

Abstract
Among the inherited ion channelopathies associated with potentially life-threatening ventricular arrhythmia syndromes in nominally structurally normal hearts are the J wave syndromes, which include the Brugada (BrS) and early repolarization (ERS) syndromes. These ion channelopathies are responsible for sudden cardiac death (SCD), most often in young adults in the third and fourth decade of life. Our principal goal in this review is to briefly outline the clinical characteristics, as well as the molecular, ionic, cellular, and genetic mechanisms underlying these primary electrical diseases that have challenged the cardiology community over the past two decades. In addition, we discuss our recently developed whole-heart experimental model of BrS, providing compelling evidence in support of the repolarization hypothesis for the BrS phenotype as well as novel findings demonstrating that voltage-gated sodium and transient outward current channels can modulate each other’s function via trafficking and gating mechanisms with implications for improved understanding of the genetics of both cardiac and neuronal syndromes.
Keywords: Sudden cardiac death, Brugada syndrome, Early Repolarization syndrome, cardiac arrhythmias, inherited cardiac arrhythmia syndromes, Langendorff-perfused hearts
Introduction
Prominent J waves in the surface electrocardiogram (ECG) and their associated risk for the development of life-threatening cardiac arrhythmias have been documented for at least 100 years in clinical conditions involving hypothermia, hypercalcemia, and acute myocardial ischemia. More recently, accentuated J waves have been associated with inherited, potentially life-threatening cardiac arrhythmia disorders including Brugada (BrS) and Early Repolarization (ERS) syndromes, the so-called J Wave Syndromes (JWS). In the case of BrS, a clinical entity associated with sudden cardiac death (SCD) first introduced by Pedro and Josep Brugada in 1992 (1), the J waves are usually limited to the right precordial leads V1-V3.
Diagnosis of JWS
BrS and ERS are both associated with vulnerability to arrhythmic SCD (pVT and VF) (1-3) in children and young adults with apparently structurally normal hearts and, rarely, to sudden infant death syndrome (4, 5).
In BrS, three distinct variations of ST segment elevation have been described (BrS Types 1, 2 and 3) (6, 7). The only form diagnostic of BrS is a Type 1 (“coved type”) ST segment elevation characterized by an ST segment elevation of ≥2 mm (0.2 mV) in ≥1 right precordial leads positioned in the 4th intercostal space (V1 and/or V2) or in more cranial positions (2nd or 3rd intercostal space). A Type 2 ST segment elevation, described as a “saddle-back” configuration with an ST segment elevation of ≥0.5 mm (commonly ≥2mm in V2) in ≥1 right precordial leads (V1-V3) and a Type 3 ST segment elevation, characterized by a “saddle-back or coved type” appearance with an ST segment elevation of <1 mm, are NOT diagnostic of BrS unless converted to a Type 1 with fever or sodium drug challenge (Figure 1). The cardiac region most affected in BrS patients is the anterior aspects of the right ventricular outflow tract (RVOT), which is facing the right precordial leads (V1-V3).
Figure 1:

Three types of ST segment elevation associated with Brugada syndrome. Only Type 1 is diagnostic of BrS. Reproduced from (111), with permission.
Early repolarization pattern (ERP) is often encountered in apparently healthy individuals, particularly in young males, black individuals, and athletes. When ERP is associated with VT/VF, the clinical condition is referred to as early repolarization syndrome (ERS).
Early repolarization is recognized with appearance of 1) an end QRS notch (J wave) or slur on the downslope of a prominent R wave (with or without ST segment elevation). The onset of the J wave is referred to as J0; 2) a distinct J wave with a peak (designated as JP) ≥ 0.1 mV in two or more contiguous ECG leads, excluding leads V1-V3; and 3) a QRS duration (measured in leads in which a notch or slur is absent) must be < 120 ms. The end of the J wave or that of the QRS slur or notch is designated as JT (Figure 2) (8). Thus, ERP is characterized by distinct J waves, J0 elevation, notch or slur of the terminal part of the QRS and ST-segment or JT elevation in the lateral (Type I), infero-lateral (Type II) or in infero-lateral + (anterior) right precordial leads (Type III).
Figure 2:

