

Abstract
The use of microphysiological systems (MPS) to support absorption, distribution, metabolism, and elimination (ADME) sciences has grown substantially in the last decade, in part driven by regulatory demands to move away from traditional animal‐based safety assessment studies and industry desires to develop methodologies to efficiently screen and characterize drugs in the development pipeline. The past decade of MPS development has yielded great user‐driven technological advances with the collective fine‐tuning of cell culture techniques, fluid delivery systems, materials engineering, and performance enhancing modifications. The rapid advances in MPS technology have now made it feasible to evaluate critical ADME parameters within a stand‐alone organ system or through interconnected organ systems. This review surveys current MPS developed for liver, kidney, and intestinal systems as stand‐alone or interconnected organ systems, and evaluates each system for specific performance criteria recommended by regulatory authorities and MPS leaders that would render each system suitable for evaluating drug ADME. Whereas some systems are more suitable for ADME type research than others, not all system designs were intended to meet the recently published desired performance criteria and are reported as a summary of initial proof‐of‐concept studies.
INTRODUCTION
Advancement of an microphysiological system (MPS) as a tool for use in the absorption, distribution, metabolism, and elimination (ADME) sciences depends on device design and relies heavily on the cell type(s) used and the response to culture conditions within the device. The ultimate goal of most MPS developers and users is to recreate a portion of an organ that retains a physiologically relevant phenotype to test and understand exposures to endogenous and xenobiotic compounds. However, each system has functional limitations. For an MPS to perform and function to support ADME‐type work, it must have multiple biochemical processes functioning to be able to deliver meaningful data and a means to demonstrate physiologic functionality. Because there are hundreds of metabolic enzymes and transporters involved in ADME it is not feasible to address the functionality of every protein therefore it is important for the user to identify which components are most relevant. If the objective is to follow the drug development pathway, the enzyme and transporter choices narrow to those that are most commonly involved in ADME and drug‐drug interactions (DDIs) as recognized by regulatory agencies; not all MPS are able to provide the needed supportive data. This survey of current liver, kidney, and intestinal MPS, either as stand‐alone organs or as multiplexed organs, is presented with an emphasis on utility toward ADME science using performance criteria suggested by regulatory and industrial experts.
Context of use (CoU) is an essential consideration for MPS use in ADME, as each system design serves a specific purpose, with its general use for ADME research limited by engineering controls, cost restraints, and the materials used to produce the device. Each MPS device is designed to recapitulate an organ system, however, their designs can vary greatly and there may be some limitations as to what questions can be answered with a specific device. The CoU can range from basic compound‐specific academic research investigations to complex organ‐to‐organ interactions with linked systems designed to provide ADME data that support regulatory filings for drugs in development. Attributes to consider when evaluating an MPS for ADME sciences include characterization of functional ADME parameters and identification of relevant pathways. Most notably, active and properly localized drug transporters, metabolic enzymes, and robust phenotypic markers are used to define the cells within the micro‐organotypic environment of an MPS.
There have been many excellent review articles on the use of MPS for work in ADME sciences in recent years that serve as a source for guidelines and performance criteria for liver, kidney, and intestinal MPS. 1 , 2 Primarily written from the pharmaceutical industry point of view, guided by the known and anticipated regulatory requirements embedded in the US Food and Drug Administration (FDA) and European Medicines Agency (EMA) ADME‐related guidance to industry documents, they provide a much‐needed framework to gauge MPS performance in the context of ADME sciences. Liver MPS development guidelines for safety risk assessments were further refined in a thorough review by pharmaceutical industry MPS leaders and provide highly detailed performance criteria for three stages of model characterization. 1 , 3 A pharmaceutical industry perspective on desired kidney MPS characteristics with an emphasis on safety and ADME applications provides a foundation of key attributes needed to support the development of new experimental therapies. 4 A review of MPS used for ADME‐related applications, again written by MPS experts, contains recommendations for intestinal MPS either as a stand‐alone organ or linked to other organ systems. 5 These review articles provide a vetted set of performance criteria for liver, kidney, and intestinal MPS devices if they are to be used for ADME‐related investigations. Each MPS organ system has its own unique set of variables and geometric complexities that determine what ADME characteristics can be reliably evaluated.
The scope of the metabolizing enzymes and transporter proteins that need to be evaluated for a new investigational drug can be daunting. In vitro test systems currently used by industry typically use microsomes and/or transfected cell lines that overexpress transporters or enzymes. Recent FDA guidance (In Vitro Drug Interaction Studies—Cytochrome P450 Enzyme‐ and Transporter‐Mediated Drug Interactions—Guidance for Industry) has listed more than seven metabolizing enzymes and nine transporter proteins for which a new drug may require evaluation as a substrate, inhibitor, or inducer. 6 These in vitro substrate determination studies typically include assessment of CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A4. Should these not appear to metabolize the investigational drug then evaluation of the following enzymes is recommended: CYP2A6, CYP2J2, CYP4F2, and CYP2E1; aldehyde oxidase (AO), carboxylesterase (CES), monoamine oxidase (MAO), flavin monooxygenase (FMO), xanthine oxidase (XO), alcohol/aldehyde dehydrogenase (ADH/ALDH) and the phase II enzymes UDP‐glucuronosyl transferases (UGTs) and sulfotransferases (SULTs). In vitro inhibition studies should be completed with CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A and initial evaluations for enzyme induction should be carried out with CYP1A2, CYP2B6, and CYP3A4 followed by an evaluation of CYP2C if CYP3A is positive for induction.
The FDA, EMA, and the Japanese Pharmaceutical and Medical Device Agency (PMDA) have required that specific transporters be evaluated either prospectively or retrospectively to support pharmaceutical regulatory filings. Table 1 lists the required transporters and which organs(s) should be evaluated for their function. To evaluate transporter‐mediated drug interactions, the regulatory agencies recommend that a new investigational drug should be tested as a substrate for: P‐glycoprotein (P‐gp) and breast cancer resistance protein (BCRP), hepatic transporters organic anion transporting polypeptide 1B1 (OATP1B1) and OATP1B3, organic anion transporter 1 (OAT1) and OAT3 and organic cation transporter 2 (OCT2), and renal transporters MATE1 and MATE2‐K. Drug developers should also be prepared to evaluate whether investigational drug or metabolites are substrates or inhibitors of the same transporters. Characterization of transport becomes a priority if the investigational drug distributes to the liver or kidneys. Conclusions about possible transporter interactions should not be made without the consideration of combined in vitro results for multiple transporters, as isolated studies may not provide the full coverage of interactions.
TABLE 1.
Required transporters and their organ location needed to be evaluated for an investigational new drug by the FDA, EMA, and PMDA
| Transporter | Gene | Liver | Kidney | Intestine |
|---|---|---|---|---|
| OAT1 | SLC22A6 | X | ||
| OAT3 | SLC22A8 | X | ||
| OCT2 | SLC22A2 | X | ||
| P‐gp | ABCB1 | X | X | X |
| BCRP | ABCG2 | X | X | X |
| OATP1B1 | SLCO1B1 | X | ||
| OATP1B3 | SLCO1B3 | X | ||
| MATE1 | SLC47A1 | X | X | |
| MATE2‐K | SLC47A2 | X | X |
Abbreviations: EMA, European Medicines Agency; FDA, US Food and Drug Administration; PMDA, Japanese Pharmaceutical and Medical Device Agency.
Routine acceptance of MPS‐derived data to support regulatory drug filings is dependent on the establishment of quality control and performance criteria between systems that will provide guidance for users to follow should they want to pursue that route. MPS characterization to support drug development has made progress with the FDA evaluating a liver MPS for use in ADME studies 7 and with the recent publications of review articles that address the topic for many organ systems. 3 , 4 , 5 Because MPS devices create an environment that is more physiological relevant than typical 2‐D cell culture techniques the evaluation of multiple CYPs and transporters within the same system is possible leading to more complex ADME evaluations.
HEPATIC MPS
Of the three MPS organ systems considered in this review, the architecture of the hepatic system is one of the more complex to recapitulate in vitro when one considers the structure of the organ microenvironments and oxygen gradients within the hepatic triad as well as the biliary efflux component. In addition to architecture constraints, there are numerous cell types present that includes hepatocytes, stellate cells, Kupffer cells, and nonparenchymal cells that can contribute to ADME characterization. MPS designs are in part governed by their context of use; some systems are intended as high‐throughput assays that allow simultaneous screening of multiple compounds for a limited set of ADME readouts while others are coupled to additional organ MPS and generate metabolites to determine coupled effects on other organs. Low‐ and medium‐throughput systems can generate a richer dataset but tend to be more labor and resource intensive thus restricting use of the evaluation of a fewer number of compounds at a time.
The pharmaceutical industry has recently published a set of liver MPS criteria for use in drug development with biological specifications relevant for ADME investigations. 3 The identified specifications provide MPS developers a development path for optimizing performance of their devices for use as tools for ADME sciences.
Stage 1 criteria of MPS model characterization focuses on basal cell health markers for hepatocytes. Cells must demonstrate that they are healthy enough to be able to produce albumin and synthesize urea at a consistent specific rate over the duration of an ADME investigation. The assessment of sustained basal mRNA expression of critical ADME genes is additional evidence that sufficient cell health is maintained to ultimately allow adequate amounts of metabolized and transported compounds to be processed.
Stage 2 of characterization criteria are more function‐based with confirmation of readouts for toxicity markers, and demonstration of basal and induced metabolic functions with an established set of inducers for metabolic enzymes. Because transport function and bile acid homeostasis are integral parts of liver function, the demonstration of transporter substrate and bile acid uptake are recommended. Visualization of the cells in MPS culture and immunohistochemistry that identify the presence and cellular localization of ADME‐related proteins support biochemical findings. Histology also allows for the comparison of hepatic‐like architecture within the MPS to normal intact organ morphology.
Stage 3 evaluates whether the liver MPS, with all relevant ADME‐related processes functioning, is able to identify whether a known hepatotoxic test compound is toxic or not. However, drug‐induced liver injury (DILI) can occur through many different mechanisms, which include direct effects of reactive metabolites, bile acid interactions, and immune‐mediated pathways. This emphasizes the need for a well‐characterized liver MPS for safety assessment with all ADME end points in mind.
Table 2 lists liver MPS systems by type and the detailed criteria for the three stages of characterization identified by Baudy et al. in the public literature. Many of these systems are proof‐of‐concept reports with varying contexts of use, so not all ADME criteria were sought; however, comparisons can be made as to which parts of the systems have been carefully characterized. In some cases, the amount of characterization for a particular category (e.g., RNA transcripts and test compounds used) is beyond current recommendations, thus not all data are listed.
TABLE 2.
Comparison of hepatic MPS systems with phase I criteria for acceptable MPS performance criteria that meet ADME standards based on recommendations from MPS experts 49 , 50
| Phase I criteria | ||||
|---|---|---|---|---|
| Representative image | System | Albumin production >37 μg/day/106 cells | Urea synthesis >56 μg/day/106 cells | Baseline mRNA quantitative gene expression profiling CYP3A4, 1A2, 2B6, 2C9, 2D6 UGT1A1, GST |
| Micropatterned hepatocytes | ||||
|
HepatoPac Hepregen MPCCs |
24–28 μg/day/106 cells days 3–18 Co‐culture w/3T3‐J2 fibroblasts ~62 μg/day/106 cells |
380–450 μg/day/106 cells Days 3–18 Co‐culture w/3T3‐J2 fibroblasts ~1150 μg/day/106 cells |
On day 42 CYP3A4, CYP2B6, CYP2C9, CYP2C19, CYP1A2, CYP2D6, CYP2C8, CYP2E1 On day 7 UGT1A1, SLC22A1, ABCB1 |

|
HμREL hepatic cultures |
Not assessed | Not assessed | CYP 1A2, CYP3A4, CYP2C19, unspecified SULT, unspecified UGT |
| Hepatocyte‐derived spheroids | ||||
|
Scaffold‐free 3D bio‐printed human liver Spheroids on skewers |
Not assessed | Not assessed | 3A4,1A2,2B6,2C9,2D6, UGT1A6, GSTT2, GSTA4 |

|
3D Spheroid cultures Fluidized bed bioreactor |
Not assessed | Not assessed | Not assessed |
|
3D Spheroids w/NPCs |
0.5–9 μg/106 cells/day to day 15 Maintained for 28 days |
30–300 μg/106 cells/day to day 15 | 3A4,1A2,2B6,2C9,2D6, UGT1A1, GSTA1 |

|
hLIMTs |
Human ~19–31 ng/day/MT to day 14 Murine‐ 19 ± 5 (day 11) to 11–44 ± 10 (day 14) μg/day/1250 cells 1.5–2.0 pg/cell/hr−35d |
Not assessed |
Human Murine CYP3A4 CYP3a11 CYP1A2 CYP1a1 CYP2C9 CYP1a2 CYP2C8 CYP2b10 CYP2C19 CYP3a11 CYP2D6 |
| 3D Bioprinted Liver | ||||
|
Organovo 3D print ExVive Liver |
~3 μg/mL/106 cells at day 14 ~1–1.5 μg/106 cells/day from days 14–28 |
Not assessed |
CYP3A4 CYP2E1 |
| Stem cell derived liver organoids | ||||
|
3D human liver tissue spheroids from stem cells | Not directly assessed | Not assessed | CYP 3A4, 1A2, 2B6, 2C9, 2D6 |
| Multiple organ chips with liver component | ||||
|
Liver sinusoid chip | 15–20 μg/106 cells/day for 28 days | 100–180 μg/106 cells/day for 28 days | Not assessed |

