

Abstract
By 24th Sep. 2021, there are more than 229 million COVID-19 cases worldwide, the researchers are tirelessly working to discover and develop an efficient drug molecule against this devastative viral infection. This study aims to evaluate the inhibitory efficiency of the organic acids and phenolic compounds present in Brassica oleracea (Tronchuda Cabbage) against spike glycoprotein in SARS-CoV-2. Thirty-seven phytocompounds are screened on the basis of their molecular weight (<500 g/mol) and 14 ligands are docked using Autodock Vina and Autodock4 (version 4.2.6). The stability of the top five docked complexes was analyzed using classical molecular dynamics (MD) simulation. ADMET analysis is performed for the top five compounds and their targets are identified using SwissTargetPrediction. Phytoactives from B. oleracea namely Astragalin, 3-p-coumaroylquinic acid, 4-p-coumaroylquinic acid and sinapoyl-D-glucoside showed high binding affinities and free energy of binding during molecular docking and MD simulation studies (∼ 8.5–9.0 kcal/mol) for the spike glycoprotein trimer of SARS-CoV2. The ADMET analysis revealed that these phytocompounds have good solubility in the aqueous phase and that they don't penetrate the blood brain barrier. Moreover, there is no P-gp substrate inhibition, CYP1A2 inhibition, CYP2C19 inhibition, CYP2C9 inhibition, CYP2D6 inhibition and CYP3A4 inhibition observed for these compounds. Additionally, zero PAINS alerts were reported. These findings from molecular docking and MD simulation studies suggest that astragalin and coumaroylquinic acids from Tronchuda cabbage possess potential inhibitory capacity against spike glycoprotein trimer of SARS-CoV-2 and could be further taken up as lead targets for drug discovery.
Keywords: SARS-CoV-2, Spike glycoprotein, Plant-bioinformatics, Antiviral drugs, Phytocompound, Brassica oleracea
Graphical abstract

1. Introduction
Coronavirus is a group of identical RNA viruses which are named after the crown-like spike structures on their surface [1]. Identification of coronavirus dates back to the 1930s in domestic chicken causing acute respiratory disease. Later human coronavirus was discovered in the mid-1960s [2]. The most common human coronaviruses that cause a mild respiratory infection are human coronavirus 229E, NL63, OC43, and HKU1, Severe Acute Respiratory Syndrome-2 (SARS-CoV-2, 2019), SARS-CoV, 2004, and Middle East Respiratory Syndrome Coronaviruses (MERS-CoV, 2012) are the recently identified human coronaviruses which initially infected animals and consequently evolved into a new form to infect humans [3]. Among these SARS-CoV-2 or the "Novel Coronavirus" was first identified recently in December 2019 in Wuhan, Hubei Province, China causing pneumonia-like illness which was later coined as Coronavirus Infection Disease (COVID-19). Since then, COVID-19 has been pandemic worldwide and the World Health organization has announced Public Health Emergency of International Concern on 30th January 2020. As of 24th Sep. 2021, the global count of confirmed cases of COVID-19 is 229,858,719 including 4713,543 deaths, reported to the World Health organization 9 [4]. Symptoms of this disease are mild and include - cough, shortness of breath, chills, muscle pain, sore throat, and loss of taste and smell [5,6]. According to WHO about 80 percent of people recover without treatment in the hospital. Severe illness from COVID-19 is experienced by older people, children and those with underlying medical concerns such as diabetes, heart problems and chronic lung disease [7]. Many Antiviral Drugs have been repurposed and used for the treatment of COVID-19 like Hydroxychloroquine, Chloroquine, Remdesivir and Favipiravir [8]. According to various clinical surveys, Remdesivir has adverse effects in liver and kidney whereas Hydroxychloroquine causes cardiovascular problems like QT prolongation [8,9]. Although Favipiravir has not yet reported to cause serious side effects, its efficacy is low [10]. Other side effects of synthetic antiviral drugs include swelling, diarrhea, nausea, rash, hypotension, anemia, constipation. Though no vaccine is available right now, more than 170 vaccines for COVID-19 are under study.
Protein inhibition is one mechanism for combating against the virus. There are basically two types of proteins in SARS-CoV-2; Non-Structural Proteins (NSP) and Structural Proteins. Several studies report that Non-structural protein of SARS CoV-2 plays a major role in the replication of corona viruses. So, this protein inhibition can help to stop the multiplication of the virus. Also, structural proteins have shown their significance in virion assembly and infection of coronavirus [11,12]. Among the structural proteins, novel coronavirus uses the spike protein present in a spike-like structure on the outer surface to enter into the human body and cause infection. Like SARS-CoV, novel coronavirus also binds with the Angiotensin Converting Enzyme (ACE-2) receptor in human cells. ACE-2 receptors occur in many organs but mostly in the cell lining of the air sacs. Hence, the infection mainly causes respiratory problems. We have chosen a spike ectodomain structure (open state) from SARS-CoV-2 (PDB ID-6VYB) for this study (Fig. 1 ). The spike glycoprotein is trimeric with two subunits- 'S1′ and 'S2′. S1 binds to the ACE 2 receptor and 'S2′ fuses the viral membrane to the cellular membrane [13]. Spike glycoprotein induces host immune response and has been recognized to be an ideal target for vaccines and drugs.
Fig. 1.

The overall structure of spike glycoprotein trimer of SARS-CoV2; (a) ribbon representation and (b) surface representations. The three chains A, B and C are depicted in red, blue and green colors respectively.
There is a direct need for a drug which should not show or would have negligible side effects. One most feasible approach could be switching to herbal medicines/phytoactives. Herbal medicine has proven to be the oldest and reliable since ancient times. In previous studies, Brassica oleacea L.var costata. (Tronchuda Cabbage) has been found to be rich in various phytocompound with medicinal properties [14,15]. Tronchuda Cabbage is one of the traditional vegetables, consumed and cultivated mostly in Northern Portuguese regions [15]. The well-balanced nutritional profile of the vegetable reveals that it has a significant amount of liposoluble (tocopherols, carotenoids) and water-soluble (vitamin C, phenolics and flavonoid) antioxidant compounds. These antioxidant compounds not only prevent oxidative stress but also prevent cardiovascular disease and are effective in cancer, specifically in stomach, colon and rectum [16]. For these reasons, the plant Brassica oleracea L. var costata were screened for potent ligands/antiviral compounds present in it. Various phenolic compounds and organic acid were identified in its internal leaves, external leaves and seeds. Fourteen ligands are chosen based on the molecular weight (<500 g/mol) and their binding capacity and interactions with the spike protein trimer are examined using molecular docking studies in Autodock. Five of the Phytoconstituents showed very high binding affinities when compared to conventional synthetic drugs are studied for their Absorption, Distribution, Metabolism, Excretion and Toxicology (ADMET) properties. Their physicochemical properties (https://pubchem.ncbi.nlm.nih.gov/) are discussed in the Table 1 . The present study, we utilized various bioinformatics approaches to discover the inhibition activities of herbal compounds from Tronchuda cabbage and their pharmacokinetic, medicinal chemistry, drug likeliness and toxicity properties were studied using SWISS ADME and Pre-ADMET servers. Autodock Vina is used to find the binding affinities using energy scores and Autodock4.2 was used to perform the energy simulations and the scores were compared to extract the best ligands from a total of 14 top scoring molecules. These findings suggest that these compounds are potential phytomolecules against SARS-CoV-2 spike protein complex. The selected phytomolecules are further subjected to MD simulation studies to understand the stability and kinetics of binding to the spike protein trimer. Inhibitory action of these phytocompounds can be further validated by using other in-vivo and in vitro analysis.
Table 1.
