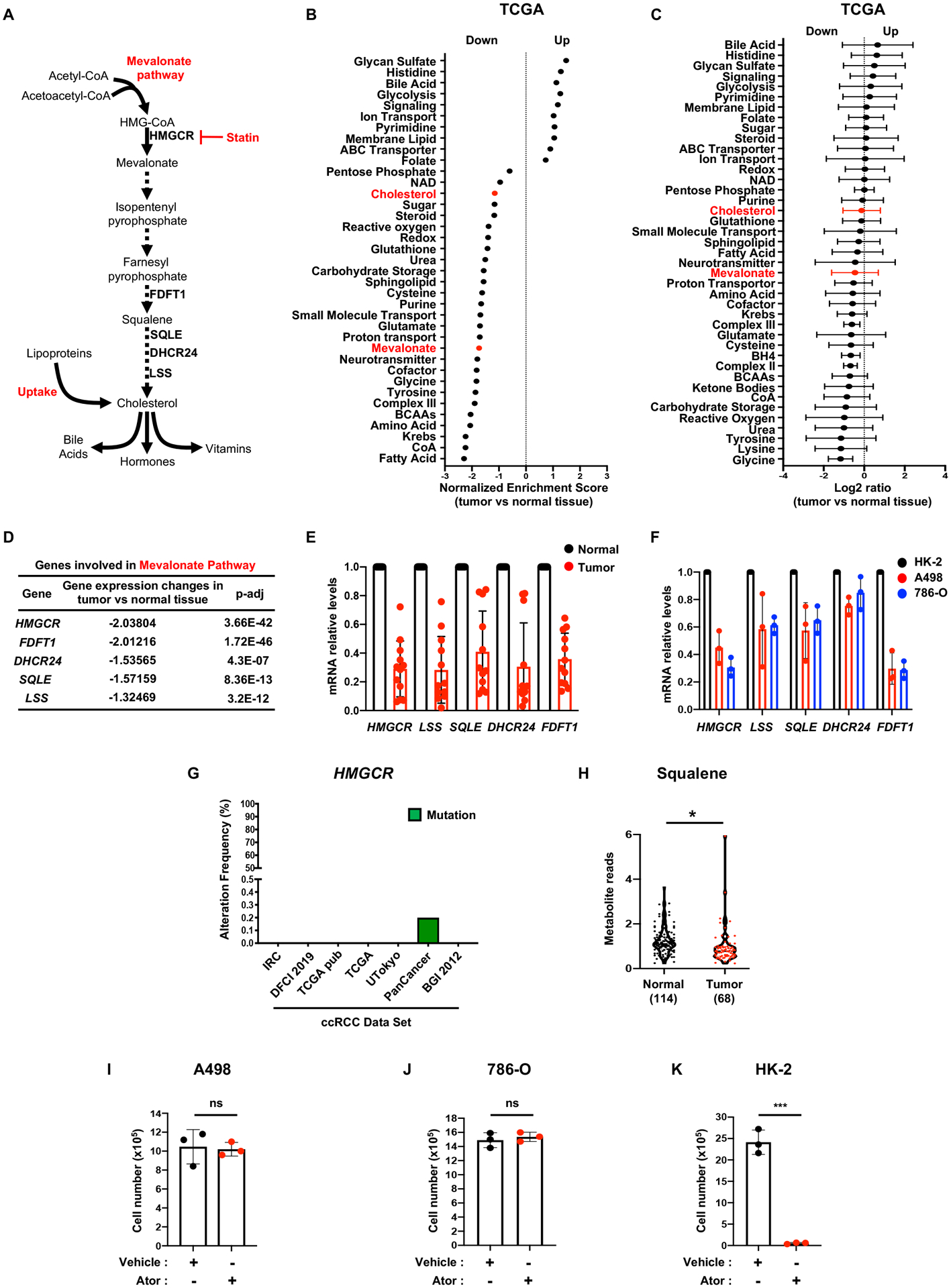
A, Simplified schematic of the mevalonate pathway. Mammalian cells maintain cholesterol homeostasis through direct synthesis, which can be inhibited by statins, or import from the extracellular environment.
B, Gene set enrichment analysis (GSEA) of RNAseq data provided by the TCGA KIRC project indicating that genes belonging to the “cholesterol” and “mevalonate” pathways have lower expression in ccRCC tumors compared to normal kidney tissue. Generated metabolic gene sets were ranked based on normalized enrichment score changes in ccRCC compared to normal tissue.
C, Metabolic gene set analysis of RNAseq data provided by the TCGA KIRC project. 538 ccRCC tumor and 72 adjacent normal tissues were included. 2,752 genes encoding all human metabolic enzymes and transporters were classified according to KEGG. Generated metabolic gene sets were ranked based on their log2 median fold expression changes in ccRCC compared to normal tissue.
D, TCGA dataset analysis shows that expression of genes involved in the “mevalonate pathway” is significantly downregulated in ccRCC tumors vs. normal tissue. HMGCR, 3-hydroxy-3-methylglutaryl-coenzyme A reductase; FDFT1, squalene synthase; DHCR24, delta 24-sterol reductase; SQLE, squalene monooxygenase; LSS, lanosterol synthase.
E, Real-time qPCR analysis performed on 12 tumor tissues and their normal counterparts, indicating mevalonate pathway gene expression is decreased in tumors compared to normal tissues.
F, Real-time qPCR analysis performed on immortalized proximal tubular renal epithelial cells (HK-2) and two ccRCC cell lines, A498 and 786-O, evaluating expression of HMGCR, LSS, SQLE, DHCR24 and FDFT1.
G, Alteration frequency of HMGCR gene in several kidney cancer genomic datasets using cBio Cancer genomic portal. IRC, Nat Genet 2012; DFCI, Science 2019; TCGA pub, firehose legacy; TCGA, Nature 2013; Utokyo, Nat Genet 2013, TCGA PanCancer Atlas; BGI, Nat Genet 2012.
H, Metabolomics analysis of squalene in 114 normal kidney tissues and 68 ccRCC tumors.
I, J and K, A498, 786-O and HK-2 cell proliferation assays showing insensitivity of ccRCC cell lines to 72h of atorvastatin (ATOR) treatment (5μM). (All experiments were performed in at least triplicates and statistical analysis was applied with *=P<0.05, **=P<0.01, ***=<0.001, n.s=non-significant).
Structural alterations in mevalonate pathway genes analyzed were found only rarely in multiple TCGA ccRCC patient data sets (cBioPortal), in contrast to VHL mutations (Figure S1D) observed in a majority of cases (Figure 1G and S1E–H). Interestingly, anaplastic large cell lymphoma (ALCL) cells with SQLE mutations accumulate squalene, which protects these cells from ferroptotic cell death (15). In contrast, pan-metabolomic analyses performed on 68 primary human ccRCC tumors and 114 normal kidney tissues from two independent cohorts (6, 16) revealed that squalene levels were underrepresented relative to corresponding normal kidney tissues (Figure 1H and S1I, J). We reasoned that because ccRCC cells are not actively synthesizing cholesterol, they should be relatively resistant to treatment with statins, which inhibit HMGCR, catalyzing the first committed step in cholesterol biosynthesis. Indeed, atorvastatin had no significant effect on ccRCC cell lines (Figure 1I, J and S1K, L), whereas it dramatically impaired HK-2 cell proliferation (Figure 1K and S1M). Taken together, these data suggest that intracellular cholesterol metabolism is deregulated in ccRCC tumor cells.
ccRCC cells depend on exogenous cholesterol levels for growth
Given that ccRCC cells do not engage in cholesterol biosynthesis, we anticipated that they depend on exogenous cholesterol and cholesterol esters imported from the microenvironment potentially in the form of low-density and high-density lipoproteins (LDLs and HDLs). To test this hypothesis, ccRCC cells (A498 and 786-O) were cultured in medium with 10% delipidated serum (DLPS), resulting in proliferation defects and increased cell death relative to cells grown in 10% fetal bovine serum (FBS) (Figure 2A–C and S2A–C). Supplementing DLPS media with exogenous cholesterol fully rescued ccRCC cell proliferation and viability (Figure 2A–C and S2A–C). Lipidomic profiles revealed that cholesterol ester (CEs) (Figure 2D) and total cellular cholesterol levels were reduced (Figure 2E and S2D) in ccRCC cells cultured in DLPS, and were restored by adding exogenous cholesterol. In contrast, viability of non-transformed human kidney cells (HK-2), which rely primarily on de novo cholesterol synthesis, was unaffected by DLPS media (Figure 2F and S2E, F). To investigate if cholesterol dependence is relevant for other kidney cancers, we evaluated Caki-2 cells which present characteristics of papillary RCC and confirmed that these are more resistant to DLPS conditions than ccRCC cell lines, at least up to day 4 (Figure S2E, G). These results indicate that ccRCC cells, and not normal kidney epithelial cells, are strikingly dependent on exogenous cholesterol for proliferation and survival, identifying cholesterol homeostasis as a metabolic vulnerability in ccRCC.
Figure 2. ccRCC cells rely on exogenous cholesterol.
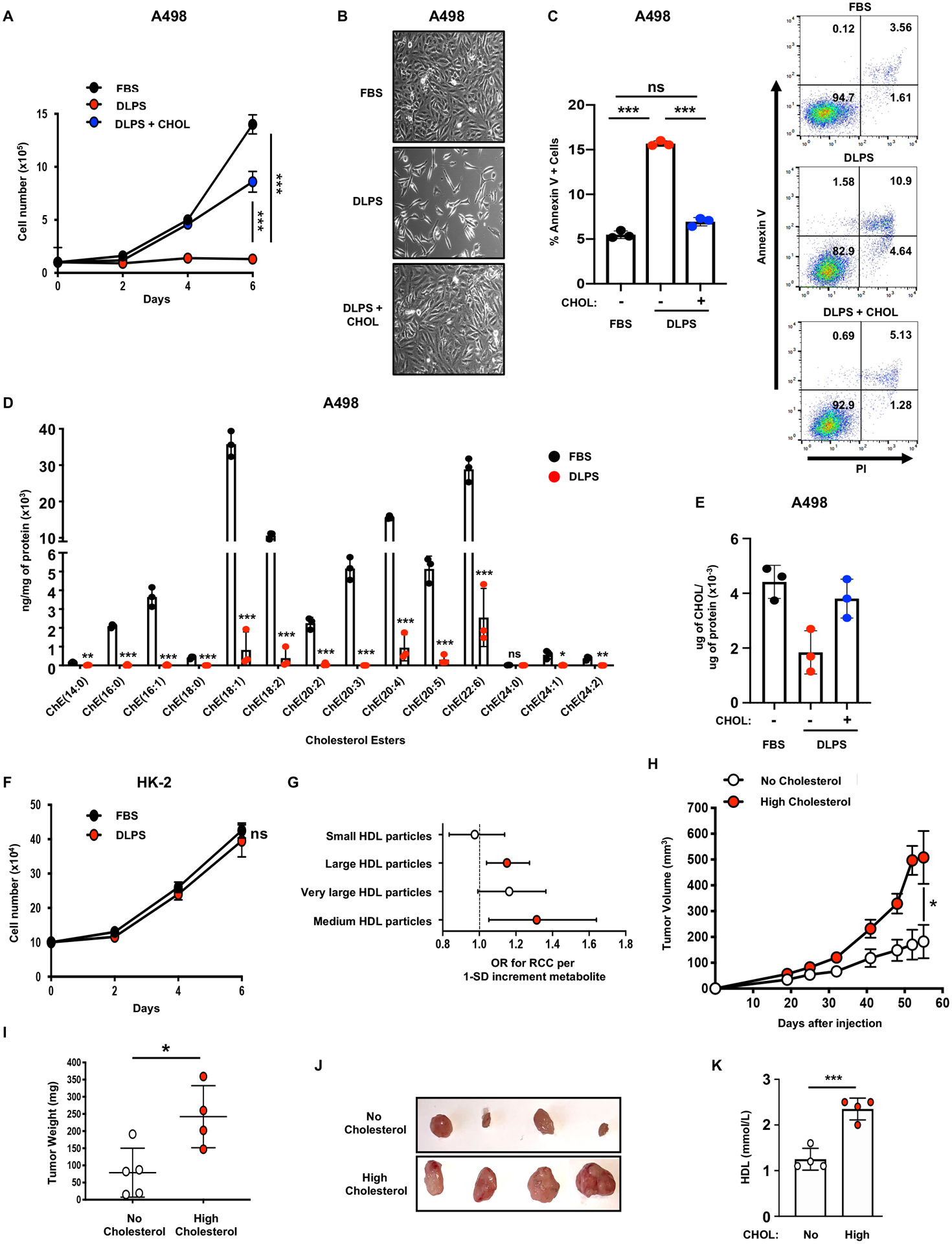
A, Proliferation assay performed on A498 cells grown in media supplemented with 10% FBS, 10% DLPS, or 10% DLPS and cholesterol (CHOL) (10μg/mL).
