

Abstract
The purpose of our study is to understand the protective effects of small molecule ligands for phosphorylated tau (p-tau) in Alzheimer’s disease (AD) progression. Many reports show evidence that phosphorylated tau is reported to be an important contributor to the formation of paired helical filaments (PHFs) and neurofibrillary tangles (NFTs) in AD neurons. In AD, glycogen synthase kinase-3 beta (GSK3β), cyclin-dependent kinase-5 and dual specificity tyrosine-phosphorylation-regulated kinase 1A (DYRK1A), are the three important kinases responsible for tau hyperphosphorylation. Currently, there are no drugs and/or small molecules that reduce the toxicity of phosphorylated tau in AD. In the present study, we rationally selected and validated small molecule ligands that bind to the phosphorylated tau at SER23 (Ser 285) and TYR44 (Tyr310). We also assessed the molecular dynamics and validated molecular docking sites for the three best ligands. Based on the best docking scores −8.09, −7.9 and −7.8 kcal/mol, we found that ligand 1 binds to key hyperphosphorylation residues of phosphorylated tau that inhibit abnormal PHF-tau, DYRK1A and GKS3β that reduce phosphorylated tau levels in AD. Using biochemical, molecular, immunoblotting, immunofluorescence and transmission electron microscopy analyses, we studied the ligand 1 inhibition as well as mitochondrial and synaptic protective effects in immortalized primary hippocampal neuronal (HT22) cells. We found interactions between NAT10-262501 (ligand 1) and phosphorylated tau at key phosphorylation sites and these ligand-based inhibitions decreased PHF-tau, DYRK1A and GSK3β levels. We also found increased mitochondrial biogenesis, mitochondrial fusion and synaptic activities and reduced mitochondrial fission in ligand 1-treated mutant tau HT22 cells. Based on these results, we cautiously conclude that phosphorylated tau NAT10-262501 (ligand 1) reduces hyperphosphorylation of tau based GKS3β and CDK5 kinase regulation in AD, and aids in the maintenance of neuronal structure, mitochondrial dynamics and biogenesis with a possible therapeutic drug target for AD.
Graphical Abstract
Graphical Abstract.
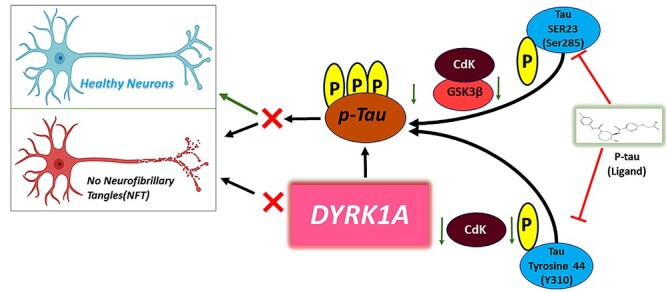
Introduction
The aberrant hyperphosphorylation of tau (p-tau) and self-aggregation within the neurons is a well-defined pathological hallmark of Alzheimer’s disease (AD) and other tauopathies (1–4). Microtubule associated protein (MAPT) plays an important role in neuronal differentiation, development and transportation of mitochondria from cell body to synapses (5,6). The balance of phosphatase and kinase activity maintains the structure of MAPT in healthy state neurons with equilibrium allowing the perseverance of neuronal structure (7–10). Disruption of the equilibrium between phosphatases and kinases causes hyperphosphorylation, which disrupts microtubule structure with radical phosphorylated tau. The self-aggregation of phosphorylated tau fibrils leads to paired helical filaments (PHFs) and neurofibrillary tangles (NFTs) observed in AD (11,12). Many reports reveal evidence that phosphorylated tau is believed to be an important contributor to the destabilization of microtubules and neuronal axon structure forming NFT’s in AD neurons (13–15). Oxidative stress and other cellular stress related events promote tau hyperphosphorylation, with decreased phosphatase functions and increased kinase actions in AD and tauopathies (16,17). In AD, the major phosphorylated kinases are glycogen synthase kinase-3 beta (GSK3β), cyclin-dependent kinase-5 (CDK5) and dual specificity tyrosine-phosphorylation-regulated kinase 1A (DYRK1A), which relay on serine/threonine/tyrosine residues of tau (18–21). Having, a nearly 80–85 long list of Ser/Thr/Tyr residues in the CNS tau isoforms, at least 48–50 have been found to be phosphorylated in AD (22,23). Hence, it is a formidable challenge to identify the hyperphosphorylated p-sites on tau and designing small molecules for AD and other tauopathies. There are a wide-range of therapeutic molecules available in the market and have been tested for phosphorylated tau inhibition to enhance phosphatase activity and microtubule stability in AD. Currently, there are no small molecules that claim to show promising results with phosphorylated tau inhibition/therapeutic modulation in AD. Designing multiple targeting small molecule inhibitors against phosphorylated tau in AD is incredibly challenging for researchers. There are no published reports on multiple phosphorylated tau site targeting therapeutic molecules (drugs) available for AD thus far.
In our earlier studies, we computationally screened ligands that specifically bind with Ser/Thr/Tyr with good docking scores (24). In the present study, we continued our bioinformatics studies and validations for our top three ligands by conducting molecular dynamic (MD) simulations and biological validation for the selected ligand 1. We validated these three ligands through simulation studies to confirm binding p-sites at SER23 (Ser 285) and TYR44 (Y310), which are key in kinase regulation of tau phosphorylation in AD (25–27). Phosphorylation of the tyrosine 310 (310Y) regulates the structure, aggregation and microtubule-stability of Tau (28–32). These phosphorylated tau sites SER 285 and TYR 310 are highly involved in kinases regulation by upregulating GSK3β, CDK5 and DYRK1A dependent phosphorylation, which are deregulated in AD (13,33–40). We evaluated three ligands for our biological validation for phosphorylated tau inhibition and neuronal protection by testing in HT22 cells transfected with mutant Tau cDNA. Hence, our proposed study is important because it unfurls the link between phosphorylated tau inhibition and kinase regulation in AD, and unravels the site-specific characterized ligand and mitochondrial and synaptic protection.
Results
MD simulations of tau
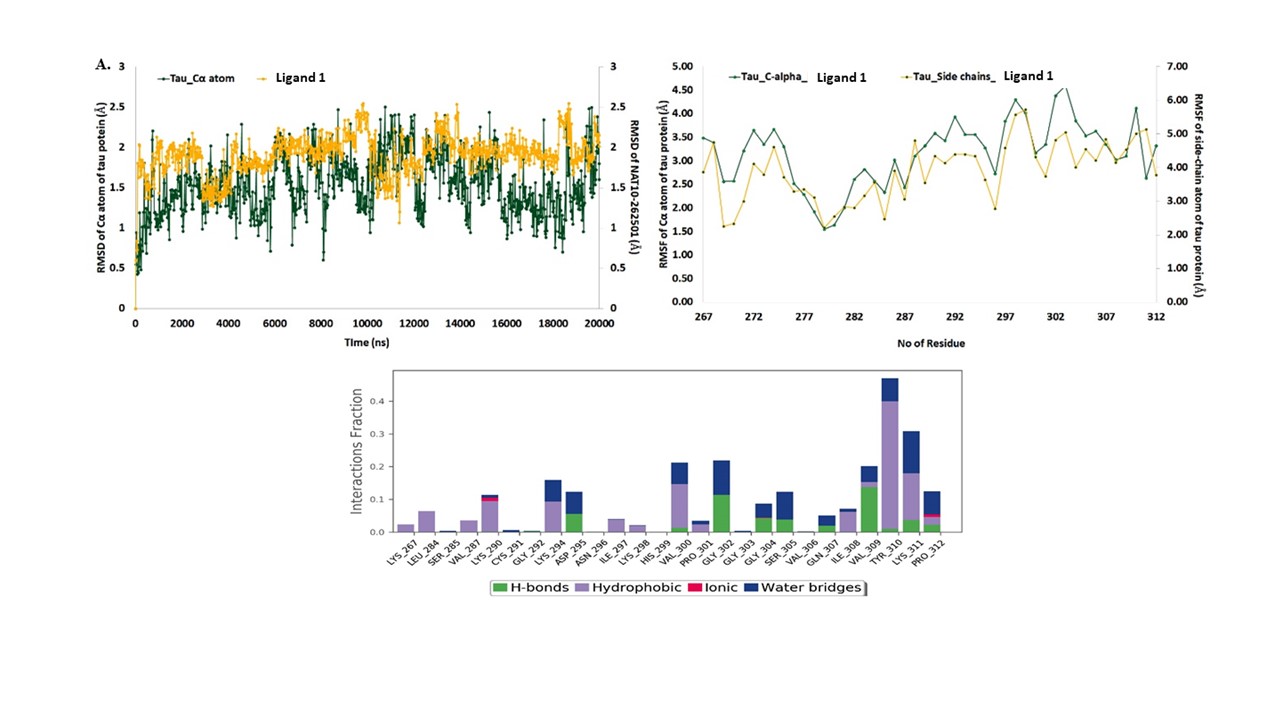
The MD simulations for the native crystal structure of phosphorylated tau protein 3-D structure are usually exercised to improve the optimum NMR stable model structure intended for the molecular docking analysis. The MD simulations results revealed that, 3-D phosphorylated tau protein was stable at 20 ns, which are stable Cartesian co-ordinate residues of 46 (residues of 267–312) and are composed with 3-helical and 12 β-strands structures (Fig. 1A). The computed total energy and potential energy remained stable from the start of the MD simulations in the case of the stable model of phosphorylated tau protein. The stable model had an average total energy of −35 704.616 kcal/mol, and potential energy of −43 589.651 kcal/mol, wherein the constant temperature and pressure were maintained at 300 K and 1 atm for phosphorylated tau protein (Supplementary Material, Fig. S1A).
Figure 1.
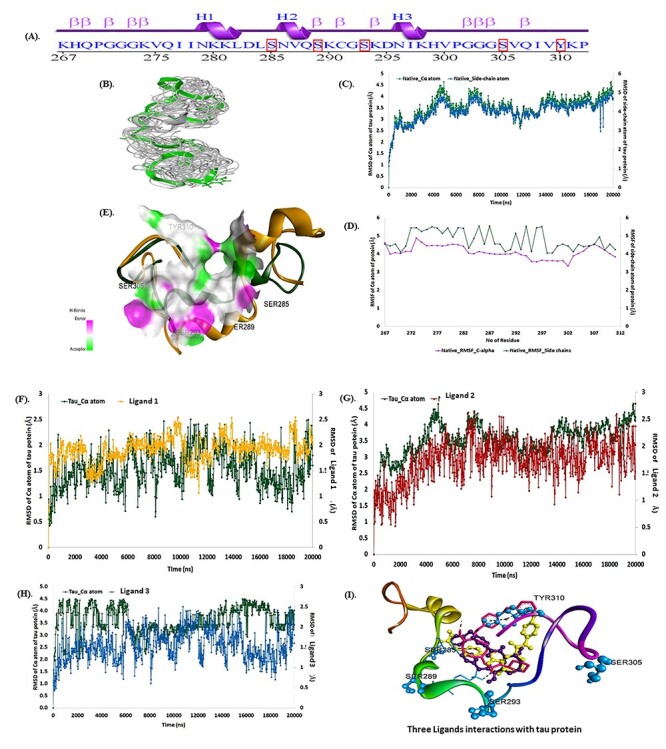
Tau protein structure stability analysis: (A) sequence of tau protein, (B) 3D sturucture of phosphorylated tau, (C) RMSD calculations of phosphorylated tau Cα atoms (green) and side-chains (blue), (D) RMSF calculations of phosphorylated tau Cα atoms (green) and side-chains (violet) and (E) superimmposition of native (yellow–orange) and post MD simulation stable structure (green) with active site of tau protein. (F). RMSD plots ligand 1, of during 20 ns period [(Green-phosphorylated tau)-Yellow-ligand 1]. (G) RMSD plots ligand 2, of during 20 ns period [(Green-phosphorylated tau)-Red-ligand 2]. (H) RMSD plots ligand 3 [(Green-phosphorylated tau)-Blue-ligand 3], of during 20 ns period. (I) ligands 1, 2 and 3 superimpose docking structure with 3-D phosphorylated tau. Ligand 1 showed best binding after simulation study at p-sites at SER23 (Ser 285) and TYR (Y) 44 (Y310).