Physicochemical Properties of fourteen selected drug candidates from Tronchuda cabbage whose molecular weights are below 500 g/mol. Their two-dimensional structures, molecular weights, molecular formula number of Hydrogen Bond Donors, Hydrogen Bond Acceptors, Total Polar Surface Area and Number of rotatable Bonds are discussed.
| S.No. | Name | Structure | Mol. Formula | PubChem ID | Mol. Weight (g/mol) | HBD | HBA | TPSA (Ų) | No. of rotatable bonds |
|---|---|---|---|---|---|---|---|---|---|
| 1. | 3-p- Coumaroyl Quinic Acid (3 PC) | 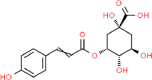 |
C16H18O8 |
9,945,785 | 338.31 |
5 | 8 | 144.52 | 5 |
| 2. | 4-p- Coumaroyl Quinic Acid (4 PC) | 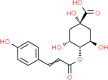 |
C16H18O8 |
5,281,766 | 338.31 |
5 | 8 | 144.52 | 5 |
| 3. | Astragalin (ASG) | 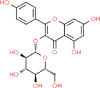 |
C21H20O11 | 5,282,102 | 448.4 |
7 | 11 | 190.28 | 4 |
| 4. | Sinapoyl-d-Glucoside (S1G) | 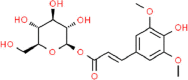 |
C17H22O10 | 13,787,030 | 386.3 | 5 | 10 | 155.14 | 7 |
| 5. | 1-Sinapoyl-d-Glucose (1SG) | 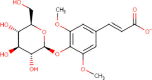 |
C17H21O10- | 25,244,357 | 385.3 |
4 | 10 | 157.97 | 7 |
| 6. | Sinapine | 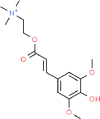 |
C16H24NO5+ | 5,280,385 | 310.36 | 1 | 6 | 65.00 | 8 |
| 7. | Sinapic Acid | 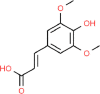 |
C11H12O5 |
637,775 | 224.21 | 2 | 5 | 76.00 | 4 |
| 8. | Quinic Acid | 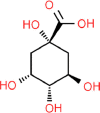 |
C7H12O6 | 6508 | 192.17 |
5 | 6 | 118.21 | 1 |
| 9. | Shikimic Acid | 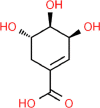 |
C7H10O5 |
8742 | 174.15 |
4 | 5 | 97.98 | 1 |
| 10. | Aconitic Acid | 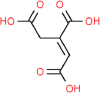 |
C6H6O6 | 309 | 174.11 |
3 | 6 | 111.90 | 4 |
| 11. | Ascorbic Acid | 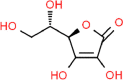 |
C6H8O6 | 54,670,067 | 176.12 |
4 | 6 | 107.22 | 2 |
| 12. | Citric Acid | 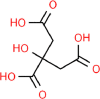 |
C6H8O7 |
311 | 192.12 | 4 | 7 | 132.12 | 5 |
| 13. | Fumaric Acid | 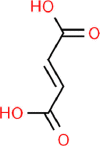 |
C4H4O4 | 444,972 | 116.07 |
2 | 4 | 74.6 | 2 |
| 14. | Malic Acid | 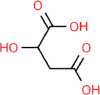 |
C4H6O5 |
525 | 134.09 | 3 | 5 | 94.83 | 3 |
2. Materials and methodology
2.1. COVID-19 targets as drug receptors
The heteroatoms present in the spike glycoprotein are removed and the receptor is visualised using LigPlot and PyMoL. The PDB coordinates of the open confirmation of the trimeric spike glycoprotein (PDB: 6VYB) was selected for this study (Fig. 1). Prior to molecular docking exercises the solvent molecules were removed, polar hydrogen atoms and Kollman's charges were added. The pdbqt files for the receptor proteins were generated to be used for molecular docking.
2.2. Molecular docking and protein-ligand preparation
Autodock Vina and Autodock4 (Ver. 4.2.6) (http://autodock.scripps.edu/downloads) are used to dock the ligands to the predicted binding site. The ligands and the protein targets are modified to perform docking using MGL Tools. The 3D structures of the 1-sinapoyl-D-glucose (1SG), sinapoyl-D-glucoside (S1G), astragalin (ASG), 3-p-coumaroylquinic acid (3 PC), 4-p-coumaroylquinic acid (4 PC), sinapine, sinapic acid, aconitic acid, malic acid, quinic acid, citric acid, ascorbic acid, shikimic acid and fumaric acid are downloaded in the sdf format from PubChem and are converted to pdbqt files using MGL tools. The conversion to pdbqt files is necessary because Autodock4 and Autodock Vina utilize pdbqt files for the docking process [17]. Gasteiger charges are added to the entities and the 14 ligands that were screened based on their molecular weights (<500 g/mol) are docked and their interactions are studied. The aromatic carbons and rotatable bonds are detected. The detected nonpolar hydrogens are merged and TORSDOF is set as per the ligand conformation. It is made sure that the receptor has no non-bonded atoms. The above protocols of ligand and target preparation are followed as a precursor for performing Autodock4 (Ver. 4.2.6) and Autodock Vina individually.
For Autodock Vina, the number of points (size) is set to 40 in x, y and z dimensions and spacing is set to 0.375 angstrom. On the other hand, the number of points (size) is set to 40 in x, y and z dimensions and spacing is set to 0.525 angstrom in Autodock4 (Ver. 4.2.6), which is based on Lamarckian genetic algorithm. The grid box is made at 209.349, 209.887 and 204.593 of x center, y center and z center respectively for spike protein. The grid information is saved as grid.txt and configuration file is saved as config.txt in which the energy range is set as 4 and exhaustiveness to 12. Affinity potential, electrostatic potential and desolvation potential are calculated for each atom of the ligand which is being docked with respect to the grid point of protein using AutoGrid4 followed by docking. Twenty million energy evaluations are performed for each ligand with a total of 10 runs.
2.3. ADMET prediction
ADMET predictions are done using SWISS-ADME (http://www.swissadme.ch/) for all the top scoring compounds from Brassica oleracea and detailed analysis of SWISS-ADME results is done using Pre-ADMET (https://preadmet.bmdrc.kr/). SWISS-ADME permits to calculate physicochemical descriptors as well as predicts pharmacokinetics properties, drug likeness and other therapeutic characteristics which eases the drug discovery process. The ligands that are saved in the sdf format from PubChem (https://pubchem.ncbi.nlm.nih.gov/) are read using these servers and predict parameters like water solubility and physicochemical properties. Pre-ADMET gives accurate and precise ADMET and drug likely nature of phytocompound including the specific violations for rules like rule of five etc. and also gives the numerical values of parameters like Blood Brain Barrier permeation and gastrointestinal absorption and also performs toxicity tests like Ames test, mouse carcinogenicity and hERG inhibition.
2.4. MD simulation using Gromacs
To get insights into the spike protein-ligand interaction stability, molecular dynamics (MD) calculations were carried out on the best docking poses. The input files for MD calculations were generated using CHARMM-GUI solution builder [18,19,20] using charmm force field parameters for protein. The topology of the ligands was generated by CHARMM General Forcefield through ParamChem server. The Charmm-gui solution builder includes five steps. In the first step, the coordinates of protein-ligand complex being read by the tool. The second step involves solvation of the protein-ligand complex as well as determining the shape and size of the system. Na+ and Cl− ions are added in this step to neutralize the system. Periodic Boundary Conditions (PBC) are set in the third step which are used for approximation of a large system by using a unit cell which is then replicated in all directions. The simulation takes place only for the atoms that are present inside the pbc box. Bad contacts are removed in this step by running short minimization. Fourth and fifth step involves equilibration of the system and production. Equilibration is done in two phases-NVT ensemble and NPT ensemble to ensure that the system has achieved the desired temperature and pressure. The input files for equilibration and production are then downloaded and desired changes were made which include number of steps of MD run, frequency of saving of trajectories and calculation of energy etc.
