

Abstract
Antibody therapeutics can be associated with unwanted immune responses resulting in the development of anti‐drug antibodies (ADA). Optimal methods to evaluate the potential effects of ADA on clinical outcomes in oncology are not well established. In this study, we assessed efficacy and safety, based on ADA status, in patients from over 10 clinical trials that evaluated the immune checkpoint inhibitor atezolizumab as a single agent or as combination therapy for several types of advanced cancers. ADA can only be observed post randomization, and imbalances in baseline prognostic factors can confound the interpretation of ADA impact. We applied methodology to account for the confounding effects of baseline clinical characteristics and survivorship bias on efficacy. Adjusted meta‐analyses revealed that despite numerical differences in overall survival and progression‐free survival between ADA‐positive and ADA‐negative patients from some studies, ADA‐positive patients from studies with an overall treatment effect derived benefit from atezolizumab, compared with their adjusted controls. Based on large, pooled populations from atezolizumab monotherapy or combination studies, unadjusted descriptive analyses did not identify a clear relationship between ADA status and frequency or severity of adverse events. Data also suggested that any ADA impact is not driven by neutralizing activity. Collectively, this exploratory analysis suggests that the potential for ADA development should not impact treatment decisions with atezolizumab.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Treatment‐emergent anti‐drug antibodies (ADA) directed against anticancer therapeutics, including atezolizumab, might affect efficacy and safety.
WHAT QUESTION DID THIS STUDY ADDRESS?
How do ADA impact overall survival, progression‐free survival, and adverse‐event frequencies with atezolizumab?
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
Based on a large data set from over 10 clinical trials, we showed that although efficacy in ADA‐positive versus ADA‐negative patients is numerically reduced in some studies, ADA‐positive patients, including those with neutralizing antibodies, can derive treatment benefit from atezolizumab. Adverse‐event frequencies were not consistently different in the presence or absence of ADA or neutralizing antibodies.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
These results underscore the importance of adjusting for baseline covariates in efficacy assessments. The impacts of ADA on atezolizumab efficacy and safety are not expected to be clinically relevant.
INTRODUCTION
Unwanted immune responses to protein therapeutics can result in the development of anti‐drug antibodies (ADA), 1 , 2 potentially affecting drug pharmacokinetics (PK), pharmacodynamics, efficacy, and safety. 3 , 4 , 5 The binding of ADA to an antibody therapeutic may result in the formation of immune complexes that are cleared from the circulation, potentially reducing drug exposure and impairing efficacy. Patients who develop ADA may have an increased risk of adverse events (AEs), most commonly infusion‐related reactions (IRRs). 6 The subset of ADA that block therapeutic target binding are termed neutralizing antibodies (NAb) 3 ; ADA that are neutralizing in vitro 7 , 8 may or may not be linked to loss of efficacy in vivo. 9 In the case of antibody therapeutics, both neutralizing and/or non‐neutralizing ADA tend to be directed against the complementarity‐determining regions (CDRs) of the molecule. 6 , 10 , 11 , 12 For example, an analysis of ADA to natalizumab in patients with multiple sclerosis revealed that NAb and non‐NAb bind to similar CDRs and CDR‐adjacent regions of this antibody therapeutic, but that NAb dissociated more slowly than non‐NAb. 10
Although ADA to protein therapeutics may cause reduced drug exposure, loss of efficacy, and/or increased AEs in other diseases (e.g., natalizumab in neurology, 9 infliximab in rheumatology, 13 and factor VIIa in hematology 14 ), it has been more difficult to determine the clinical relevance of ADA in oncology. A recent systematic assessment showed that although ADA were detected in greater than 50% of the oncology studies evaluated (mainly monoclonal antibody studies), in most cases, the clinical consequences of immunogenicity were unclear. 6 Moreover, the widely used industry standard approaches to ADA analysis and data interpretation were developed based on patients with autoimmune and neurological diseases, 15 and these may not be appropriate for oncology studies, in which a high patient attrition is often observed over time.
Reported ADA frequencies from single‐agent anti‐programmed death‐ligand 1 (anti–PD‐L1)/anti‐programmed death‐1 (PD‐1) immune checkpoint inhibitor (ICI) studies are generally low, as are NAb frequencies, and no evidence is available to date on clinical impact. Atezolizumab (anti–PD‐L1) has been approved in various types of cancers. 16 , 17 Across multiple studies, expanded analyses of ADA against atezolizumab have been conducted, showing incidence rates of 13%–54% overall and 4%–28% for NAb, 18 with no consistent trends of association among incidence rates and tumor type, line of therapy, treatment schedule, or monotherapy versus combination therapy.
ADA development may be influenced by a patient’s baseline disease characteristics. For example, tumor necrosis factor inhibitor studies have shown that C‐reactive protein (CRP) levels and erythrocyte sedimentation rate differ between patients with rheumatoid arthritis who become ADA positive (ADA+) and those who remain ADA negative (ADA–) post treatment. 19 , 20 Because ADA develop following therapeutic treatment, they are considered a postbaseline variable and therefore cannot be a stratification factor. Thus, even with randomized studies, baseline imbalances between experimental‐arm ADA‐defined subgroups can exist, rendering interpretability of clinical trial data more challenging and necessitating adjustment. 21 Any assessment of potential impacts of ADA on efficacy and/or safety in oncology must account for observable and measurable patient health and disease status at baseline, including comorbidities, tumor burden, and extent of metastases. Moreover, high mortality rates often seen in oncology, specifically with advanced or metastatic cancers, can introduce survivorship bias 22 into the study of ADA development and affect clinical outcome, underscoring the need for careful analyses to allow for meaningful comparisons. Here, to understand whether ADA to atezolizumab have consequences on efficacy and safety, we evaluated data from multiple clinical trials.
METHODS
Contributing studies
Data included in these exploratory analyses were from clinical trials in advanced or metastatic cancers with previously described study designs. 23 , 24 , 25 , 26 , 27 , 28 , 29 , 30 , 31 , 32 , 33 , 34 , 35 , 36 , 37 , 38 All trials were conducted in accordance with Good Clinical Practice guidelines and the Declaration of Helsinki. All protocols were approved by institutional ethics committees or independent review boards, and all patients provided written informed consent. Table S1 lists the contributing studies for efficacy and safety analyses. Only randomized studies (1 phase II and 10 phase III studies) were included in efficacy analyses.
ADA assessments
Baseline and up to 9 on‐treatment predose serum samples per patient were collected for ADA assessments. In agreement with industry consensus guidance and health authority guidelines on ADA analysis and data interpretation, ADA assessment was based on a tiered testing strategy, 39 which was largely the same across all trials. ADA subgroups were defined as described. 18
Efficacy endpoints and statistical analyses
Overall survival (OS) and progression‐free survival (PFS; per Response Evaluation Criteria in Solid Tumors [RECIST] version 1.1) were primary, co‐primary, or secondary endpoints in the studies included in efficacy analyses. In the experimental (atezolizumab) arms, ADA‐evaluable patients included all treated patients (i.e., those who received either ≥1 dose or any amount of study treatment, depending on the study design) who also had greater than or equal to 1 postbaseline ADA assessment. In the control arms, all treated patients were included. For IMpower130, only EGFR/KRAS wild‐type patients were included in OS and PFS analyses, and for IMpassion130, patients included in PFS analyses were derived from the PD‐L1 IC1/2/3 population (PD‐L1–expressing immune cells covering ≥1% of the tumor area [VENTANA SP142 immunohistochemistry assay]; Ventana Medical Systems).
OS and PFS analyses were based on the principal stratum estimand principle. 21 Estimands were adapted to allow for missing stratum membership, because control patients were not treated with atezolizumab and therefore ADA or NAb status was not available. 40 To correct for survivorship bias, a primary landmark time point that considered median time to ADA onset and first scheduled postbaseline ADA sampling time point (plus 1 additional week to account for delayed study visits) was chosen (4, 5, 6, or 7 weeks, depending on the study). Landmark time points were chosen closer to randomization to maximize benefit of randomization, with the small proportion of patients who experienced a PFS or OS event or who were censored prior to the landmark time point being excluded from landmark‐adjusted analyses. Atezolizumab‐arm patients were categorized into three mutually exclusive groups: landmark ADA+ (ADA+ status observed on or before the landmark time point), landmark ADA– (only ADA– status observed on or before the landmark time point), and landmark ADA missing (no ADA status available on or before the landmark time point, but ADA status observed later during study conduct). Models using time‐dependent covariates often contain mortality information during study conduct complicating interpretation and were therefore not deemed appropriate here. 40
Weighted regression imputation (WRI) 40 was used to impute ADA status at the landmark for patients without ADA status available on or before the landmark, but for whom ADA status was later observed. For these cases, missing landmark ADA status was imputed using next‐available post‐landmark ADA status and baseline covariates. To assess the robustness of WRI results, a second method was used wherein patients with a missing landmark ADA status were modeled as a separate category, in addition to ADA status, in a method called doubly weighted control (DWC). 40 For efficacy analyses based on NAb status, an NAb‐adapted version of DWC was used to handle missing ADA/NAb status at the landmark.