B, Representative photographs of A498 cells grown in media supplemented with 10% FBS, 10% DLPS, or 10% DLPS and cholesterol (CHOL) (10μg/mL) for 96h. Magnification (x100).
C, Annexin-V/PI staining and flow cytometry analysis performed on A498 cells after 96h of incubation in 10% FBS, 10% DLPS, or 10% DLPS and cholesterol (CHOL) (10μg/mL) media (left). Representative annexin-V/PI flow plots of A498 cells after 96h of incubation in 10% FBS, 10% DLPS, or 10% DLPS and cholesterol (CHOL) (10μg/mL) media (right).
D, Liquid chromatography-tandem mass spectrometry (LC/MS) analysis assessing various cholesterol ester species in A498 cells grown in 10% FBS or 10% DLPS media.
E, Cholesterol content of A498 cells grown in 10% FBS or 10% DLPS.
F, Proliferation assay performed on HK2 cells grown in media supplemented with 10% FBS, 10% DLPS, or 10% DLPS and cholesterol (CHOL) (10μg/mL).
G, Mendelian Randomization analysis using GWAS summary statistics was performed and the effect of circulating metabolites on RCC odds estimated, revealing a significant association between HDL particles and RCC risk. Estimates reflect the OR (95% CI) for RCC per SD increase in circulating metabolite concentration. (red = significant)
H, Tumor growth curves from A498 cells subcutaneously implanted in nude mice fed a no cholesterol (0%) or a high cholesterol (2%) diet. Tumor volume was assessed at the indicated timepoints using caliper measurements (n=5 mice per group, 2 tumors per mouse).
I, Tumor weight from A498 cells subcutaneously implanted in nude mice, fed a no cholesterol (0%) or a high cholesterol (2%) diet, 55 days after implantation.
J, Representative photographs of A498 tumors grown in mice fed a no cholesterol (0%) or a high cholesterol (2%) diet at day 55 after implantation.
K, Analysis of serum HDL from nude mice subcutaneously implanted with A498 cells and fed a no cholesterol (0%) or a high cholesterol (2%) diet for 70 days.
(All experiments were performed in at least triplicates and statistical analysis was applied with *=P<0.05, **=P<0.01, ***=<0.001, n.s=non-significant).
To further investigate the role of circulating cholesterol in RCC risk we employed two-sample Mendelian randomization (MR) (17) using summary association statistics from a genome wide association study (GWAS) describing 10,784 RCC cases and 20,406 controls (18–20) to estimate the causal relationship between circulating metabolites and RCC. Out of 123 measured metabolites, MR analysis revealed that the odds of developing RCC were significantly higher when genetic alleles predict elevated serum levels of HDL or HDL cholesterol (e.g. odds ratio = 1.17, 95% CI = 1.05–1.31, p = 0.005 for RCC per standard deviation (SD) increment total cholesterol in HDL, Supplementary Table 1) (Figure 2G and S3A, B). SNP heterogeneity was high for many of the metabolites (P-value range: 0.005 to 0.96, Supplementary Table 1), although not unexpected due to the many underlying biological pathways that contribute to metabolite variation. Beyond the HDL metabolite subclass, only 6 metabolites were associated with RCC risk at a nominal level of significance (Supplementary Table 1). Evidence for causality in the reverse direction (i.e. the effect of RCC on serum HDL metabolites) was weak (e.g. −0.0003 SD total cholesterol in HDL, 95% CI = −0.06–0.06 per 1 log odds of RCC, Supplemental Table 2), further supporting evidence that perturbed HDL is a cause and not a consequence of RCC risk. These data highlight circulating HDL cholesterol as a key metabolite in RCC risk.
To test the hypothesis that elevated extracellular HDL and cholesterol levels can promote ccRCC tumor growth, we injected nude mice with A498 ccRCC cell lines and provided either a cholesterol-free (NC=0% cholesterol) or high cholesterol (HC=2% cholesterol) diet. Mice receiving a HC diet exhibited larger tumor volumes compared to mice receiving a NC diet (Figure 2H–J), although neither diet impacted overall mouse body weights (Figure S3C). As anticipated, circulating HDL and total cholesterol levels were significantly higher in mice fed the HC diet compared to mice fed the NC diet (Figure 2K and S3D). These data suggest that circulating cholesterol levels promote human ccRCC tumor growth and progression.
Scavenger Receptor B1 is overexpressed in ccRCC tissues and cell lines
The apparent addiction of ccRCC cells to exogenous cholesterol, coupled with reduced expression of cholesterol biosynthetic genes, suggest that cholesterol transporters could represent therapeutic targets in ccRCC. Cells import cholesterol from the extracellular environment through a variety of receptors, including the very-low-density-lipoprotein receptor (VLDL-R), low-density-lipoprotein receptor (LDL-R), and the high-density lipoprotein (HDL) transporter scavenger receptor-B1 (SCARB1). Metabolic gene set analysis revealed that SCARB1 is significantly overexpressed in human ccRCC clinical samples relative to normal human kidney (Figure 3A, B), whereas LDLR expression is repressed (Figure 3A, C), suggesting that ccRCC tumors rely primarily on SCARB1 for cholesterol import. Elevated SCARB1 expression across all ccRCC tumor stages implies that cholesterol metabolism is deregulated early in ccRCC initiation and progression (Figure 3D).
Figure 3. Scavenger receptor 1 is overexpressed in ccRCC tissues and cell lines.
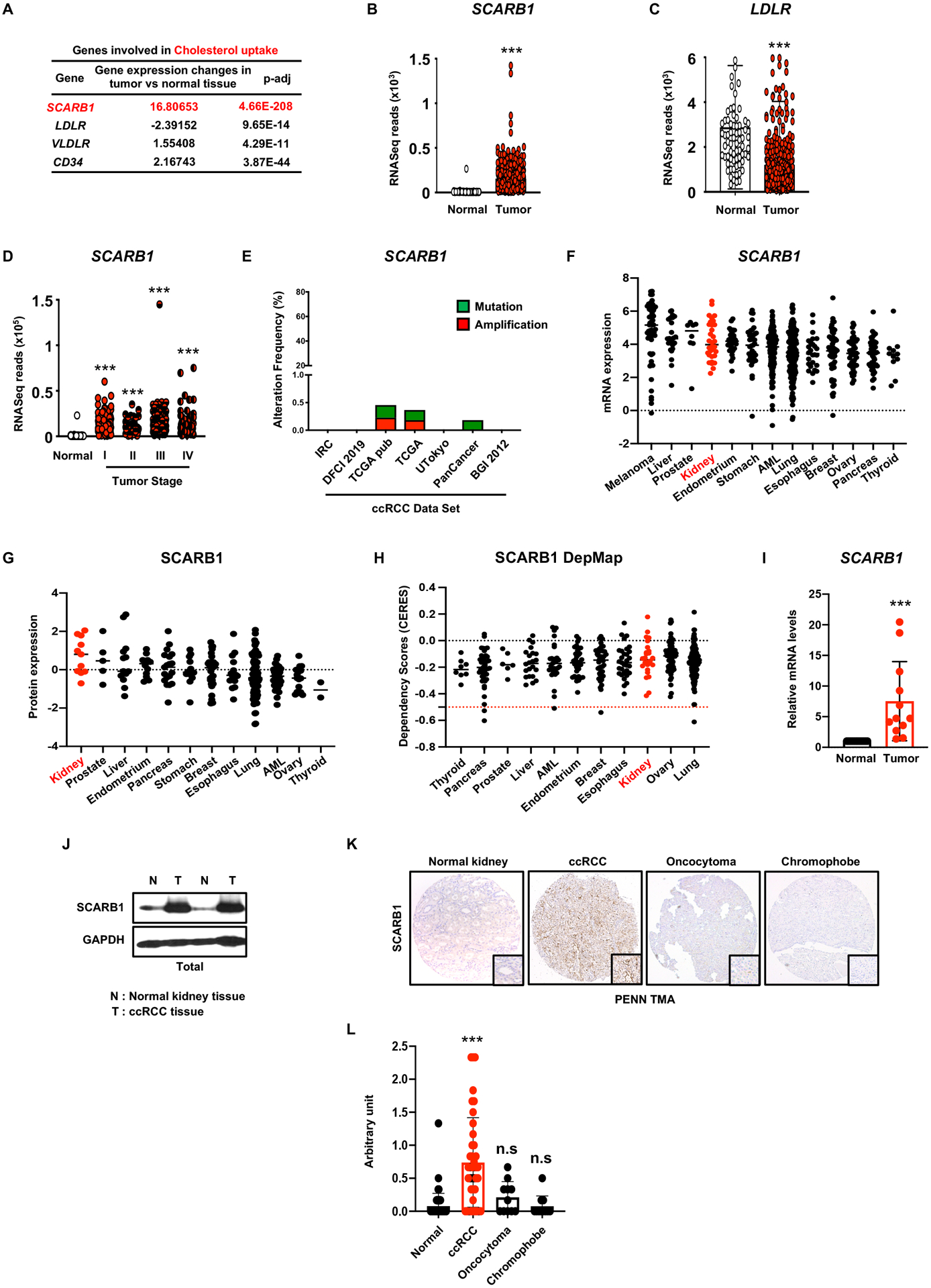
A, Expression of genes involved in “cholesterol uptake” in ccRCC tumors vs. normal tissue. SCARB1, scavenger receptor B1; LDLR, Low-density lipoproteins receptor; VLDR, very low-density lipoprotein receptor; CD36, cluster of differentiation 36.
B, Normalized RNASeq reads of SCARB1 in 72 normal kidneys and 538 ccRCC tumors (TCGA).
C, Normalized RNASeq reads of LDLR in 72 normal kidneys and 538 ccRCC tumors (TCGA).
D, Normalized RNASeq reads of SCARB1 in 72 normal kidneys and 538 ccRCC tumors grouped into stage I-IV (TCGA).
E, Alteration frequency of SCARB1 gene in several kidney cancer genomic datasets using cBio Cancer genomic portal.
F, Analysis of SCARB1 relative mRNA expression in various cancer cell lines using the Cancer Cell Line Encyclopedia.
G, Analysis of SCARB1 protein expression in various cancer cell lines using Depmap dataset.
H, Analysis of SCARB1 dependency scores across different cancer cell lines using Depmap dataset.
I, Real-time qPCR analysis performed on 12 tumor tissues and their normal counterparts. SCARB1 gene expression is highly increased in tumors compared to normal tissues.
J, SCARB1 protein expression in normal kidney tissue and ccRCC tumors assessed by immunoblots. GAPDH was used as the loading control.
K, Representative photographs of immunohistochemistry analysis of SCARB1 expression in normal kidney tissue and various RCC tumors (ccRCC, oncocytoma and chromophobe). Magnification (100X)
L, Quantification of immunohistochemistry analysis performed in J. Normal n=62, ccRCC n=40, Oncocytoma n=11, Chromophobe n=11.
(All experiments were performed in at least triplicates and statistical analysis was applied with *=P<0.05, **=P<0.01, ***=<0.001, n.s=non-significant).
Analysis of multiple TCGA data sets revealed no structural alterations in the SCARB1 gene in human ccRCCs (Figure 3E); however, previous studies identified a common single nucleotide polymorphism (SNP) within the SCARB1 locus associated with renal cancer susceptibility (12,13). Cancer cell line encyclopedia (CCLE) and Cancer dependency map (DepMap) datasets showed elevated SCARB1 mRNA and protein abundance in a variety of cell lines representing ccRCC and other cancer types (Figure 3F, G). Interestingly, no significant dependencies on SCARB1 were observed among these cancer types, suggesting that the screen was performed under different culture conditions (Figure 3H; see also discussion). SCARB1 mRNA and protein levels were also upregulated in tumors, relative to matched normal kidney tissue, in samples obtained from a cohort of 14 ccRCC patients (Figure 3I, J). Of note, human tissue microarrays (TMAs) exhibited SCARB1 accumulation in ccRCC, but not in other kidney cancer subtypes like oncocytoma, papillary and chromophobe (Figure 3K, L and S4A–C), confirming that SCARB1 expression is a particular characteristic of ccRCC and correlates to the large LDs and cholesterol ester accumulation in these cells.
