

Abstract
Carbohydrate chains are ubiquitous in the complex molecular processes of life. These highly diverse chains are recognized by a variety of protein receptors, enabling glycans to regulate many biological functions. High-resolution structures of protein–glycoligand complexes reveal the atomic details necessary to understand this level of molecular recognition and inform application-focused scientific and engineering pursuits. When experimental challenges hinder high-throughput determination of quality structures, computational tools can, in principle, fill the gap. In this work, we introduce GlycanDock—a residue-centric protein–glycoligand docking refinement algorithm developed within the Rosetta macromolecular modeling and design software suite. We performed a benchmark docking assessment using a set of 109 experimentally determined protein–glycoligand complexes as well as 62 unbound protein structures. The GlycanDock algorithm can sample and discriminate protein–glycoligand models of native-like structural accuracy with statistical reliability from starting structures of up to 7 Å root-mean-square deviation in the glycoligand ring atoms. We show that GlycanDock-refined models qualitatively replicated the known binding specificity of a bacterial carbohydrate-binding module. Finally, we present a protein–glycoligand docking pipeline for generating putative protein–glycoligand complexes when only the glycoligand sequence and unbound protein structure are known. In combination with other carbohydrate modeling tools, the GlycanDock docking refinement algorithm will accelerate research in the glycosciences.
Introduction
Carbohydrates are the most abundant and diverse biomolecules found on Earth1,2. Finite chains of carbohydrates known as glycans play numerous functional roles in all three domains of life3–9 as well as viruses10,11. Three-dimensional structures of protein–glycan complexes provide insight into how carbohydrates are recognized by proteins and mediate biological functions. For example, extensive structural analysis of lectins (carbohydrate-binding proteins) via X-ray crystallography uncovered the role of carbohydrate recognition in cell–cell interactions and pathogenic invasion12–15. The Protein Databank (PDB) serves as a global repository for experimentally determined three-dimensional structures16. Recent estimates indicate that entries containing carbohydrates make up less than 10% of the PDB17—of which only a few thousand represent a high-quality, true protein–glycoligand complex (a non-covalently attached glycan bound to a protein receptor)18. Consequently, resolved structures of protein–glycoligand complexes are relatively underrepresented compared to their ubiquity in biology. Despite technological improvements in experimental structural glycobiology19,20, the innate flexibility and chemical heterogeneity of carbohydrate chains continues to hinder high-throughput collection of high-quality structures21–24. Therefore, computational docking tools that accurately predict the conformation and interfacial interactions of protein–glycoligand complexes are needed to fill the gap in structural characterization and enable further scientific and engineering advancements25.
Computational simulations have long demonstrated utility in supplementing and deciphering experimental data on the structure of glycans and protein–glycan complexes22–24,26–35. Molecular dynamics (MD) simulations in particular are able to sample oligosaccharide conformations that are consistent with experimental data36,37 and estimate the binding free energy of protein–glycoligand complexes22,38–40. However, MD becomes too costly when simulating large systems with many atoms and degrees of freedom41, making faster (though less rigorous) computational tools for docking more practical. Protein–ligand docking software including AutoDock42, AutoDock Vina43, DOCK44, FlexX45, Glide46, and GOLD47 have all been applied to protein–glycoligand systems48–53. However, these tools work best on rigid, small-molecule (i.e. drug-like) ligands with few rotatable bonds. Accordingly, modeling and docking tools that account for the size and flexibility of glycoligands, such as AutoDock Vina-Carb54,55 and the fragment-based approach developed by Samsonov and colleagues56, are necessary.
The Rosetta macromolecular modeling and design software suite57 has been used to address diverse scientific challenges58–60. Rosetta’s protein–small molecule docking algorithm RosettaLigand61,62 has been applied to a protein–glycoligand system to capture the effects of mutations on glycoligand binding energetics63. However, like other protein–ligand docking software, RosettaLigand is only able to treat a ligand as a single residue with discrete, precomputed conformations—not as flexible oligomers. The recently developed RosettaCarbohydrate framework enabled modeling and design of glycans and glycoconjugate systems in a residue-centric (i.e., oligomeric) approach64. To that end, we sought to develop a docking refinement algorithm that leverages the RosettaCarbohydrate framework and rapid conformational sampling and optimization techniques to predict native-like, biophysically accurate models of protein–glycoligand complexes.
Here, we introduce GlycanDock—a residue-centric protein–glycoligand docking refinement algorithm available within the Rosetta software suite. In this work, we assess GlycanDock’s ability to sample and discriminate bound, native-like conformations of protein–glycoligand complexes using a benchmark target set of 109 high-resolution structures from the PDB. Targets represent protein binders of broad scientific interest, including 11 antibodies, 33 lectins, 22 enzymes, 24 carbohydrate-binding modules, and 19 viral glycan binders. These 109 proteins are bound to glycoligands of various lengths, including 24 di-, 32 tri-, 28 tetra, 14 penta-, 7 hexa-, 3 heptasaccharides, and 1 undecasaccharide. 81 are linear oligosaccharides and 28 have one or more branched connections. 17 have one or more exocyclic linkages. We also use 62 experimentally determined unbound protein structures to evaluate the effect of pre-configuration of the protein backbone on docking performance. To assess whether GlycanDock captures high-resolution structural details, we examine the counts and recovery of native-like biophysical features such as interfacial residue–residue contacts and hydrogen bonds. As a case study, we probe GlycanDock’s ability to recapitulate known glycoligand binding preferences of a carbohydrate-binding module (CtCBM6). Finally, we report on the results of a pipeline for performing “blind” glycoligand docking when only the unbound protein structure and glycoligand sequence are known. The results of the benchmark assessment presented in this work demonstrate the effectiveness and overall utility of the GlycanDock protein–glycoligand docking refinement algorithm.
Materials and Methods
GlycanDock: the Rosetta protein–glycoligand docking refinement algorithm
GlycanDock is a Monte Carlo-plus-minimization docking refinement algorithm that features a high-resolution (all-atom) sampling and refinement strategy to locally optimize a glycoligand’s conformation within a putative protein receptor pocket. GlycanDock’s sampling algorithm leverages data mined from the Protein Data Bank (PDB)16 and extracted from quantum-mechanics calculations54,55 to ensure carbohydrate-specific degrees of freedom (DoFs) fall within energetically-favorable, native-like conformational space64. The output of a GlycanDock trajectory includes the coordinates of the predicted model in PDB format and a breakdown of the model’s total Rosetta score written to a Rosetta score file. The score file additionally reports some of the docking performance metrics described in this work, such as interface score, ring-RMSD, and ring-SRMSD.
Initial protein–glycoligand complex for use as input to GlycanDock
The GlycanDock algorithm requires a pre-packed (see Stage 0 below) putative protein-glycoligand complex as input, where the protein receptor and glycoligand each have their own unique chain identifiers (e.g., protein chain A and glycoligand chain X). GlycanDock performs local docking only, meaning the input structure must have the glycoligand physically placed within the predicted the binding site and, for larger glycoligands, placed in approximately the correct rigid-body orientation. It is assumed that the protein receptor backbone is approximately correct, as it is kept fixed throughout the docking trajectory. In contrast, the initial glycosidic torsion angles can be arbitrary, as they are sampled and energetically minimized during docking; however, it is recommended that the initial glycoligand conformation provided is low energy. Protein side-chain rotamers and carbohydrate side-chain rotamers (e.g., hydroxyl and N-acetyl groups) at the protein-glycoligand interface are optimized throughout the docking trajectory to minimize clashes while searching for productive interfacial interactions. By default, carbohydrate ring conformations are not sampled, but may be included as a DoF using the command-line interface. Additional information describing the preparation of input structures is detailed in “Benchmarking and evaluation of the GlycanDock algorithm”.
Stage 0: Pre-packing the initial, putative protein–glycoligand complex
In Stage 0 of the GlycanDock algorithm, protein and carbohydrate side chains are pre-packed65 to ensure compatibility of the input complex with the Rosetta scoring function. This is performed by removing any internal clashes within the initial structure and thus establishing a low-energy conformation at non-interface regions of the protein receptor. In the case of the bound targets employed in this study, pre-packing additionally serves to erase the pre-configuration of the protein side chains at the interface to bind the glycoligand, thus reducing bias during docking. Hydrogen atoms are added, the glycoligand is separated by 1,000 Å from the protein receptor, and all non-disulfide bridge side-chain conformations are optimized by rotamer (i.e., packed) and energy-minimized. The glycoligand is then translated back to its starting position. The Stage 0 pre-packing procedure should be performed on the initial, putative protein–glycoligand complex prior to running the GlycanDock docking algorithm. Pre-packing can be performed using the following example command-line flags:
./GlycanDock.macosclangrelease -include_sugars -alternate_3_letter_codes pdb_sugar -auto_detect_glycan_connections -in:file:s target.pdb -in:file:native crystal.pdb -nstruct 1 -ex1 -ex2 -ex3 -ex4 -ex1aro -ex2aro -carbohydrates:glycan_dock:prepack_only true -docking:partners A_X -out:pdb_gz
Here, -docking:partners A_X informs the GlycanDock algorithm that the upstream protein receptor is identified by chain A and the downstream glycoligand is chain X.
Stage 1: Initialize docking trajectory by applying random perturbations to the input glycoligand configuration
Each GlycanDock trajectory begins by applying a small, random perturbation to the glycoligand’s rigid-body orientation and to all glycosidic torsion angles. The objective of Stage 1 is to increase glycoligand sampling coverage within the protein binding site by promoting additional conformational diversity to the input putative complex. A Gaussian translational perturbation centered around 0.5 Å and a rotational perturbation centered around 7.5° is applied to the center-of-mass of the glycoligand. A uniform perturbation of ± 12.5° is applied to each glycosidic torsion angle. We note that many Rosetta docking protocols typically employ an initial low-resolution (centroid) search stage in lieu of the Stage 1 initial perturbation procedure described here, but the functionality required to model carbohydrates as centroid representations has not yet been incorporated into the RosettaCarbohydrate framework 64 at the time of writing.
Stage 2: Docking and refinement of the input protein–glycoligand complex
Stage 2 of the GlycanDock algorithm focuses on exploring the local conformational space of the glycoligand through rigid-body and glycosidic torsion angle sampling and refinement. This stage consists of two sets of eight inner cycles of Monte Carlo sampling and optimization of the glycoligand conformation at the putative binding site. The inner refinement cycles are wrapped by ten outer cycles that ramp the weights of the attractive and repulsive terms in the Rosetta scoring function, similar to the approach taken in the Rosetta FlexPepDock protein–peptide docking algorithm65,66. In the first outer cycle, the weight of the repulsive Lennard–Jones term (fa_rep) is reduced to 45% of its default magnitude, and the attractive van der Waals term (fa_atr) is increased by 325%. The weights are returned to their original magnitudes incrementally over the course of the proceeding outer cycles so that the final outer cycle uses the starting weights for these two score terms.