OS and PFS efficacy analyses were adjusted for potential imbalances in baseline demographic and prognostic factors between atezolizumab‐treated and control‐arm patients. First, among atezolizumab‐treated patients, a logistic regression was estimated to model the probability of ADA+ status at the landmark, with baseline and disease characteristics as independent variables. Covariates for each study were prespecified following subject‐matter expert input based on their known prognostic value and/or predictivity for atezolizumab efficacy. To increase between‐study comparability, a common set of covariates was used across most studies; these included the following baseline characteristics: Eastern Cooperative Oncology Group performance status (ECOG PS) or Karnofsky performance status, sex (except in IMpassion130), race, number of metastatic sites, albumin, lactate dehydrogenase, sum of longest target lesion diameters, CRP, neutrophil‐to‐lymphocyte ratio, age, body weight, liver metastases, tobacco use history, and PD‐L1 expression. The common covariates were also supplemented with indication‐specific covariates (see Table S2 for details on study‐specific covariates). Next, for each control‐arm patient, the probability of ADA+ status at the landmark, had they been treated with atezolizumab, was computed by applying their baseline disease characteristics in the logistic regression. These computed probabilities served as weights in the inverse probability weighting estimation of OS and PFS. Confidence intervals (CIs) for hazard ratios (HRs) were computed using bootstrap methodology.
OS and PFS from individual trials are presented, as well as random‐effects meta‐analyses using inverse variance weighting. HRs are reported for ADA+ and ADA– subgroups versus adjusted controls (or for NAb analyses: ADA+/NAb+, ADA+/NAb–, and ADA– subgroups). Meta‐analyses evaluating OS and PFS for ADA+ experimental‐arm patients relative to control‐arm patients included studies (as referenced previously) limited to those with an overall treatment effect seen in their primary analyses. For OS meta‐analyses, only studies demonstrating OS benefit were included, and for PFS meta‐analyses, only studies demonstrating PFS benefit were included. To compare ADA and NAb subgroups, the ratios of HRs (RHRs; for ADA subgroups, HR of ADA+ vs. adjusted control and HR of ADA– vs. adjusted control; for NAb subgroups, HR of ADA+/NAb+ vs. adjusted control, and HR of ADA+/NAb– vs. adjusted control) are reported, for all studies for which ADA or NAb data were available. For RHRs close to 1, efficacy is similar in ADA+ and ADA– subgroups, compared to their respective adjusted control arms.
Safety analyses
The safety‐evaluable population comprised ADA‐evaluable patients without considering a landmark. Safety analyses were performed without adjustment for potential imbalances in baseline factors between atezolizumab‐treated and control‐arm patients. AE frequencies were reported based on the National Cancer Institute Common Terminology Criteria for Adverse Events, version 4.0, in pooled study populations as indicated below. A set of comprehensive definitions comprising Standardized MedDRA Queries, high level terms, and sponsor‐defined AE group terms based on the known mechanism of action for atezolizumab, and concerns reported with other ICIs, was used to identify and summarize AEs of special interest (AESIs) by medical concept. AE incidences by ADA and NAb status were computed in two pooled groups: patients treated with (1) atezolizumab monotherapy and (2) combination therapy, to account for potential differences in safety profiles with additional therapies.
RESULTS
Patients
Across 11 trials, 7736 patients were randomized to control (n = 3298) or experimental treatment (n = 4438). Most randomized patients (n = 4617, 60%) had non‐small cell lung cancer (NSCLC; first‐line or second‐line and beyond treatment setting). The remaining 3119 patients (40%) had small‐cell lung cancer, metastatic urothelial carcinoma, renal cell carcinoma, triple‐negative breast cancer, or hepatocellular carcinoma, across multiple treatment settings. There were 7303 randomized patients that were included in the evaluation of efficacy by ADA status (without landmark), including 3182 treated control patients, and 4121 experimental‐arm patients evaluable for treatment‐emergent ADA. Of the 4121 experimental‐arm patients, 2859 were classified as ADA− throughout the study, and 1262 had greater than or equal to 1 ADA+ assessment while on treatment. With inclusion of a landmark, there were 3081 control, 2757 ADA−, and 887 ADA+ patients; ADA status for 461 patients (11%) with no ADA status at the landmark was imputed. Additionally, 2251 patients from five nonrandomized studies contributed to ADA safety analyses (see Table S1).
Imbalances in baseline covariates, most of which were indicative of worse prognosis, were observed between ADA– and ADA+ experimental‐arm patients (Table 1). In the pooled population, characteristics with differences in median levels or frequencies among ADA+ patients (percent increase from ADA– patients >5%) included: CRP levels (+106.6% higher median value for ADA+ vs. ADA– patients), male sex (+17.2%), tumor burden (sum of target lesion diameters; +16.6% median value), ECOG PS of 1 (+9.6%), neutrophil‐to‐lymphocyte ratio (+8.4% median value), White race (+7.6%), and lactate dehydrogenase levels (+5.5% median value). Squamous histology was more common in patients with NSCLC who developed ADA (+8.3% higher). The same set of imbalances in baseline characteristics between ADA+ and ADA– patients was generally not observed between ADA+ patients who were NAb+ or NAb– (Table 1).
TABLE 1.
Baseline covariates (pooled efficacy study populations)
| Characteristic | Control (n = 3439)a | Experimental (atezolizumab) | Median difference, %a | Experimental (atezolizumab) | Median difference, %b | ||
|---|---|---|---|---|---|---|---|
| ADA– (n = 2859) | ADA+ (n = 1262) | ADA+/NAb– (n = 521) | ADA+/NAb+ (n = 618) | ||||
| Male, % | 60.6 | 62.7 | 73.5 | 17.2 | 71.6 | 79.6 | 11.2 |
| Age ≥65 years, % | 44.8 | 46.1 | 46 | −0.2 | 49.3 | 43.5 | −11.8 |
| Median weight (2.5th percentile, 97.5th percentile), kg | 70.9 (45.5, 110.2) | 72.0 (45.4, 110.6) | 73.7 (49.0, 115.8) | 2.4 | 71.0 (47.0, 111.0) | 75.7 (49.9, 124.1) | 6.6 |
| White race, % | 73.9 | 72.6 | 78.1 | 7.6 | 77.2 | 77.3 | 0.1 |
| Median albumin level (2.5th percentile, 97.5th percentile), g/L | 40.0 (27.0, 48.0) | 40.0 (27.6, 48.0) | 38.9 (25.0, 47.0) | −2.8 | 38.9 (25.0, 47.0) | 38.2 (24.0, 48.0) | −1.9 |
| Median LDH (2.5th percentile, 97.5th percentile), U/L | 227 (125, 946) | 220 (124, 854) | 232 (128, 872) | 5.5 | 231 (128, 964) | 232 (124, 858) | 0.4 |
| Median SLD (2.5th percentile, 97.5th percentile), mm | 70.0 (14.0, 217.3) | 68.0 (13.7, 205.2) | 79.3 (16.0, 224.0) | 16.6 | 80.2 (16.0, 218.0) | 79.0 (16.0, 238.0) | −1.5 |
| Median no. of metastatic sites (2.5th percentile, 97.5th percentile) | 2 (0, 5) | 2 (0, 5) | 2 (0, 5) | 0 | 2 (0, 5) | 2 (0, 5) | 0 |
| Median NLR (2.5th percentile, 97.5th percentile) | 3.51 (1.2, 15.6) | 3.46 (1.3, 14.7) | 3.75 (1.3, 17.6) | 8.4 | 3.83 (1.3, 18.5) | 3.61 (1.3, 16.8) | −5.7 |
| Median CRP (2.5th percentile, 97.5th percentile), mg/L | 11.1 (0.5, 161) | 9.8 (0.4, 148) | 20.25 (0.8, 184) | 106.6 | 21.8 (0.6, 170) | 19.5 (0.8, 200) | −10.6 |
| ECOG PS 1, % | 56.6 | 54.2 | 59.4 | 9.6 | 62 | 56.9 | −8.2 |
| Liver metastases, % | 19.6 | 16.9 | 16.4 | −3 | 20.2 | 14.4 | −28.7 |
| Squamous histology, %c | 27.8 | 28.9 | 31.3 | 8.3 | 38.1 | 29.9 | −21.5 |
| Current or previous tobacco use, %d | 85.4 | 82.8 | 84.9 | 2.5 | 85.1 | 85.1 | 0 |
Patients were pooled from the following studies unless otherwise noted (not all patients were evaluable for all measurements): POPLAR, OAK, IMpower130, IMpower133, IMpower131, IMpower150, IMpower133, IMvigor211, IMmotion151, IMpassion130 PD‐L1 IC+, and IMbrave150. ADA– refers to patients who were ADA negative and not tested for NAb. ADA+ refers to patients positive for treatment‐emergent ADA. NAb– refers to ADA+ patients who had evaluable NAb samples and for whom all post‐treatment NAb samples were negative. NAb+ refers to ADA+ patients who had greater than or equal to 1 positive post‐treatment NAb sample.
