

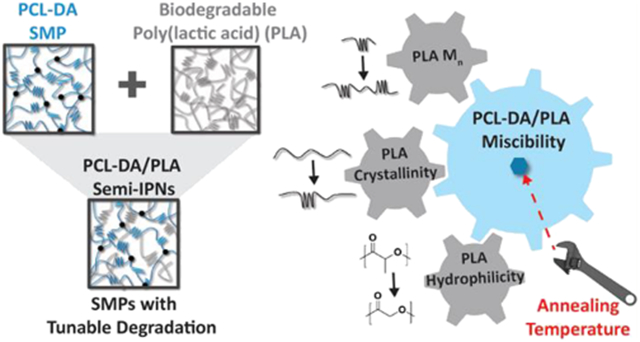
Abstract
The utility of poly(ε-caprolactone) (PCL) as a shape memory polymer (SMP) may be improved by accelerating its degradation. Recently, we have reported novel semi-interpenetrating networks (semi-IPNs) composed of cross-linked PCL diacrylate (PCL-DA) and thermoplastic poly(l-lactic acid) (PLLA) that exhibited SMP behavior, accelerated degradation, and enhanced moduli versus the PCL-DA control. Herein, we systematically varied the thermoplastic component of the PCL-based semi-IPNs, incorporating homo- and copolymers based on lactic acid of different Mn, hydrophilicity, and crystallinity. Specifically, semicrystalline PLLAs of different Mns (7.5, 15, 30, and 120 kDa) were explored as the thermoplastics in the semi-IPNs. Additionally, to probe crystallinity and hydrophilicity, amorphous (or nearly amorphous) thermoplastics of different hydrophilicities (PDLLA and PLGAs 85:15, 70:30, and 50:50, l-lactide:glycolide mole % ratio) were employed. For all semi-IPNs, the wt % ratio of the cross-linked PCL-DA to thermoplastic was 75:25. The nature of the thermoplastics was linked to semi-IPN miscibility and the trends in accelerated degradation rates.
Graphical Abstract
INTRODUCTION
Poly(ε-caprolactone) (PCL) is a thermoresponsive shape memory polymer (SMP) that has been explored as self-expanding stents,1-3 self-tightening sutures,4-6 and, recently, by our group, as self-fitting scaffolds to treat irregularly shaped bone defects.7-9 The crystalline lamellae of PCL serve as “switching segments”, actuated by a thermal melting temperature (Tm, ~55 °C). Thus, PCL solid films or porous scaffolds are able to hold a temporary shape following sequential heating (T > Tm), shaping, and cooling (T < Tm) and are able to recover their original shape upon heating (T > Tm).7-9 The slow degradation rate of PCL (>24 months) is a major limitation in its utility and is attributed to its high crystallinity (~45%) and hydrophobicity.10-12 In general, for aliphatic homopolyesters, increased rates of degradation are associated with greater hydrophilicity and lower crystallinity. For instance, compared to PCL, poly(glycolic acid) (PGA; ~45–65% crystalline) degrades more quickly (~6–12 months) as a result of its enhanced hydrophilicity. Attributed to its hydrophobicity and semicrystallinity (~25–35%), poly(l-lactic acid) (PLLA) likewise degrades slowly (>24 months), whereas amorphous poly(d,l-lactic acid) (PDLLA) degrades more quickly (2–16 months).11,12 While a lack of crystallinity is often associated with a desirably faster degradation rate, it occurs with a reduction in rigidity and strength. Moreover, retention of PCL crystallinity is essential to its shape memory behavior.
A strategy to modify PCL that would accelerate the rate of degradation but also improve mechanical properties and without loss of shape memory behavior is highly desirable. To tailor rates of degradation, most often, two or more polyesters are combined as copolymers or as blends.13-15 For instance, poly(l-lactic-co-glycolic acid) (PLGA) copolymers degrade faster than the parent homopolymers, and rates may be tuned by the lactic to glycolic acid mole % ratio, from ~1–2 (50:50) to ~5–6 months (85:15).16,17 However, since PLGA copolymers are generally amorphous, they exhibit reduced modulus values versus PGA or PLLA.12,16 Likewise, PCL has been copolymerized with d,l-lactide and glycolide, resulting in amorphous copolymers with faster rates of degradation but with a reduction in PCL crystallinity.18 PDLLA-PCL-PDLLA triblock copolymers degraded faster than the PCL homopolymer and exhibited a PCL Tm, but % crystallinity was not quantified nor were the mechanical properties.19 PCL has also been incorporated into blends, namely, for the purpose of enhancing the ductility and toughness of PLLA.20-22 By improving miscibility (i.e., reducing phase separation) with compatibilizers23-25 or reactive mixing,26,27 the mechanical properties of PCL/PLLA blends can be improved. In contrast, the immiscibility of PCL/PLLA blends contributes to enhanced rates of degradation.28 None of the aforementioned strategies utilize the cross-linked PCL network in combination with other polymers as a potential route to materials with a combination of shape memory behavior, accelerated degradation, and robust mechanical properties.
We have previously reported SMP PCL networks prepared from the photo-cross-linking of semicrystalline PCL-diacrylate (PCL-DA).7,8 Subsequently, semi-interpenetrating networks (semi-IPNs) were formed from a cross-linked PCL-DA network and thermoplastic PLLA (Mn = 15 kDa).9,29 Due to the retention of PCL crystallinity, the semi-IPNs retained their shape memory behavior. At a wt % ratio of 75:25 (PCL-DA/PLLA), semi-IPN scaffolds exhibited a superior modulus and strength but also an accelerated rate of degradation. Notably, the degradation of the semi-IPN was faster than that of the analogous blend. The accelerated degradation of the semi-IPNs was speculated to be related to phase separation.30 The ability to accelerate the degradation of PCL via a semi-IPN design, without compromising shape memory or mechanical properties, holds potential to improve the performance of numerous bioresorbable devices.
