

Abstract
Members of the PI3K signaling pathway, especially PIK3CA, the gene encoding the catalytic subunit of the PI3K complex, are highly mutated and amplified in various cancer types, including non-small cell lung cancer. Although PI3K inhibitors have been used in clinics for follicular lymphoma and chronic lymphocytic leukemia, no agents targeting PI3K aberrations in lung cancer have been approved by the FDA so far. In this study, we observed that PIK3CA-E545K, the most common mutation in lung cancer, harbored a modest induction of stem-like properties in lung epithelial cells, and drove development of adenocarcinoma autochthonously when paired with p53 loss in a murine mouse model. We also found that PIK3CA-mutant of amplified lung cancer cells were sensitive to EZH2 inhibition. EZH2 inhibition synergized with PI3K inhibition in human cancer cells in vitro and worked together efficiently in vivo. Mechanistically, EZH2 inhibition cooperated with PI3K inhibition to produce a more potent suppression of phospho-AKT downstream of PI3K. This study suggests a promising combination therapy to combat lung cancers with PIK3CA mutation or amplification. Both copanlisib, the PI3K inhibitor, and tazemetostat, the EZH2 inhibitor, are FDA-approved, which should enhance the clinical translation of this work.
Keywords: Lung cancer, Epigenetic therapy, Combination therapy, EZH2, PI3K
1. Introduction
Large scale studies of tumor genetics have uncovered that alterations in the Phosphoinositide 3-kinase (PI3K) pathway are prevalent in cancer [1]. In particular, phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA) encoding the catalytic subunit p110α of the PI3K complex is highly mutated and amplified in numerous cancer types, including lung cancer. The signaling pathways downstream of PI3K include AKT and mTOR, and genetic alterations of PIK3CA represent one of the oncogenic drivers of lung cancer, along with activating mutations in EGFR and KRAS [2]. Despite development of specific PI3K inhibitors, there is still no FDA-approved precision medicine option for this genetic subclass of lung cancer. In fact, the early clinical trials of PI3K inhibitors on lung cancers had underwhelming results and were ended early, though the patient populations included either genetically unselected cohorts, or both patients with the activation of PIK3CA and those with loss-of-function mutation in the PI3K negative regulator phosphatase and tensin homolog (PTEN) [3,4].
It is widely believed that in addition to genetic mutations, epigenetic changes may be required for maintenance of the cancer cells in proliferative and drug-resistant states. One attractive way to target tumor-related growth programs is through inhibition of epigenetic regulators of gene expression. A group of master regulators of chromatin state in stem cells and cancer are termed the Polycomb Repressive Complexes (PRCs) [5]. Research has shown that histone methylation or ubiquitination mediated by these PRCs can affect higher order chromatin structure, rendering areas in the genome inaccessible to transcriptional machineries [6]. Specifically, the complex PRC2 often contains EZH2 as the methyltransferase that tri-methylates histone H3 at lysine 27 (H3K27me3), silencing gene expression [5,7]. Recently, a small molecule inhibitor of the epigenetic histone methyltransferase EZH2, tazemetostat, was FDA-approved in solid tumors including epithelioid sarcoma and follicular lymphoma, and several others are being used in pre-clinical models and clinical trials [8]. While some data have suggested that EZH2 inhibition will synergize with immunotherapies or other chemotherapies, it is unknown if tumors mutant for the PI3K pathway would benefit from it. Furthermore, it also remains unknown if PI3K inhibitors will synergize well with EZH2 inhibitors in lung cancer.
In this study, we reveal that the PIK3CA-mutant or amplified lung cancer cells were more sensitive to EZH2 inhibition than KRAS-driven cells. PIK3CA-E545K, the most common mutation in lung cancer, presented a modest transformation capacity when combined with mutant TP53 (p53) in lung epithelial cells, and drove lung adenocarcinoma formation autochthonously when combined with Trp53 deletion. Also, we discovered that EZH2 inhibition synergized with PI3K inhibition in human cancer cells in vitro and in vivo. This study sheds light on a promising combinatorial therapy with two FDA-approved agents for patients with PIK3CA mutated or amplified lung cancer.
2. Methods and materials
2.1. Cell lines
All 16 non-small cell lung cancer cell lines and the BEAS2B cells were grown in RPMI 1640 media (Gibco) supplemented with 8–9% fetal bovine serum, penicillin/streptomycin and glutaMAX (Gibco). Plasmocin (InvivoGen) was routinely added to cultures at thaw. The human lung cancer cell lines were from Dr. Carla Kim’s laboratory at Boston Children’s Hospital, and the BEAS2B were from Dr. David Orren’s laboratory at University of Kentucky. All cell lines were authenticated by CellCheck9 assay by IDEXX BioAnalytics, and were tested and negative for mycoplasma by MycoAlert (Lonza). Mutations were previously reported [9–11]. The cells were grown on tissue culture treated dishes at 37 °C and 5% CO2 in a humidified tissue culture incubator.
2.2. Mice
The Pik3ca mouse strain was a kind gift from Dr. Susanne Baker at St. Jude Children’s Research Hospital [12,13]. The mice were crossed to produce offspring with Pik3ca-E545K LSL/+;Trp53 fl/fl cohorts [14]. Mice were genotyped and maintained in virus-free conditions with ad libitum food and water. After 6 weeks of age, mice were anesthetized with ketamine/xylazine and adeno-Cre (University of Iowa) was administered by intranasal instillation at a dosage of 2.9 × 107 PFU per mouse. Mice were monitored for any signs of lung tumor burden, including hunched appearance or difficulty breathing. Mice were sacrificed for histological lung analysis and to establish lung cancer organoid cultures. Control mouse tumor tissues from Lkb1/Pten and Kras/p53 [15,16] mouse models were from other approved studies. All mouse care and procedures were done in accordance with the University of Kentucky Institutional Animal Care and Use Committee (IACUC). For xenograft assays, 1 × 106 Calu3 cells were implanted subcutaneously into Nude (Foxn1Nu/Nu) mice using Matrigel (Corning). Tumor growth was monitored and when tumors reached an average of approximately 100 mm3, the mice were randomized to groups. Doses were EPZ6438, dosed at 250 mg/kg BID in sterile water, 0.5% sodium carboxymethylcellulose, 0.1% Tween 80 by gavage [17] and copanlisib, dosed at 14 mg/kg in sterile water with 7% mannitol by i.p. injection every other day [18,19]. Tumors were measured daily by electronic caliper and tumor volumes were calculated using the equation (length x width x width/2). Tumors invading into muscle were not included in analysis due to difficulty of measurement. Number of mice and number of tumors included in each study are reported in the figure legends.
