

Abstract
It remains uncertain whether pharmacokinetic changes following Roux‐en‐Y gastric bypass (RYGB) can be attributed to surgery‐induced gastrointestinal alterations per se and/or the subsequent weight loss. The aim was to compare short‐ and long‐term effects of RYGB and calorie restriction on CYP3A‐activity, and cross‐sectionally compare CYP3A‐activity with normal weight to overweight controls using midazolam as probe drug. This three‐armed controlled trial included patients with severe obesity preparing for RYGB (n = 41) or diet‐induced (n = 41) weight‐loss, and controls (n = 18). Both weight‐loss groups underwent a 3‐week low‐energy‐diet (<1200 kcal/day) followed by a 6‐week very‐low‐energy‐diet or RYGB (both <800 kcal/day). Patients were followed for 2 years, with four pharmacokinetic investigations using semisimultaneous oral and intravenous dosing to determine changes in midazolam absolute bioavailability and clearance, within and between groups. The RYGB and diet groups showed similar weight‐loss at week 9 (13 ± 2.4% vs. 11 ± 3.6%), but differed substantially after 2 years (−30 ± 7.0% vs. −3.1 ± 6.3%). At baseline, mean absolute bioavailability and clearance of midazolam were similar in the RYGB and diet groups, but higher compared with controls. On average, absolute bioavailability was unaltered at week 9, but decreased by 40 ± 7.5% in the RYGB group and 32 ± 6.1% in the diet group at year 2 compared with baseline, with no between‐group difference. No difference in clearance was observed over time, nor between groups. In conclusion, neither RYGB per se nor weight loss impacted absolute bioavailability or clearance of midazolam short term. Long term, absolute bioavailability was similarly decreased in both groups despite different weight loss, suggesting that the recovered CYP3A‐activity is not only dependent on weight‐loss through RYGB.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
The current literature indicates an inverse relationship between body mass index (BMI) and CYP3A‐activity, and that systemic clearance of the CYP3A‐probe midazolam increases after Roux‐en‐Y gastric bypass (RYGB). However, the knowledge regarding changes in drug disposition following weight loss in general, and bariatric surgery in particular, is sparse and inconclusive, making drug dosing challenging. In addition, it remains uncertain whether pharmacokinetic changes following bariatric surgery are attributed to the gastrointestinal alterations per se and/or the subsequent weight loss.
WHAT QUESTION DID THIS STUDY ADDRESS?
Do body weight, weight loss, and RYGB impact CYP3A‐activity in vivo?
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
This study shows that neither short‐term weight loss induced by RYGB or very‐low‐energy‐diet, nor RYGB per se, impact absolute bioavailability or systemic clearance of midazolam.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
Our results suggests that dose adjustments of CYP3A substrate drugs, 30–50% of all drugs on the market, may not be necessary following nonsurgical and surgical weight loss neither in a short‐ nor long‐term perspective.
INTRODUCTION
Obesity is a worldwide epidemic. Individuals with obesity often receive multiple medication for obesity‐related comorbidities, such as diabetes, cardiovascular diseases, and non‐alcoholic fatty liver disease (NAFLD). 1 , 2 Bariatric surgery is considered superior to nonsurgical intervention in terms of achieving long‐lasting weight loss and improvement of comorbidities. 3 , 4 The anatomic and physiological alterations in the gastrointestinal tract following Roux‐en‐Y gastric bypass (RYGB) 5 may influence oral drug bioavailability and thus have consequences for postsurgery dosing. More specifically, the total surface area available for drug absorption is reduced and the proximal intestinal segments rich in cytochrome P450 (CYP) enzymes are bypassed. 6
CYP3A is the most important drug metabolizing enzyme with regard to drug dosing, and accounts for the metabolism of 30–50% of clinically used drugs. 7 , 8 CYP3A is not only widely expressed in the liver, but also in the duodenum and proximal jejunum. 9 RYGB may therefore reduce first‐pass metabolism, resulting in an increased oral bioavailability. Besides the gastrointestinal alterations, the subsequent weight loss may also influence the expression and activity of CYP3A enzymes in both the intestine and liver. Obesity is associated with impaired CYP3A‐activity, 6 , 10 , 11 and previous studies suggest an inverse relationship between body mass index (BMI) and CYP3A‐activity, 12 , 13 possibly due to low‐grade inflammation and NAFLD. 14 , 15 , 16 Because inflammation status and NAFLD improve after RYGB or nonsurgical weight loss, 17 , 18 , 19 , 20 both intestinal and hepatic CYP3A mediated drug metabolism may also recover. 21 An increase in intestinal CYP3A‐activity is expected to decrease oral bioavailability of CYP3A substrates, unless other physiological parameters compensate for the higher activity. Clearance, however, is expected to increase with an increase in hepatic CYP3A activity, unless weight loss also results in other physiological changes that may influence clearance, such as lower hepatic blood flow. In studies investigating pharmacokinetic changes following bariatric surgery, 6 , 21 , 22 , 23 patients usually serve as their own controls and it is therefore not possible to disentangle the effect of bariatric surgery and the subsequent weight loss. To improve our understanding of the underlying mechanisms involved in restricting oral bioavailability, this is important to investigate.
RYGB may influence the pharmacokinetics of many drugs and thus have consequences for dosing recommendations. However, it remains uncertain whether such potential pharmacokinetic changes are attributed to the gastrointestinal alterations per se and/or the subsequent weight loss. To address this, we performed a three‐armed controlled trial over 2 years with a dietary control group achieving a matched short‐term weight loss as patients undergoing RYGB. 24 CYP3A activity was investigated using oral and intravenous dosing of midazolam, the clinical CYP3A probe of choice. The study objectives were to compare short‐ and long‐term effects of surgical and nonsurgical calorie restriction on CYP3A activity, and to compare CYP3A activity in a control group of normal weight to overweight individuals with patients with severe obesity.
METHODS
Patients and study design
The COCKTAIL study is an open, nonrandomized, three‐armed, single‐center controlled study performed at Vestfold Hospital Trust in Norway, as previously described in detail. 24 In short, patients with severe obesity scheduled for weight loss treatment with RYGB or a restricted calorie diet based on clinical indications were included and followed prospectively for 2 years. Additionally, a cross‐sectional control group of individuals with BMI 18.5–30 kg/m2 scheduled for cholecystectomy was included. The study was approved by the Regional Committee for Medical and Health Research Ethics (2013/2379/REK) and performed in accordance with Good Clinical Practice and the Declaration of Helsinki (NCT02386917). Written informed consent was obtained prior to study participation.