The trajectory event was observed to remain stable throughout the simulation process, which was verified by plotting the root means square deviation (RMSD) and root mean square fluctuation (RMSF) graphs during the MD production run (Fig. 1B and C). The average RMSD of the Cα atoms and side-chain atoms are observed to have minimal deviations with the 3.5 and 4.03 Å, respectively for the phosphorylated tau protein (Fig. 1D). Similarly, the RMSF plot for the phosphorylated tau protein residues of Cα atoms and side-chain atoms have a projected average RMSF of 4.10 and 4.77 Å, stable at 20 ns MDs run (Fig. 1E). The results revealed that the phosphorylated tau protein of model-8 was stable at MDs. Hence, the stabilized NMR phosphorylated tau 3D structure was desirable for the molecular docking analysis using Discovery Studio v2020. Fig. 1D depicted the superimposition (global structure–structure alignment) of native (yellow–orange) and stable (green) phosphorylated tau proteins were exploited with an RMSD of 2.48 Å with minimal fluctuations observed compared with other NMR models. The MD simulations results of RMSD, RMSF and superimposition revealed that the eighth model of NMR structure was stable, and will be further employed for molecular docking analysis.
Molecular docking
In the case of Discovery Studio, the NAT10-262501 (ligand 1) has at least a LibDock score of 156.58 and a binding energy of −7.5 kcal/mol, subsequently NAT10-367721 (ligand 2) and NAT24-392393 (ligand 3) encountered docking scores of 154.64; −6.4 kcal/mol and 153.07; −5.5 kcal/mol, respectively (Table 1). The interactions between ligand 1 and phosphorylated tau protein are highly mediated by an H-bond, π–π and π–alkyl interactions. Ligand 1 formed five H-bonds with Lys280, Asp283, Ser285 (2) and Leu284, besides π–π and π–alkyl interactions and was observed with Leu284 and Tyr310 residues of phosphorylated tau protein. Furthermore, Gln269, Asp283, Lys280, Lys281, Val287, Ile297, Asn296, Ile308 and Pro312 residues of phosphorylated tau protein formed vdW interactions within a 4 Å region of NAT10-262501 (Fig. 2).
Table 1.
Molecular docking score, interactions and bond length of selected three ligand small molecules with phosphorylated tau protein
| Compounds | Docking score (kcal/mol) | Post molecular docking interactions | |||||||
|---|---|---|---|---|---|---|---|---|---|
| AutoDock vina | Discovery Studio | ||||||||
| H-bond | H-bond length (Å) | vdW interactions | Libdock score | Binding energy (kcal/mol) | H-bond | H-bond length (Å) | vdW interactions | ||
| NAT10-262501 (Ligand 1) | −8.09 | Ser285:C=O —> OH | 2.74 | Gln269, Asp283, Leu284, Asn286, Val287, Gln288, Ser289, Lys290, Ser293, Lys294, Asp295, Lys298, His299, Val300, Pro301, Ile308, Val309, Lys311, Pro312 | 156.58 | −7.5 | Ser285:HN —> O | 2.35 | Gln269, Asp283, Lys280, Lys281, Val287, Ile297, Asn296, Ile308, Pro 312 |
| OH —> Gly292: C=O | 1.76 | Ser285: HG —> O | 2.49 | ||||||
| Asn296: NH —> CO | 2.54 | H —> O: Lys280 | 2.09 | ||||||
| Ile297: NH —> CO | 2.00 | H —> O: Asp283:O | 2.31 | ||||||
| Tyr310: NH —> CO | 1.96 | Leu284:CA —> O | 2.25 | ||||||
| π —> π Leu284 | 3.13 | ||||||||
| Tyr310 π —> π | 3.80 | ||||||||
| NAT10–367721 (Ligand 2) | −7.9 | Gly292: OH —> C=O | 2.01 | Leu284, Ser284, Asn286, Val287, Gln288, Ser289, Lys290, Cys291, Asp295, Asn296, Lys298, Val300, Gly303, Gly304, Gln307 | 154.64 | −6.4 | π —> π: Lys280 | 4.03 | Asp283, Ser285, Val287, Lys290, Asn296, Ile308, Tyr310, Pro312 |
| Ser293: NH —> C=O | 2.93 | π —> π: Lys281 | 4.32 | ||||||
| Lys294: OH —> C=O | 2.58 | π —> π: Leu284 | 3.16 | ||||||
| OH —> His299: C=O | 2.46 | H —> OD2:Asp295 | 2.73 | ||||||
| Ser305: C=O —> OH | 2.57 | Ile297: π —> π | 4.01 | ||||||
| NAT24–392393 (Ligand 3) | −7.8 | Ser293: OH —> C=O | 2.12 | Cys291, Gly292, Lys294, His299, Val300, Pro301, Gly302, Gly303, Gly304, Val306, Gln307, Ile308, Val309, Tyr310, Pro312 | 153.07 | −5.5 | H —> O: Lys281 | 2.10 | Lys280, Leu282, Asp283, Ser285, Val287, His299, Asp295, Asn296, Ile297, Ile308, Pro312, Val309 |
| Ser305: C=O —> NH | 2.77 | Leu284: π —> π | 3.09 | ||||||
| Lys311: OH —> C=O | 2.56 | Lys290: HZ3 —> O | 2.79 | ||||||
| Tyr310: π —> π | 3.63 | ||||||||
| Tyr310: π —> π | 3.62 | ||||||||
Figure 2.
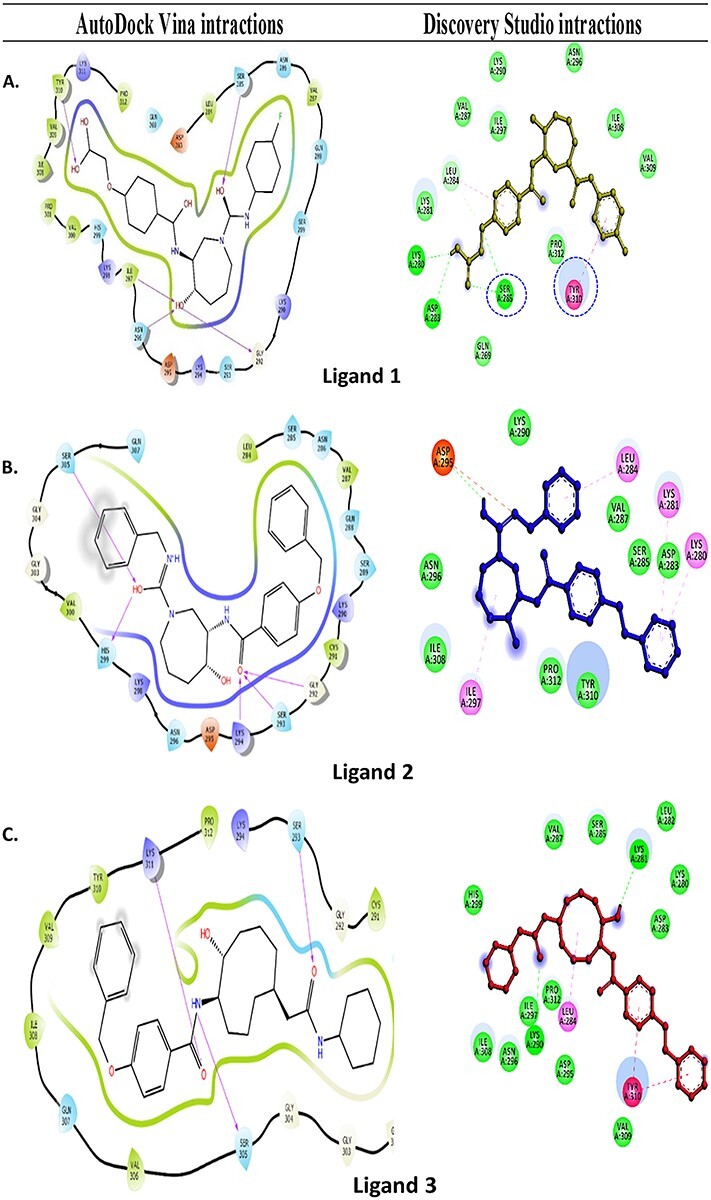
Molecular docking interactions of tau protein surrounding residues interactions with three ligands: (A) Ligand 1-phosphorylated tau with respective molecular interactions strategies like Van der Waals forces and conventional hydrogen bond (green colour), attractive charge (red colour), Alkyl and PI-Alkyl (pink colour). (B) Ligand 2-phosphorylated tau with respective molecular interactions strategies like van dalwaals and conventional hydrogen bond (green colour), attractive charge (red colour) and Alkyl and PI-Alkyl (pink colour). (C) Ligand 3-phosphorylated tau with respective molecular interactions strategies like Van der Waals and conventional hydrogen bond (green colour), attractive charge (red colour) and Alkyl and PI-Alkyl (pink colour).
NAT10-367721 (ligand 2) formed H-bonds with Asp295 and four π–π interactions with Lys280, Lys281, Leu284 and Ile297. The residues of Asp283, Ser285, Val287, Lys290, Asn296, Ile308, Tyr310 and Pro312 formed vdW interactions within a 4 Å region of ligand 2. Although the NAT24-392393 (ligand 3), also formed two H-bonds with Lys281, and Leu290 alongside, π–π interactions were observed with Leu284, and Tyr310 of the phosphorylated tau protein. The residues of Lys280, Leu282, Asp283, Ser285, Val287, His299, Asp295, Asn296, Ile297, Ile308, Pro312 and Val309 were also involved in vdW interactions with ligand 3.
In the case of AutoDock Vina, ligand 1 has the lowest docking score of −8.09 kcal/mol, when compared with NAT10-367721 (−7.9 kcal/mol) and lead 3 (7.8 kcal/mol). The observed H-bonds and vdW interactions of the active site residues are listed in (Table 1), which are pretend as releveled in Discovery Studio section. Hence, the different docking strategies revealed, that ligand 1, ligand 2 and ligand 3, are potentially bound with active site residues of tau protein. The outward of molecular docking, further we performed MD simulations to check the docked complex stability and constant interactions to each complex.
MD simulations
In order to charactarize the docking properties of the three optimized ligands, which bound with an active site pocket of phosphorylated tau protein in different docking stratagies (Fig. 1F–H). The stable average, total energy and potential energies of the docked complex were illustrated (Supplementary Material, Fig. S1) wherein the temperature and pressure were maintained at a constant of 300 K and 1 atm for phosphorylated tau protein.
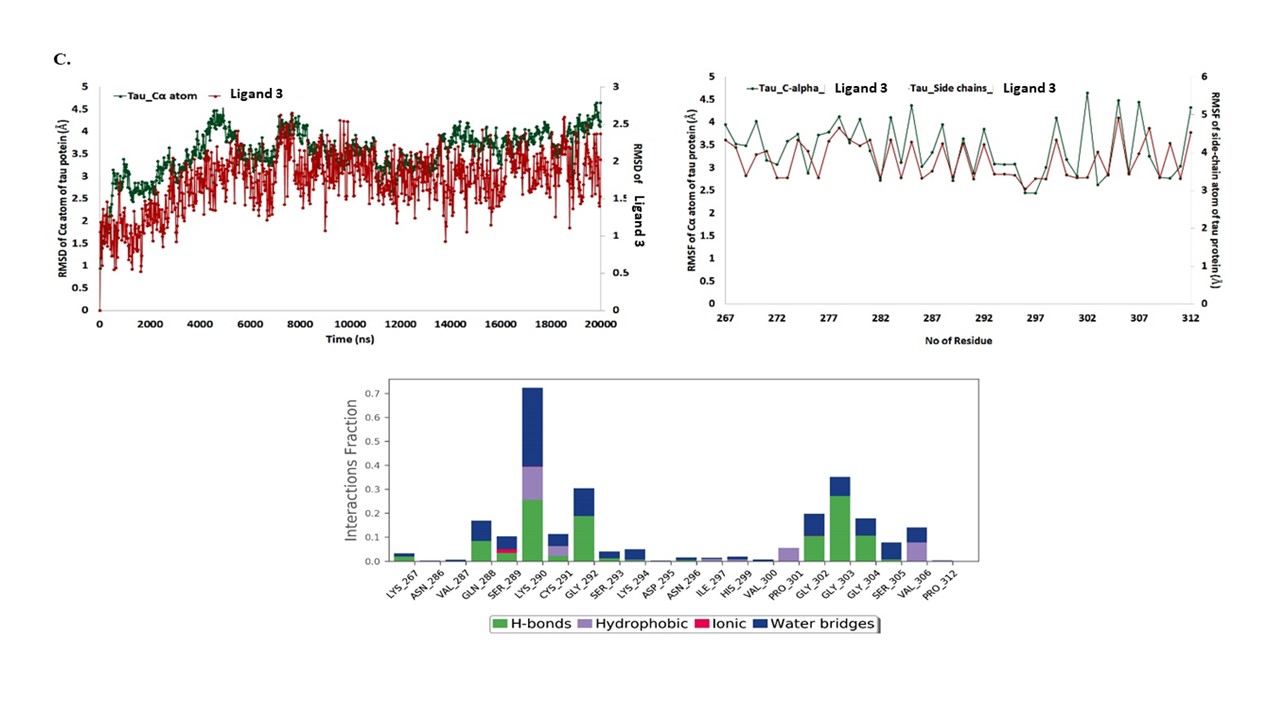
The docked complexes were used for the MD simulations analysis of a 20 ns time frame to observe the stability of the molecular interactions and additional relative structural changes by RMSD and RMSF, which might enlighten the probable molecular events behind the binding of phosphorylated tau protein with ligands i.e. ligand 1, ligand 2 and ligand 3. The RMSD graphs were plotted by assigning time in nanosecond (X-axis) and RMSD (Å) value of Cα atom of the phosphorylated tau protein and compounds in nanometer (nm), respectively in right Y-axis. (Fig. 1F–H), revealed that each phosphorylated tau protein complex with ligands were relatively stable over the MD simulation run compared with the native phosphorylated tau protein.