All complexes were initially solvated in a cubic box of TIP3P waters and then Na+ and Cl− ions were added to neutralize the net atomic charge of the whole system by random replacement of water molecules. NaCl concentration was set to the physiological value of 0.15 M [21]. The size of the solvated spike trimer-ligand complexes was > 6, 66, 186 atoms. The periodic boundary conditions (PBC) were imposed considering the system shape and size. Non-bonded interactions were treated with a 12 Å cut-off distance and the neighbor searching list were buffered with the Verlet cutoff-scheme and the long-range electrostatic interactions were treated with the particle mesh Ewald (PME) method [22,23]. CHARMM36m forcefield was applied on the protein-ligand complex. Prior to production simulation, energy minimization of the system was carried out by using steepest descent algorithm (5000 steps). The complex was then equilibrated for stabilizing its temperature and pressure by subjecting it to NVT and NPT ensemble and simulating for 125 ps at 303.15 K temperature. Finally, the complex is subjected to production simulation run for 100 ns in NPT ensemble at 303.15 K and 1 bar. To maintain the temperature Nose-Hoover thermostat was used and similarly for maintaining the pressure Parrinello-Rahman barostat was used. GROMACS 2020.4 was used for both equilibration and production run during all MD calculations [24]. LINCS algorithm was used for constraining H-bonds using the inputs provided by CHARMM-GUI. The V-rescale thermostat at 300 K with a coupling constant of 1 ps was used. Molecular geometries were relaxed with the steepest descent algorithm with 50,000,000 steps. The trajectories were stored every 2 ps. Simulations of 100 ns (with the PDB: 6VYB with bound ligands) in NPT assembly were performed for the production stage. All the molecular dynamic calculations were performed with NMRbox (https://nmrbox.org/) server platform resources [25].
2.5. Trajectory analysis
The trajectories from molecular docking and MD results and 3D plots for the receptor-ligand complex were retrieved from the VMD molecular graphics program [26]. Finally, multiple parameter analysis was carried out from the MD production stage. The root mean square deviation (RMSD) of atom position for ligand and protein was calculated by fitting protein alpha-carbon atom with the gmx_rms subprogram. Similarly, root mean square fluctuations (RMSF) of all the protein atoms were calculated using gmx_rmsf. Radius of gyration of all protein atoms was calculated with the gmx_gyrate and number of hydrogen bonds were calculated (inside the protein-ligand interface) with the gmx_hbond. PDBs for making snapshots during time course of simulation runs with every 10 ns interval were generated using gmx_ trjconv.
2.6. Binding free energy calculations
Protein-ligand complexes interactions were further analysed by performing binding free energy (BFE) calculations with the online Prodigy server and using the PDB trajectories of the complex generated at 0, 5, 10, 20, 30, 40, 50, 60, 70, 80, 90, 100 nanoseconds of the simulation run.
2.7. Target prediction
The specific targets of the drugs alter the protein activity by binding to the receptor and for this reason identification and mapping of the drug targets are essential to sort out the molecular mechanisms which are the primary cause for their bioactivity and cross-reactivity [27]. The ligand targets are predicted in five different organisms using Swiss Target Prediction (https://www.swisstargetprediction.ch) such as aldose reductase (by homology), various carbonic anhydrases, acetylcholinesterase and quinone reductase. The five top scoring ligands are loaded in sdf format to perform the analysis.
3. Results
3.1. Determination of binding affinities of phytomolecules with the spike proteins by docking analysis
The phytomolecules that were docked into the binding pocket of spike glycoprotein are studied for the interacting side chains and further analysis of docking scores revealed that the phenolic compounds present in the plant are effective phytomolecules against spike protein while organic acids didn't exhibit a high binding affinity. The Autodock4 (Ver. 4.2.6) and vina scores are correlated and studied for the top scoring compounds based on the vina scores. When the phytomolecules are analysed for their binding affinities (Table 2 ), it is observed that 4-p-coumaroylquinic acid and 3-p-coumaroylquinic acid has the highest binding affinity of -8.9 kcal/mol while astragalin has an affinity of -8.8 kcal/mol. 4 PC, 3 PC acid and ASG are the molecules that had highest binding energy or the least docking scores when compared to the total of 14 ligands. Binding with 3 PC involves AB interface of the spike trimer via three hydrogen bonds (Thr-B:1006, Gln-A:1005 and Gln-A:1002) (Fig. 3), while 4 PC forms three hydrogen bonds with Thr1006 of chain A/B/C at the interface (Fig. 3). Binding of ASG to spike trimer mainly involves AC interface via five hydrogen bonds (2 H-bond to Asp-C:950 and 1 H-bond to Arg-A:765, Glu-C:1017 and Arg-A:1019). Binding of S1G mainly involved hydrogen bonding with chain C residues namely Ser730, Lys733, Ala1056 and Gly1059 while only one hydrogen bond is seen for 1SG with Arg1019 of chain A (Fig. 3) indicating low affinity binding for the later. The interactions of 4 PC, 3 PC, ASG, 1SG and S1G are further stabilised by several low energy interactions such as vander waal forces, hydrophobic, amide bonds etc. (Fig. 2 and Fig. 3 ).
Table 2.
The fourteen ligands from Tronchuda Cabbage are screened based on their docking scores.
| Ligands | Autodock Vina Score(kcal/mol) | Autodock4Score(kcal/mol) |
|---|---|---|
| 3p Coumaroyl Quinic Acid (3PC) | -8.9 | -13.62 |
| 4p Coumaroyl Quinic Acid (4PC) | -8.9 | -13.51 |
| Astragalin (ASG) | -8.8 | -11.67 |
| Sinapoyl-D-Glucoside (S1G) | -7.6 | -11.94 |
| 1-Sinapoyl-D-Glucose (1SG) | -7.2 | -12.65 |
| Sinapine | -6.9 | -7.73 |
| Sinapic Acid | -6.9 | -8.68 |
| Quinic Acid | -6.7 | -9.67 |
| Shikimic Acid | -6.3 | -7.29 |
| Aconitic Acid | -6.2 | -6.12 |
| Ascorbic Acid | -6.2 | -8.36 |
| Citric Acid | -5.8 | -9.26 |
| Fumaric Acid | -5.1 | -5.18 |
| Malic Acid | -5.0 | -7.76 |
Fig. 3.

Binding site interactions between spike glycoprotein trimer (green ribbons) and phytocompounds (shown in orange-colored sticks) (a) Astragalin (b) 4-p-Coumaroylquinic Acid (c) 3-p-Coumaroylquinic Acid (d) Sinapoyl-D-Glucoside (e) 1-Sinapoyl-D- Glucose. Hydrogen bonds are shown in black dashes.
Fig. 2.

Ligplot representations of the binding site interactions of phytocompounds from Brassica oleracea with spike glycoprotein trimer; (a) Interaction between astragalin and spike glycoprotein. (b) Interaction between 4-p-Coumaroylquinic acid (4 PC) and spike glycoprotein. (c) Interaction between 3-p-Coumaroylquinic acid (3 PC) and spike glycoprotein (d) Interaction between Sinapoyl-1-Glucoside (S1G) and spike protein. (e) Interaction between 1-Sinapoyl Glucose (1SG) and spike protein. Ligands and interacting residues are shown in stick and ball representation. Hydrogen bonds are depicted in green dashes and hydrophobics are represented in red semicircles.
3.2. Estimations of energy by Autodock4 showing extensive binding potential
Estimated free energy of binding is defined as the sum of intermolecular energy and torsional free energy while the unbound system's energy and total internal energy cancels each other. The inhibition constant being calculated at 298.15 K varied from 362.85 μM to 1.93 mM. Vander Waals, hydrogen bonding and desolvation energy are the lowest for coumaroylquinic acids, -8.85 kcal/mol followed by astragalin with -8.31 kcal/mol. Sinapoyl-D-glucoside has -7.99 kcal/mol and the highest value was reported for 1-sinapoyl-D-glucose with -5.98 kcal/mol. Electrostatic energy was lowest for 1-sinapoyl D-glucose, -1.1 kcal/mol and coumaroylquinic acids with -0.85 kcal/mol. Astragalin and sinapoyl D-glucoside reported negligible electrostatic energy values. The widespread analysis of free binding energy, inhibition constant, intermolecular energy and torsional free energies establish these compounds as potent antiviral phytomolecules as mentioned in Table 3 .