|
Liver‐kidney coupled Nortis chips |
Not reported‐ see Nortis chip | Not reported‐ see Nortis chip | Not reported‐ see Nortis chip |

|
4‐organ chip‐kidney component iPSC organoids | Not measured | Not measured | Many genes sequenced |

|
Wyss multi‐organ | 10–18 μg/mL/106 cells for 21 days | Not measured | Not assessed |

|
Hesperos Pumpless 14 organ chip |
1–3 μg/mL from days 1–7 | ~3.5 μg/mL from days 1–7 | Not assessed |

|
Liver‐gut MPS Multi‐layer gravity fed |
Not reported | Not reported | Not reported |
| Liver chips | ||||
|
Spinning cone Rat hepatocytes Hemoshear |
Rat−70–85 μg/mL/106 cells from days 1–14 |
550–650 μg/106 cells/day for 14 days |
Human‐CYP1A1, CYP1A2, CYP2B1, CYP2B2, CYP2B6, CYP3A2, CYP3A4, CYP2C9, CYP2D6, UGT1A1, GST pi Rat‐CYP1a1, Cyp1a2, Cyp2b1, Cyp2b2, CYP3a2, GSTP1 |

|
Thermoplastic microfluidic MPS PREDICT−96 |
15–40 ng/104 cells/day from days 1–7 ~65–50 ng/device/day (100,000 cells) from days 5–13 |
1.5–0.5 μg/104 cells/day from days 1–7 | Not assessed |

|
LiverChip System CNBio Innovations and Physiome‐on‐a‐Chip |
~28–42 μg/106 cells/day up to day 6 70–120 μg/day up to day 14 |
Measured but no data reported | 3A4, 1A2, 2B6, 2C9, 2D6, UGT1A1, GST |

|
CellASIC | Not assessed | Not assessed | Not assessed |
|
SQL‐SAL sequentially layered, self‐assembly liver model Nortis chip LAMPS Nortis chips |
~2–12 μg/106 cells/day up to day 25 ~18–65 μg/106 cells/day up to day 14 ~15–20 μg/106 cells/day up to day 9 |
~12–30 μg/106 cells/day up to day 25 ~60–70 μg/106 cells/day up to day 9 |
Not reported |

|
HepaChip microplate Spheroids and monolayer |
Not reported | Not reported | Not reported |

|
VLSLL on a chip |
HepG2 cells: 20–45 ng/h/106 cells up to 14 days iPSC: 20–375 ng/h/106 cells up to 21 days |
HepG2 cells: 1.75–4 μg/h/106 cells up to 21 days iPSC: 1–2 μg/h/106 cells up to 14 days |
Not reported |
|
Perfusion Incubator Liver Chip | 40–50 μg/106 cells/day up to 9 days | 85–40 μg/106 cells/day up to 9 days | Not reported |

|
3D iPSC liver chip coupled to cardiac chip | 20–58 μg/106 cells/day up to day 9 | 80–40 μg/106 cells/day up to day 9 | Not reported |

|
Emulate Liver Chip |
~20–70 μg/106 cells/day from days 7–14 |
Not reported | BSEP |
|
3D HepTox Chip Rat cells |
1833–6619 pg/(ng‐DNA hr) up to day 3 | Not reported | CYP1A/2, UGT |
| 2 D Transwells or Plates | ||||
|
RegeneMed Liver Transwell Rat cells |
~1.5 μg/106 cells/day from days 12–77 | ~175–250 μg/106 cells/day from days 12–77 | 3A4, 1A2, 2B6, 2C9, 2D6, UGT1A1, GST |