2.3. Cell viability assay
Cell lines were dissociated by trypsin, counted and seeded in 50 μL of cell culture medium containing 2500 cells per well of 96-well plate. All wells on the edge of the plate were not used for cell culture while instead filled with 100 μL of PBS to prevent evaporation related issues. The 2000-fold chemical stock solutions were prepared in Eppendorf tubes. The working solutions were prepared on multi-channel reagent reservoir by diluting the chemical stock in cell culture medium. The working solutions were transferred to 96-well plate as 50 μl aliquots with multi-channel pipette. After 96 h, the cell viability was tested by adding 50 μL CellTiter-Glo (Promega, CellTiter-Glo® Luminescent Cell Viability Assay, #G7573) to each well, shaking 5 min, incubating at room temperature for 25 min, and chemiluminescence measurement on Cytation5 (BioTech). For dose response assays, values were normalized to vehicle controls for each drug to yield percent survival, and drug doses converted to a log scale for graphing as log(inhibitor) vs. response – variable slope (four parameters) using GraphPad Prism. Vehicle wells were set to 1E-10 and p values comparing IC50 values were extracted. Experiments with cell lines grown in an alternative vendor’s RPMI-1640 (Caisson Labs rather than Gibco) were excluded from analysis. For synergy assays, combinations of different doses of GSK126, EPZ6438 with copanlisib were added into 96-well plates as synergy matrix and measured by CellTiter-Glo. All drugs were purchased from MedChemExpress or Xcess Bio. The percentage survival for drug dose compared to the control wells, and three, six or seven independent replicate experiment results were combined into replicates and uploaded to https://synergyfinder.fimm.fi/to calculate Bliss synergy score (readout: viability; baseline correction: yes) and display 3D synergy heatmap (>1 means additive response, >10 means synergism) [20].
2.4. Matrigel colony growth assay
BEAS2B stable cells were seeded in transwells (Corning) at a concentration of 2 × 103 cells/well in a 1:1 mixture of 8–9% FBS containing RPMI 1640 medium and phenol-red free, growth factor reduced Matrigel (Corning). A volume of 500 μl of RPMI 1640 medium with 8–9% FBS was added to the bottom of the plate and the culture medium was replenished every other day. The cells were cultured for 10 days and the colony growth was imaged with Cytation5. Organoids were quantified by counting on the images and confirmed by a blinded user. Five independent replicate experiments were performed, and one was excluded due to poor growth of the control EV cells compared to the other experiments.
2.5. Soft agar transformation assay
The soft agar transformation assay was administered following the protocol described in Ref. [21] with minor modifications. Briefly, the BEAS2B cells were seeded in 6-well plate with a 1:1 mixture of 16–18% FBS containing RPMI1640 and 0.8% low melting soft agar. After seeding agar, 2 ml of RPMI 1640 containing 8–9% FBS was added to each well. The cells were maintained at 5% CO2 and 37 °C. An additional 0.5 ml of medium was added to the culture well twice a week. After 23–25 days of culture, the plates were fixed and the organoids counted by eye and also imaged with Cytation5 (BioTech). Four independent replicate experiments were performed.
2.6. Histology and immunofluorescence staining
Mouse tumors were collected and fixed in 10% neutral-buffered formalin overnight at room temperature, and then transferred to 70% ethanol, embedded in paraffin and sectioned. Hematoxylin and eosin stains were performed in the Biospecimen Procurement & Translational Pathology Shared Resource Facility (BPTP SRF) of the University of Kentucky Markey Cancer Center. Immunofluorescence staining were done as described previously [22]. The slides of tumors were immunostained with primary antibodies for CCSP (Millipore ABS1673), pro-SPC (Millipore ABS3786), Acetylated-tubulin (SIGMA T7451), MUC5ac (AbCAM ab3649), KRT5 (Biolegend PRB-160p) and SOX2 (R&D Systems AF2018), and secondary antibodies donkey anti-mouse Alexa Fluor 594, anti-rabbit Alexa Fluor 647 and anti-goat Alexa Fluor 488, and mounted with ProLong Gold Mounting media with DAPI. Fluorescence images were captured on Nikon Ti-Eclipse inverted microscope. Exposures and look-up-tables were matched for all images of each stain at each magnification, with the exception of DAPI look-up-tables that were set for best visualization. Immunohistochemistry was performed with antibody for H3K27me3 (1:500, Cell Signaling, 9733) or phospho-AKT (1:50, Cell Signaling, 4060) in a moist chamber overnight at 4 °C. Secondary antibody, biotinylated rabbit antibody (Vectastain® ABC Kit, PK-6101), was incubated for 30 min at room temperature. Tissues were incubated in ABC reagent (Vectastain® ABC Kit, PK-6101) for 30 min at room temperature, washed in PBS, then submerged in DAB (Vector Labs, SK-4100) solution and counter-stained with Harris’ Hematoxylin (Electron Microscopy Science, 26754–01).
2.7. Quantitative RT PCR
RNA from treated cell lines was extracted using Absolutely RNA kits (Agilent) and cDNA was made using the SuperScript III kit (Invitrogen). Relative gene expression was assayed with Taqman probes on the Quant Studio 3™ Real-Time PCR System (Applied Biosystems). Relative expression was calculated by Gene of Interest (Ctreference-Ctexprimental)-House-keeping gene(Ctreference-Ctexperimental) and graphed on the log2 scale. Statistics were performed on log2 data. The Taqman probe for PIK3IP1 was Hs00364627_m1. The house-keeping gene was GAPDH (426317E).
2.8. Western blotting
Cell extracts were prepared by lysing cells with RIPA buffer (0.5% Deoxycholate, 1% IGEPAL-CA630, 150 mM NaCl, 50 mM Tris-8.1) plus 1% SDS and Halt™ Protease and Phosphatase Inhibitor Cocktail (Thermo). The lysates were cleared by centrifugation, and protein concentrations were quantified with the Pierce BCA Protein Assay Kit (Thermo). For Western blotting, 20 μg (Fig. 2B) or 9 μg (Fig. 4A) of protein per sample were denatured by heating at 100 °C for 5 min in β-mercaptoethanol reducing buffer, and split into identically loaded lanes, then separated on a 4–15% acrylamide gel (BioRad) and transferred to nitrocellulose membranes (Amersham). Antibodies used for Western blotting were: EZH2 (1:1000, Cell Signaling, 5246), Histone H3 (1:5000, AbCAM, ab1791), H3K27me3 (1:1000, Cell Signaling, 9733) and phospho-AKT (1:2500, Cell Signaling, 4060), AKT (1:2500, Cell Signaling, 4691) all incubated overnight at 4 °C. Histone H3 was used as loading control for H3K27me3 and EZH2, while total AKT was used as a loading control for phospho-AKT, each run on equivalently loaded blots. Secondary antibody Anti-rabbit IgG, HRP-linked antibody (Novus, NB7160, 1:10,000) was incubated for 1 h at room temperature. After washing, chemiluminescence was visualized with West Pico PLUS Chemiluminescent Substrate and exposure onto Hyperfilm (Amersham).
