

Abstract
Purpose of Review
The ability to analyze the molecular events occurring within individual cells as opposed to populations of cells is revolutionizing our understanding of musculoskeletal tissue development and disease. Single cell studies have the great potential of identifying cellular subpopulations that work in a synchronized fashion to regenerate and repair damaged tissues during normal homeostasis. In addition, such studies can elucidate how these processes break down in disease as well as identify cellular subpopulations that drive the disease. This review highlights three emerging technologies: single cell RNA sequencing (scRNA-seq), Assay for Transposase-Accessible Chromatin using sequencing (ATAC-seq), and Cytometry by Time-Of-Flight (CyTOF) mass cytometry.
Recent Findings
Technological and bioinformatic tools to analyze the transcriptome, epigenome, and proteome at the individual cell level have advanced rapidly making data collection relatively easy; however, understanding how to access and interpret the data remains a challenge for many scientists. It is, therefore, of paramount significance to educate the musculoskeletal community on how single cell technologies can be used to answer research questions and advance translation.
Summary
This article summarizes talks given during a workshop on “Single Cell Omics” at the 2020 annual meeting of the Orthopedic Research Society. Studies that applied scRNA-seq, ATAC-seq, and CyTOF mass cytometry to cartilage development and osteoarthritis are reviewed. This body of work shows how these cutting-edge tools can advance our understanding of the cellular heterogeneity and trajectories of lineage specification during development and disease.
Keywords: Single cell RNA-seq, ATAC-seq, CyTOF, Osteoarthritis, Cartilage
Introduction
Musculoskeletal conditions, such as osteoarthritis, osteoporosis, sarcopenia, and muscular dystrophy, are characterized by a high molecular heterogeneity of cellular populations within diseased tissues as well as differences from the normal state. This heterogeneity is one of the major barriers to make progress in understanding the pathogenesis of these and other diseases. The development of next generation sequencing (NGS) technologies has revolutionized the entire field of biology. While these technologies have allowed researchers to unravel new and potentially novel biological discoveries, much of the data have been collected from bulk cell populations rather than individual cells, thus, limiting our understanding of disease processes to the majority of cells in population. New methods have emerged to refine the traditional “bulk” approaches to pave the way for single cell omics. In this review, we will discuss three of such approaches: single cell RNA-Sequencing (scRNA-seq), Assay for Transposase-Accessible Chromatin using sequencing (ATAC-seq), and Cytometry by Time-Of-Flight (CyTOF).
scRNA-seq evaluates gene signatures at a single cell level, allowing for the discovery of intermediate and/or rare sub-populations of cells within a larger population or a tissue. The single cell approach can reveal regulatory relationships between genes and track the trajectories of distinct cell lineages during development (Fig. 1). Here, Dr. Chia-Lung Wu and colleagues review how this sequencing technology and bioinformatic advances helped them define the cellular heterogeneity involved in lineage specification during chondrogenic differentiation [1].
Fig. 1.
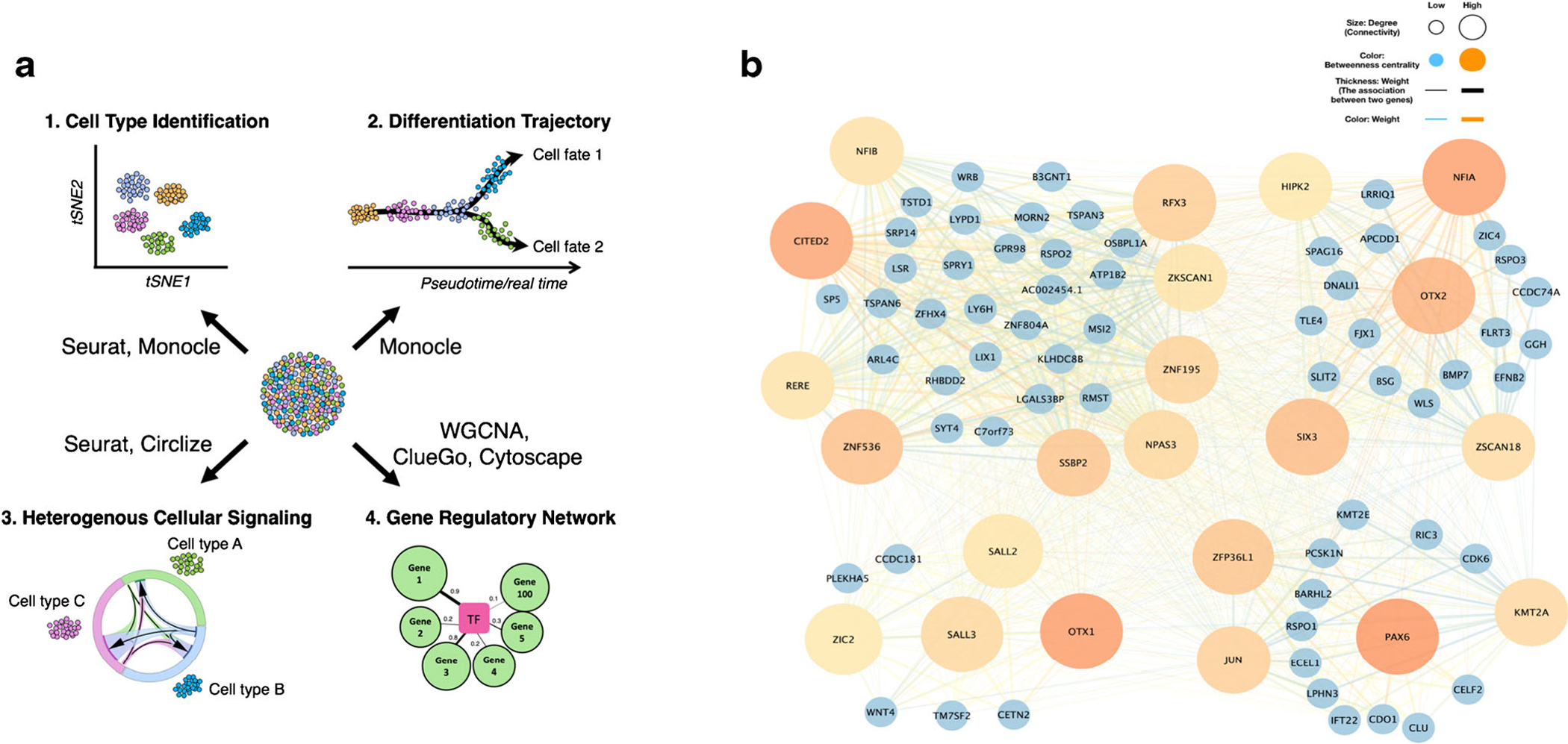
a. Representative applications that can be accomplished by scRNA-seq and bioinformatic analyses. b. A neurogenesis gene regulatory network identified by WGCNA in d28 chondrogenic pellets
ATAC-seq complements scRNA-seq as it identifies open and closed chromatin across the genome in specific cell types. Despite methodological and computational limitations, ATAC-seq can be performed at the single cell level and has great potential to find important regulatory regions in the genome as well as to characterize the epigenome of skeletal cell subtypes within and across species (Fig. 2). Below, Dr. Terrence Capellini’s team reviews their work using ATAC-seq to identify genes controlling cartilage development and osteoarthritis through evolution [2] and highlight how the use of bulk ATAC-seq methods on chondrocytes may inform future single cell ATAC-seq (scATAC-seq) studies.