In the phosphorylated tau-ligand 1 complex, stable RMSD within 3 Å was observed for the phosphorylated tau protein of Cα atom followed by an average variation of 1.5 Å RMSD at the end of the simulation interval. Although RMSD calculated for ligand 1 also showed deviation within 1.9 Å, ligand 1 heavy atoms are observed with an average variation of 3.5 Å at the end of the MD trajectories (Fig. 1F). Likewise, the protein Cα carbon in the phosphorylated tau-ligand 2 complex showed stable variations < 4.5 Å and an average of 3.5 Å RMSD, whereas protein-bound ligand 2 showed stable trajectories within 2.5 Å and an average of 1.5 Å deviations at the end of the simulation interval (Fig. 1G). However, the phosphorylated tau-ligand 3 complex also exhibited acceptable maximum deviation for the Cα atom 4.6 Å and ligand 3 showed 2.6 Å while average of deviations of 3.5 and 1.7 Å were respectively followed by the state of equilibrium until the end of the simulation (Fig. 1H).
During 20 ns of MD simulation, the RMSF analysis was employed to quantify the fluctuation in each residue over time of complex systems, consenting to identify the most flexible regions. The RMSF of the Ca atom and side-chain atoms (X-axis) of each residue (Y-axis) over different time frames was plotted to measure the dynamic changes that occur at the level of single residues and analyses the fluctuation of the interface residue of phosphorylated tau protein over the MD simulation time (Fig. 1F–H). In the phosphorylated tau-ligand 1 complex, the RMSF plot was observed for the protein of Cα atom and side-chain of residues, which has average fluctuations of 1.5 and 2.2 Å. The residue of Gly303 has larger fluctuations of Cα atoms 4.6 Å and side-chain of 5.045 Å (Fig. 1F). The active site residues of the phosphorylated tau protein showed consistent fluctuations throughout the trajectory of MD simulations. Although phosphorylated tau-ligand 2 showed that the protein residues of Cα and side chain atoms had average fluctuations of 3.6 and 4.17 Å, respectively (Fig. 1G), whereas, phosphorylated tau- ligand 3 also showed fluctuations of 3.4 Å (Cα) and 3.8 Å (side-chain; Fig. 1H). In the phosphorylated tau- ligand 2, complex of Lys274 and phosphorylated tau- ligand 3 complex of Gly302 were expressed with high fluctuations during MD simulations run. Besides, each docked complex results of RMSD and RMSF of deviations/fluctuations observed minimal fluctuations compared with native phosphorylated tau protein (Fig. 1I).
The details of molecular interactions between the phosphorylated tau and docked three ligands were shown in Supplementary Material, Fig. S2. During the MD simulations, the binding interactions between individual ligands and active site residues inside the binding pocket of phosphorylated tau protein were observed. It was observed that the ligand 1 interacted with the protein residue with Asp295, Val300, Gly302, Ser305, Gln307, Val309, Thr310, Lys311 and Pro312 H-bond (Fig. 2A). The hydrophobic interactions with Lys267, Val287, Lys290, Lys294, Ile297, Lys298, Val300, Pro301, Ile308, Thr310, Lys311, Pro312 and various water-bridges with residues of Lys294, Asp295, Val300, Gly302, Gly304, Ser305, Gln307, Val309, Tyr310, Lys311 and Pro312 carried a supportive function in binding the ligand 1 with phosphorylated tau active site (Fig. 2A). Post-molecular docking and post-dynamic simulation results revealed that, significant active site residues of Ser285 and Tyr310 persevere the continuous binding interactions with phosphorylated tau protein. The phosphorylated tau-ligand 2 complex showed, seven H-bonds by Val287, Lys290, Cys291, Ser293, Lys294, Asp295 and Asn296 (Fig. 2B) Also, the residues Val287, Lys290, Ile297 and Tyr310, Trp50 were noticed for hydrophobic interactions in the phosphorylated tau-ligand 2 complex. Besides, water bridges by Val287, Lys290, Cys291, Ser293, Lys294, Asp295, Asn296 and ionic bond by Lys290, Ser293, Lys294 and Asp295 within 4 Å regions of ligand 2 (Fig. 2B). Although, intermolecular contacts in phosphorylated tau- ligand 3 (Fig. 2C) complex exhibited nine H-bonds by the residues Lys267, Gln288, Ser289, Lys290, Cys291, Gly292, Gly302, Gly303 and Gly304. Lys290, Cys291 and Pro301, which also marked the complex for depicted hydrophobic, and water mediated bridge contacts, and Val306 indicated substantial interactions between ligand 3 and phosphorylated tau protein. Likewise, ligand 3 and phosphorylated tau protein complex showed ionic interactions by Ser289 within 4 Å regions of active site. Eventually, the MD simulation results demonstrated that each docked complex are stable during a simulation trajectory and are identically stable compared with native phosphorylated tau protein. The computational validations like molecular docking, MD simulation results, indicates that ligand 1 showed the best lead properties from the top three qualified ligands, further, biological validations proceed to test ligand 1 in mouse hippocampal neuronal cells (HT22) cultures in in vitro studies.
Cell survival and viability
To determine the protective effects of ligand 1, we assessed cell viability in four independent treatments of cells with 1) control HT22 cells; 2) HT22 cells treated with ligand 1; 3) mTauHT22 cells and 4) ligand 1 treated mTauHT22 cells. As shown in (Fig. 3), no significant cell death was observed in ligand 1 treated HT22 cells relative to ligand 1 untreated HT22 cells.
Figure 3.
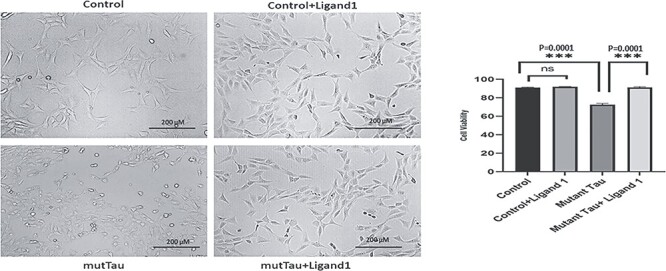
To determine the effect of ligand 1 treatments (Cell viability and MTT assay). Four different groups of cells: 1) HT22 cells (Control); 2) control + ligand 1; 3) mutant HT22 cells (Tau cDNA transfected HT22) and 4) mutant Tau+ ligand 1. Statistical significance. Increased cell viability levels in mutant Tau+ligand 1(P = 0.0001) treatment cells when compared with mTau cells. Decreased cell viability observed in mutant HT22 cells when compared with control cells (P = 0.0001). Scale bar 200 μM.
Increased cell viability levels were observed in ligand 1 treated mutant TauHT22 cells (P = 0.0001) relative ligand 1 untreated mutant TauHT22 cells. Decreased cell viability was observed in mutant HT22 cells (P = 0.0001) when compared with control HT22 cells. These observations suggest that ligand 1 increases cell viability in the presence of mutant Tau (Fig. 3).
Immunoblotting analysis
To determine the phosphorylated tau ligand 1 inhibitory effects of DYRK1A, total tau, PHF-Tau, MAP 2 and GSK3β proteins and protective effects of mitochondrial dynamics proteins (DRP1, Fis1, Mfn1, Mfn2 and Opa1); mitochondrial biogenesis proteins (PGC1α, Nrf1, Nrf2 and TFAM) and synaptic proteins PSD95 and synaptophysin, we quantified all the protein levels in four independent treatments of cells 1) control HT22 cells; 2) HT22 + ligand 1; 3) mutant Tau transfected HT22 cells and 4) ligand 1 treated mutant Tau HT22 cells.
Comparison 1: comparison with control HT22 cells versus mTauHT22 cells
Phosphorylated tau induced DYRK1A, GSK3β and CDK5: DYRK1A, total tau, Phosphorylated tau and GSK3β levels were significantly increased in mutant tau transfected HT22 (mTauHT22) cells relative to control HT22 cells; DYRK1A (P = 0.021); total tau (P = 0.0001); PHF-tau (AT8) (P = 0.0001); GSK3β (P = 0.0001) and CDK5 (P = 0.002) (Fig. 4A). In addition, decreased levels of MAP2 protein (P = 0.0001) was observed in mHT22 cells. These observations indicate that mutant Tau activates DYRK1A, GSK3β and CDK5 proteins and reduces MAP2 levels.
Figure 4.
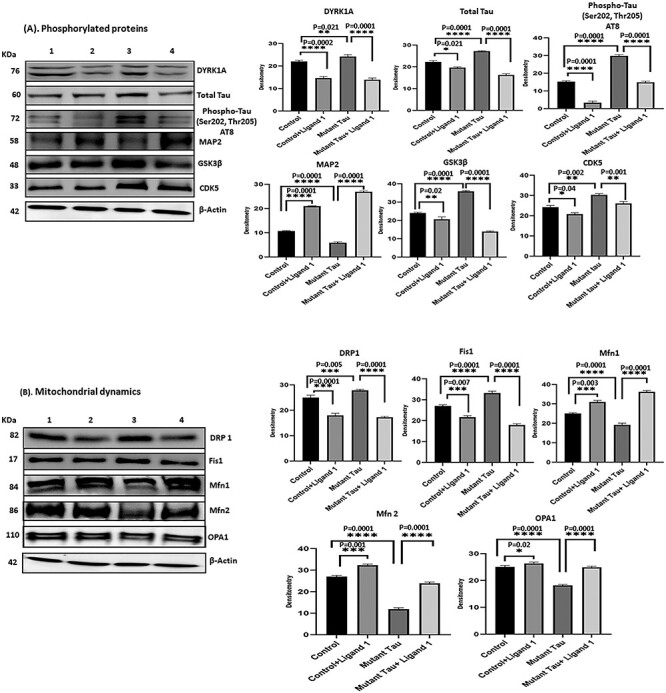
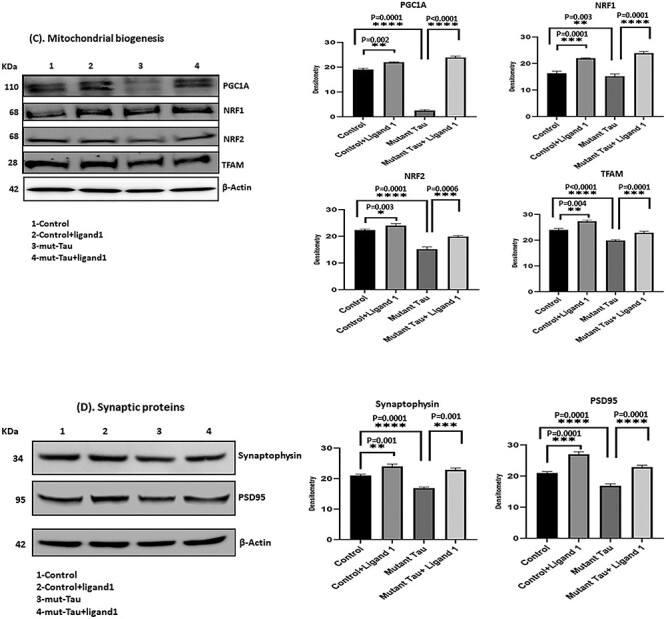
(A) Immunoblotting analysis of phosphorylated tau ligand inhibition on DYRK1A, total Tau, PHF-Tau (AT8), MAP2, GSK3β and CDK5. Immunoblotting analysis was conducted using protein lysates from four group of mouse hippocampal cells (HT22). 1) HT22 cells (Control); 2) Control +ligand 1; 3) mutant tau (Tau cDNA transfected HT22) and 4) mutant Tau+ligand 1. 1) comparison with control versus control +ligand 1 treated cells significantly decreased levels of DYRK1A, total tau, PHF-Tau (AT8), GSK3β and CDK5 proteins were found with (P = 0.0002, P = 0.0021; P = 0.0001, P = 0.0001, P = 0.02, P = 0.04) and increased MAP2 (P = 0.0001); 2) comparison with mutant tau versus mutant tau + ligand 1 treated cells, significantly reduced levels of DYRK1A, total tau and PHF-tau (AT8), and GSK3β proteins were found with (P = 0.0001, P = 0.0001; P = 0.0001, P = 0.0001, P = 0.001) and increased MAP2 (P = 0.0001). (B) Immunoblotting analysis mitochondrial dynamics proteins. Increased levels of fission proteins Drp1 and Fis1 were observed in mutant tau (Tau cDNA transfected HT22; P = 0.005, P = 0.0001) when compared with Control. Reduced Drp1 and Fis1 levels were found (ligand 1 treated control cells (P = 0.0001, P = 0.007) and mutant tau+ligand 1 treated cells (P = 0.0001; P = 0.0001). Whereas mitochondrial fusion proteins Mfn1, Mfn2 and Opa1 were decreased in mutant Tau (P = 0.0001, P = 0.0001, P = 0.0001), when compared with control, increased in mutant tau+ligand 1 (P = 0.0001, P = 0.0001, P = 0.0001). (C) Immunoblotting analysis of mitochondrial biogenesis proteins. We found decreased levels of biogenesis proteins PGC1a, NRF1, NRF2 and TFAM in cells mutant tau when compared with control (P = 0.0001, P = 0.001; P = 0.0001, P = 0.0001, P = 0.0001). Increased PGC1a, NRF1, NRF2 and TFAM levels were found in cells treated with ligand 1 (control+ ligand 1) when compared with control (P = 0.002, P = 0.0001, P = 0.003, P = 0.004) and ligand 1 treated mutant tau cells (P = 0.0001, P = 0.0001, P = 0.0006, P = 0.0001). (D) Immunoblotting analysis of synaptic proteins Synaptophysin and PSD95. We found decreased levels of synaptic proteins synaptophysin and PSD95 in cells transfected with mutant tau cells when compared with control (P = 0.0001, P = 0.0001). Synaptic proteins were increased in mutant Tau+ligand 1 cells P = 0.001; PSD95, P = 0.0001) when compared with mutant tau cells.