|
IdMOC In Vitro ADMET Lab | Not reported | Not reported | Not reported |
| Hollow‐fiber reactors | ||||
|
Microcompartment hollow fiber BAL | 5–20 μg/106 cells/day from days 1–14 |
~10–120 μg/106 cells/day from days 1–14 72–288 μg/106 cells/day from days 1–3 |
CYP1A2, CYP2B6, CYP2C9, CYP2D6, CYP3A4/5 |
Abbreviations: ADME, absorption, distribution, metabolism, and elimination; hLIMTs, human liver microtissues in sphero; iPSC, induced pluripotent stem cell; LAMPS, liver acinus microphysiological systems; MPCC, micropatterned cocultures; MPS, microphysiological systems; SQL‐SAL, sequentially layered, self‐assembled; VLSLL, very‐large scale liver lobule.
A suite of compounds should include known hepatotoxic compounds that test all known mechanisms of hepatotoxicity along with negative controls. A recommended list of test compounds has been presented by the MPS Affiliate of the International Consortium for Innovation and Quality in Pharmaceutical Development. 3 This list of compounds is presented in the heading of Table 3 as part of system comparisons based on stage 3 criteria for MPS development.
TABLE 3.
Comparison of hepatic MPS systems with phase II and III criteria for acceptable MPS performance criteria that meet ADME standards based on recommendations from MPS experts 3 , 5
| Phase II/III criteria | |||||
|---|---|---|---|---|---|
|
Liver injury biomarkers ALT, LDH, cytokines, miR122 |
Metabolic enzymes (baseline and induced) 3A4 midazolam 1A2 phenacetin 2B6 bupropion 2C9 diclofenac 2D6 dextromethorphan UGT1A1 estradiol GST |
Transporter function/bile acid homeostasis OATP/MRP2/BSEP Bile acid transport |
Histology bile canaliculi (BSEP, MRP2, AQP1) Kupffer cells (CD68) stellate cells (desmin) EM –endocells fenestrations H&E staining |
Test Compounds Sitaxsentan Pemoline Clozapine Mipomersen Diclofenac Nefazodone Zileuton Ambrisentan Fialuridine Olanzapine Tolcapone FIRU Asunaprevir Entacapon Troglitazone Pioglitazone Telithromycin Levofloxacin Trovafloxacin Buspirone (or Trazodone) |
|
| Phase | II | II | II | II | III |
| Micropatterned hepatocytes | |||||
|
HepatoPac Hepregen MPCCs |
MTT assay, ATP, and glutathione levels using Cell Titer‐Glo and GSH‐Glo luminescent kits |
CYP1A: Ethoxy‐resorufin; CYP1A2: Ethoxy‐resorufin CYP2A6: Coumarin; CYP2B6: Bupropion; CYP3A4: testosterone, luciferin ‐IPA UGTs and SULTS CYP2C9: luciferin ‐H |
Canalicular flow by CMFDA fluorescent probe |
No. of individual bile canaliculi branches Hepatocyte morphology‐polarity |
Amiodarone, clozapine, diclofenac, piroxicam, troglitazone (aspirin), dexamethasone, miconazole, prednisone, rosiglitazone |
| HμREL hepatic cultures |
MTT assay, ATP, and glutathione (GSH) levels LIVE/DEAD stain |
CYP3A4: midazolam CYP2D6: dextrorphan UGTs: 7‐hydroxycoumarin glucuronide SULTs: 7‐hydroxycoumarin sulfate |
Biliary efflux assay (LC‐MS/MS) BSEP canaliculi (CMFDA) 5(6)‐carboxy‐20,70‐dichlorofluorescein diacetate (CDCFDA) |
Morphology | Troglitazone, Nefazadone, Benzbromarone, Amiodarone, Flutamide, Bosentan, Bicalutamide, Diclofenac, Tacrine, Chlorpromazine, Cyclophosphamide, Acetaminophen, Propranolol, Rosiglitazone, Diphenhydramine, Isoproterenol, Kanamycin, Macitentan, Primadone |
| Hepatocyte‐derived spheroids | |||||
|
Scaffold‐free 3D bio‐printed human liver Spheroids on skewers |
CYP3A4 |
Bile acid secretion Genes: OAT2, OCT1, OCTN2, OATP1B1, OATP1B3, OATP2B1, NTCP, MRP3, MRP6, MDR1, MRP1, MRP2, BCRP, MATE1, BSEP |
Morphology‐H&E staining IHC‐MRP2, CD31, TUNEL OAT2/8, |
||
|
3D InSight MicrotissuesInSphero hLIMT |
ATP/MT IL−6 and TNF‐α releasing (ELISA) LDH releasing, mitochondrial activity CellTiter‐Glo 2.0 Cell Viability Assay |
CYP1A1, 2B6 2C8 2C9 2C19 2D6 CYP3A4 and 2E1 (IHC staining) |
Clozapine Diclofenac Zileuton Tolcapone Troglitazone Trovafloxacin Nefazodone Ambrisentan Entacapon Pioglitazone Levofloxacin Buspirone |
||
|
3D Spheroid cultures Fluidized‐bed bioreactor |
Live/dead staining | No data | No data | morphology | No data |
|
3D spheroids w/NPCs |
Trypan blue ATP miR‐122 GSH IL‐6 |
CYP1A: 7‐EC + induced CYP2C9: diclofenac CYP3A4: midazolam CYP1A1/CYP2D6: propranolol UGT: lamotrigine, acetaminophen SULT: salbutamol, acetaminophen GST‐acetaminophen |
HNF4α CDFDA MRP2 |
Albumin CK18 CK19 CYP 3A aPKC E cadherin MRP2 CD68 (Kupffer) Vimentin Stellate‐ α‐SMA, COL1A1 |
Diclofenac Fialuridine Pioglitazone Troglitazone |
|
hLIMTs Human Liver MicroTissues ETH Zurich Spheroids in chip |
ATP Murine IL‐6 Induction of Acta2 and H2‐Ab1 mRNA Neutral lipid accumulation |
CYP3A4‐midazolam, ritonavir, cyclophosphamide, ifosfamide CYP2B6‐bupropion, cyclophosphamide, ifosfamide Murine CYP3a11‐dexamethsone, rifampicin, Pregnenolone 16α‐carbonitrile CYP1a1‐ β naphthoflavone CYP1a2‐ β naphthoflavone CYP2b10‐Phenobarbital CYP3a11‐Phenobarbital |
Murine CMFDA accumulation BSEP |
Human Morphology Murine Kupffer‐F4/80 Stellate‐Desmin Sinusoids liver sinusoidal endothelial cells |
Murine Tetracycline Diclofenac Methotrexate Cyclophosphamide Acetaminophen Propranolol Buspirone Cyclosporine A Troglitazone |
| 3D bioprinted liver | |||||
| 3D print ExVive Liver Organovo | LDH, GSH, ATP, ALT, albumin, IL‐1β, IL‐13, IL‐6, IL‐8, MCP‐1, VEGF, Eotaxin, GMCSF, Flt‐1 |
CYP450 enzymes 3A4 CYPs 1A2, 2B6 2C9 2D6 3A4‐Rifampicin |
Many genes not specified |
Morphology Parenchymal/nonparachymal E‐cadherin, Vimentin, CD31, CD68, CD168 albumin, Stellate‐desmin, α‐SMA Fibrosis markers‐collagens I and IV H&E staining Perilipin‐lipid vesicles |
Benzbromarone Betahistine Nifedipine Chloramphenicol Trovafloxacin Levofloxacin Valproic acid Danazol Tamoxifen Tolcapone Entacapone Perhexaline Phentolamine |
| Stem cell derived liver organoids | |||||
| 3D human liver tissue spheroids from stem cells | ALT, AST, bilirubin, and ALB | CYP 3A4 | Only gene expression data and IHC: MRP1, ABCB11, OCT4, SULT1, SULT2 |
Morphology Bile caniculi IHC: MRP1, ABCB11, HNF4A, ECAD, ZO1, albumin, NANOG, FOXA2, SOX17 |
No drugs tested |
| Multiple organ chips with liver component | |||||
| Liver sinusoid chip |
Live/dead staining, CD31 AL‐1 adducts |
CYP3A4 | CMFDA |
Morphology CD31 |
|
| Liver‐kidney coupled chips | Live/dead staining, KIM‐1, ALT | No data | No data |
Morphology NQO‐1 |
Aristolochic acid |
|
4‐organ chip‐ kidney component iPSC organoids Liver spheroids |
LDH TUNEL |
No data | No data |
Morphology Albumin, ZO‐1, HNF4a, SLC10A1, CK18, Vimentin, KI67, |
|
|
Spinning cone HemoShear Rat hepatocytes |
MTT Human‐ ALT, CK18, ATP, IL‐6, Il‐8, CXCL10, MCP‐1, VEGF, VKL40, TGF‐ β, OPN, pro‐Col I, FGF‐19, apolipoproteins Rat‐MTT |
Cyp 1A1, Cyp 1B1, Cyp 3A1/2, CYP2B2, CYP2B1, GSTpi, UGT1 Rat‐CYP1a1, CYP1b1, CYP3a1/2 |
CDFDA, Rat‐IHC: MRP‐2 TEM‐bile caniculi |
Morphology IHC‐MRP‐2, HNF‐4α, E‐cadherin, TEM‐ bile caniculi, Rat‐Morphology IHC‐CYP activity,E‐cadherin, HNF‐4α TEM‐bile caniculi |
3_methylycolanthrene, dexamethasone Human‐ Morphology IHC‐Nile red, adipophilin, E‐cadherin, CD68, Reelin, SMAA |
| Wyss multi organ | Not reported | No data | Inulin uptake |
Morphology IHC MRP2 |
Not reported |
|
Hesperos 14 organ pumpless chip HepG2 |
Live/dead stain | CYP1A1, CYP3A4 | Not reported | Not reported | Not reported |
|
Liver‐gut chip Multi‐layer gravity fed |
Not reported |
CYP3A4‐BOMMC CYP2C9‐BOMMC UGT‐paracetamol |
Not reported |
Cell morphology confocal IHC‐actin |
paracetamol |
| Liver chips | |||||
|
Thermoplastic microfluidic MPS PREDICT‐96 |
Calcein AM, Albumin, VEGF, α1‐antitrypsin (ɑ1‐AT), TNF‐α, MF1, HGF, TGF‐β, IL‐6, and IL‐10 |
CYP3A4‐ LPS, linoleic acid, oleic acid | DCFDA (MRP2/3 functions) |
Morphology Lipid accumulation‐Nile red CYP activity Tdtomato‐CNA35‐ Collagen production |
Not reported |
| LiverChip System CNBio Innovations and Physiome‐on‐a‐Chip |
WST‐1 LDH Albumin Live/dead stain TNF‐α IL‐6 |
CYP‐3A4 CYP1A2‐ Phenacetin CYP2B6‐ Bupropion CYP2C9 Tolbutamide CYP2D6 Dextromethorphan CYP3A Midazolam CYP7A1 |
mRNA‐ OCT1, NTCP, OATP1B3, OATP1B1, OATP2B1, BCRP, BSEP, MRP2, MDR1 |
Morphology Albumin SEM‐bile caniculi |
Phenacetin Troglitazone Tamoxifen Digoxin Diclofenac Bupropion Tolbutamide Dextromethorphan Midazolam |
| CellAsic | LIVE/DEAD stain | Not reported | Not reported | Morphology | Diclofenac |
|
SQL‐SAL sequentially layered, self‐assembly liver model Nortis chip LAMPS Nortis chips |
LDH TNF‐α1‐antitrypsin Urea Albumin O2 sensors |
Metabolite formation: CYP450 enzymes 1A, 2C9, 3A4, CYP3A4‐Terfenadine, testosterone CYP 1A1/2 EROD CYP2C9 diclofenac UDPGT‐Phenolphthalein CYP2E1‐acetominophen protein expression |
CMFDA efflux |
Morphology α‐SMA COL1A2 Cytochrome c biosensor‐apoptosis marker IHC‐CYP2E1 |
Acetaminophen Yolcapone Trovafloxacin Caffeine Terfenadine Fexofenadine Troglitazone Rosiglitazone Pioglitazone |
|
HepaChip microplate Spheroids and monolayer |
Resazurine assay Live/dead stain |
CYP2C9‐ rifampicin CYP3A4‐ rifampicin |
Morphology IHC‐CD31, CK18 |
Diclofenac | |
| VLSLL on a chip | Live/dead staining | Not reported | CMFDA efflux |
Morphology Bile caniculi |
Not reported |
| Perfusion Incubator Liver Chip | Live/dead staining |
CYP1A2‐acetominophen CYP2B1/2‐bupropion CYP3A2‐midazolam |
Not reported | Morphology |
Acetaminophen Diclofinac |
| 3D iPSC liver chip coupled to cardiac chip | Live/dead staining | CYP 1A2, CYP 2C, CYP 2C19, CYP2D6, CYP 3A4, CYP 3A7 | OATs, OCTs, OATPs, UGTs, SULTS, P‐gp, BCRP |
Morphology Albumin, AFP |
Cisapride ketoconazole |
|
Emulate Liver Chip Rat, dog, human |
Albumin GSH ATP, ALT, AST, K18, GLDH α‐GST miRNA 122 CellROX TMRM IP‐10, IL‐6, MCP‐1 |
CYP1A‐phenacetin, bupropion, midazolam CYP2B‐cyclophosphamide CYP3A‐testosterone |
IHC‐CLF (BSEP substrate), BSEP, CDFA, MRP2 BSEP mRNA |
Morphology: H&E IHC‐BSEP, α‐SMA, CD68, Nile red, K18, MRP‐2 |
Bosetan Acetaminophen Methotrexate Fialuridine TAK‐875 |
|
3D HepTox Chip Rat cells |
Live dead: PI/SYTOX Green Albumin CYP 1A/2 UGT activities |
CYP 1A/2 UGT |
Not reported | Morphology | Acetaminophen, Diclofenac, Quinidine, Rifampin And Ketoconazole |
| 2 D Transwells or Plates | |||||
|
RegeneMed LiverTranswellRegenMed Rat cells |
ATP, GSH, LDH, ALT, caspase 3/7, live/dead protease activity. Glycogen, GM‐CSF, IFN‐γ, IL‐1β, IL‐2, IL‐6, IL‐8, IL‐10, IL 12p70, TNF‐α |
Phase I activity: CYP1A1, TCCD 2C9, rifampicin, phenobarbital 3A4 rifampicin |
Basolateral transporter uptake measured by isotope labeled tracer ‐3H‐labeled estrone‐3‐sulphate IHC: DPPIV |
Morphology IHC: albumin, ICAM‐1, F4/80, HSC |
Fenofibrate Troglitazone Pioglitazone Trovafloxacin Lovofloxacin APAP AMAP |
| IdMOC In Vitro ADMET Lab | MTT assay, ATP, GSH | Not reported | Not reported | Morphology | Cyclophosphamide |
| Hollow‐fiber reactors | |||||
| Microcompartment hollow fiber BAL | LDH, ALT, AST, glucose, lactate releasing |
CYP1A‐ phenacetin CYP2C9‐ diclofenac CYP3A‐ midazolam CYP1A2 CYP2B6 CYP2C9‐ diclofenac CYP2D6 CYP3A4/5 diclofenac |
IHC‐ MRP2, MDR1, BCRP BCRP and MRP2 expression IHC MRP2, BCRP, |
IHC‐ CK19, vW, MRP2, MDR1, BCRP, CD68 Morphology IHC‐ vimentin, BCRP, CYP2C9, CYP3A4, MRP2, CD31 TEM‐‐bile caniculi |
Diclofenac |
Abbreviations: ADME, absorption, distribution, metabolism, and elimination; ALT, alanine aminotransferase; AST, aspartate aminotransferase; H&E, hematoxylin and eosin stain; IHC, immunohistochemical; iPSC, induced pluripotent stem cell; LAMPS, liver acinus microphysiological systems; LC‐MS/MS, liquid‐chromatography tandem mass spectrometry; LDH, lactate dehydrogenase; MPCC, micropatterned cocultures; MPS, microphysiological systems; VLSLL, very‐large scale liver lobule.
With recent advances in liver MPS development and a growing acceptance of their utility in ADME sciences, there is an opportunity to fill an unmet need with the evaluation of cross‐species differences in metabolism with compounds that have been observed to be hepatotoxic in some species but not others. As with the human‐specific systems, well‐characterized hepatic MPS for test species are necessary. This presents a significant challenge as hepatic cell types from a given species may not be amenable to MPS culture and may require additional media supplements that may confound direct comparisons to human counterparts. An example of this is the relative difficulty researchers have with primary mouse hepatocyte culture for in vitro studies compared to humans and rats. Nonetheless, the evaluation of cross‐species differences (human, dog, and rat) with known hepatotoxic compounds in a liver MPS chip that cocultures hepatocytes and endothelial cells has demonstrated potential use as an interspecies ADME study platform. 8
Most current hepatic MPS chip designs are based on sandwich‐type culture formats where hepatocytes and/or other hepatic cells are plated on collagen coated surfaces or biocompatible porous membranes followed by a Matrigel or other extracellular matrix overlay to encapsulate cells and provide a biological scaffold. Functional hepatic sinusoids in some devices are formed with the addition of sequential layers of different cell types that integrate into ordered structures. Consistent in most 2D versus 3D comparisons of overall hepatocyte performance is the need for perfusion to maintain robust cell health that supports ADME‐related functions.
Transwell applications
These 3‐D coculture systems are designed for higher throughput screening applications and can provide robust metabolic functions for several months. Hepatocytes, stellate, Kupffer, and endothelial cells cocultured on a 3‐D scaffold within a removable transwell from the RegeneMed plate system can maintain CYP activity, transporter functions with robust albumin and urea production. 9 The Integrated Discrete Multiple Organ System (IdMOC) utilizes cryopreserved hepatocytes from multiple species to evaluate toxicity profiles. 10 Hepatocytes seeded into a channel are surrounded by shallow wells that can accommodate cells from other organs. Compounds are initially exposed to hepatocytes in the center channel and after a designated time the channel is flooded so media with metabolites can spill into adjacent chambers containing cells from other organs. The HμRel system uses a 96‐well plate coated with type I collagen seeded with human hepatocytes and nonparenchymal cells. 11 , 12 , 13 The plates attach to a housing system that provides flow within an incubator and is used for rapid screening of potentially hepatotoxic compounds.
Hollow fiber bioreactors
Scaled down versions of clinical 3‐D hepatic hollow fiber bioreactors have been adapted for use in ADME‐type research. Bioreactors with interwoven hollow fiber capillaries seeded with human liver cells are fitted with rotameters to control gas mixtures and exhibit robust CYP activity. 14 , 15 , 16 Transmission electron microscopic images show the formation of bile canaliculi with microvilli and immunohistochemical (IHC) staining confirms the presence of numerous drug transporters and metabolic enzymes.
Micropatterned hepatocytes
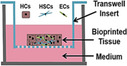
Micropatterned hepatic chips are another technique used to model liver architecture for MPS use. Hepatic micropatterned MPS designs can be simple 96‐well formats that can accommodate spheroids or multiple cell types separated by a porous membrane. Micropatterned well designs allow for the culture of different cell types to occupy a specific spatial orientation that mimics hepatic acini. 17 , 18 , 19 , 20 Bioprinted 3D liver tissues are formed by applying bioink coats of hepatocytes, stellate, and human umbilical vascular endothelial cells (HUVECs) in specific spatial orientations that provides considerable cell mass for numerous ADME applications. The Organonovo bioprinted liver tissue are formed in 24‐well transwells by the bioprinting of hepatocytes, stellate, and HUVEC cells that can grow for 28 days and possess CYP activity that allows for DILI evaluation. 17 , 21 , 22
Thermoplastic materials are used to model a hepatocellular environment that provides two‐channel flow that ensures oxygen delivery to cocultured hepatocytes and Kupffer cells on a microscale and is amenable for adaption to high‐throughput screening as a 96‐well microfluidic system. 23 , 24 , 25 Primary human hepatocytes with high CYP3A4 activity and albumin production are maintained in this system for up to 7 days. Micro‐patterned liver co‐cultures are formed by human hepatocytes seeded into extracellular matrix (ECM) domains that have optimized dimensions for stromal interactions then surrounded by 3T3‐J2 murine embryonic fibroblasts. 26 The resulting coculture displays phenotypic structure stability for several weeks and is used to predict DILI in a 96‐well format.
Spheroids
Recently, there has been significant progress in generating hepatic spheroids from multiple primary or induced pluripotent stem cell (iPSC)‐derived hepatic cell types for use in moderate‐ to high‐throughput ADME‐related investigations that includes use in MPS formats. Cells grown as spheroids tend to maintain critical hepatic phenotypes for long culture periods (5 weeks) and produce a robust number of metabolites suggesting highly functional metabolic and transporter systems. 27 Although sampling of specific tissue microcompartments (e.g., basolateral and acinus regions) is difficult with this format, proteomic, transcriptomic, and metabolite identification by liquid chromatography mass spectrometry (LC‐MS) are feasible. 28
Liver spheroids created by the hanging drop technique or from plates with U‐shaped wells with low‐adhesion surfaces can be successfully grown in static culture 28 , 29 or in microfluidic chips. Human liver microtissues (hLiMTs) by InSphero are liver spheroids comprised of hepatocytes plated into microfluidic culture chips with multiple wells and placed on a rocking platform to simulate hepatic fluid flow. 30 , 31 A multicoaxial fluidized‐bed bioreactor can culture ~ 1500 alginate encapsulated hepatocyte spheroids (4000 hepatocytes/spheroid) under fluid and oxygen control. 32 This design allows for the establishment and tuning of oxygen gradients with sampling to detect metabolic fluxes with a nuclear magnetic resonance (NMR) probe. In another modification of spheroid use, multiple liver spheroids are coaxed to form a larger liver tissue mass by placing standard‐sized spheroids on skewers in close proximity to allow fusion into a larger mass for use in ADME‐type work. 33
Liver chips
The Nortis Bio chip is used to culture liver cells as sandwich cultures with either Matrigel coated hepatocytes 34 or by sequentially coating four hepatic cell types to create a sequentially layered, self‐assembled (SQL‐SAL) MPS. 35 The next version of the SQL‐SAL termed the liver acinus MPS (LAMPS) has oxygen control capabilities that regulate oxygen tensions within designated zones as in the liver portal triad. 36 The LAMPS system uses primary or iPSC hepatocytes and endothelial, Kupffer and stellate cells to create a device that replicates the multicellular components of the liver and was found to be a robust and reproducible method compared to traditional 2‐D monolayer cultures. 37 These devices are meant for low‐ to medium‐throughput investigations as they are labor intensive.
The Emulate liver chip uses a two‐channel system with hepatocytes seeded onto an upper parenchymal channel coated within an ECM sandwich comprised of an ECM coated porous membrane that separates two parallel channels. 8 The lower channel is seeded with sinusoidal epithelial cells in addition to stellate and Kupffer cells and hepatocytes form bile canicular networks that are maintained for at least 2 weeks under flow. Cross‐species comparisons of hepatotoxicity with human, rat, and dog hepatic cells have shown species‐specific differences in response to drug treatments that simulate nonclinical safety evaluations. The HepaChip‐MP is a 24‐chamber microplate that can support culture of hepatocytes. 38 The system is housed in a chamber that allows for robotic pipetting that primes fluids, assembles cells, and performs media exchanges to cells that have been arranged in the chambers by dielectrophoresis. This system is continuously perfused and metabolically outperformed a head‐to‐head comparison with hepatocyte spheroids.
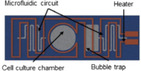
The CN Bio Innovations liver chip is a human liver‐immunocompetent MPS with an array of open well bioreactors where liver cells seeded on a 3‐D scaffold disc are inserted into reactor wells that undergo flow from a built‐in micropump. 39 , 40 , 41 , 42 The perfused microwell system maintains basal mRNA expression of many main transporters and metabolic enzymes for up to 7 days and has demonstrable metabolic activities for CYP1A2, CYP2B6, CYP2C9, CYP3A, and UGT2B4/7 enzymes. When Kupffer cells are included in the cultures, release of relevant pro‐inflammatory cytokines and chemokines occur after an LPS challenge. 43 Long‐term (28 days) culture was achieved with a two‐chambered liver sinusoid MPS chip designed to culture hepatocytes on the bottom layer cocultured with stellate cells separated by an ECM coated membrane with a Kupffer cell line populating the upper chamber. 44 HemoShear has developed a collagen gel sandwich MPS device that has controlled hemodynamics, which is generated by a spinning cone situated over the cells that produces a shear force that helps maintain the metabolic phenotype. 45 , 46 The very large scale liver lobule (VLSLL) liver chip provides users with a complex integrated network of liver‐lobule‐like hexagonal tissue‐culture chambers that mimics the central vein of the liver lobule and can accommodate either HepG2 or iPSCs for cultures up to 21 days. 47 The Perfusion Incubator liver chip contains a heater and built‐in bubble traps while providing a favorable environment for the culture of hepatocyte spheroids. 48 An integrated liver:heart chip system was used to determine DDIs using iPSCs to create both the liver MPS and cardiac MPS from the same cell line. 49
Liver organoids
As with other organ culture systems, stem cell‐derived liver organoids have found their way into MPS cultures and provide an emerging new platform for hepatic ADME study. Similar in shape as hepatocyte spheroid cultures, iPSC‐derived hepatocyte spheroids are active for up to 1 year in culture and display a wide variety of ADME‐related attributes, however, optimization of cell maturation protocols has been challenging. 50 The iPSC spheroids form in 246‐well plates then are transferred to 12‐well plates where they aggregate and can be evaluated for ADME end points. Liver organoids have been transplanted intraperitoneally into mice where they provided liver support in a liver injury model. 50 Transcriptomic analyses of hepatic iPSCs for drug‐metabolizing enzymes, transporters, and nuclear‐receptors suggest they correlate more closely to hepatocytes than human hepatoma cell lines (HepaRG, HepG2, HuH‐7, and HepG2/C3A) used to evaluate hepatic responses of hepatotoxicity. 51 Liver organoids derived from stem cells were used in the four‐organ MPS system connected with autologous organoids from the kidneys, brain, and intestine. 52 Liver iPSC spheroids fuse into organoids and express albumin, HNF4α, and SLC10A1 over a 14‐day period.
KIDNEY MPS
The kidney is a critical organ for determining many excretion parameters of ADME‐related investigations and also carries out important distribution, metabolic and reabsorption functions. Kidney MPS device development has made great strides over the last decade with MPS formats of proximal tubule epithelial cells (PTECs) seeded as monolayers on biocompatible membranes or as tubule designs with continually perfused with media that serves as surrogate urine flow. The nephron is composed of five distinct regions, each with unique ADME properties that need to be recapitulated to understand total nephron function. However, each region of the kidney has contrasting physiological environments that make the design of a physiologically relevant MPS device that accommodates all the microenvironments of each segment technically challenging. The renal cortex is highly perfused receiving 25% of the cardiac output, whereas the medullary region is poorly perfused with less than 5% of blood flow. The nephron contains many drug transporters and metabolic enzymes distributed throughout with many solely confined to certain regions which are situated at either the apical (lumenal), or basolateral sides of cells. The concentration of urine as it flows through the medullary region introduces another design variable that MPS device designers need to consider for accurate representation of the Loop of Henle or collecting ducts. To date, there is no MPS device that captures all nephron segment functionality; however, recent advances in iPSC culture techniques with kidney organoid formation have shown some promise.
Recent review papers have outlined general MPS acceptance performance criteria needed for renal ADME‐type research. 4 , 5 We have reviewed current kidney MPS applications developed in academic and commercial laboratories and compared the published performance record of each system with the recommended guidelines from MPS leaders in ADME sciences. Table 4 outlines each system by nephron segment and lists the performance criteria that exist in the publication record for each MPS system. We recognize that not every system has been developed to serve as an ADME tool and at the same stage of development, but we provide a high‐level comparison for potential use in the ADME field. Because the nephron is a complex anatomic structure, the focus of MPS developers is to target specific nephron segments. Also considered in this review section is kidney MPS linked to other systems within the same device or as part of multiplexed organ system platform. There are several PTEC kidney chips with well‐documented ADME performance, a result of many years in development with a number of publications by end‐users. This does not serve as an endorsement for use but reflects the availability of public information. In general, kidney MPS under perfusion are critical for the creation of the shear stress forces needed to facilitate cell polarization and brush border formation. Perfused media through cell‐lined lumen channels simulates urine flow and is a component of most of the kidney MPS evaluated.
TABLE 4.
Comparison of renal MPS systems with acceptable MPS performance criteria that meet ADME standards based on recommendations from MPS experts 4 , 5
| Microphysiological technologies used to model the renal proximal/distal tubule microenvironment | ||||||
|---|---|---|---|---|---|---|
| Representative image | System |
OAT1/3‐OCT2 activity on basolateral side P‐gp, BCRP, MRP2 and MATE on apical side Megalin/cubulin, SGLT2 glucose transporters, OATS 1–4 |
Relevant phenotypic markers; ZO‐1, LTL, acylated tubulin, exrin, aquaporin 1, basolateral Na+‐K+ ATPase pump | Gene Expression: AQP1, APT1A1, CDH2, SLC5A2, TJP1, Gpx3, SLC22A7, SLC22A13, |
Function‐specific cellular markers: reabsorption of 2‐NDBG, fluorescently tagged dextran, inulin or albumin, negative reabsorption of inulin, vitamin D bioactivation, GGT activity, alkaline phosphatase, leucine aminopeptidase and ACE activity |
Function changes KIM‐1, α‐GST before and after, intracellular Ca2+ changes Loss of brush border Inhibition of transporters |
| Transwell or 96‐well cultures | ||||||