Herein, the thermoplastic PLA within a PCL-based semi-IPN was systematically varied to examine the impact on semi-IPN degradation and mechanical properties (Figure 1). For Group A, semicrystalline PLLAs of varying Mns were incorporated as the thermoplastic component to assess the impact of thermoplastic Mn on semi-IPN properties. For Group B, semicrystalline PLLA as well as amorphous PDLLA and various amorphous/nearly amorphous PLGAs (85:15, 70:30, and 50:50, l-lactide:glycolide) were incorporated as the thermoplastic (Mn maintained at ~15 kDa) to assess the impact of hydrophilicity on semi-IPN properties. For all semi-IPNs, the wt % ratio of PCL-DA to thermoplastic was maintained at 75:25. The extent of phase separation was evaluated via scanning electron microscopy (SEM). Additionally, the degradation rates and accompanying erosion behavior, shape memory behavior, and mechanical properties were assessed. For select compositions, the impact of thermal annealing on miscibility and subsequent degradation rates and mechanical properties was also evaluated.
Figure 1.

Semi-IPN compositions based on a PCL-DA network and various PLA-based thermoplastics. Group A: semicrystalline PLLA of varying Mns. Group B: semicrystalline PLLA (~15 kDa), amorphous PDLLA (~15 kDa), and amorphous/nearly amorphous PLGAs (85:15, 70:30, and 50:50, ~15 kDa). The semi-IPNs are designated by the thermoplastic component.
EXPERIMENTAL SECTION
Materials.
PCL-diol (Mn = 10 kDa per manufacturer specifications), 4-(dimethylamino)pyridine (DMAP), trimethylamine (Et3N), acryloyl chloride, potassium carbonate (K2O3), anhydrous magnesium sulfate (MgSO4), (3S)-cis-3,6-dimethyl-1,4-dioxane-2,5-dione (l-lactide), 3,6-dimethyl-1,4-dioxane-2,5-dione (lactide), glycolide, tin(II) 2-ethylhexanoate [Sn(Oct)2], ethylene glycol, PLLA (Mn ~ 120 kDa), 2,2-dimethoxy-2-phenyl acetophenone (DMP), 1-vinyl-2-pyrrolidi-none (NVP), sodium hydroxide (NaOH), and common solvents were purchased from Sigma-Aldrich. All solvents and ethylene glycol were dried over 4 Å molecular sieves prior to use. All monomers and polymers were vacuum-dried prior to use.
Syntheses.
All reactions were run under a nitrogen (N2) atmosphere with a Teflon-covered stir bar. Chemical structures (including % acrylation and Mn) were confirmed with 1H NMR spectroscopy (Inova 500 MHz spectrometer operation in the FT mode with CDCl3 as the standard). The purified polymers’ thermal properties were characterized using differential scanning calorimetry (DSC, TA Instruments Q100) as described below.
PCL-diol (Mn = 10 kDa; Tg = −65 °C, Tm = 53 °C, 46.5% crystallinity) was reacted with acryloyl chloride to prepare PCL-diacrylate (PCL-DA) as previously reported.7 Briefly, PCL-diol (20.0 g, 2.0 mmol) was dissolved in CH2Cl2 (120 mL) with DMAP (6.6 mg) as the catalyst. After purging with N2, triethylamine (4.0 mmol) and acryloyl chloride (8.0 mmol) were sequentially added to the flask via dropwise incorporation, and the reaction was stirred at RT for 30 min. The crude polymer was purified to obtain PCL-DA (Mn confirmed ~10 kDa via 1H NMR end-group analysis, >90% acrylation, >80% yield).7
PLLAs [7.5, 15, and 30 kDa] were prepared via ring opening polymerization (ROP) of l-lactide (6.0 g) using ethylene glycol as the initiator and Sn(Oct)2 as the catalyst at 120 °C overnight.31 Mn was controlled via the molar equivalence ratio of monomer to initiator: [52:1] (7.5 kDa), [104:1] (15 kDa), and [208:1] (30 kDa). The crude products were each dissolved in a minimal amount of CHCl3, precipitated in methanol, filtered, and vacuum-dried. The Mns of the purified products were verified by 1H NMR end-group analysis (CH δ = 5.2 ppm and CH3 δ = 1.5 ppm in the repeat unit compared to terminal CH3 δ = 4.4 ppm). PLLA [120 kDa] was used without further purification. PLLAs exhibited the following thermal transitions: 7.5 [Tg = 45 °C, Tm = 153 °C, 49.3% crystallinity], 15 [Tg = 45 °C, Tm = 155 °C, 52.7% crystallinity], 30 [Tg = 46 °C, Tm = 159 °C, 56.2% crystallinity], and 120 kDa [Tg = 50 °C, Tm = 170 °C, 20.0% crystallinity].
PDLLA [15 kDa] was prepared via ROP of d,l-lactide (6.0 g) as above, and the Mn of the purified product was likewise verified by 1H NMR end-group analysis. The PDLLA exhibited the following thermal transitions: Tg = −28 °C, no Tm observed, 0% crystallinity.
PLGAs [15 kDa] were similarly prepared via ROP of different molar ratios of l-lactide to glycolide [85:15, 70:30, and 50:50, 6.0 g monomer total for all PLGA ROPs]. The Mns of the purified products were verified by 1H NMR end-group analysis (CH δ = 5.2 ppm and CH3 δ = 1.5 ppm in the repeat unit compared to terminal CH3 δ = 4.4 ppm). The target ratios of l-lactide to glycolide were verified using 1H NMR (CH δ = 5.2 ppm and CH3 δ = 1.5 ppm in the PLLA repeat unit compared to CH2 δ = 4.6–4.8 ppm in the PGA repeat unit). The PLGAs exhibited the following thermal transitions: 85:15 (Tg = 41 °C, Tm = 137 °C, 3.2% crystallinity), 70:30 (Tg = 37 °C, no Tm observed, 0% crystallinity), and 50:50 (Tg = 21 °C, no Tm observed, 0% crystallinity).
Film Fabrication.