Fig. 2.
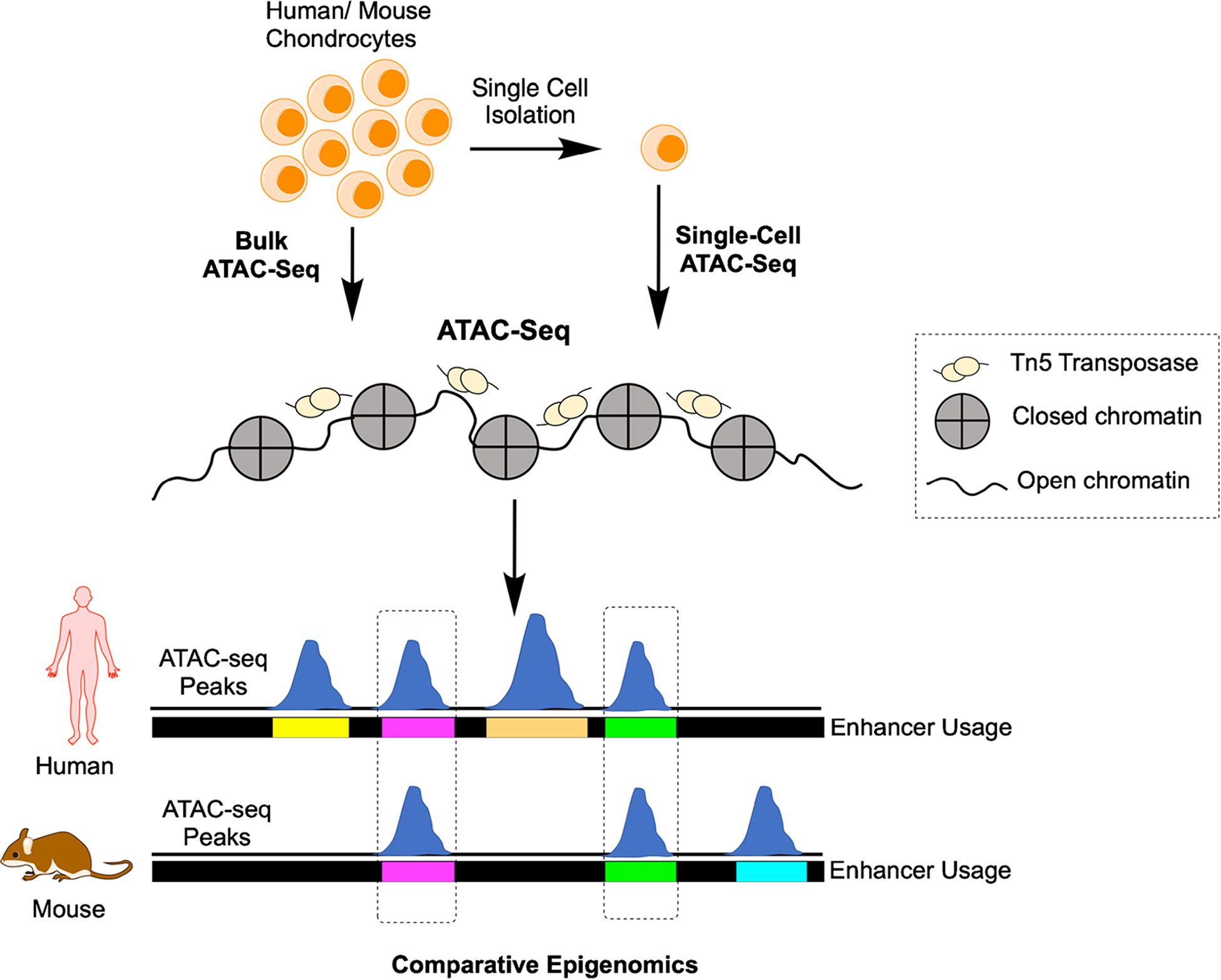
scATAC-seq identifies open chromatin and comparative epigenomics can identify evolutionarily-conserved regions contributing to disease, including osteoarthritis
Mass cytometry, also referred to as CyTOF is a multiparametric and high-dimensional analysis at a level of single cell. This technique allows for the detection of isotope-labeled antibodies, which can map about 40 to 120 protein epitopes for a single cell (Fig. 3). This high-dimensional method for proteomic analyses complements single cell transcriptomic and epigenetic techniques. Dr. Nidhi Bhutani and her team used CyTOF to reveal the heterogeneity of cellular populations in healthy and osteoarthritic skeletal tissues [3].
Fig. 3.