Fig. 2.

PIK3CA-E545K induces moderate transformation in lung epithelial cells. (A) Schematic of modifications in BEAS2B cells with indicated vector introduction. (B) Immunoblot probing phospho-AKT and total AKT protein expression from the cell lysate indicated. (C) Representative whole well images, scale bar = 1 cm, and (D) average colony counts normalized to EV control of soft agar colony formation assay with the modified BEAS2B cells indicated n = 4. (E) Representative images, scale bar = 200um, and (F) average colony counts normalized to EV control of 3D Matrigel culture with the modified BEAS2B cells indicated, n = 4, ** indicates p = 0.009, and *** indicates p < 0.0001, 2-tailed student’s T test with equal variance. (G) Bar plots of normalized enrichment scores of selected gene signatures enriched in PIK3CA-E545K/TP53 cells relative to TP53 alone, with FDR q-values indicated outside the end of bars. (H) Bar plots of normalized enrichment scores of selected gene signatures enriched in KRAS-G12V/TP53 cells relative to TP53 alone, with FDR q-values indicated outside the end of bars.
Fig. 4.

EZH2 inhibitors GSK126 and EPZ6438 synergize well with PI3K inhibitor copanlisib in vitro. (A) Immunoblots for indicated proteins in Calu3 and Sw1573 cells treated with vehicle, 2 μM GSK126 (Calu3), 5 μM GSK126 (Sw1573), 5 μM EPZ6438, 10 nM copanlisib, GSK126+co-panlisib and EPZ-6438+copanlisib for 3 days. (B) Heatmap of overall Bliss scores with 95% confidence intervals of GSK126 combined with copanlisib, EPZ-6438 combined with copanlisib in Calu3 and Sw1573, with the most synergistic area Bliss scores indicated in the plot. (C) Relative mRNA expression of PIK3IP1 in Calu3 and Sw1573 cells with the indicated treatments of 5 μM EPZ6438 and 3 nM or 10 nM copanlisib for 8–10 days, * indicates p < 0.05 with n = 3 experimental replicates.
2.9. Viruses
For BEAS2B stable cell line generation, KRAS-G12V and TP53-R175H lentiviruses and PIK3CA retroviurses were used. KRAS-G12V was a kind gift from Aveo Pharmaceuticals and TP53-R175H was a kind gift from the Kuperwasser laboratory at Tufts Medical Center [23]. PIK3CA vectors were purchased from Addgene [24]. Lentivirus and retrovirus were packaged in HEK293T or plate E cells with above vectors together with the packaging construct pCMV DR8.2Dvpr and VSV-G-pseudotyped lentiviral vectors by FuGENE 6, using established protocols (Fillmore et al., 2010). Cell lines were infected with viral-containing supernatant for a period of 4–8 h. Infected cultures were selected by puromycin (PIK3CA vectors), GFP-sorting (KRAS-G12V) or blasticidin (TP53-R175H). PCR for viral inserts was performed on isolated genomic DNA using a 59 °C annealing temperature with the following primers: PIK3CA F: 5’ – CCATACGACGTGCCAGATTA – 3′, R 5’ – CCTCACGGAGGCATTCTAAA – 3′, TP53 F: 5’ – CCTCACGGAGGCATTCTAAA – 3′, R 5’ -GCTCGACGCTAGGATCTGAC −3′, KRAS F: 5’ -TAGAGGATCCACTAGTACCACCATG −3′, R 5’ -GCTTCCTGTAGGAATCCTCTATTG −3′
2.10. RNA sequencing and gene set enrichment analysis
RNA samples from BEAS2B isogenic cell lines were sequenced by Beijing Genomics Institute. Sequencing reads were trimmed and filtered using Trimmomatic (V0.39) to remove adapters and low quality reads [25]. Reads of human samples were mapped to Ensembl GRCh38 transcripts annotation (release 98) and normalized by RSEM and R package edgeR [26,27]. Gene set enrichment analysis [28] was performed with GSEA version 4.1.0. Mouse genes were queried for Hallmarks (h.all. v7.4) and Oncogenic signatures (c6.all.v7.4). Signatures chosen and names used in figure are in Supplementary Table 1. Results were graphed by bar plots using normalized enrichment scores and FDR q-values (q < 0.05 as significant difference).
2.11. Statistics and reproducibility
All graphed data were presented as mean ± standard error of the mean (s.e.m.) unless otherwise noted. Unless indicated, p values represent 2-tailed Student’s t-test with equal variance that were used to compare continuous outcomes between two experimental groups. Statistical analyses were carried out using Excel or GraphPad Prism version 8. T tests performed in Excel were confirmed in GraphPad Prism. In Fig. 5, the copanlisib data was not normally distributed, so a Mann-Whitney U test was used to compare continuous outcomes between copanlisib and combination treatment.
Fig. 5.

EZH2 inhibitor EPZ6438 combines well with PI3K inhibitor copanlisib in vivo. (A) Percentage of tumor growth relative to Day 0 for 10 consecutive days with the indicated treatments, p values indicate 2-tailed Mann-Whitney U test on day 10, vehicle vs. combo ***p < 0.0001 and copanlisib vs. combo *p = 0.0255. Mice/tumors n are vehicle = 4/11, copanlisib = 4/14, EPZ6438 = 3/10, combo = 4/13. (B) Representative immunohistochemistry for H3K27me3 in excised tumors, scale bar = 100 μm.
2.12. Data availability
The RNA-sequencing data for study are available at the NCBI GEO database under GSE183818. The data that support the findings of this study are available from the corresponding author upon reasonable request.
