

Abstract
Oncogenic RAS signaling is an attractive target for fusion-negative rhabdomyosarcoma (FN-RMS). Our study validates the role of the ERK MAPK effector pathway in mediating RAS dependency in a panel of H/NRASQ61X-mutant RMS cells and correlates in vivo efficacy of the MEK inhibitor trametinib with pharmacodynamics of ERK activity. A screen is used to identify trametinib-sensitizing targets and combinations are evaluated in cells and tumor xenografts. We find that the ERK MAPK pathway is central to H/NRASQ61X-dependency in RMS cells, however there is poor in vivo response to clinically relevant exposures with trametinib, which correlates with inefficient suppression of ERK activity. CRISPR screening points to vertical inhibition of the RAF-MEK-ERK cascade by co-suppression of MEK and either CRAF or ERK. CRAF is central to rebound pathway activation following MEK or ERK inhibition. Concurrent CRAF suppression and MEK or ERK inhibition, or concurrent pan-RAF and MEK/ERK inhibition (pan-RAFi + MEKi/ERKi), or concurrent MEK and ERK inhibition (MEKi + ERKi) all synergistically block ERK activity and induce myogenic differentiation and apoptosis. In vivo assessment of pan-RAFi + ERKi or MEKi + ERKi potently suppress growth of H/NRASQ61X RMS tumor xenografts, with pan-RAFi + ERKi being more effective and better tolerated. We conclude that CRAF reactivation limits the activity of single agent MEK/ERK inhibitors in FN-RMS. Vertical targeting of the RAF-MEK-ERK cascade, and particularly co-targeting of CRAF and MEK or ERK, or the combination of pan-RAF inhibitors with MEK or ERK inhibitors, have synergistic activity and potently suppress H/NRASQ61X-mutant RMS tumor growth.
Keywords: Rhabdomyosarcoma, H/NRASQ61X, CRAF, RAF-MEK-ERK cascade, vertical targeting
INTRODUCTION
Rhabdomyosarcoma (RMS) is the most common soft tissue sarcoma in children, characterized by molecular and cellular features of developing muscle. While relapse-free survival for localized rhabdomyosarcoma is nearly 80%, the prognosis for patients with metastatic or relapsed disease is dismal (5-year survival rate less than 30% and 17%, respectively) [1]. Because the overall survival for patients with RMS has not improved for decades and because the current standard cytotoxic therapies cause profound lifelong health complications in children, there is a need to design novel, more effective and less-toxic therapies.
Genomic studies of RMS have identified frequent RAS genes mutations in PAX fusion-negative (FN)-RMS which comprises approximately 70 % of RMS [1, 2]. Experimental evidence further supports the importance of RAS as a driver of FN-RMS development: 1) Clonal analyses show that RAS mutations occur early in the development of FN-RMS [3]; and 2) oncogenic RAS can initiate RMS in animal models and transform myoblasts by blocking differentiation [4–6]. Studies also demonstrate that MEK or ERK inhibitors selectively inhibit RAS-mutant FN-RMS cells, cause myogenic differentiation and tumor growth delay, although single-target approaches did not result in durable responses [7]. Thus, strategies that aim to target oncogenic RAS in FN-RMS should be considered.
Currently, four strategies to interfere with oncogenic RAS are actively pursued: (1) direct pharmacologic inhibition of mutant RAS; (2) Inhibition of RAS membrane association; (3) Inhibition of RAS-associated synthetic lethal targets; and (4) Inhibition of downstream RAS-effector pathways. Direct KRAS inhibitors are under clinical evaluation [8], however these inhibitors are specific for RASG12C mutations, rarely reported in RMS. Inhibition of RAS activity through inhibition of membrane association with farnesyltransferase inhibitors (FTIs), is applicable for HRAS, but not K/NRAS-driven cancers, due to compensatory lipid modifications controlling K/NRAS membrane association. In FN-RMS, mutations in all three RAS isoforms are detected and FTIs remain to be evaluated. The third approach, to target RAS-driven synthetic lethal cancer cell dependencies, has not yet yielded tractable targets [9]. Finally, the approach to inhibit downstream RAS-effector pathways, particularly the RAF-MEK-ERK signaling cascade has emerged as the focus of most current translational and clinical research [10].
Our studies further demonstrate that the ERK MAPK pathway is a relevant target in H/NRASQ61X-driven RMS cells and tumors. However, relief of negative feedback regulatory loops and specifically ERK-mediated inhibitory phosphorylation of CRAF, limit effective pathway inhibition, ultimately leading to poor anti-tumor response to single-agent MAPK inhibitors. Vertical co-targeting of distinct nodes of the ERK MAPK pathway more effectively block pathway activity and synergistically induce cell cycle arrest, myogenic differentiation and apoptosis in H/NRASQ61X-mutant RMS cells and tumors. In vivo studies indicate that concurrent pan-RAF and ERK inhibition is better tolerated and more effective as compared to concurrent MEK and ERK inhibition, indicating wider therapeutic window and potential clinical application of pan-RAF inhibitors in FN-RMS.
MATERIALS AND METHODS
Cell lines
All RMS cell lines were maintained in high-glucose RPMI medium (Invitrogen) supplemented with 10% FBS, 37°C, 5% CO2. All cells were routinely tested for Mycoplasma using MycoAlert™ Mycoplasma Detection Kit and identity confirmed by STR analysis performed at the McDermott Center Sequencing Core, UT Southwestern Medical Center, Dallas, TX. All cell lines were used between passage 10 and 30. SMS-CTR cells were obtained in 2018 from Dr. Corinne Linardic, Duke University; JR1 cells were obtained in 2019 from Dr. Marielle Yohe, NCI; RD, Rh36, Rh18, Rh41, Rh28, Rh30 were obtained in 2018 from the Houghton laboratory, UTHSCSA.
Reagents, inhibitors and antibodies
The following antibodies were purchased from Cell Signaling Technology (CST) Inc: GAPDH (#5174), p-ERK (#4370), ERK (#9102), p-AKT (#13038), AKT (#4691), p-p90RSK1(#9355), p90RSK1(#9344), CRAF (#12552), p-CRAF(#9431), BRAF(#9433), ARAF(#4432), MEK (#4694), p-MEK (#9154), MEF2C (#5030), cleaved PARP (#5625), cleaved Caspase-3 (#9661). HRAS and NRAS antibodies were from Santa Cruz Biotechnology (# sc-520 and sc-31 respectively). KRAS antibody was from Millipore (#OP24). Myosin Heavy Chain Antibody (MF20) was from Developmental Studies Hybridoma Bank (DSHB) antibodies, University of Iowa, IA, USA (# ab-2147781). Trametinib was purchased from LC Laboratories (# T-8123), AZD8186 was from Selleck (# S7694), AZD5363 was from Selleck (# S8019), SHP099 was from MedChemExpress, # HY-100388), LY2874455 was from MedChemExpress (# HY-13304), SCH772984 was from MedChemExpress (# HY-50846); LY3009120 was from MedChemExpress (#HY-12558), Ponatinib was from MedChemExpress (# HY-12047), Doxycycline Hydrochloride, Ready Made Solution was from Millipore Sigma (# D3072). LY3214996 was provided by Eli Lilly and Company, Indianapolis, Indiana, USA.
siRNA and Plasmid Transfections
siRNA (10nM) transfection experiments were performed using Lipofectamine RNAi MAX (Life Technologies) following manufacturer instructions. Plasmid transfections were done using Lipofectamine 3000 (Life Technologies), following manufacturer instructions.
Immunoblotting and pharmacodynamics assays
Cells were lysed in 1% Triton Buffer supplemented with Halt Protease Inhibitor Cocktail (Thermo Fisher Scientific), protein concentration was determined using Bradford Assay reagent (Bio-Rad) and standard immunoblotting procedures were followed. For in vivo pharmacodynamics of ERK and p90RSK1 phosphorylation, tumors were collected and snap frozen in liquid nitrogen. Following cryo-pulverization, protein was extracted as described above.
Proliferation Assays
2 × 103 cells/well plated in 96-well plates were treated with DMSO/inhibitors. 96 h later, the media was removed and AlamarBlue reagent (Biorad) was added at 1:10 ratio in culture media and measurements were done as per manufacturer’s instructions. Following background correction, percent viability was normalized to DMSO and the concentration at which 50% of cell growth is inhibited by drug treatment (GI50) was calculated using GraphPad Prism 8. For siRNA experiments, cells were first transfected in 6-well plates, 24 h later plated in 96-well plates and viability measured in 8 days. All experiments were performed in presence of serum (10% FBS).
Colony Forming Assay
Cells were plated at low density in 6 or 12-well plates and treated with DMSO/inhibitors. In approximately 2 weeks the media was removed, cells were fixed and stained with 0.1 % crystal violet, 4% Paraformaldehyde for 20 minutes. For siRNA experiments, cells were plated 24 h after transfection.
Myogenic differentiation assay
For RAS siRNA transfections, cells were transfected in 6-well plate and 24 h later plated in Nunc® Lab-Tek® II - CC2™ Chamber Slide™ system. For drug treatments, cells were plated in Nunc® Lab-Tek® II - CC2™ Chamber Slide™ system or clear bottom 96 well plates and treated the next day. Cell were maintained in 10% FBS RPMI medium throughout the entire experiment. At the end of the transfection/treatment time point, the cells were washed in PBS, fixed and immunofluorescent staining performed.
Cell Cycle Analysis
Cells were washed with cold PBS and fixed with 70% ethanol for 24 h at −20°C, centrifuged at 200 × g, washed with PBS, and incubated with propidium iodide/TritonX-100 staining solution, supplemented with RNase A (Sigma) for 2–4 h at room temperature. DNA content was measured using Fortessa cell analyzer/ Flow Cytometer. Modfit software was used to analyze the data.