Mitochondrial dynamics: mitochondrial fission proteins Drp1, (P = 0.005) and Fis1, (P = 0.0001) were increased in mTauHT22 cells relative to control HT22 cells; in contrast, mitochondrial fusion proteins Mfn1 (P = 0.0001), Mfn2 (P = 0.0001) and Opa1 (P = 0.0001) were reduced in mTauHT22 cells relative to control HT22 cells. These observations suggest that mutant Tau impairs mitochondrial dynamics in AD cells (Fig. 4B).
Mitochondrial biogenesis: mitochondrial biogenesis proteins PGC1α, (P = 0.0001); Nrf1, (P = 0.0001); Nrf2, (P = 0.0006) and TFAM, (P = 0.0001) were reduced in mTauHT22 cells relative to control HT22 cells, indicating that phosphorylated tau reduces mitochondrial biogenesis activity in AD cells (Fig. 4C).
Synaptic proteins: the levels of synaptic proteins synaptophysin (P = 0.0001) and PSD95 (P = 0.0001), were significantly decreased in mTauHT22 cells relative to control HT22 cells (Fig. 4D), indicating that mutant Tau reduces synaptic activity.
Comparison 2: Mut Tau HT22 versus ligand 1 treated MutTauHT22 cells, and control HT22 versus ligand 1 treated HT22 cells
phosphorylated tau induced proteins: DYRK1A, total tau, phosphorylated tau, GSK3β and CDK5 levels were significantly reduced in ligand 1 treated mtauHT22 cells relative to ligand 1 untreated mTau HT22 cells; DYRK1A (P = 0.0001); total tau (P = 0.0001); PHF-tau (AT8; P = 0.0001); GSK3β (P = 0.0001) and CDK5 (P = 0.001; Fig. 4A). In addition, MAP2 protein levels were increased (P = 0.0001) in ligand 1 treated mHT22 cells relative to ligand 1 untreated mTauHT22 cells.
Significantly reduced DYRK1A (P = 0.0002), total tau (P = 0.021), Phosphorylated tau (P = 0.0001), GSK3β (P = 0.02) and CDK5 (P = 0.04) levels were found in ligand 1 treated control HT22 cells relatively ligand 1 untreated HT22 cells. In addition, MAP2 protein levels were increased (P = 0.0001) in ligand 1 treated HT22 cells relative to ligand 1 untreated HT22 cells.
Mitochondrial dynamics: mitochondrial fission proteins Drp1, (P = 0.0001); Fis1, (P = 0.0001) were decreased in ligand 1 treated mTauHT22 cells relative to ligand 1 untreated mTauHT22 cells; on the other hand, fusion proteins Mfn1 (P = 0.0001), Mfn2 (P = 0.0001) and Opa1 (P = 0.0001) were increased in ligand 1 treated mTauHT22 cells relative to ligand 1 untreated mTauHT22 cells (Fig. 4B).
In ligand 1 treated control HT22 cells relative to ligand 1 untreated HT22 cells, significantly decreased levels of mitochondrial fission proteins Drp1, (P = 0.0001) and Fis1, (P = 0.007) and increased levels of fusion proteins Mfn1 (P = 0.003), Mfn2 (P = 0.001) and Opa1 (P = 0.02) were observed (Fig. 4B).
Mitochondrial biogenesis: mitochondrial biogenesis proteins PGC1α, (P = 0.0001); Nrf1, (P = 0.0001); Nrf2, (P = 0.0006) and TFAM, (P = 0.0001) were increased in ligand 1 treated mTauHT22 cells relative to ligand 1 untreated mTauHT22 cells (Fig. 4C).
In ligand treated control HT22 cells relative to ligand 1 untreated HT22 cells, significantly increased levels of mitochondrial biogenesis proteins PGC1α, (P = 0.002); Nrf1, (P = 0.0001); Nrf2, (P = 0.003) and TFAM, (P = 0.004) were observed, indicating that ligand 1 protects mitochondrial biogenesis (Fig. 4C).
Synaptic proteins: the levels of synaptic proteins synaptophysin (P = 0.001) and PSD95 (P = 0.0001) were significantly increased in ligand 1 treated mTauHT22 cells relative to ligand 1 untreated mTauHT22 cells and (Fig. 4D).
In ligand 1 treated HT22 cells relative to ligand 1 untreated HT22 cells, significantly increased levels of synaptophysin (P = 0.001) and PSD95 (P = 0.0001) were observed (Fig. 4D). These observations indicate ligand 1 has protective properties against phosphorylated tau.
Immunofluorescence analysis of PHF-Tau (AT8), Drp1 and SNAP 25
To determine the effect of ligand 1 on PHF-Tau (AT8), and mitochondrial fission (Drp1) and synaptic activity (SNAP 25), we performed immunofluorescence analysis on four different groups of cells: 1) HT22 cells (control); 2) HT22 + ligand 1(control+ligand1); 3) mTau (mutant Tau transfected HT22 cells) and 4) mTau +1igand 1 (mHT22 cells+ligand 1).
Comparison 1: control versus mutant tau HT22 cells
Significantly increased immunoreactivities of PHF-Tau (AT8; P = 0.01; Fig. 5); Drp1 (P = 0.05; Fig. 6) were found in mHT22 cells relative to control cells and decreased levels of SNAP25 (P = 0.001) was observed (Fig. 7).
Figure 5.
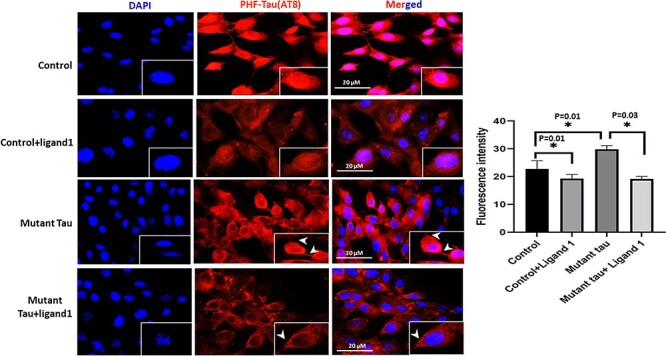
Immunofluorescence analysis of PHF-Tau (AT8). The effect of ligand 1 on phosphorylated tau using AT8 antibody immunofluorescence analysis on four different groups of cells: 1) HT22 cells (control); 2) control+ligand 1; 3) mutant Tau and 4) mutant Tau+ligand 1. Statistical significance phosphorylated tau levels were significantly increased in Mutant tau HT22cells when compared with control (P = 0.01). Reduced PHF-Tau in ligand 1 treated HT22 cells with control cells (P = 0.01). Decreased PHF-tau protein observed in mutant Tau+ligand 1 mutant Tau (P = 0.03). Scale bar 20 μM.
Figure 6.
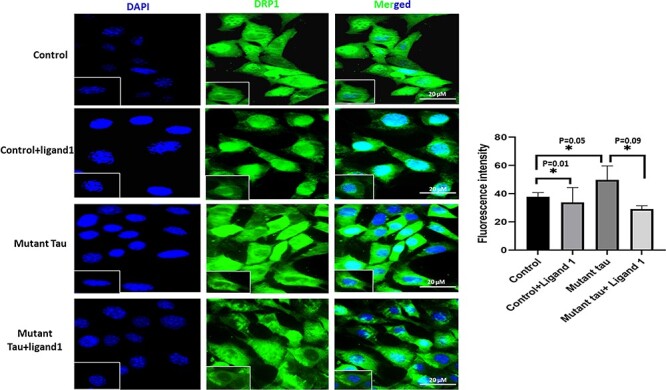
Immunofluorescence analysis of DRP1. The effect of ligand 1 on Drp1 using Drp1 antibody immunofluorescence analysis on four different groups of cells: 1) HT22 cells (control); 2) control +ligand 1; 3) mutant Tau and 4) mutant Tau+ligand 1. Drp1 levels were significantly increased in Mutant tau HT22cells when compared with control (P = 0.05). Drp1 levels were reduced in ligand 1 treated HT22 cells with control cells (P = 0.01). Reduced Drp1 protein levels were observed in mutant Tau+ligand 1 with mutant Tau (P = 0.09). Scale bar 20 μM.
Figure 7.
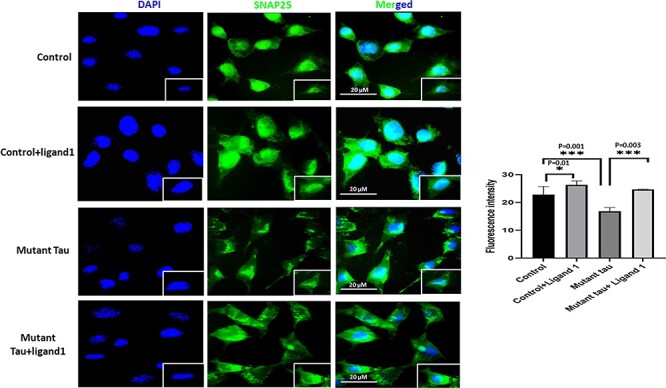
Immunofluorescence analysis of SNAP 25. The effect of ligand 1 on phosphorylated tau using SNAP 25 antibody immunofluorescence analysis on four different groups of cells: 1) HT22 cells (control); 2) control+ligand 1; 3) mutant Tau and 4) mutant Tau+ligand 1. Levels were significantly decreased in Mutant tau HT22cells when compared with control (P = 0.001). Increased SNAP25 in ligand 1 treated HT22 cells with control cells (P = 0.01). Increased SNAP25 observed in mutant Tau+ligand 1 with mutant tau (P = 0.003). Scale bar 20 μm.
Comparison 2: mutant Tau versus ligand 1+ mHT22 cells
Decreased immunoreactivity levels of PHF-Tau (AT8; P = 0.01; Fig. 5); Drp1 (P = 0.05; Fig. 6) were found in ligand 1 + mHT22 cells relative to mHT22 cells (P = 0.03; P = 0.09). Increased levels of immunoreactivities of SNAP25 (P = 0.003) were found in ligand 1 + mTauHT22 cells relative to mHT22 cells (Fig. 7).