The inner cycles perform the sampling and optimization procedures on the glycoligand. The inner cycles consist of a set of eight rigid-body perturbations and a set of eight glycosidic torsion angle perturbations (performed in either order every inner cycle). Every perturbation is followed by interfacial side-chain rotamer optimization (packing), and every other perturbation is followed by full-structure energy minimization. Rigid-body sampling consists of uniform perturbations to the glycoligand’s center-of-mass as well as occasional translation of the glycoligand toward the protein receptor’s center-of-mass. This latter “sliding” step is occasionally necessary when clashes cause large gradients that during minimization jump the glycoligand far away from the protein. Glycosidic-linkage sampling includes performing uniform and non-uniform perturbations of various magnitudes on randomly selected glycosidic torsion angles. Sampling may also include occasionally flipping an entire carbohydrate ring around with respect to the rest of the carbohydrate chain (without changing the internal conformation of the carbohydrate ring itself) for glycoligands that satisfy specific glycosidic dihedral topology requirements (see Supplemental). Further details on the sampling and optimization procedures performed in the GlycanDock algorithm are available in the Supplementary Information.
GlycanDock trajectory-level information, such as the number of inner sampling and optimization cycles performed and accepted, are reported at the bottom of the output structure file.
Command-line usage of GlycanDock
GlycanDock-specific flags are described in the Table S1. An example of the flags used for the benchmark assessment of GlycanDock is shown below:
./GlycanDock.macosclangrelease -include_sugars -maintain_links -in:file:s target-prepacked.pdb -in:file:native crystal.pdb -cst_fa_file interface.cst -nstruct 50 -n_cycles 10 -ex1 -ex2 -docking:partners A_X -out:pdb_gz
Here, -maintain_links is used rather than -auto_detect_glycan_connections because the input structure (target-prepacked.pdb) has already been processed by Rosetta and therefore has the appropriate LINK records defining the glycoligand’s carbohydrate connectivity64,67.
Benchmarking and evaluation of the GlycanDock algorithm
Metrics for evaluation of model accuracy and ranking of models
The ring-RMSD metric is used to evaluate the structural accuracy of GlycanDock models. Ring-RMSD is the root-mean-squared deviation (RMSD) of all ring atoms of the glycoligand in its predicted conformation to its native bound conformation after superposition of the protein receptor onto the native protein backbone. Ring-RMSD captures both the deviation in the shape and the orientation with respect to the binding site of the glycoligand. Models below 2 Å ring-RMSD were classified as a near-native (i.e., sufficiently representative of the native bound conformation). Ring-SRMSD was also calculated to evaluate the structural accuracy of the shape of the glycoligand irrespective of its orientation in the binding site (i.e., this metric can be calculated irrespective of the protein receptor). Ring-SRMSD is the RMSD of all ring atoms of the glycoligand after superposition of its predicted conformation onto its native bound conformation.
GlycanDock models were ranked by interface score. The interface score is calculated by taking the total Rosetta score (the weighted sum of all the terms in the Rosetta scoring function) of the model and subtracting the total score of the separated model where the glycoligand is translated 1,000 Å away from the protein receptor. The interface score approximates the binding free energy of the complex in units of REU (Rosetta Energy Units). The N5 metric is used to quantify the effectiveness of GlycanDock sampling and the discriminatory power of the calculated interface score. N5 is the count of near-native models ranked among the top-5-scoring of all predicted models for a given target.
Bootstrap statistical analysis to determine effective docking range
Bootstrap case resampling was used as described previously68,69 to determine GlycanDock’s effective docking range. Briefly, for a given target, 5,000 sets of resampled models were generated by randomly selecting 1,000 models with replacement from the original set of GlycanDock models. The observed N5 of each target from each randomly resampled set was then averaged and reported as 〈N5〉 (standard deviation σ〈N5〉). Targets that resulted in 〈N5〉 ≥1.0 were considered a docking success. The effective docking range of GlycanDock was then defined as the maximum initial ring-RMSD from which 50% or more of tested protein-glycoligand targets achieved 〈N5〉 ≥ 1.0. Bootstrap case resampling was performed utilizing the pandas70 data analysis tool (see Supplemental Information for pseudo-code example).
Analysis of biophysical features of GlycanDock models
Definition of the protein–glycoligand interface and biophysical features
A protein and carbohydrate residue are making an interfacial residue–residue contact if at least one non-hydrogen atom of a residue on one side of the interface (e.g. a protein residue) is within 5 Å of at least one non-hydrogen atom on the other side of the interface. A single protein or carbohydrate residue can make multiple unique interfacial residue–residue contacts (e.g. carbohydrate residue 1 contacts protein residue 12; carbohydrate residue 2 contacts protein residues 12 and 19). Interface residues are defined by the unique set of all residues making interfacial contacts (e.g. carbohydrate and protein residues 1, 2, 12, and 19 of the previous example are interfacial residues). The counts of interfacial residue–residue contacts and interface residues are reported at the bottom of the output structure file. In addition, the set of interface residues is reported as a PyMOL71-based residue selection at the bottom of the output structure file.
Hydrogen bonds are identified geometrically as those interactions contributing at least −0.5 energy units to the total “hbond” term of the Rosetta scoring function72,73. To be considered an interfacial hydrogen bond, a hydrogen bond must be between a carbohydrate residue of the glycoligand and a protein residue of the receptor. Interfacial hydrogen bond that pass the score cutoff are reported per carbohydrate residue in the form of a PyMOL-based residue selection at the bottom of the output structure file. The energetic filtering is performed by the HBondSelector at the end of a GlycanDock trajectory. Counting of interfacial protein-glycoligand hydrogen bonds is performed by parsing the corresponding PyMOL-based residue selections using an in-house Python script.
Measuring recovery of biophysical features
We consider an interfacial residue–residue contact or interfacial hydrogen bond recovered if the pair of interacting residues is the same pair observed in the native crystal complex. Similarly, an interfacial residue is recovered if it is present at the interface in the native crystal complex. Recovery is given as a fraction of the native biophysical feature recovered. Recovery ranges from 0.0 to 1.0, where 1.0 indicates complete native recovery. Recovery of interfacial residue–residue contacts and interfacial residues is calculated at the end of the GlycanDock trajectory and reported at the bottom of the output structure file. Recovery of interfacial hydrogen bonds is calculated using the corresponding data after parsing with the in-house Python script.
Local docking refinement of the crystal complex as a reference for GlycanDock performance All bound protein–glycoligand crystal structures employed in this benchmark were subject to GlycanDock local docking refinement to serve as a reference for the measures of docking performance (i.e. N5, 〈N5〉, and biophysical feature counts and recoveries). An example of the flags used to perform crystal refinement is shown below:
./GlycanDock.macosclangrelease -include_sugars -in:file:s crystal.pdb -in:file:native crystal.pdb -cst_fa_file interface.cst -nstruct 50 -n_cycles 10 -out:pdb_gz -maintain_links -ex1 -ex2 -docking:partners A_X -carbohydrates:glycan_dock:refine_only true
Here, the -refine_only flag makes the GlycanDock algorithm skip Stage 1 and apply only a modified version of Stage 2 in which smaller perturbations are made to the glycosidic torsion angles (see Supplementary Information).
Selection and preparation of benchmark set of protein–glycoligand complexes
A total of 109 experimentally determined bound protein–glycoligand structures were collected from the PDB16 to create the bound target benchmark set. Thirty-three of these targets were selected from the AutoDock Vina-Carb benchmark set55 while the rest were selected from protein–carbohydrate databases18,74. Some targets contain the same protein receptor sequence; however, no two proteins of the same sequence are bound to identical glycoligands. For example, Streptococcus pneumoniae endo-β-1,4-galactosidase binds three different glycoligands in PDB structures 2J1T, 2J1U, and 2J1V. We collected unbound protein structures for 62 of the bound targets to create the unbound target benchmark set. Unbound protein backbones were aligned onto the backbone of their corresponding bound protein structure. Only the coordinates of the aligned unbound protein and the glycoligand from the bound complex were kept. Alignment was performed using the align command in PyMOL and excluded hydrogens and non-protein atoms (remove hydrogens; align <unbound> and !organic, <bound> and !organic). Protein Cα-RMSD was calculated also using PyMOL (align <unbound> and name CA, <bound> and name CA, cycles=0). All benchmark targets were resolved using X-ray crystallography with a resolution of ≤ 2.0 Å. Further details on the selection and preparation of the bound and unbound protein-glycoligand benchmark sets can be found in the Supplementary Information.
Generation of increasingly perturbed starting structures used as input to GlycanDock
After the preparation procedure described above, bound and unbound target structures were pre-packed (see Stage 0 of the GlycanDock algorithm). The glycoligand was then systematically perturbed in both rigid-body and glycosidic torsion angle conformational space to generate increasingly deviated input starting structures. The glycoligand’s center-of-mass was perturbed using uniform translational perturbations of 0.25 Å, 0.5 Å, 1.0 Å, 2.0 Å, and 3.0 Å and uniform rotational perturbations of 3.75°, 7.5°, 15.0°, 30.0°, and 45.0°. Glycosidic torsion angles were perturbed using uniform perturbations of 6.25°, 15.0°, 30.0°, 60.0°, and 90.0°. Perturbed structures were binned on increasing magnitude of deviation measured using ring-RMSD (1.0 ± 0.1 Å, 2.0 ± 0.1 Å, up to 10.0 ± 0.1 Å ring-RMSD; Figure S1). For unbound targets, ring-RMSD was calculated in reference to aligned starting structures. Ten perturbed starting structures per bound and unbound target for each ring-RMSD bin were generated. This process resulted in 10,900 perturbed starting structures for the bound benchmark set and 6,200 for the unbound. All coordinates are available upon request.
Docking constraints employed during benchmarking
All GlycanDock benchmark docking trajectories employed a constraint that used a flat harmonic potential to bias the anomeric carbon atom of a specified carbohydrate residue to remain within a distance of 7.5 Å ± 2.5 Å to the Cα atom of any protein receptor residue. Constraints were split evenly among the carbohydrate residues of the glycoligand to avoid bias to the known bound conformation. For example, a tetrasaccharide glycoligand would result in 25% of all GlycanDock models in which the first carbohydrate residue was constrained, 25% in which the second carbohydrate residue was constrained, etc. The biasing effect of the docking constraint is enforced via the Rosetta scoring function.
GlycanDock benchmark run time
A single GlycanDock protein–glycoligand docking trajectory resulting in one output model took on average 316 ± 154 seconds to complete (936 ± 292 seconds for targets containing neuraminic acid). A single GlycanDock receptor-free glycoligand conformational sampling trajectory resulting in one output model took on average 68 ± 41 seconds to complete.
Rosetta Technical Details
Modeling and sampling of carbohydrates in Rosetta
Carbohydrate oligomers and their DoFs (e.g., main-chain and branch glycosidic torsion angles, internal ring torsions, side-chain torsions) are defined and modeled using the RosettaCarbohydrate framework64. In this benchmark, the internal ring torsion angles (ν) are held rigid across each docking experiment (i.e., predicted models retain the same ν values as the input starting structures). Glycosidic torsions angles (ϕ, ψ, and, if present, ω) and carbohydrate side-chain torsions (χ) are sampled and optimized throughout the GlycanDock algorithm. See Labonte et al. for more information on carbohydrate modeling in Rosetta64.