RNA sequencing
RNA was isolated from RD cells transfected with nonspecific control (48 h), two NRAS siRNA oligos (48 h each), treated with DMSO control (24 h) or 300 nM SCH772984 (24 h). Each condition was done in duplicates and RNA was isolated using RNeasy mini kit (Qiagen). Approximately 500 ng of total RNA was used for RNA sequencing (RNA-seq) library preparation by following the Illumina TruSeq stranded mRNA sample preparation guide (Illumina). RNA-seq libraries were subjected to quantification, and subsequent 50bp single-read sequencing module with Illumina HiSeq 3000 platform. After the sequencing run, demultiplexing with CASAVA (Illumina) was employed to generate the FastQ file for each sample.
RNA sequencing data processing
All RNA-seq FastQ reads were aligned with the reference genome (UCSC human genome build hg19) using TopHat2 default settings. The BAM files obtained after alignment were processed using HTSeq-count to obtain the counts per gene in all samples. The R package DEseq was used to normalize gene expression with the size factor method and perform pairwise comparisons between groups to identify differentially expressed genes (DEG). Significant DEGs were identified with BH-adjP<0.05 and FoldChange (FC) >= 2 when the gene has read abundance with rpkm>=1 within at least one treatment (samples).
CRISPR library screen
Library virus packaging and multiplicity of infection (MOI) determination was done as previously described [11]. The RD cell line was seeded in 6-well plates at 5×105 cells per well and next day virus was added at a MOI of 0.3. Following puromycin selection, a day 2 sample was collected to determine initial library representation and the remaining cells were seeded in T-225 culture flasks. Each condition (control and trametinib treatment for 3 weeks), was done in duplicates. Throughout the treatment, cells were maintained at a minimum of 500x library coverage. At the end of treatment, the cells were collected, genomic DNA was extracted with DNeasy Blood & Tissue Kit (QIAGEN) and sgRNA sequences were amplified by library specific PCR primers and an Illumina sequencing adapter with index for each sample.
Animal Studies
C.B-Igh-1b/IcrTac-Prkdcscid female mice (Envigo) 6–8 weeks of age were used throughout the studies. All animals were maintained under barrier conditions and experiments were carried out using protocols and conditions approved by the IACUC UTHSCSA. Small pieces of tumor tissue were implanted in the flank area as described previously [12]. When tumors reached 200–400 mm3 size, mice were randomized in treatment cohorts (5 mice per group) and treatment was initiated. More details are provided in Supplementary Information.
Quantification and Statistical analysis
Statistical significance between multiple groups was determined by one-way ANOVA (single variable). Statistical significance between two groups was determined by t-test. GraphPad Prism 8 software (GraphPad Software Inc.) was utilized for all statistical analyses. Quantification of immunofluorescence images (percent MHC positive cells) was done using ImageJ software.
Additional methods are described in Supplementary Methods.
Data availability
The RNA sequencing data generated in this study are available at Gene Expression Omnibus (GEO) at GSE180127.
RESULTS
ERK MAPK pathway drives H/NRASQ61X dependency in FN-RMS cells.
Experimental evidences indicate that oncogenic RAS mutations can drive RMS development [4–6]. Moreover, recent studies demonstrated that ablation of oncogenic NRAS in established RMS tumors caused profound tumor regression and increased survival of tumor-bearing mice [13]. Therefore, mutationally activated RAS is an attractive therapeutic target for FN-RMS. To further extend these findings, we examined RAS dependency in a panel of FN-RMS cell lines harboring oncogenic NRASQ61H/L mutations (most common in RMS) as well as HRASQ61K ( Supplementary table 1). Cell growth was assessed upon transfection with RAS isoform-specific siRNAs (Figures 1A and S1A). HRAS knockdown impaired the proliferation of HRAS-mutant cells, while K/NRAS knockdown had no effect. Conversely, knockdown of NRAS impaired the growth of NRAS-mutant cells only, but knockdown of K/HRAS had no or minimal effect. In the RAS-wild type RMS cell line Rh18, knockdown of each individual RAS isoform had no effect on proliferation. We concluded that mutant, but not individual wild-type RAS isoforms are essential for the proliferation of FN-RMS cells, supporting the concept of oncogene addiction.
Figure 1. ERK MAPK pathway drives H/NRAS Q61X dependency in FN-RMS cells.
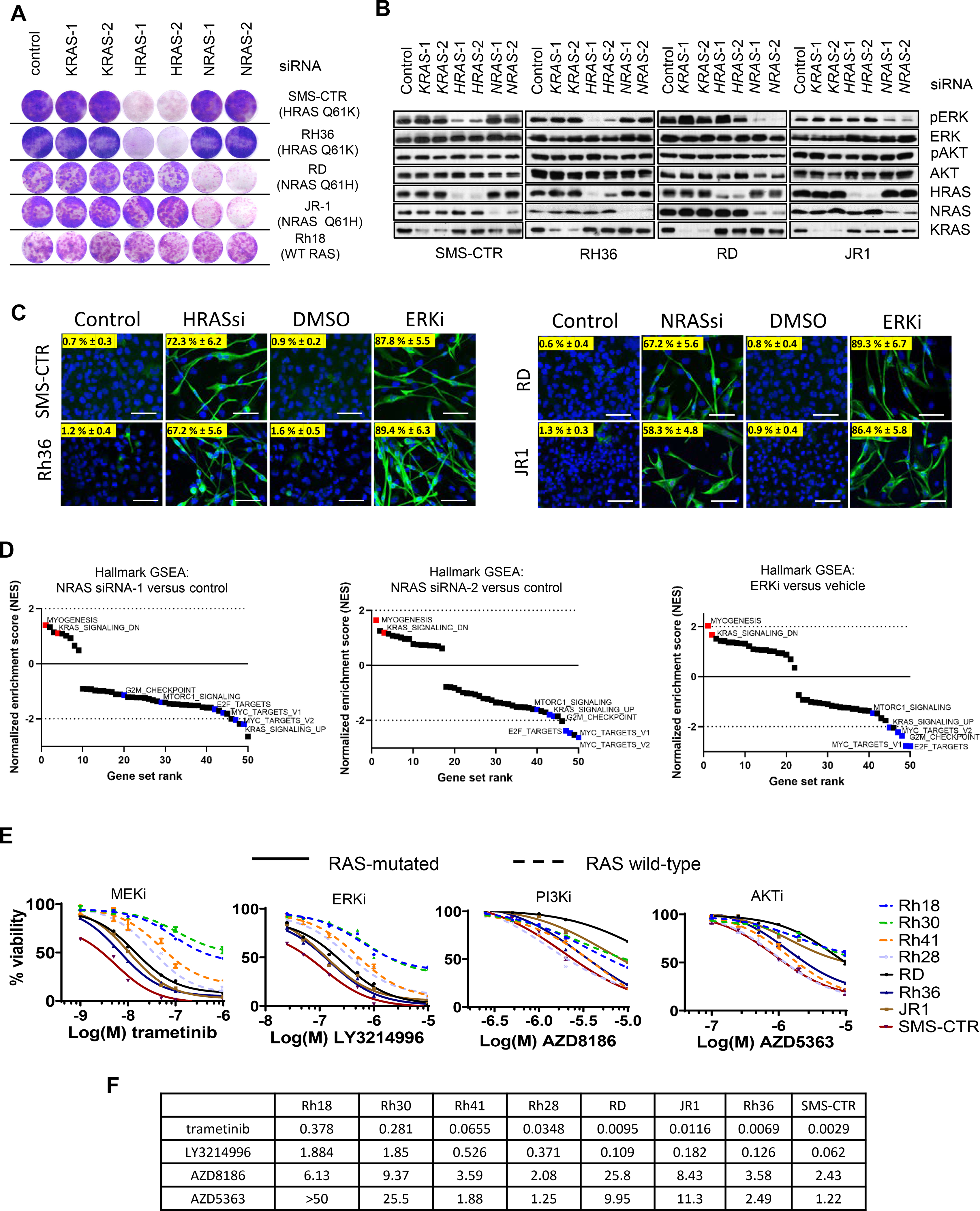
(A) Crystal violet staining images of RMS cells upon isoform-specific RAS knockdown. Indicated cells were transfected with control or two independent siRNAs against each RAS isoform and 48 h later were plated at low density in 6-well plates (remaining cells were collected for western blot analysis described in B). After >two weeks, cell growth was visualized with crystal violet staining. (B) Western blots of FN-RMS cells upon isoform-specific RAS knockdown. Indicated cells were transfected as in A and 48 h later western blots of cell lysates were done to detect phosphorylated ERK1/2 (pERK, Thr202/Tyr204), phosphorylated AKT (pAKT, Ser473), and each RAS isoform. (C) Representative immunofluorescence images of H/NRAS siRNA-transfected or ERK inhibitor-treated H/NRAS-mutant RMS cell lines. Indicated cell lines were transfected with H/NRAS siRNAs or treated with 1 μM ERKi LY3214996. After 8 days the cells were fixed and stained with DAPI or myosin heavy chain (MHC) antibody and processed for immunofluorescence. Percent MHC positive cells was calculated using ImageJ. Scale bars, 100 μm. (D) Gene set enrichment analysis (GSEA) of RNA transcripts of RD cells transfected with two independent NRAS siRNAs (NRAS siRNA-1 and NRAS siRNA-2) for 48 h or treated with 300nM ERKi SCH772984 for 24 h. Enriched or depleted hallmark gene sets compared to control siRNA (48 h) or DMSO (24 h) are shown. (E) Graphs summarizing dose-response of H/NRAS Q61X and RAS-wild type RMS cell lines to MEK, ERK, PI3K and AKT inhibitors. Indicated cell lines were treated with increasing concentrations of MEKi trametinib, ERKi LY3214996, PI3Ki AZD8186 and AKTi AZD5363. Treatment was done in 96 well-plates for 96 h and viability was measured with Alamar Blue reagent. Growth response curves were plotted using GraphPad Prism. Error bars represent SEM from three independent experiments done in triplicate. (F) Table summarizing Growth Inhibitory concentrations at 50% (GI50 μM) for each treatment and cell line shown in E. GI50 values were calculated with GraphPad Prism.