Patients were eligible for inclusion based on the following inclusion criteria: greater than or equal to 18 years, BMI greater than or equal to 18.5 kg/m2, and stable body weight (<5 kg weight change) over the last 3 months. Exclusion criteria included glomerular filtration rate less than 30 ml/min/1.73m2, previous bariatric or upper gastrointestinal surgery, or treatment with substances that may influence midazolam pharmacokinetics in close approximation to the investigations. 24
Study visits and procedures
The current analysis included data from the control group (baseline; week 0), and from the intervention groups at weeks 0, 3, and 9, and year 2. At baseline (week 0), all three groups were subjected to a 24‐h pharmacokinetic investigation one day before intervention start. Both weight loss groups were prescribed a low‐energy‐diet (LED; <1200 kcal/day) for the first 3 weeks, followed by an additional 6 weeks of an isocaloric very‐low‐energy‐diet (VLED; <800 kcal/day) or RYGB (<800 kcal/day). Thereafter, patients were treated according to local guidelines until the year 2 visit. A 24‐h pharmacokinetic investigation was repeated at all three follow‐up visits. For patients undergoing RYGB, small intestinal and hepatic biopsies were obtained on the day of surgery, as previously described. 13 , 25 Hepatic biopsies were also obtained from the control group during cholecystectomy. 13
Patients abstained from food and drugs from 10:00 p.m. the evening before all pharmacokinetic investigations. On the study day, patients met at 07:30 a.m. for blood sampling, before 1.5 mg oral midazolam syrup was administered followed by 1.0 mg intravenous midazolam 4 h later. Blood samples were collected from a peripheral venous catheter at: 0.25, 0.5, 1, 1.5, 2, 3, 4, 4.25, 4.5, 5, 5.5, 6, 8, 10, 12, 23, and 24 h. Details regarding the pharmacokinetic investigations and clinical chemistry analyses are described in the Supplement.
Outcomes
The primary outcomes were short‐ and long‐term changes in absolute bioavailability and clearance of midazolam as measures of both first‐pass and systemic CYP3A‐activity. 24 Secondary outcomes included CYP3A protein quantification in liver and intestine biopsies. Additional outcomes included full pharmacokinetic profiling; oral clearance (clearance/bioavailability), volume of distribution (Vd), terminal elimination half‐life (t1/2), maximum plasma concentration (Cmax), and time to reach maximum plasma concentration (Tmax) after oral midazolam administration as well as concentrations of the endogenous CYP3A biomarker; 4β‐hydroxycholesterol (4βOHC) formed from cholesterol via CYP3A metabolism. 26
Analytical assays
Detailed descriptions of the analytical assays are presented in the Supplement. In brief, midazolam plasma concentrations were determined by validated ultra‐high performance liquid chromatography tandem mass spectrometry (UHPLC‐MS/MS). 27 Within‐series and between‐series performance were assessed with resulting imprecision less than 12.3%, and the mean accuracy ranged from 99.3 to 104.3%. Plasma concentrations of 4βOHC was determined by an HPLC‐MS method previously described, 28 with an added filtration step, 29 at the Center for Psychopharmacology, Diakonhjemmet Hospital, Oslo, Norway. Within‐ and between‐series imprecision and inaccuracy were less than 15% at 10 ng/ml and less than 4% at 644 ng/ml. 28
Protein quantification (intestine and liver)
Proteins were extracted from small intestinal and liver biopsies in an SDS‐containing (2% w/v) lysis buffer and quantified, as previously described. 30 , 31 Details are presented in the Supplement, in short, samples were processed with the multi‐enzyme digestion filter‐aided sample preparation (MED‐FASP) protocol, using LysC and trypsin. 32 Proteomics analysis was performed with Q Exactive HF or Q Exactive HF‐X mass spectrometer in data dependent mode. MS data were processed with MaxQuant (version 1.6.10.43) 33 where proteins were identified by searching MS and MS/MS data of peptides against the human UniProtKB (UP000005640). Spectral raw intensities were normalized with variance stabilization (vsn), 34 and were subsequently used to calculate the protein concentrations using the Total Protein Approach. 35 Batch effects were removed by geometric mean centering of proteins from samples analyzed at different time points.
Data analysis
The data analysis comprised two subsequent steps. In step one, a population pharmacokinetic model was developed for characterization of individual pharmacokinetic profiles in order to retrieve individual pharmacokinetic parameters from each investigation. In step two, the individual pharmacokinetic parameters were used to assess changes over the study period using linear mixed effects modeling and visualizations.
Population pharmacokinetic modeling
Given the semisimultaneous oral and intravenous dosing of midazolam applied in this study, we developed a population pharmacokinetic model for description of individual pharmacokinetic profiles in order to retrieve individual pharmacokinetic parameters from all patients, including absolute bioavailability. A detailed description of the population pharmacokinetic model development is presented in the Supplement. In brief, a midazolam model parameterized to determine individual absolute bioavailability was developed with data from semisimultaneous administration of oral and intravenous midazolam. The modeling was performed using the nonparametric adaptive grid approach implemented in Pmetrics (version 1.5.2) for R (version 3.6.2). 36 , 37 A total of 5414 midazolam concentrations corresponding to 306 unique 24‐h pharmacokinetic profiles were available from a total of 98 patients. A catenary three‐compartment model 38 , 39 with absorption lag‐time and first‐order elimination from the central compartment (Figure S1) was developed and validated. Due to the sole interest in individual predictions of pharmacokinetic parameters and to avoid including the same potential covariate both in the pharmacokinetic population model and the statistical analysis (linear mixed effects model), no covariates were implemented in the population model. Results from the population pharmacokinetic modeling are described in the Supplement Figure S2–S4, and Table S1/S2.
Calculations
Posterior individual parameter values obtained from the final model were used to describe midazolam pharmacokinetics in each individual at each investigation. Absolute bioavailability, Cmax, and Tmax were obtained directly from the individual model predictions, whereas clearance, oral clearance, Vd, and t1/2 were calculated from the individual model parameter estimates. Equations are described in the Supplement. NAFLD score was calculated as suggested by Kotronen et al. 40
Statistical analyses and considerations
Student’s t‐tests were used for the cross‐sectional analysis between patients with obesity and controls. Linear mixed effects models, with the pharmacokinetic parameters as dependent variable and visit and group as fixed effects with individual intercepts (random effect), were used to compare delta change and 95% confidence interval (CI) from baseline at the different study visits within the RYGB and diet groups. All exploratory tests were two‐sided at the 5% level without adjustment for multiplicity. To adjust for comparison between multiple study visits in the mixed effects model, CIs were adjusted with the Tukey method. To describe the relationship between variables, linear regression analyses were performed. All statistical analyses were performed using R (version 3.6.2). 37
RESULTS
Patient characteristics
Between March 18, 2015, and May 22, 2017, 196 patients who were preparing for bariatric surgery, low calorie diet (<1200 kcal/day), or cholecystectomy, were screened for eligibility. 24 After exclusion of 88 ineligible patients, a total of 44, 44, and 20 patients were included in the RYGB, diet, and control group, respectively (Figure 1a). The first and last patient were included April 15, 2015, and June 29, 2017, respectively. Eight patients withdrew or were excluded before study start (week 0), leaving 41, 41, and 18 patients in the three groups starting treatment at week 0. Three patients did not undergo RYGB due to (1) liver cirrhosis (thus excluded from pharmacokinetic analyses), (2) intubation not possible, and (3) anaphylactic reaction. The latter two were hence excluded after the study visit at week 3. Finally, 13 patients (RYGB = 4 and diet = 9) withdrew or dropped out after week 9, leaving 34 (RYGB) and 30 (diet) patients who attended the 2‐year follow‐up. Due to technical difficulties, some patients were unable to supply evaluable pharmacokinetic profiles at all four study visits, whereas one patient in the diet group did not supply any pharmacokinetic profile. A total of 40 and 40 patients in the RYGB and diet groups, respectively, supplied at least one 24‐h pharmacokinetic profile during the study period.