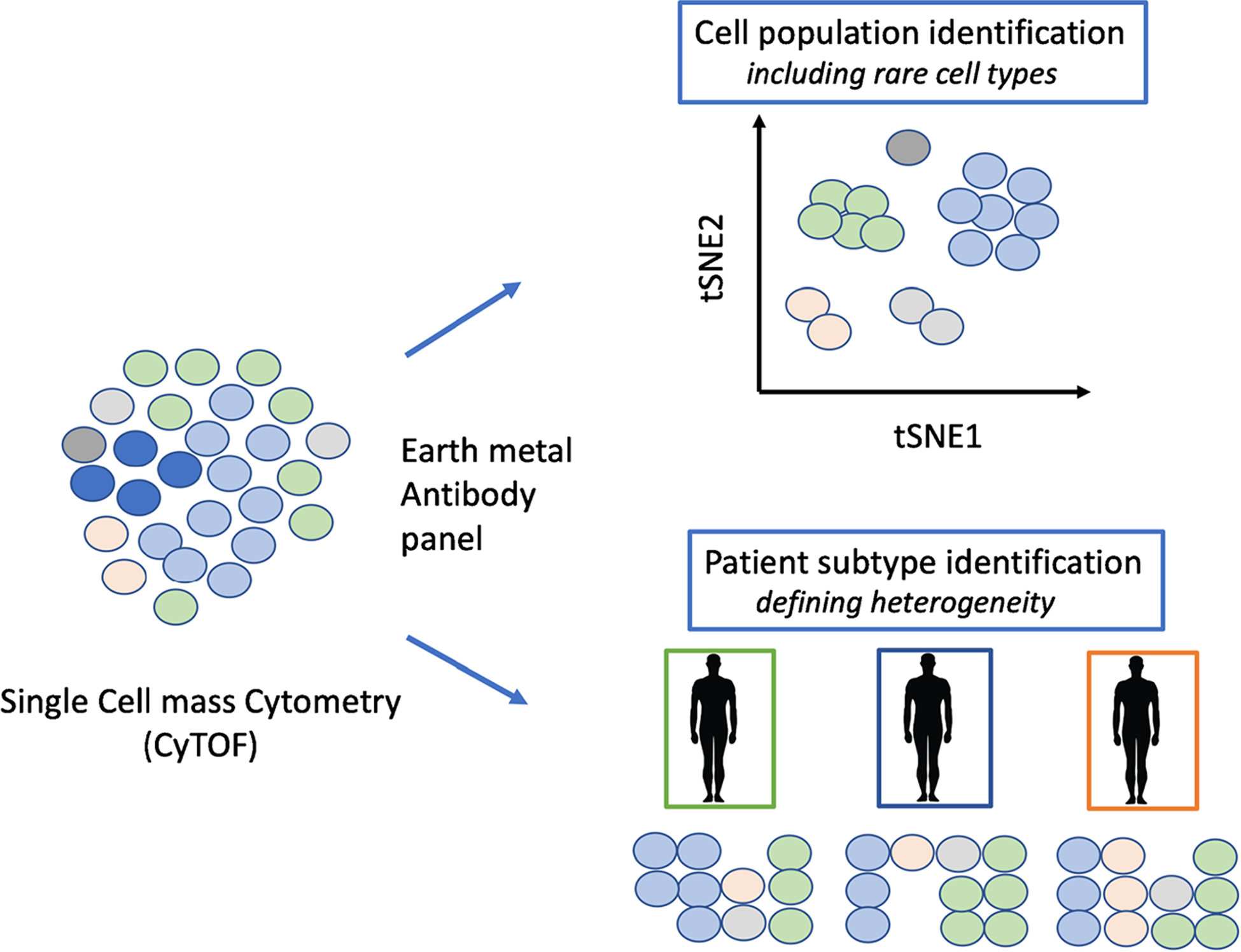
Single cell CyTOF identifies cells in tissues based on protein expression and facilitates patient OA type identification based on proteome heterogeneity
scRNA-Seq Reveals Cell Development Trajectories During Chondrogenic Differentiation
Recent advances in NGS, particularly RNA-seq, provide a high-throughput approach to investigate diverse and complex biological systems. Transcriptomic studies using “bulk” RNA-seq (i.e., average expression of a population of cells) assume that all the cells within a given tissue are homogeneous and thus they share similar gene expression profile at the given time. This approach cannot identify differences in gene expression among individual cells, particularly those that make up a small fraction of the population. The stochastic nature of gene expression can lead to heterogeneity in cellular composition in a tissue and can play a critical role in driving cell fate decisions during development. New NGS-based techniques for scRNA-seq allow for the quantification of transcriptomic profiles at the resolution of an individual cell. In the following section, using chondrogenesis of human-induced pluripotent stem cells (hiPSCs) as an example, we introduce how scRNA-seq can be applied to: (1) reveal cellular heterogeneity within a population of cells; (2) delineate differentiation trajectories; (3) construct heterogenous cellular signaling models; and (4) build gene regulatory networks (GRNs) (Fig. 1a).
Human iPSCs were induced to undergo chondrogenic differentiation using a previously established step-wise protocol of lineage specification [4]. Cells were harvested at various time points for scRNA-seq. Single cells (n = ~2500) were captured by the Chromium Controller (10x Genomics) platform and sequenced using Illumina HiSeq2500. Note that there are several other platforms, which have been developed for this purpose. The pros and cons of each platform have been reviewed elsewhere [5]. We processed sequencing reads by Cell Ranger version 2.0 (10x Genomics software), although numerous other RNA-seq mappers such as STARsolo (Spliced Transcripts Alignment to a Reference) can be employed for sequence alignment of droplet scRNA-seq data. Briefly, reads were aligned to the GRCh38 (version 90) for genome annotation, demultiplexing, barcode filtering, and gene quantification. Gene barcode matrices for each sample were generated by counting the number of unique molecular identifiers for a given gene in an individual cell.
For quality control and to determine the heterogenous composition of cell populations, gene barcode matrices were imputed into the Seurat R package [6]. Low-quality cells, defined as cells expressing <200 genes, >7000 genes, and >5% mitochondrial gene content, were removed from analysis. Moreover, genes that were detected in less than three cells were also omitted. These cutoff criteria are arbitrary and need to be adjusted for each study since gene expression levels depend on cell type. Next, to reduce the variance introduced by “unwanted” sources, we regressed out variation in gene expression driven by cell cycle stages and mitochondrial gene expression in Seurat. After the quality control, we converted Seurat objects into Monocle objects and reduced data dimensionality for further analysis [7]. Unsupervised clustering of the data was performed, and then visualized in a uniform manifold approximation and projection (UMAP) plot [8]. To determine the biological functions of the marker genes from a given cluster, we performed Gene Ontology (GO) enrichment analysis using a Database for Annotation, Visualization, and Integrated Discovery (DAVID) Gene Functional Classification Tool [9]. By comparing unique biological GO terms with the existing RNA-seq datasets and the data assembled from the literature, we were able to annotate cell clusters.