We next addressed how acute suppression of each RAS isoform effected MAPK and PI3K-AKT effector signaling in our panel of cell lines (Figure 1B). In each cell line, ERK phosphorylation at the activation loop residues Thr202/Tyr204 (pERK) was acutely inhibited only when the expression of the corresponding mutant RAS isoform was suppressed. In the HRAS-mutant cells, pERK was inhibited upon knockdown of HRAS, but not K/NRAS; in the NRAS-mutant cells, pERK was inhibited upon NRAS, but not K/HRAS knockdown. AKT phosphorylation at Thr308 (PDK1 phosphorylation site) was not affected in any cell line by either suppression of wild type or mutant RAS isoforms. These results suggested a role for the ERK MAPK effector pathway in mediating H/NRASQ61X dependency in RMS cells. Similarly, studies by Yohe et al, demonstrated through ectopic expression in myocytes of constitutively active RAS effectors, that only BRAF V600E was able to mimic the block of myogenic differentiation as seen with oncogenic RAS expression [7]. Consistently, we observed that suppression of either mutant HRAS or NRAS phenocopied ERK inhibition by causing myogenic differentiation as seen by elongated cell morphology and expression of myosin heavy chain (MHC), a marker for myogenic differentiation (Figure 1C).
To compare gene transcription changes induced by mutant RAS knockdown or ERK inhibition, we performed RNA-seq analyses in the NRASQ61H RD cells after 48 h NRAS suppression by siRNA (NRASsi) or 24 h ERK inhibitor treatment (ERKi) (Figure 1D). Consistent with suppression of ERK activity, NRASsi caused downregulation of canonical ERK transcriptional targets DUSP6 and SPRY2/4 (Figure S1B). Further, gene set enrichment analysis (GSEA) revealed significant overlap of hallmark gene signatures associated with NRASsi or ERKi. For both treatments, the strongest induction of transcription was the myogenic differentiation-related genes sets (Figures 1D and S1C), confirming the observed myogenic differentiation phenotype. In both cases, a strong suppression of KRAS-driven gene signature was also detected, consistent with a major role of the MAPK pathway in driving the RAS-gene signature. MYC-, as well as E2F-associated gene signatures, were also among the most highly suppressed gene sets in both NRASsi- and ERKi-treated cells, consistent with the well validated role of MYC and E2F transcription factors downstream of ERK. Collectively, these results indicate a major role of the ERK MAPK effector pathway in driving RAS-regulated gene expression in RMS.
Finally, we compared the sensitivity of a panel of RMS cell lines to MEK, ERK, PI3K and AKT inhibitors [14–17] (Figures 1E and 1F). All H/NRASQ61X-mutated RMS cells were selectively more sensitive to both MEK and ERK inhibitors, as compared to RAS-wild type RMS cells. Conversely, there was no correlation of RAS status and sensitivity to PI3K/AKT inhibitors. These results further supported the relevance of the MAPK pathway as therapeutic target and major effector in driving the RAS-dependent growth of RMS cells.
Single-agent MEK inhibitor treatment is ineffective in H/NRASQ61X-mutant FN-RMS tumor xenografts.
Given the relevance of the MAPK pathway in our panel of H/NRASQ61X RMS cell lines, we sought to determine the in vivo efficacy of the MEK inhibitor trametinib at clinically relevant drug exposures (Figure 2A). Mice bearing RD, Rh36 or SMS-CTR cell line-derived tumor xenografts (CDX) were treated with 1 mg/kg trametinib for 28 days. Pharmacokinetics experiments determined trametinib steady-state plasma concentrations (Figure S2A) (Cmax 13.3 ng/mL, AUC0–24hr 235.15 hr*ng/mL), which were near and did not exceed the clinical exposure in adult patients receiving the recommended 2mg trametinib tablets daily (Cmax 14 – 32.9 ng/ml, AUC0–24h 256 – 500 hr*ng/mL [18]). All xenografts progressed under treatment, with notable tumor-growth delay seen only in SMS-CTR xenografts. These responses were in sharp contrast with the response of pediatric anaplastic astrocytoma (AA) xenograft model BT-40, also driven by MAPK pathway through the BRAF-V600E activating mutation. In this model, tumors regressed, and the response was maintained during treatment.
Figure 2. Single agent MEK inhibitor treatment is ineffective in H/NRAS Q61X-mutant FN-RMS tumor xenografts.
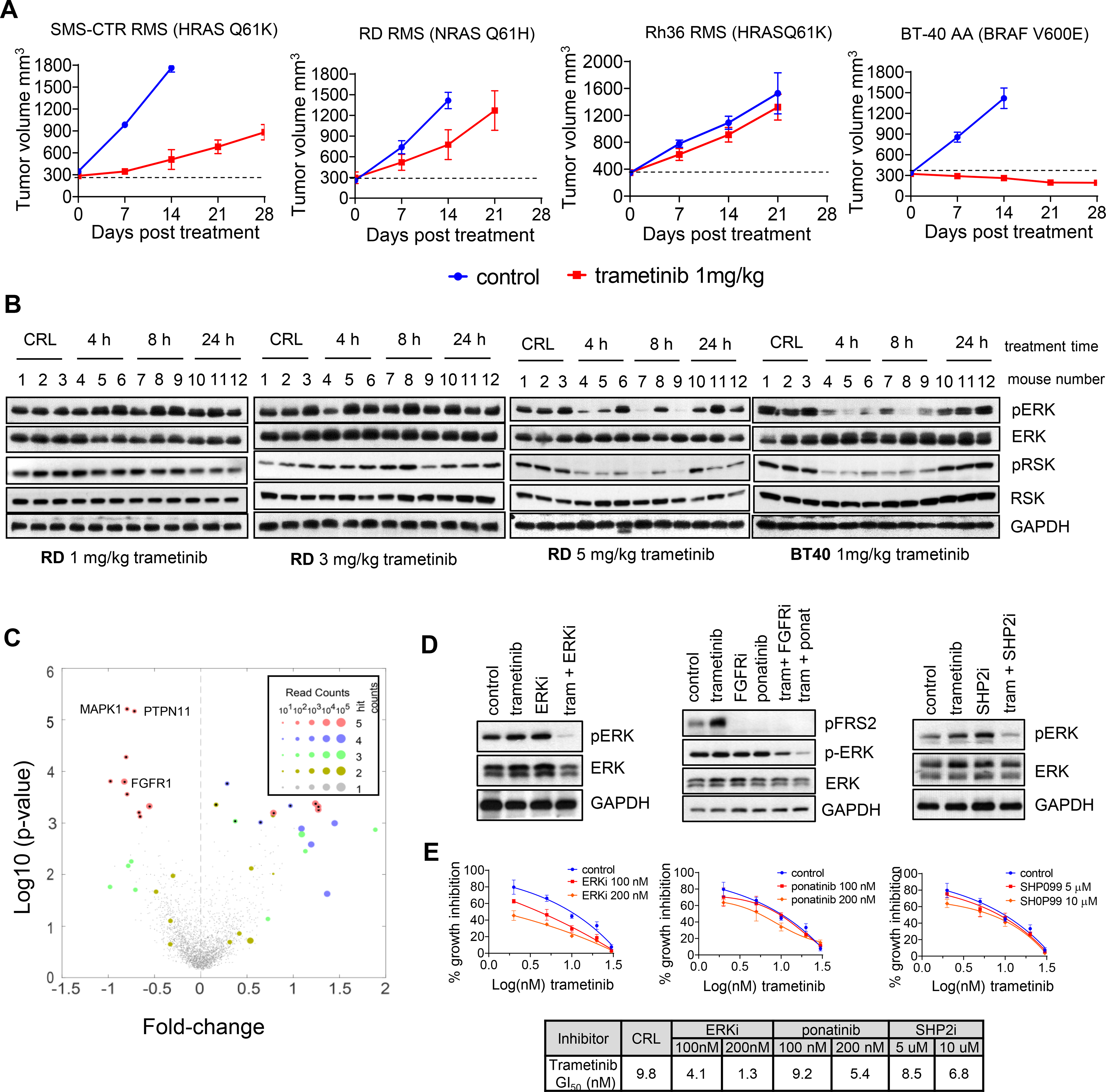
(A) Response of H/NRAS-mutant RMS tumor xenografts as compared to BRAF-V600E BT40 anaplastic astrocytoma (AA) tumor xenografts following 1 mg/kg trametinib administration. Mice bearing tumor xenografts from indicated cell lines were treated with vehicle or 1mg/kg trametinib, 7 days a week, for 28 days. Data is presented as mean tumor volume, error bars represent SEM, n=5. (B) Western blot analysis for pharmacodynamics of ERK and p90RSK1 phosphorylation in tumor xenografts from RD RMS or BT40 AA xenografts. Mice bearing RD or BT40 tumor xenografts were treated with indicated doses of trametinib, tumors were harvested at the indicated times and ERK1/2 and p90RSK1 phosphorylation was assessed by western blots. (C) Volcano plot summarizing trametinib-sensitizing CRISPR screen in RD cells. RD cells were transduced with druggable genome pLentiv2 CRISPR library. Following treatment with trametinib for 3 weeks, sgRNA region was PCR amplified and sgRNA representation determined by next-generation sequencing. Fold change for sgRNA read counts between treated and control samples was calculated and visualized using Matlab software. Y-axis: p-values of Fisher’s combined probability test of differential read counts of 5 gRNAs. Each condition was done in duplicate. (D) Western blot analysis of RD cells upon 24 h treatment with 2 nM trametinib, 200 nM pan-FGFRi LY2874455, 200 nM pan-RTKi ponatinib, 5 μM SHP2i SHP099, 50 nM ERKi LY3214996 or indicated combinations. Phosphorylated ERK1/2 (pERK, Thr202/Tyr204), phosphorylated FRS2-α (pFRS2, Tyr436) were analyzed, total ERK1/2 and GAPDH serve as controls. (E) Graphs summarizing dose-response of RD cell to increasing concentrations of trametinib alone (control, DMSO) and in combination with ERKi LY3214996 (left), pan-RTKi ponatinib (middle) or SHP2i SHP099 (right). Error bars represent SEM from three independent experiments done in triplicates. Table summary of GI50 values.