FIGURE 1.
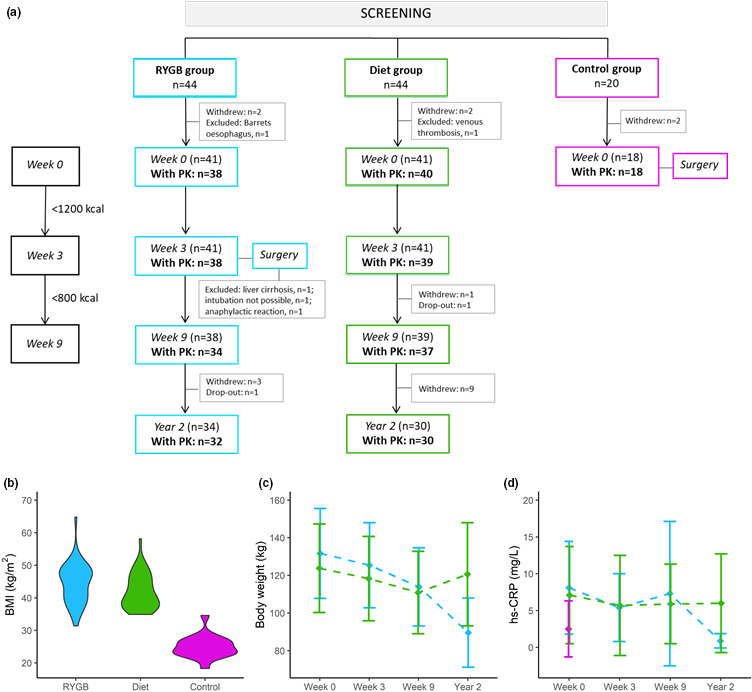
Patient flow‐chart, study design, and clinical parameters. In panel (a), patient flow chart during the study period is shown. Number of patients with PK profiles are in bold. BMI in the RYGB group (n = 41), diet group (n = 41), and control group (n = 18) at baseline (week 0) is shown in panel b. Changes in total body weight and hs‐CRP during the study period in patients with evaluable 24‐h PK profiles are shown in panels c and d, respectively. Abbreviations: BMI, body mass index; hs‐CRP, high‐sensitivity C‐reactive protein; PK, pharmacokinetic; RYGB, Roux‐en‐Y gastric bypass
At baseline, sex, age, and ethnicity did not differ between the intervention groups and controls, but the control group had lower body weight (Figure 1b), alanine aminotransferase (ALT), and high‐sensitivity C‐reactive protein (hs‐CRP) than the intervention groups (Table 1). Baseline characteristics did not differ between the intervention groups (Table 1).
TABLE 1.
Baseline (week 0) characteristics presented as mean ±SD or number of patients (%)
| RYGB n = 41 | Diet n = 41 | Control n = 18 | |
|---|---|---|---|
| Sex, n (male/female) | 14/27 | 14/27 | 3/15 |
| Age (years) | 46 ± 9 | 49 ± 10 | 42 ± 15 |
| Ethnicity, n (White/other) | 41/0 | 40/1 | 17/1 |
| Body weight (kg) | 132 ± 24 | 124 ± 23 | 71 ± 11 |
| BMI (kg/m2) | 44.5 ± 6.2 | 42.0 ± 5.4 | 25.0 ± 3.5 |
| Albumin (g/L) | 40 ± 2 | 40 ± 2 | 40 ± 2 |
| Creatinine (µmol/L) | 58 ± 11 | 59 ± 14 | 60 ± 12 |
| AST (U/L) | 29 ± 11 | 28 ± 15 | 25 ± 11 |
| ALT (U/L) | 34 ± 17 | 32 ± 18 | 22 ± 15 |
| hs‐CRP (mg/L) | 8.1 ± 6.3 | 8.2 ± 9.6 | 2.5 ± 3.8 |
Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase; BMI, body mass index; hs‐CRP, high sensitivity C‐reactive protein; RYGB, Roux‐en‐Y gastric bypass.
Changes in body weight and selected laboratory measures
Body weight decreased similarly in the RYGB and diet groups from baseline to week 3 (4.8 ± 1.1% vs. 4.4 ± 2.0%), and from baseline to week 9 (13 ± 2.4% vs. 11 ± 3.6%). Between week 9 and year 2, mean body weight decreased in the RYGB group (20 ± 9.0%) but increased in the diet group (9.0 ± 8.0%) as several patients had almost returned to baseline body weight at year 2 (Figure 1c, Table S3).
Changes in selected laboratory measures are presented in Table S3. At week 9, hs‐CRP levels were unaltered in both groups, but hs‐CRP decreased in the RYGB group at year 2 (p < 0.001; Figure 1d). ALT increased between week 0 and week 9 in the RYGB group, whereas no change was observed in the diet group.
Baseline pharmacokinetics – severe obesity versus normal weight to overweight
In total, 96 pharmacokinetic profiles (RYGB = 38, diet = 40, and control = 18) were included in the cross‐sectional analysis at baseline. Mean ±SD plots for pharmacokinetic parameters and variables in the RYGB, diet, and control groups at baseline are presented in Figure 2. Mean pharmacokinetic profiles are shown in Figure S5. Absolute bioavailability was 153% higher in patients with severe obesity (n = 78) compared with controls (n = 18, p < 0.001).
FIGURE 2.
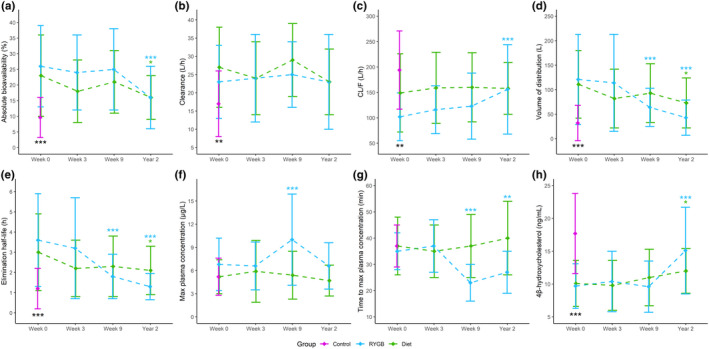
Pharmacokinetic changes during the study period. Mean ±standard deviation plots for midazolam (a) absolute bioavailability, (b) clearance, (c) oral clearance (CL/F), (d) volume of distribution, (e) elimination half‐life, (f) maximum plasma concentration (Cmax) after oral midazolam, (g) time to reach maximum plasma concentration (Tmax) after oral midazolam, and (h) 4β‐hydroxycholesterol at the different study visits. Statistically significant differences between patients with obesity versus control at baseline (from two‐sided t‐test) are given by black stars. Statistically significant differences were seen for all parameters, except from Cmax and Tmax. Statistically significant differences over time compared to baseline within the Roux‐en‐Y gastric bypass (RYGB) and diet groups (from linear mixed effects model) are given by blue and green stars, respectively. Difference within and between the two intervention groups at the different study visits are shown in Table 3. *p value <0.05; **p value ≤0.01; ***p value ≤0.001
In patients with severe obesity, 4βOHC concentrations were 44% lower compared with normal weight to overweight controls (p < 0.001). In addition, patients with severe obesity exhibited a higher systemic clearance compared with controls (46%, p = 0.003). We observed positive associations between body weight and both absolute bioavailability (β = 0.10, R2adj = 0.04) and clearance (β = 0.10, R2adj = 0.08; Figure 3a, b, respectively).