We used the Monocle 3 R package to reconstruct differentiation trajectories by computing and ordering the sequence of gene expression changes in an unsupervised manner. Quality control of scRNA-seq datasets from different timepoints were performed in Seurat version 2.4 and converted into individual Monocle objects as described in the previous section. Monocle objects were then merged into one object. The merged object was also pre-processed and reduced its dimensionality as previously described. Next, the cells were ordered based on the changes in gene expression as a function of time, and then visualized the trajectory. Using these analyses, we identified off-target differentiation (i.e., generation of neural progenitors and melanocytes) during the course of hiPSC chondrogenesis.
To investigate the ligand–receptor interaction in heterogenous multicellular signaling systems, we used a list comprising of 2557 human ligand–receptor pairs curated by the Database of Ligand-Receptor Partners, The International Union of Basic and Clinical Pharmacology (IUPHAR), and Human Plasma Membrane Receptome [10, 11]. We first quantified the percentage of the cells (i.e., neural progenitors, melanocytes, and chondrocytes) that expressed a specific WNT ligand and its associated frizzled (FZD) receptors from these scRNA-seq datasets. We used WNT3A and its potential receptor FZD1 as an example since both of these appear to be essential for neurogenic differentiation of neural stem cells [12, 13]. To ensure the ligand and receptors are uniquely expressed, we required a fold change in expression of >0.25 on a log scale. Circlize R package was then used to visualize the directions of the signaling in the cell type based on ligand–receptor pairing [14]. We noted that WNT3A was not only involved in neurogenesis of neural progenitors but also might signal through melanocytes and chondrocytes to affect cell phenotype.
To reconstruct GRNs and identify their associated hub genes that regulate cell differentiation, we used Weighted Gene Co-expression Network Analysis (WGCNA) [15]. First, the dataset of interest (e.g., a given timepoint such as day 28 chondrogenic pellets) was created in Seurat and converted into a plain matrix for a given gene (column) in an individual cell (row). The dataset was then cleaned by removing cells with too many missing values. In this case, a total of 1271 cells derived from day 28 chondrogenic pellets passed this filtering step (about 96.2% of the cells that passed the quality control). Next, we determined the proper soft-thresholding power (β) that fits the criterion of approximate scale-free topology of the network and built an adjacency matrix with β = 8. Hierarchical clustering and GRNs were constructed and modules of highly inter-associated genes were identified. Gene lists from interesting modules were extracted and submitted (i) to DAVID to retrieve GO terms (biological processes and molecular functions) and (ii) to ClueGO, a Cytoscape App, for visualization of the non-redundant biological terms for large clusters of genes in a functionally grouped network [16, 17]. For instance, we observed that one of the modules identified in day 28 chondrogenic pellets was associated with embryonic eye development and neurogenesis including forebrain and dopaminergic neuron differentiation. We next identified transcription factors (TFs) and TF regulators from the genes based on the GO terms in molecular functions. Based on the highest weight (i.e., high correlation coefficient) connected to a given TF or TF regulator, we selected the top 100 genes. Finally, the GRN based on these TFs and TF regulators underwent cluster analysis using community cluster function in clusterMaker2, also a Cytoscape App, and was then visualized using Cytoscape [18]. Hub genes for each GRN were identified as genes with high weight (summed correlation coefficients), high degree (summed connectivity; i.e., total numbers genes connected to this specific gene), and high betweenness centrality measure of the network [19]. Thus, NFIA, OTX1, OTX2, PAX6, and CITED2 were identified as hub genes of the module involved in off-target neurogenic differentiation in the course of hiPSC chondrogenesis in this study [1] (Fig. 1b).
In summary, the scRNA-seq techniques and relevant bioinformatic analysis methods are rapidly evolving, however, some relatively well-developed and extensively utilized R packages are available to analyze scRNA-seq datasets for the biological interpretation of these datasets.
Uncovering the Gene Regulatory Landscape of Cartilage Development to Understand Skeletal Disease
In the past decade, functional genomics methods have provided important insights into the molecular processes at play, including signaling pathways and transcriptional factors that control skeletal development [20, 21]. These methods have also provided additional layers of understanding, moving from individual genes to gene regulatory sequences (e.g., promoters, enhancers) to help foster deeper insights on how the skeleton is built and what causes skeletal disease. In the past 5 years, ATAC-seq [22, 23] has made it possible to epigenetically profile cell-type specific chromatin states (i.e., open or accessible versus closed or inaccessible) across the genome involved in bone development (e.g., chondrocytes, osteoblasts). Accessible regions often reflect those that are involved in gene transcriptional regulation, either from a distance (e.g., cis-acting enhancer or repressor elements) or more proximally (e.g., gene promoter elements). While this method, along with a more recent application of it in the single cell sphere i.e., scATAC-seq (see below), has great potential to identify a number of important regulatory regions in the genome as well as to characterize the epigenome of skeletal cell subtypes, there remain important methodological and computational obstacles that need to be addressed. Some of these issues are described here in the context of ATAC-seq studies on chondrocytes acquired in bulk from both mouse and human developmental skeletal samples [2]. Additionally, as scATAC-seq provides snapshots of cell–cell variability in chromatin organization by gathering data on hundreds and thousands of single cells in parallel, and is thus important for understanding cell-type specific regulatory regions, it is unfortunate that to date such methodology has not been used on chondrocytes. With this point in mind, here we also discuss how the use of bulk ATAC-seq methods on chondrocytes may inform future scATAC-seq studies.
Bulk ATAC-seq was used to epigenetically profile (i.e., identify regulatory regions of) chondrocytes extracted in vivo from mouse E15.5 and stage-matched human E59 long bone elements. At this stage, the “bone-ends” are chondrocyte models of each adult bone’s morphology. ATAC-seq was also performed on developing brain tissues and these datasets were used to remove shared open chromatin or accessible regulatory regions that were not unique to individual bone-ends (e.g., the distal femur). A method called the Irreproducible Discovery Rate (IDR) was then used to find “peaks” or “regulatory regions” that were consistently found between biological replicates per tissue per timepoint and species. IDR is a unified approach that measures the reproducibility of peaks identified from replicate experiments and provides highly stable thresholds based on reproducibility. This helps remove biological noise. For example, if starting with <100,000 unfiltered (pre-IDR) peaks for large genomes (e.g., human and mouse genomes), an IDR threshold of 0.05 is appropriate to reduce the sets to fewer number of regulatory regions but those which are identified in each replicate [24]. Finally, the regulatory and evolutionary potential of called regulatory peak sets from different bone-ends was explored using bioinformatics and comparative genomics to detect signals of anatomical evolution and the regulatory basis for skeletal disease. The comparative map of regulatory circuitry obtained through these analyses will drive an improved understanding of regulatory underpinning of cartilage biology and how such relates to skeletal development and disease.