Rosetta scoring function and the glycosidic linkage scoring term
We employed the standard Rosetta Energy Function 2015 (REF15)73 with an additional score term specific to the energetics of glycosidic torsion angles for this work. REF15 is a scoring function that includes terms for physically derived potentials, such as van der Waals attraction and Lennard–Jones repulsion, Coulombic electrostatics, and a Gaussian exclusion implicit solvation term. It also includes empirical potentials such as orientation-dependent hydrogen-bonding terms and statistically derived terms to capture the energetic preferences of backbone and side-chain torsions in proteins. All scores are expressed as a unitless Rosetta Energy Unit (REU), with negative REU values representing favorable conformations.
The additional Rosetta score term used to capture the energetic preferences of glycosidic torsion angles, deemed sugar_bb, is derived from the quantum-mechanics-based Carbohydrate Intrinsic (CHI) energy functions54,55,64. The CHI energy functions capture the energetic preferences of ϕ and ψ glycosidic torsion angles between pyranose residues. It depends on the stereochemistry of the anomeric carbon and the upstream connecting oxygen atom and not the chemical identity of the carbohydrate residue.
Recently, the sugar_bb score term was expanded to include scoring of additional glycosidic linkage types. New parameters for the CHI energy function were added for the ψ torsion of α6 and β6 linkages. Previously, parameters were only available for ψ torsions of linkages that did not include exocyclic carbons. Additionally, a new energy function was added to represent the preferences for the ω torsions of glycosidic linkages by capturing the “gauche effect”. The “gauche effect” occurs when an ω torsion angle prefers one of the two gauche orientations, instead of the expected anti configuration, when the hydroxyl group of the carbon atom two carbons previous to the exocyclic carbon is equatorial24. For example, the preferred ω angle for a residue attached in a (1→6) linkage to glucose, where O4 is equatorial, is not 180° as might be expected but rather 60° or −60°. The new energy function is essentially a set of three harmonic energy wells centered over the gauche and anti torsion angles. It effectively adds a scoring penalty to any glycosidic conformation that does not demonstrate this “gauche effect”.
In this benchmark assessment, the scoring function used included the REF15 score terms and weights, the updated sugar_bb score term with a weight of 0.5, and the fa_intra_rep_nonprotein score term with a weight of 0.55.
Rosetta version number and documentation
The GlycanDock algorithm is available as of version 61659 (weekly release #283) of the Rosetta macromolecular modeling and design software suite. See the online documentation for more information: https://rosettacommons.org/docs/latest/application_documentation/carbohydrates/GlycanDock
Results
Figure 1 outlines the Rosetta GlycanDock Monte Carlo-plus-Minimization (MCM) algorithm for docking flexible glycoligands to protein receptors. Briefly, the GlycanDock algorithm takes a pre-packed (Stage 0) putative protein–glycoligand complex as an input structure. During Stage 1 of GlycanDock, the initial glycoligand conformation is randomly perturbed in both rigid-body and glycosidic torsion angle space. Stage 1 serves to promote conformational diversity in each independent docking trajectory; therefore, this initial, random perturbation is not subject to the Metropolis criterion. During Stage 2, a set of inner refinement cycles alternates between rigid-body and glycosidic torsion angle sampling followed by protein and carbohydrate side chain optimization at the interface and full-complex energy minimization. To promote thorough sampling of local conformational space, the inner refinement cycles are wrapped in a set of outer cycles that ramp down the van der Waals attractive weight and ramp up the Lennard–Jones repulsive weight of the scoring function73. Thus, the early cycles of Stage 2 refinement allow clashes and promote diversification, while later cycles enforce rigid sterics—a strategy shown to be effective in protein–peptide docking65,66. Stages 0, 1, and 2 of the GlycanDock algorithm are performed with implicit solvent in Rosetta’s high-resolution, all-atom mode.
Figure 1:
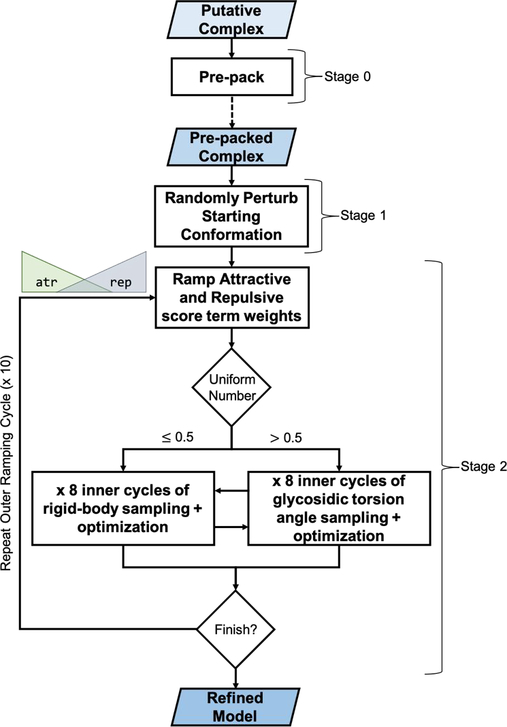
Overview of the GlycanDock algorithm. Stage 0 pre-packs the initial, putative protein–glycoligand complex. The pre-packed output structure is then given to the GlycanDock algorithm as input (indicated by the dashed arrow) to the sampling and optimization stages. Stage 1 applies a random perturbation to the glycoligand in rigid-body and glycosidic torsion angle space (without employing the Metropolis criterion). Stage 2 performs inner cycles of high-resolution rigid-body and glycosidic torsion angle sampling and optimization, where optimization includes packing and energy minimization at specific intervals. Outer cycles wrap the two sets of inner sampling cycles and control the incremental adjustment of the weights of the attractive (fa_atr) and repulsive (fa_rep) Rosetta score terms.
In this benchmark assessment, we apply an ambiguous atom-pair constraint to enforce one random carbohydrate residue of the glycoligand to remain physically close to the protein receptor throughout each independent docking trajectory. That is, this constraint ensures that the final set of models includes, for each carbohydrate residue, a portion of models where that residue contacts the protein. Bound protein–glycoligand models are ranked by a calculated interface score in REU (Rosetta Energy Units) that approximates the binding free energy of the complex. Model quality (i.e., structural accuracy) is measured by calculating the root-mean-squared deviation (RMSD) of the heavy-atoms that compose each carbohydrate ring of the glycoligand in its predicted conformation compared to its native bound state after superposition of the protein receptor backbone (ring-RMSD). We consider models under 2 Å ring-RMSD (a standard model quality cutoff in the field of molecular docking55,75) to be sufficiently representative of the native bound conformation and are thus referred to as near-native models.
Determination of effective glycoligand docking range for bound and unbound protein backbones
The effectiveness of a local docking algorithm such as GlycanDock depends on the initial quality of the putative input complex. Raveh, London, and Schueler-Furman defined an algorithm’s effective docking range as the maximum deviation of a given ligand from which near-native models can be sampled and correctly ranked66. To identify the effective docking range of the GlycanDock algorithm, we assessed docking performance on 109 bound and 62 unbound protein–glycoligand targets (Table S2) of increasing initial deviation. We generated a benchmark set of starting structures by systematically perturbing the 109 bound and 62 unbound protein–glycoligand complexes and binning the resulting conformations based on increasing ring-RMSD (1–10 Å ring-RMSD; Figure S1). We generated ten unique starting structures per target for each ring-RMSD bin to ensure diversity of input conformations. Prior to input to GlycanDock, all perturbed starting structures underwent an independent optimization of all side-chain rotamers. In the case of bound targets, this procedure erased pre-configuration of the interfacial side chains to bind the glycoligand. We then used GlycanDock to generate 2,000 models per target per ring-RMSD bin (10 input starting structures per target × 200 models each = 2,000 models). See Materials and Methods.
Figure 2 shows how GlycanDock sampled and discriminated near-native models of a branched xyloglucan oligomer bound to its receptor starting from input structures with initial glycoligand deviation of 2.0, 4.0, and 6.0 Å ring-RMSD (± 0.1 Å). For the 2.0 and 4.0 Å inputs (panels A and B, respectively), GlycanDock generated multiple low-scoring models similar to the experimental (native) crystal complex. However, in the 6.0 Å case (panel C), the top-5 lowest-scoring structures (blue diamonds) are scattered from 2.5–13 Å ring-RMSD from the native. These non-native-like models score worse (i.e., more positive REU) than the refined native models (maroon), suggesting that the scoring function is sufficient to discriminate near-native models for this target, but the sampling failed to find those low-scoring conformations.
Figure 2:
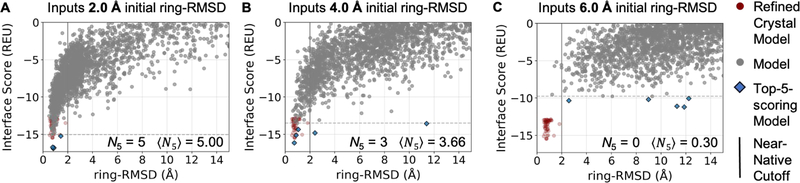
Example “funnel” plots depicting GlycanDock N5 and 〈N5〉 performance on bound target 4BJ0 at different initial ring-RMSD values. “Funnel” plots depict the relationship between interface score and ring-RMSD of a set of models (gray circles). While 〈N5〉 must be calculated, N5 can be determined directly by counting the number of near-native models (models below 2 Å ring-RMSD) within the top-5-scoring models (blue diamonds). (A) GlycanDock models predicted from 2.0 ± 0.1 Å initial ring-RMSD input structures demonstrate unambiguous (〈N5〉 = 5.00) funneling toward the refined native crystal structure (maroon circles). (B) Models from 4.0 ± 0.1 Å initial ring-RMSD input structures demonstrate acceptable funneling (〈N5〉 = 3.66) toward the refined native. (C) Models from 6.0 ± 0.1 Å initial ring-RMSD input structures demonstrate no funneling (〈N5〉 = 0.30) toward the refined native. Accordingly, the funnel plots in panels A and B demonstrate docking success while the funnel plot in panel C demonstrates docking failure.
From data like those shown in Figure 2, we quantify docking success using N5—the count of near-native models ranked among the 5-top-scoring of all predicted models. Due to the stochastic nature of any MCM sampling algorithm, we performed a bootstrap statistical analysis to quantify the variability within each benchmark docking run59,68. For each bound and unbound protein–glycoligand target across all initial ring-RMSD bins, we performed bootstrap case resampling to calculate 〈N5〉—the bootstrap average of N5 and a statistical measure of the reliability of observed docking success68,69. We defined 〈N5〉 ≥ 1.0 (i.e., sampling and discriminating at least one near-native model among the 5-top-scoring with statistical reliability) as the threshold indicating docking success. For example, 〈N5〉 = 5.00 and 〈N5〉 = 3.66 for the two successful docking cases presented in Figure 2, whereas 〈N5〉 = 0.30 for the failure case. Finally, we define the effective docking range of the GlycanDock algorithm as the maximum ring-RMSD from which 50% or more of the 109 bound and 62 unbound protein–glycoligand targets achieve 〈N5〉 ≥ 1.0.