FIGURE 3.
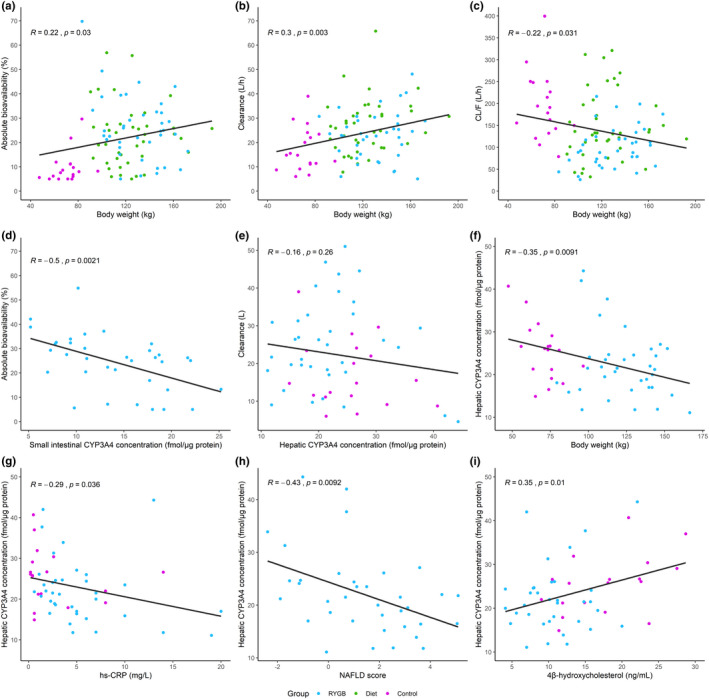
Association among body weight, CYP3A4 concentrations, and pharmacokinetics. Association between body weight and midazolam (a) absolute bioavailability, (b) clearance, and (c) oral clearance (CL/F) at baseline. Association between small intestinal CYP3A4 concentration and (d) midazolam absolute bioavailability at the time of surgery. Association between hepatic CYP3A4 concentration and (e) midazolam clearance at the time of surgery. Association among (f) body weight, (g) high sensitivity C‐reactive protein (hs‐CRP), (h) non‐alcoholic fatty liver (NAFLD) score, and (i) 4β‐hydroxycholesterol, and hepatic CYP3A concentrations at the time of surgery. R is the correlation coefficient. RYGB, Roux‐en‐Y gastric bypass
In the patients undergoing RYGB and cholecystectomy (n = 54), regression analyses revealed no associations between individual hepatic CYP3A4 concentrations and midazolam clearance at the time of surgery (Figure 3e). However, significant associations were observed among body weight and hepatic CYP3A4 concentrations (β = −0.09, R2adj = 0.11), hs‐CRP, and hepatic CYP3A4 concentrations (β = −0.48, R2adj = 0.06), NAFLD score and hepatic CYP3A4 concentrations (β = −1.7, R2adj = 0.16), and 4βOHC and hepatic CYP3A4 concentrations (β = 0.45, R2adj = 0.11; Figure 3f–i). The individual CYP3A4 concentrations in the jejunum (n = 36), information only available in the patients undergoing RYGB, showed a negative association with absolute bioavailability: β = −1.1, R2adj = 0.23 (Figure 3d).
Changes in pharmacokinetics after VLED, RYGB, and weight loss
In total, 288 pharmacokinetic profiles from 80 patients (RYGB = 40 and diet = 40) were included in the longitudinal analysis, and of these, 56 patients supplied pharmacokinetic profiles at all four study visits. At baseline, absolute bioavailability and clearance did not differ between the intervention groups, but the RYGB group had a 32% lower oral clearance than the diet group (p = 0.002). Mean ±SD pharmacokinetic parameters for all groups at the different study visits are shown in Table 2. Short‐ and long‐term pharmacokinetic outcomes are presented in Table 3.
TABLE 2.
PK variables and population pharmacokinetic model derived parameters at the different study visits in the RYGB, diet, and control group, respectively
| PPK parameter | RYGB | Diet | Control | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Mean ±SD | |||||||||
| Week 0 n = 38 | Week 3 n = 38 | Week 9 n = 34 | Year 2 n = 32 | Week 0 n = 40 | Week 3 n = 39 | Week 9 n = 37 | Year 2 n = 30 | Week 0 n = 18 | |
| Absolute bioavailability (%)a | 26 ± 13 | 24 ± 12 | 25 ± 13 | 16 ± 10 | 23 ± 13 | 18 ± 10 | 21 ± 10 | 16 ± 7 | 9.6 ± 6.4 |
| Clearance (L/h)a | 23 ± 10 | 24 ± 12 | 25 ± 9 | 23 ± 13 | 27 ± 11 | 24 ± 10 | 29 ± 10 | 23 ± 9 | 17 ± 9 |
| Oral clearance (L/h)a | 102 ± 47 | 116 ± 47 | 123 ± 65 | 156 ± 88 | 149 ± 77 | 159 ± 70 | 160 ± 68 | 158 ± 51 | 194 ± 77 |
| Volume of distribution (L)a | 121 ± 92 | 114 ± 99 | 64 ± 39 | 43 ± 36 | 111 ± 69 | 82 ± 60 | 93 ± 60 | 73 ± 51 | 32 ± 36 |
| Elimination half‐life (h)a | 3.6 ± 2.3 | 3.2 ± 2.5 | 1.8 ± 1.1 | 1.3 ± 0.65 | 3.0 ± 1.9 | 2.2 ± 1.4 | 2.3 ± 1.5 | 2.1 ± 1.2 | 1.2 ± 1.0 |
| Cmax, oral (µg/L)a | 6.8 ± 3.4 | 6.6 ± 3.1 | 10.0 ± 5.9 | 6.6 ± 3.0 | 5.2 ± 2.2 | 5.9 ± 4.0 | 5.4 ± 3.1 | 4.7 ± 2.0 | 5.2 ± 2.4 |
| 4β‐hydroxycholesterol (ng/ml) | 9.7 ± 3.4 | 10.4 ± 4.6 | 9.6 ± 3.9 | 15.1 ± 6.6 | 10.1 ± 3.5 | 9.8 ± 3.8 | 11.0 ± 4.3 | 12.0 ± 3.4 | 17.7 ± 6.1 |
Abbreviations: Cmax, maximum plasma concentration after oral midazolam; PK, pharmacokinetic; RYGB, Roux‐en‐Y gastric bypass.
