

Abstract
Mutations in superoxide dismutase 1 gene (SOD1) are linked to amyotrophic lateral sclerosis (ALS), a neurodegenerative disorder predominantly affecting upper and lower motor neurons. The clinical phenotype of ALS shows inter- and intrafamilial heterogeneity. The aim of the study was to analyze the relations between individual SOD1 mutations and the clinical presentation using in silico methods to assess the SOD1 mutations severity. We identified SOD1 causative variants in a group of 915 prospectively tested consecutive Polish ALS patients from a neuromuscular clinical center, performed molecular modeling of mutated SOD1 proteins and in silico analysis of mutation impact on clinical phenotype and survival analysis of associations between mutations and hazard of clinical end-points. Fifteen SOD1 mutations were identified in 21.1% familial and 2.3% sporadic ALS cases. Their effects on SOD1 protein structure and functioning inferred from molecular modeling and in silico analyses correlate well with the clinical data. Molecular modeling results support the hypothesis that folding intermediates rather than mature SOD1 protein give rise to the source of cytotoxic conformations in ALS. Significant associations between type of mutation and clinical end-points were found.
Subject terms: Amyotrophic lateral sclerosis, Genetic testing, Amyotrophic lateral sclerosis
Introduction
Amyotrophic lateral sclerosis (OMIM:105400) is a heterogeneous severe neurodegenerative disorder the hallmark of which is an adult-onset loss of upper and lower motor neurons. It leads to a progressive paresis and atrophy of skeletal muscles resulting in quadriplegia and fatal respiratory failure. Approximately 90–95% of patients do not have affected first-degree relatives and are described as sporadic cases (sporadic ALS, sALS)1, and ca. 10% show a familial predisposition (fALS) with Mendelian or non-Mendelian patterns of inheritance2. Since 1993, mutations in more than forty genes have been reported to associate with ALS, the most frequent are those in the SOD1 gene encoding the essential antioxidant enzyme Cu, Zn superoxide dismutase (http://alsod.iop.kcl.ac.uk/)3,4. Coding sequences (cds) SOD1 mutations have been found in ALS patients from all over the world. However, the distribution of SOD1 mutations differs markedly even among apparently similar populations (i.e., Netherlands and Belgium, Ireland and England) and the same mutations in different populations can be associated with distinct clinical presentations. The clinical phenotype is highly variable and patients with a particular SOD1 mutation show intrafamilial differences in the severity of symptoms and the speed of disease progression5. Notably, it is believed that the pathogenicity of SOD1 mutations is not due to a lack of functional protein but rather to the accumulation of its misfolded aggregates6.
It is still unclear whether all ALS-related SOD1 mutations are in fact causative, co-causative, modifying or simply accompanying variants. A prospective gene therapy targeting SOD1 expression or a pharmacotherapy aimed at elimination of misfolded SOD1 protein can only be based on a detailed understanding of the molecular mechanisms of pathogenesis of individual SOD1 mutations7.
To address the above issues, we determined SOD1 mutations in a large group of ALS patients (n = 915) and predicted their impact on SOD1 structure and functioning using molecular modeling and prioritization algorithms. These predictions were compared with the severity of ALS presentation in individual patients. A significant correspondence was found between molecular and clinical data.
Methods
Subjects
A total of 915 patients with ALS (n = 855 unrelated sALS and 57 probands from fALS families, 6.2% FALS) were diagnosed at the Department of Neurology, the Medical University of Warsaw between 2006 and 2018 and followed till 2021. The patient clinical data was prospectively analyzed and the blood was withdrawn at the time of the first clinical assessment. The patients were further regularly followed at the same out-patient clinic, and examined up to five times during the disease course. All patients were Caucasians of a Polish origin and met the El Escorial criteria for clinically possible, probable or definite ALS8.
Mutation screening and variant analysis
DNA was isolated from peripheral blood leukocytes using standard methods, and all exons with flanking intronic regions of the SOD1 gene were sequenced as described previously9.
The Ensembl Variant Effect Predictor (https://www.ensembl.org/Homo_sapiens/Tools/VEP) was used to annotate genomic variants10. Human Splicing Finder 3.0 (HSF3), EX-SKIP, and BDGP Splice Site Prediction by Neural Network webtools were used to predict the influence of detected variants on pre-mRNA splicing11–13. ConSurf (https://consurf.tau.ac.il/) was used to analyze amino acid sequence conservation14. PredictSNP (https://loschmidt.chemi.muni.cz/predictsnp/) and NetDiseaseSNP (http://www.cbs.dtu.dk/services/NetDiseaseSNP/) were used to predict the impact of mutations on the function of SOD1 protein15,16. Aggrescan (http://bioinf.uab.es/aggrescan/), TANGO (http://tango.crg.es/) and Aggrescan3D 2.0 (http://biocomp.chem.uw.edu.pl/A3D2/) software were used to predict the tendency for aggregation of mutated proteins17–19.
Molecular modeling
The crystal structure of human SOD1 was taken from the Protein Data Bank (PDB id:2C9V)20. SOD1 is a compact homodimer with each subunit of 153 amino acids forming a β-barrel structure stabilized by a C57–C146 disulfide bridge and a zinc ion in the active site. The two subunits are held together by strong hydrophobic forces making SOD1 one of the most compact and stable proteins. Each subunit also contains a copper ion undergoing alternative oxidation–reduction in the course of the dismutation of O2·− to O2 and H2O2. The metal ions are bound by the side chains of H46, H48, H63, H71, H80, D83, and H120. The force field parameters and partial charges for the metal ion binding sites were calculated following earlier quantum chemical calculations on similar systems21,22. To impose new parameters, a patch script for the topology file was constructed to model proper interactions between the metal ions and adjacent amino acid atoms. Energy minimization and molecular dynamic (MD) simulations were performed in the NAMD program version 2.10 using all-atom force field CHARMM2723. The protein dimer was simulated in the TIP3P water box with dimensions of 6.2 nm × 6.2 nm × 9.5 nm, which contained 37,000 atoms in total. Four sodium ions were also included in solution to maintain neutrality of the system. The native protein as well as its 12 mutant variants were initially subjected to 10,000 steps of energy minimization and then a 10-ns MD equilibration with temperature increasing from 20 to 298 K. For each investigated system a 20-ns MD simulation was performed. All MD simulations were conducted using Langevin (stochastic) dynamics24 used as default in the NAMD program. The friction coefficient of 5 ps−1 was used and the temperature was set to 298 K. Nonbonded interactions were dampened by employing a switching function for van der Waals and electrostatic interactions using a cutoff of 1.6 nm. All bond lengths were constrained using the SHAKE algorithm25, therefore a longer time step of 2 fs was applied. The modeled molecular structures were visualized YASARA Structure v.16.1.2.
Statistical analyses
Associations between SOD1 mutations carried by ALS patients and theirs clinical phenotype were studied with survival analysis methods. Four clinically relevant end-points were defined to estimate progression of the disease: wheelchair-bound (loss of walking capacity), bulbar involvement (speech or swallowing impairment), respiratory insufficiency and death (overall survival). All available information from patients’ records were used to find whether the chosen end-point happened (if yes the observation was considered “complete”, if no it was “censored”) and what was the time from the onset of ALS symptoms (defined as the first muscle paresis) to the achievement of the selected end-point (for complete observations) or to the end of observation (for censored observations). Kaplan–Meier curves showing survival till each end-point for patients stratified according to specific SOD1 mutations or a bioinformatics parameter common for a group of mutations were compared with log-rank test for two curves or with chi-square test for more than two curves. Cox proportional hazards regression model was used to estimate associations between survival and quantitative Consurf parameter and calculate hazard ratio (HR) and its 95% confidence interval. Only mutations found in at least four subjects with available clinical data were subjected to direct comparisons. Age of ALS onset was compared between mutations with Mann–Whitney test. Site of ALS onset and clinical phenotype were compared between mutations with Fisher exact test. P < 0.05 was considered statistically significant. Statistica 13 program was used for statistical calculations.