Targeting SCARB1 promotes ccRCC cell death in vitro and in vivo
Previous reports demonstrated that using siRNAs to inhibit SCARB1 expression in ccRCC cells reduced cholesterol import, cell growth, migration, and AKT phosphorylation in vitro (21). To assess the effect of SCARB1 inhibition on “normal” kidney cells, we targeted SCARB1 in HK-2 and RPTEC cells. Interestingly, SCARB1 is expressed at similar levels in HK-2 cells, ccRCC cell lines and tumor tissue (Figure S4D), suggesting that adaptation to tissue culture conditions explains the sensitivity of HK-2 to SCARB1 inhibition (Figure S4E–I). In the other hand, RPTEC cells, which expresses low SCARB1 levels similarly to normal healthy tissue were completely resistant to genetic and pharmacological SCARB1 inhibition (Figure S4J–M). It should be noted that established cell lines don’t always maintain expression levels of critical proteins consistently observed in our studies of a large number of primary human ccRCC (n=831) or healthy tissue (n=188). Therefore, the RPTEC cells are more representative of what would be expected for “normal” renal cells, at least in this regard. However, the functional importance of SCARB1 in human ccRCC tumor growth and progression has not been formally addressed. We generated A498 cells expressing doxycycline (dox)-inducible SCARB1 shRNAs and confirmed that SCARB1 knockdown inhibited HDL import (Figure 4A), and blocked cell proliferation (Figure 4B–D) by inducing apoptosis (Figure 4E) and cell cycle arrest (Figure 4F) in vitro. Similar results were obtained using CRISPR-Cas9 technology (see Figure S5A–D).
Figure 4. Targeting SCARB1 promotes ccRCC cell death in vitro and in vivo.
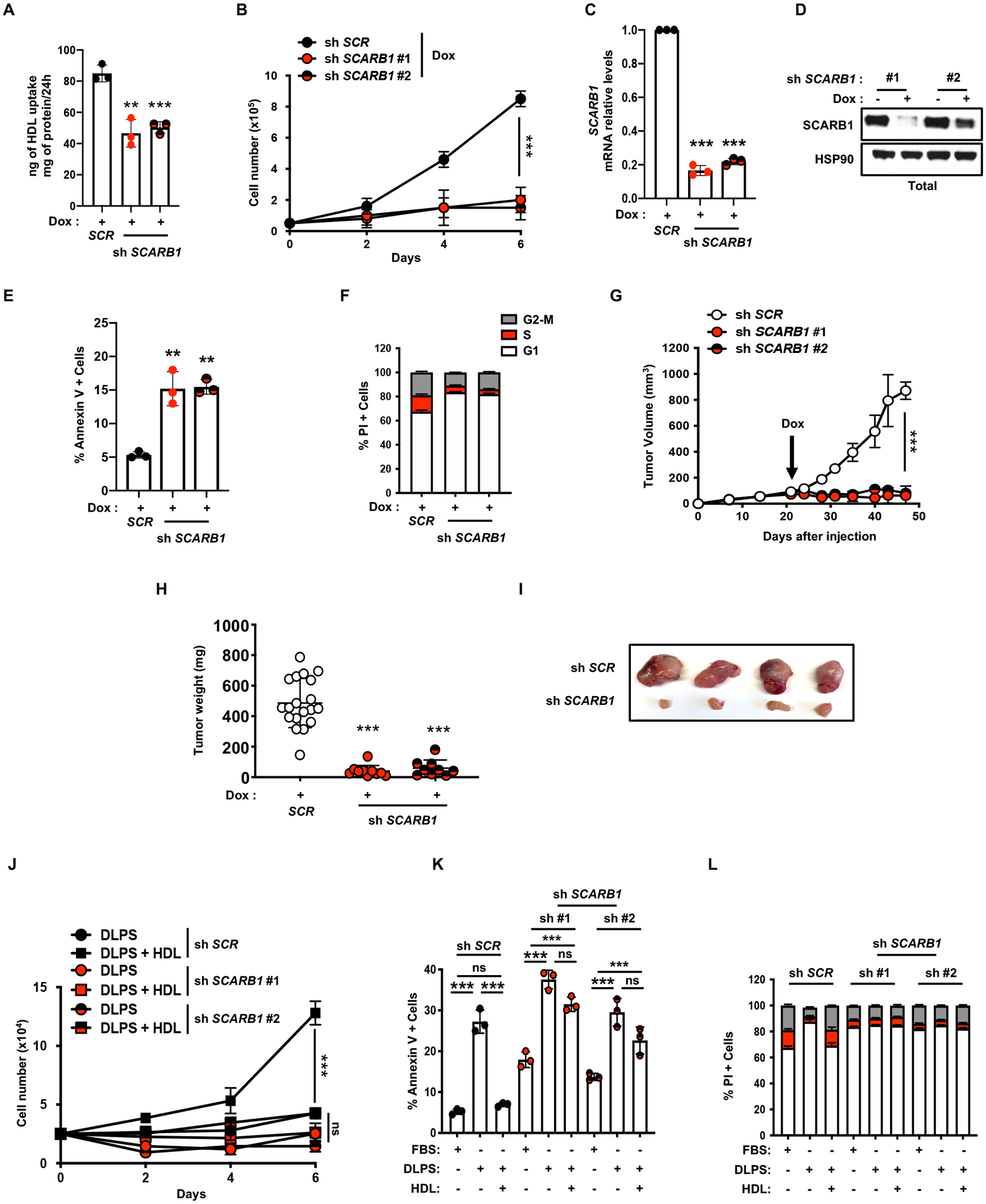
A, HDL uptake assay using shSCR and shSCARB1 A498 cells treated with doxycycline (DOX) (96h, 1μg/mL) showing reduced HDL uptake when SCARB1 is inhibited.
B, Proliferation assay performed on shSCR and shSCARB1 A498 cells grown in media with 10%FBS and supplemented with doxycycline (DOX) to induce SCARB1 knockdown.
C, Real-time qPCR analysis of SCARB1 mRNA level in A498 cells after shSCR or shSCARB1 lentiviral infection, puromycin selection (48h, 2μg/mL) and doxycycline (DOX) treatment for 4 days (1μg/mL).
D, SCARB1 protein expression assessed by immunoblots in shSCR and shSCARB1 A498 cells treated or not with doxycycline (DOX) (4 days, 1μg/mL). HSP90 was used as the loading control.
E, Annexin-V/PI staining and flow cytometry analysis performed on shSCR and shSCARB1 A498 cells after 96h of doxycycline (DOX) treatment (1μg/mL).
F, Cell cycle analysis for shSCR and shSCARB1 A498 cells after 96h of doxycycline (DOX) treatment (1μg/mL) showing increased cell cycle arrest in G1 when SCARB1 is inhibited.
G, Tumor growth curves from doxycycline-inducible shSCR and shSCARB1 A498 cells subcutaneously implanted in nude mice fed a diet containing doxycycline (DOX) (200mg/kg) when tumors reached a volume of ~100mm3. Tumor volume was assessed at the indicated timepoints using caliper measurements (n=10 mice per group).
H, Tumor weight from shSCR and shSCARB1 A498 cells subcutaneously implanted in nude mice, fed a diet containing doxycycline (DOX) (200mg/kg), 48 days after implantation.
I, Representative photographs of shSCR and shSCARB1 A498 tumors grown in nude mice fed a diet containing doxycycline (DOX) (200mg/kg), 48 days after implantation.
J, Proliferation assay performed on shSCR and shSCARB1 A498 cells grown in 10% DLPS medium with or without HDL (100μg/mL), and supplemented with doxycycline (DOX) to induce SCARB1 knockdown.
K, Annexin-V/PI staining and flow cytometry analysis performed on shSCR and shSCARB1 A498 cells treated with doxycycline (DOX) and grown in 10% FBS or 10% DLPS media supplemented with or without HDL (100μg/mL) for 4 days.
L, Cell cycle analysis for shSCR and shSCARB1 A498 cells treated with doxycycline (DOX) (1μg/mL) and grown in 10% FBS or 10% DLPS media supplemented with or without HDL (100μg/mL). Percentage of cells in G1, S and G2-M phases is displayed. (All experiments were performed in at least triplicates and statistical analysis was applied with *=P<0.05, **=P<0.01, ***=<0.001, n.s=non-significant).
To determine the uncharacterized role of SCARB1 in ccRCC tumor growth in vivo, A498 cells expressing dox-inducible SCARB1 shRNAs were used to generate palpable (100 mm3) xenograft tumors, at which time mice were fed chow containing doxycycline. shSCARB1 expression substantially reduced xenograft tumor growth, relative to controls, without affecting mouse body weight (Figure 4G–I and S5E). As validated by Western blot and immunohistochemistry, SCARB1 expression was decreased during dox treatment (Figure S5F, G) and SCARB1-depleted tumors exhibited reduced tumor proliferation and elevated cell death as indicated by Ki67 and cleaved caspase3 staining, respectively (Figure S5H, I). These data demonstrated that SCARB1 depletion compromises ccRCC tumor growth in vivo.
Whereas free cholesterol or HDLs rescued cell proliferation defects in parental A498 cells exposed to DLPS media (Figure 2A–C), HDL supplementation failed to rescue proliferation, cell death, and cell cycle arrest phenotypes in shSCARB1-expressing cells cultured in DLPS media (Figure 4J–L and S5J). Collectively, these results indicate that ccRCC cells require SCARB1 for cholesterol import, proliferation, and survival.
The SCARB1 antagonist BLT-1 impairs ccRCC cells growth in vitro and in vivo
Given the importance of SCARB1 for HDL import into ccRCC cells, we tested responses to SCARB1 antagonists (22,23), including the small molecule Block Lipid Transporter-1 (BLT-1), a potent inhibitor of SCARB1 mediated lipid transport (24). BLT-1 treated ccRCC cells exhibited proliferation defects associated with apoptosis and cell cycle arrest (Figure 5A–C and S6A–C), as well as significantly decreased intracellular cholesterol (Figure 5D and S6D). As expected, the proliferation and apoptotic phenotypes were not rescued by providing HDL to BLT-1 treated cells (Figure 5E–H and S6E–H). Treating ccRCC tumor-bearing mice with BLT-1 via oral gavage for 21 days significantly inhibited ccRCC tumor growth while increasing serum HDL levels (Figure 5I–N and S6I), indicating that ccRCC tumors rely on circulating HDL to sustain tumor growth. Surprisingly, BLT-1 treatment of ccRCC tumors only slightly lowered free cholesterol levels and actually increased cholesterol ester levels (Figure S7A, B). Cholesterol efflux through SCARB1 is a process used by cells to avoid toxic accumulation of free cholesterol. Taken together, BLT-1 is blocking both import and efflux, and HDL cholesterol uptake through SCARB1 in and of itself appears to be critical to sustain AKT signaling and control cholesterol and cholesterol ester homeostasis. Reducing HDL availability through SCARB1 inhibitors and/or dietary restriction could therefore offer a potential combination therapy for ccRCC patients.
Figure 5. The SCARB1 antagonist BLT-1 impairs ccRCC cells growth in vitro and in vivo.
A, Proliferation assay performed on A498 cells grown in media with 10%FBS and treated with the SCARB1 inhibitor, BLT-1 (5μM), or vehicle control (DMSO).
B, Annexin-V/PI staining and flow cytometry analysis performed on A498 cells after 96h of BLT-1 treatment (5μM).
C, Cell cycle analysis for A498 cells after 96h of BLT-1 treatment (5μM) showing increased cell cycle arrest in G1 when SCARB1 is inhibited.
D, HDL uptake assay using BLT-1-treated (96h, 5μM) A498 cells showing reduced HDL uptake when SCARB1 is inhibited.
E, Proliferation assay performed on A498 cells grown in media with 10%FBS or 10% DLPS media supplemented with or without HDL (100μg/mL) and treated with BLT-1 (5μM) or vehicle control (DMSO).
F, Annexin-V/PI staining and flow cytometry analysis performed on A498 cells after 96h of DMSO or BLT-1 treatment (5μM) and grown in 10% FBS or 10% DLPS media supplemented with or without HDL (100μg/mL).
G, Representative photographs of A498 cells grown in media supplemented with 10% FBS, 10% DLPS, or 10% DLPS and HDL (100μg/mL) and treated with BLT-1 (5μM) or DMSO for 96h. Magnification (100X).
H, Cell cycle analysis for A498 cells after 96h of BLT-1 (5μM) or DMSO treatment and grown in 10% FBS or 10% DLPS media supplemented with or without HDL (100μg/mL). Percentage of cells in G1, S and G2-M phases is displayed.
I, Tumor growth curves from A498 cells subcutaneously implanted in nude mice treated or not with BLT-1 (50 mg/kg) by oral gavage daily for 30 days after tumor volume reached ~100mm3. Tumor volume was assessed at the indicated timepoints using caliper measurements (n=5 mice per group).
J, Tumor weight from A498 cells subcutaneously implanted in nude mice treated or not with BLT-1 (50 mg/kg) by oral gavage daily for 30 days.
K, Representative photographs of A498 tumors grown in nude mice treated or not with BLT-1 (50 mg/kg) by oral gavage daily for 30 days.