3. Results
3.1. PIK3CA mutated or amplified cell lines have increased sensitivity to EZH2 inhibitor
We first queried the cBioportal database to understand the current landscape of PIK3CA mutations in lung cancer patients [29,30]. Leveraging data from The Cancer Genome Atlas (TCGA) pan-lung cancer dataset, we observed that 24% of lung tumors harbored PIK3CA alterations, the majority of which were gene amplifications with or without missense mutations (Fig. 1A). Strikingly, this patient subset was nearly mutually exclusive to those with KRAS genetic alterations (log2 odds ratio − 1.819, q-value <0.0001), the latter of which were mainly missense mutations. Overall, 509 (44%) out of the 1144 samples queried had genetic aberrations in either PIK3CA or KRAS, or both. Among these patients, lung adenocarcinoma (LADC) predominantly harbored genetic mutations, which were mainly KRAS mutations (Fig. 1B). On the contrary, the majority of lung squamous cell carcinoma (LSCC) samples had amplifying PIK3CA. Although LADC had a much lower frequency of PIK3CA alterations, the proportion of PIK3CA mutations were relatively higher than amplifications in LADC. The most common mutation in PIK3CA is a point mutation that yields a substitution of glutamic acid by lysine at position 545 – this specific change (PIK3CA-E545K) was observed in 29 (10.5%) of patients with PI3K alterations (Fig. 1C).
Fig. 1.

PIK3CA mutated or amplified cell lines have increased sensitivity to EZH2 inhibitor. (A) Landscape of PIK3CA and KRAS gene alterations in cBioPortal database showing 1144 non-small cell lung cancer patients. (B) Alteration frequencies of PIK3CA and KRAS in LADC and LSCC from cBioportal database. (C) Lollipop plots of each annotated mutations located in PIK3CA or KRAS coding regions. (D and E) Non-linear regression of cell line survival in response to the EZH2 inhibitor GSK126 is plotted for (D) each cell line separately and (E) by genotype, p < 0.0001 in PIK3CA-Mut/Amp vs. RAS-Mut/PIK3CA-WT.
In order to test the effects of EZH2 inhibition on lung cancer cells with differing PIK3CA and KRAS status, we screened 16 lines for sensitivity to the EZH2 inhibitor GSK126 [31]. All the cell lines with IC50 values below 7 μM were reported to be PIK3CA mutant or amplified (Fig. 1D). In contrast, the cell lines mutant for KRAS or NRAS, but without any known alteration of PIK3CA had higher IC50 values, up to 17.4 μM, for GSK126. Two cell lines, H2126 and H520, do not have mutations in either RAS or PIK3CA and those cell lines had intermediate IC50 values. Overall, PIK3CA mutant and amplified cell lines had an average IC50 value of 6.1 μM, while cell lines mutant for RAS and wild-type for PIK3CA had an average IC50 value of 10.2 μM, a 1.66-fold difference (Fig. 1E). This result suggests a window of vulnerability for PI3K-driven cancers to be targeted by EZH2 inhibition. There were two notable exceptions to the genotype-sensitivity correlation in our dataset, H460 and H1975 (Supplementary Fig. S1A). H460, which has a PIK3CA-E545K mutation, also harbors a mutation in liver kinase B1 (LKB1/STK11), which through AMPK activates downstream mTOR signaling as PI3K does. In addition, H1975, an EGFR-driven line, which is also PIK3CA mutant and LKB1 mutant, appears to be insensitive to GSK126. None of the GSK126-sensitive cell lines have LKB1 mutations, implying the combination of mutant/amplified PIK3CA and wild-type (WT) LKB1 may be the most responsive to EZH2i.
3.2. PIK3CA-E545K induces moderate transformation in lung epithelial cells
We next sought to learn about the oncogenic potential of the most common mutation of PIK3CA in lung cancer, PIK3CA-E545K. BEAS2B cells are immortalized with SV40, and therefore should have inactivation of both RB and p53 pathways [32]. We first introduced a mutated form of p53, TP53-R175H, to BEAS2B cells to both ensure that WT p53 function is completely abrogated and to possibly introduce some gain-of-function roles of mutant p53, and we used this as our base cell line [33]. We then inserted either PIK3CA-WT, mutated PIK3CA-E545K, or mutated KRAS-G12V into the base lines (Fig. 2A). The integration of the vectors was confirmed by PCR of genomic DNA with primers that included a region specific to the targeting virus (Supplementary Fig. S2A). We next tested the functional activity of PI3K pathway in these modified BEAS2B lines by detecting phospho-AKT/total AKT ratio downstream of PI3K. Immunoblots showed that BEAS2B with PIK3CA-E545K insertion had drastically increased level of phospho-AKT than the control empty vector (Fig. 2B). Introduction of PIK3CA-WT and KRAS-G12V slightly upregulated phospho-AKT. Calu3, a lung cancer cell line with PIK3CA amplification and TP53 mutation, had the highest AKT phosphorylation level of all cell lines tested.
To dissect the transformation capacity of PIK3CA-WT, PIK3CA-E545K and KRAS-G12V, we performed soft agar colony formation assay on these four lines. It showed that KRAS-G12V-transformed BEAS2B cells formed many more colonies than the other three cell lines, though due to variability of the experiments the normalized data were not significant (Fig. 2C and D). PIK3CA-E545K cells yielded slightly more colonies than the control and PIK3CA-WT BEAS2B cells, but again this result was not significant. However, in 3-dimensional (3D) Matrigel culture, both PIK3CA-E545K and KRAS-G12V lines developed significantly more organoids than the control cells (Fig. 2E and F). There were no obvious differences in organoid counts between control and PIK3CA-WT cells. In order to understand the mechanisms through which PIK3CA mutation confers modest transformation in comparison to KRAS mutation, we performed RNA-sequencing on all four of the BEAS2B isogeneic lines (Supplementary Fig. 2B). We observed that expression of mutant p53 (TP53-R175H) changed gene expression from the parental line, and interestingly increased the genes found in cells after knock-down of PTEN (Supplementary Fig. 2C). When comparing PIK3CA-E545K to EV (TP53-R175H), the only pathway significantly positively enriched was cMYC target genes, and one of the only pathways significantly negatively enriched was genes induced during rapamycin treatment (Fig. 2G). These findings are consistent with PIK3CA mutant cells having few differentially expressed genes compared to TP53-R175H (EV) alone, and with a slight increase in oncogenic programs including cMYC and mTOR. In contrast, KRAS-G12V compared to EV had numerous significantly differentially expressed genes, and enriched gene signatures, including several cMYC, KRAS signatures and genes associated with increased expression when EZH2 is knocked down. Together, these findings suggest the ability of PIK3CA-E545K mutation to drive the stem-like property of growth in Matrigel in lung epithelial cells, but when compared to transformation by oncogenic KRAS, that PIK3CA-E545K is a weak oncogene in this system.