Transmission electron microscopy
To determine the protective effects of phosphorylated tau ligand 1 on the number and length of mitochondria, we used transmission electron microscopy and assessed mitochondrial number, and length and on four different groups of cells: 1) control HT22 cells; 2) ligand 1 treated HT22 cells; 3) mutant Tau transfected HT22 cells (mTauHT22 cells) and 4) ligand 1 treated mTauHT22 cells.
Mitochondrial number and length in ligand 1-treated mHT22 cells
Comparison 1: control versus ligand 1+ HT22 cells and control versus mHT22 cells
To determine the impact of ligand 1, we studied mitochondrial number and morphology. As shown in Fig. 8, On the contrary, mitochondrial number is significantly increased in mHT22 cells (P = 0.001) when compared with control cells. However, mitochondrial number is significantly decreased in ligand 1-treated HT22 cells, relative to control cells (P = 0.01) (Fig. 8).
Figure 8.
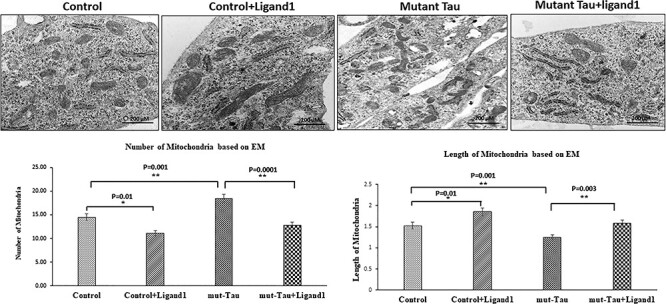
Electron microscopy of mitochondria structure of HT22 cells treated with ligand 1. We quantified mitochondrial architectures within the cell in four groups 1) HT22 cells (control); 2) control +ligand 1; 3) mutant Tau and 4) mutant Tau+ligand 1. To identify mitochondrial number and morphology. Average number of mitochondria per cell is shown in graphs. Mitochondrial number is significantly decreased in ligand 1-treated HT22 cells, relative to control (P = 0.01). On the contrary, mitochondrial number is significantly increased mutant tau (P = 0.001). Mitochondrial length is significantly increased in ligand 1-treated HT22 cells, relative to control (P = 0.01). Treated ligand 1 with mHT22 cells (mTau+ligand 1) showed increased mitochondrial length (P = 0.003) compared with mutant Tau.
Mitochondrial length is significantly decreased in mHT22 cells (P = 0.001) when compared with control cells (Fig. 8). Mitochondrial length is significantly increased in ligand 1-treated HT22 cells, relative to control cells (P = 0.01).
Comparison 2: mHT22 cells versus ligand 1 + mHT22 cells
Mitochondrial number is decreased (P = 0.0001) in mHT22 cells treated with ligand 1 relative to ligand 1 untreated mHT22 cells. Mitochondrial length increased in ligand 1 treated mHT22 cells (P = 0.003) relative to ligand 1 untreated mHT22 cells. These observations indicate that small molecule ligand 1 reduces excessive mitochondrial fragmentation and increased mitochondrial length in AD neurons.
Discussion
The long-term goal of our study is to understand the toxicity and molecular mechanism of phosphorylated tau in the progression and pathogenesis of AD. Another objective of this study is to develop ligand small molecule inhibitors referred to as ‘Ligands’ and study their protective properties against phosphorylated tau toxicities using cell cultures of primary neurons (mutant tau cDNA transfected with HT22 neurons) and transgenic Tau mice. Increasing evidence suggests that phosphorylated tau is a major contributor to the formation of PHF and NFTs in AD. Furthermore, phosphorylated tau hyperphosphorylation at S/T/Y sites are highly involved in glycogen synthase kinase-3 beta, cyclin-dependent kinase-5 and DYRK1A upregulation in AD (13,33–40). Among six isoforms of DYRK, only DYRK1A seems to be involved in AD (41–43). DYRK1A phosphorylate tau and its transcription factor cAMP response element binding (CREB), which are involved in learning and memory healthy and disease state such as AD (44,45). DYRK1A has been defined as a dual-specificity kinase because of its ability to autophosphorylate on Tyr and Ser/Thr residues (46,47). Earlier reports and our current observations support that tyrosine TYR44 (Y310) phosphorylation site of tau (28) and SER 285 involving in kinase dependent phosphorylation in AD (26,27,35). Currently, we do not have drugs and/or small molecule-based ligands that reduce phosphorylated tau-induced toxicities in AD. Therefore, our current study is focused on phosphorylated tau at SER23 (Ser 285), which is responsible for GSK3β, tyrosine phosphorylation site (Tyr-310) within the MT-binding domain (28). This residue is located in the R3 repeat peptide of tau, which has also been shown to play key roles in regulating Tau aggregation, PHF formation and membrane interaction (48,49). TYR44 (Y310) of tau involving in kinase upregulation, causes phosphorylated kinase upregulation in AD (29–31). In NFTs, tau protein is phosphorylated by several protein kinases, including GSK3β, CDK5, c-Jun N-terminal kinase, extracellular signal-regulated kinases 1/2 and p38 mitogen-activated protein kinases at deferent p-sites (50–52). As shown in Figs. 1–3, we found three ligands that showed the best docking scores −8.09 (ligand 1), −7.9 (ligand 2) and −7.8 kcal/mol (ligand 3), and our critical analysis of molecular docking revealed that ligand 1 binds to key hyperphosphorylation residues of phosphorylated tau and inhibits abnormal PHF-tau, DYRK1A, GSK3β and CDK5, which reduces phosphorylated tau levels in AD. We found interactions between NAT10-262501 (ligand 1) and phosphorylated tau at key phosphorylation sites, and these ligand-based inhibitions, decreased PHF-tau, DYRK1A, GSK3β and CDK5 levels.
Using biochemical, molecular, immunoblotting, immunofluorescence and transmission electron microscopy analyses, we studied the phosphorylated tau ligand’s protective effects against mutant tau induced synaptic and mitochondrial toxicities in HT22 cells. In mutant tau cDNA transfected HT22 cells, we found increased mitochondrial fission and reduced fusion, reduced mitochondrial biogenesis, reduced mitophagy and synaptic proteins. However, mTau-HT22 cells treated with ligand 1, showed increased mitochondrial biogenesis, mitochondrial fusion and synaptic proteins and reduced mitochondrial fission proteins. DYRK1A, total tau, PHF-tau GSK3β and CDK5 levels were significantly increased in mutant tau transfected HT22 cells relative to mTau HT22 cells. As expected, DYRK1A, total tau, PHF-tau GSK3β and CDK5 levels were significantly reduced in mTau HT22 cells treated with ligand 1. Our transmission electron microscopy analysis revealed a significant increase in mitochondrial number as well as significantly reduced mitochondrial length in mTau-HT22 cells. However, mitochondrial number is significantly reduced in ligand 1-treated mTau-HT22 cells.
Based on these results, we cautiously conclude that phosphorylated tau NAT10-262501 (ligand 1) reduces hyperphosphorylation of phosphorylated tau based GKS3β and CDK5 kinase regulation in AD, and aids in the maintenance of neuronal structure, mitochondrial dynamics and biogenesis revealing a possible therapeutic function in AD.
Ligand 1 protective properties against phosphorylated tau toxicities
It is well established that AD is associated with phosphorylated tau and NFTs in regions of the brain that are responsible for learning and memory. Increasing evidence also suggests that phosphorylated tau and NFTs are more definitive diagnostic features of AD patients. Tau is a major microtubule-associated protein that plays a large role in the outgrowth of neuronal processes and also promotes microtubule assembly, stabilizes microtubules, promotes neurite outgrowth, facilitates membrane interactions of enzyme anchoring, facilitates axonal transport of organelles to nerve terminals and affects the dynamics of microtubules in neurons (8). Tau is abundantly present in the central nervous system and is predominantly expressed in neuronal axons.
In AD, tau is hyperphosphorylated, becomes pathological, accumulates in neurons and forms PHFs (8). As a result, tau loses its capability to bind with microtubules and becomes unable to transport subcellular cellular organelles, such as mitochondria, endoplasmic reticulum, lysosomes, proteins and lipids from the soma to nerve terminals, this ultimately leads to synaptic degeneration and cognitive decline in individuals with AD.
In the current study, using mouse primary hippocampal neurons transfected with mutant tau cDNA, we studied mitochondrial dynamics, biogenesis, synaptic proteins and also three important kinases, GKS3β, CDK5 and DYRK1A (3–42). Current study findings revealed that increased levels of all kinases were observed in mTauHT22 cells. Interestingly, all kinases were significantly reduced in mTauHT22 cells treated with ligand 1. Ligand 1 reduced phosphorylated tau linked SER23 (Ser 285) activities that increased levels of GSK3β, and TYR44 (Y310) CDK5-dependent DYRK1A elevated levels in AD. Through the molecular docking validations and docking score 8.09 kcal/mol of ligand 1. Our proof-of-concept findings of ligand 1 are interesting and warrant further in vivo (mouse models of phosphorylated tau) studies, in order to understand its molecular aspects.
Protective role of ligand 1 against phosphorylated tau induced impaired mitochondrial dynamics, defective biogenesis and synaptic damage
Increased mitochondrial fission and reduced fusion, reduced mitochondrial biogenesis and reduced synaptic proteins were found in mTau-HT22 cells relative to control HT22 cells. These observations agree with our previous findings of increased mitochondrial fragmentation, reduced fusion and synaptic activities of 12-month-old transgenic Tau mice (29) and multiple lines of APP transgenic mice and mutant APP cell cultures, indicating that excessive fragmentation of mitochondria is a typical feature of AD (53–55).
However, in mTau-HT22 cells treated with ligand 1, we found reduced excessive mitochondrial fragmentation and enhanced mitochondrial fusion and synaptic activities. These observations clearly indicate that a phosphorylated tau ligand 1 inhibits phosphorylation of tau and enhances mitochondrial biogenesis and synaptic proteins in HT22 cells transfected with mutant tau cDNA.
It is well established that mutant APP/Aβ causes reduced cell survival and increased apoptotic cell death in AD state (56). In the current study, we found mTauHT22 cells showed reduced cell survival and increased apoptotic cell death. However, ligand 1 enhanced cell survival and reduced apoptotic cell death in mutant TauHT22 cells. This effect presents a strong evidence that ligand 1 is protective against phosphorylated tau-induced toxicities in AD neurons. These observations present evidence of ligand 1’s non-toxic attributes which renders it capable for further in vivo studies of its therapeutic properties.
Transmission electron microscopy based mitochondrial morphology revealed that reduced mitochondrial number and increased mitochondrial length in ligand 1 treated mTau-HTT22 cells clearly indicate that ligand 1 enhances mitochondrial function which allows the maintenance of quality mitochondria in ligand 1 treated cells.
In summary, mutant Tau transfected HT22 cells treated with ligand 1 were found to have reduced levels of PHF-tau (AT8), total tau, DYRK1A, GSK3β and CDK5 levels in AD state (mutant tau transfected HT22 cells), indicating that ligand 1 shows significant inhibition activity of phosphorylated tau induced toxicities in AD neurons. In the current study, ligand 1 treated mTauHT22 cells showed reduced levels of mitochondrial fission proteins, and increased levels of mitochondrial fusion, biogenesis and synaptic proteins. This indicates that ligand 1 is a phosphorylated tau inhibitor and protective against phosphorylated tau toxicity, mitochondrial dysfunction and synaptic damage in AD. These observations strongly suggest ligand 1 inhibits the hyperphosphorylation at particular sites, which is responsible for formation of NTFs and PHFs in AD neuronal cells.
To the best of our knowledge, this is the first small molecule reported to have effects on multiple key phosphorylated tau targeting ligands (drug) for AD and tauopathies (ligand-based phosphorylated tau, GSK3β and CDK5 targeted inhibition). Our current study ligand 1 is capable of inhibiting phosphorylation at SER 285 and TYR 310 and reducing GSK3β, CDK5 and DYRK1A dependent kinases, showing a link between the phosphorylated tau phosphorylation, which provides new clues for AD therapeutics (13,33–43). These observations strongly suggest further studies of ligand 1, in mouse models of tau using small molecule ligand(s) by conjugating with PROTACs (21).