GlycanDock’s effective docking range is 8 Å ring-RMSD for bound protein backbones and 7 Å for unbound
Figure 3A summarizes GlycanDock’s 〈N5〉 performance on the bound benchmark set as a function of the ring-RMSD of the input structures. More than 50% of the 109 bound targets achieved 〈N5〉 ≥ 1.0 (green bars) up to 8 Å ring-RMSD, suggesting an effective docking range of 8 Å initial ring-RMSD with bound protein backbones. Figure 3B summarizes the same data for the unbound benchmark set (average protein Cα-RMSD to bound 0.49 Å ± 0.48 Å, minimum 0.05 Å, maximum 3.13 Å). More than 50% of the 62 unbound targets achieved 〈N5〉 ≥ 1.0 up to 7 Å ring-RMSD, suggesting an effective docking range of 7 Å initial ring-RMSD with unbound protein backbones. To illustrate docking success, Figures 3C and D depict the near-native conformations of a top-5- and top-1-scoring model, respectively, while Figure 3E depicts a top-1-scoring, sub-angstrom model. Table S3 reports all observed N5 and bootstrap ensemble averages 〈N5〉 and standard deviations (σ〈N5〉) for the bound benchmark set; Table S4 reports docking results for the unbound benchmark set.
Figure 3:
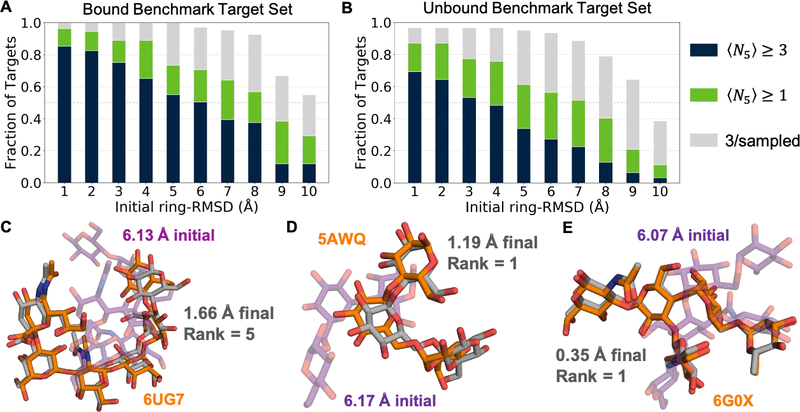
Summary of GlycanDock docking benchmark performance (A) GlycanDock 〈N5〉 performance on 109 bound protein–glycoligand targets as a function of initial ring-RMSD bin. Blue bars represent the fraction of targets that achieved 〈N5〉 ≥ 3. Green bars represent the fraction of targets that achieved 〈N5〉 ≥ 1.0 (the threshold for docking success). Gray bars represent the fraction of targets that sampled at least 3 near-native models overall but failed to rank them among the 5-top-scoring. (B) Same as (A), but on the 62 targets of the unbound benchmark set. (C–E) Glycoligand conformation from the bound crystal structure (orange) compared to conformation after the Stage 1 random perturbation (purple, transparent) and the final conformation (gray) after GlycanDock sampling for three example targets. Protein backbones omitted for clarity.
While achieving 〈N5〉 ≥ 1.0 indicates docking success, the fraction of targets surpassing the 〈N5〉 ≥ 3 threshold (i.e., sampling and discriminating at least three near-native models among the 5-top-scoring with statistical reliability) underscores the robustness of the GlycanDock algorithm. More than 50% of bound and unbound targets achieved 〈N5〉 ≥ 3 from input structures up to 5 Å and 3 Å initial ring-RMSD, respectively (Figure 3A and 3B, blue bars). For both the bound and unbound benchmark target set, we found no significant difference in the average count of near-native models among the 50-top-scoring models based on either the length of the glycoligand or whether the glycoligand was linear or branched (Figure S2).
Docking failure cases (〈N5〉 < 1.0) can be either attributed to not having sampled near-native models (i.e., sampling failure), or to the scoring function not correctly discriminating near-native models as top-scoring (i.e., scoring failure). More than 50% of bound and unbound targets sampled three or more near-native models overall from inputs up to 10 Å and 9 Å initial ring-RMSD, respectively (Figure 3A and B, gray bars). Accordingly, most GlycanDock failure cases in this benchmark assessment can be attributed to scoring failures. Similarly, the greater fraction of unbound targets exhibiting scoring failures (gray bars) compared to bound targets highlights the sensitivity of the scoring function to changes in the protein backbone induced by glycoligand binding. While additional sampling (e.g., generating more models or using more refinement cycles, re-refining top-scoring output models66, using employing alternative protein receptor backbone conformations69,76) may be sufficient to overcome cases of sampling failure, faithful discrimination of near-native models depends on the efficacy of the scoring function.
Analysis of biophysical feature recovery of top-scoring GlycanDock models
Glycan-binding proteins have evolved a variety of sequences and structures to recognize the great diversity of carbohydrates, often with impressive selectivity77,78. For example, different influenza haemagglutinin subtypes can discriminate between α2,3- and α2,6-linked terminal sialic acids, which determines if a given influenza strain can infect animals or humans or both79. To understand how proteins selectively recognize the chemical and structural diversity of carbohydrate chains, the structural model must reveal key interfacial protein-glycoligand interactions. To this end, we examined the counts and recovery rates of protein-glycoligand interfacial residue–residue contacts and hydrogen bonds by the 50-top-scoring models per target from the bound benchmark set. Explicit water molecules (involved in water-mediated hydrogen bonding or otherwise) are not modeled and thus not considered in this analysis. See Materials and Methods.
The 50-top-scoring GlycanDock models exhibited a distribution of counts of interfacial residue–residue contacts similar to that of the 50-top-scoring refined crystal structures across the initial ring-RMSD bins examined (Figure 4A). However, as initial ring-RMSD increased, more of these contacts were made between non-native pairs of residues (i.e., low contact recovery; Figure 4B). The distributions of the counts and recovery of interface residues followed a similar pattern (Figure S3). Further, top-scoring models of targets bound to short glycoligands (i.e., di- and trisaccharides) resulted in similar distributions (i.e., shifted left toward lower counts as the initial ring-RMSD bin increased) compared to targets bound to long glycoligands (Figure S4). On the other hand, top-scoring GlycanDock models did not make as many interfacial hydrogen bonds as compared to the refined crystal structures, with the recovery of native interfacial hydrogen bonds decreasing more drastically across increasing initial ring-RMSD bins (Figure 4C & 4D). Hydrogen bonds are more challenging to sample with high fidelity because they require precise atom-pair geometries to form80. For instance, native interfacial hydrogen bonding networks were difficult to identify and maintain even when the glycoligand was refined starting from its crystal conformation (Figure 4D, maroon).
Figure 4:
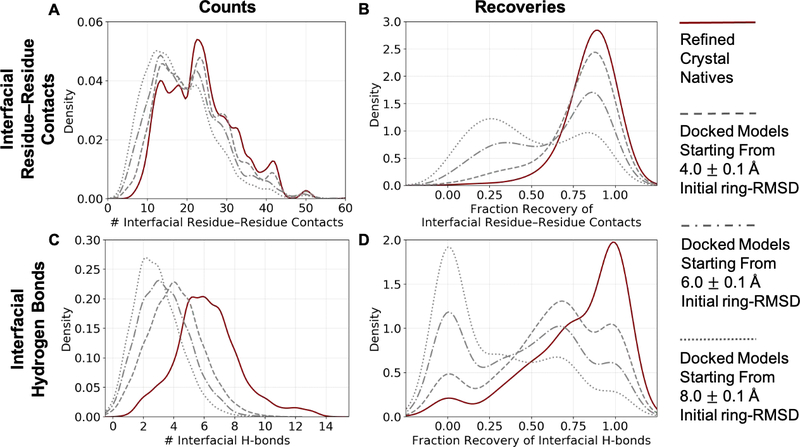
Counts and recovery of biophysical features by the 50-top-scoring GlycanDock models of each target from the bound benchmark set. (A) Distributions of the count of interfacial residue–residue contacts by the 50-top-scoring GlycanDock models per target predicted starting from input structures of 4.0, 6.0, and 8.0 Å initial ring-RMSD (gray dashed, dash-dot, and dotted lines, respectively). The distribution of the count of interfacial residue–residue contacts after GlycanDock crystal refinement (solid, maroon) serves as a reference. (B) Distributions of the recovery of native interfacial residue–residue contacts by the 50-top-scoring GlycanDock models per target. (C) Same as (A), but for the count of interfacial hydrogen bonds. (D) Same as (B), but for the recovery of interfacial hydrogen bonds. Discrete data are smoothed using kernel density fits using Seaborn81 (kdeplot), resulting in some curves extending below fractions of 0.0 and above 1.0. Bin widths of 1.0 and 0.5 were used to fit the counts of interfacial residue–residue contacts and hydrogen bonds, respectively, and a bin width of 0.1 was used to fit the recoveries.
GlycanDock’s top-scoring models sampled productive protein–glycoligand interfaces as measured by the counts of biophysical features such as interface residues and interfacial residue–residue contacts (Figures 4 and S3). However, interfacial hydrogen bonds—whether seen in the native bound structure or otherwise—were especially difficult to make (Figure 4). This analysis provides a deeper evaluation of the GlycanDock algorithm’s ability to sample biophysically realistic protein–glycoligand interfaces and highlights the challenge the Rosetta scoring function faces in correctly discriminating true, native-like interfaces. Detailed results and biophysical features for all of the 50-top-scoring models per target per initial ring-RMSD bin are available in Table S5 and S6.
GlycanDock refinement qualitatively recapitulates glycoligand specificity of CtCBM6
Carbohydrate-binding modules (CBMs) are discretely folded, non-catalytic, sugar-binding proteins82. CBMs are typically found linked to carbohydrate-active enzymes, serving to enhance catalytic efficiency by binding to carbohydrate ligands and directing the enzyme to its substrate83. While some CBMs bind to a range of different carbohydrate ligands, others display distinct binding specificities84. For example, the CBM from xylanase 10A of Clostridium thermocellum (CtCBM6) binds xylohexaose with a 100-fold higher affinity over cellohexaose85. A fast and accurate computational docking tool capable of discriminating glycoligand binders from non-binders would aid in the identification of key interfacial residues that inform protein design efforts to engineer new or improved binding behavior. Here, as a case study, we tested whether GlycanDock local docking refinement can capture the structural and energetic factors that determine the glycoligand specificity of CtCBM6.