|
Kidney derived microscaffolds (KMS) HK2 cells | No data | No data | AQPc1, ATP1B1, SLC23A1 and SLC5A2, CCL‐2, LRP‐2 | No data | Cytokine release: IL‐6, IL‐8, MCP‐1 and RANTES |
|
PTs on Intestinal submucosa hKDCs |
No data | CD13, CD24, CD44, CD73, CD34, N‐cadherin, E‐cadherin, AQP‐1, AQP‐2, LTL, DBA, Na+‐K+ ATPase pump, claudin‐2 | No data | BSA uptake | No data |
|
Decellularized kidney scaffold | Not reported | No data | No data | No data | No data |

|
Kidney organoids in 3D culture Multiple nephron segments |
No data | CDH1, PODXL, LTL, WT1, AQP1, NPHS1, UMOD | No data | No data | IHC‐KIM‐1, CDH1 |
| Hollow‐fiber systems | ||||||

|
Human proximal tubule epithelial cells cultured on hollow fiber membranes (HFM) |
OAT‐1 transport‐probenicid | ZO‐1, OCT‐2, actn | No data |
Inulin diffusion ASP uptake‐ (OCT‐2) Inulin leakage IL‐6, IL‐8, TNFa, sHLA‐1 |
No data |

|
Bioartificial Renal Tubule Assist Device (RAD) Porcine cells |
, No data | No data | No data |
Inulin leak rates Glucose transport, PAH transport GSH transport and metabolism Albumin reabsorption Ammoniagensis Vitamin D activation |
LDH leakage |
| Kidney chips | ||||||
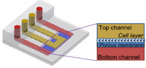
|
Perfused kidney‐on‐a‐chip | No data | ZO‐1, Occludin | No data | No data |
Transmembrane Permeability, KIM‐1, Live/dead stain |

|
Human kidney proximal tubule‐on‐a‐chip Emulate Kidney chip |
OAT1, OCT1, OCT2 inhibition MATE1, MATE2‐K P‐gp efflux transporter activity‐calcein Am |
ZO‐1, aquaporin 1, VE cadherin, beta‐catenin, occludin, Na+‐K+ ATPase pump, brush border, AQP2, acetylated tubulin |
Na+‐K+ ATPase AQP1, SGLT2 |
Albumin reabsorption Na/Pi co transporters Creatinine, PAH metformin clearance glucose reabsorption brush border alkaline phosphatase activity |
LDH, apoptosis (Annexin V) |
|
Nortis microphysiological device |
OAT 1/3‐PAH MRP 2/4 SGLT2‐glucose reabsorb |
ZO‐1, Na+/K+‐ATPase, acetylated tubulin, CD13, E‐Cadherin, AQP‐1, AQP‐2, lotus lectin, SGLT2, SLC22A11/OAT‐4, CK18 | AQP1, APT1A1, CDH2, SLC5A2, TJP1, KAP, HAVCR1, Gpx3, SLC22A7, SLC22A13, CYP24A1, PRKAG2, MT2A, MT1G, GGT1, GPX1 |
GGT‐GSH reclamation ATP production Ammonia production Bioactivation of Vitamin D |
KIM‐1 α‐SMA, AP activity, fibronectin, NGAL Heme Oxygenase‐1, live/dead stain, LCN2 |
|
PT‐ glomerulus chip PT section HK‐2 cells |
caveolae and clathrin‐coated pits | caveolin‐1, occludin, ZO‐1, clathrin or b‐catenin | No data | BSA uptake | α‐SMA, AP activity, fibronectin, NGAL |
|
Kidney Tubuloids And 40‐ leak tight tubules MIMETAS OrganoPlate |
P‐gp assay‐calcein, MRP2 MRP2/4 Efflux Inhibition SGLT2 |
proteins‐ EPCAM, KRT18 and KRT19 SLC4A1, SLC26A7, ATP6V1B1 ZO‐1, P‐gp, AQP3, Na+/K+‐ATPase, ezrin, villin2, Integrin, claudin, acetylated tubulin, SLC4A1, GATA3, CALB1, PAX8+, ITGA6, PT‐ CLDN‐3, villin, Factin CD‐ CDH1, GATA3 |
PT‐EPCAM, PAX8, ABCC1, ABCC3, ABCC4, SLC22A3, SLC40A1 collect duct‐ CDH1, AQP3; loop CLDN10, CLDN14, distal PCBD1, SLC41A3 |
No data | No data |
|
3D perfused proximal tubules‐OrganoPlate | P‐gp | ZO‐1, actin, ezrin, acetylated tubulin | No data |
SGLT2 Glucose –uptake inhib MRP2/4 Efflux Inhibition |
LDH, barrier leakage, H2A.X |
| Vascularized kidney chip | ||||||

|
Vascularized kidney chip VasPT PT char |
No data | SGLT2, Na+‐K+ ATPase pump, laminin, α‐tubulin | No data | Neg inulin, ALB transport, glucose reabsorb, permeability | Glucose reabsorb +/‐ inhib |
|
Vascularized kidney chip Nortis dual‐channeled chip VPT‐MPS |
No data | ZO‐1, Na+‐K+ ATPase pump, OAT1, OAT3, | No data | No data | MRP2, MRP4, OAT1, OAT3, PAH transport |
| Multi‐organ systems | ||||||

|
4‐organ chip‐ kidney component iPSC organoids HuMiX | No data | CK‐18, vimentin, AQP1, Na+‐K+ ATPase pump, ZO‐1, | No data | No data | LDH, KI67, TUNEL |

|
Hesperos 14 organ pumpless chip HK2 cells | No data | No data | No data | No data | No data |

