

Abstract
Baseline patient characteristics and prognostic factors are important considerations in oncology when evaluating the impact of immunogenicity on pharmacokinetics (PK) and efficacy. Here, we assessed the impact of anti‐drug antibodies (ADA) on the PK of the immune checkpoint inhibitor atezolizumab (an anti–PD‐L1 monoclonal antibody). We evaluated data from ≈ 4500 patients from 12 clinical trials across different tumor types, treatment settings, and dosing regimens. In our dataset, ~ 30% of patients (range, 13–54%) developed treatment‐emergent ADA, and in vitro neutralizing antibodies (NAb) were seen in ~ 50% of ADA‐positive (+) patients. Pooled time course data showed a trend toward lower atezolizumab exposure in ADA+ patients, which was more pronounced in ADA+/NAb+ patients. However, the atezolizumab concentration distributions overlapped, and drug concentrations exceeded 6 µg/ml, the target concentration required for receptor saturation, in greater than 95% of patients. Patients had sufficient exposure regardless of ADA status. The dose selected to allow for dosing over effects from ADA resulted in a flat exposure‐response relationship. Analysis of study results by ADA titer showed that exposure and overall survival were not affected in a clinically meaningful way. High tumor burden, low albumin, and high CRP at baseline showed the greatest association with ADA development but not with subsequent NAb development. These imbalanced factors at baseline can confound analysis of ADA impact. ADA increases atezolizumab clearance minimally (9%), and its impact on exposure based on the totality of the clinical pharmacology assessment does not appear to be clinically meaningful.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Anti‐drug antibodies (ADA) to anticancer therapeutics, including atezolizumab, might affect pharmacokinetics (PK) and drug efficacy.
WHAT QUESTION DID THIS STUDY ADDRESS?
How do treatment‐emergent ADA impact atezolizumab exposure, and is there association between baseline prognostic factors and the development of ADA?
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
Based on a dataset of ~ 4500 patients, we showed that decreased atezolizumab exposure accompanying ADA and neutralizing ADA (NAb) development does not meaningfully impact drug concentrations required for receptor saturation. Further, differences in atezolizumab concentrations seen prior to ADA development demonstrated that baseline prognostic factor imbalances other than ADA can affect the drug exposure. Notably, baseline tumor burden, albumin, and C‐reactive protein can influence ADA development.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
The traditional approach of using univariate analysis to analyze PK or efficacy by ADA status alone may be inadequate, and confounding from baseline covariates should be considered or accounted for in ADA studies. The impacts of ADA on atezolizumab exposure are not expected to be clinically relevant.
INTRODUCTION
Atezolizumab is an Fc‐engineered humanized IgG1 Monoclonal antibody that binds to programmed death‐ligand 1 (PD‐L1) and blocks its interactions with programmed death‐1 (PD‐1) and B7.1 receptors. It has been approved in the treatment of certain types of non‐small cell lung cancer (NSCLC), small cell lung cancer (SCLC), triple‐negative breast cancer, urothelial carcinoma, hepatocellular carcinoma, and melanoma. The clinical pharmacology properties of atezolizumab were described previously based on results from the dose‐finding and early clinical trials. 1 The current report provides a retrospective assessment of the impact of immunogenicity on the clinical pharmacology of atezolizumab based upon integrated data from 12 clinical trials.
Anti‐drug antibody (ADA) rates for atezolizumab in approved indications have been shown to range from 13% to 36% 2 ; these rates are higher than those reported for other PD‐L1 checkpoint inhibitors (≈ 2–38% 3 ), although direct comparison of ADA rates across molecules is difficult due to different ADA assay characteristics. In the initial dose‐finding study, low doses sometimes resulted in exposure lower than the target concentrations for patients who were ADA positive. However, at the clinical dose evaluated for efficacy (15 mg/kg), ADA did not markedly increase atezolizumab clearance (by ~ 16% 1 ). This observation infers that there is sufficient drug exposure independent of ADA development (i.e., dosing over the ADA effect is possible). A dose of 1200 mg every 3 weeks (q3w) administered intravenously was selected for numerous phase II and phase III studies to provide atezolizumab concentration greater than the receptor saturable concentration of 6 µg/ml for the vast majority of patients, regardless of their ADA status. 1 , 4
Investigating the impact of ADA in oncology is challenging because some baseline prognostic factors may influence pharmacokinetics (PK) and clinical outcomes. 5 , 6 Atezolizumab exhibits time‐dependent PK, 7 consistent with other PD‐L1 and PD‐1 checkpoint inhibitors. 8 , 9 , 10 , 11 For atezolizumab, the statistical significance of covariates for clearance ranks as follows (highest to the lowest): baseline albumin, tumor burden, body weight, ADA, sex, alkaline phosphatase (ALP), bilirubin, and neutrophil counts for clearance (CL); body weight and sex for central volume of distribution; and sex for peripheral volume of distribution. 7 None of the covariates for PK of atezolizumab were considered clinically relevant. Because some of these covariates for atezolizumab PK are also prognostic factors for overall survival (OS), deconvoluting these confounding effects is necessary to interpret exposure‐response analyses. 6 To ensure this critical confounding concept is considered in our immunogenicity assessments, we describe a novel ADA assessment framework for oncology (Figure 1).
FIGURE 1.