All semi-IPN films were prepared with a 75:25 wt % ratio of PCL-DA to the designated PLA thermoplastic. For Group A, PLLAs of varying Mns (7.5, 15, 30, and 120 kDa) were incorporated as the thermoplastic. For Group B, PDLLA (15 kDa) and PLGAs (15 kDa) [85:15, 70:30, and 50:50, molar ratios l-lactide:glycolide] were incorporated as the thermoplastic. The semi-IPNs are herein denoted according to their thermoplastic component (e.g., 7.5 k PLLA for the PCL-DA/7.5 kDa PLLA semi-IPN, PDLLA for the PCL-DA/PDLLA semi-IPN, and 85:15 PLGA for the PCL-DA/85:15 PLGA semi-IPN). A PCL-DA/PCL-diol semi-IPN control (PCL-diol) was prepared by incorporating PCL-diol (Mn ~10 kDa) as the thermoplastic component into the PCL-DA. A 100% PCL-DA only control (100% PCL-DA; i.e., no thermoplastic) was also prepared.
Semi-IPN films were prepared from polymer solutions with the designated polymer(s) (25 wt % total polymer in DCM) using 15 vol % photoinitiator solution (10 wt % DMP in NVP). The solution was transferred to a silicone mold (2 mm thickness; McMaster-Carr) secured between two glass slides and was exposed to UV light (UV-Transilluminator, 6 mW cm−2, 365 nm) for 3 min per side. The solvent-swollen discs were sequentially air-dried (RT, overnight), dried under vacuum (RT, 4 h, 30 in. Hg), soaked in EtOH on a shaker table (3 h, 150 rpm), air-dried (RT, overnight), and lastly annealed (85 °C, 1 h, 30 in. Hg). In addition, select semi-IPN films [15 k PLLA, PDLLA, and 85:15 PLGA] and the 100% PCL-DA control were also fabricated as above but with a higher annealing temperature (170 °C, 1 h, 30 in. Hg). The final thickness of films was ~1.1 mm. Blend controls were prepared at a 75:25 wt % ratio of PCL-diol to a particular PLA thermoplastic (15 k PLLA, PDLLA, and 85:15 PLGA). These blends as well as the PCL-diol only control were prepared as films (5 mm × 1.1 mm, McMaster-Carr) via simple melt casting, where the polymer(s) was heated to just above any Tm of the polymer(s), and ~35 mg of polymer mixture was cast into a silicone mold (5 mm × 1.1 mm, McMaster-Carr) on top of a glass slide. Films were removed from heat after 3 min and were allowed to set at RT for >24 h. All films were stored in a desiccator prior to use.
Thermal Transitions and % Crystallinity.
Differential scanning calorimetry (DSC; TA Instruments Q100) was utilized to determine the thermal transitions (Tg and Tm) and % crystallinity of thermoplastic polymers as well as semi-IPN films. Specimens (~10 mg; N = 3) were sealed in hermetic pans that were heated from RT to 200 °C at a heating rate of 5 °C min−1. For thermoplastic polymers, values were taken from the second DSC cycle to remove any thermal history. For semi-IPN films, values were taken from the first DSC cycle to account for the thermal history endued by the fabrication. From the endothermic PCL and PLLA melting peaks, Tm and enthalpy change (ΔHm) were measured. Percent crystallinity (% χc) of thermoplastic polymers was calculated by
| (1) |
where ΔHm is the enthalpy of fusion taken from the integral of the endothermic melt peak, ΔHc is the enthalpy of crystallization from the exothermic cold crystallization peak, and is the theoretical value for 100% crystalline PCL (139.5 J/g)32 or PLLA (93.0 J/g).33 For semi-IPN films, a correction factor was used to account for the weight percent of each polymer component
| (2) |
where w is the mass fraction of the given polymer component.
Thermal Gravimetric Analysis (TGA).
TGA (TA Instruments Q50) of semi-IPN films (~10 mg, N = 1) in a platinum pan was run under N2 from RT to 500 °C at a heating rate of 10 °C min−1. The mass of the samples throughout heating was determined to quantify percent mass loss.
Sol Content.
Discs (~5 mm × ~1.1 mm; N = 3) were immersed in 10 mL of CH2Cl2 using one specimen per scintillation vial. Vials were maintained for 48 h at 150 rpm (VWR Mini Shaker). Swollen discs were then removed, air-dried, and dried in vacuo (30 in. Hg, RT, ~24 h), and the mass of dried discs was determined to quantify percent mass loss (i.e., sol content).
Degradation Behavior.
Base-catalyzed accelerated degradation tests were performed in 1 M NaOH according to ASTM F1635. Film specimens (d ~5 mm, h ~1.1 mm) (N = 3 per time point) were immersed in 10 mL of the basic solution in a sealed glass vial and maintained at 37 °C at 60 rpm (VWR Benchtop Shaking Incubator Model 1570). At each designated time point (8, 24, 48, 72, 120, and 168 h), specimens were removed from the solution, thoroughly rinsed with DI water, blotted, and dried under vacuum (RT, overnight, 30 in. Hg). For each specimen, the initial mass and mass at the designated time point were measured with an analytical balance. Specimens were utilized for a single time point only. Surface and cross-section images of select specimens (those with a mass loss of ~20%) were obtained for SEM. Films were subjected to Au-Pt sputter coating (~7 nm), and images were obtained with a Tescan Vega 3 SEM (accelerating voltage of 10 kV).
Phase Separation.
SEM images of film surfaces and cross-sections were likewise performed to assess differences in phase separation (i.e., miscibility).
Tensile Properties.
Tensile properties of films were evaluated at RT with a tensile tester (Instron 5944). Rectangular specimens (l ~29 mm, w ~5 mm, t ~0.9 mm; N = 8) were secured with pneumatic clamps (gauge length of 7 mm) and subjected to a constant rate of strain (50 mm/min). From the resulting stress–strain curves, modulus (E) and tensile strength (TS) were determined.
Shape Memory Behavior.
Rectangular specimens (l ~20 mm × w ~5 mm × t ~0.9 mm) were subjected to the following sequence: (1) after exposure to a water bath at 85 °C (T > Tm) for 1 min, the film strip was deformed into a coiled shape with a metal mandrel and (2) cooled at RT for 3 min to fix this temporary shape. (3) The fixed coil was placed in a 85 °C water bath, and recovery was observed at t = 0 and 10 s.
Statistical Analyses.
Data were reported as mean ± standard deviation. Values were compared in GraphPad Prism via ANOVA followed by a t test where a p-value of <0.05 was considered statistically significant.
RESULTS AND DISCUSSION
Synthesis of Thermoplastic Polymers.