Table 3.
The free binding energy, inhibition constant, intermolecular energy and torsional free energies obtained by Autodock4 analysis.
| Name | Free Binding Energy (kcal/mol) (ΔG) | Inhibition constant (μM) | Intermolecular Energy (kcal/mol) | Torsional Free Energy (kcal/mol) |
|---|---|---|---|---|
| 3-p-Coumaroylquinic Acid (3PC) | -6.71 | 11.97 | -9.7 | 2.98 |
| 4-p-Coumaroylquinic Acid (4PC) | -6.69 | 12.42 | -9.68 | 2.98 |
| Astragalin (ASG) | -5.1 | 181.54 | -8.38 | 3.28 |
| Sinapoyl-D-Glucoside (S1G) | -4.69 | 362.85 | -8.27 | 3.28 |
| 1-Sinapoyl-D- Glucose (1SG) | -3.7 | 1.93 mM | -6.99 | 3.28 |
3.3. Analysing the binding stability of spike trimer-ligand interactions during MD simulations
The best 5 complexes of spike trimer with bound ligands (3-p-Coumaroylquinic Acid (3 PC), 4-p-Coumaroylquinic Acid (4 PC), Astragalin (ASG), 1-Sinapoyl-D- Glucose (1SG) and Sinapoyl-D-Glucoside (S1G)) were evaluated for their binding stability using MD calculation run for 100 ns simulations at natural room temperature conditions. Visualization of the pdb trajectories post simulation run revealed that all except the 1-Sinapoyl-D- Glucose and Sinapoyl-D-Glucoside ligand remains bound to the ligand binding groove of the spike trimer pocket. However, the binding of Spike trimer with ligand 1-Sinapoyl-D-Glucose and Sinapoyl-D-Glucoside showed very high RMSD (>10 angstrom). Visualization of the trajectory showed that the two ligands are completely out of the binding pocket after 3.5 ns and 50 ns of the simulations indicating the instability of this complex. For calculation and analysis of binding free energy and RMSD, radius of gyration and hydrogen bonds only three complexes with ligands ASG, 4 PC, 3 PC were considered. Post simulation, the root mean square deviation (RMSD), the root mean square fluctuation (RMSF), and radius of gyration values were calculated for individual ligands bound to the spike protein trimers and plotted as a function of time (0 to 100 ns). In general, at the start of the simulation run small fluctuations in the RMSD were observed (Fig. 4 (a), (b), (c)) which fall within the ideal range around 2Å (smaller RMSD values indicating the higher stability of the complex during the simulation) [28,29]. The RMSD values of the spike trimer readily increased from 0 to 15 ns, and thereafter remained stable at 7–8 Å throughout the simulation for complexes with ASG, 4 PC and 3 PC ligands (Fig. 4 (a), (b), (c)), the rmsd values for spike trimer-ASG complex showed least deviation and remained stable ∼6.0 Å throughout the simulation indicating towards better and stable binding of this complex (Fig 4 (c)). The average RMSD values of ASG, 4 PC and 3 PC ligands are in the range of 2.5–4.0 Å. The overall fluctuation (RMSF) calculated for each atom in the complete simulation run for spike-ASG, 4 PC and 3 PC complexes showed that the atoms of binding site residues do not move a lot and remain in a stable position (Fig S1).
Fig. 4.

Analysis of root mean square deviation (RMSD) of the spike trimer-ligand complexes during the time course of MD simulation run for 100 ns (A) Spike-ASG, (B) Spike-PC and (C) Spike-3 PC complexes. For each spike trimer-ligand complex the MD run parameter is shown for three replicates.
The radius of gyration (Rg) of the spike protein bound to their respective ligands lies between 4.8–5.0 nm indicative of the compact globular fold of the trimer being maintained upon complex formation. While the three ligands show average Rg value of 0.44 nm throughout the simulation run suggesting a stable interaction with spike trimer (Fig. S2). The hydrogen bonding interactions of the spike protein trimer bound with ligands were found stable throughout the simulation run. ASG forms on average of 2–4 hydrogen bonds with spike trimer while 4 PC and 3 PC interacts with spike protein by forming 3–6 hydrogen bonds throughout the time course of simulation runs suggesting a firm interaction (Fig 5 (a), (b), (c)). The binding free energy of the spike trimer-ASG, 4 PC and 3 PC, complexes were calculated using the prodigy online server using the pdb trajectory of the complexes at 0, 5,10, 20, 30, 40, 50, 60, 70, 80, 90, 100 ns of the MD calculation steps (Table 4 ). The average free energy of binding for the spike trimer complexes for ASG, 4 PC and 3 PC, are -9.75, -9.18, -9.23 kcal/mol respectively. The MD simulation analysis of selective docked complexes confirmed the substantial stability of spike trimer ligand complexes under the study.
Fig. 5.

Time course of hydrogen bond formation during the 100 ns simulation run, between spike trimer-ASG (A), 4 PC (B) & 3 PC complexes (C) where X axis corresponds to the time (ns) and Y axis corresponds to the number of hydrogen bonds. For each spike trimer-ligand complex the MD run parameter is shown for three replicates.
Table 4.
Binding free energy of the spike trimer complexes bound to ASG, 3 PC, 4 PC ligands calculated by using prodigy server at 0–100 ns of simulation with intervals of 10 ns. The binding free energy was calculated for all the three replicates of MD run for each complex.
| Ligand | ASG-run1 | ASG-Run2 | ASG-Run3 | 4PC-Run1 | 4PC-Run2 | 4PC-Run3 | 3PC-Run1 | 3PC-Run2 | 3PC-Run3 |
|---|---|---|---|---|---|---|---|---|---|
| time (ns) | ΔGnoelec (kcal/mol) |
ΔGnoelec (kcal/mol) |
ΔGnoelec (kcal/mol) |
ΔGnoelec (kcal/mol) |
ΔGnoelec (kcal/mol) |
ΔGnoelec (kcal/mol) |
ΔGnoelec (kcal/mol) |
ΔGnoelec (kcal/mol) |
ΔGnoelec (kcal/mol) |
| 0 | -10.3 | -10.5 | -10.4 | -9.6 | -9.6 | -9.4 | -9.5 | -9.4 | -9.5 |
| 10 | -9.1 | -9.2 | -9.6 | -9.1 | -9.3 | -9.1 | -9 | -9 | -9.2 |
| 20 | -9.4 | -9.3 | -9.6 | -9.2 | -9.2 | -9.2 | -8.9 | -8.9 | -9.1 |
| 30 | -9.3 | -9.1 | -9 | -9.2 | -9.1 | -8.9 | -9 | -9 | -9.3 |
| 40 | -9.3 | -9.5 | -9.7 | -9.2 | -9.3 | -9 | -9.3 | -8.9 | -9.2 |
| 50 | -9.2 | -9.6 | -9.4 | -8.8 | -9.4 | -8.9 | -9.3 | -9 | -9.2 |
| 60 | -9.2 | -9.3 | -9.4 | -9.1 | -9.4 | -9.1 | -9.2 | -9.1 | -9.3 |
| 70 | -8.7 | -9.8 | -8.9 | -8.9 | -9.4 | -9 | -9.2 | -9.1 | -9.4 |
| 80 | -8.9 | -9.6 | -9.1 | -9 | -9.4 | -9 | -9.3 | -9 | -9.5 |
| 90 | -8.3 | -9.5 | -9.3 | -9.2 | -9.3 | -9 | -9 | -8.9 | -9.4 |
| 100 | -8.4 | -9.5 | -9.4 | -9.5 | -9.4 | -9 | -9.2 | -9 | -9.5 |
| total | -9.35 | -10 | -9.9 | -9.16 | -9.35 | -9.05 | -9.17 | -9.03 | -9.50 |
| s.d. | 0.54 | 0.37 | 0.40 | 0.23 | 0.13 | 0.14 | 0.21 | 0.28 | 0.14 |
3.4. Target prediction of the screened phytomolecules
Coumaroylquinic acids shows 33.3% protease, 26.7% enzyme and 6.7% kinase, membrane receptor and electrochemical transporter activities while comparatively lower aldose reductase and leukocyte elastase activities whereas 100% Aldose reductase (by homology), Carbonic anhydrase II, Carbonic anhydrase VII, Carbonic anhydrase XII, Carbonic anhydrase IV and Acetylcholinesterase is shown by astragalin. Sinapoyl-D-Glucoside shows 20% kinase, protease and family A G protein-coupled receptor activities while 1 sinapoyl-D-glucose shows 20% oxidoreductase, electrochemical transporter activities and 13% enzyme activity. The top 5 predicted target activities of coumaroylquinic acids, astragalin, 1-sinapoyl-glucose and sinapoyl-D-glucoside support their promising application as a probable drug (Table S1) (Fig. 6 ).