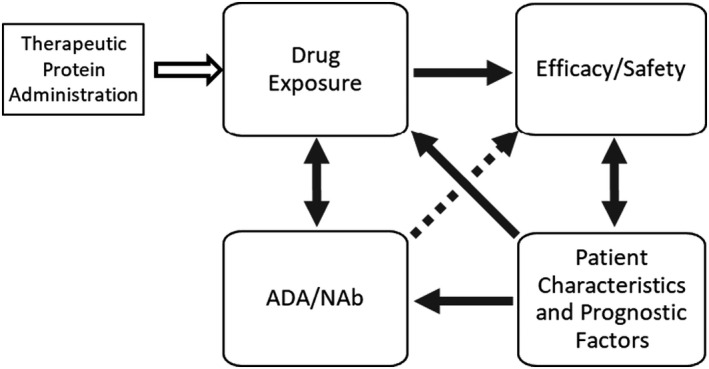
ADA assessment framework for oncology biologics. Immunogenicity assessment in oncology should consider the complex relationship of drug concentrations, ADA/NAb, efficacy, and patient characteristics and prognostic factors. The direction of the arrows indicates the direction of the impact, which can be unidirectional or bidirectional. The dotted arrow represents the hypothetical effect of ADA directly on efficacy without impacting drug exposure. ADA, anti‐drug antibody; NAb, neutralizing antibody
Figure 1 illustrates the interdependencies of several key determinants affecting immunogenicity assessment in oncology. Upon therapeutic protein administration, drug exposure drives clinical response, which can be impacted both by patient baseline characteristics and ADA status. 5 Particularly for oncology, clinical response can be impacted by patient prognostic factors; this is why stratification factors are typically incorporated into study design to achieve a balanced population between the control and treatment groups in randomized trials. The arrows between efficacy and prognostic factors are bidirectional because as a patient’s health status improves due to treatment benefit, their prognosis also changes over time, which may also impact the clearance of the drug. This is the main reason that incorporating time‐varying covariates can better describe the time‐course of drug concentrations compared to a stationary PK model for many cancer immunotherapy (CIT) treatments. 7 , 8 , 9 , 10 , 11 The dashed line between ADA and efficacy in Figure 1 infers a hypothetical relationship because a direct impact of ADA on efficacy in the absence of alterations in PK has not been published before, to our knowledge. For atezolizumab, a further decrease in exposure for neutralizing ADA was observed but the decrease was not considered clinically relevant when interpreted in the context of flat exposure‐response relationships. Furthermore, as shown in atezolizumab immunogenicity‐efficacy analyses (part 2 of this article series), neutralizing ADA did not lead to clinically relevant impact on efficacy. On the other hand, ADA can impact safety without impacting exposure through off‐target effects. 12
A concept often overlooked is the association of baseline prognostic factors with treatment‐emergent ADA development. We will describe the importance of prognostic factors that can potentially increase the chance of developing treatment‐emergent ADA. We will show that it is therefore important to adjust imbalances, if any, in prognostic factors prior to interpretation of the impact of ADA on efficacy. Detailed efficacy findings will be reported separately. In this paper, we will focus on the assessment of ADA impact on atezolizumab exposure and relationship of ADA development and baseline prognostic factors.
METHODS
A total of 12 clinical trials in five cancer types with data from over 4700 patients provided the basis of this clinical pharmacology assessment of atezolizumab immunogenicity (Table S1). All clinical trials were conducted in accordance with Good Clinical Practice Guidelines and the Declaration of Helsinki. Protocol approval was obtained from independent review boards or ethics committees at each institution, and all patients provided written informed consent. This analysis includes data from NSCLC or SCLC (POPLAR, 13 OAK, 14 IMpower130, 15 IMpower131, 16 IMpower132, 17 IMpower150, 18 and IMpower133 19 ); urothelial carcinoma (IMvigor210 20 , 21 and IMvigor211 22 ), renal cell carcinoma (IMmotion151 23 ), breast cancer (IMpassion130 24 ); and hepatocellular carcinoma (IMbrave150 25 ). Atezolizumab was administered as monotherapy or in combination with chemotherapy (carboplatin, cisplatin, pemetrexed, etoposide, paclitaxel, nab‐paclitaxel, and docetaxel) and/or with another biologic (bevacizumab).
ADA testing methods were developed and run in accordance with industry best practices and health authority guidelines. 26 , 27 , 28 , 29 , 30 All ADA samples were tested in a screening assay, and any asamples were further characterized for ADA titer and in vitro neutralizing antibody (NAb) using a tiered approach 1 ; this enabled calculation of treatment‐emergent ADA and NAb rates for each study. ADA samples were collected from patients before the first drug administration and also prior to drug retreatment (time of minimal drug concentration [Cmin]) up to nine timepoints over the course of each study while on treatment. Drug concentrations were also measured at the same timepoints as those for ADA. The lower limit of quantitation (LLOQ) for measuring atezolizumab concentrations in serum was 0.06 µg/ml. Values below the LLOQ were treated as half of the LLOQ. The PK and ADA assay performance parameters were established based on industry best practices, and industry standard definitions 31 were consistently applied to classify patients as either ADA‐negative (ADA–) or treatment‐emergent ADA+. The treatment‐emergent ADA+ group consists of two subgroups (patients with treatment‐enhanced and treatment‐induced ADA). An analysis of the impact of ADA or NAb on PK includes a descriptive analysis of atezolizumab concentration data over time by ADA and NAb status. Exposure and OS by ADA titer were evaluated. To evaluate the scenario of low exposure (<6 µg/ml at cycle 1 Cmin) and treatment‐emergent ADA positivity, data from the 12 studies were pooled to compare the clinical response (overall response rate [ORR], stable disease [SD], or progressive disease [PD]) by ADA or NAb status.
RESULTS
ADA Profiles
The ADA and NAb rates across studies are shown in Figure 2. Across the 12 studies, the ADA rates ranged from 13.1% to 54.1% with an average of ~ 30%. Approximately half of ADA+ patients developed NAb, with NAb rates ranging from 4.3% to 27.5% among all evaluable patients, with an average of ~ 15%. The median and range of percentage of patients with steady‐state concentrations (defined as Cmin at day 42) above the assay tolerance (200 ug/ml) was 4.85% (0.89 to 8.91%) following 1200 mg q3w i.v. administrations. This percentage for the one study that administered atezolizumab as 840 mg every 2 weeks (q2w) was 54%.
FIGURE 2.
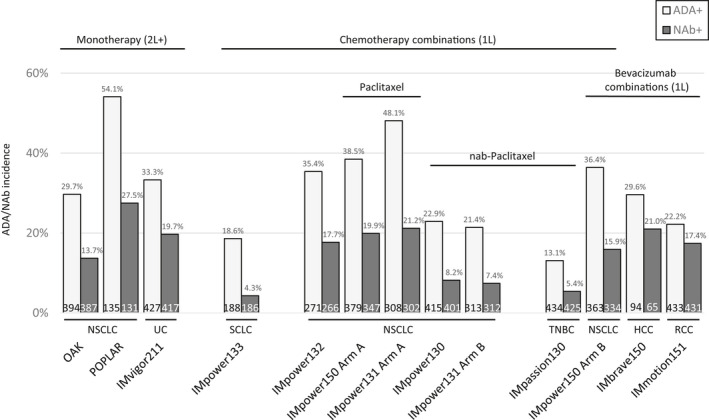
Treatment‐emergent ADA and NAb incidence rates by study. Treatment‐emergent ADA and NAb rates did not show particular trends across indications or with monotherapy versus combination therapy. Text at the bottom of each bar represents the total number of evaluable patients. 1L, first line; 2L, second line; ADA, anti‐drug antibody; HCC, hepatocellular carcinoma; NAb, neutralizing antibody; NSCLC, non‐small cell lung cancer; RCC, renal cell carcinoma; SCLC, small cell lung cancer; TNBC, triple‐negative breast cancer; UC, urothelial carcinoma
Overall, the ADA rates were variable between studies but did not show any obvious trend when atezolizumab was administered as monotherapy versus in combination, by indication, by the type of drug combination used, or by the dosing frequency (1200 mg q3w vs. 840 mg q2w).