Prior to their incorporation into semi-IPN films, all thermoplastic polymers were extensively characterized in terms of their structure and Mn (by 1H NMR) as well as their Tg, Tm, and % crystallinity (by DSC) (Figure S1). The targeted Mns were achieved as were the ratios of l-lactide to glycolide for PLGA copolymers. For PLLAs, % crystallinity was generally similar (~49 to 56%) as Mn increased (7.5 to 30 kDa, respectively) but then decreased substantially for the 120 k PLLA (~20%). This decreased % crystallinity for high Mn PLLA is consistent with a prior report.34 While 70:30 and 50:50 PLGA copolymers were amorphous, the 85:15 PLGA (i.e., the highest l-lactide content) exhibited a very low level of PLLA % crystallinity (~3%), also in agreement with the prior literature.17,35
Semi-IPN Fabrication.
Semi-IPNs composed of cross-linked PCL-DA and a PLA-based thermoplastic were prepared to assess the resulting impact on material properties. For Group A, semicrystalline PLLAs of varying Mns were incorporated as the thermoplastic component to assess the impact of thermoplastic Mn on semi-IPN properties. For Group B, semicrystalline PLLA, amorphous PDLLA, and various amorphous/nearly amorphous PLGAs (85:15, 70:30, and 50:50, l-lactide:glycolide) were incorporated as the thermoplastic (Mn maintained at ~15 kDa) to assess the impact of hydrophilicity on semi-IPN properties. The comparison of PLLA (15 k) and PDLLA (15 k) permitted evaluation of the effect of crystallinity. Additionally, comparison of PDLLA and PLGAs allowed assessment of hydrophilicity. PCL-DA/PCL-diol semi-IPN control (PCL-diol) was prepared by incorporating PCL-diol (Mn ~10 kDa) as the thermoplastic component into the PCL-DA.
The targeted 75:25 wt % ratios of PCL-DA to PLA thermoplastic (i.e., that of the precursor solutions) in the resulting semi-IPN films were confirmed with TGA experiments (Figure S2a). The thermoplastic polymers underwent a distinct onset of thermal degradation at a temperature lower than that of the PCL-DA network (~250 °C versus >400 °C). All semi-IPNs showed ~25% mass loss at temperatures below 400 °C, indicating a 75:25 wt % ratio of PCL-DA to thermoplastic. Because the PCL-diol displayed thermal stability similar to the PCL-DA, the wt % ratio of the PCL-diol semi-IPN could not be likewise confirmed.
Additionally, the sol content values of films were determined to confirm adequate cross-linking of the PCL-DA upon formation of the semi-IPNs (Figure S2b). The 100% PCL-DA control film (i.e., no thermoplastic) had a sol content value of ~12%. None of the semi-IPNs exceeded 37% sol content, a maximum value expected based on combined contributions of sol from the PCL-DA (~12%) and the thermoplastic polymer (~25%). The sol content of the 120 k PLLA (i.e., the semi-IPN composed of PCL-DA and 120 k PLLA) was particularly low (~15%) versus all other semi-IPNs, which was attributed to the diminished capacity of this high Mn PLLA to be extracted. These sol content results confirm that PCL-DA (>95% acrylation) was able to undergo cross-linking in the presence of all PLA thermoplastics, effectively forming the designated PCL-DA/PLA semi-IPN compositions.
Degradation Behavior.
PCL-DA/PLA semi-IPN degradation rates were affected by the PLA-based thermoplastic’s Mn (Group A) and hydrophilicity as well as crystallinity (Group B) (Figure 2). For Group A semi-IPNs, the rates of degradation were distinctly faster when based on lower Mn semicrystalline PLLAs (7.5 k, 15 k, and 30 k) versus the highest Mn semicrystalline PLLA (120 k). In fact, the degradation profile of the 120 k PLLA semi-IPN was similarly as slow as the 100% PCL-DA and PCL-diol controls. The 15 k PLLA semi-IPN degraded faster than the other semi-IPNs in this group. For Group B semi-IPNs, the thermoplastic Mn was maintained at 15 kDa, and they were semicrystalline (15 k PLLA) or amorphous/nearly amorphous but varied in terms of hydrophilicity (i.e., PDLLA < 85:15 PLGA < 70:30 PLGA < 50:50 PLGA). For this group, the 15 k PLLA semi-IPN degraded most rapidly, whereas the 85:15 PLGA and PDLLA semi-IPNs degraded at fast but relatively intermediate rates, and the 70:30 PLGA and 50:50 PLGA semi-IPNs degraded at slow rates, similar to the controls. The 15 k PLLA semi-IPN degraded faster than the PDLLA semi-IPN and all other semi-IPNs in this group.
Figure 2.

Mass loss under accelerated conditions (1 M NaOH, 37 °C, 60 rpm) for semi-IPNs and controls. The semi-IPNs are designated by the thermoplastic component.
The semi-IPNs and controls were subsequently grouped into two categories according to their relative degradation rates: “fast” (15 k PLLA > 30 k PLLA ≈ 7.5 k PLLA > PDLLA ≈ 85:15 PLGA) and “slow” (70:30 PLGA > 120 k PLLA≈ 50:50 PLGA > PCL-diol ≈ 100% PCL-DA) (Figure 3). Mass loss and SEM of film surfaces were acquired at different time points (8 h, 48 h, and 1 week) during degradation to reveal distinctions in degradation rates and the accompanied erosion behavior. At just 8 h, the fast degrading 15 k PLLA semi-IPN exhibited statistically greater mass loss (~20% mass loss) and visual evidence of erosion versus the 7.5 k PLLA and 30 k PLLA semi-IPNs (mass loss ~10%) (Figure 3a). By 48 h, all three of these semi-IPNs completely degraded (i.e., 100% mass loss). Among the next fast degrading semi-IPNs, substantial mass loss and erosion were apparent after 48 h, with the PDLLA and 85:15 PLGA semi-IPNs exhibiting statistically similar mass loss (~20% mass loss) (Figure 3b). At 1 week, nearly complete mass loss was observed for these two semi-IPNs. Finally, for slow degrading semi-IPNs, only at 1 week did these exhibit appreciable mass loss (Figure 3c). Here, the 70:30 PLGA semi-IPN (~20% mass loss) degraded faster versus the 120 k PLLA and 50:50 PLGA semi-IPNs (~10% mass loss), whereas the PCL-diol semi-IPN control and 100% PCL-DA controls degraded slowest (~5% mass loss). Corresponding SEM images of specimen cross sections were obtained and likewise highlight the observed trends observed in the SEM images of film surfaces (Figure S3). The fastest degrading compositions exhibited signs of surface erosion, but this may be attributed to the base-catalyzed conditions, which has been reported for the previously studied 15 k PLLA semi-IPN30 and for various PLLA-based polyesters.36
Figure 3.