Fig. 6.

Predicted target activities of Top 5 screened phytomolecules (Table S1): (a) Astragalin (b) 4-p-Coumaroylquinic acids (c) 3-p- Coumaroylquinic acids (d) Sinapoyl-D-Glucoside (e) 1-Sinapoyl-D- Glucose.
3.5. Determination of the preclinical ADME properties
The analysis of SWISS ADME and Pre-ADMET results illustrates a clear picture about the compound's bioactivity. For a drug to be effective, it should be stable at all pH, should neither penetrate the Blood Brain Barrier (BBB) nor bind the P-glycoprotein substrate while a balanced hydrophobic and hydrophilic nature is also appreciable [30]. Human Intestinal Absorption (HIA) is one of the most important ADME parameters that influences the bioavailability of drugs. Low Gastrointestinal (GI) absorption was reported while none of them reported BBB permeation, P-gp substrate inhibition, CYP1A2 inhibition, CYP2C19 inhibition, CYP2C9 inhibition, CYP2D6 inhibition and CYP3A4 inhibition. Lipophilicity and several other pharmacokinetic characteristics are discussed in the Table 5 .
Table 5.
Pharmacokinetics and lipophilicity properties of the top scoring phytomolecules:.
| Name | Pharmacokinetics |
Consensus Log Po/w | |||||
|---|---|---|---|---|---|---|---|
| Log Kp (skin permeation) (cm/s) | HIA(%) | MDCK cellpermeability(nm/sec) | in vitro Caco2 cell permeability(nm/s) | Calculated logD by SK atomic types in pH 7.4 | In vitro Plasma Protein Binding(%) | ||
| 3-p-Coumaroylquinic Acid (3 PC) | -8.9 | 32.6 | 0.13 | 5.64 | -0.0503 | 37.2 | 0.03 |
| 4-p-Coumaroylquinic acid (4 PC) | -9.45 | 31.8 | 0.62 | 16.26 | -1.38736 |
35.76 |
-0.05 |
| Astragalin (ASG) | -8.52 | 25.17 | 1.14 | 11.15 | -0.421 | 57.58 | -0.25 |
| Sinapoyl-d-glucoside (S1G) | -8.41 | 40 | 10.67 | 19.79 | -0.98966 |
46.24 | -0.34 |
| 1-sinapoyl-d- glucose (1SG) | -8.41 | 40 | 0.36 | 20.3 | -0.98966 | 49.3 | -0.94 |
3.6. Analysis on toxicology and drug likeliness
3-p-Coumaroylquinic acid, 4-p-coumaroylquinic acid, sinapoyl-D-glucoside and 1-sinapoyl-D-glucose followed Lipinski's rule but astragalin has two violations reported. Coumaroylquinic acids and sinapoyl-D-glucoside have 55% bioavailability while astragalin and 1-sinapoyl-D-glucose have less than 20% bioavailability. A number of drugs have been withdrawn from the last stage of clinical trials due to cardiotoxic effects and both the coumaroylquinic acids showed medium risk hERG inhibition while sinapoyl-D-glucoside and 1-sinapoyl-D-glucose showed low risk and astragalin showed high risk values. All the five candidates showed low levels of acute fish toxicity while negative results in two years carcinogenicity bioassay in rats. Medicinal chemistry, acute fish toxicity and drug likeliness are depicted in Table 5. Ames test predicts the mutagenicity of the compounds and it was tested negative for all compounds except astragalin. Additionally, zero alerts were reported for Pan Assay Interference Structures (PAINS) and Table 6 represents the leadlikeliness and synthetic availability of the drugs and other test results.
Table 6.
Medicinal chemistry, toxicity studies and drug likeliness studies of top five phytomolecules
| Name | Medicinal Chemistry |
Acute Fish Toxicity |
DrugLikeliness |
||
|---|---|---|---|---|---|
| Leadlikeliness | Synthetic Accessibility Score (%) | medaka | minnow | Bioavailability Score (%) | |
| 3-p-Coumaroylquinic acid (3PC) | No;1 violation | 47.3 | 9.02 | 9.85671 | 55 |
| 4-p-Coumaroylquinic acid (4PC) | No;1 violation | 46.4 | 4.5 | 3.07595 | 11 |
| Astragalin (ASG) | No;1 violation | 52.9 | 2.88 | 1.19787 | 17 |
| Sinapoyl D-glucoside (S1G) | Yes | 40.7 | 2.13 | 1.98299 | 56 |
| 1-Sinapoyl-D-Glucose (1-SG) | yes | 40.4 | 2.0 | 1.96723 | 56 |
4. Discussion
Brassica oleacea L.var costata. locally known as espigos with elevated levels of proteins, fats, β-carotene and vitamin C is nutritionally well balanced and a rich source of energy [31]. In the current pandemic situation, our group started into the investigations of the potent antiviral agents from espigos like vitamin C, which is already well studied for its active mechanism against COVID-19. The vegetable has a good ratio of phenolics, omega 3 and omega 6 fatty acids, tocopherols and flavonoids [32]. The coronavirus disease is spreading rapidly at an alarming rate and it has become a major global health crisis. It has now become an urgent need to find a cure, as soon as possible. Many drugs are already in the market like favipiravir, hydroxychloroquine and remdesivir. While remdesivir and various other drugs are approved by the FDA to be used in an emergency, it has the worst side effects in many cases. The combination of azithromycin and hydroxychloroquine that is being used in hospitals is just effective as a placebo and causes heart problems in most cases [33]. Exploring nutraceuticals and phytochemicals will be a best alternative for a potent lead drug of COVID-19. With the advances in bioinformatics, we now have many inexpensive tools for Computer Aided Drug Designing which can be relied on for the preliminary stage screening. Molecular docking studies are one of the most inexpensive and widely used tool in drug discovery, in fact has been a boon to reduce the time course of drug discovery pipeline. The pathways of SARS-CoV-2 entry to the host such as adsorption and penetration are mostly controlled by the spike protein and neutralizing antibodies target this during the onset of infection [34]. Spike proteins thus serve as a focus for drug discovery and repurposing. The docking results obtained here suggest that coumaroylquinic acids and astragalin show strong binding to the spike protein trimer of the SARS-CoV-2, this is further validated by observing their binding energy values and inhibition constant. Coumaroylquinic acids like 3-p-coumaroylquinic acid, 4-p-coumaroylquinic acid are 5-p-coumaroylquinic acid well studied for their antibacterial and anti-inflammatory activities [35,36] while astragalin reduces lipopolysaccharide-induced acute lung injury in rats, bone destruction and free radical-induced oxidative red blood cells hemolysis in humans [37,38,39]. These drug molecules reported higher efficiency when compared to other synthetic drugs like remdesivir, favipiravir and hydroxychloroquine which are currently in use against spike protein. The study thus suggests a group of potential herbal drugs which are not yet reported for the inhibitory action from Brassica oleacea L.var costata. These were first screened based on their molecular weights and availability of three-dimensional structures in PubChem. Several molecules like quercetin-3-O-sophoroside 7-O-glucoside were removed because of their higher molecular weights (<500 g/mol) because that was one of the basic criteria of a good drug for better penetration and absorption [40]. Fourteen drug candidates were selected for docking and they were astragalin, 3-p-coumaroylquinic acid, 4-p-coumaroylquinic acid, 1-sinapoyl-D-glucose, sinapoyl-D-glucoside, sinapine, sinapic acid, aconitic acid, malic acid, quinic acid, citric acid, ascorbic acid, shikimic acid and fumaric acid. Based on their docking scores i.e., those that had a binding score higher than -7.0 kcal/mol in Autodock Vina were removed and others like astragalin, 3-p-coumaroylquinic acid, 4-p-coumaroylquinic acid, 1-sinapoyl-D-glucose and sinapoyl-D-glucoside were studied for their ADMET properties and target identification.