Ethics approval and consent to participate
A written informed consent has been obtained from all study participants. The study has been approved by the Bioethics Committee of the Medical University of Warsaw, Poland (KB 157/2006, KB 52/2012, KB/163/2015), in compliance with the Declaration of Helsinki (BMJ 1991; 302:1194), national legislation and the Code of Ethical Principles for Medical Research Involving Human Subjects of the World Medical Association.
Results
Identifications of variants in SOD1
Sequencing revealed a total of 23 SOD1 variants, fifteen of which were in the protein coding sequence (for details see Supplementary Table S1). Among the 15 variants, all but one (p.W32*) have previously been described as causative of ALS. Three variants (c.-64C > T, c.-6G > T and c.*248A > C) lied in untranslated gene regions. Three other variants in the flanking intronic sequences: c.239 + 34A > C (MAF = 0.039 according to ExAC database), c.72 + 19G > A (MAF < 0.001), and a deletion of 7 bp in intron 2 previously described in families with keratoconus (MAF = 0.003)26,27. All the variants except one were in a heterozygous state: two ALS patients were homozygous and two heterozygous for the c.98G > A (p.D90A) SOD1 mutation. Variants c.10A > G (p.K3E), c.124G > A (p.G41S), c.260A > G (p.N86S), c.272A > C (p.D90A), c.418A > G (p.N139D) and c.434T > C (p.L144S) were identified in several patients with either sALS and/or fALS. All other variants but one (common c.239 + 34A > C) were identified in single patients. The most frequent mutations were K3E (5.3% fALS—3 families/57 familial index cases, 0.5% sALS—4 cases/854 sALS cases), L144S (8.8% fALS-5 families/57 familial index cases, 0.2%—2 cases/854 sALS cases), and D90A (0% fALS, 0.5%—4 cases/854 sALS cases respectively) followed by G41S, L126* and N139D. The overall frequency of SOD1 mutations in the study group was 3.5%—32/915 cases, 21.1% fALS—12 families/57 familial index cases, 2.3% sALS—30 cases/854 sALS cases).
Clinical characteristics of the patients
Fifteen different SOD1 coding-sequence (cds) mutations were identified in 12 fALS and 20 sALS cases. Based on the medical documentation and familial anamnesis we gathered clinical data on additional 32 FALS subjects from the affected families. In total we analyzed clinical data of 64 patients with SOD1 cds mutations. Their general demographics are summarized in Table 1.
Table 1.
Demographic data of patients with amyotrophic lateral sclerosis.
| ALS patients | Male:female ratio | Age at onset (years) Median; mean ± SD (range) |
Diagnosis delay (months) Median; mean ± SD (range) |
|---|---|---|---|
|
All n = 918 |
1.07 |
56 55.2 ± 12.7 (18–86) |
11 13.7 ± 10.1 (1–48) |
|
Without SOD1 mutations n = 854 |
1.1 |
57 55.6 ± 12.7 (18–86) |
11 13.6 ± 10.1 (1–48) |
|
SOD1 mutation carriers n = 64 (44 fALS, 20 sALS) |
0.7 |
51 50.2 ± 10.6 (21–75) |
10 16.6 ± 14.9 (2–72) |
Among patietns with SOD1 mutations, the classic ALS phenotype was observed in 56% of patients (both upper and lower motor neuron involvement; UMN, LMN), 41% showed progressive muscle atrophy (PMA, isolated signs of LMN), and 3% (n = 1) a mixed ALS-MSA-P (multiple system atrophy-parkinsonism) phenotype. The signs of the UMN damage included pseudobulbar syndrome, spasticity, and exaggerated reflexes and pathological signs. The LMN damage presented as muscle wasting, fasciculations, flaccid muscle tone and diminished/absent reflexes. In 88.1% of the patients the first symptoms occurred in the lower limbs, in 9.5% in the upper limbs, and in 2.4% in the bulbar-innervated muscles. The median survival was at least 84 months (mean 105.0 ± 69.4; range 12–312), while the tracheostomy-free survival was at least 36 months (mean 73.8 ± 75.8; range 11–312 months), since 17 patients were still alive at the time of analysis. Detailed clinical characteristics of the patients with an SOD1 coding-sequence mutation are presented in Table 2.
Table 2.
Clinical characteristics of ALS patients (n = 52) with SOD1 cds mutations.
| Mutation | No. of fALS families (cases)/sALS cases with identified mutations | Age at onset ± SD, range, (years) | M:F | Diagnosis delay (months) | Site of onset | Clinical phenotype | Bulbar involvement during disease course (months since onset ± SD, range, %) | Wheelchair-bound (months since onset ± SD, range, %) | IV-free survival (months since onset ± SD, range) |
|---|---|---|---|---|---|---|---|---|---|
| K3E | 3 fALS (n = 14), 4 sALS | 53.3 ± 8.1, 36–68 (n = 16) | 61% M, 39% F | 9.2 ± 3.2, 6–26 (n = 8) | 94% LL, 6% UL (n = 16) | 80% classic ALS, 20% PMA (n = 10) | 32.8 ± 25.8, 5–96 (n = 9), 78.6% yes | 33.4 ± 18.9, 12–96 (n = 12), 100% yes | 90.3 ± 68.9, 18–180 (n = 16) |
| A4V | 1 sALS | 56 | F | 10 | LL | Classic ALS | 12 | 13 | 18 |
| W32* | 1 sALS | 41 | M | 8 | LL | Classic ALS | 35 | 24 | 56 |
| G37R | 1 sALS | 40 | F | 4 | UL | Classic ALS | 8 | 14 | > 48 months |
| G41S | 1 fALS (n = 5) | 50.2 ± 11.6, 40–69 (n = 5) | 40% M 60% F | 7.0 ± 1.0, 6–8 (n = 3) | 60% LL, 40% UL (n = 5) | 75% classic ALS, 25% PMA (n = 4) | 13.0 ± 8.5, 7–19 (n = 2) | NA | 15.0 ± 3.6, 11–20 (n = 5) |
| G72S | 1 sALS | 52 | M | 20 | LL | PMA | NA | 38 | > 38 |
| N86S | 2 sALS | 45 ± 33.9, 21–70 (n = 2) | 1:1 | 15.5 ± 7.8, 10–21 (n = 2) | 50% UL, 50% LL | 100% PMA | 50% | NA | Mean 65.5 ± 41.1, 35–96, (n = 2) |
| D90A | 2 sALS heterozygote | 57.1 ± 12.7, 40–75 (n = 2) | 100% F | 8 (n = 1) | 50% LL, 50% bulbar, (n = 2) | 100% classic ALS | 48 ± 67.9, 0–96 (n = 2) | NA | 60.5 ± 50.2, 25–96 (n = 2) |
| 2 sALS homozygote | 47 (n = 1) | 100% F | 19 (n = 1) | 100% LL (n = 1) | 100% classic ALS (LMN > UMN) | 48 (n = 1) | NA | > 132 (n = 1) | |
| G93C | 1 sALS | 32 | F | 72 | LL | PMA | 96 | 96 | > 96 |
| S105L | 1 sALS | 42 | M | 12 | LL | PMA | 10 | Yes | 36 |
| D109Y | 1 sALS | 59 | M | 33 | LL | ALS-MSA-P | No | No | > 52 |
| C111Y | 1 sALS | 43 | M | 8 | UL | Classic ALS | NA | NA | NA |
| L126* | 1 fALS (n = 4) | 49.7 ± 14.3, 34–62 (n = 3) | 75% F, 25% M | 19.0 ± 14.8, 9–36 (n = 3) | 100% LL | 75% PMA, 25% classic ALS | 50% | 26 (n = 1) | 132.0 ± 135.8, 36–228 (n = 2) |
| N139D | 2 fALS (n = 4) | 57.0 ± 7.0, 49–67 (n = 2) | 100% F | 9 (n = 1) | 100% LL (n = 3) | 100% PMA (n = 3) | 33% (n = 3) | 12 (n = 1) | 99 ± 43.6, 24–144 (n = 3) |
| L144S | 5 fALS families (n = 17), 2 sALS | 45.8 ± 9.8, 29–63 (n = 14) | 84% F, 16%M | 23.9 ± 14.7, 2–45, (n = 7) | 100% LL, n = 9 | 50% classic ALS, 50% PMA (n = 8) | 67.5 ± 6.3, 63–72 (n = 2/14, 14%) | 87.2.2 ± 50.3, 36–156 (n = 6) | 125.0. ± 68.4, 36–219 (n = 14) |
Molecular modeling
The structures of the SOD1 wild-type dimer and 14 mutant proteins were subjected to 20-ns all-atom MD simulations in a water environment, which is enough to equilibrate the system and form new interactions after a mutation is introduced. A comparison of the obtained mutant structures with the WT one, revealed how the mutation affected the structure of the SOD1 dimer and also how it could alter potential interactions with other proteins (Table 3, Fig. 1, Supplementary Figs. S1–S15).