Abbreviations: ADA, anti‐drug antibody; CRP, C‐reactive protein; ECOG PS, Eastern Cooperative Oncology Group performance status; LDH, lactate dehydrogenase; NAb, neutralizing antibody; NLR, neutrophil‐to‐lymphocyte ratio; NSCLC, non‐small cell lung cancer; PD‐L1, programmed death‐ligand 1; SCLC, small cell lung cancer; SLD, sum of the longest diameters; UC, urothelial cancer.
Refers to difference between ADA– to ADA+.
Refers to difference between ADA+ NAb– to ADA+ NAb+.
In patients with NSCLC only (n = 1757; POPLAR, OAK, IMpower130, IMpower131, IMpower132, and IMpower150).
In patients with NSCLC (POPLAR, OAK, IMpower130, IMpower131, IMpower132, and IMpower150), SCLC (IMpower133), and UC (IMvigor211) only (n = 2397).
Efficacy
For each randomized study, we evaluated OS and PFS in patients who did or did not develop ADA compared with their respective adjusted control‐arm patients (Figure S1). For most studies, HR point estimates for ADA+ patients (vs. adjusted control patients) were similar to those for ADA– patients versus adjusted control (<0.1 difference), with corresponding CIs generally wide and overlapping. OS and PFS meta‐analyses comparing ADA+ versus ADA– patients across all 11 randomized studies, reported as RHRs (atezolizumab group vs. adjusted control), are shown in Figure 1. When comparing ADA+ patients specifically to their adjusted controls (Figure 2 and Figure S1)—and limiting meta‐analyses to studies that showed a treatment effect in the intention‐to‐treat (ITT) or overall analysis population (Figure 2)—we observed that OS and PFS HR point estimates favored ADA+ atezolizumab‐arm versus adjusted control‐arm patients (i.e., <1), with the upper bound of 95% CIs less than 1. Efficacy results were generally consistent when a DWC method for handling missing ADA status at the landmark was applied (Figures S1–S3).
FIGURE 1.
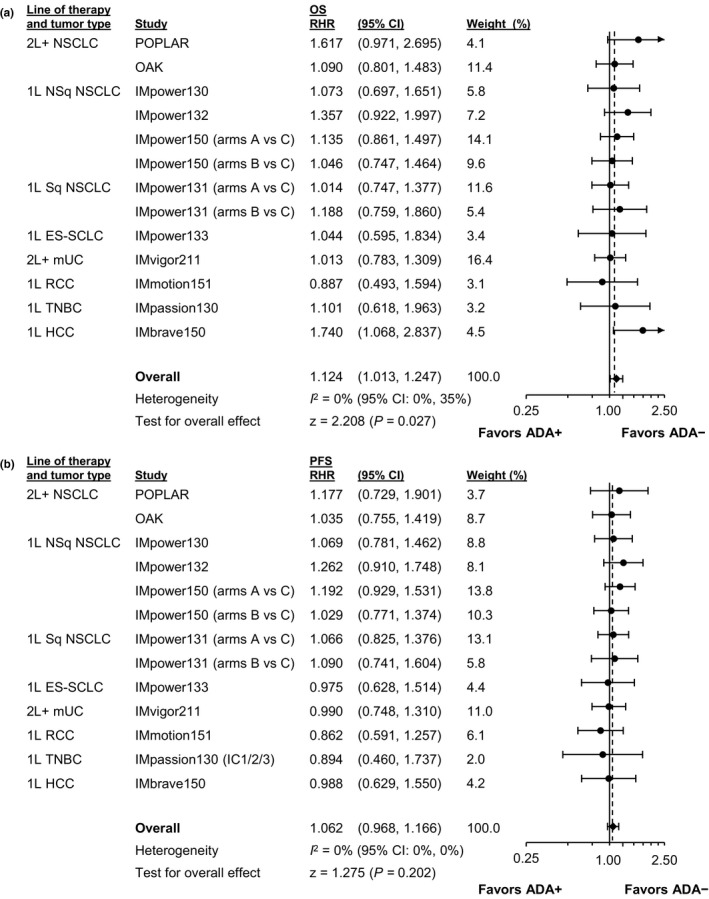
Meta‐analysis evaluating RHRs in ADA+ patients (vs. their adjusted controls) with ADA experimental‐arm patients (vs. their adjusted controls). Forest plots for (a) OS and (b) PFS are shown for all studies. 1L, first line; 2L+, second line and beyond; ADA, anti‐drug antibody; CI, confidence interval; ES‐SCLC, extensive‐stage small cell lung cancer; HCC, hepatocellular carcinoma; mUC, metastatic urothelial carcinoma; NSCLC, non‐small cell lung cancer; NSq, nonsquamous; OS, overall survival; PFS, progression‐free survival; RCC, renal cell carcinoma; RHR, ratio of hazard ratios; Sq, squamous; TNBC, triple‐negative breast cancer
FIGURE 2.
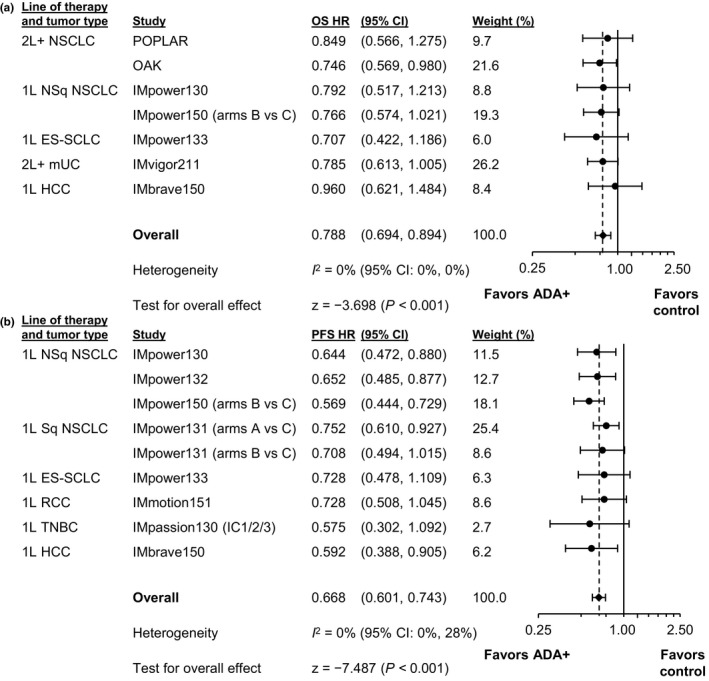
Meta‐analysis comparing ADA+ experimental‐arm patients to adjusted control‐arm patients. Forest plots for (a) OS and (b) PFS include only studies with an overall treatment effect. 1L, first line; 2L+, second line and beyond; ADA, anti‐drug antibody; CI, confidence interval; ES‐SCLC, extensive‐stage small cell lung cancer; HCC, hepatocellular carcinoma; HR, hazard ratio; mUC, metastatic urothelial carcinoma; NSCLC, non‐small cell lung cancer; NSq, nonsquamous; OS, overall survival; PFS, progression‐free survival; RCC, renal cell carcinoma; SCLC, small cell lung cancer; Sq, squamous; TNBC, triple‐negative breast cancer
To evaluate whether the type of ADA (namely, neutralizing or not) affected efficacy, we compared ADA+/NAb+ and ADA+/NAb– patients in studies with sufficient patient numbers in NAb subgroups (Figure S4). HR point estimates were similar for most studies (<0.15 difference) between the ADA+/NAb+ and ADA+/NAb– subgroups. Individual studies and meta‐analytic estimates (Figure 3) showed that RHRs were generally dispersed around 1 for ADA+/NAb+ versus ADA+/NAb– patients, with wide overlapping CIs crossing 1, both for OS and PFS. In meta‐analyses using studies with an overall treatment effect (Figure 4), the meta‐analytic OS and PFS HR point estimates favored ADA+/NAb+ atezolizumab‐arm patients relative to adjusted control‐arm patients.