The duration of ADA appears to be transient. Approximately 70% of treatment‐emergent ADA positivity was based on only one ADA+ timepoint among the multiple timepoints that were analyzed. The first ADA+ sample for a patient was typically detected early during treatment (pretreatment of cycle 2), whereas NAb were typically detected later (cycles 2–4).
Atezolizumab exposure by ADA status
Peak and trough atezolizumab concentrations (Cmax and Cmin) were derived from samples collected in the clinical trials. The pooled time‐course of atezolizumab concentrations by ADA and NAb status is shown in Figure 3a. Sampling for Cmax was collected at cycle 1 in all studies, and additional Cmax samples were collected at cycle 3 in three of the studies. When comparing the median exposure, a trend of decreasing exposure was observed from ADA– to binding‐only ADA+ (ADA+NAb–) to NAb‐positive (ADA+NAb+) across different timepoints. However, the population distributions of atezolizumab concentrations from the subgroups largely overlap, and the majority of concentrations in all three subgroups are above the receptor saturable target concentration of 6 µg/ml (shown as a dashed line in Figure 3a) across the timepoints. Atezolizumab has a half‐life of 27 days. 2 With an accumulation ratio of approximately two to threefold following 1200 mg q3w or 840 mg q2w i.v. administrations, the majority of patients (>95%) maintain drug exposure that is above the target concentration, regardless of ADA status.
FIGURE 3.
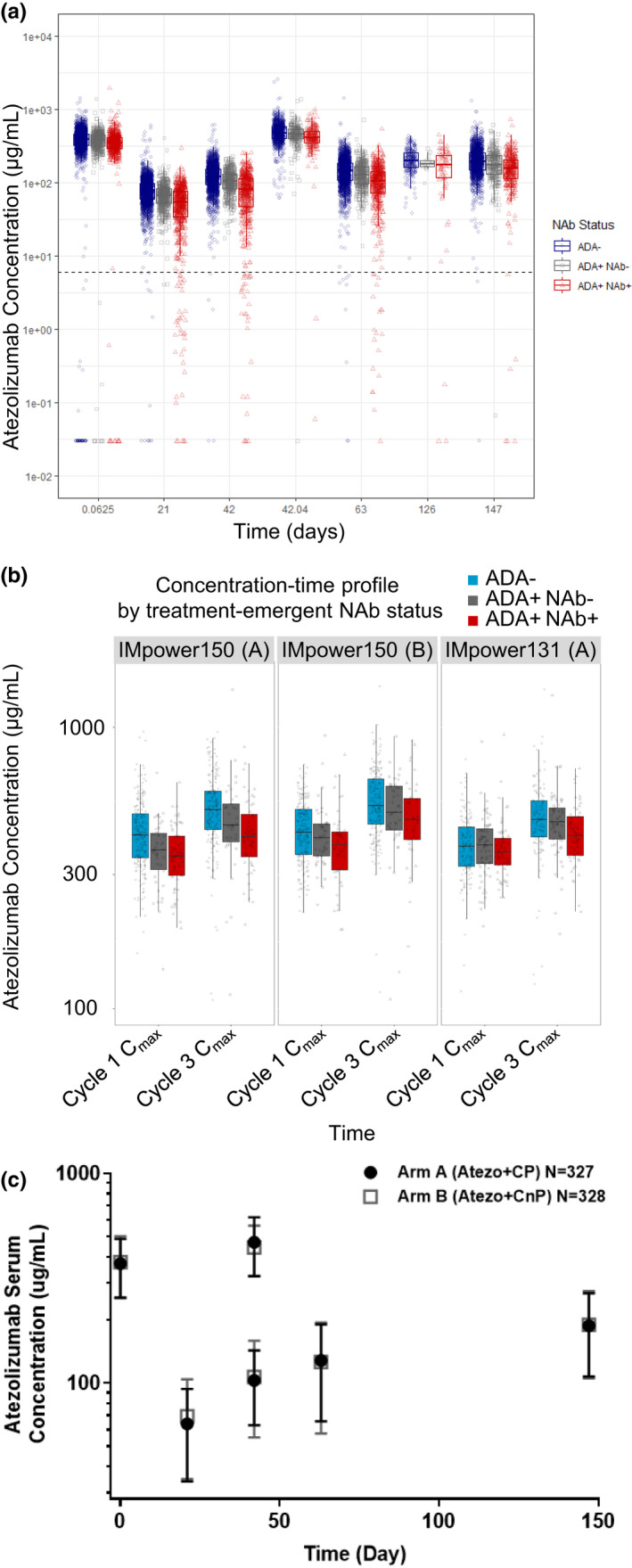
Atezolizumab exposure by ADA status. (a) Peak and trough atezolizumab concentrations over time by ADA and NAb status based on pooled data. (b) Differences in exposure immediately after the first dose and third dose (Cmax before and after ADA formation). (c) Atezolizumab concentrations in IMpower131 study arms with a twofold difference in the ADA rate. As seen in panel a, a trend of lower exposure in ADA+ and NAb+ patients was observed when compared to the exposure of ADA– patients but the distribution of the subgroups largely overlaps. The horizontal dash line represents receptor saturable concentration of 6 µg/ml. Greater than 95% of exposure data were above the receptor saturable concentration. As seen in panel b, factors other than ADA may contribute to the differences in exposure in treatment‐emergent ADA/NAb subgroups. This difference did not increase before (cycle 1 Cmax) or after (cycle 3 Cmax) ADA development. Panel c shows a study in which two atezolizumab treatment arms were included (IMpower131); doubling of ADA rates (48% in arm A vs. 22% in arm B) did not impact the overall atezolizumab concentrations; peak and trough concentrations of atezolizumab over time were superimposable. ADA, anti‐drug antibody; Cmax, maximum plasma concentration; NAb, neutralizing antibody
Atezolizumab concentrations from samples collected 30 min after the first dose (cycle 1 Cmax), in the absence of any circulating ADA, are shown in Figure 3b. Prior to ADA development, a difference of ~ 10% between treatment‐emergent ADA+ and ADA– subgroups was observed. ADA would not be present at the cycle 1 Cmax timepoint, because ADA would not develop immediately upon first drug exposure. Hence, the difference is likely due to imbalances in baseline covariates. This difference between ADA+ and ADA– patients did not increase at cycle 3 Cmax (42 days after the first dose) when ADA development is physiologically plausible.