A synthetic CtCBM6–cellopentaose starting structure was created by manually adding the atoms of glucose’s exocyclic hydroxymethyl moiety to each carbohydrate residue of the native CtCBM6–xylopentaose crystal structure (PDB 1UXX). A synthetic CtCBM6U-cellopentaose starting structure (where CtCBM6U distinguishes the unbound protein backbone, PDB 1GMM) was created using the same approach on the aligned unbound CtCBM6U–xylopentaose structure from the unbound benchmark target set. RMSD calculations for cellopentaose-bound models used the input starting structure as the reference structure, whereas xylopentaose-bound models used the native bound crystal structure (PDB 1UXX). We then applied the GlycanDock algorithm as described earlier on all four complexes, including the pre-packing in Stage 0 and the random perturbations in Stage 1. Despite these perturbations, we might expect the bound CtCBM6–xylopentaose case to have some memory of the crystal interface (PDB 1UXX) that favors it enough to provide a lower score. But the unbound docking case (starting with PDB 1GMM) will not have this memory and will provide a balanced comparison when docking different substrates.
Figure 5A depicts the resulting funnel plot for the 50-top-scoring CtCBM6–xylopentaose GlycanDock models (orange circles) versus the 50-top-scoring CtCBM6U–xylopentaose models (blue triangles) versus the 2,000 CtCBM6–cellopentaose models (gray circles) versus the 2,000 CtCBM6U–cellopentaose models (gray triangles). As expected, the Rosetta scoring function clearly favored the native xylopentaose glycoligand docked to the bound conformation of CtCBM6 (orange). More importantly, the docking of xylopentaose was favored in the unbound docking case (blue) over the 100-fold weaker cellopentaose binder docked to either conformation of CtCBM6 (gray).
Figure 5:
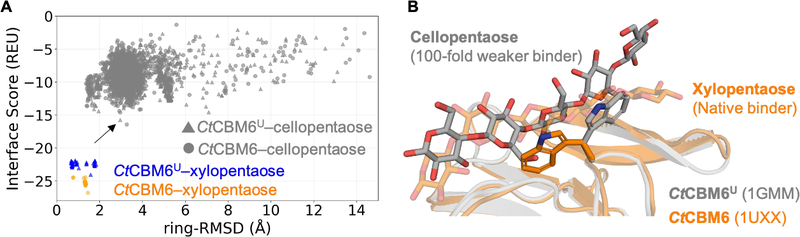
Results of GlycanDock local docking refinement qualitatively discriminate between a native glycoligand binder versus a 100-fold weaker binder to CtCBM6. (A) Funnel plots depicting results of GlycanDock local docking refinement of the native CtCBM6–xylopentaose complex (orange circles) versus CtCBM6U-xylopentaose (blue triangles; where CtCBM6U distinguishes the unbound protein backbone) versus CtCBM6-cellopentaose (gray circles) versus CtCBM6U–cellopentaose (gray triangles). The top-scoring CtCBM6U-cellopentaose model containing the 100-fold weaker cellopentaose binder is marked with an arrow. (B) Comparison of the conformation of the native bound CtCBM6–xylohexaose crystal structure (orange, transparent; PDB code 1UXX) to the top-scoring CtCBM6U–cellopentaose model marked in panel A (gray). The rearranged receptor tryptophan is shown in sticks.
It has been suggested that the striking difference in binding affinity between xylopentaose and cellopentaose is due to steric clashes with the distinguishing exocyclic hydroxymethyl moiety of glucose in two subsites of CtCBM6’s binding site85. The effect of these proposed steric clashes can be seen in Figure 5B where GlycanDock’s top-scoring CtCBM6U cellopentaose model (gray) is unable to bury as deeply in the binding site as the native xylopentaose glycoligand (orange). Further, these steric clashes led to a rearrangement of a receptor tryptophan in the binding site, disrupting an important CH–π “stacking” interaction86 with a carbohydrate residue of the glycoligand (Figure 5B). These results suggest that cellopentaose would not bind as well as xylopentaose; accordingly, the results of GlycanDock local docking refinement qualitatively match the experimentally determined glycoligand specificity of CtCBM6.
Combination of FTMap and RosettaLigand produces putative protein–glycoligand models within GlycanDock’s effective docking range starting from “blind”-like inputs In real molecular docking cases, the bound conformation of the target complex is unknown. In fully “blind” docking cases without experimental data, the location of the binding site on the protein receptor is also unknown. In this work, we demonstrated that the GlycanDock algorithm’s effective glycoligand docking range is up to 7 Å initial ring-RMSD when using unbound protein backbones (Figure 3). Accordingly, GlycanDock requires as input a putative protein–glycoligand complex where the glycoligand is placed near the binding site and in approximately the correct orientation. When only the unbound protein structure and glycoligand sequence is known, an approach to effectively generate this putative complex is necessary. Here, we report the results of a pipeline for “blind” glycoligand docking using FTMap87 and RosettaLigand61,62. We used FTMap to predict the glycoligand binding site and RosettaLigand to generate an initial protein–glycoligand structure. Details on the setup and usage of FTMap and RosettaLigand can be found in the Supplemental Information.
While various ligand binding site prediction software exist88, including some specific to carbohydrate ligands89,90, we chose FTMap for its speed and ease of use via its online webserver91. FTMap predicts ligand-binding “hot spots” by extensively sampling the surface of a macromolecular receptor using various small organic molecules as probes. Among the probes used is cyclohexane (CHX)—a compound structurally similar to that of a carbohydrate. We hypothesized that the FTMap “hot spots” predicted with a CHX probe would map to glycoligand binding sites. In 36 of the 62 unbound targets, the FTMap server resulted in a CHX probe within the known glycoligand binding site as determined by visual inspection (Figure 6A; see Table S2 for list of the 36 targets). Accordingly, we used the 36 unbound protein structures and the Cartesian coordinate of the center-of-mass of the CHX probe to produce putative protein–glycoligand models using RosettaLigand.
Figure 6:
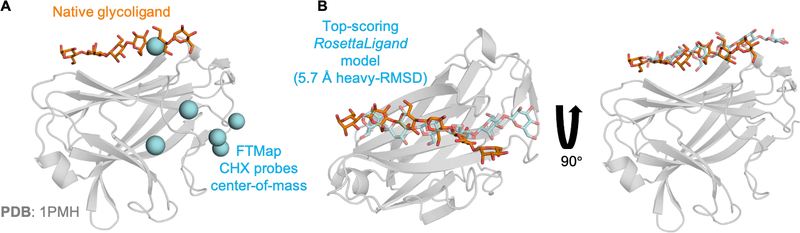
Example results of FTMap binding site prediction and RosettaLigand docking. (A) Results of FTMap ligand binding “hot spot” prediction using the unbound structure of CsCBM27–1 (PDB 1PMJ) as input. Cyan spheres represent the center-of-mass of each site prediction identified by the cyclohexane (CHX) probe. The native bound crystal structure (PDB 1PMH) shows that one carbohydrate residue of the glycoligand (orange sticks) overlaps directly on one of the FTMap CHX probe predictions. (B) A 5.7 Å heavy-RMSD RosettaLigand model ranked among the 5-top-scoring models. The same native bound crystal structure from (A) is shown. The predicted conformation of the glycoligand (transparent cyan sticks) overlaps reasonably well with the native conformation (orange sticks) in the binding site but is shifted over by one carbohydrate unit. This RosettaLigand predicted conformation (transparent cyan sticks) is within the effective docking range of the GlycanDock algorithm.
RosettaLigand requires pre-generated conformations of ligands that have multiple rotatable bonds. We used the Rosetta GlycanSampler algorithm (Jared Adolf-Bryfogle, unpublished) to generate 200 initial conformations for each of the 36 unbound target glycoligands (see Supplemental Information for details and for other methods of generating initial glycoligand conformations). GlycanSampler glycoligand models had an average heavy-SRMSD of 1.48 Å ± 0.65 Å (minimum 0.73 Å, maximum 4.98 Å), where heavy-SRMSD is calculated using all glycoligand heavy-atoms after superposition of the model glycoligand onto the native bound glycoligand. Using the center-of-mass of the CHX probe predicted within the known glycoligand binding site as the starting coordinates, we generated 2,000 docked models per target using RosettaLigand. Models were ranked by interface score, and glycoligand RMSD was calculated using all glycoligand heavy-atoms after alignment of the protein receptor (heavy-RMSD). RosettaLigand sampled one or more models below 7 Å heavy-RMSD within the 5-top-scoring models for 25 of the 36 unbound targets (Figure 6B). Three of the eleven failure targets were carbohydrate binding modules that resulted in top-scoring RosettaLigand models where the glycoligand was docked in the reverse direction compared to the native structure (i.e., the non-reducing-end carbohydrate of the model aligned with the reducing end of the native structure). RosettaLigand sampled near-native models (here, below 2 Å heavy-RMSD) among the 5-top-scoring for only seven of the 36 unbound targets. Taken together, we have shown that the combination of FTMap and RosettaLigand can produce putative protein–glycoligand models within the effective docking range of the GlycanDock algorithm starting from “blind”-like docking conditions.
Discussion
We have developed and evaluated GlycanDock (Figure 1), a new, residue-centric protein-glycoligand docking refinement algorithm within the Rosetta macromolecular modeling and design software suite. GlycanDock treats carbohydrate chains as flexible oligomers, allowing for extensive conformational sampling of the glycoligand. Further, conformations with glycosidic linkages that fall within pre-determined, energetically-favorable torsion space are rewarded during sampling to ensure biophysically-realistic carbohydrate structures54,55,64. The RosettaCarbohydrate framework64,67 enables the ability to handle both simple and complex glycoligands including variations in length, composition, ring shape, glycosidic connectivity, and branching, making GlycanDock a robust carbohydrate modeling tool. With continued efforts to improve the capabilities of the RosettaCarbohydrate framework and expand the Rosetta chemical database57 with natural and non-natural monosaccharides and chemical modifications, GlycanDock will be able to simulate systems that represent the carbohydrate diversity observed in nature92 and beyond.
In this work, we described the results of a benchmark assessment of the Rosetta GlycanDock protein–glycoligand docking refinement algorithm. We evaluated GlycanDock performance on 109 bound and 62 unbound protein–glycoligand targets using input structures of systematically increasing initial ring-RMSD. Docking performance was measured per target using N5—the count of near-native models ranked among the five top-scoring models (Figure 2). We used bootstrap case resampling to calculate 〈N5〉, setting a threshold of 〈N5〉 ≥ 1.0 to define docking success. Bootstrap statistical analysis indicated that GlycanDock’s effective docking range is 8 Å ring-RMSD with bound protein backbones and 7 Å for unbound (Figure 3). Notably, benchmarking of GlycanDock included modeling side-chain flexibility at the protein–glycoligand interface and examination of glycoligand docking performance on 62 unbound protein structures (~58% of the 109 bound benchmark targets).
We sought to measure the GlycanDock algorithm’s ability to recapitulate some of the biophysical features that drive glycoligand binding. Hydrogen bonding, for instance, is an important interaction at protein–glycoligand interfaces24, but forming productive hydrogen bonds requires precise alignment of local atomic geometries80. Sampling these exact interactions can be challenging, as evidenced by the relatively poor interfacial hydrogen bonding recovery of top-scoring GlycanDock models (Figures 4C & D). Future work on the RosettaCarbohydrate framework will address additional carbohydrate modeling considerations such as CH–π “stacking” interactions86,93–98 and water-mediated hydrogen bonding99–101, which may improve GlycanDock sampling of native-like biophysical features at protein–glycoligand interfaces.