|
Wyss Mulitple chip componet Kidney section |
No data | ZO‐1 | No data |
Albumin reabsorption Inulin uptake |
No data |
Abbreviations: ADME, absorption, distribution, metabolism, and elimination; BSA, body surface area; MPS, microphysiological systems.
Glomerular interface
The glomerulus serves as the first component of the nephron unit that is encountered by a circulating xenobiotic that is destined for renal excretion. Among the design attributes for artificial glomeruli for MPS use is to maintain native physiological structural integrity and the net negative charge formed by the glomerular basement membrane and glycocalyx. The negative charge reduces filtration of anionic substances as it selects which compounds are more likely to be filtered based on charge. Perhaps the most important functional attribute of the glomerulus is its filtering capacity rendered by podocyte‐formed pores that allow macromolecules less than ~ 69 kD to enter the nephron. The complex architecture of glomeruli has been technically challenging to reconstruct in MPS devices as it contains many cell types (endothelial, mesangial, and podocytes) that need to be cocultured and structurally aligned to maintain relevant filtering characteristics. A consortium of industry leaders have compiled guidance for glomerular attributes needed for use in MPS devices if they are to be used for ADME and safety applications. 4 The endothelial cells should have immunohistochemical evidence of Von Willebrand factor (vWf), CD31, VEGFR‐2, and VE‐cadherin proteins; mesangial cells should contain αSMA, fibronectin, and PDGFβ‐R; whereas the podocytes should be nephrin, podocin+, WT1+, and PAX2‐, with demonstrable basement‐membrane collagen IV production. Differential clearance performance is measured with albumin and inulin serving as model compounds for retention and excretion, respectively.
Microphysiological systems device development with glomerular interfaces has been attempted by several groups with considerably different design approaches based on the context of use. Most of these efforts were challenged by the lack of the ability of the cells to form proper sized pores on their own and were aided with membranes that simulated the appropriate filtering size cutoff. A glomerulus‐on‐a‐chip was developed wherein glomerular endothelial cells were grown on top of a membrane separating podocytes cultured on the underside and interrogated with varying flow rates to test barrier permeability. 53 A chip designed to simulate glomerular filtering with the use of filters of varying pore sizes coated with glomerular endothelial cells was fitted on the upstream side of a chip that contained proximal tubule cells. 54 , 55 The use of membrane filters to provide passive filtering of serum proteins before they enter a PTEC channel provides a simple means to simulate molecular sieving in MPS. A glomerular‐intestine hybrid chip was designed with cocultures of primary glomerular endothelial cells, podocytes, and mesangial growing on one side of a porous membrane while intestinal origin cells grew on the other. 56 Drug absorption is tested by exposure of the intestinal cells followed by nephrotoxicity assessment of the underlying glomeruli‐origin cells. A glomerulus chip has been developed with human iPSCs to be directly differentiated into mature kidney podocytes, which can mimic the kidney glomerular capillary wall. 57 The podocyte cells that display many podocyte‐specific markers and develop complex foot processes, are cultured on porous laminin imbedded polydimethylsiloxane (PDMS) membranes serving to simulate the glomerular basement membrane as a barrier to primary glomerular endothelial cell layer on the capillary compartment side. The two‐channel chip is used as a stand‐alone system or can be multiplexed with organ chips using a programmable vacuum regulator system. Nephron organoids derived from human iPSCs have been successfully grown in 2D and 3D cultures to form nephron‐like structures that self‐organize and includes glomerular‐like structures with podocyte phenotypes. 58 This promising cell culture technique is still in early stages of development and efforts are underway to adapt these cultures to MPS that could lead to the incorporation of an appropriate glomerular interface for ADME‐type investigations.
Proximal/distal tubules
The proximal tubule has been by far the most developed segment of the nephron for use in MPS devices. A key site for reabsorption, metabolism, and secretion, it is the most common site for compound‐specific kidney injury and because of this trait has been the focus of many MPS developers. Kidney MPS for ADME investigations have been developed in many different formats, including stand‐alone devices, vascularized stand‐alone devices, stand‐alone devices that can be coupled to other organ chips, kidney‐intestinal devices, and multiplexed devices interconnected with many other organ systems.
Kidney tubule MPS devices have been the major focus of many device developers driven partly by industry and regulatory needs, but also by ADME scientists who desire an in vitro investigative tool that has fully functioning transporters, metabolic enzymes, and cell morphology to accomplish the task. MPS leaders have identified critical baseline guidelines for ADME investigators who work in the kidney tubule MPS field to ensure that their systems are functional within their intended context of use and what developers can expect to characterize for general acceptance use. 4 , 5 It is not practical to test every parameter listed; however, key components should be identified that are integral parts of the ADME question at hand and confirmed in the MPS to establish that the device is mechanistically qualified to perform the desired ADME function. Criteria components are met by either the confirmation of presence and location of the ADME‐related protein in cells by immunohistochemistry, baseline mRNA transcript confirmation, and a battery of functional assays.
Because the proximal tubule contains numerous important transporters, at the minimum, the activity of OAT1/3 and OCT2 on the basolateral side and P‐gp, BCRP, MRP2, and MATE on the apical surface should be demonstrated as well as their cellular positions by IHC staining. In addition, functional transport by megalin/cubilin, SGLT2 glucose transporters, and OATs 1–4 would support the characterization of critical ADME transporters. IHC of PTEC phenotypic markers should identity the presence and/or location of ZO‐1, LTL, acetylated tubulin, ezrin, aquaporin 1, and the basolateral Na+‐K+ ATPase pump. Baseline mRNA characterization should confirm the presence of AQP1, APT1A1, CDH2, SLC5A2, TJP1, Gpx3, SLC22A7, and SLC22A13. Function‐specific assays should include reabsorption of 2‐NDBG and fluorescently tagged dextran, inulin, or albumin, and the negative re‐absorption of inulin. In addition, demonstration of vitamin D bioactivation and GGT, alkaline phosphatase, leucine aminopeptidase, and ACE activities will confirm PTEC functionality. Functional changes that influence ADME parameters, many safety‐related, should demonstrate measurable changes in extracellular KIM‐1 and αGST concentrations, transporter inhibition with appropriate inhibitors, intracellular Ca+ changes, and loss of the brush border. Assays that measure general cell health, such as fluorescent live/dead stains with calcein AM and ethidium homodimer‐1 and kits that stain for apoptotic cells, are commonly used in the MPS field. Distal tubule cells are more problematic to culture as they quickly lose phenotypic markers that distinguish them from proximal tubule cells.
Stand‐alone proximal tubule chips
There are many designs for stand‐alone kidney chips or kidney chips coupled to other organ chips. Because primary PTEC cells are capable of propagating and will polarize and exhibit metabolic and transporter functions under flow, they are commonly used to populate kidney MPS. The most common format for PTEC MPS cultures is sandwich cultures housed within a chip where PTECs (or equivalent) are grown, under flow, on ECM‐coated porous membranes with channels on either side to allow sampling from the apical and basal membrane sides. 59 The sandwich style format also allows for the culture of other cell types adjacent to the basolateral region of the PTECs either with endothelial cells to simulate exchanges with the vasculature or with intestinal or hepatic cells. The use of sandwich culture in an MPS device under flow has been implemented with MDCK cell, 60 primary PTECs, 59 , 61 and with a glomerular interface consisting of an upstream porous membrane. 54
The MPS chip by Nortis Bio has a cell culture format that mimics a renal tubular environment as PTEC cells adhere to the wall of a 120 μm wide cylindrical channel that traverses a collagen‐filled compartment. 62 , 63 , 64 Under flow provided by syringe or pneumatic pumps, the PTECs polarize and retain many of the phenotypic characteristics necessary for ADME studies. The Nortis chip has the capability of linkage to other Nortis chips that contain cells from other organ systems as was demonstrated with a liver:kidney linked system used to investigate the toxic effects of hepatic aristolochic acid metabolism on nephrotoxicity. 34 The Nortis kidney chip system was evaluated by a National Center for Advancing Translational Sciences sponsored independent MPS testing laboratory (Texas A&M and MIT) for robustness and reproducibility and the MPS technology was found to be successfully transferable with the greatest source of variability being the cell source. 65 , 66
The Emulate kidney chip is a multilayer microfluidic device that can accommodate sandwich‐style seeded cells while operated by a syringe pump. 61 Proximal tubule cells seeded on ECM‐coated porous membranes allows for access to the interstitial fluid compartment beneath the basolateral side of the PTECs. Another type of kidney chip that uses a sandwich‐type configuration with an upper and lower channel separated by a ECM‐coated porous membrane was fitted with a filter with a molecular weight cutoff similar to the glomerulus that was coated with HUVEC cells and glomerular basal membrane extracellular matrix proteins to serve as a surrogate glomerulus. 54 Passive filtration of “serum” proteins in the perfused media provides a more realistic exposure scenario to the proximal tubule cells.
The three‐channel OrganoPlate chip (Mimetas) platform, which has 40 separate microfluidic cultures and uses a rocker platform to simulate flow, was used to culture primary PTECs that formed confluent polarized tubules and displayed SGLT2 and MRP2/4 efflux activity. 67 The same OrganoPlate chip system was used to culture kidney tubuloids derived from PSCs isolated from patient’s urine that differentiated into proximal and distal tubule cells. 68 The kidney tubuloids also displayed epithelial transport functions and correctly positioned phenotypic markers on the apical and basolateral sides of the polarized cells.
Kidney MPS are included in several multi‐organ MPS systems as prototypes for human on a chip system designs that allow connectivity of multiple organs simultaneously. These systems are extremely complex as they contain multiple cell types that require organ‐specific media, differential oxygenation, and microarchitecture to mimic specific physiological environments.
The Wyss Institute has designed a custom MPS system with eight vascularized, two‐channeled organ chips (intestine, liver, kidney, heart, lung, skin, blood‐brain barrier, and brain) that allows for robotic control of fluid flow, sampling and in situ microscopy. 69 The kidney subunit in this system uses a semipermeable membrane barrier to separate a layer of kidney‐derived endothelial cells on the lower channel with an upper channel lined with proximal tubule cells. Albumin re‐absorption and barrier integrity was demonstrated with inulin.
The iPSCs were used to create a four‐organ‐chip (intestine, liver, brain, and kidney) organoid system with autologous cells that allows for organ crosstalk with the kidney tubule equivalent outfitted with a glomerular unit designed to house podocytes on one side of a membrane with endothelial cells growing on the other side. 52 The kidney organoids are a mixture of epithelial and mesenchymal cells and the presence of transporter Na+/K+‐ATPase in the cell membranes of select cells was demonstrated.
A pumpless 14‐compartment MPS body‐on‐a‐chip was created to simulate fluid exchanges between organ compartments and uses solid and barrier tissue gaskets that serve to isolate certain organs and to provide a means to have direct access to select tissues. 70 Fluid motion is created by a rocking platform and the kidney subunit was represented by HK2 cells.
Vascularized kidney chips
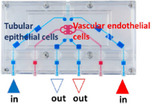
Kidney MPS devices come in many formats and utilize different cell types with some more conducive to ADME work than others. Stand‐alone vascularized kidney chips allow the study of tubular‐vascular exchanges within closed perfusion systems. Two types of vascularized kidney chips with adjacent independent parallel channels containing either PTECs or endothelial cells model the capillary filled environment of the proximal tubule and can measure the transport of compounds between compartments. Epithelium‐endothelium crosstalk is capable of being studied with a 3D vascularized proximal tubule by the Wyss Institute that features a colocalized vascular channel with a proximal tubule. 71 Using a Nortis manufactured dual‐channel chip (VPT‐MPS), with two parallel proximal tubule and vascular channels separated by 1 mm, Chapron et al. demonstrated basolateral uptake from the endothelial channel into the PTEC channel. 72
Hollow‐fiber culture techniques
Bioartificial kidneys with hollow‐fiber membranes cultured with renal epithelial cells that maintain transporter function provide a means to treat patients with advanced kidney disease and were not originally designed to be used for ADME research; however, scaled‐down versions could serve as an MPS. Cultures of renal epithelial cells in hollow fiber membranes demonstrate differential transport of organic cation compounds with scaled up versions of this format intended to serve to treat patients with uremic syndrome. 73 , 74 , 75 The device demonstrates OCT2 and OAT1 transporter functions with correct cellular localization. A similar bioartificial kidney is under development at the University of Michigan to treat patients with acute kidney failure.
Organoid culture kidney chips
Advances in kidney organoid cultures from induced pluripotent stem cells have led to the publication of many promising proof‐of‐concept studies that have or can be adapted to MPS chip formats. Proximal and distal tubuloid cultures cultured and grown in sandwich culture within an MPS have evidence of transporter function. 68 A four‐organ MPS with autologous differentiated stem cells for intestine, liver, kidneys, and neurons provides a means to test organ crosstalk with cells from the same patient. 52 Organoids that self‐organize into continuous segmental arrangements resemble nephrons that include podocytes and cells from the Loop of Henle. 58
Kidney scaffold systems
Kidney‐derived micro‐scaffolds are created by the decellularization of rat kidney sections followed by the seeding of HK‐2 cells onto the 3‐D proteinaceous kidney skeleton that can lead to the formation of glomerular and tubule sections. 76 , 77 Cells cultured in the micro‐scaffold demonstrated higher expression of key ADME genes compared to their 2‐D plastic grown counterparts and released a wide array of cytokines upon stimulation. Proximal tubule fragment cultures are formed from harvested intact murine proximal tubules that are enzymatically isolated from surrounding tissue then cultured in 3D ECM gels while retaining many proximal tubule transporter activity and RNA transcript characteristics. 78 These tubule fragments are cultured in 96‐well plates and successfully demonstrated OAT1, OAT3, and OCT2 activity with fluorescent dyes.
INTESTINAL MPS
There has been a rapid evolution of intestinal MPS in the last 5 years that has led to systems that are more complex and with increased utility for ADME investigations. The major challenge for MPS developers has been to achieve a gut‐lining model that functions metabolically with relevant transporter functions. Most early intestinal MPS designs used Caco‐2 cells monolayers to represent the gut epithelia as this cell line can evaluate the absorption capabilities of orally administered drugs. The absorptive properties of drugs are important determinant for bioavailability properties of orally administered drugs therefore the integrity of the epithelial wall must be robust enough to allow for meaningful absorptive property data. Caco‐2 cells can achieve tight junctions; however, permeability is not the only ADME parameter that needs to be evaluated at the gut‐lining interface.
Changes in Caco‐2 culture conditions, made more achievable with MPS designs, can result in cells that are more phenotypically relevant for ADME studies. Traditionally, monolayers of Caco‐2 cells are the accepted in vitro model for the small intestine, but they lack the cytoarchitecture and robust metabolic/transporter protein profiles needed to predict human responses. The use of Caco‐2 cells in the MPS bridges a more physiologically relevant environment by which cells respond forming a polarized columnar sheet with enhanced metabolic and transporter activities compared to monolayer counterparts. Although Caco‐2 and MDCK cells transfected to overexpress transporters are currently used to estimate human absorptive properties, they do not have the full complement of metabolic activity or transporter function necessary to address all ADME‐relevant functions. In addition, the gut lining is comprised of multiple cell types (goblet, enteroendocrine, and Paneth cells) as well as immune cells (PBMC) that can influence tissue function and provide a more realistic environment to study ADME properties. Most intestinal MPS systems with Caco‐2 cells have two independent channels that when under flow, simulate fluid movements within the gut lining and blood flow in the underlying vasculature. This design allows for access and sampling of the intestinal epithelial channel (Caco‐2, iPSCs, and PBMCs, +/‐ microbiota), and the vascular endothelial channel (HUVECs and HIMECs), which are separated by a porous membrane coated with ECM constituents. Most cell types in these systems, as with other organs in MPS, respond favorably to flow conditions and maintain phenotypes that are more conducive for ADME study.
In addition to permeability assessments, there is a need to evaluate facilitated absorption and first pass metabolism effects, as most drugs need to be highly absorbed with minimal first pass metabolism to be efficacious. As with the liver and kidneys, leaders in intestinal MPS development have compiled recommended baseline characteristics for intestinal MPS use in ADME work. 1 , 5 , 79 To that end, MPS should have demonstrable carboxylesterase 2 (CES2), CYP3A4, and UGT activities, whereas PEPT1, OATP2B1, P‐gp, and BCRP activities would also add value. Baseline characterization of an intestinal MPS should include a TEER value of 50–100 Ω cm2 and should be viable for up to a week with access to apical and basolateral regions for sampling purposes. 5 In some cases, the evaluation of TEER values is not possible due to MPS designs or with the type of cell format used (e.g., organoids).
Compounds used to measure and characterize first pass metabolism within intestinal MPS should include those that test different sets of important metabolic enzymes and drug transporters. Metabolic activity is necessary to capture first pass metabolism effects and CYP3A4 activity tested with compounds with relatively low, medium, and high substrate activity, such as lovastatin, midazolam, and repaglinide, respectively, are recommended as test compounds for these measures. UGT activity can be evaluated with naloxifene and CES2 activity with irinotecan. P‐gp and BCRP activities are desirable and rosuvastatin and topotecan exposure can be used to test their activities, respectively.
Compared to liver and kidney MPS, characterization of the metabolic enzyme and drug transporters within intestinal MPS devices is not as far along with respect to first‐pass metabolism functions with test sets of drugs with known wide ranges of intestinal availability (Fg). This is due, in part, to the limitations of systems that solely use Caco‐2 cells to represent the intestinal lining as these cells have limited metabolic and transporter activity capabilities even when subjected to flow and with an adjacent underlying endothelial cell layer. Recent advances in intestinal MPS design shows some promise for filling in this gap but these systems are complex and require multiple cell types and/or introduced mechanical forces to maintain phenotypes. Table 5 lists intestinal MPS, either as a single organ or incorporated in multi‐organ systems, reported in the literature with identification of any desired performance criteria suggested by Fowler et al.
TABLE 5.
Comparison of intestinal MPS systems for acceptable MPS performance criteria that meet ADME standards based on recommendations from MPS experts 5
| Microphysiological technologies used to model the intestinal microenvironment 5 | ||||||
|---|---|---|---|---|---|---|
| System | Cell types used |
Barrier function TEER 50–100 Ω m2 |
Morphology Appropriate cellular positions of key transporters |
Transit for CES2 (irinotecan) CYP3A4, lovastatin, midazolam & repaglinide UGT (raloxifene) substrates | Transporters P‐gp (digoxin), rosuvastatin ortopotecan (BCRP), PEPT1, OATP2B1 | |
| Stand‐Alone Intestinal MPS | ||||||
|
Microfluidic gut on chip | Caco2 cells |
Not done Difficult to measure in device‐used fluorescent dye |
Villi scaffold | Aminopeptidase and cytochrome P450 (CYP) 3A4 | No data |