Several findings from this study elucidate the general regulatory biology of chondrocytes between bone-ends as well as between species. Moreover, they shed light on issues important for future scATAC-seq applications (see below). In each species, tens of thousands of (IDR-filtered) chromatin-accessible regulatory regions were found that are shared between chondrocytes extracted from different anatomical locations (i.e., from the developing proximal and distal bone ends of the skeleton). These regions are likely general chondrocyte regulatory elements. In addition, hundreds to thousands of bone-end-specific chondrocyte regulatory regions (e.g., chromatin-accessible regions unique to distal femur as compared to the proximal femur or proximal tibia), ranging from 5 to 15% of each peak set, were discovered likely reflecting modularized regulatory control of specific anatomical parts. scATAC-seq will be important application to further identify for each bone-end the diversity of chondrocyte (and nonchondrocyte) cell types that in part underlie unique as well as shared signals, and what role differences in differentiation states between bone-ends drive some of the regulatory divergence we observed. Importantly, use of IDR on each species tissue replicates caused pre-IDR filtered peak sets to be reduced by 50–80%, resulting in much smaller conservative regulatory element sets per tissue, providing some evidence that by removing extraneous noise (in part), there exists a real signal of regulatory divergence between bone ends. Yet, it must be emphasized that these methods on in vivo extracted tissues reveal considerable inter-individual variation, some of which is likely experimental noise, but some reflecting individual differences in differentiation, growth, and overall development of different bone-ends. These inter-individual differences and the “noise” they generate could be important drivers of disease mechanism, especially if genetic risk variants fall within them. Given these findings, the use of scATAC-seq on similar in vivo extracted tissues may require greater number of biological replicates and greater numbers of sequenced cells per replicate to find reproducible regions.
Comparisons of ATAC-seq peaks between developmental stage- and anatomical-site-matched mouse and human samples also revealed only partial regulatory region overlap (~25–45%) between species at orthologous loci or sequences. These findings indicate that in many cases, the presence of regulatory elements at a given locus or sequence in the mouse may not reflect the same regulatory use or potential in the orthologous human region (and vice versa). Therefore, investigations into the regulatory control of chondrocyte biology for the purpose of human disease studies should attempt to demonstrate one-to-one functional regulatory orthology when using the mouse as model. This can be accomplished by performing ATAC-seq on the homologous tissue of each species (e.g., the distal femur), then mapping sequencing reads, calling peaks or regulatory regions, and finally identifying regulatory regions that reside on the same orthologous sequence in each species. Moreover, if scATAC-seq is to be used to compare and characterize species differences in cellular heterogeneity in a given tissue, larger number of biological replicates and cells should be sequenced in order to better determine real (species-specific) biological differences versus those resulting from sampling issues and ascertainment bias. We also were able to perform evolutionary analyses on human/mouse sequences using the bulk ATAC-seq data and revealed that regulatory regions unique to distinct bone ends (e.g., the distal femur as compared to the proximal femur) display substantial evidence of ancient natural selection in humans and modern human sequence constraint (i.e., reductions of human genetic variation), reflecting functional demands of joint sites during the evolution of human bipedalism [2]. In turn, we also found evidence that this process of natural selection and constraint on human knee regulatory elements has shaped knee osteoarthritis disease prevalence and heritability (see below and [2]). These insights reflect the power of bulk ATAC-seq when used with other computational bioinformatic and comparative genomic methodologies.
Both general chondrocyte as well as bone-end-specific chondrocyte regulatory region sets also displayed important overlaps with bone-end-specific anatomy and disease risk. We found that chondrocyte ATAC-seq sets, which on average were 500 nucleotide in size, exhibited enrichments for cartilage diseases, most notably osteoarthritis. Moreover, knee-specific ATAC-seq regulatory regions showed stronger enrichments for knee osteoarthritis Genome-wide Association Study (GWAS) genetic variants, along with regulatory regions being located near genes with disease annotations of osteoarthritis and joint-related pathologies (see Richard et al., 2020) [2]. In the context of understanding disease-pathogenic mechanism, genetic variants located within knee regulatory regions were also likely to alter important transcription factor binding sites (e.g., KLF5, FOXP1/2, PITX1/2) involved in both chondrocyte biology as well as osteoarthritis. To carry out these analyses, computational transcription factor binding site analyses were performed on bulk ATAC-seq regulatory region sets for specific bone ends to identify enriched binding sites for chondrocytes and transcription factors that might promote anatomical site specificity or disease activity. The results of these studies, as reported in detail in Richard et al., 2020 [2], indicate that at the regulatory sequence level, as well as at the level of the proteins that bind to them, there are signals of osteoarthritis risk. In other words, the genetic circuitry involved in forming a knee also appears to have been disrupted to cause osteoarthritis later in life.
Importantly, these studies also inform on the potential use of scATAC-seq for addressing these specific questions. For example, since there are overall fewer copies of DNA present in selected single cells for scATAC-seq, and there is inherent per-cell data sparsity, in which only 1–10% of expected chromatin accessible regions are argued to be detectable by scATAC-seq compared to say 10–45% of expressed genes detected in single cells from scRNA-seq data [25, 26], we anticipate that these issues would drastically reduce the number of detected regulatory regions, called transcription factor binding motifs within peaks, and most importantly variants/motif intersections (i.e., given the lower numbers of called regions, important peak/variant intersections would be missed), thereby impacting transcription factor binding site detection for sets of motif-altering variants. Thus, the discrete, sparse, and noisy data obtained from scATAC-seq poses significant data analysis challenges in terms of understanding single-cell heterogeneity and regulation. Moreover, there is also the notion that the computational tools developed for bulk ATAC-seq will not be able to effectively analyze data obtained from scATAC-seq, and as a result, there is an increasing need for the development of new data analysis tools for single cell regulome analysis. Recently, several single-cell regulome analysis methods have been developed including chromVar, SCRAT, BROCKMAN, Dr.seq2, cicero, scasat, Destin, scABC, PRISM, cisTopic, and SCATE [27–37]. SCATE consistently outperformed all the other methods in terms of providing more accurate identification of cis-regulatory element activities and transcription factor binding sites using inherently sparser data from scATAC-seq [37]. However, all these mapping technologies can only get a snapshot of a cell regulatory activity at one time point, and they cannot accurately describe the continuous steady state regulatory activity in a cell. Future efforts will need to develop tools to study regulatory activities along continuous pseudotemporal trajectories at a single-cell resolution and also develop new methods that utilize improved regulatory activity estimation from scATAC-seq data to more accurately reconstruct gene regulatory networks. This will also be important in order to examine how genetic regulatory variants impact disease risk on the cell-type specific level.