L, Tumor volume fold change over the course of 30 day-vehicle or 30 day-BLT-1 (50 mg/kg) treatments.
M, Analysis of serum HDL from nude mice subcutaneously implanted with A498 cells and treated or not with BLT-1 (50 mg/kg) by oral gavage daily for 30 days.
N, Body weight average of mice treated by oral gavage daily for 30 days with either vehicle control or BLT-1 (50 mg/kg). (All experiments were performed in at least triplicates and statistical analysis was applied with *=P<0.05, **=P<0.01, ***=<0.001, n.s=non-significant)
Enhanced cholesterol uptake promotes cell growth through the PI3K/AKT signaling pathway
TCGA analysis revealed frequent alterations in the PI3K/AKT signaling pathway (Figure 6A) in human ccRCC, consistent with its designation as a metabolic disease (25). We hypothesized that our observed cholesterol-dependent proliferation phenotype was at least partially due to an impaired PI3K/AKT signaling axis in ccRCC. AKT phosphorylation was decreased in ccRCC cells grown in DLPS and restored by supplementation with HDL (Figure 6B, C). We further observed that SCARB1-deficient ccRCC cells also showed reduced AKT phosphorylation (Figure 6D), consistent with a previous report (21). Moreover, providing HDL to BLT-1 treated ccRCC cells did not fully restore AKT phosphorylation (Figure 6E). Treatment with PI3K and AKT inhibitors (LY-294002 and GSK-690693, respectively) also led to ccRCC proliferation defects that were not rescued by HDL (Figure 6F–H), confirming the importance of the PI3K signaling downstream of SCARB1 to sustain ccRCC proliferation. In macrophages and endothelial cells, the PDZ domain containing 1 (PDZK1) protein has been shown to link SCARB1, HDL import, and AKT activation (26,27); however, initial analysis indicates that AKT activation appears to be insensitive to PDZK1 inhibition in ccRCC cells (Figure S7C). Therefore, how SCARB1 is connected to AKT signaling will required more investigation in the future.
Figure 6. Enhanced cholesterol uptake promotes cell growth through the PI3K/AKT signaling pathway.
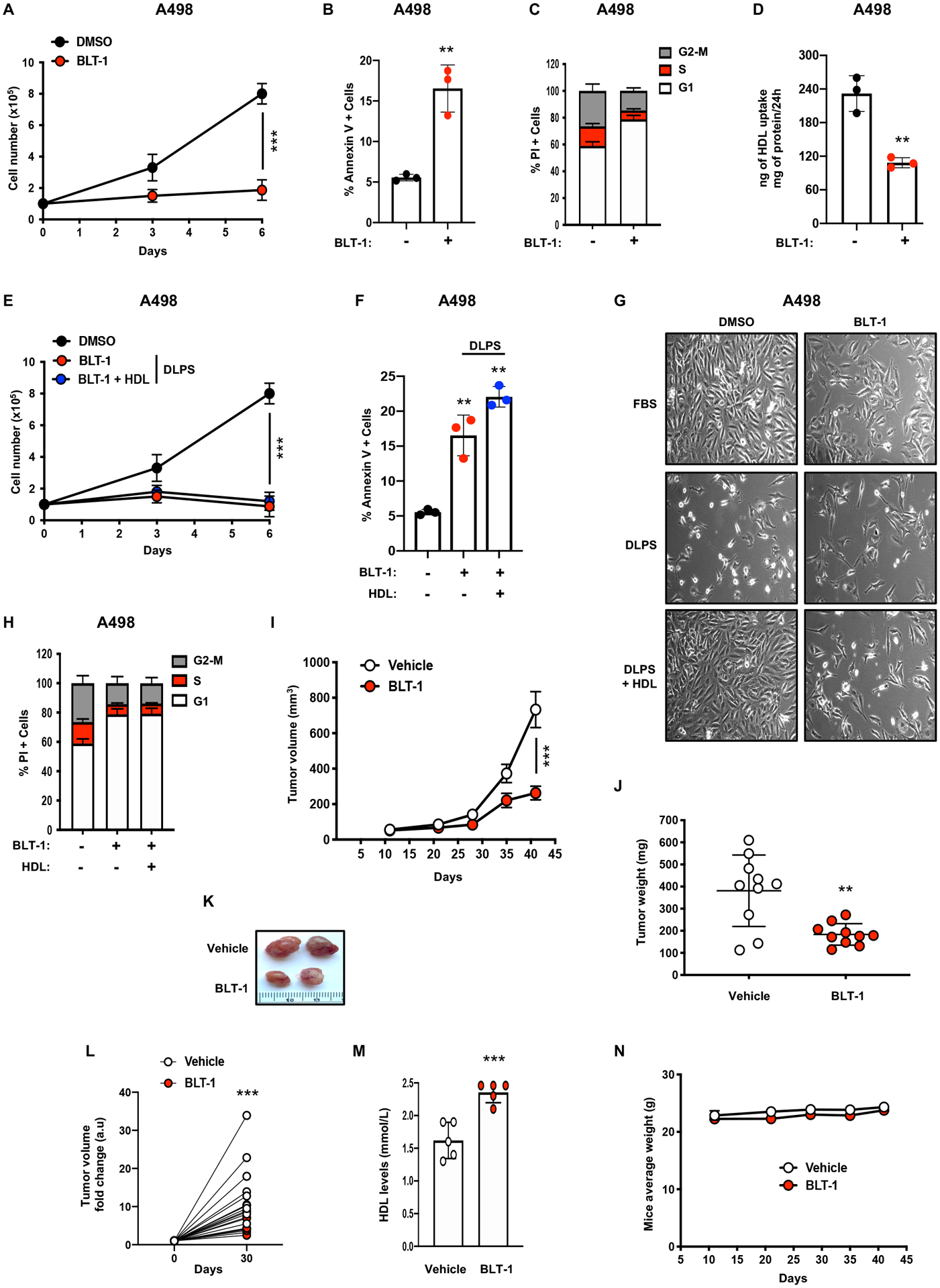
A, Simplified schematic representing interactions between the PI3K/AKT pathway, lipid rafts and cell proliferation. Edelfosine is a synthetic alkyl-lysophospholipid accumulating in membrane lipid rafts and disrupting their compositions and functions, LY294002 is a flavonoid derivative and potent PI3K inhibitor, GSK690693 is an ATP competitive, potent pan-AKT inhibitor.
B and C, AKT phosphorylation and AKT protein expression assessed by immunoblots in A498 and 786-O cells cultured for 4 days in 10% FBS or 10% DLPS media supplemented with or without HDL (100μg/mL). GAPDH was used as the loading control.
D, AKT phosphorylation and AKT protein expression assessed by immunoblots in A498 control shRNA and SCARB1 knockdown cells treated with doxycycline. GAPDH was used as the loading control.
E, AKT phosphorylation, AKT and SCARB1 protein expression assessed by immunoblots in A498 cells treated with the SCARB1 inhibitor, BLT-1, for 4 days in 10% FBS or 10% DLPS medium supplemented with or without HDL (100μg/mL). GAPDH was used as the loading control.
F, Representative photographs of A498 cells grown in media supplemented with 10% FBS and treated with LY-294002 (10μM) or GSK690693 (10 μM) for 4 days. Magnification (x100).
G, Representative photographs of A498 cells grown in 10% FBS or 10% DLPS media supplemented or not with HDL (100μg/mL) and treated with LY-294002 (10μM) for 4 days. Magnification (x100).
H, Representative photographs of 786-O cells grown in media supplemented with 10% FBS and treated with LY-294002 (10μM) or GSK690693 (10 μM) for 4 days. Magnification (x100).
I and J, Representative photographs of A498 and 786-O cells grown in media supplemented with 10% FBS and treated with Edelfosine (5μM) for 4 days. Magnification (x100).
K and L, AKT phosphorylation and AKT protein expression assessed by immunoblots in A498 and 786-O cells cultured for 4 days in 10% FBS medium and treated with Edelfosine (5μM). GAPDH was used as the loading control.
M and N, Annexin-V/PI staining and flow cytometry analysis performed on A498 and 786-O cells after 24h of Edelfosine treatment (5μM).
O, AKT, PDK1, SCARB1 protein expression assessed by immunoblots in A498 cells cultured for 4 days in 10% FBS or 10% DLPS media supplemented with or without HDL (100μg/mL). Cytosolic and membrane fractionation indicating that DLPS conditions decrease PDK1 membrane localization relative to the cytosol, which is rescued by HDL addition. GAPDH was used as the loading control. (All experiments were performed in at least triplicates and statistical analysis was applied with *=P<0.05, **=P<0.01, ***=<0.001, n.s = non-significant)
Cholesterol is a critical component of biological membranes and an important constituent of lipid rafts (LRs). LRs serve as critical membrane platforms to amplify signaling cascades, including the PI3K/AKT pathway (28,29). Moreover, SCARB1 clusters have been found in caveolin-enriched membrane domains that may be required for HDL binding and lipid uptake (30). To investigate whether cholesterol regulates LR-mediated PI3K/AKT signaling pathways, ccRCC cell lines were treated with edelfosine, an alkyl-lysophospholipid cholesterol analogue that accumulates in LRs and alters cellular signaling (31–33). Edelfosine treatment decreased AKT phosphorylation and cell growth in vitro accompanied by increased apoptosis (Figure 6I–N). Elevated levels of PDK1, a kinase that activates AKT and localizes to LR-like domains, were detected in cytosolic versus membrane fractions from cells cultured in 10% DLPS compared to 10% FBS (Figure 6O), and increased membrane fractionation was observed upon HDL supplementation. Collectively, our findings indicate that cholesterol import through SCARB1 might contribute to lipid raft homeostasis and particularly PI3K/AKT pathway activation.
Enhanced cholesterol uptake contributes to ROS homeostasis
Squalene and other cholesterol pathway intermediates can protect cells from excess reactive oxygen species (ROS) mediated oxidative stress (15) that also promotes development and progression of multiple cancer types (34,35). Previous analyses indicated that ccRCC cells express elevated levels of antioxidant proteins (36), suggesting that regulating ROS levels is critical for maintaining cellular redox homeostasis. Cholesterol depletion may therefore produce toxic levels of intracellular ROS in ccRCC cells.
2’,7’-dichlorodihydrofluorescein diacetate (DCFDA) fluorescence analysis was used to evaluate ROS levels in ccRCC cells. As predicted, lipid-deprived cells exhibited significantly higher ROS levels compared to cells in complete media (Figure 7A). Supplementing DLPS medium with HDL returned intracellular ROS levels to baseline (Figure 7B), as did treatment with the well-established reducing agents α-tocopherol or N-acetyl-cysteine (NAC) (Figure 7C and S7D). Of note, NAC was unable to rescue cell proliferation mediated by DLPS media, explained by the inability to rescue AKT phosphorylation (Figure S7E, F). Depleting SCARB1 in ccRCC cells grown in replete medium was sufficient to elevate ROS to similar levels as control ccRCC cells grown in DLPS media (Figure 7D, E). Interestingly, whereas NAC and α-tocopherol rescued ROS levels in SCARB1-deficient cells (Figure S7G, H), HDL did not (Figure 7F), confirming that ROS homeostasis in ccRCC cells depends on cholesterol availability and import mediated by SCARB1.
Figure 7. Enhanced cholesterol uptake promotes cell growth by maintaining ROS homeostasis.
A, ROS levels assessed by flow cytometry measuring DCFDA fluorescence in A498 cells cultured in 10% FBS or 10% DLPS media for 72h. Representative plots (left) and mean fluorescence intensity quantifications are shown (right).
B, ROS levels assessed by flow cytometry measuring DCFDA fluorescence in A498 cells cultured in 10% FBS or 10% DLPS media supplemented with or without HDL (100μg/mL) for 72h. Representative plots (left) and mean fluorescence intensity quantifications are shown (right).
C, ROS levels assessed by flow cytometry measuring DCFDA fluorescence in A498 cells cultured in 10% FBS or 10% DLPS media supplemented with or without α-tocopherol (0.5mM) for 72h. Representative plots (left) and mean fluorescence intensity quantifications are shown (right).
D and E, ROS levels assessed by flow cytometry measuring DCFDA fluorescence in shSCR and shSCARB1 A498 cells treated with doxycycline (DOX) (1μg/mL) and grown in 10% FBS or 10% DLPS media for 72h.
F, ROS levels assessed by flow cytometry measuring DCFDA fluorescence in shSCR and shSCARB1 A498 cells treated with doxycycline (DOX) (1μg/mL) and grown in 10% FBS or 10% DLPS media supplemented with or without HDL (100μg/mL) for 72h.