Figure 3c shows the concentration‐time profiles of the two experimental study arms from an NSCLC trial (IMpower131 study). The sample sizes for the two study arms were similar (327 vs. 328) but the ADA rate in the atezolizumab combined with cisplatin and paclitaxel arm was 48%, whereas the ADA rate in the atezolizumab combined with cisplatin and nab‐paclitaxel arm was 21%. Figure 3c shows that despite an approximately twofold difference in ADA rates for the two arms of the study, the overall exposures were superimposable.
Low exposure population
To evaluate the potential clinical impact of reduced atezolizumab exposure in the ADA+ subgroup, objective responses in ADA+ and ADA– patients who had low exposure (≤6 µg/ml in a pooled analysis) were compared to responses in the same ADA subgroups of patients with exposure greater than 6 µg/ml (Table S2). The pooled analysis was based on cycle 1 Cmin exposures, which generally represent the timepoint of lowest trough exposure because this is prior to drug accumulation. The results show similar rates of objective response (OR), SD and PD, regardless of level of exposure at cycle 1 Cmin or treatment‐emergent ADA status. Similar results were observed between exposure groups according to NAb status (Table S3).
ADA titer
The titer of each ADA+ sample was determined by analyzing serial dilutions of the sample and finding the dilution, expressed as the base 10 log, from which the signal crossed the ADA assay cut point or decision threshold. Across the trials, the ADA titer values ranged from 1.21 to 4.9 titer units (TU), with a median of 2.11 TU. A scatter plot of ADA titer versus drug concentrations at cycle 1 Cmin showed a random distribution without any trend (data not shown). To further evaluate the potential impact of ADA titer on exposure and efficacy, the pooled PK data were divided into two groups using the median titer value (2.11 TU) as a cutoff. Because the analysis required both ADA titer value and cycle 1 Cmin data, this criterion was met by 27% of ADA+ patients, including 484 patients in the high‐ADA titer subgroup and 493 patients in the low‐ADA titer subgroup. Figure 4a shows the pooled median cycle 1 Cmin of the high‐ADA titer subgroup lowered by a minimal 9.06% compared to the low‐ADA titer subgroup.
FIGURE 4.
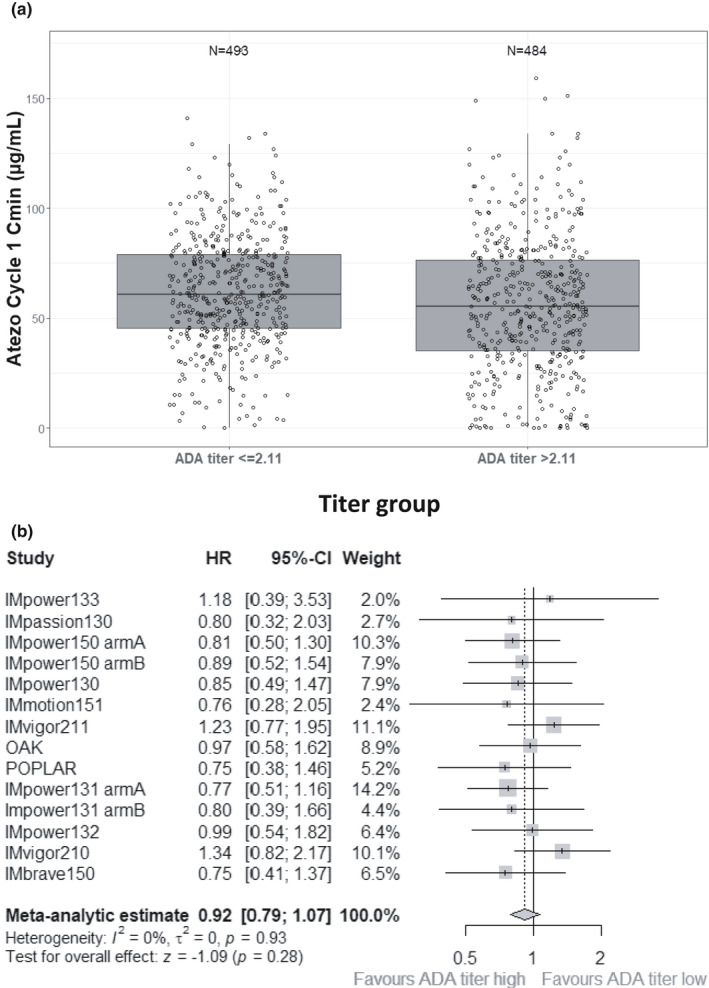
ADA titer analyses. (a) Atezolizumab concentration by ADA titer and (b) overall survival HR of high versus low ADA titer. In panel a, a pooled analysis of cycle 1 Cmin by ADA titer values at the same timepoint showed an ~ 10% decrease in exposure in the high ADA titer subgroup compared to the low ADA titer subgroup. The difference was statistically significant (p < 0.05), but the distributions of the two groups highly overlapped. In panel b, meta‐analysis HR of overall survival from the high ADA titer versus low ADA titer was 0.92 with 90% confidence interval crossing 1 (95% CI, 0.79 to 1.07). ADA, anti‐drug antibody; CI, confidence interval; Cmin, minimum plasma concentration; HR, hazard ratio
OS by ADA titer status was also evaluated. The demographic profiles between patients with high‐ versus low‐ADA titer samples were mostly balanced but slightly unfavorable C‐reactive protein (CRP), tumor burden, and ALP values were observed in the high‐ADA titer patients (Table S4). A hazard ratio (HR) value was computed for each study based on Kaplan‐Meier curves of high ADA titer vs low ADA titer, as shown in Figure 4b, such that an HR value greater than 1 would suggest that high ADA titer has negative effect on OS compared to low ADA titer. A meta‐analysis of the HR across the studies was performed with weighting based on trial size and confidence interval. This overall HR was 0.93 with a 95% confidence interval of 0.79 to 1.09, suggesting that no decrease in efficacy was observed in patients with high ADA titer compared to low ADA titer.
Novel relationship between prognostic factors and ADA development
The association of prognostic factors with drug disposition and ADA development was evaluated using data pooled from the 12 clinical trials. The baseline prognostic factors evaluated were age, albumin, body weight, CRP, lactate dehydrogenase, neutrophil lymphocyte ratio (NLR), and tumor burden. The median baseline CRP in the ADA+ subgroup was ~ 100% higher than the median baseline CRP for the ADA– subgroup, but the distribution was highly variable with large overlaps (Figure 5a). A breakdown of baseline CRP into six tiles revealed a trend of increasing prevalence of treatment‐emergent ADA development with increasing baseline CRP values (Figure 5b). This pattern was also observed for baseline NLR and albumin, such that patients with worse baseline prognostic factors had a higher likelihood of developing ADA compared to those with favorable baseline factors. At the individual study level, similar trends of higher likelihood of developing ADA with poorer baseline prognostic factors were observed for all studies, regardless of indication, combination therapy or monotherapy, and line of therapy. However, when similar analyses were applied to ADA+NAb– versus ADA+NAb+, this trend was not consistently observed (data not shown).