|
Microbiome gut chip |
Caco2 cells 8 bacteria strains PBMCs |
3000–4500 Ω cm2 | F‐actin (microvilli) | No data | No data |
|
Microbiome gut chip |
HeLa S3 2 bacterial strains |
Not done | No data | No data | No data |

|
Gut chip with microbes HuMiX |
Caco2 2 bacterial strains |
1000 Ω cm2 | occludin | No data | No data |

|
Complex microbiome gut chip Emulate chip |
Caco2, HIMEC, 11 bacterial genera |
Permeability dye | Villin, ZO‐1, Factin, Muc2 | No data | No data |

|
3D immunocompetent int chip MOTiF chip |
Caco2, HUVECs, PBMCs 2 bacterial strains |
Not done Difficult to measure in device‐used fluorescent dye |
Villin, ZO‐1, occluding, b‐catenin, CYP3A4, a defensing, mucin‐2, E‐cadherin, vWf, f‐Actin | No data |
Gene expression‐ (MDR1, BCRP, MRP2, MRP3, PepT1, OATP2B1, OCT1, SLC40A1) |
|
Emulate Gut Chip Intestinal organoids |
primary human intestinal epithelium and HIMECS |
F‐actin, Muc5AC, Integrin b4, Na/K‐ATPase, NHE3, villin, E‐cadherin, ZO‐1, VE‐cadherin, chromogranin A, lysozyme, Sucrase activity, MUC2, MDR1, BCRP, PEPT1 |
CYP3A4 induction CYP3A4 metabolism‐ testosterone, VitD3 |
MDR1 efflux assay | |

|
iPSCs derived organoids | iPSCs ‐> Paneth cells, goblet cells, enterocytes, and enteroendocrine cells | Not characterized |
ZO‐1, E‐cadherin, vimentin, CDX2, villin, MUC2, LYZ, FABP2, CHGA, LGR5, WDR43 |
No data |
IDO1 and GBP1 induced by IFNg PLA2G2a, MUC4, LYZ |
|
Xenogenic‐free organoid system | Organoids | No data | IHC ‐Villin, LGR5, CDX2, ECAD, CGA, MUC2, DEFA6, SMA, PGP9.5, serotonin, EGFP, CFTR, actin |
CFTR‐FIS assay Dipeptide uptake |
ECAD, CDX2, villin, LGR5, SOX9, CGA, lactase, SMA, CD34, CKIT, PGP9.5, lysozyme, PEPT1, ABCB1, ABCG2 |
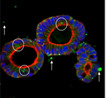
|
Rat Duodenal organoids | Organoids | No data | F‐actin, Muc2, lysozyme | Cyp3a1 midazolam, Cyp2j3 astemizole, Ces2 irinotecan, Ugt1a1 7‐hydroxycoumarin, Sn−38 | |
| Kidney‐Gut MPS HepaRG/ | ||||||
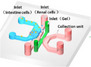
|
Glomerulus‐Intestine chip |
Caco2 | Max 1400 Ω cm2 | ZO‐1 | ALP activity | P‐gP:Digoxin, verapamil, colestyramin |
| Liver‐Gut MPS | ||||||
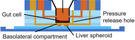
|
Liver‐gut MPS Modified transwell |
HepG2/Caco‐2 | No data | Morphology: F‐actin | Paracetamol | No data |
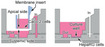
|
Liver‐Intestinal chip |
Caco‐2/ | 383–1132 Ω cm2 | No data | CYP3A‐ triazolam | No data |
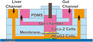
|
Gut‐liver chip | Caco‐2/HepaRG | fluorescein | Morphology: F‐actin | Apigenin – sulfation and glucuronide conjugates | No data |

|
Liver‐gut MPS GuMi Griffith |
Goblet cells/hepatocytes‐Kupffer cells/Caco2 | 450 Ω cm2 | Morphology | Diclofenac, hydrocortisone | No data |
| Multi‐organ systems | ||||||

|
4‐organ chip‐ kidney component TissUse |
iPSC organoids | No data | Morphology:CDX2, Na+/K+‐ATPase, Cytokeratin 8/18 Vimentin, Zo‐1 | No data | No data |
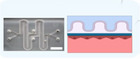
|
Wyss multi organ | Caco‐2/HUVEC/ | Inulin leakage | ZO‐1 | No data | No data |