G, Schematic representing SCARB1, the HDL receptor, as a central receptor in ccRCC cells for cholesterol import compensated by diminished biosynthetic mevalonate pathway. High intracellular cholesterol levels allow ccRCC cells to maintain PI3K/AKT pathway activation, control ROS homeostasis and store cholesterol surplus in lipid droplets (CE: Cholesterol ester, TG: Triglycerides).
(All experiments were performed in at least triplicates and statistical analysis was applied with *=P<0.05, **=P<0.01, ***=<0.001, n.s=non-significant).
Collectively, our findings reveal that ccRCC cells require the import of exogenous cholesterol through SCARB1 to maintain viability, proliferation, and PI3K/AKT signaling, and to prevent ROS mediated damage (Figure 7G). Our data also demonstrate that elevated HDL levels are associated with increased risk of ccRCC and limiting cholesterol availability, or inhibiting SCARB1 genetically or pharmacologically, reduces the growth of extant ccRCC tumors in murine models. These results suggest new therapeutic strategies that, either singly or in combination, could benefit ccRCC patients.
DISCUSSION
The clear cell phenotype observed in ccRCC is associated with intracellular accumulation of neutral lipids, including triglycerides and esterified sterols. In particular, ccRCCs exhibit 8-fold and 35-fold higher levels of cholesterol and cholesterol esters, respectively, compared to normal kidney tissue (11). In mammals, cholesterol is considered a non-essential metabolite, the majority of which is synthesized de novo from acetyl-coenzyme A (Figure 1A). Gene expression, genomic and metabolomic data reveal that ccRCC cells exhibit a global suppression of the cholesterol biosynthetic pathway not associated with mutations in mevalonate pathway genes. Of note, the biosynthetic pathway is not suppressed via the VHL/HIF system, based on careful evaluation of RNA, proteomic, and lipidomic publicly available datasets for ccRCC cells and tumors with HIF-2α inhibited pharmacologically or genetically (37–41). Using functional in vitro and in vivo studies, we demonstrate that ccRCC cells behave as cholesterol auxotrophs. Future studies will explore the mechanistic basis of the loss of cholesterol biosynthesis in ccRCC. Additional cancers are also dependent on exogenous cholesterol, including breast cancers and glioblastomas (42,43), although a SCARB1 dependency was not revealed by DepMap in Figure 3H. We are unsure why DepMap is not reflecting our findings other than their culture conditions may differ from ours. Interestingly, prostate tumors appear to rely more on intracellular cholesterol than on exogenous uptake (44), suggesting that extracellular cholesterol dependency varies among cancer types. More recently, Garcia-Bermudez et al. reported that ALK+ anaplastic large cell lymphomas (ALCLs) are auxotrophs for cholesterol, due to a deficiency in squalene monooxygenase that results in accumulation of the anti-oxidant squalene (15). In contrast, squalene and other pathway intermediates are less abundant in ccRCC compared to healthy kidney tissue, consistent with general suppression of the mevalonate biosynthetic pathway (45).
We show here that ccRCC cells require extracellular cholesterol to proliferate and survive. Providing ccRCC tumor-bearing mice with a high-cholesterol diet, which increased serum HDL levels as expected, was sufficient to increase xenograft tumor growth. This was surprising, as low HDL levels have been associated previously with increased risk of various cancer types including myeloma, lymphoma, breast, lung, and brain (46). As HDL has reported antioxidant, anti-inflammatory, anti-thrombotic, anti-apoptotic, and vascular protective effects (47), we hypothesized that HDL fulfills one or more cytoprotective functions for ccRCC cells. Our preclinical results mesh with human data describing 31,190 individuals analyzed by two-sample Mendelian randomization, which demonstrated that genetic variants associated with elevated levels of circulating HDL are also positively associated with increased RCC risk. In contrast, genetic predisposition to RCC does not cause increased HDL levels, indicating that the causal pathway is clearly from HDL to RCC. High serum HDL levels may promote chronic kidney disease (CKD) by damaging renal tubular epithelial cells, although CKD has not been firmly established as a risk factor for renal cancer.
Our data reveal that the HDL receptor SCARB1 is essential for ccRCC cell proliferation and survival and represents a novel therapeutic target. SCARB1 is typically expressed in liver and adrenal tissues, where it contributes to the reciprocal loading and unloading of HDL particles by forming plasma membrane channels. Previous genome-wide association studies (GWAS) identified an association between a SCARB1 gene polymorphism (chr12q24.31 - rs4765623) and increased ccRCC risk (12,48), although our analysis of available TCGA gene expression data failed to uncover a significant correlation between SCARB1 transcript levels and patient prognosis or survival, in contrast to a previous report (21).
SCARB1 mRNA and protein expression are highly elevated in ccRCC tumors and kidney cancer cell lines compared to normal controls, although the molecular basis of increased SCARB1 expression remain unclear. The index RCC-associated SCARB1 polymorphism is adjacent to a functional HIF binding sequence motif, suggesting that SCARB1 may be transcriptionally regulated by the HIFs. However, re-expression of pVHL and subsequent HIF suppression, or treatment with a HIF-2α inhibitor (38,49), had no effect on SCARB1 expression in ccRCC cells or xenograft tumors, respectively (37–41). More recent work demonstrated that the chr12q24.31-rs4765623 SCARB1 polymorphism is contained within a super enhancer characterized by elevated H3K27 acetylation (50), and treatment of ccRCC cell lines with the BRD4 inhibitor JQ1 significantly decreased SCARB1 expression. In aggregate, these data support a model in which high HDL levels and elevated SCARB1 expression contribute directly to ccRCC tumorigenesis and growth, apparently independent of the pVHL tumor suppressor.
The underlying signaling events linking cholesterol metabolism to ccRCC cell proliferation and survival are likely to be complex. Previous TCGA analysis revealed that approximately 30% of ccRCC tumors harbor genetic alterations in the PI3K/AKT/mTOR pathway, consistent with an important role in the initiation and/or progression of the disease (25,51). HDL is known to activate the PI3K/AKT, Src and AMPK signaling pathways, which could reinforce oncogenic signaling (52) in ccRCC cells. We demonstrate that exposure to delipidated serum or SCARB1 inhibition dramatically reduces AKT activation in ccRCC cells, which is restored in a SCARB1-dependent manner by addition of HDL or cholesterol. Interestingly, SCARB1 has also been implicated in promoting HDL efflux (53), consistent with our data indicating that BLT-1 treatment of ccRCC tumors surprisingly increased cholesterol ester levels. Therefore, BLT-1 appears to block both import and efflux. Overall, HDL cholesterol uptake through SCARB1 sustains AKT signaling and controls cholesterol and cholesterol ester homeostasis. Cholesterol is also required for cell membrane fluidity and protein trafficking and is a central component of lipid rafts (LRs), specialized membrane microdomains that serve as organizing centers to promote signal transduction. We demonstrate that the LR-disrupting agent edelfosine suppresses ccRCC growth and induces apoptosis while inhibiting AKT signaling, suggesting that SCARB1 may interact with LR enriched plasma membrane domains and facilitate the incorporation of cholesterol to maintain PI3K/AKT pathway signaling. Further investigation is needed to fully elucidate the underlying signaling mechanisms linking SCARB1 and HDL to oncogenic signaling in ccRCC.
Our data also indicate that HDL import contributes to ROS homeostasis in ccRCC cells. Enhanced metabolic activity of cancer cells results in ROS overproduction leading to changes in many processes necessary for tumor initiation, growth, and progression (54). On the other hand, permanently elevated ROS levels have cytotoxic effects and maintaining redox homeostasis is a challenging feature for many cancer types. ccRCC tumors express high levels of antioxidant pathways, such as reduced glutathione (GSH) and glutathione peroxidases (GPXs) to prevent multiple ROS accumulation and the induction of ferroptosis (36,55). Therefore, HDL import contributes to redox balance, but other mechanisms are critical as well underscoring the importance of redox balance in this disease. Additionally, in non-cancerous liver cells, excess cholesterol is detoxified in part by conversion to bile acid, the initial step of which is hydroxylation of the cholesterol 7α-position. This hydroxylation is rate-limiting and catalyzed by the enzyme CYP7A1; however, cholesterol oxidation also occurs non-enzymatically in a ROS-dependent manner to produce oxysterols that are ultimately converted to bile acids. We found that CYP7A1 gene expression was not significantly upregulated in ccRCC tumors compared to surrounding normal tissue in the TCGA dataset. In contrast, the enzyme HSD3B7, which is required for subsequent conversion and detoxification of oxysterols, is significantly overexpressed in ccRCC tumors. These observations suggest that catabolism of cholesterol by reactivating the bile acid pathway may contribute to ROS homeostasis in ccRCC cells.
In addition to its critical role in HDL uptake, SCARB1 has very recently been reported to contribute to SARS-CoV-2/COVID-19 and HCV virus entry in cells (56,57). This feature makes SCARB1 an attractive antiviral target, leading to the development of a SCARB1 inhibitors (58) including ITX-5061, which has entered phase 1 clinical trials in HCV-infected humans (NCT01292824, NCT01560468, NCT01165359) and been well tolerated with no significant drug interactions. Future studies will be required to assess the efficacy and clinical utility of repurposing ITX-5061 to inhibit SCARB1 in ccRCC. It should also be noted that deregulated cholesterol metabolism has recently been implicated in regulating tumor-infiltrating T lymphocyte activity and tumor-associated macrophage (TAM) polarization (59,60). Elucidating how cholesterol availability impacts the tumor microenvironment, tumor-infiltrating immune cells and immune checkpoint function, could suggest new combination therapies. SCARB1 may also be a valuable therapeutic target for ccRCC patients whose tumors exhibit either inherent or acquired resistance to the FDA-approved HIF-2α inhibitor, who may also benefit from dietary HDL and cholesterol restriction.
METHODS
LEAD CONTACT AND MATERIALS AVAILIBITY
Further information and request for resources and reagents should be directed to and will be fulfilled by the lead contact, M. Celeste Simon (celeste2@pennmedicine.upenn.edu). This study did not generate new unique reagents.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Primary patient samples and gene expression data.
Matched tumor/normal samples were obtained from the Cooperative Human Tissue Network (CHTN). Tumors were homogenized in TRIZOL (see quantitative real-time PCR) or whole cell elution buffer (see western blot) and analyzed for SCARB1 mRNA and protein expression. RNA-seq data for 538 ccRCC and 72 normal kidney samples were downloaded from TCGA in April, 2020. Differential gene expression analysis of tumor and normal samples was performed using DeSeq (Bioconductor Version 2.12). TCGA mutation and copy number data were downloaded from cBioPortal for Cancer Genomics.
Mendelian randomization studies.
Mendelian randomization (MR) uses genetic variants to estimate causal associations, reducing the potential for confounding and reverse causation that limit observational studies (61). We conducted two sample MR to estimate the causal relationship between 123 circulating metabolites and RCC risk (62). Summary statistics from a genome wide association study of circulating metabolites (n=24,925) (63) and renal cell carcinoma (RCC, n=31,190) (64) were obtained for the purpose of this analysis. We identified SNPs that were independently associated (r2 < 0.001 and P < 5×10−8) with 119 of 123 metabolites detected by targeted nuclear magnetic resonance (NMR) metabolomics to instrument circulating metabolites; these included lipoprotein subclass-specific lipids, amino acids, fatty acids, inflammatory glycoproteins and others (63). For each metabolite, we estimated the unconfounded odds ratio for RCC per standard deviation increment in metabolite using the inverse variance weighted method under a random-effects model (Supplementary Table 2) (65). As a genetic variant in the HDL receptor (SCARB1) had been previously identified in an RCC GWAS (12), we wanted to confirm this was not sorely responsible for driving the causal association. To test the “exchangeability” and “exclusion restriction” assumptions (that the instrument does not share a common cause with the outcome and that any effect of the instrument on the outcome is exclusively through its potential effect on the exposure, respectively) (66), we conducted a sensitivity analysis excluding a SCARB1 variant contained in a number of the metabolite instruments (Supplementary Table 3). F-statistics for genetic instruments indicated sufficient strength to satisfy the ‘relevance’ assumption (Supplementary Tables 1–2) (67). Cochrane’s Q-statistics was used to assess heterogeneity of SNP effects (smaller P-values were interpreted to indicate higher heterogeneity, suggesting directional pleitropy, Supplementary Tables 1–3) (68). All analyses were performed in R version 3.6.1 (R Core Team (2019). R: A language and environment for statistical computing) using the TwoSampleMR package (69) and (70).
Mice.