Table 3.
Impact of mutations on SOD1 protein.


Alternative splicing analysis—probability of altering splicing by mutated allel vs. reference (+ higher; −no changes) (details Supplementary Table S4) HSF Human Splice Finder interperatation, E-S EX-SKIP, BDGP BDGP splice site, Aggregation prediction the tendency for aggregation of mutated allel vs reference (+ higher, − lower) (details Supplementary Table S5, Supplementary File 2, Supplementary File 3) AGG AGGRESCAN, TAN TANGO, A3D Aggrescan3D.
Figure 1.
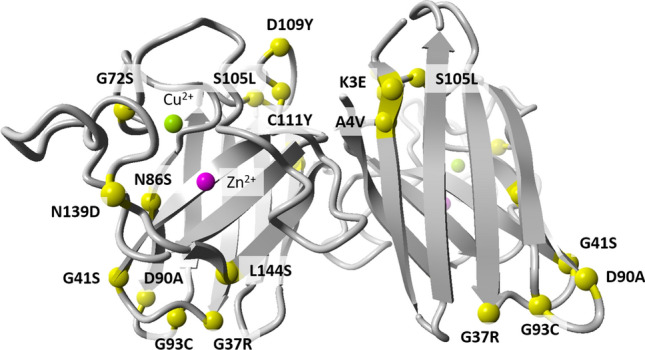
Comparison of protein structure of reference and mutant SOD1. The following mutants were modelled basing on the reference structure (PDB id:2C9V): K3E, A4V, G41S, N86S, D90A, G93C, S105L, D109Y, C111Y, L126*, N139D, and L144S.
Analysis of coding sequence conservation
Most of the identified mutations affected evolutionarily conserved residues of SOD1. Positions 32 and 109 are evolutionarily variable, and two other variable positions, D90 and L144, were the most frequently mutated in the analyzed group in both familial and sporadic cases. For details see Supplementary Table S2.
Predicting the functional impact of mutation
To infer probable functional consequences of the SOD1 mutations we analyzed them with PredictSNP software, which is a consensus classifier of eight commonly used tools: MAPP, nsSNPAnalyzer, PANTHER, PhD-SNP, PolyPhen-1, PolyPhen-2, SIFT, and SNAP. The most common mutations identified in the analyzed group (s/fALS) were K3E, D90A and L144S predicted as neutral (accuracy 63%). No statistical association was identified for results of analysis with NetDiseaseSNP. For details see Supplementary Table S3.
Analysis of alternative splicing potential
An analysis of the possible impact of the 15 mutations on alternative splicing with three web tools (HSF 3.0, EX-SKIP, and BGBD) showed inconsistent results. HSF 3.0 showed that most of the mutations could alter splicing, while BDGE found that only S105L potentially created a new acceptor site. For details see Supplementary Table S4.
Analysis of SOD1 protein aggregation potential
Analysis of SOD1 protein aggregation potential Aggrescan, TANGO, and Aggrescan3D 2.0 software were used to predict the tendency for aggregation of mutated protein. Aggrescan software used to predict hot spots of aggregation indicated four mutations (K3E, G38R, N139D, and L144S) as less aggregation-prone than the wild-type (WT) SOD1 sequence.
The Aggrescan and Aggrescan3D indicated nearly no mutation-induced changes in the aggregation potential for other mutations. For details see Supplementary Table S5, Supplementary File 2 and Supplementary File 3. Aggrescan3D indicate lower aggregation propensity for region of mutation D109Y in mutated dimers (D109Y/D109Y and D109Y/ref) compared to reference dimer. Tango, based on the physico-chemical principles of beta-sheet formation, extended by the assumption that the core regions of an aggregate are fully buried indicate K3E, A4V and S105L as more aggregation prone, whereas L126*, L144S as less aggregation prone comparing to WT sequence.
Associations of clinical end-points with mutations and their molecular modeling results
Due to a limited number of available samples, the statistical analyses were available only for five mutation: K3E, L144S, G41S, L126*, and N139D.
The overall survival differed significantly between the K3E, L144S, G41S, L126* and N139D mutation carriers (p = 0.00028, chi2 test, Supplementary Figure S16). It was significantly shorter for G41S compared to other four mutations (p < 0.025). The survival in L144 mutation carriers was significantly longer compared to patients harboring G41S, K3E and N139D mutations (p < 0.05). Molecular predictors associated with longer survival included: neutral mutation according to PredictSNP software (p = 0.0034, Supplementary Fig. S17), neutral or mild potential consequence of mutation according to molecular modeling (p = 0.024, Supplementary Fig. S18), decrease aggregation propensity by TANGO (p = 0.012, Supplementary Fig. S19) and mutation of variable residues by Consurf (HR 0.43, 95% CI 0.23–0.81, p = 0.0086). The multivariate Cox regression model adjusted for patients’ gender and age of ALS onset showed that the deleterious mutation predicted by PredictSNP software was the strongest independent predictor of death (HR 4.47, 95% CI 1.98–10.11, p = 0.00032).
The time since the first ALS symptoms onset to respiratory insufficiency significantly differed between patients carrying K3E, L144S and G41S mutations (p = 0.00022, chi2 test, Supplementary Fig. S20). It was the longest for L144S (no patient reached the end-point), the shortest for G41S (all patients reached the end-point within 2 years) and medium for K3E; the differences were significant for all 3 pairs of mutations (p < 0.02). A longer time to respiratory insufficiency was associated with neutral mutation according to PredictSNP software (p = 0.0027, Supplementary Fig. S21), not altered splicing predicted with HSF 3.0 software (p = 0.0040, Supplementary Fig. S22), neutral or mild potential consequence of mutation according to molecular modeling (p = 0.0036, Supplementary Fig. S23), decrease aggregation propensity by TANGO software (p = 0.012, Supplementary Fig. S24) and mutation of variable residues by Consurf (higher value) (HR 0.28, 95% CI 0.11–0.68, p = 0.0052). The multivariate Cox regression model adjusted for patients’ gender and age of ALS onset showed that the deleterious mutation predicted by PredictSNP software was the strongest independent predictor of the development of respiratory insufficiency (HR 23.76, 95% CI 4.87–115.86, p = 0.00009).