Materials and Methods
Retrieval of 3D structure and stability analysis
The crystal structure of the microtubule-associated protein tau was obtained from the protein data bank (https://www.rcsb.org) using PDB ID: 2MZ7 and visualized in Schrodinger Maestro v12.7 (57). The phosphorylated tau protein was solved by the nuclear magnetic resonance (NMR), which consists of 20 conformer models with 267–312 residues (58). The 20 lowest energy conformations models of phosphorylated tau protein bound to the similar residues of microtubules (MTs) play a crucial role in cell organization and function. Indeed, the different conformers of the NMR models may not be appropriate for drug discovery owing to distant active site Cartesian co-ordinates of the phosphorylated tau protein (59). In ordered to determine which NMR model has stable potential energy, lattice active site and stable structural coordinates in the state of water versus definitive time-dependent through the MD simulations as a potential protein target for molecular docking (60,61). We performed MD simulations for obtained 20 ns phosphorylated tau protein models and employed Desmond v2019-4. The Desmond package (Schrodinger v2019-4) was utilized to run a dynamics simulation that was employed for the native phosphorylated tau protein models to find stability and catalytic interactions throughout a simulation’s trajectory.
Initially, built a systematic illumination using the model system builder wizard to carry out the dynamics simulations with a run duration of 20 ns (nano seconds). By extending the specifications, a startup process with the system builder as TIP3P (water model) was built, and a water model as a solvent within the periodic boundary condition of the orthorhombic box set at 10 Å × 10 Å × 10 Å for the complex of concern was also built (62–65). The system was neutralized by ions of Na+ and Cl− at a final concentration of 0.15 M (66). The system was minimized and pre-equilibrated using the standard equilibration protocol implemented in Desmond, which contains several steps before the final production run. After building the solvated system to each model, minimization and relaxation were performed under the constant pressure and temperature (NPT) ensemble using Desmond’s minimization & relaxation protocol that consists of nine stages (61). The Nose–Hoover Chain thermostat and Martyna–Tobias–Klein barostat were used to maintain constant temperature and pressure, respectively (62,63). The behavior of each MD simulation trajectory file was calculating the potential energies; RMSD and RMSF, to check each model of the structural stability. These parameters qualitatively validate the system stability throughout the 20 ns run and protein changes at the simulated length of chemical time for the given temperature, pressure and volume of the total simulation box.
Molecular docking
The stable model of the phosphorylated tau protein was further subjected to various docking strategies of Discovery Studio v2020 (Accelrys, San Diego, USA) of LibDock and AutoDock Vina of PyRx protocols under the protein-compound interactions (66,67).
In the Discovery Studio, a high-throughput algorithm of LibDock consisting the polar and non-polar features as ‘Hotspots’. The amino acids found in the active site pockets of phosphorylated tau protein residues are Ser285, Ser289, Ser293, Ser305 and Tyr310 (21,24). The compounds of NAT10-262501 (ligand 1), NAT10-367721 (ligand 2) and NAT24-392393 (ligand 3) were obtained from NATx Synthetic ligand library. Before docking, the ligands were prepared using the ‘prepare ligand’ module. The compounds were primarily positioned in the binding site by using LibDock, which is a high throughput docking algorithm that positions catalyst generated compound conformations in the protein active site based on polar interaction sites [hotspots (68)]. Protein-compound interactions were further optimized by short MD using CHARMM and clean geometry of Discovery Studio. Force fields applied in CHARMM are energies and forces on each particle of the system, which will define the positional relationship between atoms that determine potential energy. Each docked poses of the phosphorylated tau protein-compound complexes of binding energy and H-bond interactions was visualized by Schrodinger Maestro v12.7 (69,70).
In the PyRx, the obtained 2D/3D compound structures were prepared with default protocol of Open Babel molecule format convert SDF to PDBQT file. The force field of mmff94 and conjugate gradients optimization algorithm applied to minimize the obtained compounds energy using PyRx-Python prescription 0.8 for 200 steps. AutoDock Tools 1.5.6 (ADT) was used for the docking of the protein and compounds. Gestgeiger partial charges were allocated and torsions were introduced to the obtained compounds. Using ADT, Kollman charges, polar hydrogen atoms and solvation parameters were applied. The grid box was generated within the active site residues of Ser285, Ser289, Ser293, Ser305 and Tyr310 with the cube box dimension X = 8.95, Y = −15.18 and Z = −15.74 center box size of X = 20.65, Y = 18.45 and Z = 16.95, respectively (65). The exhaustiveness of 100 was used to optimize the magnificent complex of protein-compound. Each docking poses with binding energy and H-bond interactions were evaluated through the Discovery Studio visualizer (64).
Further, to optimize the protein-compound complex stability, MD simulations were performed for 20 ns using Desmond v2019-4 as the aforementioned section of protein stability. The behavior of each MD simulations trajectory file was analyzed by using Simulation Quality Analysis, Simulation Event Analysis tools of Desmond modules to calculate the potential energies, RMSD, RMSF, a radius of gyration, Solvent-Accessible Surface Area and total intramolecular H-bonds contributing to the complex structural stability (65,70).
Cell culture work
Chemicals and reagents
Ligand 1 was purchased from TimTech, LCC Chemical Company (Newark, DE, USA); Tau (P301L)-VC: we used mutant Tau (P301L) cDNA clone developed by Arubala Reddy (unpublished). Briefly, mutant tau cDNA clone (P301L) was purchased from Addgene https://www.addgene.org and verified expression of mutations P301L and further sub-cloned into a mammalian expression vector; and HT22 cells were purchased from American Type Culture Collection (ATCC; Manassas, VA, USA). Dulbecco’s Modified Eagle Medium (DMEM) and Minimum Essential Medium (MEM), penicillin/streptomycin, Trypsin–EDTA and fetal bovine serum were purchased from GIBCO (Gaithersberg, MD, USA).
The HT22 cells were grown for 6 days in a serum-free medium (1:1 mixture of DMEM and OptiMEM), plus penicillin and streptomycin (Invitrogen, Carlsbad, CA, USA) until the cells developed neuronal processes. As shown in (Fig. 3), these cells were used for 4 groups—one control group and three treatment groups. 1) HT22 cells (the control group); 2) HT22 cells treated with ligand 1 for 24 h (HT22 + ligand 1); 3) HT22 cells transfected mTau cDNA with the (lipofectamine; referred as mTauHT22; 20 μM final concentration for 6 h; mTau) and 4) HT22 cells transfected with mTau cDNA for 24 h, then treated with ligand 1 for 6 h (mTau+ligand 1 treatment group). We performed 4 independent cell cultures and treatments for all experiments (n = 4; Fig. 9).
Figure 9.
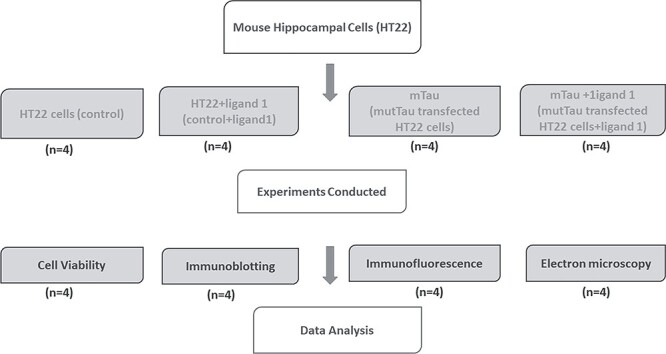
Flow chart of experimental design and conducted experiments for testing biological validation of phosphorylated tau ligand 1 in HT22 cells. Scale bar 200 μM.
Cell survival and MTT assay
Phosphorylated tau ligand 1 treated HT22 cells; cell-based apoptosis assay was performed using Cellometer Vision CBA Image Cytometry System (Nexcelom Bioscience LLC, Lawrence, MA) with two fluorophore Annexin V-FITC and Propidium Iodide (PI) staining solution, according to manufacturer’s instructions. Briefly, cells were harvested using trypsin, then spun down at 300 g for 3 min and pellets were washed with 1XPBS, cells were counted using hematocytometer. Collected 100 000 to 150 000 cells and cells/pellet were resuspended in 40 μl of Annexin V binding buffer.
Immunoblotting analysis
To determine whether phosphorylated tau ligand 1 and mutant tau alters the protein levels of PHF-tau, total tau, DYRK1A, GSK3β, mitochondrial fission (Drp1 and Fis1) and fusion (Mfn1, Mfn2 and Opa1), biogenesis (PGC1α, NRF1, NRF2 and TFAM), dendritic protein, MAP2. We performed immunoblotting analyses of protein lysates from four different groups of cells as described previously: 1) untreated HT22 cells versus; 2) HT22 + ligand 1; 3); cells transfected with mTau and 4) mTau +ligand 1. We also performed immunoblot analysis for the above proteins. Details of the antibody dilutions are given in (Table 2). Twenty micrograms of protein lysates were resolved on a 4–12% Nu-PAGE gel (Invitrogen). The resolved proteins were transferred to nylon membranes (Novax Inc., San Diego, CA, USA) and then incubated for 1 h at room temperature with a blocking buffer (5% dry milk dissolved in a TBST buffer). The membranes were washed with a TBST buffer three times at 10 min intervals and were then incubated for 2 h with appropriate secondary antibodies (1: 5000 dilutions of Anti-mouse IgG HRP; Anti-rabbit IgG HRP, GE Health Care life Sciences, Pittsburgh, PA), followed by three additional washes at 10 min intervals. Proteins were detected with chemiluminescence reagents (Pierce Biotechnology, Rockford, IL), and the bands from immunoblots were visualized.
Table 2.
Summary of antibody dilutions and conditions used in the immunoblotting analysis of PHF-Tau (AT8), total tau, DYRK1A, MAP2, GSK3β, cdk5 and mitochondrial biogenesis and synaptic proteins in HT22 cells, HT22 cells treated with phosphorylated tau ligand 1, cells transfected with cDNA mutant Tau, mutant Tau cDNA transfected HT22 cells treated with phosphorylated tau ligand 1
| Marker | Primary antibody— species and dilution | Purchased from company, city & state | Secondary antibody, dilution | Purchased from company, city & state |
|---|---|---|---|---|
| DYRK1A | Rabbit Polyclonal 1:500 | ABCAM, Cambridge, MA 02139–1517, USA | Goat 1:10 000 anti-rabbit | ABCAM, Cambridge, MA 02139–1517, USA |
| Total tau | Mouse Monoclonal 1:200 | Thermo Fisher Scientific, Waltham, MA | Goat anti-mouse, HRP 1:60 000 | Thermo Fisher Scientific, Waltham, MA |
| Tau-pSer202/ Thr205 (AT8) | Mouse Monoclonal 1:500 | Thermo Fisher Scientific, Waltham, MA | Donkey anti-mouse 1:10 000 | Thermo Fisher Scientific, Waltham, MA |
| MAP2 | Mouse Monoclonal Antibody 1:500 | Thermo Fisher Scientific, Waltham, MA | Goat anti-mouse, HRP 1:4000 | Thermo Fisher Scientific, Waltham, MA |
| GSK-3β (D5C5Z) | Rabbit Monoclonal Antibody 1:1000 | Cell Signaling Technology, MA 01923 | Donkey anti-rabbit HRP 1:10 000 | Cell Signaling Technology, MA 01923 |
| CDK5 | Rabbit Polyclonal Antibody 1:1000 | Cell Signaling Technology, MA 01923 | Goat 1:10 000 anti-rabbit | Cell Signaling Technology, MA 01923 |
| PGC1α | Rabbit Polyclonal 1:500 | Novus Biological, Littleton, CO | Donkey anti-rabbit HRP 1:10 000 | GE Healthcare Amersham, Piscataway, NJ |
| Nrf1 | Rabbit Polyclonal 1:300 | Novus Biological, Littleton, CO | Donkey anti-rabbit HRP 1:10 000 | GE Healthcare Amersham, Piscataway, NJ |
| Nrf2 | Rabbit Polyclonal 1:300 | Novus Biological, Littleton, CO | Donkey anti-rabbit HRP 1:10 000 | GE Healthcare Amersham, Piscataway, NJ |
| TFAM | Rabbit Polyclonal 1:300 | Novus Biological, Littleton, CO | Donkey anti-rabbit HRP 1:10 000 | GE Healthcare Amersham, Piscataway, NJ |
| Mfn1 | Rabbit Polyclonal 1:300 | Novus Biological, Littleton, CO | Donkey anti-rabbit HRP 1:10 000 | GE Healthcare Amersham, Piscataway, NJ |
| Mfn2 | Rabbit Polyclonal 1:300 | Novus Biological, Littleton, CO | Donkey anti-rabbit HRP 1:10 000 | GE Healthcare Amersham, Piscataway, NJ |
| OPA1 | Rabbit Polyclonal 1:300 | Novus Biological, Littleton, CO | Donkey anti-rabbit HRP 1:10 000 | GE Healthcare Amersham, Piscataway, NJ |
| DRP1 | Rabbit Polyclonal 1:300 | Novus Biological, Littleton, CO | Donkey anti-rabbit HRP 1:10 000 | GE Healthcare Amersham, Piscataway, NJ |
| FIS1 | Rabbit Polyclonal 1:300 | Novus Biological, Littleton, CO | Donkey anti-rabbit HRP 1:10 000 | GE Healthcare Amersham, Piscataway, NJ |
| PSD95 | Rabbit Monoclonal 1:300 | Novus Biological, Littleton, CO | Donkey anti-rabbit HRP 1:10 000 | GE Healthcare Amersham, Piscataway, NJ |
| SYN | Rabbit Monoclonal 1:400 | Novus Biological, Littleton, CO | Donkey anti-rabbit HRP 1:10 000 | GE Healthcare Amersham, Piscataway, NJ |
| B-Actin | Mouse Monoclonal 1:500 | Sigma-Aldrich, St Luis, MO | Sheep anti-mouse HRP 1:10 000 | GE Healthcare Amersham, Piscataway, NJ |
Immunofluorescence analysis of PHF tau (AT8), Drp1 and SNAP 25
To determine PHF-tau (AT8), Drp1 and SNAP 25 inhibition, mitochondrial network, immunofluorescence analysis was performed using mutant transfected HT22 cells and phosphorylated tau ligand 1 treated mutant transfected HT22 cells as methods described in (47). Cells were plated onto coverslips at 4 × 102 cells/mm2, allowed to recover for 3 days, then washed and fixed with 4% paraformaldehyde. After subsequent washing, cells were treated with 0.1% Triton X-100 and blocked with 5% BSA, which were both in PBS. Cells were then washed, incubated in Alexa Fluor 488-conjugated mouse secondary antibody for 1 h at room temperature and washed again. Finally, before mounting, cells were incubated with nuclear stain DAPI. Photographs were taken with a multiphoton laser scanning microscope system (Olympus IX83).