In conclusion, insights gained from the ATAC-seq studies performed on chondrocytes acquired in bulk from developing mouse and human bone-ends will serve as a valuable resource for future scATAC-seq benchmark studies and could guide development of computational tools aimed at solving the remaining challenges associated with analysis of scATAC-seq datasets.
Single Cell CyTOF Analyses to Map Cellular Heterogeneity in Healthy and Diseased Skeletal Tissues
Mass cytometry was initially applied to immune cells in the blood that have a well-characterized cellular identity, lineage, and function [38]. CyTOF, is a mass-spectrometry-based method for single-cell detection of isotope-labeled antibodies [39, 40]. The use of rare metal isotopes instead of fluorophores greatly expands the number of antibodies that can be utilized thereby making it possible to map as many as 40 to 120 protein epitopes for a single cell [40]. This high-dimensional method is therefore extremely useful for proteomic analyses, and compliments single cell transcriptomic and epigenetic techniques. CyTOF can also provide a snapshot of post-translational regulation and active signaling events at a single cell level. Since the protein panel to be interrogated is preselected for CyTOF studies, this approach is limited as compared to the unbiased genome-wide interrogation that is made possible by single cell transcriptomics. It is however possible to study a large number of cells with CyTOF (a few millions) in a cost-effective manner as compared to the higher costs associated with scRNA-seq that prohibit studying more than a few thousand cells at best. CyTOF studies therefore have an advantage in terms of identifying and validating rare cell populations. The availability of the multiple and complimentary single cell techniques for gene expression and proteomic analyses now makes it possible to really identify the single cell landscape of skeletal tissues and to study the diverse cell populations that drive development, regeneration, and disease.
Recent studies have now begun to expand the repertoire to other tissues including a few skeletal tissues [41–43]. In recent studies, healthy and osteoarthritic cartilage samples were profiled using mass cytometry to establish a single-cell atlas for cartilage [3]. In contrast to readily accessible blood cells, the method for isolating chondrocytes from the cartilage tissue is often tricky due to the abundance of extracellular matrix (ECM) surrounding the chondrocytes. After testing multiple protease digestion strategies, we utilized a collagenase-based protocol for optimal isolation and later staining of chondrocytes. After a detailed study of the literature and some preliminary analyses, a panel of 33-markers were labeled with rare-earth metal isotopes and optimized for profiling chondrocytes. This panel included cell surface receptors, adhesion molecules, signaling mediators and cell cycle and transcription factors that are known to be important for cartilage homeostasis.
Although cartilage is a relatively simple tissue, with only one cell type, chondrocytes, being embedded in its secreted ECM, it has a zonal organization that differentiates cellular identity. Multiple studies have interrogated the presence of stem and progenitor like cells in adult cartilage but the precise identity and characteristics of these cells remain controversial as opposed to the unequivocal data in support of muscle or neural stem cells, for example [44]. Using putative markers for cartilage progenitor cells (CPC) in Sox9 and CD44 positive cells, including CD105/CD90/Notch/Stro/CD151, distinct subpopulations were identified [3]. These data suggest the existence of multiple CPC subsets. It will be interesting in future studies, however, to isolate and study the precise function of each of these subsets especially their contribution to cartilage regeneration and repair.
Single cell CyTOF analyses further revealed the presence of various “rare” cell populations that constituted only a small percentage (2–5%) of the total chondrocytes [3]. These populations were however persistent in all the patients profiled and were reminiscent of other small populations like senescent cells that are found in aged cartilage. Previous studies have demonstrated that senescent cells secrete a specific senescence-associated secretory phenotype that can increase tissue aging and damage. Removal of the senescent cells led to cartilage protection in a mouse model of post-traumatic osteoarthritis, showing the significance of rare disease-associated cell populations [45]. The mass cytometry data revealed two novel inflammation modulating subpopulations in osteoarthritic cartilage—an inflammation amplifying (Inf-A) chondrocyte population and an inflammation dampening (Inf-D) population [3]. Inhibition of the Inf-D population led to an overall decrease in the inflammatory secretome of osteoarthritic chondrocytes. However, a pharmacological strategy targeting both Inf-A and Inf-D cells showed a significantly higher decrease in inflammation in osteoarthritic chondrocytes, highlighting the relevance as well as the cross-talk between small cellular sub-populations in osteoarthritis. Histologically, a few atrophic cells can be identified in OA cartilage along with some expanded clusters that likely result from proliferation although it is difficult to ascertain the timescale of formation of these clusters. For the CyTOF studies, we explicitly sorted live cells, hence putatively atrophic cells would have been excluded. Insignificant numbers of Ki67+ cells were observed, hence actively proliferating cells were negligible in OA chondrocytes.
Finally, the single cell proteomic data allowed for the stratification of osteoarthritis patients into three groups based on the relative proportions of inflammatory to regenerative cells [3]: increased in osteoarthritis; unchanged between osteoarthritis and normal; decreased in osteoarthritis. The different proportions of chondrocyte subtypes likely reflect the variable degree of degeneration associated with each osteoarthritis patient as well as their immune and metabolic profiles. Understanding cartilage (and other joint tissues in the future) at a single cell level can therefore provide a higher resolution and novel insights into the onset and progression of osteoarthritis pathology.
In summary, single cell proteomic studies have a great potential to provide a deeper understanding of healthy and diseased skeletal tissues as well as identify the patient-to-patient differences that may be critical in determining precision medicine approaches for effective therapies.