The time since ALS onset to bulbar involvement differed significantly between the K3E, L144S and G41S mutation carriers (p = 0.0048, chi2 test, Supplementary Fig. S25). Harboring the L144S was associated with a significantly longer time to bulbar involvement than K3E (p = 0.0015, log-rank test) and with borderline significance longer time than G41S (p = 0.050), without significant difference between K3E and G41S (p = 0.44). The time to bulbar involvement was significantly longer in familial compared to sporadic ALS cases (p = 0.0095, Supplementary Fig. S26). The longer time to bulbar involvement was also associated with decrease aggregation propensity by TANGO software prediction when compared to neutral or increase aggregation propensities (p = 0.00043, Supplementary Fig. S27) and with potentially not altered splicing when compared to potentially altered splicing according to HSF 3.0 (p = 0.00062, Supplementary Fig. S28).
Time since ALS onset to a wheelchair use was significantly longer for L144S compared to K3E (p = 0.0039, Supplementary Fig. S29). A longer time to wheelchair use was also associated with a decrease aggregation propensity by TANGO software prediction, when compared to neutral or increase aggregation propensities (p = 0.0031, Supplementary Fig. S30).
We have found that a more advanced age at disease onset was linked to a shorter survival (HR 1.046) with a growing risk of death of 4.6% at every additional year at onset. Further association analysis of clinical data revealed that the age of ALS onset was significantly lower in L144S compared to patients carrying the K3E mutation (45.8 ± 9.8 vs 53.3 ± 8.1 years, p = 0.019). The G41S was associated with upper limbs onset as compared to other frequent mutations (K3E, L144S, L126* and N139D, p = 0.017). The PMA phenotype was significantly more frequent among the N139D mutation carriers as compared to K3E (100% vs 19%, p = 0.021).
Discussion
From over 185 SOD1 mutations identified to date, only some (e.g., H46R, D90A, and R115G) cause ALS with a defined clinical phenotype, including characteristic age of onset, survival time and/or site of onset (lower limbs in D90A and H46R). Other mutations present a more varied course or have only been identified in individual patients/families, making a comprehensive analysis of the genotype–phenotype relation difficult.
The frequency of SOD1 mutations in fALS patients varies between populations from 13 to 20% and in sALS patients from 1 to 2%28–30. Thus the mutation frequency in the present study (21.1% in fALS, 2.3% in sALS) is relatively high. The L144S and K3E variants were the most frequent among the Polish ALS patients (but not in other populations, except for L144S among Brazilian patients31–33. Contrary, D90A, the most frequent European SOD1 mutation, was only present in 0.4% of cases (4.1% of fALS). From the mutations with the highest number of representing individuals the L144S was associated with the earliest disease onset and the slowest progression, while the G41S was characterized by a particularly aggressive progression and a short survival, comparable with the phenotype reported for A4V in North American population34. Although the number of affected individuals were not sufficient for statistical analysis, based on the clinical observation, the individuals with G37R, N86S, homozygous D90A or G93C mutations presented with a relatively less severe, while A4V, G72S, and S105L with more severe phenotypes (based on time for reaching clinical end-points), similarly to previous reports from other populations9,35–38. The L126* mutation, previously described as aggressive, showed a highly variable survival in our study, ranging from 36 to 228 months39. As for the clinical phenotypes, the statistical analysis showed that N139D mutation was linked to the most homogenous clinical presentation of PMA, as opposed to the K3E characterized by the prevalence of classic ALS. From the clinical observations: beside the N139 D, the isolated lower motor neuron involvement (PMA) was observed in the G72S, N86S, G93C, S105L and L126*, while the classic phenotype prevailed among patients with the K3E, G41S, D90A, and L144S mutations, and was also shown in A4V, W32* and G37R represented by single individuals. The D90A homozygotes shared the classic phenotype with prevalent lower motor neuron involvement and lower limbs onset, whereas one of the patients heterozygous for D90A had a bulbar onset and a short survival of 25 months. We cannot exclude that the MSA-P in the patient with the D109Y mutation was an accompanying condition, as the mutation had previously been described in classic ALS with prevalent UMN involvement, bulbar onset and long survival40. In contrast to other studies we found a highly infrequent onset in the upper limbs among patients harboring SOD1 mutations.
All the cds mutations identified in our study group were classed as disease-causing according to the Human SOD1 non synonymous SNP analysis (http://bioinfogroup.com/sod1/snp/), which is consistent with our prioritization results41. However, according to the SNP analysis database none of the mutations was predicted to influence the aggregation tendency or amyloid propensity. Our protein modeling results (Table 3, Supplementary Figs. S1–S15) in which we compared WT and mutated SOD1 structures after the all-atom MD simulations of a SOD1 dimer in a water environment imitating mammalian cytosol conditions42 showed that only two mutations, D109Y and C111Y, had an increased aggregation potential. However, these mutations were not considered aggregation-prone by SNP analysis, Aggrescan, TANGO or Aggrescan3d (Supplementary Table S5). This is concordant with previous observations that SOD1 in fALS has a reduced propensity to form aggregates, while soluble heterodimers and trimeric SOD1 complexes may be more toxic as compared to large aggregates43,44.
A slightly higher aggregation potential was predicted for the A4V mutant. The movement of loops adjacent to residue after the A > V mutation is considerable (Supplementary Fig. S3), which can facilitate aggregation. After the mutation, the valine side chain is directed towards the center of the b-barrel rather than exposed but it still could promote movements of adjacent fragments. Ours result appears to support the hypothesis that folding intermediates of SOD1 are an important source of cytotoxic conformations in ALS pathology6. Indeed, it was previously proposed that A4V mutation destabilizes SOD1 monomer and weaken the dimer interface45. Recently Brasil et al. observed low levels of SOD1 monomers in cells co-expressing WT and A4V SOD1, and the predominant formation of heteromeric species46. Based on this they suggested that WT SOD1 might exist primarily as unfolded monomeric intermediates and then fully active dimers. On the other hand, unfolded and misfolded monomers might be the predominant mutant SOD1 form.
We found L126* to be the most damaging to the protein structure. The truncation of a substantial fragment of the polypeptide (loop 126–141 and β-thread 142–151) rearranges both the dimer interface and the active site. The reported differences between the results concerning SOD1 protein stability obtained with different methods (bioinformatics, molecular modeling, in vitro, in vivo) suggest the existence of as yet unidentified factors involved in the formation of pathogenic SOD1 conformations in vivo43,47. SOD1 gene variants undergoing alternative splicing have already been described in fALS patients48,49. In the present study potential for alternative splicing due to the cds mutations was disputable, as only some of programs used indicated a small probability of such an effect. Nevertheless, the effects of two of those mutations, W32* and S105L seems sufficiently likely to deserve an experimental verification. Potentially altered splicing predicted with HSF 3.0 software was significantly associated with bulbar involvemenhuman spt in sALS patients and respiratory insufficiency in familial and sporadic ALS patients.