Transmission electron microscopy of mitochondria
To determine the protective effects of ligand 1 in neuronal cell culture treatments and morphology of mitochondria, the rescue effects of ligand 1 were assessed on mTau induced mitochondrial damage. We conducted transmission electron microscopy (TEM) of HT22 cells from control and experimental treatments (n = 4) methods described in (48). Treated and untreated HT22 cells were fixed in 100 mm sodium cacodylate (pH 7.2), 2.5% glutaraldehyde, 1.6% paraformaldehyde, 0.064% picric acid and 0.1% ruthenium red. They were gently washed and post-fixed for 1 h in 1% osmium tetroxide plus 08% potassium ferricyanide, in 100 mm sodium cacodylate, pH 7.2. After a thorough rinsing in water, the HT22 cells were dehydrated, infiltrated overnight in 1:1 acetone:Epon 812, infiltrated for 1 h with 100% Epon 812 resin and embedded in the resin. After polymerization, 60- to 80 nm thin sections were cut on a Reichert ultramicrotome and stained for 5 min in lead citrate. They were then rinsed and post-stained for 30 min in uranyl acetate, and then rinsed again and allowed to dried. EM was performed at 60 kV on a Philips Morgagne TEM, equipped with a CCD, and images were collected at original magnifications of 1000–37 000×.
Quantification and statistical analyses
Statistical analyses were conducted in three ways: 1) control versus mutant cDNA transfected HT22 cells (mHT22), 2) transfected ligand 1 treated HT22 cells versus mutant transfected HT22 cells and 3) cells transfected mTau versus mTau+ligand 1 for protein levels, cell viability and mitochondrial structural and biogenesis using appropriate statistical analysis.
Data and software availability
The PDB files that support the findings of this study have been available in Protein Data Bank under accession codes 2mz7.
Conflict of Interest statement. None declared.
Funding
National Institutes of Health grants (AG042178, AG047812, NS105473, AG060767, AG069333 and AG066347 to P.H.R.), Alzheimer’s Association through a Sex and Gender in Alzheimer’s, Garrison Family Foundation Grant and National Institutes of Health grant (AG063162 to A.P.R.).
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Contributor Information
Jangampalli Adi Pradeepkiran, Department of Internal Medicine, Texas Tech University Health Sciences Center, Lubbock, TX 79430, USA.
Manne Munikumar, Clinical Division, ICMR-National Institute of Nutrition, Hyderabad, Telangana 500007, India.
Arubala P Reddy, Nutritional Sciences Department, College of Human Sciences, Texas Tech University, Lubbock TX 79409, USA.
P Hemachandra Reddy, Department of Internal Medicine, Texas Tech University Health Sciences Center, Lubbock, TX 79430, USA; Department of Pharmacology and Neuroscience, Texas Tech University Health Sciences Center, Lubbock, TX 79430, USA; Department of Neurology, Texas Tech University Health Sciences Center, Lubbock, TX 79430, USA; Department of Public Health, Graduate School of Biomedical Sciences, Texas Tech University Health Sciences Center, Lubbock, TX 79430, USA; Department of Speech, Language, and Hearing Sciences, Texas Tech University Health Sciences Center, Lubbock, TX 79430, USA.
References
- 1. Mandelkow, E.M. and Mandelkow, E. (2012) Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harb Perspect Med., 2, a006247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wang, J.Z., Xia, Y.Y., Grundke-Iqbal, I. and Iqbal, K. (2013) Abnormal hyperphosphorylation of tau: sites, regulation, and molecular mechanism of neurofibrillary degeneration. J. Alzheimers Dis., 33(Suppl 1), S123–S139. [DOI] [PubMed] [Google Scholar]
- 3. Schneider, A., Biernat, J., von Bergen, M., Mandelkow, E. and Mandelkow, E.M. (1999) Phosphorylation that detaches tau protein from microtubules (Ser262, Ser214) also protects it against aggregation into Alzheimer paired helical filaments. Biochemistry, 38, 3549–3558. [DOI] [PubMed] [Google Scholar]
- 4. Reddy, P.H. and Oliver, D.M. (2019) Amyloid beta and phosphorylated tau-induced defective autophagy and mitophagy in Alzheimer's disease. Cell, 8, 488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Guillaud, L., El-Agamy, S.E., Otsuki, M. and Terenzio, M. (2020) Anterograde axonal transport in neuronal homeostasis and disease. Front. Mol. Neurosci., 13, 556175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chen, Y.C., Huang, H.R., Hsu, C.H. and Ou, C.Y. (2021) CRMP/UNC-33 organizes microtubule bundles for KIF5-mediated mitochondrial distribution to axon. PLoS Genet., 17, e1009360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Liu, X.A., Rizzo, V. and Puthanveettil, S.V. (2012) Pathologies of axonal transport in neurodegenerative diseases. Transl. Neurosci., 3, 355–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Guo, T., Noble, W. and Hanger, D.P. (2017) Roles of tau protein in health and disease. Acta Neuropathol., 133, 665–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shimojo, M., Takuwa, H., Takado, Y., Tokunaga, M., Tsukamoto, S., Minatohara, K., Ono, M., Seki, C., Maeda, J., Urushihata, T. et al. (2020) Selective disruption of inhibitory synapses leading to neuronal hyperexcitability at an early stage of tau pathogenesis in a mouse model. J. Neurosci., 40, 3491–3501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Utton, M.A., Connell, J., Asuni, A.A., van Slegtenhorst, M., Hutton, M., de Silva, R., Lees, A.J., Miller, C.C. and Anderton, B.H. (2002) The slow axonal transport of the microtubule-associated protein tau and the transport rates of different isoforms and mutants in cultured neurons. J. Neurosci., 22, 6394–6400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pérez, M., Cuadros, R. and Medina, M. (2018) Tau assembly into filaments. Methods Mol. Biol., 1779, 447–461. [DOI] [PubMed] [Google Scholar]
- 12. Oakley, S.S., Maina, M.B., Marshall, K.E., Al-Hilaly, Y.K., Harrington, C.R., Wischik, C.M. and Serpell, L.C. (2020) Tau filament self-assembly and structure: tau as a therapeutic target. Front. Neurol., 11, 590754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mietelska-Porowska, A., Wasik, U., Goras, M., Filipek, A. and Niewiadomska, G. (2014) Tau protein modifications and interactions: their role in function and dysfunction. Int. J. Mol. Sci., 15, 4671–4713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cowan, C.M., Bossing, T., Page, A., Shepherd, D. and Mudher, A. (2010) Soluble hyper-phosphorylated tau causes microtubule breakdown and functionally compromises normal tau in vivo. Acta Neuropathol., 120, 593–604. [DOI] [PubMed] [Google Scholar]
- 15. Fujiwara, H., Watanabe, S., Iwata, M., Ueda, S., Nobuhara, M., Wada-Kakuda, S., Misonou, H. and Miyasaka, T. (2020) Inhibition of microtubule assembly competent tubulin synthesis leads to accumulation of phosphorylated tau in neuronal cell bodies. Biochem. Biophys. Res. Commun., 521, 779–785. [DOI] [PubMed] [Google Scholar]
- 16. Falzone, T.L., Gunawardena, S., McCleary, D., Reis, G.F. and Goldstein, L.S. (2010) Kinesin-1 transport reductions enhance human tau hyperphosphorylation, aggregation and neurodegeneration in animal models of tauopathies. Hum. Mol. Genet., 19, 4399–4408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sherman, M.A., LaCroix, M., Amar, F., Larson, M.E., Forster, C., Aguzzi, A., Bennett, D.A., Ramsden, M. and Lesné, S.E. (2016) Soluble conformers of Aβ and tau alter selective proteins governing axonal transport. J. Neurosci., 36, 9647–9658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Engmann, O. and Giese, K.P. (2009) Crosstalk between Cdk5 and GSK3beta: implications for Alzheimer's disease. Front. Mol. Neurosci., 2, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wen, Y., Planel, E., Herman, M., Figueroa, H.Y., Wang, L., Liu, L., Lau, L.F., Yu, W.H. and Duff, K.E. (2008) Interplay between cyclin-dependent kinase 5 and glycogen synthase kinase 3 beta mediated by neuregulin signaling leads to differential effects on tau phosphorylation and amyloid precursor protein processing. J. Neurosci., 28, 2624–2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Coutadeur, S., Benyamine, H., Delalonde, L., de Oliveira, C., Leblond, B., Foucourt, A., Besson, T., Casagrande, A.S., Taverne, T., Girard, A., Pando, M.P. and Désiré, L. (2015) A novel DYRK1A (dual specificity tyrosine phosphorylation-regulated kinase 1A) inhibitor for the treatment of Alzheimer's disease: effect on tau and amyloid pathologies in vitro. J. Neurochem., 133, 440–451. [DOI] [PubMed] [Google Scholar]
- 21. Jangampalli Adi, P. and Reddy, P.H. (1867) Phosphorylated tau targeted small-molecule PROTACs for the treatment of Alzheimer's disease and tauopathies. Biochim. Biophys. Acta Mol. basis Dis., 2021, 166162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sontag, J.M. and Sontag, E. (2014) Protein phosphatase 2A dysfunction in Alzheimer's disease. Front. Mol. Neurosci., 7, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Perluigi, M., Barone, E., Di Domenico, F. and Butterfield, D.A. (2016) Aberrant protein phosphorylation in Alzheimer disease brain disturbs pro-survival and cell death pathways. Biochim. Biophys. Acta, 1862, 1871–1882. [DOI] [PubMed] [Google Scholar]
- 24. Pradeepkiran, J.A. and Reddy, P.H. (2019) Structure based design and molecular docking studies for phosphorylated tau inhibitors in Alzheimer's disease. Cell, 8, 260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mendoza, J., Sekiya, M., Taniguchi, T., Iijima, K.M., Wang, R. and Ando, K. (2013) Global analysis of phosphorylation of tau by the checkpoint kinases Chk1 and Chk2 in vitro. J. Proteome Res., 12, 2654–2665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Park, S., Lee, J.H., Jeon, J.H. and Lee, M.J. (2018) Degradation or aggregation: the ramifications of post-translational modifications on tau. BMB Rep., 51, 265–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ait-Bouziad, N., Chiki, A., Limorenko, G., Xiao, S., Eliezer, D. and Lashuel, H.A. (2020) Phosphorylation of the overlooked tyrosine 310 regulates the structure, aggregation, and microtubule- and lipid-binding properties of tau. J. Biol. Chem., 295, 7905–7922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Williamson, R., Scales, T., Clark, B.R., Gibb, G., Reynolds, C.H., Kellie, S., Bird, I.N., Varndell, I.M., Sheppard, P.W., Everall, I. and Anderton, B.H. (2002) Rapid tyrosine phosphorylation of neuronal proteins including tau and focal adhesion kinase in response to amyloid-beta peptide exposure: involvement of Src family protein kinases. J. Neurosci., 22, 10–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Derkinderen, P., Scales, T.M., Hanger, D.P., Leung, K.Y., Byers, H.L., Ward, M.A., Lenz, C., Price, C., Bird, I.N., Perera, T. et al. (2005) Tyrosine 394 is phosphorylated in Alzheimer’s paired helical filament tau and in fetal tau with c-Abl as the candidate tyrosine kinase. J. Neurosci., 25, 6584–6593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Scales, T.M., Derkinderen, P., Leung, K.Y., Byers, H.L., Ward, M.A., Price, C., Bird, I.N., Perera, T., Kellie, S., Williamson, R., Anderton, B.H. and Reynolds, C.H. (2011) Tyrosine phosphorylation of tau by the SRC family kinases lck and fyn. Mol. Neurodegener., 6, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kimura, T, Sharma, G, Ishiguro, K, Hisanaga, S.I. (2018) Phospho-tau bar code: analysis of phosphoisotypes of tau and its application to tauopathy. Front. Neurosci. 12, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bhat, R.V., Shanley, J., Correll, M.P., Fieles, W.E., Keith, R.A., Scott, C.W. and Lee, C.M. (2000) Regulation and localization of tyrosine216 phosphorylation of glycogen synthase kinase-3beta in cellular and animal models of neuronal degeneration. Proc. Natl. Acad. Sci. U. S. A., 26, 11074–11079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang, Q.M., Fiol, C.J., DePaoli-Roach, A.A. and Roach, P.J. (1994) Glycogen synthase kinase-3 beta is a dual specificity kinase differentially regulated by tyrosine and serine/threonine phosphorylation. J. Biol. Chem., 269, 14566–14574. [PubMed] [Google Scholar]