3.3. PIK3CA-E545K with p53 loss drives lung adenocarcinomas in a novel mouse model
Next, in order to study the role of Pik3ca-E545K mutation in the tumorigenesis in vivo, we obtained the genetically engineered mouse model (GEMM) that harbored floxed alleles of Trp53 [14] and a Lox--stop-Lox conditional activating allele encoding Pik3ca-E545K knocked into the endogenous Pik3ca [12,13] (called Pik3ca/p53) (Fig. 3A). We administered adeno-Cre virus via intranasal instillation to cohorts of mice and monitored mice for signs of tumor burden. At more than 52-weeks post adeno-Cre, five out of seven Pik3ca/p53 mice analyzed (71.4%) had frank lung tumors that were uniformly adenocarcinoma by histological examination (Fig. 3B and C and Supplementary Fig. S3A). These tumors had high expression of pro-surfactant protein C (SPC) and no detectable expression of acetylated tubulin, keratin 5 (KRT5), sex determining region Y-box 2 (SOX2) or club cell secretory protein (CCSP), suggesting that the tumors were adenocarcinoma that likely originated from alveolar type 2 (AT2) progenitor cells. Some tumors had light and sporadic staining for mucin 5AC (MUC5AC), but no obvious goblet cell differentiation. The antibodies used were validated on tracheal and esophagus sections (Fig. S3B). To understand if the PI3K pathway was active in the tumors, we utilized immunohistochemistry of phosphorylation of AKT at serine 473 (Fig. 3D). Consistent with reports that activation of PI3K is required for nuclear translocation of phospho-AKT [34], all four Pik3ca tumors sampled showed punctate nuclear staining (Fig. 3D). This punctate nuclear phospho-AKT staining pattern was observed in the Lkb1/Pten control tumor, but not Kras/p53 tumors. The prolonged latency of this Pik3ca/p53 model compared with Kras-G12D/Trp53-null mouse model, which is typically 100–150 days [35,36], is consistent with our results in the BEAS2B cell line, suggesting that PIK3CA-E545K is a relatively weak oncogene relative to oncogenic KRAS. It was also accordant with the observation that PIK3CA mutations were more frequently seen than amplified PIK3CA in LADC, in spite of the prevalence of PIK3CA alterations, mainly amplifications, in LSCC.
Fig. 3.
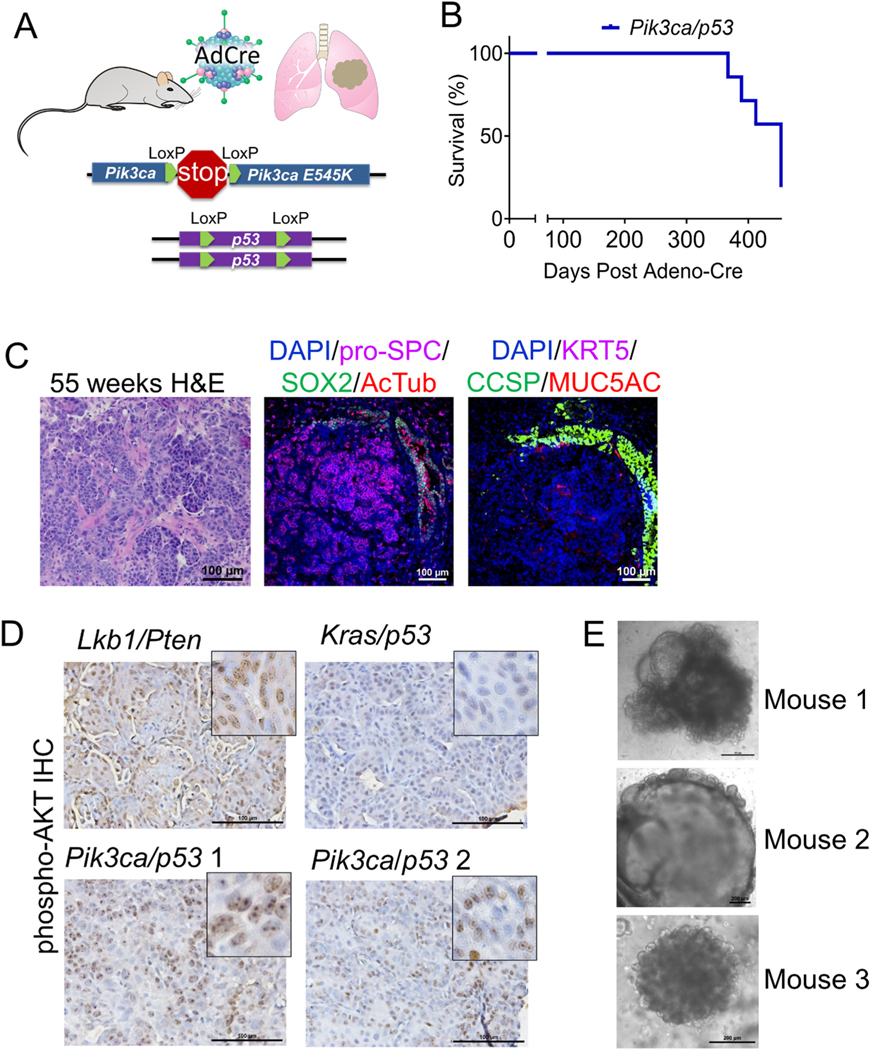
Pik3ca-E545K with Trp53 loss drives lung adenocarcinomas in a novel mouse model. (A) Schematic of Pik3ca-E545K LSL/+;Trp53 fl/fl (Pik3ca/p53) cohorts of mouse models. (B) Tumor-free survival of PI3K/p53 mice after adeno-Cre instillation. (C) H&E and IF for the indicated markers on PI3K/p53 murine tumor sections 55 weeks post infection, scale bar = 100 μm. (D) Immunohistochemistry for phospho-AKT on tumors from mice with biallelic deletion of Lkb1 and Pten (Lkb1/Pten), activation of Kras with p53 deletion (Kras/p53) or activation of Pik3ca with p53 deletion (Pik3ca/p53), scale bar = 100μm. ((E) Microscopic images of 3D organotypic cultures of PI3K/p53 murine tumoroids from three different mice with neonatal lung mesenchymal cells mixed into the Matrigel cultures, scale bar = 200 μm.