We observed that the least evolutionarily conserved positions in SOD1 (D90 and L144) were also the most frequently mutated in our study group, and the D90A and L144S carriers suffered from a slowly progressing ALS. Mutations of conserved residues of SOD1 were significantly associated with shorter survival times and shorter time between the disease onset and respiratory failure in ALS patients. Similarly, the PredictSNP (a consensus classifier for prediction of disease-related amino acid mutations) classified the K3E, D90A, D109Y, and L144S mutations as neutral and their carriers’ symptoms were relatively less severe in terms of age of onset and survival times.
The pathogenicity of at least some ALS-related SOD1 mutations seems to involve the formation of amyloid-like aggregates. However, Aggrescan classified WT SOD1 and nearly all its mutated versions studied here as of low aggregation propensity (highly negative Na4vSS scores); still, specific nucleation points from which an ordered fibrillary structure could spread under certain conditions would nevertheless make the mutated protein amyloidogenic. Notably, the K3E, L144S, and N139D variants were predicted to be even less aggregate-prone than the WT SOD1, while A4V was the only mutant with a markedly enhanced aggregation potential. Algorithms that have been derived from and used to predict amyloid fibril formation in the absence of other biological factors also offer a considerable degree of accuracy for predicting amyloid-aggregation propensity in vivo remains to be improve to extend this prediction to disease manifestation and pathology50. Statistical analyses indicate some ALS clinically relevant end-points (bulbar involvement, wheelchair-bound, respiratory insufficiency, survival time) with increase aggregation propensity but also neutral by TANGO software prediction. To sum up, while the prediction programs and molecular modeling could not define consistently the pathogenicity of each and every SOD1 mutation, there was a good agreement between those predictions and the disease severity for the both ends of the ALS spectrum, i.e., the least and the most severe cases.
A caveat of our study is that the only experimentally-derived SOD1 structure available is a dimer of the mature molecule, whereas the prion-like ALS pathomechanism is most probably associated with an immature or misfolded protein6. For instance it was shown recently that both WT and mutant SOD1 form dimers and oligomers, but only mutant SOD1 aggregates and form intracellular inclusions. Moreover, co-expression of WT and mutant SOD1 in various cell models resulted in the formation of a larger number of inclusions, as compared to cells expressing WT or mutated SOD1 separately51. Taking into consideration the dysfunction of numerous cellular pathways observed in ALS, aggregation of SOD1 does not seem to be the only cause of ALS. According to the multistep hypothesis of ALS52, single SOD1 mutations may influence more than one step leading to ALS onset.
The major effect of SOD1 mutations in ALS is linked to the protein aggregation and a prion-like propagation of misfolded molecules. These mutations may also lead to a loss of function of SOD1 by affecting its structure and/or interactions pattern. The loss of function involves not only the dismutase enzymatic activity, e.g., associated with the N86S mutation53, but may also involve a loss of the nuclear function where SOD1 acts as a transcription factor54. In one sporadic ALS patient we identified a nonsense mutation at codon 32 (p.W32*), which was absent from the whole exome/genome databases (1000 GP, gnomAD)55. Since the W32* was also found in the patient’s asymptomatic mother, age > 70, we were not able to prove the mutation was pathogenic. We further found that SOD1 W32* was associated with a dismutation activity in erythrocytes reduced by half53, which might point at the loss of SOD1 function56,57. The premature stop codon could result in the shortest reported truncated SOD1 protein, but most likely the nonsense-mediated mRNA decay prevents the synthesis of such abnormal protein58.
A putative loss of SOD1 function in ALS was reported in previous studies. For instance, the mutation V30Dfs*859 should produce a very short, non-functional truncated SOD1 protein. G28_P29del caused by alternative splicing of exon 2 of SOD1 leads to reduced transcription and a low level of SOD1 protein in the mutation carriers60. A similar result was reported for mutation S108Lfs*1561, as the authors observed ca. 50% reduction of the SOD1 protein level and could not detect the truncated SOD1 (a protein with the predicted molecular weight) by Western blotting. The above mentioned SOD1 mutations, as well as other pathogenic variants including D90A, G41S, and I112M62,63, showed a reduced penetrance.
Interestingly, a recent in vitro study has shown that the tryptophan residue at position 32 (W32) is necessary for the formation of a competent seed for aggregation allowing the prion-like propagation of SOD1 misfolding from cell to cell, and the W32S substitution blocked this phenomenon64. Also a study on SOD1 single copy/knock-in models of ALS in C. elegans suggests an involvement of both the loss and gain of function of SOD1 in ALS development65. The contribution of the loss and gain of function mechanisms vary in different neuronal populations. In the studied model, a glutamatergic neuron degeneration was induced by oxidative stress due to the loss of SOD1 function, a phenomenon also observed in a significant fraction of ALS patients. Also recent reports on children with a homozygous truncation mutation (p.C112Wfs*11) with no SOD1 activity and severe symptoms during infancy suggest that the loss of SOD1 enzymatic activity contributes to motor neuron disorders66,67.
To sum up, the SOD1 haploinsufficiency with all its consequences might be one of the factors in an oligogenic etiology of ALS. It is most likely that many cases of ALS are due to the presence of multiple gene variants with different pathogenicity. Understanding the input of such variants to the development of neurodegeneration and their interactions with diverse environmental factors (e.g., toxins or the microbiome) is critical for the development of efficient therapies, especially in regards to potential gene therapy4,68.
Conclusions
We found L144S and K3E to be the most frequent SOD1 mutations among Polish ALS patients. Carrying L144S mutation was linked to the longest, while G41S to the shortest overall survival. Despite intrafamilial heterogeneity, L144S was significantly associated with the least severe, K3E with medium and the G41S the most aggressive disease progression. In silico analysis and molecular modeling of SOD1 variants allowed to identify relationsbetween SOD1 mutations and the ALS clinical phenotype. The time to the clinically relevant endpoints including walking loss, bulbar involvement, respiratory insufficiency and overall survival, were significantly associated with the in silico predictions results including substantial disturbance of SOD1 structure. However, our analyses indicate that the in silico prediction of mutation consequences might be incompatible with disease course (i.e. N86S-less severe phenotype and severe mutation consequences predicted by used software).
Supplementary Information
Author contributions
M.B.—design and conceptualization of study, analysis and interpretation of data, drafting manuscript; K.S.—design and conceptualization of study, analysis and interpretation of data, drafting manuscript; C.Ż.—design and conceptualization of study, analysis and interpretation of data, drafting manuscript; P.M.—design and conceptualization of study, analysis and interpretation of data; P.M.A.—revising manuscript for intellectual content; M.M.—acquisition of data, analysis of data, interpretation of the data, revising the manuscript for intellectual content; S.F.—design and conceptualization of study, acquisition of data, interpretation of data, revising manuscript for intellectual content; M.K.K.—design and conceptualization of study; acquisition of clinical data; analysis and interpretation of clinical data; drafting the manuscript. All authors have read and approved the final manuscript.
Funding
This work was partially supported by the National Science Center, Poland (NCN) project SONATA9 [UMO-2015/17/D/NZ2/03712] and EU Joint Programme—Neurodegenerative Disease Research (JPND) projects SOPHIA (5/SOPHIA/JPND/2012), OnWEBDUALS (DZP/2/JPND-III/2015) and ERA-NET-E-Rare-3/IV/Maxomod/11/2020.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Mariusz Berdyński, Przemysław Miszta and Krzysztof Safranow.
Contributor Information
Mariusz Berdyński, Email: m.berdynski@imdik.pan.pl.