We next asked how effectively trametinib was able to inhibit ERK activity in RMS tumor xenografts as compared to the responsive BT-40 AA xenografts. We used pharmacodynamic assays to assess the in vivo inhibition of pERK and phosphorylation of its direct substrate p90RSK (pRSK), following administration of trametinib (Figure 2B). Treatment of mice bearing RD xenografts with single dose 1 or 3 mg/kg trametinib did not lead to detectable inhibition of pERK or pRSK. Inhibition of pERK/pRSK was only detected when trametinib was increased to 5 mg/kg. In sharp contrast, when mice bearing BRAF-V600E AA xenografts were treated with 1 mg/kg trametinib, inhibition of pERK/pRSK was readily detected. These results indicated ineffective ERK inhibition in RMS tumors, which could account for the poor anti-tumor activity of trametinib.
To identify possible mechanisms of resistance and trametinib-sensitization targets in RMS cells, we performed screen with a ~2400 gene CRISPR/Cas9 library designed to target druggable proteins or proteins in druggable pathways [11]. We screened the RD cells with continuous dosing of trametinib at concentrations near GI50 values. We identified genes whose suppression resulted in increased or decreased sensitivity to trametinib (Figures 2C and S2B). From the top five trametinib-sensitizing hits, three were genes encoding proteins associated with regulation of MAPK pathway activity, such as MAPK1 (ERK2), PTPN11 (SHP2) and FGFR1. These results are consistent with the well-known notion that common mechanisms of resistance to single-agent MEKi/ERKi in RAS-driven cancers involve reactivation of the MAPK pathway due to the loss of multiple negative feedbacks [19, 20]. Inhibition of either SHP2, FGFR1, EKR or RAF have been all shown to potentiate MEK inhibitor treatments in KRAS or BRAF-driven adult cancers [21–26].
We tested how combining trametinib with SHP2, FGFR or ERK1/2 inhibitors affected MAPK pathway activity and growth inhibition in our RAS-mutant RMS cell lines. As expected, all combinations inhibited pERK more potently as compared to single treatments (Figures 2D and S2C). However, the combination of trametinib with ERKi had better effect than the combinations with SHP2i or FGFRi, as seen by more potent shift in trametinib GI50 values (Figures 2E, S2D, S2E) and acute growth suppression in long-term treatment colony forming assays (Figures S2F and S2G). Figure S2H summarizes reported Kd (or Ki) and biochemical IC50 values, as well as Cmax concentrations of recommended clinical doses (if reported) for the respective inhibitors. We concluded that in H/NRASQ61X-mutant RMS cells, the MEKi + ERKi combination is more potent than the MEKi + SHP2i or MEKi + FGFRi combinations.
CRAF suppression sensitizes H/NRASQ61X-mutant FN-RMS cells and tumor xenografts to MEK or ERK inhibition
Previous studies have shown that the requirement for individual RAF isoforms (A/B/CRAF) in RAS-driven cancers is context specific [27–29]. We asked if, in our panel of H/NRAS-mutant RMS cell lines, specific RAF isoforms mediated resistance and pathway reactivation upon MEK/ERK inhibition. In our CRISPR screen, we looked at the depletion rate of sgRNAs targeting each RAF isoform in the presence or absence of trametinib. When sgRNA representation in day 2 library infected cells was compared to day 14 infected untreated cells, overall CRAF-specific sgRNAs were depleted significantly, while depletion of A/BRAF-specific sgRNAs were either undetected or detected to a lesser extent (Figure S3A). When day 14 infected untreated cells were compared to day 14 infected trametinib-treated cells, there was further depletion of CRAF-specific sgRNAs after trametinib treatment, while significantly less depletion was detected in A/BRAF-specific sgRNAs (Figure S3A). These results indicated that CRAF suppression in RD cells: 1) inhibits growth in the absence of treatment, and 2) further sensitizes to trametinib treatment. To confirm these conjectures, we suppressed each RAF isoform by stable knockdown using previously validated RAF-isoform selective shRNA lentiviral vectors [30] and assessed the growth of our panel of RMS cells with or without trametinib/ERKi treatment. In the absence of treatment, acute suppression of CRAF significantly inhibited the growth of all RAS-mutant cell lines in long-term assays, while A/BRAF suppression had no or minimal effect (Figures S3B and S3C). When RAF-suppressed cells were treated with trametinib or ERKi, suppression of CRAF contributed to dramatic shift of GI50 values in all RAS-mutant cell lines, while A/BRAF suppression had little to no effect (Figure S3D). In the WT RAS cell line Rh18, knockdown of individual RAF isoforms did not have significant effect. Further, ablation of CRAF, but not A/BRAF, synergized with trametinib to inhibit pERK in both HRAS and NRAS-mutant cells (Figure 3A).
Figure 3. CRAF reactivation mediates resistance to MEK or ERK inhibitors.
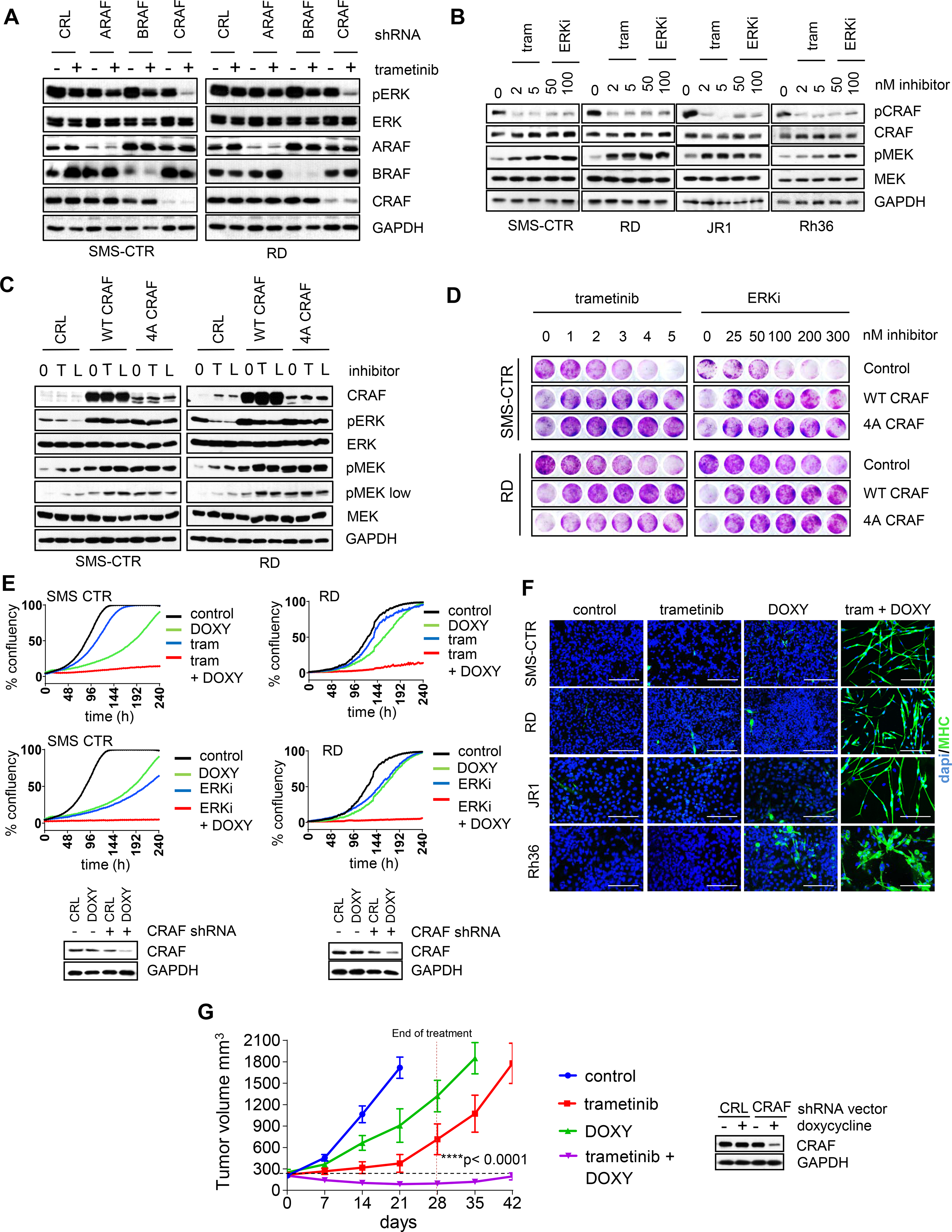
(A) Western blot images of H/NRAS-mutant RMS cells following A/B/CRAF knockdown ± trametinib. SMS-CTR and RD cells were transduced with PLKO.1 lentiviral constructs expressing shRNA against GFP (CRL) or A/B/CRAF, selected with puromycin for 24 h, then treated with 2 nM trametinib for 72 h. (B) Western blots of FN-RMS cells treated with trametinib or ERKi LY3214996. FN-RMS cells were treated with indicated concentrations of trametinib or LY3214996 for 72 h and phosporylated CRAF (pCRAF, S289/296/301) and phosphorylated MEK1/2 (pMEK, Ser217/221) were detected by western blots. (C) Western blots of RMS cells expressing wild-type or phosphorylation deficient CRAF constructs and treated with trametinib or ERKi LY3214996. SMS-CTR and RD cells were transduced with retroviral vectors expressing wild-type CRAF (WT CRAF) or mutant CRAF deficient for phosphorylation at SER 29, 289, 296, 301 (4A CRAF). Control is empty vector (CRL). After puromycin selection for 2 days, the cells were treated with trametinib (T) or ERKi LY3214996 (L) for 72 h and cell lysates analyzed by western blots. (D) Crystal violet staining images of FN-RMS cells expressing empty vector, WT CRAF or 4A CRAF constructs and treated with increasing concentrations of trametinib or LSN3214996. Cells were plated at low density in 24-well plates and next day treated with increasing concentrations of trametinib or LY3214996. After two weeks, cell growth was visualized with crystal violet staining. (E) Top, growth curves of FN-RMS cells expressing doxycycline-inducible shRNA against CRAF. SMS-CTR or RD cells were transduced with pSMART lentiviral vector expressing doxycycline-inducible CRAF shRNA, selected with puromycin for 2 days and plated in 96 well plates. Next day, the cells were treated with DMSO, 200 ng/ml doxycycline, 2 nM trametinib +/− doxycycline or 100 nM LY3214996 +/− doxycycline. Percent confluency was determined by imaging with Incucyte every 2 h for 240 h. Bottom, western blot images of SMS-CTR and RD cells expressing pSMART CRAF shRNA vector and treated with DMSO or 200 ng/ml doxycycline for 72 h. (F) Immunofluorescence staining images for MHC of FN-RMS cell lines expressing dox-inducible shRNA against CRAF. Indicated cells were plated in 8-well slide chambers and treated with DMSO, 200 ng/ml doxycycline, 2 nM trametinib or both for 8 days. Scale bars, 200 μm. (G) Response to trametinib of SMS-CTR xenografts expressing dox-inducible CRAF shRNA. SMS-CTR cells transduced with pSMART vector expressing dox-inducible CRAF shRNA were subcutaeously injected in the flank area of SCID mice and indicated treatment cohorts were generated. N=5 mice per group. 1mg/kg trametinib was administered PO, 7 days a week for 28 days. Doxycycline was delivered in diet at 1 g/kg for 28 days. Error bars represent SEM. p-values were calculated using one-way ANOVA for single treatments vs combinatorial treatment at the end of the treatment (day 28).