Absolute bioavailability and Cmax were obtained directly from the individual model predictions, whereas clearance, oral clearance, volume of distribution, and elimination half‐life were calculated from model derived values.
TABLE 3.
Short‐ and long‐term outcomes in PK parameters and variables from baseline to week 3, week 9, and year 2 in the RYGB group and diet group, respectively
| PK parameter | RYGB | Diet | Difference between groupsb | |||||
|---|---|---|---|---|---|---|---|---|
| Estimated mean difference (∆) [95% CI]a | ||||||||
| Week 0 – Week 3 | Week 0 – Week 9 | Week 0 – Year 2 | Week 0 – Week 3 | Week 0 – Week 9 | Week 0 – Year 2 | Week 9 | Year 2 | |
| Absolute bioavailability (%) | −2.0 [−7.7, 3.7] | −1.0 [−6.9, 4.9] | −10.0 *** [−16.1, −4.0] | −4.4 [−10.0, 1.1] | −2.1 [−7.8, 3.6] | −7.0* [−13.1, −0.97] | 4.3 [−0.99, 9.6] | 0.22 [−5.4, 5.8] |
| Clearance (L/h) | 1.6 [−3.9, 7.1] | 1.4 [−4.3, 7.1] | −0.66 [−6.5, 5.2] | −3.0 [−8.4, 2.4] | 1.7 [−3.8, 7.2] | −3.6 [−9.4, 2.3] | −4.2 [−9.0, 0.69] | −0.93 [−6.1, 4.2] |
| Oral clearance, CL/F (L/h) | 13 [−18, 45] | 19 [−14, 51] | 54 *** [21, 87] | 10 [−21, 40] | 13 [−18, 44] | 12 [−21, 45] | −41** [−71, −10] | −4.5 [−37, 28] |
| Volume of distribution (L) | 5.1 [−41, 30] | −58*** [−95, −21] | −77*** [−114, −39] | −29 [−64, 5.9] | −20 [−55, 15] | −41* [−79, −3.3] | −29 [−60, 3.2] | −26 [−60, 7.3] |
| Elimination half‐life, t½ (h) | −0.38 [−1.2, 0.49] | −1.8*** [−2.7, −0.88] | −2.2*** [−3.1, −1.3] | −0.75 [−1.6, 0.10] | −0.72 [−1.6, 0.14] | −1.0* [−1.9, −0.09] | −0.49 [−1.3, 0.30] | −0.65 [−1.5, 0.20] |
| Cmax, oral (µg/L) | −0.28 [−1.9, 1.3] | 3.2*** [1.6, 4.9] | −0.20 [−1.9, 1.5] | 0.70 [−0.89, 2.3] | 0.45 [−1.2, 2.1] | −0.33 [−2.1, 1.4] | 4.4*** [2.7, 6.0] | 1.7 [0.0, 3.5] |
| 4β‐hydroxycholesterol (ng/ml) | 0.69 [−1.1, 2.5] | −0.25 [−2.1, 1.6] | 5.3*** [3.4, 7.1] | −0.34 [−2.1, 1.4] | 1.2 [−0.59, 2.9] | 2.2* [0.26, 4.1] | −1.7 [−3.7, 0.28] | 2.8** [0.70, 4.9] |
Abbreviations: CI, confidence interval; CL/F, oral clearance; Cmax, maximum plasma concentration after oral midazolam; PK, pharmacokinetic; RYGB, Roux‐en‐Y gastric bypass.
Bold values show significant differences.
The p values are calculated from ∆ at week 3, week 9, and year 2 compared to baseline (week 0) within both groups and between groups at week 9 and year 2 (without adjustment for multiplicity). *p value <0.05; **p value ≤0.01; ***p value ≤0.001.
Linear mixed model was used to estimate mean difference in change (∆).
Difference between groups is calculated with RYGB as reference group.
Short‐term effect of RYGB and diet on midazolam pharmacokinetics
Three weeks of LED did not change midazolam pharmacokinetics in the two intervention groups (Table 3). After an additional 6 weeks of VLED, absolute bioavailability and clearance were still unaltered in both groups and did not show difference between groups (Figure 2a,b). In the RYGB group, a 35 ± 2.6% shorter Tmax and a 52 ± 15% higher Cmax was, however, observed 6 weeks after surgery (week 9; Figure 2f,g).
Long‐term effect of RYGB and diet on midazolam pharmacokinetics
At year 2, absolute bioavailability was decreased in both the RYGB and diet groups by 40 ± 7.5% (p < 0.001) and 32 ± 6.1% (p = 0.016), respectively (Table 3, Figure 2a). In addition, both groups exhibited increases in 4βOHC concentrations: 60 ± 22% in the RYGB group (p < 0.001) versus 23 ± 6.8% in the diet group (p = 0.018; Table 3, Figure 2h). Mean difference between the groups at year 2 was 20% (p = 0.009). No difference in clearance was observed over time, nor between the groups (Table 3, Figure 2b). In the RYGB group, Cmax after oral midazolam administration showed similar values as prior to surgery, but Tmax was still shorter (−28%, p < 0.001; Table 3, Figure 2f,g).
Inter‐ and intra‐individual variability
A large inter‐individual variability was observed for all pharmacokinetic parameters and variables (Figure 2). Patients with severe obesity also exhibited a substantial intra‐individual variability over time, as illustrated in Figure S6. The intra‐individual variability was assessed as the coefficient of variation (CV) in patients with pharmacokinetic profiles at all of the three first study visits (n = 69). Of these patients with pharmacokinetic profiles at weeks 0, 3, and 9, 61% and 41% had a CV greater than 30% for absolute bioavailability and clearance, respectively.
DISCUSSION
The main finding of this three‐armed, controlled study with 98 participants was that RYGB per se did not have any significant effect on absolute bioavailability or systemic clearance of midazolam short‐term (week 9). Considering the rearrangement of the gastrointestinal tract leading to both loss of absorptive area and bypass of the most CYP3A rich part of the intestine, we expected to observe an increased oral bioavailability following RYGB, which has also been suggested in previous studies. 21 , 22 , 41 However, the unaltered absolute bioavailability and clearance of midazolam 6 weeks postsurgery suggests that neither the surgery induced gastrointestinal alterations, nor a moderate weight loss, influences midazolam pharmacokinetics in the short‐term perspective. The unique dietary control group included in this study showing matched weight loss with the surgery group short‐term (11% vs. 13%), further substantiates that such a significant weight loss seems to have limited clinical relevance considering dosing of CYP3A substrates, such as midazolam. Nevertheless, these findings may reduce the uncertainty for clinicians regarding the need for dose adjustments of CYP3A substrates in the early period after RYGB, but also following a moderate weight loss after nonsurgical calorie restriction.