FIGURE 5.
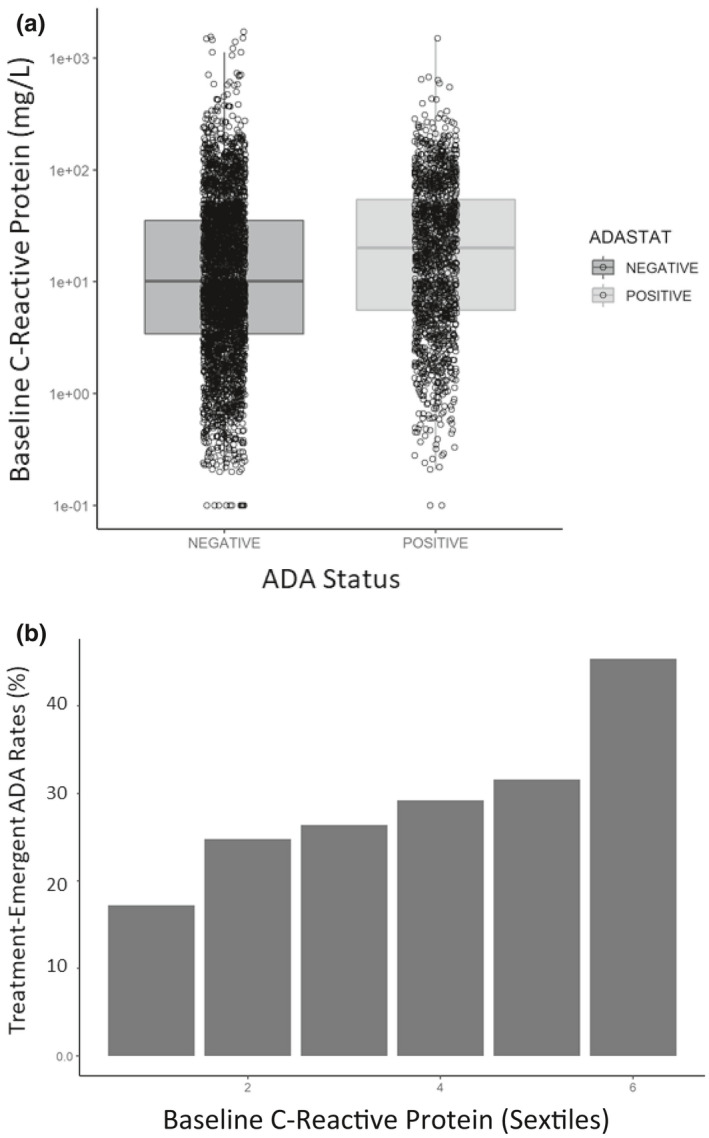
Pooled baseline prognostic factor association with ADA status. (a) Baseline CRP by ADA status and (b) ADA incidence rate by sextile of baseline CRP. As seen in panel a, patients who tested positive for treatment‐emergent ADA tended to have higher baseline CRP (worse health status). As seen in panel b, dividing the baseline CRP values into six tiles revealed an increasing chance of developing ADA after treatment. ADA, anti‐drug antibody; CRP, C‐reactive protein
DISCUSSIONS/CONCLUSIONS
The traditional persistent and transient definitions for ADA that were established by an American Association of Pharmaceutical Scientists expert group 31 to describe the duration of an ADA response can be useful for non‐oncology patients, but these definitions have limitations when applied to oncology because of early death and censorship. The Shankar paper 31 defined a “persistent” ADA response as an ADA response that is detectable for at least 16 weeks or wherein the last ADA sampling timepoint is positive. In oncology, aggressive disease can lead to early discontinuation or death, often in a substantial proportion of patients, resulting in a higher chance of defining a patient as having persistent ADA based on just one ADA+ timepoint. Furthermore, to define persistent ADA as having at least 16 weeks with an ADA+ result also generates survival bias in cancer types with short median survival. Hence, these persistent and transient ADA definitions should be used with caution in oncology. With that said, the duration of ADA effect is an important indicator of the relevant clinical impact of ADA. 32 The atezolizumab studies were designed with systematic ADA testing plans, including ADA analysis from cycle 1 to 16 with each cycle defined as 2 or 3 weeks, depending on whether 840 mg q2w or 1200 mg q3w of atezolizumab was administered. Using this regular ADA monitoring for atezolizumab trials, the treatment‐emergent ADA rates ranged from 13 to 54%, but ~ 70% of ADA+ patients had only a single ADA+ result at cycles 2 or 3 (Table 1). It is possible that these early transient occurrences of ADA are primary immunoglobulin M (IgM) responses rather than the long‐lasting IgG responses, with IgM responses less likely to lead to detrimental clinical impact. 33
TABLE 1.
Frequency of ADA positivity
| Study | Frequency of ADA positivity for indicated number of timepoints (in treatment‐emergent ADA+ patients), n (%) | |||||
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | |
| OAK | 71 (56) | 31 (25) | 12 (10) | 8 (6) | 3 (2) | 1 (1) |
| POPLAR | 32 (44) | 18 (25) | 10 (14) | 8 (11) | 4 (5) | 1 (1) |
| IMpower130 | ||||||
| Arm A (Atezolizumab + CnP) | 72 (66) | 26 (24) | 6 (6) | 4 (4) | 1 (1) | 0 |
| Arm B (CnP) | 13 (57) | 3 (13) | 3 (13) | 3 (13) | 1 (4) | 0 |
| IMpower132 | ||||||
| Arm A (Atezolizumab + carboplatin + pemetrexed combined) | 49 (80) | 8 (13) | 2 (3) | 2 (3) | 1 (1) | 0 |
| Arm B (Atezolizumab + cisplatin + pemetrexed combined) | 27 (71) | 8 (21) | 3 (9) | 0 | 0 | 0 |
| IMpower150 | ||||||
| Arm A (Atezolizumab + CP) | 103 (65) | 35 (22) | 14 (9) | 4 (3) | 3 (2) | 0 |
| Arm B (Atezolizumab + bevacizumab + CP) | 86 (61) | 37 (26) | 14 (10) | 2 (1) | 2 (1) | 1 (1) |
| IMpower131 | ||||||
| Arm A (Atezolizumab + CP) | 80 (57) | 41 (29) | 13 (9) | 5 (4) | 1 (1) | 0 |
| Arm B (Atezolizumab + CnP) | 43 (72) | 9 (15) | 6 (10) | 2 (3) | 0 | 0 |
| IMpower133 | 30 (77) | 7 (18) | 2 (5) | 0 | 0 | 0 |
| IMvigor210 | 102 (56) | 39 (21) | 27 (15) | 12 (7) | 2 (1) | 0 |
| IMvigor211 | 88 (59) | 34 (23) | 13 (9) | 7 (5) | 6 (4) | 0 |
| IMmotion151 | 77 (64) | 33 (28) | 7 (6) | 3 (3) | 0 | 0 |
| IMbrave150 | 63 (72) | 14 (16) | 9 (10) | 2 (2) | 0 | 0 |
| IMpassion130 | 35 (66) | 9 (17) | 3 (6) | 3 (6) | 2 (4) | 1 (2) |
Majority of studies show treatment‐emergent ADA as a one‐time event over the course of the study.