As previously described, CRAF is negatively regulated through a direct feedback phosphorylation by ERK [31]. Consistently, treatment of RMS cells with trametinib or ERKi led to reduced phosphorylation of CRAF at ERK-regulated phosphorylation sites (Figure 3B). This coincided with increased MEK phosphorylation (pMEK) at activation loop residues Ser217/212 (phosphorylated by RAF kinases), indicating increased MEK activity. Overexpression of wild type CRAF or CRAF deficient for phosphorylation at four ERK sites (4A CRAF) increased basal pMEK and pERK as well as prevented suppression of pERK following trametinib/ERKi treatments (Figure 3C). Of note, while overexpression of both wild type and 4A CRAF caused increased basal MEK phosphorylation, treatment with trametinib/ERKi caused further increase in pMEK only in wild type CRAF- overexpressing cells. WT or 4A CRAF overexpression also caused resistance to trametinib or ERKi treatment as indicated by lack of growth suppression in long-term colony forming assays (Figure 3D). The observed growth delay in CRAF-overexpressing cells is likely due to increased ERK activity, which can be toxic for RAS-mutant cells [32].
We further validated that suppression of CRAF expression sensitized RMS cells to MEK and ERK inhibition by using tet-regulatable CRAF shRNA with partial CRAF suppression (Figure 3E). With single treatment doxycycline (DOXY), the cells continued to proliferate, although at a slower rate. Similarly, single treatments with low concentrations of trametinib/ERKi did not impair proliferation significantly. However, combined treatment with DOXY + trametinib/ERKi completely blocked cell proliferation (Figure 3E). In the wells with combined treatments, dramatic morphological changes were seen characterized by elongation and the appearance of multinucleated cells, indicating myogenic differentiation (Figure S3E). These effects were not seen in cells expressing control vector (Figures S3F and S3G). Myogenic differentiation was confirmed by immunostaining for MHC, with more than 85% positivity in DOXY + trametinib treated cells, but almost absent in single treatments (Figure 3F). Finally, we validated if suppression of CRAF would sensitize SMS-CTR tumor xenografts in vivo to trametinib treatment. Tumors expressing tet-regulatable CRAF shRNA, progressed upon administration of trametinib or DOXY alone, however co-administration of DOXY + trametinib caused significant tumor regression, which was maintained at least two weeks after treatment was ended (Figure 3G). These results confirmed that CRAF inhibition significantly potentiated trametinib anti-tumor activity.
Vertical inhibition of MAPK pathway synergistically inhibits ERK activity and H/NRASQ61X-mutant FN-RMS cells growth.
Our results indicated that co-targeting of MEK and ERK or co-inhibition of CRAF and MEK/ERK would achieve more efficient pathway suppression and growth inhibition in H/NRASQ61X-mutant RMS cells. While currently, there are no CRAF selective inhibitors, type II pan-RAF inhibitors potently inhibit RAF isoforms without causing paradoxical pathway activation as seen with type I RAF inhibitors [33]. Moreover, recent studies indicated the use of type II pan-RAF inhibitors to target distinct nodes of the MAPK cascade in KRAS-driven adult cancers [23, 26]. Thus, we sought to examine the activity of MEKi + ERKi and pan-RAFi + MEKi/ERKi in our panel of H/NRASQ61X RMS cells.
Immunoblot analyses determined that following long-term treatment (72 h) of H/NRASQ61X FN-RMS cells with either combinations of MEKi + ERKi (trametinib + LY3214996) or MEKi + pan-RAFi (trametinib + LY3009120) as compared to single treatments retained inhibition of ERK signaling as measured by pERK and pRSK. This was not observed in the wild type RAS cell line Rh18, where both combinations did not provide further advantage in inhibiting pERK/pRSK. In addition, in all H/NRAS-mutant cell lines, there was evidence of increased apoptosis (cleaved PARP) with the combinations (Figure 4A). Next, we measured extent of growth inhibition upon these combinations. In all RAS-mutant cell lines, low concentrations of single agents modestly reduced, or had no effect on the proliferation rate, while the combinations completely abrogated proliferation. Consistent with lack of ERK activity suppression, the combinations did not block proliferation of the RAS wild type Rh18 cell line (Figure 4B). The potent synergy of these combinations was further confirmed in long-term treatment colony forming assay, where single treatments had either no or minor effects on cell growth, while the combinations caused acute growth suppression of all RAS-mutant, but not the RAS-wild type cell lines (Figure 4C). 5×5-dose Bliss matrix analyses indicated significant Bliss synergy positivity in RAS-mutant, but almost none in the RAS-wild type cells. Interestingly, the combination pan-RAFi + MEKi had overall higher synergy scores across all RAS-mutant cell lines (Figures S4A and S4B).
Figure 4. Vertical inhibition of MAPK pathway synergistically inhibits ERK activity and H/NRAS Q61X-mutant FN-RMS cells growth.
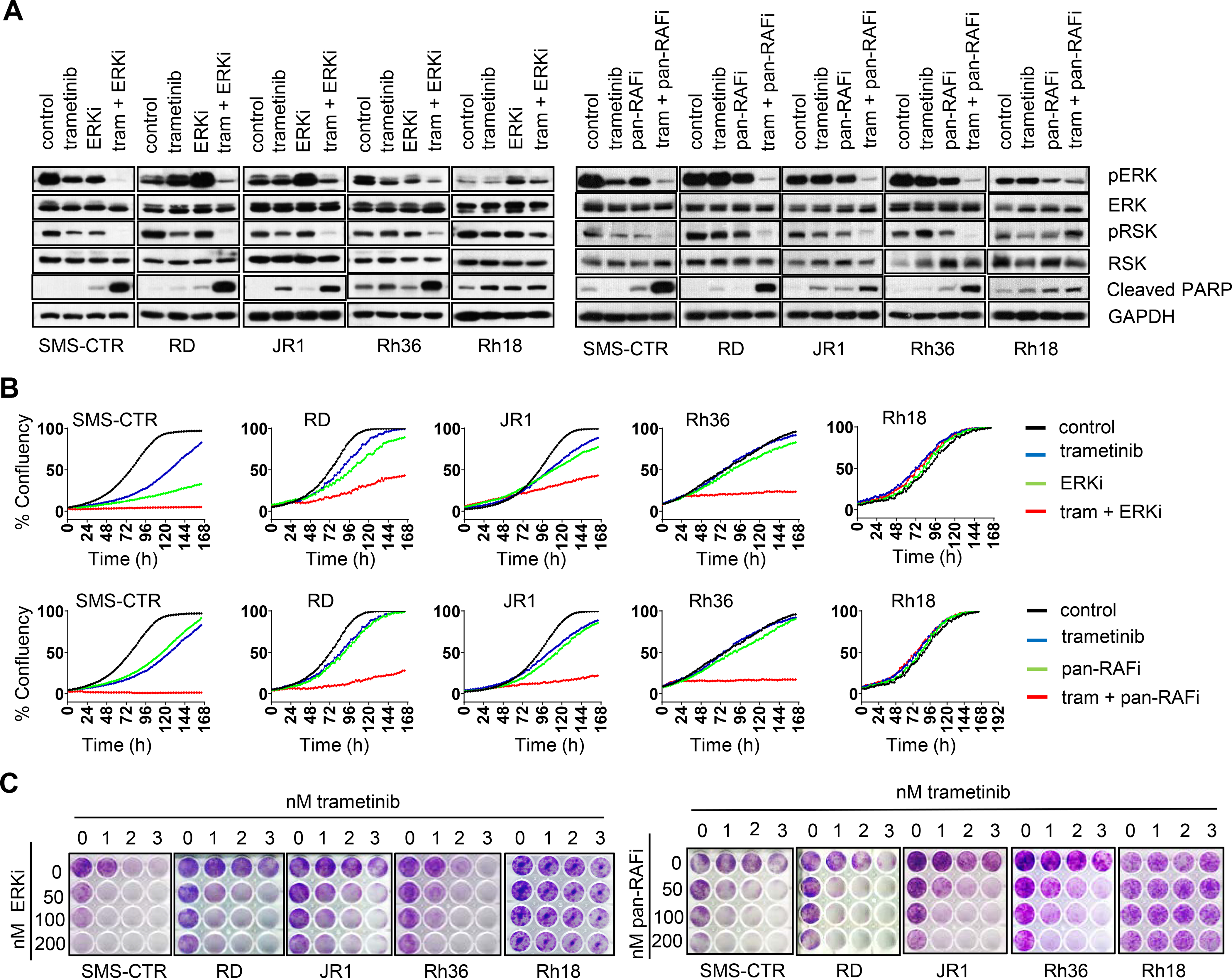
(A) Effect of vertical targeting of MAPK pathway on ERK activity as measured by phosphorylated ERK (pERK, Thr202/Tyr204)) and phosphorylated p90RSK (pRSK, Thr573). Apoptosis marker is also shown (cleaved PARP). Cells were treated with DMSO (control), 2 nM trametinib, 100 nM ERKi LY3214996, 100 nM pan-RAFi LY3009120 or combinations for 72 h and cell lysates analyzed by western blots. (B) Growth curves of RMS cells following treatment with DMSO, trametinib, ERKi LY3214996, pan-RAFi LY3009120 or combinations. Cells were plated in 96 well plates and next day treated as in A. Percent confluency was determined by imaging with Incucyte every 2 h for 168 h. (C) Crystal violet staining images of RAS-mutant RMS cell lines and one RAS wild-type (Rh18) upon treatment with trametinib, ERKi LY3214996, pan-RAFi LY3009120 or combinations. Cells were plated at low density in 12-well plates and next day treated with DMSO or increasing concentrations of the indicated drugs/combinations. After two weeks, cell growth was visualized with crystal violet staining.