Experiments were approved by the Institutional Animal Care and Use Committee at the University of Pennsylvania (Philadelphia, PA). NIH-III nude mice (Charles River Laboratories, 4–6 weeks old) were injected in each flank with 2×106 A498, or A498 shcontrol (shSCR) or A498 shSCARB1 cells. Prior to injection, cells were grown in complete media (DMEM containing 10% FBS) in 15-cm dishes. Cells were then resuspended in ice-cold PBS and were mixed 1:1 with Matrigel (BD Biosciences, cat. 356234) in a final volume of 200μL per injection. For the experiments using a no cholesterol diet (Teklad Global 2018C) or high cholesterol diet (Teklad Custom Diet TD.01383 2018 + 2% Cholesterol), mice were fed the appropriate diet for 2 weeks before cell injection. Tumor volumes were recorded at the indicated timepoints using caliper measurements, calculated by the formula V=(π/6)(L)(W2), where L was the longer measurement and W was the shorter measurement. For the mice bearing xenografts and receiving vehicle 2-hydroxypropyl-β-cyclodextrin (20%) or BLT-1 (50mg/kg) by oral gavage, tumor growth was closely monitored and when tumor volume reached 100mm3, mice were randomized and divided into groups showing similar tumor volumes. At the end of the experiment, mice were sacrificed by CO2 inhalation, and tumors were dissected for further analyses. Total cholesterol and HDL levels were assessed using a Lipidocare Analyzer following manufacturer’s instructions.
Cell Culture.
Human ccRCC cell lines (786-O, A498), Caki-2, hTERT immortalized RPTEC and 293T cells were obtained from the American Type Culture Collection (ATCC) and were cultured in DMEM (ThermoFisher Scientific, cat. 11965092) supplemented with 10% FBS (Gemini Bio-Products, cat. 900–108) or 10% delipidated FBS, referenced as DLPS, (Gemini Bio-Products, cat. 900–123) supplemented with either cholesterol (10μg/ml) or HDL (100μg/ml). ATCC carries out routine authentication via short tandem repeat (STR) analysis. Immortalized proximal tubular renal epithelial cells (HK2) were obtained from ATCC and cultured in Keratinocyte Serum Free Media with appropriate supplements (ThermoFisher Scientific, cat. 17005042). Cells were routinely tested to for mycoplasma (MycoAlert) and experiments performed with early passages.
Constructs and Viral Transduction.
Human single-guide RNAs (sgRNA) targeting SCARB1 #1 (ctccgtggatctctccgtaa) and control gRNA targeting mouse Rosa26 locus (aagatgggcgggagtcttct) were cloned into LentiCRISPRv2 plasmid. Mature antisense human SCARB1 shRNA #1 sequence (clone ID: TRCN0000001673) along with scrambled (SCR) control were cloned into a doxycycline-inducible pLKO lentiviral plasmid (AddGene, cat. 21915). Lentivirus was prepared by co-transfection of 293T cells with shRNA or CRISPR plasmid of interest along with packaging plasmids pMD2.G (AddGene, cat. 12259), psPAX2 (AddGene, cat. 12260) and Lipofectamine PLUS and LTX transfection reagent (Invitrogen, cat. 15338). Lentivirus-containing media was collected from plates at 48h post-transfection, filtered using a 0.45μm filter, and stored at −80°C. For viral transduction, cells were incubated with lentivirus-containing medium and 8 μg/mL polybrene for 6h. Cells were allowed to recover for another 48h before selection with puromycin. All experiments were performed with cells that survived puromycin selection and displayed knockdown/knockout of SCARB1 as assayed by western blot.
Immunohistochemistry.
Xenograft tumors were dehydrated, embedded in paraffin, and sectioned for staining. Immunohistochemistry was performed as previously described (71) using 1:200 SCARB1 (Abcam, cat. Ab52629), 1:100 Ki67 (Abcam, cat. Ab15580), and 1:400 Cleaved Caspase-3 (Cell Signaling Technology, cat. 9661). Briefly, xenograft slides or tissue microarray slides (US Biomax KD20812 or Penn TMA) were deparaffinized by baking slides at 60°C for 30min. The slides were rehydrated in series of ethanol solutions and endogenous peroxidase activities were quenched by 1% H2O2 in distilled water for 20 min. After three washes in TT buffer (500mM NaCl, 10mM Trizma, and 0.05% Tween-20), antigen retrieval was performed by boiling slides for 20 min in a citrate-based antigen unmasking solution (Vector labs, H3300). After cooling down to room temperature, slides were blocked in 2% normal goat serum and 4% BSA in TT buffer for 1h. Next, tissue slides were incubated with various primary antibodies at 4°C overnight. After three washes in TT buffer, biotinylated secondary antibody was added onto these slides for 1h, following by 1h treatment of the Vectastain Elite ABC reagents (Vector Labs, PK-6100). After three TT washes, the slides were processed with DAB peroxidase substrate kit (Vector Labs, SK-4100), and hematoxylin solutions for immunohistochemistry staining, dehydrated in a standard ethanol/xylenes series, and mounted in 75%v/v Permount (Fischer Scientific, SP15–500) in xylenes.
Quantitative real-time PCR (qRT-PCR).
Total RNA was processed and extracted with TRIzol reagent (Thermo Fisher Scientific, catalog no. 15596026) and RNeasy Mini Kit (Qiagen, catalog no. 74104). cDNAs were synthesized using a High-Capacity RNA-to-cDNA kit (Applied Biosystems, 4368814). qRT-PCR were then performed using TaqMan Master Mix (Life Technologies) and a ViiA7 Real-Time PCR Instrument (Applied Biosystems). Pre-designed Taqman primers were obtained from Life Technologies for the following genes:
B2M (Hs0187842_m1), SQLE (Hs01123768_m1), HMGCR (Hs00168352_m1), SCARB1 (Hs00969821), LSS (Hs01552331_m1), DHCR24 (Hs00207388_m1), FDFT1 (Hs00926054_m1), 18S (Hs03928985_g1)
Western Blot Analysis.
Cells were washed with PBS and lysed using lysis buffer (150mM NaCl, 10mM Tris pH7.6, 0.1% SDS and 5mM EDTA) containing Halt Protease and Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific, 78445). For western blots of xenograft tissue, approximately 10–20mg of tissue was suspended in 500μl lysis buffer and homogenized on ice using a Tissue-Tearor (Biospec, 985370). Samples were centrifuged at 12,000rpm for 20min at 4°C. Protein lysates were resolved by SDS-PAGE and were transferred to nitrocellulose membranes (Bio-Rad, 162–0115, 0.45μm pore size for all experiments). All membranes were incubated with the indicated primary antibodies overnight at 4 °C and were diluted in TBS-T (20mM Tris pH7.5, 150mM NaCl, 0.1% Tween-20) supplemented with 5% bovine serum albumin (BSA, Sigma-Aldrich, A7906). The next day and after TBS-T washes, membranes were incubated with secondary antibody and Western Lightning Plus-ECL, Enhanced Chemiluminescence Substrate (PerkinElmer, cat. NEL103E001EA) was used to visualize proteins.
Annexin V-PI apoptosis assay.
Cell death was determined using the FITC–Annexin V, PI Kit (catalog no. 556547) from BD Biosciences according to the manufacturer’s instructions. Briefly, 20,000 cells of each cell line were plated in triplicate in 6-well plates under the indicated conditions. Four days later, cells were prepared using the FITC–Annexin V, PI Kit (BD Biosciences, cat. 556547) according to the manufacturer’s instructions. For the drug treatment, 24h after plating, cells were treated or not for 48h with either BLT-1. Flow cytometry was performed using the BD FACS Calibur instrument, with dead cells represented as Annexin V-positive population.
Cell-cycle analysis.
Cells were plated in 6-cm plates in duplicates and harvested at 80% confluency. Cells were then resuspended in 1× PBS and fixed with 70% cold ethanol, and incubated at −20°C 1h. Then, cells were washed with ice-cold PBS and resuspended in 1mL PBS. They were treated with RNAse (100μg/mL), stained with PI (20μg/mL), and then analyzed by flow cytometry using the BD FACSCalibur instrument.
Proliferation assays.
20,000 cells of each cell line were plated in triplicate on 6-well plates and supplemented with either DMEM containing 10% FBS, 10% delipidated FBS, 10% delipidated FBS and cholesterol, or 10% delipidated FBS and HDL. Every other day and as indicated on the figures, one set of cultures was collected and counted. Cells were trypsinized and counted using a cell counter (Countess II Life Technologies) with Trypan blue (Gibco, Cat: 15250–061).
ROS staining.
20,000 cells of each cell line were plated in triplicate on 6-well plates. Three days later, live cells were washed twice in PBS and incubated in DCFDA (Abcam, cat113851) for 1h at 37°C. After staining and trypsinization, cells were washed twice in PBS and resuspended in PBS, passed through a cell strainer, and flow cytometry was performed on a BD Accuri C6 instrument under FL-1.
Subcellular Fractionation.
Cytosolic and membrane fractionation of indicated cells were performed using Mem-PER Plus membrane protein extraction kit (Thermo Fisher Scientific, 89842) by following the manufacturer’s protocol.
HDL uptake.
20,000 cells of each cell line were plated in triplicate on 6-well plates. HDL-cholesterol uptake assay was performed using HDL Uptake Assay Kit (Fluorometric) (Abcam, Cat: ab204717). Assays were performed according to the manufacturer’s protocols. Briefly, ccRCC cell lines were incubated in the assay media containing fluorescently-labeled HDL for 3h, then were washed, and the fluorescence was measured in a microplate reader at Ex/Em = 540/575 nm. Unlabeled-HDL included in the kit is used for assay validation. The cell uptake was normalized by total protein amounts of each well.
Intracellular cholesterol quantification.
Cholesterol content in ccRCC cells was measured with a Cholesterol Quantitation kit (Sigma, Cat: MAK043), following manufacturer’s instructions. Briefly, samples (106 cells) were extracted with 200μl of chloroform:isopropanol:IGEPAL CA-630 (7:11:0.1). After centrifugation at 13,000g for 10min to remove insoluble material, the organic phase of samples was transferred to a new tube and air dried in an incubator at 50°C. Then organic solvent residue was removed in a SpeedVac for 1h. Dried lipids were then dissolved with 200μl of the Cholesterol Assay Buffer. Fifty microlitres of samples and standards (1–5ng) were added to 50μl of reaction mixture and absorbance at 570 nm measured after 60 min incubation at 37 °C.
Metabolomics.
Optima grade methanol (MeOH), water, acetonitrile, methyl tert-butyl ether (MTBE) and 2-propanol were from Thermo Fisher Scientific (Pittsburg, PA). Gasses were supplied by Airgas (Philadelphia, PA). Glassware and HPLC vials were from Waters Corp (Milford, MA). Lipids extraction was done according to Matyash et al (72). For the HRMS analysis, a recently calibrated QE Exactive-HF mass spectrometer (Thermo Fisher Scientific) was used in positive ion mode with a HESI source. The operating conditions were: spray voltage at 3.5kV; capillary temperature at 285°C; auxiliary temperature 370°C; tube lens 45. Nitrogen was used as the sheath gas at 45 units, the auxiliary gas at 10 units and sweep gas was 2 units. Same MS conditions were used in negative ionization mode, but with a spray voltage at 3.2kV. Control extraction blanks were made in the same way using just the solvents instead of the cell homogenate. The control blanks were used for the exclusion list with a threshold feature intensity set at 1e10^5. Untargeted analysis and targeted peak integration were conducted using LipidsSearch 4.2 (Thermo Fisher Scientific). An external mass calibration was performed using the standard calibration mixture approximately every three days. All samples were analyzed in a randomized order in full scan MS that alternated with MS2 of top 20, with HCD scans at 30, 45 or 60 eV. Full scan resolution was set to 120,000 in the scan range between m/z 250–1800. The pool sample was run every 10 samples. The coefficient of variation of less than 10% of the pooled sample was used to assess a robust signal for cholesterols esters. Calibration curves were prepared in the range 2ng to 800ng for cholesterol, and were plotted against the area ratio of the dehydration product (m/z 369.3513) to the corresponding internal standard. The top 14 abundant CEs were identified based on their MS2 data from the LipidsSearch library and quantification was done from the full scan in positive mode. The areas were normalized based on the 3500ng of CE internal standard (18:1-D7) added per sample (as found in the SPLASH® LIPIDOMIX®). All amounts were normalized to the original protein amount per plate.
Plasma cholesterol assays.
During blood sample collection, 0.5ml was collected through retro-orbital bleeding. After the blood was prepared in the heparin-coated tubes, plasma was collected by centrifugation at 2000rpm for 10min. The plasma levels of triglycerides (TG), total cholesterol (TC) and high-density lipoprotein (HDL) were examined using a plasma lipid profile kit (Standard LipidoCare from SD Biosensor) according to manufacturer’s instructions.
Statistical analysis.
Data are expressed as mean±SD. The results were analyzed by a Student’s t test, and p values less than 0.05 were considered to be statistically significant (*P<0.05, **P<0.01, and ***P<0.001).
Supplementary Material
STATEMENT OF SIGNIFICANCE.