We found that the results of GlycanDock local docking refinement qualitatively corresponded with the experimentally determined binding specificity of CtCBM6 to xylopentaose and cellopentaose glycoligands (Figure 5). While further study is needed, initial results indicate GlycanDock can be used to predict binder from non-binder glycoligands.
The performance of other tools for protein–glycoligand docking such as AutoDock Vina-Carb54,55 and the fragment-based approach by Samsonov and colleagues56 has also been published. However, both assessments employed different benchmark target sets and docking success definitions, making direct performance comparison difficult. Further, both AutoDock Vina-Carb and the method by Samsonov et al. include full rigid-body rotations and translations using a grid box sampling approach, whereas our GlycanDock docking refinement algorithm does not perform such extensive sampling of rigid-body space. Accordingly, we presented a possible “blind” docking pipeline that utilizes FTMap87,91 and RosettaLigand61,62 (the latter of which performs grid box sampling) to generate putative protein–glycoligand complexes that are within the effective docking range of the GlycanDock docking refinement algorithm (Figure 6). Models from this FTMap-RosettaLigand docking pipeline, AutoDock Vina-Carb, or the method by Samsonov et al. could then be refined by the GlycanDock algorithm for further evaluation. The scientific community therefore has a selection of useful tools to address a variety of modeling and prediction challenges in glycoscience research.
Protein–carbohydrate interactions modulate many cellular and molecular processes that are fundamental to all life. High-resolution models of protein–glycoligand complexes help us understand how proteins recognize carbohydrates and how glycan structure can bring about such diversity in observable function. While experimental limitations continue to hinder high-quality protein–glycoligand structure determination, computational modeling tools have served to fill this gap. We developed the GlycanDock docking refinement algorithm to model, dock, and refine protein–glycoligand complexes and serve as a tool to reveal the atomic details behind the molecular roles carbohydrates play. Also, GlycanDock refinement can be combined with the design algorithms of the Rosetta software suite. With the ubiquity of glycans in biology and biotechnology, the expanded suite of computational tools for glycans has the potential to aid in innovations in human health and disease, glycomimetic drug design, pathogen detection and defense, plant-based renewable bioenergy, and more.
Supplementary Material
Acknowledgements
MLN and JWL thank members of the Gray Lab for their helpful discussions. MLN thanks Rahel Frick and Jaime Ransohoff for providing additional manuscript review. The authors thank Sergey Lyskov and Matthew Mulqueen for maintaining relevant computational resources. The authors acknowledge the work of the large community of developers in the RosettaCommons (http://www.rosettacommons.org) for continued research and development of the Rosetta software package. This work is supported by NIH R01-GM078221 (MLN, JJG) and NIH R01-GM127578 (JWB, JJG).
Footnotes
Conflict-of-interest declarations
Dr. Jeffrey J. Gray is an unpaid board member of the Rosetta Commons. Under institutional participation agreements between the University of Washington, acting on behalf of the RosettaCommons, Johns Hopkins University may be entitled to a portion of revenue received on licensing Rosetta software including methods developed in this study. As a member of the Scientific Advisory Board, JJG has a financial interest in Cyrus Biotechnology. Cyrus Biotechnology distributes the Rosetta software, which may include methods developed in this study. These arrangements have been reviewed and approved by the Johns Hopkins University in accordance with its conflict-of-interest policies.
References
- (1).Werz DB; Ranzinger R; Herget S; Adibekian A; Von der Lieth CW; Seeberger PH Exploring the Structural Diversity of Mammalian Carbohydrates (“glycospace”) by Statistical Databank Analysis. ACS Chem. Biol. 2007, 2 (10), 685–691. 10.1021/cb700178s. [DOI] [PubMed] [Google Scholar]
- (2).Ajit V; Sharon N Chapter 1 Historical Background and Overview. Essentials Glycobiol. 2009, 1–21. 10.1101/glycobiology.3e.001. [DOI] [Google Scholar]
- (3).Lauc G; Krištić J; Zoldoš V Glycans - the Third Revolution in Evolution. Front. Genet 2014. 10.3389/fgene.2014.00145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Corfield AP; Berry M Glycan Variation and Evolution in the Eukaryotes. Trends in Biochemical Sciences. 2015. 10.1016/j.tibs.2015.04.004. [DOI] [PubMed] [Google Scholar]
- (5).Eichler J; Koomey M Sweet New Roles for Protein Glycosylation in Prokaryotes. Trends in Microbiology. 2017. 10.1016/j.tim.2017.03.001. [DOI] [PubMed] [Google Scholar]
- (6).Schmidt MA; Riley LW; Benz I Sweet New World: Glycoproteins in Bacterial Pathogens. Trends in Microbiology. 2003. 10.1016/j.tim.2003.10.004. [DOI] [PubMed] [Google Scholar]
- (7).Jarrell KF; Ding Y; Meyer BH; Albers S-V; Kaminski L; Eichler J N-Linked Glycosylation in Archaea: A Structural, Functional, and Genetic Analysis. Microbiol. Mol. Biol. Rev. 2014. 10.1128/mmbr.00052-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Varki A Biological Roles of Glycans. Glycobiology 2017, 27 (1), 3–49. 10.1093/glycob/cww086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).van Kooyk Y; Rabinovich GA Protein-Glycan Interactions in the Control of Innate and Adaptive Immune Responses. Nature Immunology. 2008. 10.1038/ni.f.203. [DOI] [PubMed] [Google Scholar]
- (10).Watanabe Y; Bowden TA; Wilson IA; Crispin M Exploitation of Glycosylation in Enveloped Virus Pathobiology. Biochimica et Biophysica Acta - General Subjects. 2019. 10.1016/j.bbagen.2019.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Vigerust DJ; Shepherd VL Virus Glycosylation: Role in Virulence and Immune Interactions. Trends in Microbiology. May 2007, pp 211–218. 10.1016/j.tim.2007.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Weis WI; Drickamer K Structural Basis of Lectin-Carbohydrate Recognition. Annual Review of Biochemistry. 1996. 10.1146/annurev.bi.65.070196.002301. [DOI] [PubMed] [Google Scholar]
- (13).Sharon N; Lis H History of Lectins: From Hemagglutinins to Biological Recognition Molecules. Glycobiology. 2004. 10.1093/glycob/cwh122. [DOI] [PubMed] [Google Scholar]
- (14).Collins BE; Paulson JC Cell Surface Biology Mediated by Low Affinity Multivalent Protein-Glycan Interactions. Current Opinion in Chemical Biology. 2004. 10.1016/j.cbpa.2004.10.004. [DOI] [PubMed] [Google Scholar]
- (15).Imberty A; Mitchell EP; Wimmerová M Structural Basis of High-Affinity Glycan Recognition by Bacterial and Fungal Lectins. Current Opinion in Structural Biology. 2005. 10.1016/j.sbi.2005.08.003. [DOI] [PubMed] [Google Scholar]
- (16).Berman HM; Westbrook J; Feng Z; Gilliland G; Bhat TN; Weissig H; Shindyalov IN The Protein Data Bank (Www.Rcsb.Org). Nucleic Acids Res. 2000. 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).De Meirelles JL; Nepomuceno FC; Peña-García J; Schmidt RR; Pérez-Sánchez H; Verli H Current Status of Carbohydrates Information in the Protein Data Bank. J. Chem. Inf. Model. 2020. 10.1021/acs.jcim.9b00874. [DOI] [PubMed] [Google Scholar]
- (18).Copoiu L; Torres PHM; Ascher DB; Blundell TL; Malhotra S ProCarbDB: A Database of Carbohydrate-Binding Proteins. Nucleic Acids Res. 2020. 10.1093/nar/gkz860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Buda S; Nawój M; Mlynarski J Recent Advances in NMR Studies of Carbohydrates. In Annual Reports on NMR Spectroscopy; 2016. 10.1016/bs.arnmr.2016.04.002. [DOI] [Google Scholar]
- (20).Malhotra S; Ramsland PA Editorial Overview: Carbohydrates – Structural Glycobiology Catches the Wave of Rapid Progress. Current Opinion in Structural Biology. Elsevier Ltd; 2020. 10.1016/j.sbi.2020.04.004. [DOI] [PubMed] [Google Scholar]
- (21).Imberty A Oligosaccharide Structures: Theory versus Experiment. Curr. Opin. Struct. Biol. 1997. 10.1016/S0959-440X(97)80069-3. [DOI] [PubMed] [Google Scholar]
- (22).DeMarco ML; Woods RJ Structural Glycobiology: A Game of Snakes and Ladders. Glycobiology. 2008. 10.1093/glycob/cwn026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Woods RJ; Tessier MB Computational Glycoscience: Characterizing the Spatial and Temporal Properties of Glycans and Glycan-Protein Complexes. Current Opinion in Structural Biology. NIH Public Access October 2010, pp 575–583. 10.1016/j.sbi.2010.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Woods RJ Predicting the Structures of Glycans, Glycoproteins, and Their Complexes; American Chemical Society, 2018; Vol. 118, pp 8005–8024. 10.1021/acs.chemrev.8b00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).National Research Council (US). Transforming Glycoscience: A Roadmap for the Future; National Academies Press: Washington, DC, 2012. 10.17226/13446. [DOI] [PubMed] [Google Scholar]
- (26).Imberty A; H. Prestegard J Structural Biology of Glycan Recognition; 2015. [PubMed]
- (27).Xiong X; Chen Z; Cossins BP; Xu Z; Shao Q; Ding K; Zhu W; Shi J Force Fields and Scoring Functions for Carbohydrate Simulation. Carbohydrate Research. Elsevier Ltd; January 12, 2015, pp 73–81. 10.1016/j.carres.2014.10.028. [DOI] [PubMed] [Google Scholar]
- (28).Copoiu L; Malhotra S The Current Structural Glycome Landscape and Emerging Technologies. Current Opinion in Structural Biology. Elsevier Ltd; June 1, 2020, pp 132–139. 10.1016/j.sbi.2019.12.020. [DOI] [PubMed] [Google Scholar]