Vertical targeting of MAPK pathway synergistically induces cell cycle arrest, myogenic differentiation and apoptosis in H/NRASQ61X-mutant FN-RMS cells
To determine the effect of co-targeting distinct nodes of the MAPK cascade on cell cycle regulation we treated the cells with low concentrations of MEKi, ERKi, pan-RAFi, or the combinations MEKi + ERKi, MEKi + pan-RAFi and pan-RAFi + ERKi (Figures 5A and S5A). In all RAS-mutant cells, single treatments with low drug concentrations did not cause significant changes, while all combinations caused arrest in the G1 phase of the cell-cycle at levels significantly higher than the combined effect of single treatments. No significant changes were detected in the wild type cells. In addition, all three combinations, unlike single treatments, induced cell death preferentially in the RAS-mutant cells as demonstrated by an increased percentage of cells in the sub-G1 phase. Notably, both combinations involving the pan-RAF inhibitor increased apoptosis at higher rates than the MEKi + ERKi combination, consistent with the higher synergy scores.
Figure 5. Vertical targeting of MAPK pathway synergistically induces cell cycle arrest, myogenic differentiation and apoptosis in H/NRAS Q61X-mutant FN-RMS cells.

(A) Cell cycle distribution and sub-G1 quantification in RMS cells upon treatment with 2 nM trametinib, 100 nM ERKi LY3214996, 100 nM pan-RAFi LY3009120 or combinations for 72 h, followed by propidium iodide staining, flow cytometry measurements and analysis with ModFit software. SMS-CTR (HRAS Q61K) and Rh18 (RAS wild type) are shown (see additional cell lines in supplementary figure 4). (B) Immunofluorescence staining images for MHC of indicated cell lines following treatment with 2 nM trametinib, 100 nM ERKi LY3214996, 100nM pan-RAFi LY3009120 or combinations for 8 days. Scale bars, 200 μm.
We next assessed myogenic differentiation of the cells treated with low concentrations of each inhibitor alone or the combinations of trametinib + ERKi, trametinib + pan-RAFi and pan-RAFi + ERKi (Figures 5B and S5B). Similar to the effect on cell cycle distribution, low concentrations of single agent treatments did not induce MHC or MEF2C expression. In contrast, in RAS-mutant cells, all three combinations caused dramatic cellular elongation and acute positivity for both MHC and MEF2C. In the wild type cell line, the combinations did not induce detectable MHC or MEF2C expression.
Vertical targeting of MAPK pathway demonstrates enhanced in vivo efficacy in H/NRASQ61X-mutant FN-RMS xenograft models.
The in vivo efficacy of targeting distinct nodes of the MAPK cascade was assessed in two subcutaneous tumor xenografts in mice: SMS-CTR (HRASQ61K) and RD (NRASQ61H) (Figures 6A–B). Mice bearing the xenografts were treated with trametinib alone (1mg/kg PO), the ERKi LY3214996 alone at half of the maximum tolerated dose (MTD) (50mg/kg PO) [16], pan-RAFi LY3009120 at half the dose previously shown to be well tolerated (15 mg/kg intra-peritoneal) [23] or the combinations of trametinib + LY3214996 (MEKi + ERKi) or LY3009120 + LY3214996 (pan-RAFi + ERKi). In SMS-CTR xenografts, single agent trametinib or ERKi slowed tumor progression, however the combination of both agents caused complete tumor regression, which was maintained even 2 weeks after treatment was ended. Pan-RAF inhibitor, at the doses used, had no effect on SMS-CTR tumor growth; however, co-treatment with ERKi caused rapid and complete regression, also maintained 2 weeks after treatment ended. In RD xenografts, single treatments had minimal or no effect and the combination of MEKi + ERKi caused significant tumor growth delay, however regression was not observed. In contrast, pan-RAFi + ERKi combination caused more than partial RD tumor regressions and tumors remained below initial size 2 weeks after treatment ended. Both combinatorial treatments were well tolerated, with MEKi + ERKi causing on average 5–10% body weight loss and pan-RAFi + ERKi causing no significant body weight loss (Figures S6A and S6B). In pharmacodynamic assays for ERK inhibition in SMS-CTR xenografts, similar to what was observed in RD xenografts (Figure 2B), single treatments had no effect on pERK or pRSK (Figure 6C). However, the combinations of trametinib + ERKi or ERKi + pan-RAFi both inhibited pERK/pRSK (Figure 6C). In addition, immunohistochemistry analyses indicated acute MHC and caspase-3 positivity in tumors treated with both combinations, while almost undetectable with single treatments (Figure 6D).
Figure 6. Vertical targeting of MAPK pathway demonstrates enhanced in vivo efficacy in H/NRAS Q61X-mutant FN-RMS xenograft models.

(A) SCID mice bearing SMS-CTR tumor xenografts were treated with 1 mg/kg trametinib per os qd, 50 mg/kg ERKi LY3214996 per os qd or combination (left); or 15 mg/kg pan-RAFi LY3009120 ip qd, 50 mg/kg ERKi LY3214996 per os qd or combination (right). Treatment duration was 28 days. N=5 mice per group. The relative tumor volume was calculated and plotted +/− SEM. p-values were calculated using one-way ANOVA for single treatments vs combinatorial treatment at the end of the treatment (day 28). (B) SCID mice bearing RD tumor xenografts were treated as in A for SMS-CTR xenografts and tumor volume calculated and plotted as in A. p-values were calculated using one-way ANOVA for single treatments vs combinatorial treatment at the end of the treatment (day 28). (C) Western blot analysis for pharmcodynamics of MAPK pathway inhibition in SMS-CTR tumor xenografts treated as in A for the indicated time points. (D) Representative immunohistochemical staining images for cleaved caspase-3 and myosin heavy chain (MHC) and hematoxylin and eosin (H&E) staining of tumor tissue treated as in A for 4 days.
DISCUSSION
The high incidence of RAS mutations in FN-RMS makes RAS signaling an attractive therapeutic target for this childhood cancer. However, there is still very limited preclinical data to demonstrate potential anti-RAS therapeutic approaches for RAS-mutated FN-RMS.
The two key RAS effector pathways PI3K-AKT-mTOR and RAF-MEK-ERK are the most attractive targets for RAS-driven cancers and have received the highest interest in both academia and industry [10]. The potency of effector activation and effector pathway dependency is dictated by specific cellular contexts, type of RAS mutation, or co-mutations [34]. In FN-RMS, activating mutations in all three RAS isoforms are detected, with NRAS mutations at codon Q61 being the most frequent [2, 35].
Extending recent studies [7, 13], our results further indicate that FN-RMS cells are addicted to oncogenic H/NRAS (in particular Q61X) and that the MAPK effector pathway is central driver of this dependency. A recent study indicated that, in non-small cell lung cancer, KRASQ61H preferentially signals through RAF [36], suggesting that MAPK preference in H/NRASQ61X RMS cells could be attributed the Q61 codon mutations. At this moment it is not clear if similar conclusions can be applied to KRAS-mutant RMS, or mutations at different codons such as G12 or G13. Future studies will address these points.
We also find that treatment of H/NRASQ61X RMS tumors with single-agent MEKi will likely not yield durable clinical responses. Similarly, therapeutic targeting of MEK in RAS-driven adult cancers has shown limited activity [37]. Multiple mechanisms of pathway reactivation are likely responsible for inefficient pathway inhibition and poor response to single-agent treatments [38]. The better response to trametinib treatment of the BRAF V600E AA xenografts is likely due to the more effective MAPK pathway inhibition. Since BRAF V600E mutants signal as monomers, they are less influenced by feedbacks along the pathway. Our CRISPR screen pointed to four potential strategies of vertical inhibition of the MAPK cascade that could be applied to FN-RMS, such as combining trametinib with SHP2, FGFR, ERK or CRAF inhibition. However, in regard to long-term growth effect, we found that co-targeting distinct nodes of the ERK MAPK cascade downstream of RAS was more potent than co-targeting upstream signaling, such as FGFR or SHP2, suggesting that this approach is likely of better therapeutic value.