(a–c) Mass loss under accelerated conditions (1 M NaOH, 37 °C, 60 rpm) for semi-IPNs and controls and (d) corresponding SEM images of film surfaces at the noted time points. Scale bar = 250 μm.
These differences in semi-IPN degradation rates were assessed based on the following considerations: PCL % crystallinity, PLLA % crystallinity, and finally the extent of phase separation (i.e., immiscibility) observed in the semi-IPNs. First, PCL % crystallinity was considered since a reduction would be expected to produce accelerated degradation rates. However, this was statistically similar among all the semi-IPNs (~40%; corrected for relative mass percent of PCL-DA in the semi-IPN) (Figure S4a). Next, the PLLA % crystallinity of semi-IPNs was assessed in cases where semi-IPNs were prepared with semicrystalline thermoplastics (Figure S4b). Among Group A, the PLLA % crystallinity of the 120 k PLLA semi-IPN (~33%) was significantly reduced versus those of 7.5 k, 15 k, and 30 k PLLA semi-IPNs (~45%). The relative rates of hydrolytic degradation of PLLA homopolymers are known to increase with an increase in PLLA Mn, provided that there is a concomitant decrease in % crystallinity that promotes water diffusion.34 In this study, the 120 k PLLA semi-IPN degraded slowly despite it being the least crystalline of the PLLAs investigated. For Group B, it was noted that the 85:15 PLGA thermoplastic exhibited minor % crystallinity (~3%), but for the 85:15 PLGA semi-IPN, the PLLA % crystallinity increased (~20%) (Figure S4b). Interestingly, in this group, the 15 k PLLA semi-IPN degraded the most quickly followed by the 85:15 PLGA and PDLLA semi-IPNs, while the degradation rates of the 70:30 PLGA and 50:50 PLGA semi-IPNs were slow, similar to the PCL-diol and PCL-DA controls. Thus, in these PCL-based semi-IPNs, the incorporation of relatively more crystalline and hydrophobic PLA-based polymers ultimately resulted in faster rates. This is contrary to the trends of hydrolysis rates of homo- or copolymers based on crystallinity and hydrophilicity. Therefore, the extent of phase separation (i.e., immiscibility) was finally considered to explain the relative degradation rates of the semi-IPNs.
It has been previously shown that the degree of phase separation in polymer blends can influence degradation as well as mechanical properties.20,21,37 SEM imaging has been previously used to categorize the degree of immiscibility in polymer systems, where phase separation is marked by coalescence, or defined circular regions where one polymer has separated from the other.38,39 Prior to degradation, SEM images of the controls and semi-IPN film surfaces (Figure 4) and cross sections (Figure S5) revealed distinct phase morphologies. Each composition was characterized as “miscible”, “partially miscible”, or “immiscible” corresponding to slow, fast, or slow degradation rates, respectively. The PCL-diol semi-IPN and 100% PCL-DA control, the slowest degrading films, exhibited the highest degree of miscibility as evidenced by their uniform morphologies. This was expected based on their chemical homogeneity. Slow degrading 120 k PLLA (Group A) and 70:30 PLGA and 50:50 PLGA (Group B) semi-IPNs exhibited the highest degree of immiscibility as evidenced by distinct domains indicative of polymer coalescence. Semi-IPNs 15 k PLLA, 7.5 k PLLA, and 30 k PLLA (Group A) and PDLLA and 85:15 PLGA (Group B) were deemed as partially miscible based on their intermediate morphologies; these corresponded to the fastest degradation rates. In the case of the 15 k PLLA, 7.5 k PLLA, and 30 k PLLA semi-IPNs, their partial miscibility (rather than immiscibility like that of the 120 k PLLA semi-IPN) could be attributed to similar Mns of these PLLAs versus that of the PCL-DA (10 kDa). The partial miscibility of the PDLLA and 85:15 PLGA semi-IPNs (versus the immiscibility of 70:30 PLGA and 50:50 PLGA semi-IPNs) could be attributed to their lack of hydrophilicity that renders them more similar to the hydrophobic PCL-DA. In total, the degree of phase separation of the semi-IPNs could be correlated to the degradation rate. Partial miscibility led to the fastest rates of degradation, presumably by reducing the barrier to water penetration. Immiscible semi-IPNs degraded more slowly due to domains of greater homogeneity that effectively acted like individual miscible phases, thereby slowing water penetration and subsequent degradation.
Figure 4.

SEM images of semi-IPNs and control film surfaces prior to degradation. Categorization of miscibility (miscible, partially miscible, or immiscible) based on the extent of phase separation and corresponding relative rates of degradation (slow or fast). Scale bar = 50 μm.
Tensile Properties.
Tensile testing was performed to determine the modulus and TS values of the semi-IPNs and controls (Figure 5). The modulus values of all PLA-containing semi-IPNs were significantly improved compared to those of the 100% PCL-DA and the PCL-diol semi-IPN controls. This is notable given that phase separation, observed to varying degrees for all semi-IPNs, is known to negatively impact mechanical properties such as modulus.14,21 While all semi-IPNs retained similar PCL % crystallinity versus the PCL-DA control (Figure S4), their increased moduli are also notable given the substitution of 25 wt % cross-linked PCL-DA for a thermoplastic. Not unexpectedly, the greatest increases in modulus were for semi-IPNs based on PLLAs of greatest crystallinity (e.g., 7.5 k PLLA, 15 k PLLA, and 30 k PLLA). The TS values of the PLA-containing semi-IPNs did not always surpass but did remain within 25% of that of the PCL-DA control. TS values were likewise the highest for semi-IPNs prepared from semicrystalline thermoplastics.
Figure 5.