- 34. Giese, K.P. (2009) GSK-3: a key player in neurodegeneration and memory. IUBMB Life, 61, 516–521. [DOI] [PubMed] [Google Scholar]
- 35. Martin, L., Page, G. and Terro, F. (2011) Tau phosphorylation and neuronal apoptosis induced by the blockade of PP2A preferentially involve GSK3β. Neurochem. Int., 59, 235–250. [DOI] [PubMed] [Google Scholar]
- 36. Pradeepkiran, J.A., Reddy, A.P. and Reddy, P.H. (2019) Pharmacophore-based models for therapeutic drugs against phosphorylated tau in Alzheimer's disease. Drug Discov. Today, 24, 616–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dhavan, R. and Tsai, L.H. (2001) A decade of CDK5. Nat. Rev. Mol. Cell. Biol., 2, 749–759. [DOI] [PubMed] [Google Scholar]
- 38. Li, T., Hawkes, C., Qureshi, H.Y., Kar, S. and Paudel, H.K. (2006) Cyclin-dependent protein kinase 5 primes microtubule-associated protein tau site-specifically for glycogen synthase kinase 3beta. Biochemistry, 45, 3134–3145. [DOI] [PubMed] [Google Scholar]
- 39. Wegiel, J., Gong, C.X. and Hwang, Y.W. (2011) The role of DYRK1A in neurodegenerative diseases. FEBS J., 278, 236–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chen, B., McCuaig-Walton, D., Tan, S., Montgomery, A.P., Day, B.W., Kassiou, M., Munoz, L. and Recasens, A. (2021) DYRK1A negatively regulates CDK5-SOX2 pathway and self-renewal of glioblastoma stem cells. Int. J. Mol. Sci., 22, 4011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ferrer, I., Barrachina, M., Puig, B., Martinez de Lagran, M., Marti, E., Avila, J. and Dierssen, M. (2005) Constitutive Dyrk1A is abnormally expressed in Alzheimer disease, down syndrome, pick disease, and related transgenic models. Neurobiol. Dis., 20, 392–400. [DOI] [PubMed] [Google Scholar]
- 42. Ferrer, I., Blanco, R., Carmona, M., Ribera, R., Goutan, E., Puig, B., Rey, M.J., Cardozo, A., Vinals, F. and Ribalta, T. (2001) Phosphorylated map kinase (ERK1, ERK2) expression is associated with early tau deposition in neurones and glial cells, but not with increased nuclear DNA vulnerability and cell death, in Alzheimer disease, Pick’s disease, progressive supranuclear palsy and corticobasal degeneration. Brain Pathol., 11, 144–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kimura, R., Kamino, K., Yamamoto, M., Nuripa, A., Kida, T., Kazui, H., Hashimoto, R., Tanaka, T., Kudo, T., Yamagata, H. et al. (2007) The DYRK1A gene, encoded in chromosome 21 down syndrome critical region, bridges between beta-amyloid production and tau phosphorylation in Alzheimer disease. Hum. Mol. Genet., 16, 15–23. [DOI] [PubMed] [Google Scholar]
- 44. Woods, Y.L., Cohen, P., Becker, W., Jakes, R., Goedert, M., Wang, X. and Proud, C.G. (2001) The kinase DYRK phosphorylates protein-synthesis initiation factor eIF2Bepsilon at Ser539 and the microtubule-associated protein tau at Thr212: potential role for DYRK as a glycogen synthase kinase 3-priming kinase. Biochem. J., 355, 609–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yang, E.J., Ahn, Y.S. and Chung, K.C. (2001) Protein kinase Dyrk1 activates cAMP response element-binding protein during neuronal differentiation in hippocampal progenitor cells. J. Biol. Chem., 276, 39819–39824. [DOI] [PubMed] [Google Scholar]
- 46. Himpel, S., Tegge, W., Frank, R., Leder, S., Joost, H.G. and Becker, W. (2000) Specificity determinants of substrate recognition by the protein kinase DYRK1A. J. Biol. Chem., 275, 2431–2438. [DOI] [PubMed] [Google Scholar]
- 47. Himpel, S., Panzer, P., Eirmbter, K., Czajkowska, H., Sayed, M., Packman, L.C., Blundell, T., Kentrup, H., Grötzinger, J., Joost, H.G. and Becker, W. (2001) Identification of the autophosphorylation sites and characterization of their effects in the protein kinase DYRK1A. Biochem. J., 359, 497–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Von Bergen, M., Friedhoff, P., Biernat, J., Heberle, J., Mandelkow, E.M. and Mandelkow, E. (2000) Assembly of t protein into Alzheimer paired helical filaments depends on a local sequence motif (306VQIVYK311) forming β structure. Proc. Natl. Acad. Sci. U. S. A., 97, 5129–5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pérez, M., Arrasate, M., Montejo de Garcini, E., Muñoz, V. and Ávila, J. (2001) In vitro assembly of tau protein: mapping the regions involved in filament formation. Biochemistry, 40, 5983–5991. [DOI] [PubMed] [Google Scholar]
- 50. Planel, E., Yasutake, K., Fujita, S.C. and Ishiguro, K. (2001) Inhibition of protein phosphatase 2A overrides tau protein kinase I / glycogen synthase kinase 3beta and Cyclin-dependant kinase 5 inhibition and results in tau hyperphosphorylation in the hippocampus of starved mouse. J. Biol. Chem., 276, 34298–34306. [DOI] [PubMed] [Google Scholar]
- 51. Nygaard, H.B. (2018) Targeting Fyn Kinase in Alzheimer’s disease. Biol. Psychiatry, 83, 369–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Martin, L., Latypova, X., Wilson, C.M., Magnaudeix, A., Perrin, M.L., Yardin, C. and Terro, F. (2013) Tau protein kinases: involvement in Alzheimer’s disease. Ageing Res. Rev., 12, 289–309. [DOI] [PubMed] [Google Scholar]
- 53. Reddy, P.H., Yin, X., Manczak, M., Kumar, S., Pradeepkiran, J.A., Vijayan, M. and Reddy, A.P. (2018) Mutant APP and amyloid beta-induced defective autophagy, mitophagy, mitochondrial structural and functional changes and synaptic damage in hippocampal neurons from Alzheimer's disease. Hum. Mol. Genet., 27, 2502–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Manczak, M., Kandimalla, R., Yin, X. and Reddy, P.H. (2018) Hippocampal mutant APP and amyloid beta-induced cognitive decline, dendritic spine loss, defective autophagy, mitophagy and mitochondrial abnormalities in a mouse model of Alzheimer's disease. Hum. Mol. Genet., 27, 1332–1342. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 55. Wang, W., Yin, J., Ma, X. and Zhao, F/, Siedlak, S.L., Wang, Z., Torres, S., Fujioka, H., Xu, Y., Perry, G., Zhu, X. (2017) Inhibition of mitochondrial fragmentation protects against Alzheimer's disease in rodent model. Hum. Mol. Genet., 26, 4118–4131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zhang, H., Wei, W., Zhao, M., Ma, L., Jiang, X., Pei, H., Cao, Y. and Li, H. (2021) Interaction between Aβ and tau in the pathogenesis of Alzheimer's disease. Int. J. Biol. Sci., 17, 2181–2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kadavath, H., Jaremko, M., Jaremko, J., Biernat, J., Mandelkow, E. and Zweckstetter, M. (2015) Folding of the tau protein on microtubules. Angew. Chemie - Int. Ed. 10.1002/anie.201501714. [DOI] [PubMed] [Google Scholar]
- 58. Barbier, P., Zejneli, O., Martinho, M., Lasorsa, A., Belle, V., Smet-Nocca, C., Tsvetkov, P.O., Devred, F. and Landrieu, I. (2019) Role of tau as a microtubule-associated protein: structural and functional aspects. Front. Aging Neurosci. 10.3389/fnagi.2019.0020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zhang, L., Bouguet-Bonnet, S. and Buck, M. (2012) Combining NMR and molecular dynamics studies for insights into the allostery of small GTPase-protein interactions. Methods Mol. Biol., 796, 235–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Krepl, M., Cléry, A., Blatter, M., Allain, F.H. and Sponer, J. (2016) Synergy between NMR measurements and MD simulations of protein/RNA complexes: application to the RRMs, the most common RNA recognition motifs. Nucl. Acids Res., 44, 6452–6470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Li, D.W. and Brüschweiler, R. (2014) Protocol to make protein NMR structures amenable to stable long time scale molecular dynamics simulations. J. Chem. Theory Comput., 10, 1781–1787. [DOI] [PubMed] [Google Scholar]
- 62. Kalikka, J. and Akola, J. (2011) Steered molecular dynamics simulations of ligand-receptor interaction in lipocalins. Eur. Biophys. J., 40, 181–194. [DOI] [PubMed] [Google Scholar]
- 63. Munikumar, M., Krishna, V.S., Reddy, V.S., Rajeswari, B., Sriram, D. and Rao, M.V. (2018) In silico design of small peptides antagonist against leptin receptor for the treatment of obesity and its associated immune-mediated diseases. J. Mol. Graph. Model., 82, 20–36. [DOI] [PubMed] [Google Scholar]
- 64. Munikumar, M., Natarajan, P., Amineni, U. and Radha Krishna, K.V. (2019) Discovery of potential lumazine synthase antagonists for pathogens involved in bacterial meningitis: in silico study. Informatics Med. Unlocked., 15, 100187. [Google Scholar]
- 65. Naik, V.R., Munikumar, M., Ramakrishna, U., Srujana, M., Goudar, G., Naresh, P., Kumar, B.N. and Hemalatha, R. (2020) Remdesivir (GS-5734) as a therapeutic option of 2019-nCOV main protease - in silico approach. J. Biomol. Struct. Dyn., 39, 4701–4714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Mark, P. and Nilsson, L. (2001) Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J. Phys. Chem. A, 105, 9954–9960. [Google Scholar]
- 67. Adi, P.J., Yellapu, N.K. and Matcha, B. (2016) Modeling, molecular docking, probing catalytic binding mode of acetyl-CoA malate synthase G in Brucella melitensis 16M. Biochem. Biophys. Rep., 8, 192–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Vilar, S., Karpiak, J., Berk, B. and Costanzi, S. (2011) In silico analysis of the binding of agonists and blockers to the β2-adrenergic receptor. J. Mol. Graph. Model., 29, 809–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ram, T.S., Munikumar, M., Raju, V.N., Devaraj, P., Boiroju, N.K., Hemalatha, R., Prasad, P.V.V., Gundeti, M., Sisodia, B.S., Pawar, S. et al. (2021) In silico evaluation of the compounds of the ayurvedic drug, AYUSH-64, for the action against the SARS-CoV-2 main protease. J Ayurveda Integr Med. 10.1016/j.jaim.2021.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Diller, D.J. and Merz, K.M. (2001) High throughput docking for library design and library prioritization. Proteins Struct. Funct. Genet., 43, 113–124. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The PDB files that support the findings of this study have been available in Protein Data Bank under accession codes 2mz7.
Conflict of Interest statement. None declared.