The RAF kinases are the RAS effectors that initiate the activation of MAPK cascade downstream of RAS. Of the three RAF isoforms (A/B/CRAF), CRAF has been implicated as most important for initiation of KRAS-driven lung carcinoma [27, 28]. In another study, CRAF was dispensable for KRAS-driven oncogenesis of pancreatic cancer, thus indicating a lineage-specific requirement for the RAF isoforms [29]. Our screen and subsequent validation indicated that CRAF suppression was essential for the growth of all four H/NRASQ61X RMS cell lines. Further, suppression of CRAF, but to a lesser extent A/BRAF, synergized with low doses of trametinib or ERKi treatment by enabling long-term MAPK pathway suppression, profound myogenic differentiation, and xenograft tumor regression. Similarly, the central role for CRAF in mediating resistance to MEK inhibitors has been described for KRAS-driven lung cancer [39]. Mechanistically, relief of ERK-mediated negative feedback regulation of CRAF limits the ability of MEK/ERK inhibitors to inhibit ERK signaling in RMS cells.
While CRAF specific inhibitors are currently not available, type II pan-RAF inhibitors potently inhibit all three or BRAF + CRAF isoforms [33, 40, 41]. We found that the combinations pan-RAFi + MEKi, pan-RAFi + ERKi or MEKi + ERKi synergistically induced G1 cell cycle arrest, myogenic differentiation, and apoptosis, demonstrating the ability of these combinations to block tumor growth and cause tumor regression of RMS. Similar to our findings, recent studies in KRAS-driven adult cancers demonstrated the efficacy of vertical inhibition of the RAF-MEK-ERK cascade by either MEKi + ERKi, pan-RAFi + MEKi or pan-RAFi + ERKi [23, 25, 26]. However, normal tissue toxicity with such treatments is a potential concern, underscored by the fact that in a phase 1 combination clinical trial, a tolerable MEKi + ERKi regime could not be established [42]. Our studies demonstrated higher synergy scores and induction of apoptosis with combinations using pan-RAFi + MEKi/ERKi as compared to MEKi + ERKi in H/NRAS Q61X-mutant RMS cells. It is possible that the relief of ERK-mediated inhibitory CRAF phosphorylation remains a limiting factor in MEKi + ERKi treatment. Further, our in vivo results indicated that, compared to MEKi + ERKi, the pan-RAFi + ERKi combination was better tolerated, yet had greater synergy and anti-tumor effect in H/NRAS Q61X RMS cells and xenografts. These results suggest that combining pan-RAF inhibitor with either MEK or ERK inhibitor would likely have wider therapeutic window.
One limitation of our studies is that currently we are unable to contextualize our combinatorial in vivo experiments in terms of clinically-relevant drug exposures. Clinical pharmacokinetics for the ERK inhibitor LY3214996 are not available, and clinical development of the pan-RAF inhibitor LY3009120 was discontinued due to short half-life in patients. Therefore, use of LY3009120 is limited to a tool. Two other pan-RAF inhibitors (belvarafenib and LXH254) were recently reported to have favorable clinical pharmacokinetics and antitumor activities in adult patients [43, 44]. Combination studies with MEK/ERK inhibitors are ongoing (NCT02974725, NCT04835805) and encouraging preliminary clinical data in NRAS-mutant melanoma was reported [45]. Another pan-RAF inhibitor Day101 recently showed very encouraging activity in pediatric patients with BRAF-fusion positive low grade gliomas [46]. However, clinical PK studies for recommended doses are still not available for any of these inhibitors, making it difficult to conduct precise preclinical evaluations. Clinical and preclinical studies with the multi-kinase inhibitor sorafenib (also targeting B/CRAF) did not indicate activity in RMS, although RAS-mutant RMS was not the focus in these studies [47, 48]. In addition, sorafenib might target RAF kinases in a mechanism distinct from new generation pan-RAF inhibitors since in a clinical trial with pediatric astrocytomas, sorafenib caused unprecedented acceleration of tumor growth, irrespective of BRAF status, likely due to paradoxical pathway activation [49]. Sorafenib might not be appropriate for RAS-driven RMS and future studies need to focus on evaluating the preclinical efficacy of Belvarafenib, LXH254 or Day101 as single agents and in combination with MEK/ERK inhibitors in H/NRAS-mutant RMS.
In summary, our study demonstrated a central role for the ERK MAPK pathway in H/NRASQ61X FN-RMS cells, and that relief of CRAF negative regulation by ERK is limiting the response to single agent MEK or ERK inhibitors. Vertical targeting of the MAPK pathway, and specifically the use of a pan-RAF inhibitor in combination with MEK or ERK inhibitors, could have therapeutic value. Given the central role of CRAF described in our studies, development of a CRAF-specific inhibitors could also be an effective and possibly less-toxic approach for vertical inhibition of the MAPK cascade in H/NRASQ61X-mutant RMS.
Supplementary Material
Acknowledgments
This work was funded by Max and Minnie Tomerlin Voelcker Fund (AVV), 1U01CA199297-01 (PJH), 7P01CA165995-03 (PJH), R01CA207083 (KCW) and U54CA231630 (KCW). We are grateful to: Dr. Abhik Bandyopadhyay and Samson Ghilu (GCCRI) for help with in vivo studies; Raehum Paik and Selika Garza (UTHSCSA Mouse Genome Engineering and Transgenic Facility) for generating CRAF 4A construct; Daniel Robledo (GGCRI histology lab) for performing IHC; Kevin Lin (Duke University) for the original design, synthesis, and cloning of CRISPR library; Dr. Shripad Bhagwat (Eli Lilly and Company) for advice on in vivo use of LY3214996 and review of manuscript; Dr. Channing Der (University of North Carolina at Chapel Hill) for review of manuscript; Genome Sequencing Facility/Bioinformatics (supported by NIH P30 CA054174, NIH S10 1S10OD021805-01, CPRIT Core Facility Award RP160732, and NIH 1UL1 TR002645-01); UTHSCSA Flow Cytometry Shared Resource Facility (supported by NIH P30 CA054174 and NIH UL1 TR002645).
Footnotes
Conflict of interest: The authors declare no potential conflicts of interest.
References:
- 1.Shern JF, Yohe ME, and Khan J, Pediatric Rhabdomyosarcoma. Crit Rev Oncog, 2015. 20(3–4): p. 227–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shern JF, Chen L, Chmielecki J, Wei JS, Patidar R, Rosenberg M, et al. , Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer Discov, 2014. 4(2): p. 216–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen L, Shern JF, Wei JS, Yohe ME, Song YK, Hurd L, et al. , Clonality and evolutionary history of rhabdomyosarcoma. PLoS Genet, 2015. 11(3): p. e1005075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kirsch DG, Dinulescu DM, Miller JB, Grimm J, Santiago PM, Young NP, et al. , A spatially and temporally restricted mouse model of soft tissue sarcoma. Nat Med, 2007. 13(8): p. 992–7. [DOI] [PubMed] [Google Scholar]
- 5.Hettmer S, Liu J, Miller CM, Lindsay MC, Sparks CA, Guertin DA, et al. , Sarcomas induced in discrete subsets of prospectively isolated skeletal muscle cells. Proc Natl Acad Sci U S A, 2011. 108(50): p. 20002–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Langenau DM, Keefe MD, Storer NY, Guyon JR, Kutok JL, Le X, et al. , Effects of RAS on the genesis of embryonal rhabdomyosarcoma. Genes Dev, 2007. 21(11): p. 1382–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yohe ME, Gryder BE, Shern JF, Song YK, Chou HC, Sindiri S, et al. , MEK inhibition induces MYOG and remodels super-enhancers in RAS-driven rhabdomyosarcoma. Sci Transl Med, 2018. 10(448). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Janes MR, Zhang J, Li LS, Hansen R, Peters U, Guo X, et al. , Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell, 2018. 172(3): p. 578–589 e17. [DOI] [PubMed] [Google Scholar]
- 9.Downward J, RAS Synthetic Lethal Screens Revisited: Still Seeking the Elusive Prize? Clin Cancer Res, 2015. 21(8): p. 1802–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cox AD, Fesik SW, Luo J, Kimmelman AC and Der CJ Drugging the undruggable Ras: mission possible?. Nat Rev Drug Discov, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lin KH, Rutter JC, Xie A, Pardieu B, Winn ET, Bello RD, et al. , Using antagonistic pleiotropy to design a chemotherapy-induced evolutionary trap to target drug resistance in cancer. Nat Genet, 2020. 52(4): p. 408–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kolb EA, Gorlick R, Houghton PJ, Morton CL, Neale G, Keir ST, et al. , Initial testing (stage 1) of AZD6244 (ARRY-142886) by the Pediatric Preclinical Testing Program. Pediatr Blood Cancer, 2010. 55(4): p. 668–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Phelps MP, Yang H, Patel S, Rahman MM, McFadden G, and Chen E, Oncolytic Virus-Mediated RAS Targeting in Rhabdomyosarcoma. Mol Ther Oncolytics, 2018. 11: p. 52–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barlaam B, Cosulich S, Degorce S, Fitzek M, Green S, Hancox U, et al. , Discovery of (R)-8-(1-(3,5-difluorophenylamino)ethyl)-N,N-dimethyl-2-morpholino-4-oxo-4H-chromene-6-carboxamide (AZD8186): a potent and selective inhibitor of PI3Kβ and PI3Kδ for the treatment of PTEN-deficient cancers. J Med Chem, 2015. 58(2): p. 943–62. [DOI] [PubMed] [Google Scholar]