(a) Tensile modulus of semi-IPNs and controls; **p < 0.01 versus the PCL-DA control. (b) Tensile strength of semi-IPNs and controls. The gray bar represents ±25% of the tensile strength of the PCL-DA.
Shape Memory Properties.
SMP properties of the semi-IPNs were analyzed qualitatively. Like the 100% PCL-DA control, all compositions were able to fix (“hold”) a temporary coil shape following sequential heating, shaping, and cooling (Figure S6a). After exposure to water (85 °C), all compositions also recovered their original shape within 10 s (Figure S6b). This result was expected since the PCL lamellae serve as the switching segments, and the PCL crystallinity was retained for all semi-IPNs (Figure S4a).
Effect of Increased Annealing Temperature.
In the aforementioned experiments, the semi-IPNs were annealed at Tanneal = 85 °C (i.e., greater than Tm,PCL ~55 °C) in the final step of their fabrication. In our previous work, 15 k PLLA was prepared as porous scaffolds using Tanneal = 85 °C as well as 170 °C (i.e., greater than Tm,PLLA ~155 °C); the latter higher temperature induced greater shrinking and a concomitant reduction in pore size.9 Herein, it was hypothesized that a higher annealing temperature would impact relative degradation rates. Thus, select semi-IPNs were chosen due to their partial miscibility and relatively fast rates of degradation (decreasing in order 15 k PLLA > PDLLA ≈ 85:15 PLGA) and their equivalent thermoplastic Mn (~15 kDa) (Figure S7). Analogous melt-casted blends were also included to determine distinct degradation behaviors versus semi-IPNs.
The relative degradation rates of the 100% PCL-DA control were not substantially impacted by the annealing temperature attributed to its chemical homogeneity and miscibility (Figure 6a). For the 15 k PLLA semi-IPNs, annealing at the higher temperature resulted in a decrease in the rate of degradation, with semi-IPNs annealed at 85 °C fully degrading within 72 h while those annealed at 170 °C lasting beyond 1 week (Figure 6b). For the PDLLA semi-IPNs, degradation also slowed when annealed at the higher temperature, but mass loss differences were not significant until the one-week time point (Figure 6c). Finally, for the 85:15 PLGA semi-IPNs, the higher annealing temperature dramatically reduced the rate of degradation, and the blend degraded similarly slowly (Figure 6d). In the case of analogous blends, the 100% PCL-diol blend lost significantly less mass at 1 week versus the PCL-DA controls. Analogous blends of 15 k PLLA and 85:15 PLGA semi-IPNs degraded more slowly. However, the blend analogue of the PDLLA semi-IPN degraded somewhat more quickly. PCL % crystallinity was dramatically increased for blends by >50% for all compositions including the control (Figure S8a). PLLA % crystallinity was somewhat similar or slightly lower (Figure S8b). Despite the lack of cross-linking, the higher PCL % crystallinity of blends may contribute to their differing degradation profiles versus the corresponding semi-IPNs.
Figure 6.

Mass loss under accelerated conditions (1 M NaOH, 37 °C, 60 rpm) for compositions annealed at 85 °C, at 170 °C, and analogous melt-casted blends: (a) 100% PCL-DA control and 100% PCL-diol blend, (b) 15 k PLLA semi-IPNs and blend, (c) PDLLA semi-IPNs and blend, and (d) 85:15 PLGA semi-IPNs and blend. *p < 0.05 and **p < 0.01 versus the corresponding compositions annealed at 85 °C.
Irrespective of annealing temperature, semi-IPNs exhibited similar PCL % crystallinity (Figure S8A) and a similar or slight increase in PLLA % crystallinity (for 15 k PLLA and 85:15 PLGA semi-IPNs, respectively) (Figure S8B). Thus, % crystallinity does not appear to be a major factor in the relatively slower degradation rates of the semi-IPNs annealed at the higher temperature. So, for nondegraded films, phase separation of the surfaces (Figure 7a) and cross sections (Figure S9) was evaluated. For all semi-IPNs, the extent of phase separation was reduced when annealed at 170 °C, which likely resulted in the observed slower rates of degradation.
Figure 7.

(a) SEM of semi-IPN and control film surfaces (annealed at 85 or 170 °C) prior to degradation; scale bar = 50 μm, (b) modulus and (c) TS. Statistics noted in “red” indicate comparisons between the semi-IPNs and 100% PCL-DA control annealed at 170 °C; statistics noted in “black” indicate comparisons between the designated film annealed at 170 °C and the analogous film annealed at 85 °C. *p < 0.05 and **p < 0.01.
As noted above, these selected PCL-DA/PLA semi-IPNs exhibited higher modulus values and maintained TS values versus the PCL-DA control when annealed at 85 °C (Figure 5). To determine the impact of the higher annealing temperature, the semi-IPNs and control annealed at 170 °C were likewise evaluated (Figure 7b,c). When annealed at this higher temperature, the semi-IPNs all exhibited higher modulus and TS values versus the PCL-DA controls. For PDLLA and 85:15 PLGA semi-IPNs, the higher annealing temperature resulted in a statistically significant increase in modulus and TS values versus when annealed at the lower temperature. This may be attributed to their decreased phase separation. Moreover, when annealed at 170 °C, all of these semi-IPNs all exhibited significantly higher modulus and significantly higher TS values versus the 100% PCL-DA controls. Finally, shape fixity and shape recovery were shown to be maintained for semi-IPNs annealed at 170 °C, attributed to the maintenance of PCL % crystallinity (Figure S10).
CONCLUSIONS
This study demonstrated that the degradation rates of PCL-based SMPs can be accelerated and tuned with a semi-IPN design composed of cross-linked PCL-DA and a PLA-based thermoplastic of varying Mn, crystallinity, and hydrophilicity. It was shown that the extent of phase separation observed for the semi-IPNs was linked to the relative rates of degradation. PCL-DA/PLA semi-IPNs that resulted in partial miscibility exhibited relatively fast rates of degradation, whereas greater immiscibility led to relatively slow rates of degradation (Figure 8). However, all semi-IPNs degraded faster than the PCL-DA control. Among partially miscible semi-IPNs, fabrication that included a higher annealing temperature resulted in increased miscibility and a slower rate of degradation. Due to their retention in PCL crystallinity, all semi-IPNs exhibited shape memory behavior. Finally, the modulus increased for all semi-IPNs versus the PCL-DA control. Annealing at higher temperatures can reduce phase separation, thereby reducing the rate of degradation and potentially improving mechanical properties. This work points to the utility of a semi-IPN design to create PCL-based semi-IPNs with tunable degradation rates and improved rigidity.