Magdalena Kuźma-Kozakiewicz, Email: mkuzma@wum.edu.pl.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-021-03891-8.
References
- 1.Byrne S, Walsh C, Lynch C, Bede P, Elamin M, Kenna K, McLaughlin R, Hardiman O. Rate of familial amyotrophic lateral sclerosis: A systematic review and meta-analysis. J. Neurol. Neurosurg. Psychiatry. 2011;82:623–627. doi: 10.1136/jnnp.2010.224501. [DOI] [PubMed] [Google Scholar]
- 2.Andersen PM, Al-Chalabi A. Clinical genetics of amyotrophic lateral sclerosis: What do we really know? Nat. Rev. Neurol. 2011;7:603–615. doi: 10.1038/nrneurol.2011.150. [DOI] [PubMed] [Google Scholar]
- 3.Abel O, Powell JF, Andersen PM, Al-Chalabi A. ALSoD: A user-friendly online bioinformatics tool for amyotrophic lateral sclerosis genetics. Hum. Mutat. 2012;33:1345–1351. doi: 10.1002/humu.22157. [DOI] [PubMed] [Google Scholar]
- 4.Ghasemi M, Brown RH. Genetics of amyotrophic lateral sclerosis. Cold Spring Harb. Perspect. Med. 2018;8:a024125. doi: 10.1101/cshperspect.a024125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perrone B, Conforti FL. Common mutations of interest in the diagnosis of amyotrophic lateral sclerosis: How common are common mutations in ALS genes? Expert. Rev. Mol. Diagn. 2020;20:703–714. doi: 10.1080/14737159.2020.1779060. [DOI] [PubMed] [Google Scholar]
- 6.Culik RM, Sekhar A, Nagesh J, Deol H, Rumfeldt JAO, Meiering EM, Kay LE. Effects of maturation on the conformational free-energy landscape of SOD1. Proc. Natl. Acad. Sci. U. S. A. 2018;115:E2546–E2555. doi: 10.1073/pnas.1721022115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Andersen PM. Extensive heterogeneity in patients with ALS with mutations in SOD1 in France. J. Neurol Neurosurg. Psychiatry. 2021 doi: 10.1136/jnnp-2021-326553. [DOI] [PubMed] [Google Scholar]
- 8.Brooks BR, Miller RG, Swash M, Munsat TL. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Mot. Neuron Disord. Off. Publ. World Fed. Neurol. Res. Group Mot. Neuron Dis. 2000;1:293–299. doi: 10.1080/146608200300079536. [DOI] [PubMed] [Google Scholar]
- 9.Berdyński M, Kuźma-Kozakiewicz M, Ricci C, Kubiszewska J, Millecamps S, Salachas F, Łusakowska A, Carrera P, Meininger V, Battistini S, Kwieciński H, Zekanowski C. Recurrent G41S mutation in Cu/Zn superoxide dismutase gene (SOD1) causing familial amyotrophic lateral sclerosis in a large Polish family. Amyotroph. Lateral Scler. Off. Publ. World Fed. Neurol. Res. Group Mot. Neuron Dis. 2012;13:132–136. doi: 10.3109/17482968.2011.600316. [DOI] [PubMed] [Google Scholar]
- 10.McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GRS, Thormann A, Flicek P, Cunningham F. The ensembl variant effect predictor. Genome Biol. 2016;17:122. doi: 10.1186/s13059-016-0974-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Desmet F-O, Hamroun D, Lalande M, Collod-Béroud G, Claustres M, Béroud C. Human Splicing Finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009;37:e67. doi: 10.1093/nar/gkp215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Raponi M, Kralovicova J, Copson E, Divina P, Eccles D, Johnson P, Baralle D, Vorechovsky I. Prediction of single-nucleotide substitutions that result in exon skipping: Identification of a splicing silencer in BRCA1 exon 6. Hum. Mutat. 2011;32:436–444. doi: 10.1002/humu.21458. [DOI] [PubMed] [Google Scholar]
- 13.Reese MG, Eeckman FH, Kulp D, Haussler D. Improved splice site detection in Genie. J. Comput. Biol. J. Comput. Mol. Cell Biol. 1997;4:311–323. doi: 10.1089/cmb.1997.4.311. [DOI] [PubMed] [Google Scholar]
- 14.Ashkenazy H, Abadi S, Martz E, Chay O, Mayrose I, Pupko T, Ben-Tal N. ConSurf 2016: An improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016;44:W344–W350. doi: 10.1093/nar/gkw408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bendl J, Stourac J, Salanda O, Pavelka A, Wieben ED, Zendulka J, Brezovsky J, Damborsky J. PredictSNP: Robust and accurate consensus classifier for prediction of disease-related mutations. PLoS Comput. Biol. 2014;10:e1003440. doi: 10.1371/journal.pcbi.1003440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Johansen MB, Izarzugaza JMG, Brunak S, Petersen TN, Gupta R. Prediction of disease causing non-synonymous SNPs by the Artificial Neural Network Predictor NetDiseaseSNP. PloS One 2013;8:e68370. [DOI] [PMC free article] [PubMed]
- 17.Conchillo-Solé O, de Groot NS, Avilés FX, Vendrell J, Daura X, Ventura S. AGGRESCAN: A server for the prediction and evaluation of ‘hot spots’ of aggregation in polypeptides. BMC Bioinform. 2007;8:65. doi: 10.1186/1471-2105-8-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Linding R, Schymkowitz J, Rousseau F, Diella F, Serrano L. A comparative study of the relationship between protein structure and β-aggregation in globular and intrinsically disordered proteins. J. Mol. Biol. 2004;342:345–353. doi: 10.1016/j.jmb.2004.06.088. [DOI] [PubMed] [Google Scholar]
- 19.Kuriata A, Iglesias V, Pujols J, Kurcinski M, Kmiecik S, Ventura S. Aggrescan3D (A3D) 2.0: Prediction and engineering of protein solubility. Nucleic Acids Res. 2019;47:W300–W307. doi: 10.1093/nar/gkz321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Strange RW, Antonyuk SV, Hough MA, Doucette PA, Valentine JS, Hasnain SS. Variable metallation of human superoxide dismutase: Atomic resolution crystal structures of Cu-Zn, Zn-Zn and as-isolated wild-type enzymes. J. Mol. Biol. 2006;356:1152–1162. doi: 10.1016/j.jmb.2005.11.081. [DOI] [PubMed] [Google Scholar]
- 21.Shen J, Wong CF, Subramaniam S, Albright TA, McCammon JA. Partial electrostatic charges for the active center of Cu, Zn superoxide dismutase. J. Comput. Chem. 1990;11:346–350. [Google Scholar]
- 22.Branco RJF, Fernandes PA, Ramos MJ. Molecular dynamics simulations of the enzyme Cu, Zn superoxide dismutase. J. Phys. Chem. B. 2006;110:16754–16762. doi: 10.1021/jp056855l. [DOI] [PubMed] [Google Scholar]
- 23.MacKerell AD, Banavali N, Foloppe N. Development and current status of the CHARMM force field for nucleic acids. Biopolymers. 2000;56:257–265. doi: 10.1002/1097-0282(2000)56:4<257::AID-BIP10029>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 24.Kubo, R., Toda, M., Hashitsume, N. Statistical Physics II: Nonequilibrium Statistical Mechanics. 2nd edn. (Springer, 1991). 10.1007/978-3-642-58244-8.