FIGURE 3.
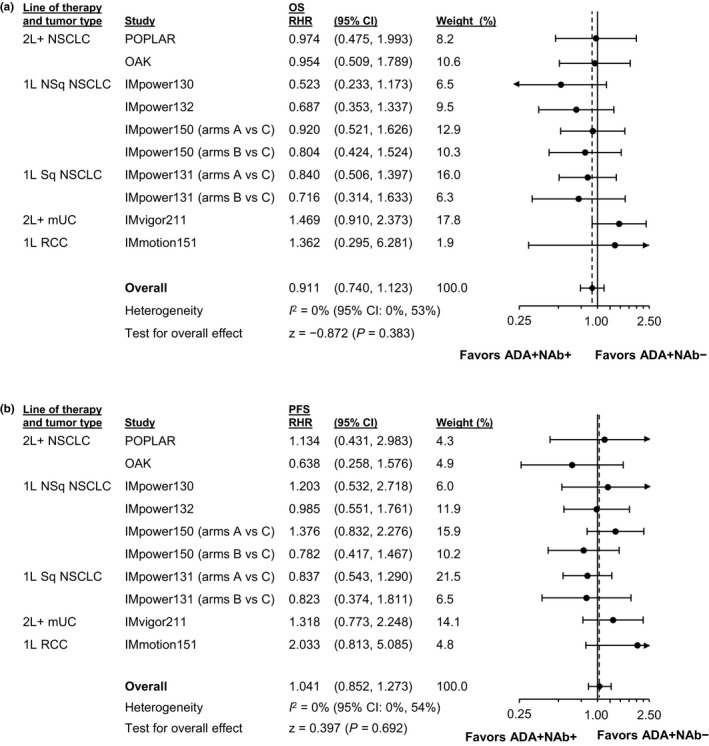
Meta‐analysis evaluating RHRs in ADA+/NAb+ patients (vs. their adjusted controls) with ADA+/NAb– experimental‐arm patients (vs. their adjusted controls). Forest plots for (a) OS and (b) PFS are shown for all studies. IMpower133, IMpassion130, and IMbrave150 (OS and PFS plots) are not included based on the small number of patients in some of the NAb subgroups from these studies. 1L, first line; 2L+, second line and beyond; ADA, anti‐drug antibody; CI, confidence interval; HCC, hepatocellular carcinoma; mUC, metastatic urothelial carcinoma; NAb, neutralizing (anti‐drug) antibody; NSCLC, non‐small cell lung cancer; NSq, nonsquamous; OS, overall survival; PFS, progression‐free survival; RCC, renal cell carcinoma; RHR, ratio of hazard ratios; SCLC, small cell lung cancer; Sq, squamous
FIGURE 4.
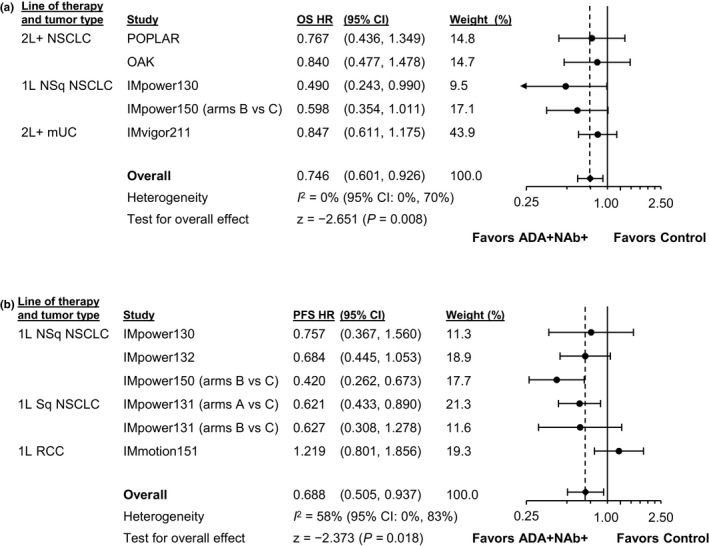
Meta‐analysis comparing ADA+/NAb+ experimental‐arm patients to adjusted control‐arm patients. Forest plots for (a) OS and (b) PFS are shown only for studies with an overall treatment effect. 1L, first line; 2L+, second line and beyond; ADA, anti‐drug antibody; CI, confidence interval; HCC, hepatocellular carcinoma; HR, hazard ratio; mUC, metastatic urothelial carcinoma; NAb, neutralizing (anti‐drug) antibody; NSCLC, non‐small cell lung cancer; NSq, nonsquamous; OS, overall survival; PFS, progression‐free survival; RCC, renal cell carcinoma; SCLC, small cell lung cancer; Sq, squamous
Safety
Safety summaries are shown in Tables 2 and 3. Results are unadjusted for baseline covariates. Regardless of attribution, all‐grade AE frequencies were 96.4%–98.8% across ADA– and ADA+ subgroups included in either monotherapy or combination‐therapy study pools. Most overall categories of AEs occurred at similar frequencies for ADA– and ADA+ patients (see Table 3). Somewhat higher rates were seen for ADA+ versus ADA– patients for all‐cause grade 3/4 AEs in monotherapy studies (49.3% vs. 44.4%), serious AEs (monotherapy: 42.4 vs. 37.7%; combination: 43.5% vs. 36.6%) and all‐cause AEs leading to atezolizumab withdrawal in combination studies (13.2% vs. 9.5%; see Table 2). Differences in treatment‐related serious AEs were more pronounced in combination vs monotherapy studies. Trends in AE rates were more variable based on NAb status, with some AE frequencies higher in NAb+ patients, and others higher in NAb– patients (see Table 3).
TABLE 2.
Safety summary by ADA status (pooled safety study populations)
| Pooled atezolizumab monotherapya (n = 2706) | Pooled atezolizumab combination therapyb (n = 3666) | |||
|---|---|---|---|---|
| ADA– (n = 1704) | ADA+ (n = 1002) | ADA– (n = 2638) | ADA+ (n = 1028) | |
| Total number of AEs, n | 18,621 | 11,057 | 38,623 | 14,018 |
| Patients with ≥1 indicated AE, n (%) | ||||
| All AEs | 1643 (96.4) | 973 (97.1) | 2602 (98.6) | 1016 (98.8) |
| Treatment related | 1216 (71.4) | 705 (70.4) | 2447 (92.8) | 962 (93.6) |
| Atezolizumab related | 1216 (71.4) | 705 (70.4) | 1998 (75.7) | 790 (76.8) |
| Grade 3/4 AE | 757 (44.4) | 494 (49.3) | 1590 (60.3) | 635 (61.8) |
| Treatment related | 271 (15.9) | 156 (15.6) | 1306 (49.5) | 526 (51.2) |
| Atezolizumab related | 271 (15.9) | 156 (15.6) | 675 (25.6) | 286 (27.8) |
| Grade 5 AE | 52 (3.1) | 32 (3.2) | 90 (3.4) | 49 (4.8) |
| Treatment related | 2 (0.1) | 3 (0.3) | 26 (1.0) | 17 (1.7) |
| Atezolizumab related | 2 (0.1) | 3 (0.3) | 19 (0.7) | 12 (1.2) |
| Serious AE | 642 (37.7) | 425 (42.4) | 965 (36.6) | 447 (43.5) |
| Treatment related | 182 (10.7) | 111 (11.1) | 496 (18.8) | 254 (24.7) |
| Atezolizumab related | 182 (10.7) | 111 (11.1) | 340 (12.9) | 166 (16.1) |
| AE leading to any study treatment withdrawal | 114 (6.7) | 61 (6.1) | 507 (19.2) | 236 (23.0) |
| AE leading to atezolizumab withdrawal | 114 (6.7) | 61 (6.1) | 251 (9.5) | 136 (13.2) |
| AE leading to any dose modification or study treatment interruption | 470 (27.6) | 308 (30.7) | 1564 (59.3) | 641 (62.4) |
| AE leading to atezolizumab interruption | 470 (27.6) | 307 (30.6) | 1191 (45.1) | 491 (47.8) |
| Total number of AESIs, n | 1157 | 737 | 3005 | 1232 |
| Patients with ≥1 indicated AESI, n (%) | ||||
| All AESIs | 590 (34.6) | 365 (36.4) | 1410 (53.4) | 562 (54.7) |
| Treatment related | 431 (25.3) | 264 (26.3) | 1160 (44.0) | 463 (45.0) |
| Atezolizumab related | 431 (25.3) | 264 (26.3) | 1063 (40.3) | 428 (41.6) |
| Grade 3/4 AESI | 124 (7.3) | 80 (8.0) | 282 (10.7) | 149 (14.5) |
| Treatment related | 91 (5.3) | 53 (5.3) | 223 (8.5) | 107 (10.4) |
| Atezolizumab related | 91 (5.3) | 53 (5.3) | 201 (7.6) | 102 (9.9) |
| Grade 5 AESI | 1 (<0.1) | 1 (<0.1) | 12 (0.5) | 4 (0.4) |
| Treatment related | 0 | 1 (<0.1) | 10 (0.4) | 4 (0.4) |
| Atezolizumab related | 0 | 1 (<0.1) | 10 (0.4) | 4 (0.4) |
| Serious AESI | 83 (4.9) | 43 (4.3) | 180 (6.8) | 90 (8.8) |
| Treatment related | 73 (4.3) | 33 (3.3) | 146 (5.5) | 72 (7.0) |
| Atezolizumab related | 73 (4.3) | 33 (3.3) | 138 (5.2) | 70 (6.8) |
| AESI leading to any study treatment withdrawal | 34 (2.0) | 16 (1.6) | 127 (4.8) | 52 (5.1) |
| AESI leading to atezolizumab withdrawal | 34 (2.0) | 16 (1.6) | 108 (4.1) | 48 (4.7) |
| AESI leading to any dose modification or study treatment interruption | 109 (6.4) | 78 (7.8) | 335 (12.7) | 146 (14.2) |
| AESI leading to atezolizumab interruption | 109 (6.4) | 78 (7.8) | 292 (11.1) | 127 (12.4) |
| AESI requiring the use of systemic corticosteroids | 123 (7.2) | 84 (8.4) | 365 (13.8) | 161 (15.7) |
Abbreviations: ADA, anti‐drug antibodies; AE, adverse event; AESI, adverse event of special interest.