|
Hesperos | Caco‐2 | No data | No data | No data | No data |
Abbreviations: ADME, absorption, distribution, metabolism, and elimination; iPSC, induced pluripotent stem cell; MPS, microphysiological systems.
A common feature used by most intestinal MPS devices is the incorporation of an underlying channel of endothelial cells to serve as the blood vasculature compartment. A modification of this design is a kidney‐intestinal MPS hybrid chip where a layer of Caco‐2 cells and primary glomerular cells are separated by a porous membrane that allows compounds and metabolites formed by compound exposed Caco‐2 cells to interact with the glomerular cells. 56 A more common hybrid intestinal chip design type is the intestinal‐liver chip with Caco‐2 cells as the gut lining with either HepaRG cells 80 , 81 , 82 or primary hepatocytes mixed with Kupffer cells serving to represent the liver. 83 , 84 These hybrid gut‐liver systems allow for interrogation of inter‐MPS communication that is dependent on metabolic and transporter functions of each organ system.
An intestinal MPS component is part of several multi‐organ systems ranging from the four organ TissUse system (intestine, liver, brain, and kidney) that uses iPSCs for each organ component 52 to the Wyss Institute’s eight vascularized‐organ system that uses robotic fluid coupling to interconnect and sample from each organ chip. 69 Whereas a pumpless 14 compartment MPS uses Caco‐2 cells to serve as the intestinal organ and uses a rocking motion to simulate fluid flow. 70 These complex multi‐organ systems are attempts at creating a human on a chip and the desired performance criteria for the intestinal component applies to these as well. Two organ MPS systems with intestinal‐liver combinations have also used to model first pass metabolism effects with these devices designed to accommodate microbiomes to incorporate their absorptive and metabolic influencing properties into the model.
Multicellular intestinal MPS and chips with microbiomes
Mechanical forces on PTECs and hepatic cells caused by shear stress with perfused media results in augmented cell polarization and morphology characteristics that more closely resemble in vivo organs. This phenomenon also occurs with human Caco‐2 epithelial cells as the application of mechanical strain, which simulates peristaltic motion results in the formation of intestinal villi with cell differentiation into goblet, enteroendocrine, Paneth, and absorptive cells within villi crypt regions. 85 With these four cell types in place in one chamber and an underlying chamber with endothelial cells, there is measurable mucus formation and increases in specific CYP3A4 activity localized to the villi. The introduction of microbiomes to this complex model demonstrated how bacterial growth rates were associated with peristaltic motion and that inflammatory states could be triggered by the presence of pathogenic bacteria and PBMCs. 86
The introduction of microbiomes into intestinal MPS chips introduces an important variable to consider for ADME characteristic evaluation as the microbiome can directly affect drug pharmacokinetics and efficacy through biotransformation processes. 86 , 87 , 88 Microbiota‐drug interactions with specific bacteria species may alter first pass metabolism profiles and microbiome seeded MPS chips would be able to capture this important interface and provide a more physiologically relevant scenario for studying intestinal absorption.
Intestinal organoids
Another approach for studying intestinal absorption and first pass metabolism in MPS is the use of intestinal organoids to serve as the representative intestinal tissue compartment. Intestinal organoids, derived from human biopsy samples 89 , 90 or induced iPSCs, 91 can represent all regions of the intestine, have four major intestinal epithelial subtypes, and possess improved CYP3A4 activity and induction compared to Caco‐2 cells. Access to the organoid lumens presents a challenge but was overcome with the disassociation and seeding of intestinal organoids into a chip containing a vascular channel that, in combination with introduced cyclic mechanical strain, developed a microvilli surface with multiple cell types and measurable MDR1 transport function. 90 Fine‐tuning of iPSC differentiating protocols along with a patterned substrate resulted in the formation of gut organoids with cells from three germ‐layer lineages that exhibited gut‐like peristalsis and transporter function. 92 Successful development of rat intestinal organoids by Hedrich et al. from duodenum stem cells provides a means to investigate cross species ADME investigations with a system that exhibits phase I and II metabolism and transporter functions. 93
FUTURE DIRECTIONS
Although much progress has been made in implementing conditions that favor ADME‐related phenotypes in MPS cultures, there are still gaps to be filled that would pave the way for general acceptance of the MPS to provide meaningful data for ADME sciences. One of the goals of the MPS is to recapitulate the physiological function of an intact organ that retains a physiologically relevant phenotype to test and understand exposures to endogenous and xenobiotic compounds, however, there needs to be a standardization of methodologies and a consensus of expected performance criteria for its broader acceptance in the field.
The ultimate goal for MPS technologies is to complete the 3R’s of toxicology (reduce/refine/replace) research entailing animals. The MPS has shown some promise for drug safety testing and has the potential to reduce and/or replace animal use in safety assessment studies. Once fully validated, the MPS may serve as a tool for drug screening, allowing cross‐referencing of animal‐based MPS data with in vivo animal data that can be further translated to human‐based MPS data and clinical outcomes. It is worth noting that DARPA funded two groups at Harvard/Wyss Institute and MIT to create a “human on a chip” within 5 years (https://www.darpa.mil/program/microphysiological‐systems). This program ran in partnership with NCATS and resulted in many successes. However, the goal of a “human on a chip” was not achieved. Why is that the case? In closing, the reasons are multifarious but, suffice it to say, that each individual organ chip system is quite complex and linking them is an even greater challenge. A linked system requires the establishment of appropriate oxygen gradients in each organ, maintained sterility, physiologically relevant microarchitectures throughout, appropriate organ‐specific flow rates, maintaining simultaneous cocultures of all possible organ cell types, including immune cells and sampling accessibility. Cell sourcing ranges with established cell lines such as the Caco‐2 or HepG2 cell lines gives consistent performance but they have their limitations as far as ADME capabilities. Of great importance for establishing the robustness of the MPS to support ADME studies is the access to reliable and diverse sources of cells to seed the MPS. Sources for commercial primary hepatocytes are far more abundant than for kidney and gastrointestinal cells as good performing cryopreserved hepatocytes from a number of donors and species are currently available allowing for investigations of donor and genetic background variability, whereas commercially available sources of gut and kidney primary cells are limited. Equally challenging is optimization of primary cell culture media so that primary cells from different species can be successfully cultured using the same media. Currently, primary cell culture media needs to be supplemented with species‐specific factors to maintain desirable phenotypes and differences in these factors can be substantial as some cells require serum while others are fine without it. Primary cells that proliferate, such as kidney epithelial cells, tend to transition into mesenchymal cells after a few passages and can quickly lose their characteristic PTEC phenotypes that includes ADME attributes. The ability of epithelial cells to transition to mesenchymal cells is variable and donor‐dependent and thus, it is important to carefully monitor PTEC cultures for transitional morphologies before seeding kidney chips. The iPSCs cultured into various organoids provides users cells from the same donor but requires complicated cell culture manipulations to achieve the desired cell maturation phenotypes. There is also the consideration of high costs and labor/resource commitments for these very advanced cell culture methods. There have been a number of groups, including ours, that have successfully linked several organ chip systems. However, including all the requirements listed above, to achieve a fully integrated human on a chip system will necessitate vascularization of the individual components and creation of a “universal media,” which supports proper physiological and ADME function.
CONFLICT OF INTEREST
Drs. Kelly and Yeung are consultants for Nortis, Inc., Woodinville, WA. All other authors declared no competing interests for this work.
AUTHOR CONTRIBUTIONS
All authors wrote the manuscript.
Supporting information
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Van Ness KP, Cesar F, Yeung CK, Himmelfarb J, Kelly EJ. Microphysiological systems in absorption, distribution, metabolism, and elimination sciences. Clin Transl Sci. 2022;15:9–42. 10.1111/cts.13132
[Correction added on 9 September 2021, after first online publication: adsorption has been corrected to absorption through the article.]
Funding information
NCATS: Jonathan Himmelfarb UG3TR002158; NCATS: Jonathan Himmelfarb UG3TR003288; NCATS: Jonathan Himmelfarb UG3TR003288 NIGMS: Catherine (K) Yeung R01GM121354; NIEHS: Edward (J) Kelly P30ES007033; NIEHS: Edward (J) Kelly R21ES031359. Also supported by an unrestricted gift from the Northwest Kidney Centers to the Kidney Research Institute.
REFERENCES
- 1. Ishida S. Organs‐on‐a‐chip: Current applications and consideration points for in vitro ADME‐Tox studies. Drug Metab Pharmacokinet. 2018;33(1):49‐54. [DOI] [PubMed] [Google Scholar]
- 2. Caetano‐Pinto P, Stahl SH. Perspective on the application of microphysiological systems to drug transporter studies. Drug Metab Dispos. 2018;46(11):1647‐1657. [DOI] [PubMed] [Google Scholar]
- 3. Baudy AR, Otieno MA, Hewitt P, et al. Liver microphysiological systems development guidelines for safety risk assessment in the pharmaceutical industry. Lab Chip. 2020;20(2):215‐225. [DOI] [PubMed] [Google Scholar]
- 4. Phillips JA, Grandhi TSP, Davis M, et al. A pharmaceutical industry perspective on microphysiological kidney systems for evaluation of safety for new therapies. Lab Chip. 2020;20(3):468‐476. [DOI] [PubMed] [Google Scholar]
- 5. Fowler S, Chen WLK, Duignan D, et al. Microphysiological systems for ADME‐related applications: current status and recommendations for system development and characterization. Lab Chip. 2020;20(3):446‐467. [DOI] [PubMed] [Google Scholar]
- 6. US Food and Drug Administration . In Vitro Drug Interaction Studies — Cytochrome P450 Enzyme‐ and Transporter‐Mediated Drug Interactions Guidance for Industry. U.S. Department of Health and Human Services, Editor. Center for Drug Evaluation and Research (CDER); 2020. https://www.fda.gov/regulatory‐information/search‐fda‐guidance‐documents/vitro‐drug‐interaction‐studies‐cytochrome‐p450‐enzyme‐and‐transporter‐mediated‐drug‐interactions. [Google Scholar]
- 7. Rubiano A, Indapurkar A, Yokosawa R, et al. Characterizing the reproducibility in using a liver microphysiological system for assaying drug toxicity, metabolism and accumulation. Clin Transl Sci. 2021;14:1049‐1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jang K‐J, Otieno MA, Ronxhi J, et al. Reproducing human and cross‐species drug toxicities using a Liver‐Chip. Sci Transl Med. 2019;11(517):eaax5516. [DOI] [PubMed] [Google Scholar]
- 9. Kostadinova R, Boess F, Applegate D, et al. A long‐term three dimensional liver co‐culture system for improved prediction of clinically relevant drug‐induced hepatotoxicity. Toxicol Appl Pharmacol. 2013;268(1):1‐16. [DOI] [PubMed] [Google Scholar]
- 10. Li AP. Evaluation of adverse drug properties with cryopreserved human hepatocytes and the integrated discrete multiple organ co‐culture (IdMOC(TM)) system. Toxicol Res. 2015;31(2):137‐149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Novik E, Maguire TJ, Chao P, Cheng KC, Yarmush ML. A microfluidic hepatic coculture platform for cell‐based drug metabolism studies. Biochem Pharmacol. 2010;79(7):1036‐1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hultman I, Vedin C, Abrahamsson A, Winiwarter S, Darnell M. Use of HμREL human coculture system for prediction of intrinsic clearance and metabolite formation for slowly metabolized compounds. Mol Pharm. 2016;13(8):2796‐2807. [DOI] [PubMed] [Google Scholar]
- 13. Novik EI, Dwyer J, Morelli JK, et al. Long‐enduring primary hepatocyte‐based co‐cultures improve prediction of hepatotoxicity. Toxicol Appl Pharmacol. 2017;336:20‐30. [DOI] [PubMed] [Google Scholar]
- 14. Zeilinger K, Schreiter T, Darnell M, et al. Scaling down of a clinical three‐dimensional perfusion multicompartment hollow fiber liver bioreactor developed for extracorporeal liver support to an analytical scale device useful for hepatic pharmacological in vitro studies. Tissue Eng Part C Methods. 2011;17(5):549‐556. [DOI] [PubMed] [Google Scholar]
- 15. Hoffmann SA, Müller‐Vieira U, Biemel K, et al. Analysis of drug metabolism activities in a miniaturized liver cell bioreactor for use in pharmacological studies. Biotechnol Bioeng. 2012;109(12):3172‐3181. [DOI] [PubMed] [Google Scholar]
- 16. Gerlach JC, Mutig K, Sauer IM, et al. Use of primary human liver cells originating from discarded grafts in a bioreactor for liver support therapy and the prospects of culturing adult liver stem cells in bioreactors: a morphologic study. Transplantation. 2003;76(5):781‐786. [DOI] [PubMed] [Google Scholar]
- 17. Nguyen DG, Funk J, Robbins JB, et al. Bioprinted 3D primary liver tissues allow assessment of organ‐Level response to clinical drug induced toxicity in vitro. PLoS One. 2016;11(7):e0158674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Davidson MD, Pickrell J, Khetani SR. Physiologically inspired culture medium prolongs the lifetime and insulin sensitivity of human hepatocytes in micropatterned co‐cultures. Toxicology. 2021;449:152662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lee PJ, Hung PJ, Lee LP. An artificial liver sinusoid with a microfluidic endothelial‐like barrier for primary hepatocyte culture. Biotechnol Bioeng. 2007;97(5):1340‐1346. [DOI] [PubMed] [Google Scholar]
- 20. Toh YC, Lim TC, Tai D, et al. A microfluidic 3D hepatocyte chip for drug toxicity testing. Lab Chip. 2009;9(14):2026‐2035. [DOI] [PubMed] [Google Scholar]
- 21. Crogan‐Grundy C, Smith TR, Smith RC, Hardwick RN, Nguyen DG. Utilization of the ExVive human liver tissue model to assess drug‐induced liver injury across a diverse set of chemical classes, in Society of Toxicology, Organovo, Editor. Organovo; 2017. [Google Scholar]
- 22. Norona LM, Nguyen DG, Gerber DA, Presnell SC, LeCluyse EL. Editor's highlight: modeling compound‐induced fibrogenesis in vitro using three‐dimensional bioprinted human liver tissues. Toxicol Sci. 2016;154(2):354‐367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bale SS, Manoppo A, Thompson R, et al. A thermoplastic microfluidic microphysiological system to recapitulate hepatic function and multicellular interactions. Biotechnol Bioeng. 2019;116(12):3409‐3420. [DOI] [PubMed] [Google Scholar]