Abbreviations: ADA, anti‐drug antibody; CP, carboplatin + paclitaxel; CnP, carboplatin + nab‐paclitaxel.
Decreases in atezolizumab concentrations by ADA status have been observed in both univariate and multivariate analyses. First, the univariate analysis was based on observed atezolizumab concentrations in serum and post hoc clearance estimates by ADA status. The decrease in observed cycle 1 Cmin was ~ 20% in the overall mean in the ADA+ subgroup compared to the ADA– subgroup. When only comparing the post hoc clearance values between the ADA subgroups with no covariate adjustment, the difference averaged 22% (range, 18 to 49%). 2 However, not all the differences in the observed exposure were due to ADA. This was apparent from evaluating descriptive atezolizumab concentration data and model‐based multivariate analyses (see below). By evaluating the exposure at a timepoint that is prior to ADA development (cycle 1 Cmax, collected 30 min after the end of the first atezolizumab infusion), a difference in mean exposure could be observed when subsetting the data by treatment‐emergent ADA status at later timepoints. Because the typical time for the immune system to develop an initial antibody response is ~ 5 to 10 days, 34 the difference in exposure at 30 min after the first dose could not be due to ADA and is likely due to the underlying patient disease characteristics and health status prior to atezolizumab treatments.
Second, model‐based multivariate analysis included both stationary and time‐varying population PK models. A time‐varying PK model that incorporates longitudinal prognostic factors to evaluate the ADA impact is the most appropriate. 32 After accounting for important time‐varying covariates of exposure, the impact of treatment‐emergent ADA on atezolizumab clearance was 9%. 7 This impact is less than that seen in the stationary population PK model (16%), suggesting that fluctuations in other factors (e.g., albumin level) during the treatment period are more important for clearance than is ADA development. 1
Finally, atezolizumab exhibits a flat exposure‐response relationship over a wide range of drug concentrations (~ 2‐fold). 1 , 35 For a drug with a large therapeutic window that exhibits a flat exposure‐response relationship, an increase of 9% in clearance due to ADA is not expected to be clinically meaningful. Likewise, the 20% clearance difference observed at cycle 1 Cmin is unlikely to be meaningful when there is almost a 200% difference in cycle 1 Cmin values across the study population and a flat exposure‐response relationship.
The flat exposure‐response relationship of atezolizumab and the minimal increase in atezolizumab clearance observed in ADA+ patients confirm the effectiveness of the strategy of dosing over any potential ADA effect on exposure. As observed from the pooled concentration‐time plot by ADA status, adequate exposure was achieved regardless of ADA or NAb status in cycle 1 Cmin for over 95% of the population. Overall, the totality of the clinical pharmacology data show that the impact of ADA on atezolizumab PK does not appear to be clinically meaningful.
NAb often develop in patients with multiple sclerosis (MS) after interferon‐beta (IFN‐β) treatment. In a small study of IFN‐β–treated patients with MS, Hegen et al. 36 found that 13% of patients developed in vitro NAb after 24 months of therapy, and that the subset of ADA+ patients with the highest ADA titers at 3 months went on to develop NAb. However, this study did not assess the impact of ADA on drug exposure or on clinical outcomes. For atezolizumab, the incidence rates of NAb+ were approximately half of those in patients who were ADA+ (4–28% of ADA‐evaluable patients, median 17.4%), and patients with NAb+ had a further 20% decrease in atezolizumab exposure compared to ADA+NAb– patients. However, the same principle of dosing over the ADA effects should apply to the NAb subgroup, in which atezolizumab concentrations were also maintained above the receptor saturable concentration. Furthermore, efficacy (OS) was not reduced in the high ADA titer subgroup compared with the low ADA titer subgroup.
Emerging oncology data suggest that there may be a relationship between some baseline prognostic factors and the potential to develop treatment‐emergent ADA. In oncology, particularly with CIT drugs, prognostic factors not only impact clinical outcome, but also impact PK and treatment‐emergent ADA development (see Figure 5). These confounding effects are well known for exposure‐response analyses. 5 , 6 In our analysis, we observed that the sets of covariates that are linked to efficacy, PK, and ADA development only partially overlap. For example, in NSCLC, well known prognostic factors for OS include baseline tumor burden, liver metastasis status, albumin, sex, smoking status, histology marker, and Eastern Cooperative Oncology Group performance status. In the same indication, the main covariates for atezolizumab PK are albumin and baseline tumor burden. In NSCLC, we have observed that baseline CRP, NLR, albumin, and tumor burden are associated with ADA development. The shared covariates, if not addressed, would confound the interpretation of any univariate analysis (i.e., direct evaluation of efficacy outcome by ADA status). Patients with the poorest baseline prognostic factors would likely have a reduced response to treatment, shorter OS, lower exposure, and increased likelihood of developing ADA. However, whereas this general association is observed, there is no particular prognostic factor or a set of prognostic factors that can reliably predict ADA development.
Once any imbalance(s) in covariates between ADA subgroups are addressed, the true impact of ADA can be assessed. For atezolizumab, the impact of ADA on exposure (9–16%) was inconsequential after the confounding effects of prognostic factors were addressed. Hence, only multivariate analysis or adjustment method analysis should be applied to interpret the true impact of ADA on exposure in oncology. In essence, our findings indicate that conclusions about ADA impact on exposure should not be drawn based on simply plotting concentration by ADA status, but rather would require a multivariate model‐based approach.