- 25.Ryckaert J-P, Ciccotti G, Berendsen HJC. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 1977;23:327–341. [Google Scholar]
- 26.Udar N, Atilano SR, Brown DJ, Holguin B, Small K, Nesburn AB, Kenney MC. SOD1: A candidate gene for keratoconus. Investig. Ophthalmol. Vis. Sci. 2006;47:3345–3351. doi: 10.1167/iovs.05-1500. [DOI] [PubMed] [Google Scholar]
- 27.De Bonis P, Laborante A, Pizzicoli C, Stallone R, Barbano R, Longo C, Mazzilli E, Zelante L, Bisceglia L. Mutational screening of VSX1, SPARC, SOD1, LOX, and TIMP3 in keratoconus. Mol. Vis. 2011;17:2482–2494. [PMC free article] [PubMed] [Google Scholar]
- 28.Renton AE, Chiò A, Traynor BJ. State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 2014;17:17–23. doi: 10.1038/nn.3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Radunović A, Leigh PN. ALSODatabase: Database of SOD1 (and other) gene mutations in ALS on the Internet. European FALS Group and ALSOD Consortium. Amyotroph. Lateral Scler. Mot. Neuron Disord. Off. Publ. World Fed. Neurol. Res. Group Mot. Neuron Dis. 1999;1:45–49. doi: 10.1080/146608299300080040. [DOI] [PubMed] [Google Scholar]
- 30.Marangi G, Traynor BJ. Genetic causes of amyotrophic lateral sclerosis: New genetic analysis methodologies entailing new opportunities and challenges. Brain Res. 2015;1607:75–93. doi: 10.1016/j.brainres.2014.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kuźma-Kozakiewicz M, Berdyński M, Morita M, Takahashi Y, Kawata A, Kaida K-I, Kaźmierczak B, Lusakowska A, Goto J, Tsuji S, Zekanowski C, Kwieciński H. Recurrent K3E mutation in Cu/Zn superoxide dismutase gene associated with amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2013;14:608–614. doi: 10.3109/21678421.2013.812119. [DOI] [PubMed] [Google Scholar]
- 32.Alavi A, Nafissi S, Rohani M, Zamani B, Sedighi B, Shamshiri H, Fan J-B, Ronaghi M, Elahi E. Genetic analysis and SOD1 mutation screening in Iranian amyotrophic lateral sclerosis patients. Neurobiol. Aging. 2013;34(1516):e1–8. doi: 10.1016/j.neurobiolaging.2012.09.006. [DOI] [PubMed] [Google Scholar]
- 33.Chadi G, Maximino JR, de Jorge FMH, de Borba FC, Gilio JM, Callegaro D, Lopes CG, Santos SND, Rebelo GNS. Genetic analysis of patients with familial and sporadic amyotrophic lateral sclerosis in a Brazilian Research Center. Amyotroph. Lateral Scler. Front. Degener. 2017;18:249–255. doi: 10.1080/21678421.2016.1254245. [DOI] [PubMed] [Google Scholar]
- 34.Saeed M, Yang Y, Deng H-X, Hung W-Y, Siddique N, Dellefave L, Gellera C, Andersen PM, Siddique T. Age and founder effect of SOD1 A4V mutation causing ALS. Neurology. 2009;72:1634–1639. doi: 10.1212/01.wnl.0000343509.76828.2a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kuźma-Kozakiewicz M, Andersen PM, Elahi E, Alavi A, Sapp PC, Morita M, Żekanowski C, Berdyński M. Putative founder effect in the Polish, Iranian and United States populations for the L144S SOD1 mutation associated with slowly uniform phenotype of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2020;22:1–6. doi: 10.1080/21678421.2020.1803359. [DOI] [PubMed] [Google Scholar]
- 36.Cudkowicz ME, McKenna-Yasek D, Sapp PE, Chin W, Geller B, Hayden DL, Schoenfeld DA, Hosler BA, Horvitz HR, Brown RH. Epidemiology of mutations in superoxide dismutase in amyotrophic lateral sclerosis. Ann. Neurol. 1997;41:210–221. doi: 10.1002/ana.410410212. [DOI] [PubMed] [Google Scholar]
- 37.Gamez J, Corbera-Bellalta M, Nogales G, Raguer N, García-Arumí E, Badia-Canto M, Lladó-Carbó E, Álvarez-Sabín J. Mutational analysis of the Cu/Zn superoxide dismutase gene in a Catalan ALS population: Should all sporadic ALS cases also be screened for SOD1? J. Neurol. Sci. 2006;247:21–28. doi: 10.1016/j.jns.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 38.Hou L, Jiao B, Xiao T, Zhou L, Zhou Z, Du J, Yan X, Wang J, Tang B, Shen L. Screening of SOD1, FUS and TARDBP genes in patients with amyotrophic lateral sclerosis in central-southern China. Sci. Rep. 2016;6:32478. doi: 10.1038/srep32478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Byström R, Andersen PM, Gröbner G, Oliveberg M. SOD1 mutations targeting surface hydrogen bonds promote amyotrophic lateral sclerosis without reducing apo-state stability. J. Biol. Chem. 2010;285:19544–19552. doi: 10.1074/jbc.M109.086074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Naini A, Mehrazin M, Lu J, Gordon P, Mitsumoto H. Identification of a novel D109Y mutation in Cu/Zn superoxide dismutase (sod1) gene associated with amyotrophic lateral sclerosis. J. Neurol. Sci. 2007;254:17–21. doi: 10.1016/j.jns.2006.12.009. [DOI] [PubMed] [Google Scholar]
- 41.Moreira LGA, Pereira LC, Drummond PR, De Mesquita JF. Structural and functional analysis of human SOD1 in amyotrophic lateral sclerosis. PLoS One. 2013 doi: 10.1371/journal.pone.0081979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lodish H, Berk A, Zipursky SL, Matsudaira P, Baltimore D, Darnell J. Molecular Cell Biology. 4. Freeman; 2000. [Google Scholar]
- 43.Prudencio M, Hart PJ, Borchelt DR, Andersen PM. Variation in aggregation propensities among ALS-associated variants of SOD1: Correlation to human disease. Hum. Mol. Genet. 2009;18:3217–3226. doi: 10.1093/hmg/ddp260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhu C, Beck MV, Griffith JD, Deshmukh M, Dokholyan NV. Large SOD1 aggregates, unlike trimeric SOD1, do not impact cell viability in a model of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. U. S. A. 2018;115:4661–4665. doi: 10.1073/pnas.1800187115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim J, Lee H, Lee JH, Kwon D, Genovesio A, Fenistein D, Ogier A, Brondani V, Grailhe R. Dimerization, oligomerization, and aggregation of human amyotrophic lateral sclerosis copper/zinc superoxide dismutase 1 protein mutant forms in live cells. J. Biol. Chem. 2014;289:15094–15103. doi: 10.1074/jbc.M113.542613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.de Brasil AA, de Carvalho MDC, Gerhardt E, Queiroz DD, Pereira MD, Outeiro TF, Eleutherio ECA. Characterization of the activity, aggregation, and toxicity of heterodimers of WT and ALS-associated mutant Sod1. Proc. Natl. Acad. Sci. 2019;116:25991–26000. doi: 10.1073/pnas.1902483116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sannigrahi A, Chowdhury S, Das B, Banerjee A, Halder A, Kumar A, Saleem M, Naganathan AN, Karmakar S, Chattopadhyay K. The metal cofactor zinc and interacting membranes modulate SOD1 conformation-aggregation landscape in an in vitro ALS model. Elife. 2021;10:e61453. doi: 10.7554/eLife.61453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Birve A, Neuwirth C, Weber M, Marklund SL, Nilsson A-C, Jonsson PA, Andersen PM. A novel SOD1 splice site mutation associated with familial ALS revealed by SOD activity analysis. Hum. Mol. Genet. 2010;19:4201–4206. doi: 10.1093/hmg/ddq338. [DOI] [PubMed] [Google Scholar]