The semisimultaneous administration of oral and intravenous midazolam revealed an almost three‐fold higher absolute bioavailability in patients with severe obesity compared with normal weight to overweight individuals. Similar results for absolute bioavailability of midazolam have been shown in a smaller study by Brill et al. comparing patients with severe obesity and healthy normal weight controls. 10 We also observed that patients with severe obesity had an almost 50% higher clearance compared to normal weight with overweight controls. Considering previous literature on CYP3A‐activity in patients with obesity, 10 , 11 , 21 the inverse association between hepatic CYP3A concentration and body weight (Figure 3f), and that patients with obesity had lower 4βOHC concentrations compared with controls, this was unexpected. However, patients with severe obesity have higher hepatic blood flow, possibly induced by a combination of increased blood volume, cardiac output, and liver volume. 42 , 43 Given that midazolam is a medium‐to‐high extraction ratio drug, hepatic blood flow will affect midazolam clearance and potentially, based on the present results, have a more significant role than CYP3A activity on clearance estimates in this patient population. 11 , 44 Regardless, this finding is in contrast to what Brill et al. observed in their study. 10 We can only speculate on the reasons for this discrepancy in midazolam clearance. In our study, the age of the patients in the different groups were comparable, whereas, in the study of Brill et al., the mean age of the patients with obesity was similar to our patients (44 years), but their controls were considerable younger (mean age of 22 years). Younger individuals will typically have higher liver blood flow resulting in higher clearance of midazolam compared with older individuals. 11 , 44 Further, the present study supports previous findings of an inverse relationship between BMI and oral clearance of CYP3A substrates (Figure 3c). 10 , 12 , 13 Given that we assessed both absolute bioavailability and clearance, we can argue that the negative association between BMI and oral clearance is explained by both higher bioavailability and clearance with higher body weight, but that the effect on bioavailability was greater.
The protein quantification in the present study supports previous findings of a lower hepatic CYP3A expression and activity in patients with severe obesity 12 , 13 and in patients with NAFLD (Figure 3f,h). 15 , 16 The lack of short‐term changes in absolute bioavailability and clearance of midazolam with body weight reduction in our study indicates that 9 weeks is not sufficient time for CYP3A activity to recover. Given the suggested mechanism of reduced CYP3A activity due to low‐grade inflammation and NAFLD this appears plausible. 14 , 15 , 16 This also seems reasonable considering the hepatic CYP3A4 enzyme turnover half‐life, 45 and that hs‐CRP did not change in this period. The decreased absolute bioavailability in the RYGB group at year 2 (40%) supports the hypothesis that CYP3A activity is recovered following weight loss, given enough time, considering a sustained mean weight loss of 30%. However, other simultaneous physiological alterations, such as changed splanchnic and hepatic blood flow during substantial body weight loss, limit the true mechanistic understanding of the effects. The diet group also showed decreased bioavailability (32%) despite the fact that many of these patients had returned to their baseline body weight at year 2 (mean weight loss of 3% from baseline). Overall, this suggests that other mechanisms known to influence midazolam pharmacokinetics, such as gut microbiota, 46 may also be involved.
The lack of association between clearance and hepatic CYP3A4 expression observed in the present study (Figure 3e), further supports that hepatic blood flow may have a larger role in determining clearance of midazolam. Thus, CYP3A activity may be recovered, although clearance was unaltered at the 2‐year study visit. The observed increase in 4βOHC concentrations in the present study supports a recovery of CYP3A activity. Additionally, this endogenous CYP3A biomarker was positively associated with hepatic CYP3A4 concentrations and negatively correlated with BMI, which has also been shown previously. 47
The major strength of this study is the matched short‐term weight loss between a group undergoing RYGB and a dietary control group, enabling us to separate the surgery effect per se on drug disposition from the subsequent weight loss effect. To our knowledge, this is the first study to achieve this. In addition, we were not limited to investigating the surrogate variable oral clearance as we assessed both absolute bioavailability and clearance using a semisimultaneous oral and intravenous administration of midazolam, the standard CYP3A in vivo probe. Additionally, our study has a relatively large sample size considering the study design, and it maintains a high quality compared to other studies with similar objectives. 10 , 21 , 23 , 48 In addition, the rich pharmacokinetic data (18 concentrations per 24‐h profile) combined with population pharmacokinetic analyses, the high completion rate and the long‐term follow‐up, form a strong foundation for this thorough in‐depth investigation of both short‐ (9 weeks) and long‐term (2 years) effects.
The results in this study should, however, be interpreted in light of some important limitations. Although midazolam (a moderate to high extraction drug) is the preferred probe drug to study CYP3A activity in vivo, other mechanisms, such as alterations in blood flow and/or unbound fraction, will also impact midazolam pharmacokinetics. However, no other in vivo probes available will bypass this problem (e.g., a low extraction drug would bypass a hepatic blood flow effect but not provide detailed information on absolute oral bioavailability). In addition, the midazolam absolute bioavailability observed in the control group in our study (~10%) was lower than the large proportion of estimates reported in the literature, ranging from 20 to 70% in different populations and a range of doses. 10 , 27 , 49 , 50 We also observed a high intra‐individual variability in midazolam pharmacokinetics, in addition to the expected high inter‐individual variability. 21 , 23 The reason for the high intra‐individual variability is unknown, although the pharmacokinetic population modeling analyses indicate that the initial volume of distribution may be a main contributor. It may be speculated that this might be due to the rapid changes in body composition in the early study period, resulting in very high midazolam concentrations immediately after intravenous dosing and thus low distribution volume estimates in some patients. We therefore applied model predicted area under the curve (AUC) to determine clearance, instead of deriving clearance from the individual model parameter estimates of elimination rate constant and distribution volume.
In conclusion, neither RYGB per se nor the subsequent weight loss impacted absolute bioavailability or systemic clearance of midazolam short term. Thus, our results suggest that, despite the extensive rearrangement of the gastrointestinal tract by RYGB, dose adjustments may not be necessary for the extensive group of drugs that is substrates for CYP3A metabolism in the early period after RYGB. In the long‐term perspective, absolute bioavailability decreased in the RYGB group, suggesting that CYP3A activity is recovered following substantial weight loss. This is also supported by an increased concentration of 4βOHC at year 2. A decreased absolute bioavailability and an increased concentration of 4βOHC were also observed in the diet group, although the majority of these patients had returned to their baseline weight at year 2, suggesting that other unknown processes also have influenced the CYP3A activity. The large inter‐ and intra‐individual pharmacokinetic variability should be kept in mind when initiating CYP3A therapies within a broad range of BMI, and warrants further investigation in future clinical trials.
CONFLICT OF INTEREST
S.A., C.K., and R.J.‐L. are AstraZeneca employees and own shares in AstraZeneca. T.B.A. and C.W. are former AstraZeneca employees. All other authors have no conflict of interest to declare.
AUTHOR CONTRIBUTIONS
K.E.K., I.R., and A.Å. wrote the manuscript. J.H., A.Å., S.A., C.K., T.B.A., H.C., E.S., and R.S. designed the research. I.R., V.K., L.K.J., P.C., B.M.W., K.H., P.A., C.W., and R.J. performed the research. K.E.K., I.R., A.Å., E.S., C.W., and P.A. analyzed the data.