We next isolated tumor cells from the Pik3ca/p53 GEMMs and grew the cells in specialized air-liquid-interface 3D Matrigel cultures. These types of cultures have allowed for successful propagation of every murine lung cancer model so far studied, including Kras-G12D/Trp53-null, Kras-G12D/Lkb1-null, and Lkb1-null/Pten-null tumor models [15, 37]. Initially the Pik3ca/p53 tumor cells grew as tumoroids that resembled alveolar lung organoids [38]. Upon passaging, it was clear that the model did not have the robust growth capacity observed from our other tumor models. Given the alveolar structure of the tumoroids, we reasoned that they may grow best with neonatal lung mesenchymal cells, which help to support alveolar cells [38,39]. Co-culturing with these mesenchymal cells, either as a monolayer on the bottom of the transwell or mixed into the Matrigel, allowed for improved growth of the tumoroids so that several models could be established (Fig. 3E). These models will be important for future drug studies. Altogether, Pik3ca-E545K mutation with Trp53 loss drove lung adenocarcinoma in vivo with moderate oncogenic properties.
3.4. EZH2 inhibition synergizes with PI3K inhibition in lung cancer
Given the significant increase in sensitivity of PIK3CA mutant/amplified lung cancer cells to EZH2 inhibitor GSK126, we reasoned that a combination of the PI3K inhibitors, such as copanlisib, with the EZH2 inhibitors could allow for a more potent therapeutic response in lung cancer with PIK3CA mutation/amplification. During the course of our studies, the EZH2 inhibitor tazemetostat (EPZ6438) became FDA approved [8,40] and therefore, we included this EZH2 inhibitor in our drug synergy studies. We identified that both GSK126 and EPZ6438 were very effective at reducing the PRC2 histone mark, H3K27me3, and that copanlisib was very effective at reducing the levels of phospho-AKT in both Calu3 and Sw1573 lung cancer cells in short term (3 day) cultures (Fig. 4A). Of note, Sw1573 cells have PIK3CA-K111E and KRAS-G12C mutations, while Calu3 cells have PIK3CA amplification but no RAS mutations and showed substantially higher phospho-AKT than Sw1573. Intriguingly, copanlisib combined with EPZ6438 led to a lower level of phospho-AKT than copanlisib alone or copanlisib combined with GSK126.
In order to examine the synergy of EZH2 inhibitor and PI3K inhibition, we performed dose synergy matrix experiments and analyzed the data with SynergyFinder2.0. The results showed that copanlisib had synergistic or potent additive effect with EZH2 inhibitors GSK126 and EPZ6438 in both Calu3 and Sw1573 cells (Fig. 4B). Next, we studied drug synergy in BEAS2B cells with control empty vector, PIK3CA-WT, PIK3CA-E545K mutation, or mutated KRAS-G12V. Results showed that PIK3CA-WT or mutation did not change the sensitivity to GSK126, while KRAS mutation conferred resistance to GSK126 in BEAS2B cells (Supplementary Fig. S4A). Moreover, copanlisib synergized with GSK126 and EPZ6438 in all the modified BEAS2B cells except for copanlisib with EPZ6438 in KRAS-G12V line, which showed an antagonistic effect (Supplementary Fig. S4B). Mechanistically, EZH2 inhibitors and PI3K inhibitor all raised the mRNA expression of PI3K interacting protein 1 (PIK3IP1), a negative regulator of PI3K that works by binding to the p110 catalytic subunit of PI3K and suppressing its activity [41]. Combining copanlisib with GSK126 or EPZ6438 induced a higher expression of PIK3IP1 in Calu3, Sw1573 and all the BEAS2B lines (Fig. 4C and Supplementary Fig. S4C).
To validate the use of this drug combination in vivo, we administered four treatment arms including vehicle, copanlisib, EPZ6438 and copanlisib combined with EPZ6438 (combo) to mice harboring Calu3 xenografts. After 10 days of treatment, tumor growth was significantly lower in copanlisib and combo treated mice compared with vehicle (Fig. 5A). Combo treated xenograft tumors were also significantly smaller than the copanlisib alone, demonstrating a collaborative effect of these two drugs in vivo. Immunohistochemistry for the EZH2 catalytic mark H3K27me3 showed a reduction of H3K27me3 staining in tumor nuclei of xenografts from mice that received either EPZ6438 or combo treatment (Fig. 5B). Interestingly, we performed the xenograft experiment in both old and young Nude mice (mice were aged due to COVID19 restrictions). EPZ6438 appears to work significantly better in the old Nude mouse cohort (Supplementary Figs. S5A–B). This suggests that age may play a role in whether EPZ6438 and copanlisib synergize in vivo and that some of the effects may be due to host immune cell interactions. For toxicity, we monitored the mouse weight and found that both copanlisib and combination reduced mouse body weight by an average of 6%, but the difference between placebo and combination treatment was not significant (Supplementary Fig. S5C). Together, these results reveal a promising combination therapy of EZH2 inhibition with PI3K inhibition to target lung cancers with PIK3CA genetic alterations.
4. Discussion
Although PI3K pathway mutations are prevalent in lung cancer, efforts to target this genetic subclass of tumor have been limited. This study shows that PIK3CA mutation is a moderate oncogene with weak transformation capacity compared with KRAS mutation. These results suggest that lung cancer cells may not heavily rely on the PI3K pathway, especially when negative regulators such as PTEN are intact or when bypass activation of PI3K downstream targets exist. Previous studies have shown that Pik3ca-H1047R under the control of the TetO promoter can be oncogenic in the lung [42], and that Pik3ca-H1047R is more efficient at driving tumors that Pik3ca-E545K when expressed transgenically in murine breast cells [43]. Similarly, a study utilizing Pik3ca-H1047R knocked into the endogenous mouse Pik3ca locus showed lack of tumors in multiple organs, but that mutant Pik3ca could cooperate with other lesions such as partial loss of the tumor suppressor APC [44]. Our research is the first time to describe the oncogenic property of Pik3ca-E545K, the most prevalent mutation in lung cancer, in lung epithelium using an inducible genetically engineered knock-in model. This model solidifies that Pik3ca-E545K, when combined with Trp53 loss, can be oncogenic, and will be an important tool to understand this genetic subclass of tumors going forward. Furthermore, our syngeneic genetically engineered human cell lines suggest that in addition to mTOR, cMYC is a predominant pathway activated by PIK3CA-E545K and this observation adds to our understanding of the oncogenic properties of PIK3CA mutations.