Conclusions
Single cell “omics” technologies allow researchers to identify rare and transient cell populations in normal and diseased tissues. These single cell technologies are being increasingly embraced by the musculoskeletal community. Successful completion of these projects requires teams of basic scientists, clinicians, and bioinformaticians because tools are advancing rapidly. While becoming an expert on all new tools is challenging, it is of paramount significance that bench scientists and clinicians or surgeons in the musculoskeletal community understand how single cell technologies can be used to answer research questions and identify new treatment targets or avenues.
This review article summarizes the work of three teams who used different yet complementary aspects of single cell “omics” to understand cartilage development and the osteoarthritic tissues. Wu et al. showed that scRNA-seq analysis can unravel the cellular heterogeneity and trajectories of lineage specification of hiPSC to chondrocytes [1]. They also identified ligand–receptor pairs that modulate chondrocyte lineage commitment. The Capellini team described how ATAC-seq and RNA-seq data can be combined to understand evolution of gene signatures during cartilage development and disease [2]. Finally, the work of the Bhutani team reviewed how high-content single-cell analysis (CyTOF, mass cytometry) can reveal rare cell populations in osteoarthritic cartilage [3]. These three studies highlight the emerging role of single cell “omics” technologies in musculoskeletal research but are just a few examples of what has been done. Other single transcriptomic studies have been published [46–48] and we anticipate many more in the future.
The generation of these large single cell “omics” datasets has revealed many new insights into musculoskeletal biology and disease pathogenesis, but translation into advanced diagnostic tools and treatments has yet to be realized. The size and complexity of the datasets and bioinformatics processes pose many challenges that must be overcome to reach this goal. One challenge is the integration of all omics data into a user-friendly interface that is easy to access and available to the entire research community. With this goal in mind, a working group from the International Federation of Musculoskeletal Research Societies (IFMRS) has teamed with the Broad Institute to generate a Musculoskeletal Knowledge Portal (mskkp.org) [49]. The majority of the data on the MSKKP are genetic association studies at this time, but work is ongoing to integrate transcriptomic, epigenomic, and proteomic data to the genetic studies on a single multi-omics platform. A second challenge of having many large datasets is reconciling the results from different studies, and unlimited combinations of experimental designs and analysis tools. Machine learning and artificial intelligence tools will be needed to decipher the similarities and difference of rare and transient or intermediate cell states over time, and to identify disease-causing genes that could be targets of intervention.
In conclusion, rapid advances in single cell sequencing and proteomic analyses are revealing new and exciting information on musculoskeletal cell development and disease pathogenesis. This review summarizes some of the first high impact work in cartilage development and osteoarthritis. The future will be exciting as the data sets are reconciled and new therapeutic targets are identified for musculoskeletal conditions that cause disability across the globe.
Acknowledgements
This work was supported by grants from the National Institutes of Health (AR075899 to CLW, AG15768 to F Guilak, AR070139 to TC, AR070864 and AR070865 to NB) and National Science Foundation (Graduate Research Fellowship to F. Grandi).
Footnotes
Conflict of Interest MFR, TC, PM, FG, NB, and JJW declare no conflicts of interest.
CLW, ARD, and FG have a patent pending on compositions and methods discussed within.
Human and Animal Rights and Informed Consent This article does not contain any primary studies with human or animal subjects performed by any of the authors.
References
- 1.Wu CL, Dicks A, Steward N, Tang R, Katz DB, Choi YR, et al. Single cell transcriptomic analysis of human pluripotent stem cell chondrogenesis. Nat Commun. 2021;12(1):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Richard D, Liu Z, Cao JX, Kiapour AM, Willen J, Yarlagadda S, et al. Evolutionary selection and constraint on human knee chondrocyte regulation impacts osteoarthritis risk. Cell. 2020;181(2): 362–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grandi FC, Baskar R, Smeriglio P, Murkherjee S, Indelli PF, Amanatullah DF, et al. Single-cell mass cytometry reveals cross-talk between inflammation-dampening and inflammation-amplifying cells in osteoarthritic cartilage. Sci Adv. 2020;6(11): eaay5352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Adkar SS, Wu CL, Willard VP, Dicks A, Ettyreddy A, Steward N, et al. Step-wise chondrogenesis of human induced pluripotent stem cells and purification via a reporter allele generated by CRISPRCas9 genome editing. Stem Cells. 2019;37(1):65–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Valihrach L, Androvic P, Kubista M. Platforms for single-cell collection and analysis. Int J Mol Sci. 2018;19(3):807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol. 2018;36(5):411–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cao J, Spielmann M, Qiu X, Huang X, Ibrahim DM, Hill AJ, et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature. 2019;566(7745):496–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Becht E, McInnes L, Healy J, Dutertre CA, Kwok IWH, Ng LG, et al. Dimensionality reduction for visualizing single-cell data using UMAP. Nat Biotechnol. 2019;37:38–44. [DOI] [PubMed] [Google Scholar]
- 9.Huang d W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. [DOI] [PubMed] [Google Scholar]
- 10.Hrvatin S, Hochbaum DR, Nagy MA, Cicconet M, Robertson K, Cheadle L, et al. Single-cell analysis of experience-dependent transcriptomic states in the mouse visual cortex. Nat Neurosci. 2018;21(1):120–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ramilowski JA, Goldberg T, Harshbarger J, Kloppmann E, Lizio M, Satagopam VP, et al. A draft network of ligand-receptor-mediated multicellular signalling in human. Nat Commun. 2015;6:7866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mardones MD, Andaur GA, Varas-Godoy M, Henriquez JF, Salech F, Behrens MI, et al. Frizzled-1 receptor regulates adult hippocampal neurogenesis. Mol Brain. 2016;9:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lie DC, Colamarino SA, Song HJ, Desire L, Mira H, Consiglio A, et al. Wnt signalling regulates adult hippocampal neurogenesis. Nature. 2005;437(7063):1370–5. [DOI] [PubMed] [Google Scholar]
- 14.Gu Z, Gu L, Eils R, Schlesner M, Brors B. Circlize implements and enhances circular visualization in R. Bioinformatics. 2014;30(19): 2811–2. [DOI] [PubMed] [Google Scholar]