Supporting information
Supplementary Material
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to express their gratitude to the participants, the surgical staff, and the study personnel working on the COCKTAIL study at Vestfold Hospital Trust. We are also grateful for the laboratory assistance provided by Grete Hasvold and master students (Anders Ulvmoen, Peter Huy Vo Le, Martin Vu, Ingeborg Karbø, Maria Ilyas, and Louise Borgenhard) at the Department of Pharmacy. The authors thank Matthew McGee for proofreading the manuscript. The authors thank Sabry Razick and Frode Strømsvåg at the University Center Information Technology (USIT), University of Oslo for performing optimization of Pmetrics. The optimization of Pmetrics was funded and carried out as an advanced user support from UNINETT Sigma2, the National Infrastructure for High Performance Computing and Data Storage in Norway (project NN9736K). The authors also thank the Swedish Research Council, approval numbers 5715 and 01951 (C.W., T.B.A., and P.A.) for supporting the proteomics analyses.
Kvitne KE, Robertsen I, Skovlund E, et al. Short‐ and long‐term effects of body weight loss following calorie restriction and gastric bypass on CYP3A‐activity – a non‐randomized three‐armed controlled trial. Clin Transl Sci. 2022;15:221–233. 10.1111/cts.13142
Funding information
This study was funded by the Vestfold Hospital Trust, Norway; Department of Pharmacy, University of Oslo, Norway; Swedish Research Council; and AstraZeneca, Sweden.
REFERENCES
- 1. Guh DP, Zhang W, Bansback N, Amarsi Z, Birmingham CL, Anis AH. The incidence of co‐morbidities related to obesity and overweight: a systematic review and meta‐analysis. BMC Public Health. 2009;9:88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Machado M, Marques‐Vidal P, Cortez‐Pinto H. Hepatic histology in obese patients undergoing bariatric surgery. J Hepatol. 2006;45:600‐606. [DOI] [PubMed] [Google Scholar]
- 3. Colquitt JL, Pickett K, Loveman E, Frampton GK. Surgery for weight loss in adults. Cochrane Database Sys Rev. 2014;8:CD003641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jakobsen GS, Småstuen MC, Sandbu R, et al. Association of bariatric surgery vs medical obesity treatment with long‐term medical complications and obesity‐related comorbidities. JAMA. 2018;319:291‐301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kral JG, Näslund E. Surgical treatment of obesity. Nat Clin Pract Endocrinol Metab. 2007;3:574‐583. [DOI] [PubMed] [Google Scholar]
- 6. Angeles PC, Robertsen I, Seeberg LT, et al. The influence of bariatric surgery on oral drug bioavailability in patients with obesity: A systematic review. Obes Rev. 2019;20:1299‐1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rendic S, Guengerich FP. Survey of human oxidoreductases and cytochrome P450 enzymes involved in the metabolism of xenobiotic and natural chemicals. Chem Res Toxicol. 2015;28:38‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Guengerich FP. Cytochrome P‐450 3A4: regulation and role in drug metabolism. Annu Rev Pharmacol Toxicol. 1999;39:1‐17. [DOI] [PubMed] [Google Scholar]
- 9. Paine MF, Hart HL, Ludington SS, Haining RL, Rettie AE, Zeldin DC. The human intestinal cytochrome P450 "pie". Drug Metab Dispos. 2006;34:880‐886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Brill MJ, van Rongen A, Houwink API, et al. Midazolam pharmacokinetics in morbidly obese patients following semi‐simultaneous oral and intravenous administration: a comparison with healthy volunteers. Clin Pharmacokinet. 2014;53:931‐941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. van Rongen A, Brill MJE, Vaughns JD, et al. Higher midazolam clearance in obese adolescents compared with morbidly obese adults. Clin Pharmacokinet. 2018;57:601‐611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ulvestad M, Skottheim IB, Jakobsen GS, et al. Impact of OATP1B1, MDR1, and CYP3A4 expression in liver and intestine on interpatient pharmacokinetic variability of atorvastatin in obese subjects. Clin Pharma Ther. 2013;93:275‐282. [DOI] [PubMed] [Google Scholar]
- 13. Krogstad V, Peric A, Robertsen I, et al. Correlation of body weight and composition with hepatic activities of cytochrome P450 enzymes. J Pharm Sci. 2021;110(1):432‐437. [DOI] [PubMed] [Google Scholar]
- 14. Jover R, Bort R, Gómez‐Lechón MJ, Castell JV. Down‐regulation of human CYP3A4 by the inflammatory signal interleukin‐6: molecular mechanism and transcription factors involved. FASEB J. 2002;16:1799‐1801. [DOI] [PubMed] [Google Scholar]
- 15. Kolwankar D, Vuppalanchi R, Ethell B, et al. Association between nonalcoholic hepatic steatosis and hepatic cytochrome P‐450 3A activity. Clin Gastroenterol Hepatol. 2007;5:388‐393. [DOI] [PubMed] [Google Scholar]
- 16. Woolsey SJ, Mansell SE, Kim RB, Tirona RG, Beaton MD. CYP3A activity and expression in nonalcoholic fatty liver disease. Drug Metab Dispos. 2015;43:1484‐1490. [DOI] [PubMed] [Google Scholar]
- 17. Rao SR. Inflammatory markers and bariatric surgery: a meta‐analysis. Inflamm Res. 2012;61:789‐807. [DOI] [PubMed] [Google Scholar]
- 18. Schwenger KJP, Fischer SE, Jackson T, Okrainec A, Allard JP. In nonalcoholic fatty liver disease, Roux‐en‐Y gastric bypass improves liver histology while persistent disease is associated with lower improvements in waist circumference and glycemic control. Surg Obes Relat Dis. 2018;14:1233‐1239. [DOI] [PubMed] [Google Scholar]
- 19. Nicklas JM, Sacks FM, Smith SR, et al. Effect of dietary composition of weight loss diets on high‐sensitivity c‐reactive protein: the Randomized POUNDS LOST trial. Obesity (Silver Spring). 2013;21:681‐689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Vilar‐Gomez E, Martinez‐Perez Y, Calzadilla‐Bertot L, et al. Weight loss through lifestyle modification significantly reduces features of nonalcoholic steatohepatitis. Gastroenterology. 2015;149:367‐78.e5:quiz e14–e15. [DOI] [PubMed] [Google Scholar]
- 21. Brill MJ, van Rongen A, van Dongen EP, et al. The pharmacokinetics of the CYP3A substrate midazolam in morbidly obese patients before and one year after bariatric surgery. Pharma Res. 2015;32:3927‐3936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Skottheim IB, Stormark K, Christensen H, et al. Significantly altered systemic exposure to atorvastatin acid following gastric bypass surgery in morbidly obese patients. Clin Pharmacol Ther. 2009;86:311‐318. [DOI] [PubMed] [Google Scholar]