- 15.Addie M, Ballard P, Buttar D, Crafter C, Currie G, Davies BR, et al. , Discovery of 4-Amino-N-[(1S)-1-(4-chlorophenyl)-3-hydroxypropyl]-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidine-4-carboxamide (AZD5363), an Orally Bioavailable, Potent Inhibitor of Akt Kinases. Journal of Medicinal Chemistry, 2013. 56(5): p. 2059–2073. [DOI] [PubMed] [Google Scholar]
- 16.Bhagwat SV, McMillen WT, Cai S, Zhao B, Whitesell M, Shen W, et al. , ERK Inhibitor LY3214996 Targets ERK Pathway-Driven Cancers: A Therapeutic Approach Toward Precision Medicine. Mol Cancer Ther, 2020. 19(2): p. 325–336. [DOI] [PubMed] [Google Scholar]
- 17.Yamaguchi T, Yoshida T, Kurachi R, Kakegawa J, Hori Y, Nanayama T, et al. , Identification of JTP-70902, a p15(INK4b)-inductive compound, as a novel MEK1/2 inhibitor. Cancer Sci, 2007. 98(11): p. 1809–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Infante JR, Fecher LA, Falchook GS, Nallapareddy S, Gordon MS, Becerra C, et al. , Safety, pharmacokinetic, pharmacodynamic, and efficacy data for the oral MEK inhibitor trametinib: a phase 1 dose-escalation trial. Lancet Oncol, 2012. 13(8): p. 773–81. [DOI] [PubMed] [Google Scholar]
- 19.Samatar AA and Poulikakos PI, Targeting RAS-ERK signalling in cancer: promises and challenges. Nat Rev Drug Discov, 2014. 13(12): p. 928–42. [DOI] [PubMed] [Google Scholar]
- 20.Morrison DK, MAP kinase pathways. Cold Spring Harb Perspect Biol, 2012. 4(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fedele C, Ran H, Diskin B, Wei W, Jen J, Geer MJ, et al. , SHP2 Inhibition Prevents Adaptive Resistance to MEK Inhibitors in Multiple Cancer Models. Cancer Discov, 2018. 8(10): p. 1237–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Manchado E, Weissmueller S, Morris J.P.t., Chen CC, Wullenkord R, Lujambio A, et al. , A combinatorial strategy for treating KRAS-mutant lung cancer. Nature, 2016. 534(7609): p. 647–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yen I, Shanahan F, Merchant M, Orr C, Hunsaker T, Durk M, et al. , Pharmacological Induction of RAS-GTP Confers RAF Inhibitor Sensitivity in KRAS Mutant Tumors. Cancer Cell, 2018. 34(4): p. 611–625 e7. [DOI] [PubMed] [Google Scholar]
- 24.Xue Y, Martelotto L, Baslan T, Vides A, Solomon M, Mai TT, et al. , An approach to suppress the evolution of resistance in BRAF(V600E)-mutant cancer. Nat Med, 2017. 23(8): p. 929–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Merchant M, Moffat J, Schaefer G, Chan J, Wang X, Orr C, et al. , Combined MEK and ERK inhibition overcomes therapy-mediated pathway reactivation in RAS mutant tumors. PLoS One, 2017. 12(10): p. e0185862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ozkan-Dagliyan I, Diehl JN, George SD, Schaefer A, Papke B, Klotz-Noack K, et al. , Low-Dose Vertical Inhibition of the RAF-MEK-ERK Cascade Causes Apoptotic Death of KRAS Mutant Cancers. Cell Rep, 2020. 31(11): p. 107764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Blasco RB, Francoz S, Santamaria D, Canamero M, Dubus P, Charron J, et al. , c-Raf, but not B-Raf, is essential for development of K-Ras oncogene-driven non-small cell lung carcinoma. Cancer Cell, 2011. 19(5): p. 652–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karreth FA, Frese KK, DeNicola GM, Baccarini M, and Tuveson DA, C-Raf is required for the initiation of lung cancer by K-Ras(G12D). Cancer Discov, 2011. 1(2): p. 128–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eser S, Reiff N, Messer M, Seidler B, Gottschalk K, Dobler M, et al. , Selective requirement of PI3K/PDK1 signaling for Kras oncogene-driven pancreatic cell plasticity and cancer. Cancer Cell, 2013. 23(3): p. 406–20. [DOI] [PubMed] [Google Scholar]
- 30.Freeman AK, Ritt DA, and Morrison DK, The importance of Raf dimerization in cell signaling. Small GTPases, 2013. 4(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dougherty MK, Muller J, Ritt DA, Zhou M, Zhou XZ, Copeland TD, et al. , Regulation of Raf-1 by direct feedback phosphorylation. Mol Cell, 2005. 17(2): p. 215–24. [DOI] [PubMed] [Google Scholar]
- 32.Unni AM, Harbourne B, Oh MH, Wild S, Ferrarone JR, Lockwood WW, et al. , Hyperactivation of ERK by multiple mechanisms is toxic to RTK-RAS mutation-driven lung adenocarcinoma cells. Elife, 2018. 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Peng SB, Henry JR, Kaufman MD, Lu WP, Smith BD, Vogeti S, et al. , Inhibition of RAF Isoforms and Active Dimers by LY3009120 Leads to Anti-tumor Activities in RAS or BRAF Mutant Cancers. Cancer Cell, 2015. 28(3): p. 384–98. [DOI] [PubMed] [Google Scholar]
- 34.Yuan TL, Amzallag A, Bagni R, Yi M, Afghani S, Burgan W, et al. , Differential Effector Engagement by Oncogenic KRAS. Cell Rep, 2018. 22(7): p. 1889–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen X, Stewart E, Shelat AA, Qu C, Bahrami A, Hatley M, et al. , Targeting oxidative stress in embryonal rhabdomyosarcoma. Cancer Cell, 2013. 24(6): p. 710–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou ZW, Ambrogio C, Bera AK, Li Q, Li XX, Li L, et al. , KRAS(Q61H) Preferentially Signals through MAPK in a RAF Dimer-Dependent Manner in Non-Small Cell Lung Cancer. Cancer Res, 2020. 80(17): p. 3719–3731. [DOI] [PubMed] [Google Scholar]
- 37.Blumenschein GR Jr., Smit EF, Planchard D, Kim DW, Cadranel J, De Pas T, et al. , A randomized phase II study of the MEK1/MEK2 inhibitor trametinib (GSK1120212) compared with docetaxel in KRAS-mutant advanced non-small-cell lung cancer (NSCLC)dagger. Ann Oncol, 2015. 26(5): p. 894–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lake D, Correa SA, and Muller J, Negative feedback regulation of the ERK1/2 MAPK pathway. Cell Mol Life Sci, 2016. 73(23): p. 4397–4413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lito P, Saborowski A, Yue J, Solomon M, Joseph E, Gadal S, et al. , Disruption of CRAF-mediated MEK activation is required for effective MEK inhibition in KRAS mutant tumors. Cancer Cell, 2014. 25(5): p. 697–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hatzivassiliou G, Song K, Yen I, Brandhuber BJ, Anderson DJ, Alvarado R, et al. , RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature, 2010. 464(7287): p. 431–5. [DOI] [PubMed] [Google Scholar]
- 41.Ramurthy S, Taft BR, Aversa RJ, Barsanti PA, Burger MT, Lou Y, et al. , Design and Discovery of N-(3-(2-(2-Hydroxyethoxy)-6-morpholinopyridin-4-yl)-4-methylphenyl)-2-(trifluorom ethyl)isonicotinamide, a Selective, Efficacious, and Well-Tolerated RAF Inhibitor Targeting RAS Mutant Cancers: The Path to the Clinic. J Med Chem, 2019. [DOI] [PubMed] [Google Scholar]
- 42.Weekes C, Lockhart A, LoRusso P, Murray E, Park E, Tagen M, et al. , A Phase Ib Study to Evaluate the MEK Inhibitor Cobimetinib in Combination with the ERK1/2 Inhibitor GDC-0994 in Patients with Advanced Solid Tumors. Oncologist, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Janku F, Iyer G, Spreafico A, Yamamoto N, Bang Y-J, Elez E, et al. , A phase I study of LXH254 in patients (pts) with advanced solid tumors harboring MAPK pathway alterations. Journal of Clinical Oncology, 2018. 36(15_suppl): p. 2586–2586. [Google Scholar]
- 44.Kim TW, Lee J, Shin SJ, Kim J-S, Kim YJ, Han HS, et al. , Belvarafenib, a novel pan-RAF inhibitor, in solid tumor patients harboring BRAF, KRAS, or NRAS mutations: Phase I study. Journal of Clinical Oncology, 2019. 37(15_suppl): p. 3000–3000.31557067 [Google Scholar]
- 45.Shin SJ, Lee J, Kim TM, Kim J-S, Kim YJ, Hong YS, et al. , A phase Ib trial of belvarafenib in combination with cobimetinib in patients with advanced solid tumors: Interim results of dose-escalation and patients with NRAS-mutant melanoma of dose-expansion. Journal of Clinical Oncology, 2021. 39(15_suppl): p. 3007–3007. [Google Scholar]
- 46.Kilburn LB, Jabado N, Franson A, Chi SN, Fisher MJ, Hargrave DR, et al. , FIREFLY-1: A phase 2 study of the pan-RAF inhibitor DAY101 in pediatric patients with low-grade glioma. Journal of Clinical Oncology, 2021. 39(15_suppl): p. TPS10056–TPS10056. [Google Scholar]
- 47.Keir ST, Maris JM, Lock R, Kolb EA, Gorlick R, Carol H, et al. , Initial testing (stage 1) of the multi-targeted kinase inhibitor sorafenib by the pediatric preclinical testing program. Pediatr Blood Cancer, 2010. 55(6): p. 1126–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim A, Widemann BC, Krailo M, Jayaprakash N, Fox E, Weigel B, et al. , Phase 2 trial of sorafenib in children and young adults with refractory solid tumors: A report from the Children’s Oncology Group. Pediatr Blood Cancer, 2015. 62(9): p. 1562–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Karajannis MA, Legault G, Fisher MJ, Milla SS, Cohen KJ, Wisoff JH, et al. , Phase II study of sorafenib in children with recurrent or progressive low-grade astrocytomas. Neuro Oncol, 2014. 16(10): p. 1408–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA sequencing data generated in this study are available at Gene Expression Omnibus (GEO) at GSE180127.