The pooled atezolizumab monotherapy population comprised patients enrolled in the atezolizumab monotherapy cohorts or experimental arms of the following studies: POPLAR, OAK, IMvigor211, FIR, BIRCH, IMvigor210, and PCD4989g.
The pooled atezolizumab combination therapy population comprised patients enrolled in the atezolizumab‐containing experimental cohorts or arms of the following studies: IMpower130, IMpower132, IMpower131, IMpower150, IMpower133, IMmotion151, IMpassion130, IMbrave150, and GO30140.
TABLE 3.
Safety summary by NAb status (pooled safety study populations)
| Pooled atezolizumab monotherapya (n = 1103) | Pooled atezolizumab combination therapyb (n = 3345) | |||||
|---|---|---|---|---|---|---|
| ADA+/NAb– (n = 173) | ADA+/NAb+ (n = 190) | ADA– or NAb– (n = 913) | ADA+/NAb– (n = 401) | ADA+/NAb+ (n = 463) | ADA– or NAb– (n = 2882) | |
| Total number of AEs, n | 1509 | 2004 | 8659 | 5605 | 6465 | 42,918 |
| Patients with ≥1 indicated AE, n (%) | ||||||
| All AEs | 163 (94.2) | 186 (97.9) | 876 (95.9) | 394 (98.3) | 461 (99.6) | 2848 (98.8) |
| Treatment related | 116 (67.1) | 146 (76.8) | 621 (68.0) | 383 (95.5) | 436 (94.2) | 2721 (94.4) |
| Atezolizumab related | 116 (67.1) | 146 (76.8) | 621 (68.0) | 310 (77.3) | 366 (79.0) | 2209 (76.6) |
| Grade 3/4 AE | 73 (42.2) | 87 (45.8) | 386 (42.3) | 269 (67.1) | 276 (59.6) | 1806 (62.7) |
| Treatment related | 24 (13.9) | 34 (17.9) | 157 (17.2) | 237 (59.1) | 219 (47.3) | 1507 (52.3) |
| Atezolizumab related | 24 (13.9) | 34 (17.9) | 157 (17.2) | 121 (30.2) | 134 (28.9) | 777 (27.0) |
| Grade 5 AE | 13 (7.5) | 8 (4.2) | 41 (4.5) | 18 (4.5) | 27 (5.8) | 104 (3.6) |
| Treatment related | 1 (0.6) | 1 (0.5) | 3 (0.3) | 5 (1.2) | 9 (1.9) | 30 (1.0) |
| Atezolizumab related | 1 (0.6) | 1 (0.5) | 3 (0.3) | 3 (0.7) | 6 (1.3) | 21 (0.7) |
| Serious AE | 63 (36.4) | 83 (43.7) | 326 (35.7) | 173 (43.1) | 213 (46.0) | 1099 (38.1) |
| Treatment related | 16 (9.2) | 33 (17.4) | 103 (11.3) | 98 (24.4) | 123 (26.6) | 574 (19.9) |
| Atezolizumab related | 16 (9.2) | 33 (17.4) | 103 (11.3) | 63 (15.7) | 83 (17.9) | 389 (13.5) |
| AE leading to any study treatment withdrawal | 12 (6.9) | 16 (8.4) | 69 (7.6) | 91 (22.7) | 115 (24.8) | 581 (20.2) |
| AE leading to atezolizumab withdrawal | 12 (6.9) | 16 (8.4) | 69 (7.6) | 48 (12.0) | 72 (15.6) | 288 (10.0) |
| AE leading to any dose modification or study treatment interruption | 39 (22.5) | 62 (32.6) | 245 (26.8) | 266 (66.3) | 292 (63.1) | 1786 (62.0) |
| AE leading to atezolizumab interruption | 39 (22.5) | 61 (32.1) | 245 (26.8) | 201 (50.1) | 234 (50.5) | 1367 (47.4) |
| Total number of AESIs, n | 98 | 170 | 549 | 410 | 644 | 3232 |
| Patients with ≥1 indicated AESI, n (%) | ||||||
| All AESIs | 56 (32.4) | 83 (43.7) | 301 (33.0) | 204 (50.9) | 275 (59.4) | 1529 (53.1) |
| Treatment related | 39 (22.5) | 67 (35.3) | 215 (23.5) | 180 (44.9) | 223 (48.2) | 1283 (44.5) |
| Atezolizumab related | 39 (22.5) | 67 (35.3) | 215 (23.5) | 169 (42.1) | 205 (44.3) | 1177 (40.8) |
| Grade 3/4 AESI | 8 (4.6) | 18 (9.5) | 70 (7.7) | 53 (13.2) | 74 (16.0) | 315 (10.9) |
| Treatment related | 6 (3.5) | 11 (5.8) | 55 (6.0) | 43 (10.7) | 52 (11.2) | 254 (8.8) |
| Atezolizumab related | 6 (3.5) | 11 (5.8) | 55 (6.0) | 42 (10.5) | 49 (10.6) | 233 (8.1) |
| Grade 5 AESI | 0 | 1 (0.5) | 0 | 0 | 2 (0.4) | 11 (0.4) |
| Treatment related | 0 | 1 (0.5) | 0 | 0 | 2 (0.4) | 9 (0.3) |
| Atezolizumab related | 0 | 1 (0.5) | 0 | 0 | 2 (0.4) | 9 (0.3) |
| Serious AESI | 3 (1.7) | 12 (6.3) | 47 (5.1) | 34 (8.5) | 45 (9.7) | 198 (6.9) |
| Treatment related | 2 (1.2) | 12 (6.3) | 42 (4.6) | 29 (7.2) | 34 (7.3) | 163 (5.7) |
| Atezolizumab related | 2 (1.2) | 12 (6.3) | 42 (4.6) | 28 (7.0) | 33 (7.1) | 156 (5.4) |
| AESI leading to any study treatment withdrawal discontinuation | 2 (1.2) | 4 (2.1) | 25 (2.7) | 19 (4.7) | 26 (5.6) | 137 (4.8) |
| AESI leading to atezolizumab withdrawal | 2 (1.2) | 4 (2.1) | 25 (2.7) | 17 (4.2) | 25 (5.4) | 118 (4.1) |
| AESI leading to any dose modification or study treatment interruption | 9 (5.2) | 18 (9.5) | 59 (6.5) | 56 (14.0) | 75 (16.2) | 379 (13.2) |
| AESI leading to atezolizumab interruption | 9 (5.2) | 18 (9.5) | 59 (6.5) | 48 (12.0) | 66 (14.3) | 331 (11.5) |
| AESI requiring the use of systemic corticosteroids | 13 (7.5) | 24 (12.6) | 78 (8.5) | 59 (14.7) | 85 (18.4) | 409 (14.2) |
NAb+ refers to ADA+ patients who had ≥1 positive post‐treatment NAb sample. NAb– refers to ADA+ patients who had evaluable NAb samples and were negative for all post‐treatment NAb samples. ADA– refers to patients who were ADA negative and were not tested for NAb.