We demonstrate that ccRCC cells are auxotrophic for exogenous cholesterol to maintain PI3K/AKT signaling pathway and ROS homeostasis. Blocking cholesterol import through the HDL transporter SCARB1 compromises ccRCC cell survival and tumor growth, suggesting a novel pharmacological target for this disease.
ACKNOLEDGEMENTS
We acknowledge the National Institutes of Health grants P01CA104838 and R35CA197602 to M.C. Simon, which support N. Skuli, and Damon Runyon postdoctoral fellowship DRG2326-18 to R. Riscal. Mendelian Randomization and analysis were performed by E.E. Vincent and C.J. Bull supported by Diabetes UK (17/0005587), the Wellcome Trust (202802/Z/16/Z), the CRUK Integrative Cancer Epidemiology Programme (C18281/A29019) and the World Cancer Research Fund (WCRF UK), as part of the World Cancer Research Fund International grant program (IIG_2019_2009). Mass spectrometry and analysis were performed by E.S. Ho, J.P. Xu and C. Mesaros supported by the National Institute of Environmental Health Sciences grant P30ES013508. We are grateful to J. Tobias for help with processing the human TCGA data and P. Lal for providing the tissue microarrays. We thank members of the Simon laboratory for helpful comments on the manuscript. Illustrations were generated using Servier medical art.
Footnotes
DECLARATION OF INTERESTS
The authors declare no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
REFERENCES
- 1.Rini BI, Campbell SC, Escudier B. Renal cell carcinoma. Lancet [Internet] 2009. [cited 2019 Oct 16];373:1119–32. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19269025 [DOI] [PubMed] [Google Scholar]
- 2.Keith B, Johnson RS, Simon MC. HIF1α and HIF2α: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer [Internet] 2011. [cited 2019 Nov 7];12:9–22. Available from: http://www.nature.com/articles/nrc3183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Linehan WM, Srinivasan R, Schmidt LS. The genetic basis of kidney cancer: A metabolic disease [Internet]. Nat. Rev. Urol 2010. [cited 2020 Apr 3]. page 277–85. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20448661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ward PS, Thompson CB. Metabolic Reprogramming: A Cancer Hallmark Even Warburg Did Not Anticipate. Cancer Cell 2012. page 297–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.DeBerardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv [Internet]. Sci Adv; 2016. [cited 2020 Dec 1];2:e1600200. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27386546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li B, Qiu B, Lee DSM, Walton ZE, Ochocki JD, Mathew LK, et al. Fructose-1,6-bisphosphatase opposes renal carcinoma progression. Nature [Internet] 2014. [cited 2019 Nov 6];513:251–5. Available from: http://www.nature.com/articles/nature13557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ochocki JD, Khare S, Hess M, Ackerman D, Qiu B, Daisak JI, et al. Arginase 2 Suppresses Renal Carcinoma Progression via Biosynthetic Cofactor Pyridoxal Phosphate Depletion and Increased Polyamine Toxicity. Cell Metab [Internet] Cell Press; 2018. [cited 2020 Apr 3];27:1263–1280.e6. Available from: http://www.ncbi.nlm.nih.gov/pubmed/29754953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Valera VA, Merino MJ. Misdiagnosis of clear cell renal cell carcinoma. Nat Rev Urol [Internet] 2011. [cited 2019 Sep 5];8:321–33. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21587224 [DOI] [PubMed] [Google Scholar]
- 9.Fujimoto T, Parton RG. Not just fat: The structure and function of the lipid droplet. Cold Spring Harb Perspect Biol [Internet] 2011. [cited 2020 Apr 3];3:1–17. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21421923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qiu B, Ackerman D, Sanchez DJ, Li B, Ochocki JD, Grazioli A, et al. HIF2α-Dependent Lipid Storage Promotes Endoplasmic Reticulum Homeostasis in Clear-Cell Renal Cell Carcinoma. Cancer Discov [Internet] American Association for Cancer Research Inc.; 2015. [cited 2020 Apr 3];5:652–67. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25829424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gebhard RL, Clayman RV, Prigge WF, Figenshau R, Staley NA, Reesey C, et al. Abnormal cholesterol metabolism in renal clear cell carcinoma. J Lipid Res [Internet] 1987. [cited 2019 Jul 15];28:1177–84. Available from: http://www.ncbi.nlm.nih.gov/pubmed/3681141 [PubMed] [Google Scholar]
- 12.Purdue MP, Johansson M, Zelenika D, Toro JR, Scelo G, Moore LE, et al. Genome-wide association study of renal cell carcinoma identifies two susceptibility loci on 2p21 and 11q13.3. Nat Genet [Internet] 2011. [cited 2019 Dec 6];43:60–5. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21131975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grampp S, Schmid V, Salama R, Lauer V, Kranz F, Platt JL, et al. Multiple renal cancer susceptibility polymorphisms modulate the HIF pathway. Linehan M, editor. PLOS Genet [Internet] 2017. [cited 2019 Sep 5];13:e1006872. Available from: https://dx.plos.org/10.1371/journal.pgen.1006872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clark DJ, Dhanasekaran SM, Petralia F, Pan J, Song X, Hu Y, et al. Integrated Proteogenomic Characterization of Clear Cell Renal Cell Carcinoma. Cell [Internet] 2019. [cited 2019 Nov 6];179:964–983.e31. Available from: http://www.ncbi.nlm.nih.gov/pubmed/31675502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garcia-Bermudez J, Baudrier L, Bayraktar EC, Shen Y, La K, Guarecuco R, et al. Squalene accumulation in cholesterol auxotrophic lymphomas prevents oxidative cell death. Nature [Internet] 2019. [cited 2019 Jul 15];567:118–22. Available from: http://www.ncbi.nlm.nih.gov/pubmed/30760928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hakimi AA, Reznik E, Lee CH, Creighton CJ, Brannon AR, Luna A, et al. An Integrated Metabolic Atlas of Clear Cell Renal Cell Carcinoma. Cancer Cell. Cell Press; 2016;29:104–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu L, Lin SL, Schooling CM. A Mendelian randomization study of the effect of calcium on coronary artery disease, myocardial infarction and their risk factors. Sci Rep [Internet] Nature Publishing Group; 2017. [cited 2020 Feb 7];7:42691. Available from: http://www.nature.com/articles/srep42691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scelo G, Purdue MP, Brown KM, Johansson M, Wang Z, Eckel-Passow JE, et al. Genome-wide association study identifies multiple risk loci for renal cell carcinoma. Nat Commun [Internet] Nature Publishing Group; 2017. [cited 2020 Feb 7];8:15724. Available from: http://www.nature.com/articles/ncomms15724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Locke AE, Kahali B, Berndt SI, Justice AE, Pers TH, Day FR, et al. Genetic studies of body mass index yield new insights for obesity biology. Nature [Internet] 2015. [cited 2020 Feb 7];518:197–206. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25673413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kettunen J, Tukiainen T, Sarin A-P, Ortega-Alonso A, Tikkanen E, Lyytikäinen L-P, et al. Genome-wide association study identifies multiple loci influencing human serum metabolite levels. Nat Genet [Internet] Nature Publishing Group; 2012. [cited 2020 Feb 7];44:269–76. Available from: http://www.nature.com/articles/ng.1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.hua Xu G, Lou N, chuan Shi H, chen Xu Y, long Ruan H, Xiao W, et al. Up-regulation of SR-BI promotes progression and serves as a prognostic biomarker in clear cell renal cell carcinoma. BMC Cancer [Internet] BioMed Central Ltd.; 2018. [cited 2020 Apr 11];18:88. Available from: http://www.ncbi.nlm.nih.gov/pubmed/29357836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nieland TJF, Penman M, Dori L, Krieger M, Kirchhausen T. Nonlinear partial differential equations and applications: Discovery of chemical inhibitors of the selective transfer of lipids mediated by the HDL receptor SR-BI. Proc Natl Acad Sci [Internet] 2002. [cited 2019 Jul 26];99:15422–7. Available from: http://www.pnas.org/cgi/doi/10.1073/pnas.222421399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rowe IA, Tully DC, Armstrong MJ, Parker R, Guo K, Barton D, et al. Effect of scavenger receptor class B type I antagonist ITX5061 in patients with hepatitis C virus infection undergoing liver transplantation. Liver Transpl [Internet] NIH Public Access; 2016. [cited 2019 Jul 26];22:287–97. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26437376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nieland TJF, Penman M, Dori L, Krieger M, Kirchhausen T. Discovery of chemical inhibitors of the selective transfer of lipids mediated by the HDL receptor SR-BI. Proc Natl Acad Sci U S A 2002;99:15422–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guo H, German P, Bai S, Barnes S, Guo W, Qi X, et al. The PI3K/AKT Pathway and Renal Cell Carcinoma. J. Genet. Genomics Institute of Genetics and Developmental Biology; 2015. page 343–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Al-Jarallah A, Chen X, González L, Trigatti BL. High density lipoprotein stimulated migration of macrophages depends on the scavenger receptor class B, type I, PDZK1 and Akt1 and is blocked by sphingosine 1 phosphate receptor antagonists. PLoS One [Internet]. PLoS One; 2014. [cited 2020 Nov 17];9:e106487. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25188469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seetharam D, Mineo C, Gormley AK, Gibson LL, Vongpatanasin W, Chambliss KL, et al. High-density lipoprotein promotes endothelial cell migration and reendothelialization via scavenger receptor-B type I. Circ Res [Internet]. Circ Res; 2006. [cited 2020 Nov 17];98:63–72. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16339487 [DOI] [PubMed] [Google Scholar]
- 28.Simons K, Toomre D. Lipid rafts and signal transduction. Nat Rev Mol Cell Biol [Internet] 2000. [cited 2019 Jul 25];1:31–9. Available from: http://www.ncbi.nlm.nih.gov/pubmed/11413487 [DOI] [PubMed] [Google Scholar]
- 29.Zhuang L, Lin J, Lu ML, Solomon KR, Freeman MR. Cholesterol-rich lipid rafts mediate akt-regulated survival in prostate cancer cells. Cancer Res [Internet] 2002. [cited 2019 Jul 25];62:2227–31. Available from: http://www.ncbi.nlm.nih.gov/pubmed/11956073 [PubMed] [Google Scholar]
- 30.Babitt J, Trigatti B, Rigotti A, Smart EJ, Anderson RGW, Xu S, et al. Murine SR-BI, a high density lipoprotein receptor that mediates selective lipid uptake, is N-glycosylated and fatty acylated and colocalizes with plasma membrane caveolae. J Biol Chem. J Biol Chem; 1997;272:13242–9. [DOI] [PubMed] [Google Scholar]
- 31.van der Luit AH, Budde M, Ruurs P, Verheij M, van Blitterswijk WJ. Alkyl-lysophospholipid Accumulates in Lipid Rafts and Induces Apoptosis via Raft-dependent Endocytosis and Inhibition of Phosphatidylcholine Synthesis. J Biol Chem [Internet] 2002. [cited 2019 Jul 25];277:39541–7. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12183451 [DOI] [PubMed] [Google Scholar]
- 32.Mollinedo F, de la Iglesia-Vicente J, Gajate C, Estella-Hermoso de Mendoza A, Villa-Pulgarin JA, Campanero MA, et al. Lipid raft-targeted therapy in multiple myeloma. Oncogene [Internet] 2010. [cited 2019 Jul 25];29:3748–57. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20418917 [DOI] [PubMed] [Google Scholar]
- 33.Gajate C, Gonzalez-Camacho F, Mollinedo F. Involvement of Raft Aggregates Enriched in Fas/CD95 Death-Inducing Signaling Complex in the Antileukemic Action of Edelfosine in Jurkat Cells. Bauer JA, editor. PLoS One [Internet] 2009. [cited 2019 Jul 25];4:e5044. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19352436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kumari S, Badana AK, G MM, G S, Malla R. Reactive Oxygen Species: A Key Constituent in Cancer Survival. Biomark Insights [Internet] SAGE Publications; 2018. [cited 2019 Sep 4];13. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5808965/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim J, Kim J, Bae J-S. ROS homeostasis and metabolism: a critical liaison for cancer therapy. Exp Mol Med [Internet]. Korean Society for Biochemistry and Molecular Biology; 2016. [cited 2019 Sep 4];48:e269. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27811934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bansal A, Sanchez DJ, Nimgaonkar V, Sanchez D, Riscal R, Skuli N, et al. Gamma-Glutamyltransferase 1 Promotes Clear Cell Renal Cell Carcinoma Initiation and Progression. Mol Cancer Res [Internet] 2019. [cited 2019 Sep 4];17:1881–92. Available from: http://www.ncbi.nlm.nih.gov/pubmed/31151999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sun N, Petiwala S, Lu C, Hutti JE, Hu M, Hu M, et al. VHL Synthetic Lethality Signatures Uncovered by Genotype-Specific CRISPR-Cas9 Screens. Cris J [Internet] Mary Ann Liebert, Inc., publishers 140 Huguenot Street, 3rd Floor New Rochelle, NY: 10801 USA; 2019. [cited 2021 Jun 1];2:230–45. Available from: https://www.liebertpub.com/doi/10.1089/crispr.2019.0018 [DOI] [PubMed] [Google Scholar]