Determining the preclinical ADME properties and target prediction have increased the chances of development of potential drug molecules. Most of the studied drugs follow Lipinski's rules and don't penetrate the BBB while 55% of the coumaroylquinic acids have the potential to reach the target organ. They are expected to exhibit 33.3% protease activity on the spike protein while astragalin shows 100% aldose reductase and carbonic anhydrase actions. A highly negative value of Log P was reported for the compounds which means that they had higher affinity for the aqueous phase. PAIN alerts were given by none of the five molecules. They do not have any adverse effect on synthesis of cholesterol or other lipids, epoxygenase, drug metabolism, NADPH-dependent electron transport pathway and hydroxylation of antimalarial drug quinine because they don't inhibit CYP1A2, CYP2C19, CYP2D6, CYP2D9 and CYP3A4 respectively [41]. In addition to the higher binding affinity, extensive Autodock4 simulations are also carried out to analyze the interactions between the spike protein and ligands. Astragalin and Coumaroylquinic acids forms strong hydrogen bonds with several amino acid residues of the spike protein chain A namely, Glu1010, Arg765, Ile1013, Arg1019, Asp950, Glu773, Lys776, Thr1006, Gln1005, Gln1002.
Interestingly, these compounds show the significant binding with S2 domain of spike protein unlike the interactions of S1/RBD interface with most of the drugs which have been reported in previous research. The S2 domain of spike protein plays crucial role in the fusion of viral and host cell membrane by generating a six-helical bundle via the two-heptad repeat domain [12,13,42]. In the present study, we have demonstrated the substantial accessibility of selected drug candidates to the interior of spike protein and their interaction with S2 domain of spike protein. The binding pattern of Astragalin and Coumaroylquinic acids with S2 domain reveals newer insights to the virus entry. Based on our in-silico binding studies we here propose a plausible scheme (Fig.1) showing the ways to inhibit the fusion of viral membrane with the host cell surface, thereby reducing the internalization of viral particles into the host cells.
In SARS-CoV-2, SARS-CoV and MERS-CoV proteins, crucial active sites are situated in a shallow groove between the two beta-sheets that is composed of His 235, His 250, Lys 290, Thr 341, Tyr 343, and Ser 294. In our study Astragalin is observed to form a hydrogen bond with the Arg765 and Glu773 as well as two hydrogen bonds with Asp950, this is reflected on the lowest binding free energy values of -9.55 kcal/mol observed for spike trimer-ASG complex further confirming the docking studies and stability of the complex). According to a recent study [43], three residues i.e., His 374, His 378, Glu 402 were reported as key amino acids at the active site pocket of spike glycoprotein. Similar binding mode is seen for Astragalin which interacts strongly with Glu773 and Asp950 via hydrogen bonds (Fig. 2a, 3a). Strong binding is also seen in 3p-Coumaroylquinic acid, with Glu 661 via two hydrogen bonds as seen earlier for other ligands [44]. The smaller fluctuations in RMSD values <2.0 Å during the time course of simulation runs confirms the substantial stability of the spike trimer complexes when bound to representative ligands. The binding free energy of the spike trimer glycoprotein complexes obtained post simulation runs are in good agreement with the docking calculations indicating that all the three ligands ASG, 4 PC and 3 PC, forms a tight binding complex by forming consistent hydrogen bonds involving key residues in the binding groove of the spike trimer (Fig 5). The spike trimer complex with ligands 3-p-Coumaroylquinic Acid and Astragalin in this case showed lower values of RMSD values and better binding free energy suggesting that these ligands strongly interact with the trimeric spike glycoprotein. The binding free energy of spike trimer-ASG, 4 PC and 3 PC bound complexes at 0, 10, 20, 30, 40, 50, 60, 70, 80, 90, 100 ns interval of the MD sampling run indicates stable interactions of the ligands in the binding groove (Table 4). Although, there have been recent studies showing that density functional tight binding (DFTB) may also provide more accurate binding interactions than MD along with some additional insights and could also scale well to the large systems [45,46]. This is new heterogeneous CPU+GPU-enhanced DFTB approach to carry out the tedious and competent simulation of large chemical and biological systems.
In silico toxicity studies reveal that the top scoring drugs are safe for their extensive use either as a therapeutic or as a prophylactic measure but due to lockdown situations in India, we are not able to carry out in vitro and in vivo validations and effectiveness of these drugs. However, we understand that these findings may help the researchers who are tirelessly working to develop or discover an effective drug against SARS-CoV-2.
5. Conclusion
The study discovered that several natural phytomolecules like phenolics and organic acids from Tronchuda cabbage (B. oleracea) are active nontoxic molecules with diverse predicted enzyme, protease and kinase activities are effective against SARS-CoV-2 spike protein. The binding affinities, energy estimations and ADMET analysis resulted in the discovery of potential molecules like coumaroylquinic acids, astragalin, sinapoyl-D-glucoside and 1-sinapoyl-D-glucose which have binding energies less than -7 kcal/mol. Coumaroylquinic acids and astragalin are better in action and cause fewer toxic effects than market available drugs like remdesivir and hydroxychloroquine. Coumaroylquinic acids with lower numerical values of rotatable bonds, total polar surface area and accurate number of hydrogen bond acceptors and hydrogen bond donors (as per Lipinski's rule) make them suitable drug molecules with good skin penetration and solubility, average gastrointestinal tract absorption and non-blood brain barrier permeant. When compared to other molecules like Cangrelor and Dpnh (NADH) with binding affinities of -7.23 kcal/mol and -7.03 kcal/mol respectively, these molecules are much better in their two dimensional and three-dimensional structural properties, binding scores and free intermolecular energies [47]. The inhibitor constant, Ki, is a value suggesting exactly how effective an inhibitor is; it is the concentration of the drug obligatory to yield half maximum inhibition and the coumaroylquinic acids gave the least Ki value which shows the ability to inhibit even at the concentration of about 11 μM while astragalin requires 181.54 μM. Although these in silico approaches give positive response regarding use and safety of coumaroylquinic acids, astragalin, sinapoyl-D-glucoside and 1-sinapoyl-D-glucose for inhibiting SARS-CoV-2, further in vitro and in vivo experimental assays are suggested to evaluate the clinical efficacy.
Funding
Not applicable.
Conflicts of interest/Competing interests
Authors wish to confirm that there are no known conflicts of interest associated with this publication and there has been no significant financial support for this work that could have influenced its outcome.
Ethics approval
Not applicable.
Consent to participate
All authors have given their consent to participate in this work.
Consent for publication
All authors have given their consent to publish this work.
Availability of data and material
Not applicable.
Code availability
Not applicable.
Author's contribution
MD, SJ and MG and have designed and conceptualized the work. Experiments were done by SJ and MG. US prepared the input files for molecular dynamics simulation runs. US and JQS performed molecular dynamics calculations. US and MD analyzed the simulation results. Manuscript is written by MD, SJ, MG and US. MD and US are co-senior authors.