- 23. Chan LN, Lin YS, Tay‐Sontheimer JC, et al. Proximal Roux‐en‐Y gastric bypass alters drug absorption pattern but not systemic exposure of CYP3A4 and P‐glycoprotein substrates. Pharmacotherapy. 2015;35:361‐369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hjelmesaeth J, Åsberg A, Andersson S, et al. Impact of body weight, low energy diet and gastric bypass on drug bioavailability, cardiovascular risk factors and metabolic biomarkers: protocol for an open, non‐randomised, three‐armed single centre study (COCKTAIL). BMJ Open. 2018;8:e021878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Krogstad V, Peric A, Robertsen I, et al. A comparative analysis of cytochrome P450 activities in paired liver and small intestinal samples from patients with obesity. Drug Metab Dispos. 2020;48:8‐17. [DOI] [PubMed] [Google Scholar]
- 26. Bodin K, Andersson U, Rystedt E, et al. Metabolism of 4 beta ‐hydroxycholesterol in humans. J Biol Chem. 2002;277:31534‐31540. [DOI] [PubMed] [Google Scholar]
- 27. Egeland EJ, Witczak BJ, Zaré HK, Christensen H, Åsberg A, Robertsen I. Chronic inhibition of CYP3A is temporarily reduced by each hemodialysis session in patients with end‐stage renal disease. Clin Pharmacol Ther. 2020;108(4):866‐873. [DOI] [PubMed] [Google Scholar]
- 28. Gjestad C, Huynh DK, Haslemo T, Molden E. 4β‐hydroxycholesterol correlates with dose but not steady‐state concentration of carbamazepine: indication of intestinal CYP3A in biomarker formation? Br J Clin Pharmacol. 2016;81:269‐276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Størset E, Hole K, Midtvedt K, Bergan S, Molden E, Åsberg A. The CYP3A biomarker 4β‐hydroxycholesterol does not improve tacrolimus dose predictions early after kidney transplantation. Br J Clin Pharmacol. 2017;83:1457‐1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wegler C, Garcia LP, Klinting S, et al. Proteomics‐informed prediction of rosuvastatin plasma profiles in patients with a wide range of body weight. Clin Pharmacol Ther. 2021;109(3):762‐771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wegler C, Ölander M, Wiśniewski JR, et al. Global variability analysis of mRNA and protein concentrations across and within human tissues. NAR Genom Bioinformatics. 2019;2(1):lqz010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wiśniewski JR, Mann M. Consecutive proteolytic digestion in an enzyme reactor increases depth of proteomic and phosphoproteomic analysis. Anal Chem. 2012;84:2631‐2637. [DOI] [PubMed] [Google Scholar]
- 33. Tyanova S, Temu T, Cox J. The MaxQuant computational platform for mass spectrometry‐based shotgun proteomics. Nat Protoc. 2016;11:2301‐2319. [DOI] [PubMed] [Google Scholar]
- 34. Huber W, von Heydebreck A, Sültmann H, Poustka A, Vingron M. Variance stabilization applied to microarray data calibration and to the quantification of differential expression. Bioinformatics. 2002;18(Suppl 1):S96‐S104. [DOI] [PubMed] [Google Scholar]
- 35. Wiśniewski JR, Rakus D. Multi‐enzyme digestion FASP and the ‘Total Protein Approach'‐based absolute quantification of the Escherichia coli proteome. J Proteomics. 2014;109:322‐331. [DOI] [PubMed] [Google Scholar]
- 36. Neely MN, van Guilder MG, Yamada WM, Schumitzky A, Jelliffe RW. Accurate detection of outliers and subpopulations with Pmetrics, a nonparametric and parametric pharmacometric modeling and simulation package for R. Ther Drug Monit. 2012;34:467‐476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. R Foundation for Statistical Computing . R: A language and environment for statistical computing. R Foundation for Statistical Computing; 2018. [Google Scholar]
- 38. Świętaszczyk C, Jødal L. Three‐compartment pharmacokinetic models of radiotracers used in the GFR‐determination ‐ estimation of their parameters using the time‐concentration curves. Nucl Med Rev Cent East Eur. 2019;22:60‐68. [DOI] [PubMed] [Google Scholar]
- 39. de Biasi J. Four open mammillary and catenary compartment models for pharmacokinetics studies. J Biomed Eng. 1989;11:467‐470. [DOI] [PubMed] [Google Scholar]
- 40. Kotronen A, Peltonen M, Hakkarainen A, et al. Prediction of non‐alcoholic fatty liver disease and liver fat using metabolic and genetic factors. Gastroenterology. 2009;137:865‐872. [DOI] [PubMed] [Google Scholar]
- 41. Darwich AS, Pade D, Ammori BJ, Jamei M, Ashcroft DM, Rostami‐Hodjegan A. A mechanistic pharmacokinetic model to assess modified oral drug bioavailability post bariatric surgery in morbidly obese patients: interplay between CYP3A gut wall metabolism, permeability and dissolution. J Pharm Pharmacol. 2012;64:1008‐1024. [DOI] [PubMed] [Google Scholar]
- 42. Sung‐Joon CI‐S, Dae‐Duk K. Obesity‐related physiological changes and their pharmacokinetic consequence. J Pharma Investig. 2013;43:161‐169. [Google Scholar]
- 43. Smith A, Henriksen B, Cohen A. Pharmacokinetic considerations in Roux‐en‐Y gastric bypass patients. Am J Health‐system Pharmacy. 2011;68:2241‐2247. [DOI] [PubMed] [Google Scholar]
- 44. Salem F, Abduljalil K, Kamiyama Y, Rostami‐Hodjegan A. Considering age variation when coining drugs as high versus low hepatic extraction ratio. Drug Metab Dispos. 2016;44:1099‐1102. [DOI] [PubMed] [Google Scholar]
- 45. Yang J, Liao M, Shou M, et al. Cytochrome p450 turnover: regulation of synthesis and degradation, methods for determining rates, and implications for the prediction of drug interactions. Curr Drug Metab. 2008;9:384‐394. [DOI] [PubMed] [Google Scholar]
- 46. Togao M, Kawakami K, Otsuka J, Wagai G, Ohta‐Takada Y, Kado S. Effects of gut microbiota on in vivo metabolism and tissue accumulation of cytochrome P450 3A metabolized drug: Midazolam. Biopharm Drug Dispos. 2020;41:275‐282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hole K, Heiberg PL, Gjestad C, Mehus LL, Rø Ø, Molden E. Elevated 4β‐hydroxycholesterol/cholesterol ratio in anorexia nervosa patients. Pharmacol Res Perspect. 2018;6:e00430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Brill MJ, Välitalo P, Darwich A, et al. Semiphysiologically based pharmacokinetic model for midazolam and CYP3A mediated metabolite 1‐OH‐midazolam in morbidly obese and weight loss surgery patients. CPT Pharmacometrics Syst Pharmacol. 2016;5:20‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Heizmann P, Eckert M, Ziegler WH. Pharmacokinetics and bioavailability of midazolam in man. Br J Clin Pharmacol. 1983;16(Suppl 1):43s‐s49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hohmann N, Kocheise F, Carls A, Burhenne J, Haefeli WE, Mikus G. Midazolam microdose to determine systemic and pre‐systemic metabolic CYP3A activity in humans. Br J Clin Pharmacol. 2015;79:278‐285. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Supplementary Material