Discovery of novel combinatorial strategies may improve the efficacy of PI3K inhibitors in lung cancer. Although PI3K inhibitors in clinical trials did not show robust anti-tumor effect in lung cancer [3,45–47], there are some promising combinations, including combinations of PI3K inhibitors with receptor tyrosine kinase inhibitors, endocrine therapies, mTOR inhibitors, MEK inhibitors and DNA damaging chemotherapies [48]. Our data show that EZH2 inhibition with either GSK126 or EPZ6438 has good synergism with the PI3K inhibition copanlisib when PIK3CA is amplified and KRAS is wild-type. Mechanistically, we showed that PI3K inhibition or EZH2 inhibition alone could increase the expression of PI3K negative regulator PIK3IP1, and that combining these two strategies induced a more robust upregulation of PIK3IP1. This is consistent with the immunoblot result which showed copanlisib combined with EPZ6438 led to lower phospho-AKT compared to copanlisib alone. However, such decrease in phospho-AKT was not observed in copanlisib-GSK126 combined treatment. Thus, we cannot exclude the possibility of GSK126-induced toxicity in this scenario. The mechanisms of PI3K inhibition synergizing with EZH2 inhibition may include several other pathways. In the future, we propose to perform RNA-sequencing on the cells treated with the four arms to help discover other novel mechanisms to explain the synergy combining EZH2 inhibitor with PI3K inhibitor.
The reason why Pik3ca-E545K mutation generated LADC but not LSCC in mouse models remains unknown. The result of adenocarcinoma formation was consistent with the cBioportal results that LADC harbored more PIK3CA mutations than amplified PIK3CA, while most of the LSCC samples had PIK3CA amplification, not PIK3CA mutations. PIK3CA mutation or amplification may have distinct molecular targets driving different programs of tumor initiation and progression. Such difference was also reported in HER-2 positive lung cancer [49]. Mutation or amplification of PIK3CA may also transform different cells-of-origin, like AT2 cells or basal cells respectively, which requires further investigation in other systems [50]. PI3K pathway was also found to have important roles in embryonic development and cell fate determination [51]. Therefore, understanding of such differences will help define the appropriate target patients for PI3K inhibitor, potentially combined with EZH2 inhibition.
In conclusion, we report here a new model of Pik3ca-E545K; Trp53-null lung adenocarcinoma that may become an important tool for the field. We also identified EZH2 inhibition as a promising therapy to combine with PI3K inhibition to combat lung cancers with PIK3CA mutation or amplification. Both copanlisib, the PI3K inhibitor, and tazemetostat, the EZH2 inhibitor, are FDA-approved, which should enhance the clinical translation of this work.
Supplementary Material
Acknowledgements
This work was supported in part by NCI K22 CA201036, Kentucky Lung Cancer Research Program, V Foundation Scholar Award, American Cancer Society Institutional Research Grant IRG-85-001-25, NCI R01 CA237643, NIGMS P20 GM121327-03, American Cancer Society Research Scholar Grant 133123-RSG-19-081-01-TBG and American Association for Cancer Research-Bayer Innovation and Discovery Grant (CFB), NIEHS T32 5T32ES007266-30 (TJD and ALB) and NHLBI F31 HL151111-01 (ALB). This research was also supported by the Biostatistics & Bioinformatics Shared Resource Facility, Biospecimen Procurement & Translational Pathology Shared Resource Facility and Flow Cytometry & Immune Monitoring Shared Resource Facility of the University of Kentucky Markey Cancer Center (P30CA177558). We also thank D. Gilbreath in the Markey Cancer Center Research Communications Office for graphic designs.
Abbreviations:
- PI3K
Phosphoinositide 3-kinase
- LSCC
Lung squamous cell carcinoma
- LADC
Lung adenocarcinoma
- H3K27me3
Histone H3 lysine 27 tri-methylation
- AT2
Alveolar type II cells
- SV40
Simian vacuolating virus 40
Footnotes
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.canlet.2021.10.010.
References
- [1].Samuels Y, et al. , High frequency of mutations of the PIK3CA gene in human cancers, Science 304 (5670) (2004) 554. [DOI] [PubMed] [Google Scholar]
- [2].Massacesi C, et al. , PI3K inhibitors as new cancer therapeutics: implications for clinical trial design, OncoTargets Ther. 9 (2016) 203–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Vansteenkiste JF, et al. , Safety and efficacy of buparlisib (BKM120) in patients with PI3K pathway-activated non-small cell lung cancer: results from the phase II BASALT-1 study, J. Thorac. Oncol 10 (9) (2015) 1319–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Tan AC, Targeting the PI3K/Akt/mTOR pathway in non-small cell lung cancer (NSCLC) 11 (3) (2020) 511–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Simon JA, Kingston RE, Mechanisms of Polycomb gene silencing: knowns and unknowns, Nat. Rev. Mol. Cell Biol 10 (10) (2009) 697–708. [DOI] [PubMed] [Google Scholar]
- [6].Simon JA, Lange CA, Roles of the EZH2 histone methyltransferase in cancer epigenetics, Mutat. Res. Fund Mol. Mech. Mutagen 647 (1–2) (2008) 21–29. [DOI] [PubMed] [Google Scholar]
- [7].Bracken AP, Helin K, Polycomb group proteins: navigators of lineage pathways led astray in cancer, Nat. Rev. Cancer 9 (11) (2009) 773–784. [DOI] [PubMed] [Google Scholar]
- [8].Hoy SM, Tazemetostat: first approval, Drugs 80 (5) (2020) 513–521. [DOI] [PubMed] [Google Scholar]
- [9].Yamamoto H, et al. , PIK3CA mutations and copy number gains in human lung cancers, Cancer Res. 68 (17) (2008) 6913–6921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Bamford S, et al. , The COSMIC (catalogue of somatic mutations in cancer) database and website, Br. J. Cancer 91 (2) (2004) 355–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Barretina J, et al. , The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity, Nature 483 (7391) (2012), 603–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Roy A, et al. , Mouse models of human PIK3CA-related brain overgrowth have acutely treatable epilepsy, eLife 4 (2015) e12703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Robinson G, et al. , Novel mutations target distinct subgroups of medulloblastoma, Nature 488 (7409) (2012) 43–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Marino S, et al. , Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum, Genes Dev. 14 (8) (2000) 994–1004. [PMC free article] [PubMed] [Google Scholar]
- [15].Xu C, et al. , Loss of Lkb1 and pten leads to lung squamous cell carcinoma with elevated PD-L1 expression, Cancer Cell 25 (5) (2014) 590–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lau AN, et al. , Tumor-propagating cells and Yap/Taz activity contribute to lung tumor progression and metastasis, EMBO J. 33 (2014) 468–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Knutson SK, et al. , Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2, Proc. Natl. Acad. Sci. Unit. States Am 110 (19) (2012) 7922–7927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Liu N, et al. , BAY 80–6946 is a highly selective intravenous PI3K inhibitor with potent p110α and p110δ activities in tumor cell lines and xenograft models, Mol Cancer Ther 12 (11) (2013) 2319–2330. [DOI] [PubMed] [Google Scholar]