- (29).Imberty A; Hardman KD; Carver JP; Perez S Molecular Modelling of Protein-Carbohydrate Interactions. Docking of Monosaccharides in the Binding Site of Concanavalin A. Glycobiology 1991. 10.1093/glycob/1.6.631. [DOI] [PubMed] [Google Scholar]
- (30).Pérez S; Meyer C; Imberty A Practical Tools for Molecular Modeling of Complex Carbohydrates and Their Interactions with Proteins. Mol. Eng 1995. 10.1007/BF00999595. [DOI] [Google Scholar]
- (31).Woods RJ Computational Carbohydrate Chemistry: What Theoretical Methods Can Tell Us. Glycoconjugate Journal. 1998. 10.1023/A:1006984709892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Rockey WM; Laederach A; Reilly PJ Automated Docking of α-(1→4)- and α-(1→6)-Linked Glucosyl Trisaccharides and Maltopentaose into the Soybean β-Amylase Active Site. Proteins Struct. Funct. Genet. 2000. . [DOI] [PubMed] [Google Scholar]
- (33).Fadda E; Woods RJ Molecular Simulations of Carbohydrates and Protein-Carbohydrate Interactions: Motivation, Issues and Prospects. Drug Discov. Today 2010, 15 (15–16), 596–609. 10.1016/j.drudis.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Sapay N; Nurisso A; Imberty A Simulation of Carbohydrates, from Molecular Docking to Dynamics in Water; Humana Press, Totowa, NJ, 2013; pp 469–483. 10.1007/978-1-62703-017-5_18. [DOI] [PubMed] [Google Scholar]
- (35).Grant OC; Woods RJ Recent Advances in Employing Molecular Modelling to Determine the Specificity of Glycan-Binding Proteins; NIH Public Access, 2014; Vol. 0, p 47. 10.1016/j.sbi.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Vliegenhardt JFG NMR Spectroscopy and Computer Modeling of Carbohydrates: Recent Advances; 2006. [Google Scholar]
- (37).Kirschner KN; Yongye AB; Tschampel SM; González-Outeiriño J; Daniels CR; Foley BL; Woods RJ GLYCAM06: A Generalizable Biomolecular Force Field. Carbohydrates. J. Comput. Chem. 2008. 10.1002/jcc.20820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Bryce RA; Hillier IH; Naismith JH Carbohydrate-Protein Recognition: Molecular Dynamics Simulations and Free Energy Analysis of Oligosaccharide Binding to Concanavalin A. Biophys. J. 2001. 10.1016/S0006-3495(01)75793-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Hadden JA; Tessier MB; Fadda E; Woods RJ Calculating Binding Free Energies for Protein–Carbohydrate Complexes. Methods Mol. Biol 2015. 10.1007/978-1-4939-2343-4_26. [DOI] [PubMed] [Google Scholar]
- (40).Koppisetty CAK; Frank M; Lyubartsev AP; Nyholm PG Binding Energy Calculations for Hevein-Carbohydrate Interactions Using Expanded Ensemble Molecular Dynamics Simulations. J. Comput. Aided. Mol. Des. 2015. 10.1007/s10822-014-9792-5. [DOI] [PubMed] [Google Scholar]
- (41).Dror RO; Jensen MØ; Borhani DW; Shaw DE Exploring Atomic Resolution Physiology on a Femtosecond to Millisecond Timescale Using Molecular Dynamics Simulations. J. Gen. Physiol. 2010. 10.1085/jgp.200910373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Morris Garrett M.; Huey Ruth; Lindstrom William; Sanner Michel F.; Belew RIchard K.; Goodsell David S; Olson. Arthur J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009. 10.1002/jcc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Trott O; Olson AJ AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2009. 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Lang PT; Brozell SR; Mukherjee S; Pettersen EF; Meng EC; Thomas V; Rizzo RC; Case DA; James TL; Kuntz ID DOCK 6: Combining Techniques to Model RNA-Small Molecule Complexes. RNA 2009. 10.1261/rna.1563609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Claussen H; Gastreich M; Apelt V; Greene J; Hindle S; Lemmen C The FlexX Database Docking Environment - Rational Extraction of Receptor Based Pharmacophores. Curr. Drug Discov. Technol 2006. 10.2174/1570163043484815. [DOI] [PubMed] [Google Scholar]
- (46).Friesner RA; Murphy RB; Repasky MP; Frye LL; Greenwood JR; Halgren TA; Sanschagrin PC; Mainz DT Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein-Ligand Complexes. J. Med. Chem. 2006. 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- (47).Hartshorn MJ; Verdonk ML; Chessari G; Brewerton SC; Mooij WTM; Mortenson PN; Murray CW Diverse, High-Quality Test Set for the Validation of Protein-Ligand Docking Performance. J. Med. Chem. 2007. 10.1021/jm061277y. [DOI] [PubMed] [Google Scholar]
- (48).Nurisso A; Kozmon S; Imberty A Comparison of Docking Methods for Carbohydrate Binding in Calcium-Dependent Lectins and Prediction of the Carbohydrate Binding Mode to Sea Cucumber Lectin CEL-III. Mol. Simul 2008, 34 (4), 469–479. 10.1080/08927020701697709. [DOI] [Google Scholar]
- (49).Adam J; Kříž Z; Prokop M; Wimmerová M; Koča J In Silico Mutagenesis and Docking Studies of Pseudomonas Aeruginosa PA-IIL Lectin - Predicting Binding Modes and Energies. J. Chem. Inf. Model 2008, 48 (11), 2234–2242. 10.1021/ci8002107. [DOI] [PubMed] [Google Scholar]
- (50).Agostino M; Jene C; Boyle T; Ramsland PA; Yuriev E Molecular Docking of Carbohydrate Ligands to Antibodies: Structural Validation against Crystal Structures. J. Chem. Inf. Model. 2009. 10.1021/ci900388a. [DOI] [PubMed] [Google Scholar]
- (51).Agostino M; Yuriev E; Ramsland PA A Computational Approach for Exploring Carbohydrate Recognition by Lectins in Innate Immunity. Front. Immunol 2011, 2 (June). 10.3389/fimmu.2011.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Mishra SK; Adam J; Wimmerová M; Koča J In Silico Mutagenesis and Docking Study of Ralstonia Solanacearum RSL Lectin: Performance of Docking Software to Predict Saccharide Binding. J. Chem. Inf. Model. 2012, 52 (5), 1250–1261. 10.1021/ci200529n. [DOI] [PubMed] [Google Scholar]
- (53).Agostino M; Ramsland PA; Yuriev E Docking of Carbohydrates into Protein Binding Sites. Struct. Glycobiol. 2012, 111–138. [Google Scholar]
- (54).Nivedha AK; Makeneni S; Foley BL; Tessier MB; Woods RJ Importance of Ligand Conformational Energies in Carbohydrate Docking: Sorting the Wheat from the Chaff. J. Comput. Chem. 2014, 35 (7), 526–539. 10.1002/jcc.23517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Nivedha AK; Thieker DF; Makeneni S; Hu H; Woods RJ Vina-Carb: Improving Glycosidic Angles during Carbohydrate Docking. J. Chem. Theory Comput. 2016, 12 (2), 892–901. 10.1021/acs.jctc.5b00834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Samsonov SA; Zacharias M; Chauvot de Beauchene I Modeling Large Protein-Glycosaminoglycan Complexes Using a Fragment-Based Approach. J. Comput. Chem. 2019, 40 (14), 1429–1439. 10.1002/jcc.25797. [DOI] [PubMed] [Google Scholar]
- (57).Leaver-Fay A; Tyka M; Lewis SM; Lange OF; Thompson J; Jacak R; Kaufman K; Renfrew PD; Smith CA; Sheffler W; Davis IW; Cooper S; Treuille A; Mandell DJ; Richter F; Ban YEA; Fleishman SJ; Corn JE; Kim DE; Lyskov S; Berrondo M; Mentzer S; Popović Z; Havranek JJ; Karanicolas J; Das R; Meiler J; Kortemme T; Gray JJ; Kuhlman B; Baker D; Bradley P Rosetta3: An Object-Oriented Software Suite for the Simulation and Design of Macromolecules. In Methods in Enzymology; 2011. 10.1016/B978-0-12-381270-4.00019-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Kaufmann KW; Lemmon GH; DeLuca SL; Sheehan JH; Meiler J Practically Useful: What the R OSETTA Protein Modeling Suite Can Do for You. Biochemistry 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Bender BJ; Cisneros A; Duran AM; Finn JA; Fu D; Lokits AD; Mueller BK; Sangha AK; Sauer MF; Sevy AM; Sliwoski G; Sheehan JH; DiMaio F; Meiler J; Moretti R Protocols for Molecular Modeling with Rosetta3 and RosettaScripts. Biochemistry 2016. 10.1021/acs.biochem.6b00444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Leman JK; Weitzner BD; Lewis SM; Adolf-Bryfogle J; Alam N; Alford RF; Aprahamian M; Baker D; Barlow KA; Barth P; Basanta B; Bender BJ; Blacklock K; Bonet J; Boyken SE; Bradley P; Bystroff C; Conway P; Cooper S; Correia BE; Coventry B; Das R; De Jong RM; DiMaio F; Dsilva L; Dunbrack R; Ford AS; Frenz B; Fu DY; Geniesse C; Goldschmidt L; Gowthaman R; Gray JJ; Gront D; Guffy S; Horowitz S; Huang PS; Huber T; Jacobs TM; Jeliazkov JR; Johnson DK; Kappel K; Karanicolas J; Khakzad H; Khar KR; Khare SD; Khatib F; Khramushin A; King IC; Kleffner R; Koepnick B; Kortemme T; Kuenze G; Kuhlman B; Kuroda D; Labonte JW; Lai JK; Lapidoth G; Leaver-Fay A; Lindert S; Linsky T; London N; Lubin JH; Lyskov S; Maguire J; Malmström L; Marcos E; Marcu O; Marze NA; Meiler J; Moretti R; Mulligan VK; Nerli S; Norn C; Ó’Conchúir S; Ollikainen N; Ovchinnikov S; Pacella MS; Pan X; Park H; Pavlovicz RE; Pethe M; Pierce BG; Pilla KB; Raveh B; Renfrew PD; Burman SSR; Rubenstein A; Sauer MF; Scheck A; Schief W; Schueler-Furman O; Sedan Y; Sevy AM; Sgourakis NG; Shi L; Siegel JB; Silva DA; Smith S; Song Y; Stein A; Szegedy M; Teets FD; Thyme SB; Wang RYR; Watkins A; Zimmerman L; Bonneau R Macromolecular Modeling and Design in Rosetta: Recent Methods and Frameworks. Nature Methods. 2020. 10.1038/s41592-020-0848-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Combs SA; Deluca SL; Deluca SH; Lemmon GH; Nannemann DP; Nguyen ED; Willis JR; Sheehan JH; Meiler J Small-Molecule Ligand Docking into Comparative Models with Rosetta. Nat. Protoc 2013, 8 (7), 1277–1298. 10.1038/nprot.2013.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).DeLuca S; Khar K; Meiler J Fully Flexible Docking of Medium Sized Ligand Libraries with Rosettaligand. PLoS One 2015, 10 (7), 1–19. 10.1371/journal.pone.0132508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Bolia A; Woodrum BW; Cereda A; Ruben MA; Wang X; Ozkan SB; Ghirlanda G A Flexible Docking Scheme Efficiently Captures the Energetics of Glycan-Cyanovirin Binding. Biophys. J. 2014, 106 (5), 1142–1151. 10.1016/j.bpj.2014.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Labonte JW; Adolf-Bryfogle J; Schief WR; Gray JJ Residue-Centric Modeling and Design of Saccharide and Glycoconjugate Structures. J. Comput. Chem. 2017, 38 (5), 276–287. 10.1002/jcc.24679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Raveh B; London N; Zimmerman L; Schueler-Furman O Rosetta FlexPepDock Ab-Initio: Simultaneous Folding, Docking and Refinement of Peptides onto Their Receptors. PLoS One 2011, 6 (4), e18934. 10.1371/journal.pone.0018934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Raveh B; London N; Schueler-Furman O Sub-Angstrom Modeling of Complexes between Flexible Peptides and Globular Proteins. Proteins Struct. Funct. Bioinforma 2010, 78 (9), NA–NA. 10.1002/prot.22716. [DOI] [PubMed] [Google Scholar]