- 38.Chen W, Hill H, Christie A, Kim MS, Holloman E, Pavia-Jimenez A, et al. Targeting renal cell carcinoma with a HIF-2 antagonist. Nature [Internet] Nature Publishing Group; 2016. [cited 2020 Dec 10];539:112–7. Available from: http://www.nature.com/articles/nature19796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Courtney KD, Ma Y, Diaz de Leon A, Christie A, Xie Z, Woolford L, et al. HIF-2 Complex Dissociation, Target Inhibition, and Acquired Resistance with PT2385, a First-in-Class HIF-2 Inhibitor, in Patients with Clear Cell Renal Cell Carcinoma. Clin Cancer Res [Internet] American Association for Cancer Research; 2020. [cited 2021 Jun 1];26:793–803. Available from: http://www.ncbi.nlm.nih.gov/pubmed/31727677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gao Y-H, Wu Z-X, Xie L-Q, Li C-X, Mao Y-Q, Duan Y-T, et al. VHL deficiency augments anthracycline sensitivity of clear cell renal cell carcinomas by down-regulating ALDH2. Nat Commun [Internet] Nature Publishing Group; 2017. [cited 2021 Jun 1];8:15337. Available from: http://www.nature.com/articles/ncomms15337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zou Y, Palte MJ, Deik AA, Li H, Eaton JK, Wang W, et al. A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nat Commun [Internet] Nature Publishing Group; 2019. [cited 2021 Jun 1];10:1617. Available from: http://www.nature.com/articles/s41467-019-09277-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gallagher EJ, Zelenko Z, Neel BA, Antoniou IM, Rajan L, Kase N, et al. Elevated tumor LDLR expression accelerates LDL cholesterol-mediated breast cancer growth in mouse models of hyperlipidemia. Oncogene [Internet] Nature Publishing Group; 2017. [cited 2021 Jun 1];36:6462–71. Available from: http://www.nature.com/articles/onc2017247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Villa GR, Hulce JJ, Zanca C, Bi J, Ikegami S, Cahill GL, et al. An LXR-Cholesterol Axis Creates a Metabolic Co-Dependency for Brain Cancers. Cancer Cell [Internet] NIH Public Access; 2016. [cited 2019 Dec 4];30:683–93. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27746144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stopsack KH, Gerke TA, Andrén O, Andersson S-O, Giovannucci EL, Mucci LA, et al. Cholesterol uptake and regulation in high-grade and lethal prostate cancers. Carcinogenesis [Internet] Oxford Academic; 2017. [cited 2021 Jun 1];38:806–11. Available from: https://academic.oup.com/carcin/article/38/8/806/3862699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rajamani K, Thirugnanasambandan SS, Natesan C, Subramaniam S, Thangavel B, Aravindan N. Squalene deters drivers of RCC disease progression beyond VHL status. Cell Biol Toxicol [Internet] Springer; 2020. [cited 2020 Dec 9];1–21. Available from: http://link.springer.com/10.1007/s10565-020-09566-w [DOI] [PubMed] [Google Scholar]
- 46.Pedersen KM, Çolak Y, Bojesen SE, Nordestgaard BG. Low high-density lipoprotein and increased risk of several cancers: 2 population-based cohort studies including 116,728 individuals. J Hematol Oncol [Internet]. BioMed Central; 2020. [cited 2020 Dec 9];13:129. Available from: https://jhoonline.biomedcentral.com/articles/10.1186/s13045-020-00963-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ben-Aicha S, Badimon L, Vilahur G. Advances in HDL: Much More than Lipid Transporters. Int J Mol Sci [Internet] Multidisciplinary Digital Publishing Institute (MDPI); 2020. [cited 2020 Dec 9];21. Available from: http://www.ncbi.nlm.nih.gov/pubmed/31979129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Grampp S, Schmid V, Salama R, Lauer V, Kranz F, Platt JL, et al. Multiple renal cancer susceptibility polymorphisms modulate the HIF pathway. Linehan M, editor. PLOS Genet [Internet] 2017. [cited 2019 Dec 6];13:e1006872. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28715484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.C H, D X, R JP, L E, C AA, G W, et al. On-target efficacy of a HIF-2α antagonist in preclinical kidney cancer models. Nature [Internet]. Nature; 2016. [cited 2020 Dec 10];539. Available from: https://pubmed.ncbi.nlm.nih.gov/27595393/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gusev A, Spisak S, Fay AP, Carol H, Vavra KC, Signoretti S, et al. Allelic imbalance reveals widespread germline-somatic regulatory differences and prioritizes risk loci in Renal Cell Carcinoma. bioRxiv [Internet] Cold Spring Harbor Laboratory; 2019. [cited 2020 Dec 10];631150. Available from: https://www.biorxiv.org/content/10.1101/631150v1 [Google Scholar]
- 51.Sato Y, Yoshizato T, Shiraishi Y, Maekawa S, Okuno Y, Kamura T, et al. Integrated molecular analysis of clear-cell renal cell carcinoma. Nat Genet [Internet] Nature Publishing Group; 2013. [cited 2020 Nov 17];45:860–7. Available from: http://www.nature.com/articles/ng.2699 [DOI] [PubMed] [Google Scholar]
- 52.Nofer J-R. Signal Transduction by HDL: Agonists, Receptors, and Signaling Cascades Springer, Cham; 2015. [cited 2021 Jan 5]. page 229–56. Available from: http://link.springer.com/10.1007/978-3-319-09665-0_6 [DOI] [PubMed] [Google Scholar]
- 53.Shen W-J, Azhar S, Kraemer FB. SR-B1: A Unique Multifunctional Receptor for Cholesterol Influx and Efflux. Annu Rev Physiol [Internet]. Annual Reviews; 2018. [cited 2021 Jun 2];80:95–116. Available from: http://www.annualreviews.org/doi/10.1146/annurev-physiol-021317-121550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sullivan LB, Chandel NS. Mitochondrial reactive oxygen species and cancer. Cancer Metab [Internet]. BioMed Central; 2014. [cited 2021 Jun 1];2:17. Available from: https://cancerandmetabolism.biomedcentral.com/articles/10.1186/2049-3002-2-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Miess H, Dankworth B, Gouw AM, Rosenfeldt M, Schmitz W, Jiang M, et al. The glutathione redox system is essential to prevent ferroptosis caused by impaired lipid metabolism in clear cell renal cell carcinoma. Oncogene [Internet] Europe PMC Funders; 2018. [cited 2021 Jun 1];37:5435–50. Available from: http://www.ncbi.nlm.nih.gov/pubmed/29872221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Catanese MT, Ansuini H, Graziani R, Huby T, Moreau M, Ball JK, et al. Role of scavenger receptor class B type I in hepatitis C virus entry: kinetics and molecular determinants. J Virol [Internet]. J Virol; 2010. [cited 2020 Dec 2];84:34–43. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19828610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wei C, Wan L, Yan Q, Wang X, Zhang J, Yang X, et al. HDL-scavenger receptor B type 1 facilitates SARS-CoV-2 entry. Nat Metab [Internet]. Nat Metab; 2020. [cited 2020 Dec 2]; Available from: http://www.ncbi.nlm.nih.gov/pubmed/33244168 [DOI] [PubMed] [Google Scholar]
- 58.M D, K M, I M, L CJ, M SG, K BD, et al. Increased HDL cholesterol and apoA-I in humans and mice treated with a novel SR-BI inhibitor. Arterioscler Thromb Vasc Biol [Internet]. Arterioscler Thromb Vasc Biol; 2009. [cited 2021 Jan 5];29. Available from: https://pubmed.ncbi.nlm.nih.gov/19815817/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Goossens P, Rodriguez-Vita J, Etzerodt A, Masse M, Rastoin O, Gouirand V, et al. Membrane Cholesterol Efflux Drives Tumor-Associated Macrophage Reprogramming and Tumor Progression. Cell Metab [Internet] 2019. [cited 2019 Jul 1];29:1376–1389.e4. Available from: http://www.ncbi.nlm.nih.gov/pubmed/30930171 [DOI] [PubMed] [Google Scholar]
- 60.Ma X, Bi E, Lu Y, Su P, Huang C, Liu L, et al. Cholesterol Induces CD8+ T Cell Exhaustion in the Tumor Microenvironment. Cell Metab 2019;30:143–156.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Davey Smith G, Ebrahim S. ‘Mendelian randomization’: can genetic epidemiology contribute to understanding environmental determinants of disease?*. Int J Epidemiol [Internet] Oxford Academic; 2003. [cited 2020 Nov 13];32:1–22. Available from: https://academic.oup.com/ije/article-lookup/doi/10.1093/ije/dyg070 [DOI] [PubMed] [Google Scholar]
- 62.Lawlor D, Richmond R, Warrington N, McMahon G, Davey Smith G, Bowden J, et al. Using Mendelian randomization to determine causal effects of maternal pregnancy (intrauterine) exposures on offspring outcomes: Sources of bias and methods for assessing them. Wellcome open Res [Internet]. The Wellcome Trust; 2017. [cited 2020 Nov 13];2:11. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28405635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kettunen J, Demirkan A, Würtz P, Draisma HHM, Haller T, Rawal R, et al. Genome-wide study for circulating metabolites identifies 62 loci and reveals novel systemic effects of LPA. Nat Commun [Internet] Nature Publishing Group; 2016. [cited 2020 Nov 13];7:11122. Available from: http://www.nature.com/articles/ncomms11122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Scelo G, Purdue MP, Brown KM, Johansson M, Wang Z, Eckel-Passow JE, et al. Genome-wide association study identifies multiple risk loci for renal cell carcinoma. Nat Commun [Internet] Nature Publishing Group; 2017. [cited 2019 Sep 5];8:15724. Available from: http://www.nature.com/articles/ncomms15724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Richmond RC, Davey Smith G. Commentary: Orienting causal relationships between two phenotypes using bidirectional Mendelian randomization. Int J Epidemiol [Internet] Oxford Academic; 2019. [cited 2021 Feb 10];48:907–11. Available from: https://academic.oup.com/ije/article/48/3/907/5531241 [DOI] [PubMed] [Google Scholar]
- 66.Labrecque J, Swanson SA. Understanding the Assumptions Underlying Instrumental Variable Analyses: a Brief Review of Falsification Strategies and Related Tools. Curr Epidemiol Reports [Internet] Springer; 2018. [cited 2021 Feb 10];5:214–20. Available from: http://link.springer.com/10.1007/s40471-018-0152-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Burgess S, Thompson SG, Collaboration CCG. Avoiding bias from weak instruments in Mendelian randomization studies. Int J Epidemiol [Internet] Oxford Academic; 2011. [cited 2021 Feb 10];40:755–64. Available from: https://academic.oup.com/ije/article/40/3/755/745918 [DOI] [PubMed] [Google Scholar]
- 68.Bowden J, Hemani G, Davey Smith G. Invited Commentary: Detecting Individual and Global Horizontal Pleiotropy in Mendelian Randomization-A Job for the Humble Heterogeneity Statistic? Am J Epidemiol [Internet]. Am J Epidemiol; 2018. [cited 2021 Feb 10];187:2681–5. Available from: http://www.ncbi.nlm.nih.gov/pubmed/30188969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hemani G, Zheng J, Elsworth B, Wade KH, Haberland V, Baird D, et al. The MR-Base platform supports systematic causal inference across the human phenome. Elife [Internet] 2018. [cited 2020 Nov 13];7. Available from: https://elifesciences.org/articles/34408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hemani G, Tilling K, Davey Smith G. Orienting the causal relationship between imprecisely measured traits using GWAS summary data. Li J, editor. PLOS Genet [Internet] Public Library of Science; 2017. [cited 2020 Nov 13];13:e1007081. Available from: https://dx.plos.org/10.1371/journal.pgen.1007081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sanchez DJ, Steger DJ, Skuli N, Bansal A, Simon MC. PPARγ is dispensable for clear cell renal cell carcinoma progression. Mol Metab [Internet] Elsevier; 2018. [cited 2021 Jan 18];14:139–49. Available from: http://www.ncbi.nlm.nih.gov/pubmed/29866440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Matyash V, Liebisch G, Kurzchalia TV, Shevchenko A, Schwudke D. Lipid extraction by methyl-tert-butyl ether for high-throughput lipidomics. J Lipid Res [Internet]. J Lipid Res; 2008. [cited 2021 Jan 20];49:1137–46. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18281723 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.