Abbrevitions
Severe Acute Respiratory Syndrome, SARS-CoV; Middle East Respiratory Syndrome related Coronaviruses, MERS-CoV; CoronaVirus Infection Disease, COVID-19; World Health organization, WHO; Non-Structural Proteins, NSP; Angiotensin Converting Enzyme, ACE; Absorption, Distribution, Metabolism, Excretion and Toxicology, ADMET; Blood Brain Barrier, BBB; Human Intestinal Absorption, HIA; Gastrointestinal, GI absorption; Pan Assay Interference Structures, PAINS, 3-p-Coumaroylquinic Acid; 3 PC, 4-p-Coumaroylquinic Acid; 4 PC, Astragalin; ASG, 1-Sinapoyl-D- Glucose; 1SG) and Sinapoyl-D-Glucoside; S1G.
CRediT authorship contribution statement
Sandra Jose: Software, Methodology, Investigation, Writing – original draft, Validation. Megha Gupta: Software, Investigation. Urvashi Sharma: Software, Investigation, Formal analysis, Validation, Writing – review & editing. Jorge Quintero-Saumeth: Software, Investigation, Formal analysis. Manish Dwivedi: Conceptualization, Methodology, Formal analysis, Writing – original draft, Writing – review & editing, Supervision.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgment
MD acknowledge the DST-INSPIRE Faculty award, 2017 (IFA17-LSPA82), Govt. of India. US acknowledges DBT, Govt. of India for Ramalingaswami re-entry fellowship. The authors acknowledge the VMs access from the NMR box platform. Gerard Weatherby from NMR support team is acknowledged for the help with High performance computer.
Footnotes
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.molstruc.2022.132369.
Appendix. Supplementary materials
References
- 1.Rabi F.A., Al Zoubi M.S., Kasasbeh G.A., Salameh D.M., Al-Nasser A.D. SARS-CoV-2 and coronavirus disease 2019: what we know so far. Pathogens. 2020;9(3):231. doi: 10.3390/pathogens9030231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shereen M.A., Khan S., Kazmi A., Bashir N., Siddique R. COVID-19 infection: origin, transmission, and characteristics of human coronaviruses. J. Adv. Res. 2020;16(24):91–98. doi: 10.1016/j.jare.2020.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xia S., Liu M., Wang C., Xu W., Lan Q., Feng S., Qi F., Bao L., Du L., Liu S., Qin C. Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 2020;30(4):343–355. doi: 10.1038/s41422-020-0305-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a.https://www.who.int/emergencies/diseases/novel-coronavirus-2019/situation-reports
- 5.Singh K., Mittal A.K., Gujral A. The Epidemiology, Evolution, Transmission and Therapeutics of COVID-19 Outbreak: an Update on the Status. Coronaviruses. 2021;2(11):9. doi: 10.2174/2666796702666210210140840. [DOI] [Google Scholar]
- 6.Minga I.G., Golemi L., Tafur A., Pursnani A. The Novel Coronavirus Disease (COVID-19) and Its Impact on Cardiovascular Disease. Cardiol. Rev. 2020;28(4):163–176. doi: 10.1097/CRD.0000000000000317. [DOI] [PubMed] [Google Scholar]
- 7.b.https://www.who.int/indonesia/news/detail/08-03-2020-knowing-the-risk-for-covid-19.
- 8.Costanzo M., De Giglio M.A.R., Roviello G.N. SARS-CoV-2: recent Reports on Antiviral Therapies Based on Lopinavir/Ritonavir, Darunavir/Umifenovir, Hydroxychloroquine, Remdesivir, Favipiravir and other Drugs for the Treatment of the New Coronavirus. Curr. Med. Chem. 2020;27(27):4536–4541. doi: 10.2174/0929867327666200416131117. [DOI] [PubMed] [Google Scholar]
- 9.Food and Drug Administration, 2020. FDA cautions against use of hydroxychloroquine or chloroquine for COVID-19 outside of the hospital setting or a clinical trial due to risk of heart rhythm problems.
- 10.Yanai H. Favipiravir: a Possible Pharmaceutical Treatment for COVID-19. J. Clin. Endocrinol. Metab. 2020;10(2):33–34. doi: 10.14740/jem645. [DOI] [Google Scholar]
- 11.Walls A.C., Park Y.J., Tortorici M.A., Wall A., McGuire A.T., Veesler D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell. 2020;16(2):281–292.e6. doi: 10.1016/j.cell.2020.02.058. 181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ke Z., Oton J., Qu K., Cortese M., Zila V., McKeane L., Nakane T., Zivanov J., Neufeldt C.J., Cerikan B., Lu J.M., Peukes J., Xiong X., Kräusslich H.G., Scheres S.H.W., Bartenschlager R., Briggs J.A.G. Structures and distributions of SARS-CoV-2 spike proteins on intact virions. Nature. 2020;588(7838):498–502. doi: 10.1038/s41586-020-2665-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cai Y., Zhang J., Xiao T., Peng H., Sterling S.M., Walsh R.M., Jr., Rawson S., Rits-Volloch S., Chen B. Distinct conformational states of SARS-CoV-2 spike protein. Science. 2020;25(6511):1586–1592. doi: 10.1126/science.abd4251. 369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aires A., Fernandes C., Carvalho R., Bennett R.N., Saavedra M.J., Rosa E.A. Seasonal effects on bioactive compounds and antioxidant capacity of six economically important Brassica vegetables. Molecules. 2011;16(8):6816–6832. doi: 10.3390/molecules16086816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Batista C., Barros L., Carvalho A.M., Ferreira I.C. Nutritional and nutraceutical potential of rape (Brassica napus L. var. napus) and “tronchuda” cabbage (Brassica oleraceae L. var. costata) inflorescences. Food Chem. Toxicol. 2011;49(6):1208–1214. doi: 10.1016/j.fct.2011.02.023. [DOI] [PubMed] [Google Scholar]
- 16.Vrchovská V., Sousa C., Valentão P., Ferreres F., Pereira J.A., Seabra R.M., Andrade P.B. Antioxidative properties of tronchuda cabbage (Brassica oleracea L. var. costata DC) external leaves against DPPH, superoxide radical, hydroxyl radical and hypochlorous acid. Food Chem. 2006;98(3):416–425. doi: 10.1016/j.foodchem.2005.06.019. [DOI] [Google Scholar]
- 17.Morris G.M., Huey R., Lindstrom W., Sanner M.F., Belew R.K., Goodsell D.S., Olson AJ. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 2009;30(16):2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jo S., Kim T., Iyer V.G., Im W. CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput. Chem. 2008;29(11):1859–1865. doi: 10.1002/jcc.20945. [DOI] [PubMed] [Google Scholar]
- 19.Lee J., Cheng X., Swails J.M., Yeom M.S., Eastman P.K., Lemkul J.A., Wei S., Buckner J., Jeong J.C., Qi Y., Jo S., Pande V.S., Case D.A., Brooks C.L., 3rd, MacKerell A.D., Jr., Klauda J.B., Im W. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016;12(1):405–413. doi: 10.1021/acs.jctc.5b00935. 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brooks B.R., Brooks C.L., 3rd, Mackerell A.D., Jr., Nilsson L., Petrella R.J., Roux B., Won Y., Archontis G., Bartels C., Boresch S., Caflisch A., Caves L., Cui Q., Dinner A.R., Feig M., Fischer S., Gao J., Hodoscek M., Im W., Kuczera K., Lazaridis T., Ma J., Ovchinnikov V., Paci E., Pastor R.W., Post C.B., Pu J.Z., Schaefer M., Tidor B., Venable R.M., Woodcock H.L., Wu X., Yang W., York D.M., Karplus M. CHARMM: the biomolecular simulation program. J. Comput. Chem. 2009;30(10):1545–1614. doi: 10.1002/jcc.21287. 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hammad S., Bouaziz-Terrachet S., Meghnem R., Meziane D. Pharmacophore development, drug-likeness analysis, molecular docking, and molecular dynamics simulations for identification of new CK2 inhibitors. J. Mol. Model. 2020;26(6):160. doi: 10.1007/s00894-020-04408-2. [DOI] [PubMed] [Google Scholar]