- (67).Frenz B; Rämisch S; Borst AJ; Walls AC; Adolf-Bryfogle J; Schief WR; Veesler D; DiMaio F Automatically Fixing Errors in Glycoprotein Structures with Rosetta. Structure 2019, 27 (1), 134–139.e3. 10.1016/j.str.2018.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Chaudhury S; Berrondo M; Weitzner BD; Muthu P; Bergman H; Gray JJ Benchmarking and Analysis of Protein Docking Performance in Rosetta v3.2. PLoS One 2011, 6 (8), e22477. 10.1371/journal.pone.0022477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Marze NA; Roy Burman SS; Sheffler W; Gray JJ Efficient Flexible Backbone Protein–Protein Docking for Challenging Targets. Bioinformatics 2018, 34 (20), 3461–3469. 10.1093/bioinformatics/bty355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Reback J; McKinney W; jbrockmendel; den Bossche J. Van; Augspurger T; Cloud P; gfyoung; Sinhrks; Klein A; Roeschke M; Hawkins S; Tratner J; She C; Ayd W; Petersen T; Garcia M; Schendel J; Hayden A; MomIsBestFriend; Jancauskas V; Battiston P; Seabold S; chris-b1; h-vetinari; Hoyer S; Overmeire W; alimcmaster1; Dong K; Whelan C; Mehyar M Pandas-Dev/Pandas: Pandas 1.0.3. Zenodo March 2020. 10.5281/zenodo.3715232. [DOI] [Google Scholar]
- (71).The PyMOL Molecular Graphics System, Version 2.3.5 Schrödinger, LLC. [Google Scholar]
- (72).Kortemme T; Morozov AV; Baker D An Orientation-Dependent Hydrogen Bonding Potential Improves Prediction of Specificity and Structure for Proteins and Protein-Protein Complexes. J. Mol. Biol. 2003. 10.1016/S0022. [DOI] [PubMed] [Google Scholar]
- (73).Alford RF; Leaver-Fay A; Jeliazkov JR; O’Meara MJ; DiMaio FP; Park H; Shapovalov MV; Renfrew PD; Mulligan VK; Kappel K; Labonte JW; Pacella MS; Bonneau R; Bradley P; Dunbrack RL; Das R; Baker D; Kuhlman B; Kortemme T; Gray JJ The Rosetta All-Atom Energy Function for Macromolecular Modeling and Design. J. Chem. Theory Comput. 2017, 13 (6), 3031–3048. 10.1021/acs.jctc.7b00125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Bonnardel F; Mariethoz J; Salentin S; Robin X; Schroeder M; Perez S; Lisacek FDSF; Imberty A Unilectin3d, a Database of Carbohydrate Binding Proteins with Curated Information on 3D Structures and Interacting Ligands. Nucleic Acids Res. 2019, 47 (D1), D1236–D1244. 10.1093/nar/gky832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Yuriev E; Agostino M; Ramsland PA Challenges and Advances in Computational Docking: 2009 in Review. J. Mol. Recognit. 2011, 24 (2), 149–164. 10.1002/jmr.1077. [DOI] [PubMed] [Google Scholar]
- (76).Harmalkar A; Gray JJ Advances to Tackle Backbone Flexibility in Protein Docking. Current Opinion in Structural Biology. Elsevier Ltd; April 1, 2021, pp 178–186. 10.1016/j.sbi.2020.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Taylor ME; Drickamer K; Schnaar RL; Etzler ME; Varki A Discovery and Classification of Glycan-Binding Proteins; 2015. [PubMed] [Google Scholar]
- (78).Cummings RD; Schnaar RL; Esko JD; Drickamer K; Taylor ME Principles of Glycan Recognition; 2015. [Google Scholar]
- (79).Kosik I; Yewdell JW Influenza Hemagglutinin and Neuraminidase: Yin–Yang Proteins Coevolving to Thwart Immunity. Viruses. 2019. 10.3390/v11040346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).O’Meara MJ; Leaver-Fay A; Tyka MD; Stein A; Houlihan K; Dimaio F; Bradley P; Kortemme T; Baker D; Snoeyink J; Kuhlman B Combined Covalent-Electrostatic Model of Hydrogen Bonding Improves Structure Prediction with Rosetta. J. Chem. Theory Comput. 2015. 10.1021/ct500864r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Waskom M; the seaborn development team. Mwaskom/Seaborn. Zenodo September 2020. 10.5281/zenodo.592845. [DOI] [Google Scholar]
- (82).Boraston AB; Bolam DN; Gilbert HJ; Davies GJ Carbohydrate-Binding Modules: Fine-Tuning Polysaccharide Recognition. Biochemical Journal. Portland Press Ltd; September 15, 2004, pp 769–781. 10.1042/BJ20040892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Shoseyov O; Shani Z; Levy I Carbohydrate Binding Modules: Biochemical Properties and Novel Applications. Microbiol. Mol. Biol. Rev. 2006. 10.1128/mmbr.00028-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).The CAZypedia Consortium. Ten Years of CAZypedia: A Living Encyclopedia of Carbohydrate-Active Enzymes. Glycobiology 2018. 10.1093/glycob/cwx089. [DOI] [PubMed] [Google Scholar]
- (85).Pires VMR; Henshaw JL; Prates JAM; Bolam DN; Ferreira LMA; Fontes CMGA; Henrissat B; Planas A; Gilbert HJ; Czjzek M; R Pires VM; Henshaw JL; M Prates JA; Bolam DN; A Ferreira LM; G A Fontes CM; Henrissat B; Planas A; Gilbert HJ; Czjzek M; Pires VMR; Henshaw JL; Prates JAM; Bolam DN; Ferreira LMA; Fontes CMGA; Henrissat B; Planas A; Gilbert HJ; Czjzek M The Crystal Structure of the Family 6 Carbohydrate Binding Module from Cellvibrio Mixtus Endoglucanase 5A in Complex with Oligosaccharides Reveals Two Distinct Binding Sites with Different Ligand Specificities. J. Biol. Chem. 2004. 10.1074/jbc.M401599200. [DOI] [PubMed] [Google Scholar]
- (86).Spiwok V CH/π Interactions in Carbohydrate Recognition. Molecules 2017, 22 (7). 10.3390/molecules22071038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Brenke R; Kozakov D; Chuang GY; Beglov D; Hall D; Landon MR; Mattos C; Vajda S Fragment-Based Identification of Druggable “hot Spots” of Proteins Using Fourier Domain Correlation Techniques. Bioinformatics 2009, 25 (5), 621–627. 10.1093/bioinformatics/btp036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Zhao J; Cao Y; Zhang L Exploring the Computational Methods for Protein-Ligand Binding Site Prediction. Computational and Structural Biotechnology Journal. Elsevier B.V. January 1, 2020, pp 417–426. 10.1016/j.csbj.2020.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Zhao H; Taherzadeh G; Zhou Y; Yang Y Computational Prediction of Carbohydrate-Binding Proteins and Binding Sites. Curr. Protoc. Protein Sci. 2018, 94 (1), e75. 10.1002/cpps.75. [DOI] [PubMed] [Google Scholar]
- (90).Gattani S; Mishra A; Hoque MT StackCBPred: A Stacking Based Prediction of Protein-Carbohydrate Binding Sites from Sequence. Carbohydr. Res. 2019. 10.1016/j.carres.2019.107857. [DOI] [PubMed] [Google Scholar]
- (91).Kozakov D; Grove LE; Hall DR; Bohnuud T; Mottarella SE; Luo L; Xia B; Beglov D; Vajda S The FTMap Family of Web Servers for Determining and Characterizing Ligand-Binding Hot Spots of Proteins. Nat. Protoc. 2015, 10 (5), 733–755. 10.1038/nprot.2015.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Gagneux P; Aebi M; Varki A Evolution of Glycan Diversity. In Essentials of Glycobiology; 2015. 10.1101/glycobiology.3e.020. [DOI] [Google Scholar]
- (93).Muraki M; Morikawa M; Jigami Y; Tanaka H The Roles of Conserved Aromatic Amino-Acid Residues in the Active Site of Human Lysozyme: A Site-Specific Mutagenesis Study. Biochim. Biophys. Acta (BBA)/Protein Struct. Mol. 1987. 10.1016/0167-4838. [DOI] [PubMed] [Google Scholar]
- (94).Fernández-Alonso MDC; Cañada FJ; Jiménez-Barbero J; Cuevas G Molecular Recognition of Saccharides by Proteins. Insights on the Origin of the Carbohydrate-Aromatic Interactions. J. Am. Chem. Soc. 2005. 10.1021/ja051020+. [DOI] [PubMed] [Google Scholar]
- (95).Kerzmann A; Neumann D; Kohlbacher O SLICK - Scoring and Energy Functions for Protein - Carbohydrate Interactions. J. Chem. Inf. Model. 2006. 10.1021/ci050422y. [DOI] [PubMed] [Google Scholar]
- (96).Kerzmann A; Fuhrmann J; Kohlbacher O; Neumann D BALLDock/SLICK: A New Method for Protein-Carbohydrate Docking. J. Chem. Inf. Model. 2008, 48 (8), 1616–1625. 10.1021/ci800103u. [DOI] [PubMed] [Google Scholar]
- (97).Hsu C-H; Park S; Mortenson DE; Foley BL; Wang X; Woods RJ; Case DA; Powers ET; Wong C-H; Dyson HJ; Kelly JW The Dependence of Carbohydrate-Aromatic Interaction Strengths on the Structure of the Carbohydrate. J. Am. Chem. Soc. 2016, 138 (24), 7636–7648. 10.1021/jacs.6b02879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Stanković IM; Blagojević Filipović JP; Zarić SD Carbohydrate – Protein Aromatic Ring Interactions beyond CH/π Interactions: A Protein Data Bank Survey and Quantum Chemical Calculations. Int. J. Biol. Macromol. 2020, 157, 1–9. 10.1016/j.ijbiomac.2020.03.251. [DOI] [PubMed] [Google Scholar]
- (99).Tschampel SM; Woods RJ Quantifying the Role of Water in Protein-Carbohydrate Interactions. 2003. 10.1021/JP035027U. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (100).Nurisso A; Blanchard B; Audfray A; Rydner L; Oscarson S; Varrot A; Imberty A Role of Water Molecules in Structure and Energetics of Pseudomonas Aeruginosa Lectin I Interacting with Disaccharides. J. Biol. Chem. 2010. 10.1074/jbc.M110.108340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (101).Ruvinsky AM; Aloni I; Cappel D; Higgs C; Marshall K; Rotkiewicz P; Repasky M; Feher VA; Feyfant E; Hessler G; Matter H The Role of Bridging Water and Hydrogen Bonding as Key Determinants of Noncovalent Protein–Carbohydrate Recognition. ChemMedChem 2018. 10.1002/cmdc.201800437. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.