For efficacy, these findings also suggest that the impact of ADA on drug efficacy cannot be accurately estimated by plotting OS or progression‐free survival curves according to ADA status. Instead, an adjustment method using the appropriate control population is more appropriate.
In summary, this paper provided cross‐study comparison of immunogenicity analysis that is important to evaluate given the high variability of ADA incidence rates (ranges from 13 to 54%). This paper also included the novel ADA framework for oncology that can be applied to other therapeutic proteins, atezolizumab ADA titer, neutralizing ADA data, and the novel relationships of prognostic factor and ADA development that were not published previously. The authors believe having extensive immunogenicity data in oncology with more than 10 registrational trials is unusual and, thus, provides an important learning opportunity for the scientific community. The novel relationships of baseline CRPs and tumor burden and ADA development could lead to additional research to understand the biological effects of these complex relationships that may not be specific to atezolizumab.
Specifically for atezolizumab, a time‐varying PK model was best suited to evaluate the impact of ADA on atezolizumab PK, 32 which showed an impact of 9% on clearance based on monotherapy trials. 7 The flat exposure‐response relationship, 1 , 35 together with clinical exposure that results in target saturation in greater than 95% of population regardless of ADA or NAb status, supports the successful strategy of dosing over ADA for atezolizumab. An ADA titer analysis did not find a clinically meaningful relationship between ADA titer and atezolizumab PK or OS outcomes (not adjusted for imbalances in baseline prognostic factors). PK, clinical efficacy, and ADA development all have their unique covariates as well as shared covariates. It is this complex relationship with prognostic factors that makes immunogenicity assessment in oncology particularly challenging. Given the confounding relationships, the traditional univariate analysis of PK or efficacy by ADA status alone is inadequate. Baseline albumin, tumor burden, and CRP levels showed the greatest association with the development of an ADA response after treatment, but no one factor or a set of prognostic factors could adequately predict ADA status. Emerging advanced quantitative tools, such as machine learning approaches, could be explored to further understand the association of factors determining ADA development. For atezolizumab, based on the lack of pharmacologically relevant impact of ADA on exposure, ADA would not be expected to have a clinically meaningful impact on efficacy.
CONFLICT OF INTEREST
B.W., N.S., P.A., J.S., S.V., R.B., M.B., C.B., P.C., J.J., S.G., A.J., and V.Q. are employees of Genentech Inc/Roche and Roche stockholders. J.R. is a former employee of Roche and Roche stockholder.
AUTHOR CONTRIBUTIONS
B.W., V.Q., R.B., and P.C. wrote the manuscript. B.W., J.R., and S.G. designed the research. S.V., P.A., and J.S. performed the research. B.W., N.S., M.B., C.B., J.J., S.G., A.A., R.B., and P.C. analyzed the data. B.W. and J.R. contributed new reagents/analytical tools.
Supporting information
Supplementary Material
ACKNOWLEDGEMENT
The authors would like to thank Ash Jindal (Genentech Inc.,) for generating the pooled prognostic factors by ADA figures. Editorial assistance for this manuscript was provided by Ashley J. Pratt, PhD, of Health Interactions, and funded by F. Hoffmann‐La Roche, Ltd.
Wu B, Sternheim N, Agarwal P, et al. Evaluation of atezolizumab immunogenicity: Clinical pharmacology (part 1). Clin Transl Sci. 2022;15:130–140. 10.1111/cts.13127
Funding information
This study was funded by F. Hoffmann‐La Roche, Ltd and Genentech, Inc.
REFERENCES
- 1. Stroh M, Winter H, Marchand M, et al. Clinical pharmacokinetics and pharmacodynamics of atezolizumab in metastatic urothelial carcinoma. Clin Pharmacol Ther. 2017;102:305‐312. [DOI] [PubMed] [Google Scholar]
- 2. Genentech, Inc . TECENTRIQ (atezolizumab) [prescribing information]. Genentech, Inc.; 2021. [Google Scholar]
- 3. Davda J, Declerck P, Hu‐Lieskovan S, et al. Immunogenicity of immunomodulatory, antibody‐based, oncology therapeutics. J Immunother Cancer. 2019;7:105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Deng R, Bumbaca D, Pastuskovas CV, et al. Preclinical pharmacokinetics, pharmacodynamics, tissue distribution, and tumor penetration of anti‐PD‐L1 monoclonal antibody, an immune checkpoint inhibitor. MAbs. 2016;8:593‐603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dai HI, Vugmeyster Y, Mangal N. Characterizing exposure‐response relationship for therapeutic monoclonal antibodies in immuno‐oncology and beyond: challenges, perspectives, and prospects. Clin Pharmacol Ther. 2020;108:1156‐1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kawakatsu S, Bruno R, Kagedal M, et al. Confounding factors in exposure‐response analyses and mitigation strategies for monoclonal antibodies in oncology. Br J Clin Pharmacol. 2021;87(6):2493‐2501. [DOI] [PubMed] [Google Scholar]
- 7. Marchand M, Zhang R, Chan P, et al. Time‐dependent population PK models of single agent atezolizumab in patients with cancer. Cancer Chemother Pharmacol. 2021;88:211‐221. [DOI] [PubMed] [Google Scholar]
- 8. Li H, Yu J, Liu C, et al. Time dependent pharmacokinetics of pembrolizumab in patients with solid tumor and its correlation with best overall response. J Pharmacokinet Pharmacodyn. 2017;44:403‐414. [DOI] [PubMed] [Google Scholar]
- 9. Liu C, Yu J, Li H, et al. Association of time‐varying clearance of nivolumab with disease dynamics and its implications on exposure response analysis. Clin Pharmacol Ther. 2017;101:657‐666. [DOI] [PubMed] [Google Scholar]
- 10. Baverel PG, Dubois VFS, Jin CY, et al. Population pharmacokinetics of durvalumab in cancer patients and association with longitudinal biomarkers of disease status. Clin Pharmacol Ther. 2018;103:631‐642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wilkins JJ, Brockhaus B, Dai H, et al. Time‐varying clearance and impact of disease state on the pharmacokinetics of avelumab in Merkel cell carcinoma and urothelial carcinoma. CPT Pharmacometrics Syst Pharmacol. 2019;8:415‐427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Guinn D, Madabushi R, Wang YM, Zineh I, Maxfield K. Elucidating the impact of immunogenicity assessment postapproval: a targeted analysis of immunogenicity postmarketing requirements and commitments. Clin Pharamacol Ther. 2021;109(3):697‐704. [DOI] [PubMed] [Google Scholar]