- [19].Fang B, et al. , Inhibition of PI3K by copanlisib exerts potent antitumor effects on Merkel cell carcinoma cell lines and mouse xenografts, Sci. Rep 10 (1) (2020) 8867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ianevski A, Giri AK, Aittokallio T, SynergyFinder 2.0: visual analytics of multi- drug combination synergies, Nucleic Acids Res. 48 (W1) (2020) W488–W493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Wang Z, et al. , Reversal and prevention of arsenic-induced human bronchial epithelial cell malignant transformation by microRNA-200b, Toxicol. Sci 121 (1) (2011) 110–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Lee JH, et al. , Lung stem cell differentiation in mice directed by endothelial cells via a BMP4-NFATc1-thrombospondin-1 axis, Cell 156 (3) (2014) 440–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Proia TA, et al. , Genetic predisposition directs breast cancer phenotype by dictating progenitor cell fate, Cell Stem Cell 8 (2) (2011) 149–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Zhao JJ, et al. , The oncogenic properties of mutant p110alpha and p110beta phosphatidylinositol 3-kinases in human mammary epithelial cells, Proc. Natl. Acad. Sci. U. S. A 102 (51) (2005) 18443–18448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Bolger AM, Lohse M, Usadel B, Trimmomatic: a flexible trimmer for Illumina sequence data, Bioinformatics 30 (15) (2014) 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Li B, Dewey CN, RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome, BMC Bioinf. 12 (1) (2011) 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Robinson MD, McCarthy DJ, Smyth GK, edgeR: a Bioconductor package for differential expression analysis of digital gene expression data, Bioinformatics 26 (1) (2010) 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Subramanian A, et al. , Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles, Proc. Natl. Acad. Sci. Unit. States Am 102 (43) (2005) 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Cerami E, et al. , The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data, Cancer Discov. 2 (5) (2012) 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Gao J, et al. , Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal, Sci. Signal 6 (269) (2013) pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].McCabe MT, et al. , EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations, Nature 492 (7427) (2012) 108–112. [DOI] [PubMed] [Google Scholar]
- [32].Reddel RR, et al. , SV40-induced immortalization and ras-transformation of human bronchial epithelial cells, Int. J. Cancer 61 (2) (1995) 199–205. [DOI] [PubMed] [Google Scholar]
- [33].Polireddy K, et al. , Mutant p53(R175H) promotes cancer initiation in the pancreas by stabilizing HSP70, Cancer Lett. 453 (2019) 122–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Xuan Nguyen TL, et al. , Akt phosphorylation is essential for nuclear translocation and retention in NGF-stimulated PC12 cells, Biochem. Biophys. Res. Commun 349 (1) (2006) 789–798. [DOI] [PubMed] [Google Scholar]
- [35].Oliver TG, et al. , Chronic cisplatin treatment promotes enhanced damage repair and tumor progression in a mouse model of lung cancer, Genes & Development 24 (8) (2010) 837–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Serresi M, et al. , Polycomb repressive complex 2 is a barrier to KRAS-driven inflammation and epithelial-mesenchymal transition in non-small-cell lung cancer, Cancer Cell 29 (1) (2016) 17–31. [DOI] [PubMed] [Google Scholar]
- [37].Zhang H, et al. , Lkb1 inactivation drives lung cancer lineage switching governed by Polycomb Repressive Complex 2, Nat. Commun 8 (2017), 14922–14922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Lee J-H, et al. , Lung stem cell differentiation in mice directed by endothelial cells via a BMP4-NFATc1-thrombospondin-1 Axis, Cell 156 (3) (2014) 440–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Dost AFM, et al. , Organoids model transcriptional Hallmarks of oncogenic KRAS activation in lung epithelial progenitor cells, Cell Stem Cell 27 (4) (2020) 663–678, e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Italiano A, et al. , Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: a first-in-human, open-label, phase 1 study, Lancet Oncol. 19 (5) (2018) 649–659. [DOI] [PubMed] [Google Scholar]
- [41].Bitler BG, et al. , Synthetic lethality by targeting EZH2 methyltransferase activity in ARID1A-mutated cancers, Nat. Med 21 (3) (2015) 231–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Engelman JA, et al. , Effective use of PI3K and MEK inhibitors to treat mutant K- ras G12D and PIK3CA H1047R murine lung cancers, Nat. Med 14 (12) (2008) 1351–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Meyer DS, et al. , Expression of PIK3CA mutant E545K in the mammary gland induces heterogeneous tumors but is less potent than mutant H1047R, Oncogenesis 2 (9) (2013) e74–e74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Berenjeno IM, et al. , Oncogenic PIK3CA induces centrosome amplification and tolerance to genome doubling, Nat. Commun 8 (1) (2017) 1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Chen X, et al. , Low-pass whole-genome sequencing of circulating cell-free DNA demonstrates dynamic changes in genomic copy number in a squamous lung cancer clinical cohort, Clin. Cancer Res 25 (7) (2019) 2254–2263. [DOI] [PubMed] [Google Scholar]
- [46].Yamamoto N, et al. , Phase Ia/Ib study of the pan-class I PI3K inhibitor pictilisib (GDC-0941) administered as a single agent in Japanese patients with solid tumors and in combination in Japanese patients with non-squamous non-small cell lung cancer, Invest. N. Drugs 35 (1) (2017) 37–46. [DOI] [PubMed] [Google Scholar]
- [47].Tan AC, Targeting the PI3K/Akt/mTOR pathway in non-small cell lung cancer (NSCLC), Thorac Cancer 11 (3) (2020) 511–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Castel P, et al. , Rationale-based therapeutic combinations with PI3K inhibitors in cancer treatment, Mol Cell Oncol 1 (3) (2014) e963447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Li BT, et al. , HER2 amplification and HER2 mutation are distinct molecular targets in lung cancers, J. Thorac. Oncol 11 (3) (2016) 414–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Heng WS, Gosens R, Kruyt FAE, Lung cancer stem cells: origin, features, maintenance mechanisms and therapeutic targeting, Biochem. Pharmacol 160 (2019) 121–133. [DOI] [PubMed] [Google Scholar]
- [51].Yu JS, Cui W, Proliferation, survival and metabolism: the role of PI3K/AKT/mTOR signalling in pluripotency and cell fate determination, Development 143 (17) (2016) 3050–3060. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-sequencing data for study are available at the NCBI GEO database under GSE183818. The data that support the findings of this study are available from the corresponding author upon reasonable request.