Figure 8.

(a) Key properties of PLA-based thermoplastics as well as annealing temperature may be considered in preparing PCL-DA/PLA semi-IPNs with accelerated rates of degradation. (b) The relationship between degradation rates and miscibility observed for PCL-DA/PLA semi-IPNs; details about the PLA thermoplastic components are listed.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by NIH NIDCR 1R01DK095101-01A1. The use of the Texas A&M Microscopy and Imaging Center is acknowledged. The FE-SEM acquisition was supported in part by the National Science Foundation under Grant No. DBI-0116835.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.biomac.0c00454.
1H NMR and DSC data for all polymers, 1H NMR showing PCL acrylation, 1H NMR showing PLLA with increasing Mn, and 1H NMR showing PLGA with increasing glycolide content. TGA and sol content to verify film compositions and cross-linking. SEM images showing cross sections of degraded films at selected time points. PCL and PLLA % crystallinity in control and semi-IPN films. SEM images showing cross sections of nondegraded films to analyze relative polymer miscibility. Qualitative analyses of SMP properties in semi-IPN films. Schematic summarizing the study to assess the impact of annealing temperature of selected semi-IPNs and the analogous blend controls. PCL and PLLA % crystallinity in semi-IPNs annealed at 85 and 170 °C and those of analogous blends. SEM of semi-IPN and control film cross sections annealed at 85 or 170 °C prior to degradation. Qualitative analyses of SMP properties in semi-IPN films annealed at 170 °C (PDF)
The authors declare no competing financial interest.
Contributor Information
Michaela R. Pfau, Department of Biomedical Engineering, Texas A&M University, College Station, Texas 77843, United States
Kelly G. McKinzey, Department of Biomedical Engineering, Texas A&M University, College Station, Texas 77843, United States
Abigail A. Roth, Department of Biomedical Engineering, Texas A&M University, College Station, Texas 77843, United States
Melissa A. Grunlan, Department of Biomedical Engineering, Department of Materials Science and Engineering, and Department of Chemistry, Texas A&M University, College Station, Texas 77843, United States.
REFERENCES
- (1).Ansari M; Golzar M; Baghani M; Soleimani M Shape memory characterization of poly(ε-caprolactone) (PCL)/polyurethane (PU) in combined torsion-tension loading with potential applications in cardiovascular stent. Polym. Test 2018, 68, 424–432. [Google Scholar]
- (2).Yang C-S; Wu H-C; Sun J-S; Hsiao H-M; Wang T-W Thermo-Induced Shape-Memory PEG-PCL Copolymer as a Dual-Drug-Eluting Biodegradable Stent. ACS Appl. Mater. Interfaces 2013, 5, 10985–10994. [DOI] [PubMed] [Google Scholar]
- (3).Xue L; Dai S; Li Z Biodegradable shape-memory block copolymers for fast self-expandable stents. Biomaterials 2010, 31, 8132–8140. [DOI] [PubMed] [Google Scholar]
- (4).Lendlein A; Langer R Biodegradable, Elastic Shape-Memory Polymers for Potential Biomedical Applications. Science 2002, 296, 1673. [DOI] [PubMed] [Google Scholar]
- (5).Jing X; Mi H-Y; Huang H-X; Turng L-S Shape memory thermoplastic polyurethane (TPU)/poly(ε-caprolactone) (PCL) blends as self-knotting sutures. J. Mech. Behav. Biomed. Mater 2016, 64, 94–103. [DOI] [PubMed] [Google Scholar]
- (6).Duarah R; Singh YP; Gupta P; Mandal BB; Karak N Smart self-tightening surgical suture from a tough bio-based hyperbranched polyurethane/reduced carbon dot nanocomposite. Biomed. Mater 2018, 13, No. 045004. [DOI] [PubMed] [Google Scholar]
- (7).Nail LN; Zhang D; Reinhard JL; Grunlan MA Fabrication of a Bioactive, PCL-based “Self-fitting” Shape Memory Polymer Scaffold. J. Visualized Exp 2015, 104, No. e52981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Zhang D; George OJ; Petersen KM; Jimenez-Vergara AC; Hahn MS; Grunlan MA A bioactive “self-fitting” shape memory polymer scaffold with potential to treat cranio-maxillo facial bone defects. Acta Biomater. 2014, 10, 4597–4605. [DOI] [PubMed] [Google Scholar]
- (9).Woodard LN; Kmetz KT; Roth AA; Page VM; Grunlan MA Porous Poly(ε-caprolactone)–Poly(l-lactic acid) Semi-Interpenetrating Networks as Superior, Defect-Specific Scaffolds with Potential for Cranial Bone Defect Repair. Biomacromolecules 2017, 18, 4075–4083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Sun H; Mei L; Song C; Cui X; Wang P The in vivo degradation, absorption and excretion of PCL-based implant. Biomaterials 2006, 27, 1735–1740. [DOI] [PubMed] [Google Scholar]
- (11).Doppalapudi S; Jain A; Khan W; Domb AJ Biodegradable polymers–an overview. Polym. Adv. Technol 2014, 25, 427–435. [Google Scholar]
- (12).Gunatillake P; Mayadunne R; Adhikari R Recent developments in biodegradable synthetic polymers. Biotechnol. Annu. Rev 2006, 12, 301–347. [DOI] [PubMed] [Google Scholar]
- (13).Woodard LN; Grunlan MA Hydrolytic Degradation and Erosion of Polyester Biomaterials. ACS Macro Lett. 2018, 7, 976–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Imre B; Pukánszky B Compatibilization in bio-based and biodegradable polymer blends. Eur. Polym. J 2013, 49, 1215–1233. [Google Scholar]