- 13. Fehrenbacher L, Spira A, Ballinger M, et al. Atezolizumab versus docetaxel for patients with previously treated non‐small‐cell lung cancer (POPLAR): a multicentre, open‐label, phase 2 randomised controlled trial. Lancet. 2016;387:1837‐1846. [DOI] [PubMed] [Google Scholar]
- 14. Rittmeyer A, Barlesi F, Waterkamp D, et al. Atezolizumab versus docetaxel in patients with previously treated non‐small‐cell lung cancer (OAK): a phase 3, open‐label, multicentre randomised controlled trial. Lancet. 2017;389:255‐265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. West H, McCleod M, Hussein M, et al. Atezolizumab in combination with carboplatin plus nab‐paclitaxel chemotherapy compared with chemotherapy alone as first‐line treatment for metastatic non‐squamous non‐small‐cell lung cancer (IMpower130): a multicentre, randomised, open‐label, phase 3 trial. Lancet Oncol. 2019;20:924‐937. [DOI] [PubMed] [Google Scholar]
- 16. Jotte R, Cappuzzo F, Vynnychenko I, et al. Atezolizumab in combination with carboplatin and nab‐paclitaxel in advanced squamous NSCLC (IMpower131): results from a randomized phase III trial. J Thorac Oncol. 2020;15:1351‐1360. [DOI] [PubMed] [Google Scholar]
- 17. Nishio M, Barlesi F, West H, et al. Atezolizumab plus chemotherapy for first‐line treatment of non‐squamous non‐small cell lung cancer: results from the randomized phase III IMpower132 trial. J Thorac Oncol. 2021;16:653‐664. [DOI] [PubMed] [Google Scholar]
- 18. Reck M, Mok TSK, Nishio M, et al. Atezolizumab plus bevacizumab and chemotherapy in non‐small‐cell lung cancer (IMpower150): key subgroup analyses of patients with EGFR mutations or baseline liver metastases in a randomised, open‐label phase 3 trial. Lancet Respir Med. 2019;7:387‐401. [DOI] [PubMed] [Google Scholar]
- 19. Horn L, Mansfield AS, Szczęsna A, et al. First‐line atezolizumab plus chemotherapy in extensive‐stage small‐cell lung cancer. N Engl J Med. 2018;379:2220‐2229. [DOI] [PubMed] [Google Scholar]
- 20. Rosenberg JE, Hoffman‐Censits J, Powles T, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum‐based chemotherapy: a single‐arm, multicentre, phase 2 trial. Lancet. 2016;387:1909‐1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Balar AV, Galsky MD, Rosenberg JE, et al. Atezolizumab as first‐line treatment in cisplatin‐ineligible patients with locally advanced and metastatic urothelial carcinoma: a single‐arm, multicentre, phase 2 trial. Lancet. 2017;389:67‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Powles T, Durán I, van der Heijden MS, et al. Atezolizumab versus chemotherapy in patients with platinum‐treated locally advanced or metastatic urothelial carcinoma (IMvigor211): a multicentre, open‐label, phase 3 randomised controlled trial. Lancet. 2018;391:748‐757. [DOI] [PubMed] [Google Scholar]
- 23. Rini BI, Powles T, Atkins MB, et al. Atezolizumab plus bevacizumab versus sunitinib in patients with previously untreated metastatic renal cell carcinoma (IMmotion151): a multicentre, open‐label, phase 3, randomised controlled trial. Lancet. 2019;393:2404‐2415. [DOI] [PubMed] [Google Scholar]
- 24. Schmid P, Adams S, Rugo HS, et al. Atezolizumab and nab‐paclitaxel in advanced triple‐negative breast cancer. N Engl J Med. 2018;379:2108‐2121. [DOI] [PubMed] [Google Scholar]
- 25. Finn RS, Qin S, Ikeda M, et al. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med. 2020;382:1894‐1905. [DOI] [PubMed] [Google Scholar]
- 26. Mire‐Sluis AR, Barrett YC, Devanarayan V, et al. Recommendations for the design and optimization of immunoassays used in the detection of host antibodies against biotechnology products. J Immunol Methods. 2004;289:1‐16. [DOI] [PubMed] [Google Scholar]
- 27. Gupta S, Devanarayan V, Finco D, et al. Recommendations for the validation of cell‐based assays used for the detection of neutralizing antibody immune responses elicited against biological therapeutics. J Pharm Biomed Anal. 2011;55:878‐888. [DOI] [PubMed] [Google Scholar]
- 28. Gupta S, Indelicato SR, Jethwa V, et al. Recommendations for the design, optimization, and qualification of cell‐based assays used for the detection of neutralizing antibody responses elicited to biological therapeutics. J Immunol Methods. 2007;321:1‐18. [DOI] [PubMed] [Google Scholar]
- 29.US Food and Drug Administration (FDA) . FDA Guidance for Industry: Immunogenicity Assessment for Therapeutic Protein Products. 2014. https://www.fda.gov/regulatory‐information/search‐fda‐guidance‐documents/immunogenicity‐assessment‐therapeutic‐protein‐products. Accessed March 24, 2021.
- 30.US Food and Drug Administration (FDA) . Immunogenicity Testing of Therapeutic Protein Products —Developing and Validating Assays for Anti‐Drug Antibody Detection. 2019. https://www.fda.gov/regulatory‐information/search‐fda‐guidance‐documents/immunogenicity‐testing‐therapeutic‐protein‐products‐developing‐and‐validating‐assays‐anti‐drug. Accessed March 24, 2021.
- 31. Shankar G, Arkin S, Cocea L, et al. Assessment and reporting of the clinical immunogenicity of therapeutic proteins and peptides‐harmonized terminology and tactical recommendations. AAPS J. 2014;16:658‐673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang YM, Wang J, Hon YY, Zhou L, Fang L, Ahn HY. Evaluating and reporting the immunogenicity impacts for biological products–a clinical pharmacology perspective. AAPS J. 2016;18:395‐403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Krishna M, Nadler SG. Immunogenicity to biotherapeutics ‐ the role of anti‐drug immune complexes. Front Immunol. 2016;7:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. van Brummelen EM, Ros W, Wolbink G, Beijnen JH, Schellens JH. Antidrug antibody formation in oncology: clinical relevance and challenges. Oncologist. 2016;21:1260‐1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Morrissey KM, Marchand M, Patel H, et al. Alternative dosing regimens for atezolizumab: an example of model‐informed drug development in the postmarketing setting. Cancer Chemother Pharmacol. 2019;84:1257‐1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hegen H, Millonig A, Bertolotto A, et al. Early detection of neutralizing antibodies to interferon‐beta in multiple sclerosis patients: binding antibodies predict neutralizing antibody development. Mult Scler. 2014;20:577‐587. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material