- 15.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11): 2498–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bindea G, Mlecnik B, Hackl H, Charoentong P, Tosolini M, Kirilovsky A, et al. ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics. 2009;25(8):1091–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morris JH, Apeltsin L, Newman AM, Baumbach J, Wittkop T, Su G, et al. clusterMaker: a multi-algorithm clustering plugin for Cytoscape. BMC Bioinformatics. 2011;12:436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barthelemy M Betweenness centrality in large complex networks. Eur Phys J B. 2004;38(2):163–8. [Google Scholar]
- 20.Karsenty G, Wagner EF. Reaching a genetic and molecular understanding of skeletal development. Dev Cell. 2002;2(4):389–406. [DOI] [PubMed] [Google Scholar]
- 21.Hojo H, McMahon AP, Ohba S. An emerging regulatory landscape for skeletal development. Trends Genet. 2016;32(12):774–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Buenrostro JD, Wu B, Chang HY, Greenleaf WJ. ATAC-seq: a method for assaying chromatin accessibility genome-wide. Curr Protoc Mol Biol. 2015;109:21 9 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleo-some position. Nat Methods. 2013;10(12):1213–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li Q, Brown JB, Huang H, Bickel PJ. Measuring reproducibility of high-throughput experiments. Ann Appl Stat. 2011;5(3): 1752–79. [Google Scholar]
- 25.Mereu E, Lafzi A, Moutinho C, Ziegenhain C, McCarthy DJ, Alvarez-Varela A, et al. Benchmarking single-cell RNA-sequencing protocols for cell atlas projects. Nat Biotechnol. 2020;38(6): 747–55. [DOI] [PubMed] [Google Scholar]
- 26.Ding J, Adiconis X, Simmons SK, Kowalczyk MS, Hession CC, Marjanovic ND, et al. Systematic comparison of single-cell and single-nucleus RNA-sequencing methods. Nat Biotechnol. 2020;38(6):737–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schep AN, Wu B, Buenrostro JD, Greenleaf WJ. chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat Methods. 2017;14(10):975–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ji Z, Zhou W, Ji H. Single-cell regulome data analysis by SCRAT. Bioinformatics. 2017;33(18):2930–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Boer CG, Regev A. BROCKMAN: deciphering variance in epigenomic regulators by k-mer factorization. BMC Bioinformatics. 2018;19(1):253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao C, Hu S, Huo X, Zhang Y. Dr.seq2: a quality control and analysis pipeline for parallel single cell transcriptome and epigenome data. PLoS One. 2017;12(7):e0180583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pliner HA, Packer JS, McFaline-Figueroa JL, Cusanovich DA, Daza RM, Aghamirzaie D, et al. Cicero predicts cis-regulatory DNA interactions from single-cell chromatin accessibility data. Mol Cell. 2018;71(5):858–71. e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baker SM, Rogerson C, Hayes A, Sharrocks AD, Rattray M. Classifying cells with Scasat, a single-cell ATAC-seq analysis tool. Nucleic Acids Res. 2019;47(2):e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Urrutia E, Chen L, Zhou H, Jiang Y. Destin: toolkit for single-cell analysis of chromatin accessibility. Bioinformatics. 2019;35(19): 3818–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zamanighomi M, Lin Z, Daley T, Chen X, Duren Z, Schep A, et al. Unsupervised clustering and epigenetic classification of single cells. Nat Commun. 2018;9(1):2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cai S, Georgakilas GK, Johnson JL, Vahedi G. A cosine similarity-based method to infer variability of chromatin accessibility at the single-cell level. Front Genet. 2018;9:319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bravo Gonzalez-Blas C, Minnoye L, Papasokrati D, Aibar S, Hulselmans G, Christiaens V, et al. cisTopic: cis-regulatory topic modeling on single-cell ATAC-seq data. Nat Methods. 2019;16(5): 397–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ji Z, Zhou W, Hou W, Ji H. Single-cell ATAC-seq signal extraction and enhancement with SCATE. Genome Biol. 2020;21(1):161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bendall SC, Nolan GP, Roederer M, Chattopadhyay PK. A deep profiler’s guide to cytometry. Trends Immunol. 2012;33(7):323–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bendall SC, Simonds EF, Qiu P, el AD A, Krutzik PO, Finck R, et al. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science. 2011;332(6030):687–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hartmann FJ, Simonds EF, Vivanco N, Bruce T, Borges L, Nolan GP, et al. Scalable conjugation and characterization of Immunoglobulins with stable mass isotope reporters for single-cell mass cytometry analysis. Methods Mol Biol. 1989;2019:55–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Severe N, Karabacak NM, Gustafsson K, Baryawno N, Courties G, Kfoury Y, et al. Stress-induced changes in bone marrow stromal cell populations revealed through single-cell protein expression mapping. Cell Stem Cell. 2019;25(4):570–83. e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Porpiglia E, Samusik N, Ho ATV, Cosgrove BD, Mai T, Davis KL, et al. High-resolution myogenic lineage mapping by single-cell mass cytometry. Nat Cell Biol. 2017;19(5):558–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Croft AP, Campos J, Jansen K, Turner JD, Marshall J, Attar M, et al. Distinct fibroblast subsets drive inflammation and damage in arthritis. Nature. 2019;570(7760):246–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jiang Y, Tuan RS. Origin and function of cartilage stem/progenitor cells in osteoarthritis. Nat Rev Rheumatol. 2015;11(4):206–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jeon OH, Kim C, Laberge RM, Demaria M, Rathod S, Vasserot AP, et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat Med. 2017;23(6):775–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang F, Wei K, Slowikowski K, Fonseka CY, Rao DA, Kelly S, et al. Defining inflammatory cell states in rheumatoid arthritis joint synovial tissues by integrating single-cell transcriptomics and mass cytometry. Nat Immunol. 2019;20(7):928–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu Z, Shou L, Wang J, Xu X. Identification of the key gene and pathways associated with osteoarthritis via single-cell RNA sequencing on synovial fibroblasts. Medicine (Baltimore). 2020;99(33):e21707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ji Q, Zheng Y, Zhang G, Hu Y, Fan X, Hou Y, et al. Single-cell RNA-seq analysis reveals the progression of human osteoarthritis. Ann Rheum Dis. 2019;78(1):100–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kiel DP, Kemp JP, Rivadeneira F, Westendorf JJ, Karasik D, Duncan E, et al. The musculoskeletal knowledge portal: making Omics data useful to the broader scientific community. J Bone Miner Res. 2020;35(9):1626–33. [DOI] [PMC free article] [PubMed] [Google Scholar]