- 22.Darden T., York D., Pedersen L. Particle mesh Ewald: an N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993;98(12):10089. doi: 10.1063/1.464397. [DOI] [Google Scholar]
- 23.Essmann U., Perera L., Berkowitz M.L., Darden T., Lee H., Pedersen L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995;103(19):8577. doi: 10.1063/1.470117. [DOI] [Google Scholar]
- 24.Abraham M.J., Murtola T., Schulz R., Páll S., Smith J.C., Hess B., Lindahl E. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1–2:19–25. doi: 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- 25.Maciejewski M.W., Schuyler A.D., Gryk M.R., Moraru I.I., Romero P.R., Ulrich E.L., Eghbalnia H.R., Livny M., Delaglio F., Hoch J.C. NMRbox: a Resource for Biomolecular NMR Computation. Biophys. J. 2017;112:1529–1534. doi: 10.1016/j.bpj.2017.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14(1):33–38. doi: 10.1016/0263-7855(96)00018-5. Feb;27-8. [DOI] [PubMed] [Google Scholar]
- 27.Gfeller D., Grosdidier A., Wirth M., Daina A., Michielin O., Zoete V. SwissTargetPrediction: a web server for target prediction of bioactive small molecules. Nucleic Acids Res. Spec. Publ. 2014;42(W1):W32–W38. doi: 10.1093/nar/gku293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gohlke H., Hendlich M., Klebe G. Knowledge-based scoring function to predict protein-ligand interactions. J. Mol. Biol. 2000;295(2):337–356. doi: 10.1006/jmbi.1999.3371. [DOI] [PubMed] [Google Scholar]
- 29.Kramer B., Rarey M., Lengauer T. Evaluation of the FLEXX incremental construction algorithm for protein-ligand docking. Proteins. 1. 1999;37(2):228–241. doi: 10.1002/(sici)1097-0134(19991101)37:2<228::aid-prot8>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 30.Lin J.H., Yamazaki M. Role of P-glycoprotein in pharmacokinetics: clinical implications. Clin. Pharmacokinet. 2003;42(1):59–98. doi: 10.2165/00003088-200342010-00003. [DOI] [PubMed] [Google Scholar]
- 31.Ferreres F., Sousa C., Vrchovská V., Valentão P., Pereira J.A., Seabra R.M., Andrade P.B. Chemical composition and antioxidant activity of tronchuda cabbage internal leaves. Eur. Food Res. Technol. 2006;222(1–2):88–98. doi: 10.1021/jf072740y. [DOI] [Google Scholar]
- 32.Charles, D.J., 2012. Sources of natural antioxidants and their activities. In: Antioxidant properties of spices, Herbs and Other Sources. Springer, New York, NY, pp. 65–138. https://doi.org/10.1007/978-1-4614-4310-0_4
- 33.Molina J.M., Delaugerre C., Le Goff J., Mela-Lima B., Ponscarme D., Goldwirt L., de Castro N. No evidence of rapid antiviral clearance or clinical benefit with the combination of hydroxychloroquine and azithromycin in patients with severe COVID-19 infection. Med. Mal. Infect. 2020;50(384):30085–30088. doi: 10.1016/j.medmal.2020.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu, X., Gao, F., Gou, L., Chen, Y., Gu, Y., Ao, L., Shen, H., Hu, Z., Guo, X. and Gao, W., 2020. Neutralizing Antibodies Isolated by a site-directed Screening have Potent Protection on SARS-CoV-2 Infection. bioRxiv. https://doi.org/10.1101/2020.05.03.074914.
- 35.Bazylko A., Granica S., Filipek A., Piwowarski J., Stefańska J., Osińska E., Kiss A.K. Comparison of antioxidant, anti-inflammatory, antimicrobial activity and chemical composition of aqueous and hydroethanolic extracts of the herb of Tropaeolum majus L. Ind. Crops Prod. 2013;50:88–94. doi: 10.1016/j.indcrop.2013.07.003. [DOI] [Google Scholar]
- 36.Ștefănescu B.E., Szabo K., Mocan A., Crişan G. Phenolic compounds from five Ericaceae species leaves and their related bioavailability and health benefits. MOLEFW. 2019;24(11):2046. doi: 10.3390/molecules24112046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zheng D., Liu D., Liu N., Kuang Y., Tai Q. Astragalin reduces lipopolysaccharide-induced acute lung injury in rats via induction of heme oxygenase-1. Arch. Pharm. Res. 2019;42(8):704–711. doi: 10.1007/s12272-019-01171-8. [DOI] [PubMed] [Google Scholar]
- 38.Jia Q., Wang T., Wang X., Xu H., Liu Y., Wang Y., Shi Q., Liang Q. Astragalin suppresses inflammatory responses and bone destruction in mice with collagen-induced arthritis and in human fibroblast-like synoviocytes. Front. Pharmacol. 2019;10:94. doi: 10.3389/fphar.2019.00094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Choi J., Kang H.J., Kim S.Z., Kwon T.O., Jeong S.I., Jang S.I. Antioxidant effect of astragalin isolated from the leaves of Morus alba L. against free radical-induced oxidative hemolysis of human red blood cells. Arch. Pharm. Res. 2013;36(7):912–917. doi: 10.1007/s12272-013-0090-x. [DOI] [PubMed] [Google Scholar]
- 40.Abdullahi M., Adeniji S.E. In-silico Molecular Docking and ADME/Pharmacokinetic Prediction Studies of Some Novel Carboxamide Derivatives as Anti-tubercular Agents. Chem. Afr. 2020;3:989–1000. doi: 10.1007/s42250-020-00162-3. [DOI] [Google Scholar]
- 41.Gopisankar M.G. CYP2D6 pharmacogenomics. Egypt. J. Med. Hum. Genet. 2017;18(4):309–313. doi: 10.1016/j.ejmhg.2017.03.001. [DOI] [Google Scholar]
- 42.Huang Y., Yang C., Xu X.F., Xu W., Liu SW. Structural and functional properties of SARS-CoV-2 spike protein: potential antivirus drug development for COVID-19. Acta Pharmacol. Sin. 2020;41:1141–1149. doi: 10.1038/s41401-020-0485-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Prasanth D.S.N.B.K., Murahari M., Chandramohan V., Panda S.P., Atmakuri L.R., Guntupalli C. In silico identification of potential inhibitors from Cinnamon against main protease and spike glycoprotein of SARS CoV-2. J. Biomol. Struct. Dyn. 2021;39(13):4618–4632. doi: 10.1080/07391102.2020.1779129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sinha N., Balayla G. Hydroxychloroquine and COVID-19. Postgrad. Med. J. 2020;96(1139):550–555. doi: 10.1136/postgradmedj-2020-137785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Allec S.I., Sun Y., Sun J., Chang C.A., Wong B.M. Heterogeneous CPU+GPU-Enabled Simulations for DFTB Molecular Dynamics of Large Chemical and Biological Systems. J. Chem. Theory Comput. 2019;14(5):2807–2815. doi: 10.1021/acs.jctc.8b01239. 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yoshikawa T., Komoto N., Nishimura Y., Nakai H. GPU-Accelerated Large-Scale Excited-State Simulation Based on Divide-and-Conquer Time-Dependent Density-Functional Tight-Binding. J. Comput. Chem. 2019;5(31):2778–2786. doi: 10.1002/jcc.26053. 40. [DOI] [PubMed] [Google Scholar]
- 47.Hall D.C., Jr., Ji HF. A search for medications to treat COVID-19 via in silico molecular docking models of the SARS-CoV-2 spike glycoprotein and 3CL protease. Travel Med. Infect. Dis. 2020;35 doi: 10.1016/j.tmaid.2020.101646. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Not applicable.