- (15).Luckachan GE; Pillai CKS Biodegradable Polymers-A Review on Recent Trends and Emerging Perspectives. J. Polym. Environ 2011, 19, 637–676. [Google Scholar]
- (16).Sabir MI; Xu X; Li L A review on biodegradable polymeric materials for bone tissue engineering applications. J. Mater. Sci 2009, 44, 5713–5724. [Google Scholar]
- (17).Miller RA; Brady JM; Cutright DE Degradation rates of oral resorbable implants (polylactates and polyglycolates): Rate modification with changes in PLA/PGA copolymer ratios. J. Biomed. Mater. Res 1977, 11, 711–719. [DOI] [PubMed] [Google Scholar]
- (18).Liu Y; Bai X; Liang A Synthesis, Properties, and In Vitro Hydrolytic Degradation of Poly(d,l-lactide-co-glycolide-co-ε-caprolactone). Int. J. Polym. Sci 2016, 2016, 1–9. [Google Scholar]
- (19).Huang M-H; Li S; Vert M Synthesis and degradation of PLA–PCL–PLA triblock copolymer prepared by successive polymerization of ε-caprolactone and dl-lactide. Polymer 2004, 45, 8675–8681. [Google Scholar]
- (20).Urquijo J; Guerrica-Echevarráa G; Eguiazábal JI Melt processed PLA/PCL blends: Effect of processing method on phase structure, morphology, and mechanical properties. J. Appl. Polym. Sci 2015, 132 (), 10.1002/app.42641. [DOI] [Google Scholar]
- (21).Ostafinska A; Fortelny I; Nevoralova M; Hodan J; Kredatusova J; Slouf M Synergistic effects in mechanical properties of PLA/PCL blends with optimized composition, processing, and morphology. RSC Adv. 2015, 5, 98971–98982. [Google Scholar]
- (22).López-Rodráguez N; López-Arraiza A; Meaurio E; Sarasua JR Crystallization, morphology, and mechanical behavior of polylactide/poly(ε-caprolactone) blends. Polym. Eng. Sci 2006, 46, 1299–1308. [Google Scholar]
- (23).Maglio G; Migliozzi A; Palumbo R; Immirzi B; Volpe MG Compatibilized poly(ϵ-caprolactone)/poly(L-lactide) blends for biomedical uses. Macromol. Rapid Commun. 1999, 20, 236–238. [Google Scholar]
- (24).Chavalitpanya K; Phattanarudee S Poly(Lactic Acid)/Polycaprolactone Blends Compatibilized with Block Copolymer. Energy Procedia 2013, 34, 542–548. [Google Scholar]
- (25).Vilay V; Mariatti M; Ahmad Z; Pasomsouk K; Todo M Improvement of microstructures and properties of biodegradable PLLA and PCL blends compatibilized with a triblock copolymer. Mater. Sci. Eng. A 2010, 527, 6930–6937. [Google Scholar]
- (26).Wang L; Ma W; Gross RA; McCarthy SP Reactive compatibilization of biodegradable blends of poly(lactic acid) and poly(ε-caprolactone). Polym. Degrad. Stab 1998, 59, 161–168. [Google Scholar]
- (27).Harada M; Iida K; Okamoto K; Hayashi H; Hirano K Reactive compatibilization of biodegradable poly(lactic acid)/poly(ε-caprolactone) blends with reactive processing agents. Polym. Eng. Sci 2008, 48, 1359–1368. [Google Scholar]
- (28).Tsuji H; Ikada Y Blends of aliphatic polyesters. II. Hydrolysis of solution-cast blends from poly(L-lactide) and poly(E-caprolactone) in phosphate-buffered solution. J. Appl. Polym. Sci 1998, 67, 405–415. [Google Scholar]
- (29).Woodard LN; Page VM; Kmetz KT; Grunlan MA PCL–PLLA Semi-IPN Shape Memory Polymers (SMPs): Degradation and Mechanical Properties. Macromol. Rapid Commun 2016, 37, 1972–1977. [DOI] [PubMed] [Google Scholar]
- (30).Woodard LN; Grunlan MA Hydrolytic Degradation of PCL–PLLA Semi-IPNs Exhibiting Rapid, Tunable Degradation. ACS Biomater. Sci. Eng 2019, 5, 498–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Kaihara S; Matsumura S; Mikos AG; Fisher JP Synthesis of poly(L-lactide) and polyglycolide by ring-opening polymerization. Nat. Protoc 2007, 2, 2767–2771. [DOI] [PubMed] [Google Scholar]
- (32).Pitt CG; Chasalow FI; Hibionada YM; Klimas DM; Schindler A Aliphatic polyesters. I. The degradation of poly(ϵ-caprolactone) in vivo. J. Appl. Polym. Sci 1981, 26, 3779–3787. [Google Scholar]
- (33).Fukushima K; Tabuani D; Dottori M; Armentano I; Kenny JM; Camino G Effect of temperature and nanoparticle type on hydrolytic degradation of poly(lactic acid) nanocomposites. Polym. Degrad. Stab 2011, 96, 2120–2129. [Google Scholar]
- (34).Cam D; Hyon S-H; Ikada Y Degradation of high molecular weight poly(l-lactide) in alkaline medium. Biomaterials 1995, 16, 833–843. [DOI] [PubMed] [Google Scholar]
- (35).Gilding DK; Reed AM Biodegradable polymers for use in surgery—polyglycolic/poly(actic acid) homo- and copolymers: 1. Polymer 1979, 20, 1459–1464. [Google Scholar]
- (36).von Burkersroda F; Schedl L; Göpferich A Why degradable polymers undergo surface erosion or bulk erosion. Biomaterials 2002, 23, 4221–4231. [DOI] [PubMed] [Google Scholar]
- (37).Arias V; Höglund A; Odelius K; Albertsson A-C Tuning the Degradation Profiles of Poly(l-lactide)-Based Materials through Miscibility. Biomacromolecules 2014, 15, 391–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Lyu S-P; Bates FS; Macosko CW Coalescence in polymer blends during shearing. AIChE J. 2000, 46, 229–238. [Google Scholar]
- (39).Van Hemelrijck E; Van Puyvelde P; Velankar S; Macosko CW; Moldenaers P Interfacial elasticity and coalescence suppression in compatibilized polymer blends. J. Rheol 2004, 48, 143–158. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.