Abbreviations: ADA, anti‐drug antibody; AE, adverse event; AESI, adverse events of special interest; NAb, neutralizing antibody.
The pooled atezolizumab monotherapy population comprised patients enrolled in the experimental arms of the following studies: POPLAR, OAK, and IMvigor211 (Arm A).
The pooled atezolizumab combination therapy cohort comprised patients enrolled in the atezolizumab‐containing experimental arms of the following studies: IMpower130, IMpower132, IMpower131, IMpower150, IMpower133, IMmotion151, IMpassion130, and IMbrave150.
AESIs of potential immune etiology by ADA and NAb status are detailed in Table S3 and Table S4. AESI profiles for atezolizumab‐treated patients were similar regardless of ADA and/or NAb development, with few specific toxicities varying between subgroups. AESI frequencies were generally similar across subgroups, although ADA+ patients given atezolizumab as combination therapy had numerically higher frequencies of grade 3/4 AESIs (14.5% vs. 10.7%), serious AESIs (8.8% vs. 6.8%), and AESIs requiring corticosteroids (15.7% vs. 13.8%; see Table 2). Hepatitis (diagnosis and laboratory abnormalities) was numerically higher in combination‐therapy study ADA+ patients than in ADA– patients (19.7% vs. 16.6%), as were IRRs (5.5% vs. 2.6%) (see Table S3). IRRs were also numerically higher in combination‐therapy study ADA+ patients who developed NAb+ (8.0% in ADA+/NAb+ patients vs. 2.7% in ADA+/NAb– patients vs. 2.6% in ADA– or ADA+/NAb– patients); the majority of these events were of low grade of severity (grade 1 or 2), and the overall impact was not considered to be clinically relevant (see Table S4).
DISCUSSION
ICIs have advanced the treatment of multiple cancers, and potential impacts on clinical outcomes associated with immunogenicity require careful consideration. Initial descriptive analyses of treatment‐emergent ADA from atezolizumab clinical trial data—conducted using industry‐standard approaches to ADA data interpretation 15 without the use of landmark analysis or adjustment for baseline characteristics—had suggested a trend of lower atezolizumab exposure in treatment‐emergent ADA+ patients, which was not clinically relevant as most patients were still expected to have trough concentrations above levels required for target saturation. 18 Atezolizumab also exhibits a flat exposure‐response relationship, 39 , 41 with the impact of ADA on clearance being only 9% as estimated by a time‐varying population PK model. 42 Shorter survival in ADA+ patients compared with ADA– patients was also previously reported but was confounded by imbalances in baseline prognostic factors. Here, we evaluated clinical outcomes per ADA status using clinical trial data from 7303 patients across several cancer types who were treated with either atezolizumab monotherapy or combination therapy. We applied methodology in efficacy analyses to address the limitation that ADA status is a postbaseline variable and is unavailable for control arms and to address the vulnerability of analyses to survivorship bias. 40 Our adjusted meta‐analyses showed that atezolizumab efficacy in ADA+ and ADA– patients was similar for both PFS and OS after adjusting for covariates, with wide overlapping CIs. However, the direction of the point estimates of the RHRs suggested that some attenuation of OS benefit in ADA+ patients compared with ADA– patients remains. Meta‐analytic comparisons of ADA+ versus adjusted control patients indicated that ADA+ patients derive clinically relevant OS and PFS benefits from atezolizumab. We found no difference in efficacy based on NAb+ status, suggesting that any ADA impact is not driven by neutralizing activity. We showed that the overall safety profile of atezolizumab was generally consistent between ADA– and ADA+ patients (and between NAb+ and NAb– patients), with no specific pattern of AEs seen based on ADA status, suggesting that management of AEs is not expected to be affected by ADA status.
A novel observation was the trend of prognostically worse baseline clinical characteristics among patients who developed ADA to atezolizumab while on treatment compared with those who did not. These characteristics included higher frequencies of ECOG PS of 1 versus 0, 43 greater baseline tumor burden, and higher levels of the inflammatory response protein CRP. 44 Notably, these imbalances stood out in a pooled, heterogeneous population across tumor types. To a lesser extent, higher median neutrophil‐lymphocyte ratios, higher frequencies of squamous histology (in patients with lung cancer), and more men were seen among ADA+ versus ADA– patients. For neutralizing versus non‐neutralizing ADA, results were less clear, with most factors listed previously differing by ADA status but not differing (or differing with an inverse trend) in NAb+ versus NAb– patients. These observations suggest that the likelihood of ADA development after atezolizumab treatment is influenced by pre‐existing pathophysiology. For example, systemic inflammation in patients with greater disease burden at baseline may elevate the chance of ADA development. Similar observations have been made in patients with rheumatoid arthritis treated with tumor necrosis factor inhibitors; patients who developed ADA had higher baseline disease activity and CRP levels, longer disease duration, and more often erosive disease. 19 , 20
Adjusted analyses are the most scientifically appropriate and statistically sound methods to compare outcomes between treatment arms and are consistent with International Council for Harmonisation guidelines. 21 Further, survivorship bias necessitates the need for landmark analyses, which introduces the challenge of missing data at the landmark time point. Assessing the clinical significance of an unwanted immune response to a biologic in oncology is a complex subject for which the science is still evolving. This analysis took these challenges into consideration directly. Whereas the wide, overlapping CIs between ADA subgroups precluded identification of definitive patterns of efficacy for ADA+ compared with ADA– patients, results from modeling that addressed the limitations of unadjusted analyses suggested some attenuation of OS efficacy in ADA+ patients versus adjusted control compared with ADA– patients versus adjusted control. This attenuation was less evident with respect to PFS. Meta‐analysis results comparing ADA+ with adjusted control‐arm patients showed that ADA+ patients appear to derive OS and PFS benefit from atezolizumab treatment across studies where an ITT treatment effect was observed.
No major trends in efficacy with ADA were seen with respect to monotherapy versus combination therapy, and our main findings were generally consistent across studies, with a few study‐specific exceptions. For instance, POPLAR and IMbrave150 showed improved OS in ADA– versus ADA+ patients. It is notable that a smaller efficacy difference between ADA+ versus ADA– patients was observed for the larger phase III OAK study relative to the smaller phase II POPLAR study, despite nearly identical study designs and target populations. Results for IMbrave150, a study with co‐primary endpoints of OS and PFS, were notable for the inconsistency of ADA subgroup differences: whereas ADA+ patients had similar OS benefit with atezolizumab plus bevacizumab compared with sorafenib, PFS benefit of atezolizumab plus bevacizumab versus sorafenib was clinically meaningful and similar between ADA subgroups. Although it is possible that OS may be more affected by baseline prognostic or post‐treatment variables than PFS, differences were not consistently seen across studies. Therefore, it is important to consider these individual findings in the context of the totality of the data, including meta‐analyses, when evaluating potential impacts of ADA status on efficacy.
Available safety data do not allow conclusions to be drawn on clinically relevant patterns of adverse drug reactions attributable to atezolizumab immunogenicity. Imbalances exist in the incidence of various AE categories between ADA subgroups in both directions, with certain types of AEs more frequent in ADA– patients and others more frequent in ADA+ patients. Imbalances in health and disease characteristics were observed at baseline (pretreatment) between ADA+ and ADA– patients. Due to difficulties to adjust for baseline covariates in a safety analysis, no formal analysis was performed. However, it is plausible that these imbalances may confound the safety data and contribute to higher incidences of grade 3–4 AEs and serious AEs in ADA+ patients. Numerically higher frequencies of IRRs occurred in combination therapy‐study ADA+ patients who developed NAb+; most events were of low grade of severity, and the overall impact was not considered clinically relevant. Overall, clinical management of toxicities in atezolizumab‐treated patients should not be affected by patient ADA status. This is based on the approach taken in completed and ongoing atezolizumab clinical trials wherein management of adverse reactions is independent of patient ADA status. This analysis used a conservative approach for safety, with all events considered while assessing the impact of ADA/NAb on safety irrespective of temporal association or biological plausibility. It is notable that our observations of a typical atezolizumab safety profile in both ADA subgroups is consistent with pharmacologically active drug exposure regardless of ADA status.
It is important to acknowledge the limitations and exploratory nature of the study. Despite the large overall study size, in some cases, the ADA and/or NAb subgroups were small. In addition, in the majority of cases, HR CIs were wide and overlapping between ADA subgroups (vs. adjusted control), precluding definitive conclusions on the impact of ADA/NAb on efficacy in individual studies. Further, it cannot be ruled out that some confounding variables were excluded from the analyses.