- 24. Bale SS, Borenstein JT. Microfluidic cell culture platforms to capture hepatic physiology and complex cellular interactions. Drug Metab Dispos. 2018;46(11):1638‐1646. [DOI] [PubMed] [Google Scholar]
- 25. Tan K, Keegan P, Rogers M, et al. A high‐throughput microfluidic microphysiological system (PREDICT‐96) to recapitulate hepatocyte function in dynamic, re‐circulating flow conditions. Lab Chip. 2019;19(9):1556‐1566. [DOI] [PubMed] [Google Scholar]
- 26. Khetani SR, Kanchagar C, Ukairo O, et al. Use of micropatterned cocultures to detect compounds that cause drug‐induced liver injury in humans. Toxicol Sci. 2013;132(1):107‐117. [DOI] [PubMed] [Google Scholar]
- 27. Bell CC, Hendriks DF, Moro SM, et al. Characterization of primary human hepatocyte spheroids as a model system for drug‐induced liver injury, liver function and disease. Sci Rep. 2016;6:25187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bell CC, Dankers ACA, Lauschke VM, et al. Comparison of hepatic 2D sandwich cultures and 3D spheroids for long‐term toxicity applications: a multicenter study. Toxicol Sci. 2018;162(2):655‐666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bell CC, Chouhan B, Andersson LC, et al. Functionality of primary hepatic non‐parenchymal cells in a 3D spheroid model and contribution to acetaminophen hepatotoxicity. Arch Toxicol. 2020;94(4):1251‐1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lohasz C, Bonanini F, Hoelting L, Renggli K, Frey O, Hierlemann A. Predicting metabolism‐related drug‐drug interactions using a microphysiological multitissue system. Advanced Biosystems. 2020;4(11):2000079. [DOI] [PubMed] [Google Scholar]
- 31. Nudischer R, Renggli K, Hierlemann A, Roth AB, Bertinetti‐Lapatki C. Characterization of a long‐term mouse primary liver 3D tissue model recapitulating innate‐immune responses and drug‐induced liver toxicity. PLoS One. 2020;15(7):e0235745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tikunov AP, Shim Y‐S, Bhattarai N, et al. Dose‐response in a high density three‐dimensional liver device with real‐time bioenergetic and metabolic flux quantification. Toxicol In Vitro. 2017;45(Pt 1):119‐127. [DOI] [PubMed] [Google Scholar]
- 33. Kizawa H, Nagao Eri, Shimamura M, Zhang G, Torii H. Scaffold‐free 3D bio‐printed human liver tissue stably maintains metabolic functions useful for drug discovery. Biochem Biophys Rep. 2017;10:186‐191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chang SY, Weber EJ, Sidorenko VS, et al. Human liver‐kidney model elucidates the mechanisms of aristolochic acid nephrotoxicity. JCI Insight. 2017;2(22):e95978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Vernetti LA, Senutovitch N, Boltz R, et al. A human liver microphysiology platform for investigating physiology, drug safety, and disease models. Exp Biol Med (Maywood). 2016;241(1):101‐114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lee‐Montiel FT, George SM, Gough AH, et al. Control of oxygen tension recapitulates zone‐specific functions in human liver microphysiology systems. Exp Biol Med (Maywood). 2017;242(16):1617‐1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sakolish C, Reese CE, Luo Y‐S, et al. Analysis of reproducibility and robustness of a human microfluidic four‐cell liver acinus microphysiology system (LAMPS). Toxicology. 2021;448:152651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Busche M, Tomilova O, Schütte J, et al. HepaChip‐MP ‐ a twenty‐four chamber microplate for a continuously perfused liver coculture model. Lab Chip. 2020;20(16):2911‐2926. [DOI] [PubMed] [Google Scholar]
- 39. Vivares A, Salle‐Lefort S, Arabeyre‐Fabre C, et al. Morphological behaviour and metabolic capacity of cryopreserved human primary hepatocytes cultivated in a perfused multiwell device. Xenobiotica. 2015;45(1):29‐44. [DOI] [PubMed] [Google Scholar]
- 40. Long TJ, Cosgrove PA, Dunn RT, et al. Modeling therapeutic antibody‐small molecule drug‐drug interactions using a three‐dimensional perfusable human liver coculture platform. Drug Metab Dispos. 2016;44(12):1940‐1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kostrzewski T, Cornforth T, Snow SA, et al. Three‐dimensional perfused human in vitro model of non‐alcoholic fatty liver disease. World J Gastroenterol. 2017;23(2):204‐215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hughes DJ, Kostrzewski T, Sceats EL. Opportunities and challenges in the wider adoption of liver and interconnected microphysiological systems. Exp Biol Med (Maywood). 2017;242(16):1593‐1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sarkar U, Ravindra KC, Large E, et al. Integrated assessment of diclofenac biotransformation, pharmacokinetics, and omics‐based toxicity in a three‐dimensional human liver‐immunocompetent coculture system. Drug Metab Dispos. 2017;45(7):855‐866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Prodanov L, Jindal R, Bale SS, et al. Long‐term maintenance of a microfluidic 3D human liver sinusoid. Biotechnol Bioeng. 2016;113(1):241‐246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dash A, Simmers MB, Deering TG, et al. Hemodynamic flow improves rat hepatocyte morphology, function, and metabolic activity in vitro. Am J Physiol Cell Physiol. 2013;304(11):C1053‐C1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Feaver RE, Cole BK, Lawson MJ, et al. Development of an in vitro human liver system for interrogating nonalcoholic steatohepatitis. JCI Insight. 2016;1(20):e90954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Banaeiyan AA, Theobald J, Paukštyte J, Wölfl S, Adiels Caroline B, Goksör M. Design and fabrication of a scalable liver‐lobule‐on‐a‐chip microphysiological platform. Biofabrication. 2017;9(1):015014. [DOI] [PubMed] [Google Scholar]
- 48. Yu F, Deng R, Hao Tong W, et al. A perfusion incubator liver chip for 3D cell culture with application on chronic hepatotoxicity testing. Sci Rep. 2017;7(1):14528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lee‐Montiel FT, Laemmle A, Charwat V, et al. Integrated isogenic human induced pluripotent stem cell‐based liver and heart microphysiological systems predict unsafe drug‐drug interaction [published online ahead of print May 7, 2021]. Front Pharmacol. 10.3389/fphar.2021.667010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rashidi H, Luu N‐T, Alwahsh SM, et al. 3D human liver tissue from pluripotent stem cells displays stable phenotype in vitro and supports compromised liver function in vivo. Arch Toxicol. 2018;92(10):3117‐3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gao X, Liu Y. A transcriptomic study suggesting human iPSC‐derived hepatocytes potentially offer a better in vitro model of hepatotoxicity than most hepatoma cell lines. Cell Biol Toxicol. 2017;33(4):407‐421. [DOI] [PubMed] [Google Scholar]
- 52. Ramme AP, Koenig L, Hasenberg T, et al. Autologous induced pluripotent stem cell‐derived four‐organ‐chip. Future Sci OA. 2019;5(8):FSO413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhou M, Zhang X, Wen X, et al. Development of a functional glomerulus at the organ level on a chip to mimic hypertensive nephropathy. Sci Rep. 2016;6(1):31771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sakolish CM, Mahler GJ. A novel microfluidic device to model the human proximal tubule and glomerulus. RSC Advances. 2017;7(8):4216–4225. [Google Scholar]
- 55. Sakolish CM, Philip B, Mahler GJ. A human proximal tubule‐on‐a‐chip to study renal disease and toxicity. Biomicrofluidics. 2019;13(1):014107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Li Z, Su W, Zhu Y, et al. Drug absorption related nephrotoxicity assessment on an intestine‐kidney chip. Biomicrofluidics. 2017;11(3):034114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Musah S, Dimitrakakis N, Camacho DM, et al. Directed differentiation of human induced pluripotent stem cells into mature kidney podocytes and establishment of a Glomerulus Chip. Nat Protoc. 2018;13(7):1662‐1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Morizane R, Lam AQ, Freedman BS, Kishi S, Valerius MT, Bonventre JV. Nephron organoids derived from human pluripotent stem cells model kidney development and injury. Nat Biotechnol. 2015;33(11):1193‐1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hoppensack A, Kazanecki CC, Colter D, et al. A human in vitro model that mimics the renal proximal tubule. Tissue Eng Part C Methods. 2014;20(7):599‐609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kim S, LesherPerez SC, Kim B C, et al. Pharmacokinetic profile that reduces nephrotoxicity of gentamicin in a perfused kidney‐on‐a‐chip. Biofabrication. 2016;8(1):015021. [DOI] [PubMed] [Google Scholar]
- 61. Jang KJ, Mehr AP, Hamilton GA, et al. Human kidney proximal tubule‐on‐a‐chip for drug transport and nephrotoxicity assessment. Integr Biol (Camb). 2013;5(9):1119‐1129. [DOI] [PubMed] [Google Scholar]
- 62. Weber EJ, Chapron A, Chapron BD, et al. Development of a microphysiological model of human kidney proximal tubule function. Kidney Int. 2016;90(3):627‐637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Adler M, Ramm S, Hafner M, et al. A quantitative approach to screen for nephrotoxic compounds in vitro. J Am Soc Nephrol. 2016;27(4):1015‐1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kelly EJ, Wang Z, Voellinger JL, et al. Innovations in preclinical biology: ex vivo engineering of a human kidney tissue microperfusion system. Stem Cell Res Ther. 2013;4(Suppl 1):S17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sakolish C, Weber EJ, Kelly EJ, et al. Technology transfer of the microphysiological systems: a case study of the human proximal tubule tissue chip. Sci Rep. 2018;8(1):14882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Maass C, Sorensen NB, Himmelfarb J, et al. Translational assessment of drug‐induced proximal tubule injury using a kidney microphysiological system. CPT Pharmacometrics Syst Pharmacol. 2019;8(5):316‐325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Vormann MK, Gijzen L, Hutter S, et al. Nephrotoxicity and kidney transport assessment on 3D perfused proximal tubules. AAPS J. 2018;20(5):90. [DOI] [PubMed] [Google Scholar]
- 68. Schutgens F, Rookmaaker MB, Margaritis T, et al. Tubuloids derived from human adult kidney and urine for personalized disease modeling. Nat Biotechnol. 2019;37(3):303‐313. [DOI] [PubMed] [Google Scholar]
- 69. Novak R, Ingram M, Marquez S, et al. Robotic fluidic coupling and interrogation of multiple vascularized organ chips. Nat Biomed Eng. 2020;4(4):407‐420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Miller PG, Shuler ML. Design and demonstration of a pumpless 14 compartment microphysiological system. Biotechnol Bioeng. 2016;113(10):2213‐2227. [DOI] [PubMed] [Google Scholar]
- 71. Lin NYC, Homan KA, Robinson SS, et al. Renal reabsorption in 3D vascularized proximal tubule models. Proc Natl Acad Sci U S A. 2019;116(12):5399‐5404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Chapron A, Chapron BD, Hailey DW, et al. An Improved vascularized, dual‐channel microphysiological system facilitates modeling of proximal tubular solute secretion. ACS Pharmacol Transl Sci. 2020;3(3):496‐508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Jansen J, De Napoli IE, Fedecostante M, et al. Human proximal tubule epithelial cells cultured on hollow fibers: living membranes that actively transport organic cations. Sci Rep. 2015;5:16702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Chevtchik NV, Mihajlovic M, Fedecostante M, et al. A bioartificial kidney device with polarized secretion of immune modulators. J Tissue Eng Regen Med. 2018;12(7):1670‐1678. [DOI] [PubMed] [Google Scholar]
- 75. Humes HD, Mackay SM, Funke AJ, Buffington DA. Tissue engineering of a bioartificial renal tubule assist device: in vitro transport and metabolic characteristics. Kidney Int. 1999;55(6):2502‐2514. [DOI] [PubMed] [Google Scholar]
- 76. Finesilver G, Bailly J, Kahana M, Mitrani E. Kidney derived micro‐scaffolds enable HK‐2 cells to develop more in‐vivo like properties. Exp Cell Res. 2014;322(1):71‐80. [DOI] [PubMed] [Google Scholar]
- 77. Fedecostante M, Onciu OG, Westphal KGC, Masereeuw R. Towards a bioengineered kidney: recellularization strategies for decellularized native kidney scaffolds. Int J Artif Organs. 2017;40(4):150‐158. [DOI] [PubMed] [Google Scholar]
- 78. Diekjürgen D, Grainger DW. Drug transporter expression profiling in a three‐dimensional kidney proximal tubule in vitro nephrotoxicity model. Pflugers Arch. 2018;470(9):1311‐1323. [DOI] [PubMed] [Google Scholar]
- 79. Ashammakhi N, Nasiri R, de Barros NR, et al. Gut‐on‐a‐chip: current progress and future opportunities. Biomaterials. 2020;255:120196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Lee DW, Ha SK, Choi I, Sung JH. 3D gut‐liver chip with a PK model for prediction of first‐pass metabolism. Biomed Microdevices. 2017;19(4):100. [DOI] [PubMed] [Google Scholar]
- 81. Arakawa H, Sugiura S, Kawanishi T, et al. Kinetic analysis of sequential metabolism of triazolam and its extrapolation to humans using an entero‐hepatic two‐organ microphysiological system. Lab Chip. 2020;20(3):537‐547. [DOI] [PubMed] [Google Scholar]
- 82. Choe A, Ha SK, Choi I, et al. Microfluidic Gut‐liver chip for reproducing the first pass metabolism. Biomed Microdevices. 2017;19(1):4. [DOI] [PubMed] [Google Scholar]
- 83. Tsamandouras N, Chen WLK, Edington CD, Stokes CL, Griffith LG, Cirit M. Integrated gut and liver microphysiological systems for quantitative in vitro pharmacokinetic studies. AAPS J. 2017;19(5):1499‐1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Maurer M, Gresnigt MS, Last A, et al. A three‐dimensional immunocompetent intestine‐on‐chip model as in vitro platform for functional and microbial interaction studies. Biomaterials. 2019;220:119396. [DOI] [PubMed] [Google Scholar]
- 85. Kim HJ, Ingber DE. Gut‐on‐a‐Chip microenvironment induces human intestinal cells to undergo villus differentiation. Integr Biol (Camb). 2013;5(9):1130‐1140. [DOI] [PubMed] [Google Scholar]
- 86. Kim HJ, Li H, Collins JJ, Ingber DE. Contributions of microbiome and mechanical deformation to intestinal bacterial overgrowth and inflammation in a human gut‐on‐a‐chip. Proc Natl Acad Sci U S A. 2016;113(1):E7‐E15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Shah P, Fritz JV, Glaab E, et al. A microfluidics‐based in vitro model of the gastrointestinal human‐microbe interface. Nat Commun. 2016;7:11535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Jalili‐Firoozinezhad S, Gazzaniga FS, Calamari EL, et al. A complex human gut microbiome cultured in an anaerobic intestine‐on‐a‐chip. Nat Biomed Eng. 2019;3(7): 520‐531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Kasendra M, Tovaglieri A, Sontheimer‐Phelps A, et al. Development of a primary human small intestine‐on‐a‐chip using biopsy‐derived organoids. Sci Rep. 2018;8(1):2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Kasendra M, Luc R, Yin J, et al. Duodenum intestine‐chip for preclinical drug assessment in a human relevant model. eLife. 2020;9:e50135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Workman MJ, Gleeson JP, Troisi EJ, et al. Enhanced utilization of induced pluripotent stem cell‐derived human intestinal organoids using microengineered chips. Cell Mol. Gastroenterol Hepatol. 2018;5(4):669‐677 e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Uchida H, Machida M, Miura T, et al. A xenogeneic‐free system generating functional human gut organoids from pluripotent stem cells. JCI Insight. 2017;2(1):e86492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Hedrich WD, et al. Development and characterization of rat duodenal organoids for ADME and toxicology applications. Toxicology. 2020;446:152614. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material