- 49.Valdmanis PN, Belzil VV, Lee J, Dion PA, St-Onge J, Hince P, Funalot B, Couratier P, Clavelou P, Camu W, Rouleau GA. A mutation that creates a pseudoexon in SOD1 causes familial ALS. Ann. Hum. Genet. 2009;73:652–657. doi: 10.1111/j.1469-1809.2009.00546.x. [DOI] [PubMed] [Google Scholar]
- 50.Belli M, Ramazzotti M, Chiti F. Prediction of amyloid aggregation in vivo. EMBO Rep. 2011;12:657–663. doi: 10.1038/embor.2011.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brasil AA, Magalhães RSS, De Carvalho MDC, Paiva I, Gerhardt E, Pereira MD, Outeiro TF, Eleutherio ECA. Implications of fALS mutations on Sod1 function and oligomerization in cell models. Mol. Neurobiol. 2018;55:5269–5281. doi: 10.1007/s12035-017-0755-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chiò A, Mazzini L, D’Alfonso S, Corrado L, Canosa A, Moglia C, Manera U, Bersano E, Brunetti M, Barberis M, Veldink JH, van den Berg LH, Pearce N, Sproviero W, McLaughlin R, Vajda A, Hardiman O, Rooney J, Mora G, Calvo A, Al-Chalabi A. The multistep hypothesis of ALS revisited: The role of genetic mutations. Neurology. 2018;91:e635–e642. doi: 10.1212/WNL.0000000000005996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Keskin I, Birve A, Berdynski M, Hjertkvist K, Rofougaran R, Nilsson TK, Glass JD, Marklund SL, Andersen PM. Comprehensive analysis to explain reduced or increased SOD1 enzymatic activity in ALS patients and their relatives. Amyotroph. Lateral Scler. Front. Degener. 2017;18:457–463. doi: 10.1080/21678421.2017.1301481. [DOI] [PubMed] [Google Scholar]
- 54.Pansarasa O, Bordoni M, Diamanti L, Sproviero D, Gagliardi S, Cereda C. SOD1 in amyotrophic lateral sclerosis: ‘Ambivalent’ behavior connected to the disease. Int. J. Mol. Sci. 2018 doi: 10.3390/ijms19051345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Genomes Project Consortium A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Saccon RA, Bunton-Stasyshyn RKA, Fisher EMC, Fratta P. Is SOD1 loss of function involved in amyotrophic lateral sclerosis? Brain J. Neurol. 2013;136:2342–2358. doi: 10.1093/brain/awt097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sau D, De Biasi S, Vitellaro-Zuccarello L, Riso P, Guarnieri S, Porrini M, Simeoni S, Crippa V, Onesto E, Palazzolo I, Rusmini P, Bolzoni E, Bendotti C, Poletti A. Mutation of SOD1 in ALS: A gain of a loss of function. Hum. Mol. Genet. 2007;16:1604–1618. doi: 10.1093/hmg/ddm110. [DOI] [PubMed] [Google Scholar]
- 58.Hug N, Longman D, Cáceres JF. Mechanism and regulation of the nonsense-mediated decay pathway. Nucleic Acids Res. 2016;44:1483–1495. doi: 10.1093/nar/gkw010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hu J, Chen K, Ni B, Li L, Chen G, Shi S. A novel SOD1 mutation in amyotrophic lateral sclerosis with a distinct clinical phenotype. Amyotroph. Lateral Scler. Off. Publ. World Fed. Neurol. Res. Group Mot. Neuron Dis. 2012;13:149–154. doi: 10.3109/17482968.2011.621437. [DOI] [PubMed] [Google Scholar]
- 60.Zinman L, Liu HN, Sato C, Wakutani Y, Marvelle AF, Moreno D, Morrison KE, Mohlke KL, Bilbao J, Robertson J, Rogaeva E. A mechanism for low penetrance in an ALS family with a novel SOD1 deletion. Neurology. 2009;72:1153–1159. doi: 10.1212/01.wnl.0000345363.65799.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Canosa A, De Marco G, Lomartire A, Rinaudo MT, Di Cunto F, Turco E, Barberis M, Brunetti M, Casale F, Moglia C, Calvo A, Marklund SL, Andersen PM, Mora G, Chiò A. A novel p.Ser108LeufsTer15 SOD1 mutation leading to the formation of a premature stop codon in an apparently sporadic ALS patient: Insights into the underlying pathomechanisms. Neurobiol. Aging. 2018;72:189.e11–189.e17. doi: 10.1016/j.neurobiolaging.2018.08.014. [DOI] [PubMed] [Google Scholar]
- 62.Gamez J, Caponnetto C, Ferrera L, Syriani E, Marini V, Morales M, Bordo D, Pirro C, Garre C, Origone P. I112M SOD1 mutation causes ALS with rapid progression and reduced penetrance in four Mediterranean families. Amyotroph. Lateral Scler. Off. Publ. World Fed. Neurol. Res. Group Mot. Neuron Dis. 2011;12:70–75. doi: 10.3109/17482968.2010.487906. [DOI] [PubMed] [Google Scholar]
- 63.Khoris J, Moulard B, Briolotti V, Hayer M, Durieux A, Clavelou P, Malafosse A, Rouleau GA, Camu W. Coexistence of dominant and recessive familial amyotrophic lateral sclerosis with the D90A Cu, Zn superoxide dismutase mutation within the same country. Eur. J. Neurol. 2000;7:207–211. doi: 10.1046/j.1468-1331.2000.00028.x. [DOI] [PubMed] [Google Scholar]
- 64.Pokrishevsky E, McAlary L, Farrawell NE, Zhao B, Sher M, Yerbury JJ, Cashman NR. Tryptophan 32-mediated SOD1 aggregation is attenuated by pyrimidine-like compounds in living cells. Sci. Rep. 2018;8:15590. doi: 10.1038/s41598-018-32835-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Baskoylu SN, Yersak J, Ohern P, Grosser S, Simon J, Kim S, Schuch K, Dimitriadi M, Yanagi KS, Lins J, Hart AC. Single copy/knock-in models of ALS SOD1 in C. elegans suggest loss and gain of function have different contributions to cholinergic and glutamatergic neurodegeneration. PLoS Genet. 2018;14:e1007682. doi: 10.1371/journal.pgen.1007682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Park JH, Elpers C, Reunert J, McCormick ML, Mohr J, Biskup S, Schwartz O, Rust S, Grüneberg M, Seelhöfer A, Schara U, Boltshauser E, Spitz DR, Marquardt T. SOD1 deficiency: A novel syndrome distinct from amyotrophic lateral sclerosis. Brain J. Neurol. 2019;142:2230–2237. doi: 10.1093/brain/awz182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Andersen PM, Nordström U, Tsiakas K, Johannsen J, Volk AE, Bierhals T, Zetterström P, Marklund SL, Hempel M, Santer R. Phenotype in an infant with SOD1 homozygous truncating mutation. N. Engl. J. Med. 2019;381:486–488. doi: 10.1056/NEJMc1905039. [DOI] [PubMed] [Google Scholar]
- 68.Amado DA, Davidson BL. Gene therapy for ALS: A review. Mol. Ther. 2021 doi: 10.1016/j.ymthe.2021.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.