Here, we applied statistically sound methods to assess clinical consequences of ADA development in atezolizumab clinical trials; we anticipate that application of this methodology may be helpful in future assessments of the impact of ADA or other intercurrent events. Collectively, the minimal impact of ADA on atezolizumab clearance, 42 flat exposure‐response relationship, 39 , 41 and lack of dose‐limiting toxicities or maximum tolerated dose 38 support the favorable efficacy and safety findings for atezolizumab. An assessment of ADA/NAb impact on PK has revealed that ADA/NAb do not have a clinically meaningful impact on exposure. Interestingly, there appear to be unique and shared covariates associated with efficacy, PK, and ADA development. 18
In conclusion, this is the first comprehensive ADA analysis of atezolizumab trial data, using both study‐by‐study and integrated analyses. To our knowledge, this is one of the largest and most comprehensive ADA analyses completed for a therapeutic protein. We used approaches to account for factors relevant to oncology that are not appropriately addressed by standard ITT subgroup analyses. Although some studies suggested that ADA could attenuate the efficacy benefit of atezolizumab, impacts were not seen consistently across indications, and meta‐analysis results showed that ADA+ patients derive benefit from atezolizumab over control. The efficacy findings in ADA+ patients are consistent with the lack of clinically meaningful decrease in exposure and the strategy for dosing over any potential ADA effect. 18 Some numerical differences in AE frequencies were seen between ADA/NAb subgroups, possibly due to the underlying baseline differences in patient health status between the subgroups; no adjustment was possible on these data. Collectively, a positive benefit‐risk balance is observed with atezolizumab treatment for approved indications regardless of ADA status.
CONFLICT OF INTEREST
S.P. reports personal fees for speaking or attending advisory boards, and has participated in investigation in trials for Amgen, AstraZeneca, Boehringer‐Ingelheim, Bristol Myers Squibb, Clovis, F. Hoffmann‐La Roche, Illumina, Merck Sharp & Dohme, Merck Serono, Novartis, and Pfizer; and personal fees for speaking or attending advisory boards for AbbVie, Bayer, Biocartis, Bioinvent, Daiichi Sankyo, Debiopharm, ecancer, Foundation Medicine, Janssen, Lilly, Medscape, Merrimack, Pharma Mar, Regeneron, Sanofi, Seattle Genetics, and Takeda (all fees to institution). P.R.G. reports a consulting or advisory role and received honoraria from AdaptImmune, AstraZeneca, Bayer, Bristol Myers Squibb, Eisai, Ipsen, Lilly, Merck Sharp & Dohme, Roche, and Sirtex; has been on a speakers bureau for AstraZeneca, Bayer, Bristol Myers Squibb, Eisai, Ipsen, Lilly, Merck Sharp & Dohme, Roche, and Sirtex; has received research funding from Bayer and Roche; has provided expert testimony for Lilly; and has received travel or accommodation expenses from AstraZeneca, Bayer, Bristol Myers Squibb, Eisai, Ipsen, Lilly, and Roche. C.A.B., M.B., R.B., V.Q., A.V., B.W., and N.S. are employees of Genentech, Inc/Roche and Roche stockholders. J.R. is a former employee of Genentech, Inc/Roche and Roche stockholder. M.R. reports honoraria for consultancy and lecture roles from Amgen, AstraZeneca, Boehringer Ingelheim, Bristol Myers Squibb, Lilly, Merck, Mirati, Merck Sharp & Dohme, Novartis, and Pfizer.
AUTHOR CONTRIBUTIONS
All authors wrote the manuscript. C.A.B., N.S., M.B., B.W., and R.B. designed the research. S.P., P.R.G., and M.R. performed the research. C.A.B., N.S., M.B., B.W., and A.V. analyzed the data. C.A.B., B.W., J.R., and V.Q. contributed new reagents/analytical tools.
Supporting information
Supplementary Material
ACKNOWLEDGEMENTS
This study was sponsored by F. Hoffmann‐La Roche, Ltd./Genentech, Inc., a member of the Roche Group. We thank the patients and their families. We thank Veronica Craine and Dominik Heinzmann of Roche as well as Shengchun Kong and Joel Laxamana of Genentech for their contributions to the statistical analyses. Medical writing assistance for this manuscript was provided by Ashley J. Pratt, PhD, of Health Interactions and funded by F. Hoffmann‐La Roche, Ltd.
Peters S, Galle PR, Bernaards CA, et al. Evaluation of atezolizumab immunogenicity: Efficacy and safety (Part 2). Clin Transl Sci. 2022;15:141–157. 10.1111/cts.13149
Funding information
This study was sponsored by F. Hoffmann‐La Roche, Ltd./Genentech, Inc., a member of the Roche Group.
DATA AVAILABILITY STATEMENT
Qualified researchers may request access to individual patient‐level data through the clinical study data request platform (https://vivli.org/). Further details on Roche’s criteria for eligible studies are available here (https://vivli.org/members/ourmembers/). For further details on Roche’s Global Policy on the Sharing of Clinical Information and how to request access to related clinical study documents, see here (https://www.roche.com/research_and_development/who_we_are_how_we_work/clinical_trials/our_commitment_to_data_sharing.htm).
References
- 1. Mire‐Sluis AR, Barrett YC, Devanarayan V, et al. Recommendations for the design and optimization of immunoassays used in the detection of host antibodies against biotechnology products. J Immunol Methods. 2004;289:1‐16. [DOI] [PubMed] [Google Scholar]
- 2. Schellekens H. Immunogenicity of therapeutic proteins: clinical implications and future prospects. Clin Ther. 2002;24:1720‐1740. [DOI] [PubMed] [Google Scholar]
- 3. US Food and Drug Administration . Immunogenicity testing of therapeutic protein products — developing and validating assays for anti‐drug antibody detection. 2019. https://www.fda.gov/regulatory‐information/search‐fda‐guidance‐documents/immunogenicity‐testing‐therapeutic‐protein‐products‐developing‐and‐validating‐assays‐anti‐drug. Accessed February 2, 2021.
- 4. Gunn GR 3rd, Sealey DC, Jamali F, Meibohm B, Ghosh S, Shankar G. From the bench to clinical practice: understanding the challenges and uncertainties in immunogenicity testing for biopharmaceuticals. Clin Exp Immunol. 2016;184:137‐146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wang YM, Wang J, Hon YY, Zhou L, Fang L, Ahn HY. Evaluating and reporting the immunogenicity impacts for biological products—a clinical pharmacology perspective. AAPS J. 2016;18:395‐403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. van Brummelen EM, Ros W, Wolbink G, Beijnen JH, Schellens JH. Antidrug antibody formation in oncology: clinical relevance and challenges. Oncologist. 2016;21:1260‐1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gupta S, Devanarayan V, Finco D, et al. Recommendations for the validation of cell‐based assays used for the detection of neutralizing antibody immune responses elicited against biological therapeutics. J Pharm Biomed Anal. 2011;55:878‐888. [DOI] [PubMed] [Google Scholar]
- 8. Gupta S, Indelicato SR, Jethwa V, et al. Recommendations for the design, optimization, and qualification of cell‐based assays used for the detection of neutralizing antibody responses elicited to biological therapeutics. J Immunol Methods. 2007;321:1‐18. [DOI] [PubMed] [Google Scholar]
- 9. Calabresi PA, Giovannoni G, Confavreux C, et al. The incidence and significance of anti‐natalizumab antibodies: results from AFFIRM and SENTINEL. Neurology. 2007;69:1391‐1403. [DOI] [PubMed] [Google Scholar]
- 10. Cassotta A, Mikol V, Bertrand T, et al. A single T cell epitope drives the neutralizing anti‐drug antibody response to natalizumab in multiple sclerosis patients. Nat Med. 2019;25:1402‐1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Enrico D, Paci A, Chaput N, Karamouza E, Besse B. Antidrug antibodies against immune checkpoint blockers: impairment of drug efficacy or indication of immune activation? Clin Cancer Res. 2020;26:787‐792. [DOI] [PubMed] [Google Scholar]
- 12. Harding FA, Stickler MM, Razo J, DuBridge RB. The immunogenicity of humanized and fully human antibodies: residual immunogenicity resides in the CDR regions. MAbs. 2010;2:256‐265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mehta P, Manson JJ. What is the clinical relevance of TNF inhibitor immunogenicity in the management of patients with rheumatoid arthritis? Front Immunol. 2020;11:589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lamberth K, Reedtz‐Runge SL, Simon J, et al. Post hoc assessment of the immunogenicity of bioengineered factor VIIa demonstrates the use of preclinical tools. Sci Transl Med. 2017;9:eaag1286. [DOI] [PubMed] [Google Scholar]
- 15. Shankar G, Arkin S, Cocea L, et al. Assessment and reporting of the clinical immunogenicity of therapeutic proteins and peptides‐harmonized terminology and tactical recommendations. AAPS J. 2014;16:658‐673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. TECENTRIQ (atezolizumab) [prescribing information] . 2021. Genentech, Inc., South San Francisco, CA. https://www.gene.com/download/pdf/tecentriq_prescribing.pdf. Accessed April 4, 2021.
- 17. TECENTRIQ (atezolizumab) [summary of product characteristics] 2021. Roche Registration Limited, Welwyn Garden City, UK. https://www.ema.europa.eu/en/documents/product‐information/tecentriq‐epar‐product‐information_en.pdf. Accessed April 4, 2021.
- 18. Wu B, Sternheim N, Agarwal P, et al. Evaluation of atezolizumab immunogenicity: clinical pharmacology (part 1). Clin Transl Sci. 2021;1–11. 10.1111/cts.13127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bartelds GM, Krieckaert CL, Nurmohamed MT, et al. Development of antidrug antibodies against adalimumab and association with disease activity and treatment failure during long‐term follow‐up. JAMA. 2011;305:1460‐1468. [DOI] [PubMed] [Google Scholar]
- 20. Garces S, Antunes M, Benito‐Garcia E, da Silva JC, Aarden L, Demengeot J. A preliminary algorithm introducing immunogenicity assessment in the management of patients with RA receiving tumour necrosis factor inhibitor therapies. Ann Rheum Dis. 2014;73:1138‐1143. [DOI] [PubMed] [Google Scholar]
- 21. European Medicines Agency ICH E9 (R1) . ICH E9 (R1) addendum on estimands and sensitivity analysis in clinical trials to the guideline on statistical principles for clinical trials. 2020. https://www.ema.europa.eu/en/documents/scientific‐guideline/ich‐e9‐r1‐addendum‐estimands‐sensitivity‐analysis‐clinical‐trials‐guideline‐statistical‐principles_en.pdf. Accessed February 3, 2021.
- 22. Gleiss A, Oberbauer R, Heinze G. An unjustified benefit: immortal time bias in the analysis of time‐dependent events. Transpl Int. 2018;31:125‐130. [DOI] [PubMed] [Google Scholar]
- 23. Fehrenbacher L, Spira A, Ballinger M, et al. Atezolizumab versus docetaxel for patients with previously treated non‐small‐cell lung cancer (POPLAR): a multicentre, open‐label, phase 2 randomised controlled trial. Lancet. 2016;387:1837‐1846. [DOI] [PubMed] [Google Scholar]
- 24. Rittmeyer A, Barlesi F, Waterkamp D, et al. Atezolizumab versus docetaxel in patients with previously treated non‐small‐cell lung cancer (OAK): a phase 3, open‐label, multicentre randomised controlled trial. Lancet. 2017;389:255‐265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. West H, McCleod M, Hussein M, et al. Atezolizumab in combination with carboplatin plus nab‐paclitaxel chemotherapy compared with chemotherapy alone as first‐line treatment for metastatic non‐squamous non‐small‐cell lung cancer (IMpower130): a multicentre, randomised, open‐label, phase 3 trial. Lancet Oncol. 2019;20:924‐937. [DOI] [PubMed] [Google Scholar]
- 26. Jotte R, Cappuzzo F, Vynnychenko I, et al. Atezolizumab in combination with carboplatin and nab‐paclitaxel in advanced squamous NSCLC (IMpower131): results from a randomized phase III trial. J Thorac Oncol. 2020;15:1351‐1360. [DOI] [PubMed] [Google Scholar]
- 27. Nishio M, Barlesi F, West H, et al. Atezolizumab plus chemotherapy for first‐line treatment of non‐squamous non‐small cell lung cancer: results from the randomized phase III IMpower132 trial. J Thorac Oncol. 2021;16:653‐664. [DOI] [PubMed] [Google Scholar]
- 28. Reck M, Mok TSK, Nishio M, et al. Atezolizumab plus bevacizumab and chemotherapy in non‐small‐cell lung cancer (IMpower150): key subgroup analyses of patients with EGFR mutations or baseline liver metastases in a randomised, open‐label phase 3 trial. Lancet Respir Med. 2019;7:387‐401. [DOI] [PubMed] [Google Scholar]
- 29. Horn L, Mansfield AS, Szczesna A, et al. First‐line atezolizumab plus chemotherapy in extensive‐stage small‐cell lung cancer. N Engl J Med. 2018;379:2220‐2229. [DOI] [PubMed] [Google Scholar]
- 30. Powles T, Duran I, van der Heijden MS, et al. Atezolizumab versus chemotherapy in patients with platinum‐treated locally advanced or metastatic urothelial carcinoma (IMvigor211): a multicentre, open‐label, phase 3 randomised controlled trial. Lancet. 2018;391:748‐757. [DOI] [PubMed] [Google Scholar]
- 31. Rini BI, Powles T, Atkins MB, et al. Atezolizumab plus bevacizumab versus sunitinib in patients with previously untreated metastatic renal cell carcinoma (IMmotion151): a multicentre, open‐label, phase 3, randomised controlled trial. Lancet. 2019;393:2404‐2415. [DOI] [PubMed] [Google Scholar]
- 32. Schmid P, Adams S, Rugo HS, et al. Atezolizumab and nab‐paclitaxel in advanced triple‐negative breast cancer. N Engl J Med. 2018;379:2108‐2121. [DOI] [PubMed] [Google Scholar]
- 33. Finn RS, Qin S, Ikeda M, et al. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med. 2020;382:1894‐1905. [DOI] [PubMed] [Google Scholar]
- 34. Peters S, Gettinger S, Johnson ML, et al. Phase II trial of atezolizumab as first‐line or subsequent therapy for patients with programmed death‐ligand 1‐selected advanced non‐small‐cell lung cancer (BIRCH). J Clin Oncol. 2017;35:2781‐2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Spigel DR, Chaft JE, Gettinger S, et al. FIR: efficacy, safety, and biomarker analysis of a phase II open‐label study of atezolizumab in PD‐L1‐selected patients with NSCLC. J Thorac Oncol. 2018;13:1733‐1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rosenberg JE, Hoffman‐Censits J, Powles T, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum‐based chemotherapy: a single‐arm, multicentre, phase 2 trial. Lancet. 2016;387:1909‐1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Balar AV, Galsky MD, Rosenberg JE, et al. Atezolizumab as first‐line treatment in cisplatin‐ineligible patients with locally advanced and metastatic urothelial carcinoma: a single‐arm, multicentre, phase 2 trial. Lancet. 2017;389:67‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Herbst RS, Soria JC, Kowanetz M, et al. Predictive correlates of response to the anti‐PD‐L1 antibody MPDL3280A in cancer patients. Nature. 2014;515:563‐567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Stroh M, Winter H, Marchand M, et al. Clinical pharmacokinetics and pharmacodynamics of atezolizumab in metastatic urothelial carcinoma. Clin Pharmacol Ther. 2017;102:305‐312. [DOI] [PubMed] [Google Scholar]
- 40. Kong S, Heinzmann D, Lauer S, Lu T. Weighted approach for estimating effects in principal strata with missing data for a categorical post‐baseline variable in randomized controlled trials. Preprint posted online January 21, 2021. https://arxiv.org/abs/2101.04263. Accessed February 3, 2021.
- 41. Morrissey KM, Marchand M, Patel H, et al. Alternative dosing regimens for atezolizumab: an example of model‐informed drug development in the postmarketing setting. Cancer Chemother Pharmacol. 2019;84:1257‐1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Marchand M, Zhang R, Chan P, et al. Time‐dependent population PK models of single agent atezolizumab in patients with cancer. Cancer Chemother Pharmacol. 2021;88:211‐221. [DOI] [PubMed] [Google Scholar]
- 43. Oken MM, Creech RH, Tormey DC, et al. Toxicity and response criteria of the Eastern Cooperative Oncology Group. Am J Clin Oncol. 1982;5:649‐655. [PubMed] [Google Scholar]
- 44. Hart PC, Rajab IM, Alebraheem M, Potempa LA. C‐reactive protein and cancer—diagnostic and therapeutic insights. Front Immunol. 2020;11: 595835. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Data Availability Statement
Qualified researchers may request access to individual patient‐level data through the clinical study data request platform (https://vivli.org/). Further details on Roche’s criteria for eligible studies are available here (https://vivli.org/members/ourmembers/). For further details on Roche’s Global Policy on the Sharing of Clinical Information and how to request access to related clinical study documents, see here (https://www.roche.com/research_and_development/who_we_are_how_we_work/clinical_trials/our_commitment_to_data_sharing.htm).