Different manifestations of Early Repolarization. A. The J wave may be distinct or appear as a slur. In the latter case, part of the J wave is buried inside the QRS, resulting in an elevation of J0. Patients with a very prominent J waves have a worse prognosis. B. The ST segment may be upsloping, horizontal or descending. Horizontal and descending ST segments are associated with a worse prognosis. Reproduced from (111), with permission.
Pharmacological provocative tests
When diagnosis is suspected, a provocative drug test using a sodium channel blocker is recommended, which involves administration of either ajmaline (1 mg/kg over 10 min, IV), flecainide (2 mg/kg over 10 min, IV, or 200-300 mg over more than 1 h when administrated orally), procainamide (10 mg/kg over 10 min, IV), or pilsicainide (1 mg/kg over 10 min, IV) (www.brugadadrugs.org). The test is considered positive if a Type 1 ECG pattern is obtained (9).
In contrast to the effect of sodium channel blockers to unmask or exacerbate the BrS Type 1 phenotype, these drugs have been shown to reduce the amplitude of the J wave in patients with ERS (10). However, Nakagawa et al. reported that J waves recorded using unipolar LV-anterior epicardial leads in ERS patients are augmented following provocative drug testing, while the J waves recorded in the lateral precordial leads are diminished indicating that the J waves in the surface leads are reduced because they are engulfed by the wider QRS (10, 11). Further support for the notion that the pathophysiological basis of ERS and BrS are closely related derives from reports of cases in which ERS transitions into ERS plus BrS phenotypes (12, 13).
Tables 1A and 1B show the Shanghai Score system proposed for the diagnosis of BrS and ERS (9). Validation of the scoring system for BrS was performed by Kawada and co-workers using 393 patients evaluated for BrS. The authors concluded that the Shanghai Score System is useful for diagnosis as well as risk stratification of BrS (14).
Table 1A.
Shanghai Score system for Diagnosis of Brugada Syndrome
| Points | |
|---|---|
| I. ECG (12-Lead/Ambulatory) | |
| A. Spontaneous type 1 Brugada ECG pattern at nominal or high leads | 3.5 |
| B. Fever-induced type 1 Brugada ECG pattern at nominal or high leads | 3 |
| C. Type 2 or 3 Brugada ECG pattern that converts with provocative drug challenge | 2 |
| *Only award points once for highest score within this category. One item from this category must apply. | |
| II. Clinical History* | |
| A. Unexplained cardiac arrest or documented VF/polymorphic VT | 3 |
| B. Nocturnal agonal respirations | 2 |
| C. Suspected arrhythmic syncope | 2 |
| D. Syncope of unclear mechanism/unclear etiology | 1 |
| E. Atrial flutter/fibrillation in patients < 30 years without alternative etiology | 0.5 |
| *Only award points once for highest score within this category. | |
| III. Family History | |
| A. First- or second-degree relative with definite BrS | 2 |
| B. Suspicious SCD (fever, nocturnal, Brugada aggravating drugs) in a first- or second-degree relative | 1 |
| C. Unexplained SCD < 45 years in first- or second- degree relative with negative autopsy | 0.5 |
| *Only award points once for highest score within this category. | |
| IV. Genetic Test Result | |
| A. Probable pathogenic mutation in BrS susceptibility gene | 0.5 |
| Score (requires at least 1 ECG finding) | |
| ≥3.5 points: Probable/definite BrS | |
| 2–3 points: Possible BrS | |
| < 2 points: Nondiagnostic | |
BrS = Brugada syndrome; SCD = sudden cardiac death; VF = ventricular fibrillation; VT = ventricular tachycardia.
Reproduced from (111), with permission.
Table 1B.
Shanghai Score system for Diagnosis of Early Repolarization Syndrome
| Points | |
|---|---|
| 1. Clinical History | |
| A. Unexplained cardiac arrest, documented VF or polymorphic VT | 3 |
| B. Suspected arrhythmic syncope | 2 |
| C. Syncope of unclear mechanism/unclear etiology | 1 |
| *Only award points once for highest score within this category | |
| II. Twelve-Lead ECG | |
| A. ER ≥0.2 mV in ≥2 inferior and/or lateral ECG leads with horizontal/descending ST segment | 2 |
| B. Dynamic changes in J-point elevation (≥0.1 mV) in ≥2 inferior and/or lateral ECG leads | 1.5 |
| C. ≥0.1 mV J-point elevation in at least 2 inferior and/or lateral ECG leads | 1 |
| *Only award points once for highest score within this category | |
| III. Ambulatory ECG Monitoring | |
| A. Short-coupled PVCs with R on ascending limb or peak of T wave | 2 |
| IV. Family History | |
| A. Relative with definite ERS | 2 |
| B. ≥2 first-degree relatives with a II.A. ECG pattern | 2 |
| C. First-degree relative with a II.A. ECG pattern | 1 |
| D. Unexplained sudden cardiac death <45 years in a first- or second-degree relative | 0.5 |
| *Only award points once for highest score within this category | |
| V. Genetic Test Result | |
| A. Probable pathogenic ERS susceptibility mutation | 0.5 |
| Score (requires at least 1 ECG finding) | |
| ≥5 points: Probable/definite ERS | |
| 3–4.5 points: Possible ERS | |
| <3 points: Nondiagnostic |
ER = early repolarization; ERS = early repolarization syndrome; PVC = premature ventricular contraction; VF = ventricular fibrillation; VT = ventricular tachycardia.
Reproduced from (111), with permission.
Risk Assessment
J waves with rapidly ascending ST segments particularly in the lateral ECG leads have a high prevalence but are associated with a relatively low arrhythmic risk. In contrast, J waves appearing in the inferior leads or infero-lateral leads are associated with a much higher risk, particularly when displaying a flat or descending ST segment (15). J waves appearing globally in the ECG have a very low prevalence, but are associated with a very high level of arrhythmic risk as are subjects resuscitated from cardiac arrest (9). The presence of a notched or fragmented QRS is likewise associated with high risk for development of life-threatening arrhythmias. (16-19) Figure 3 summarizes the prevalence and arrhythmic risk associated with the various presentations of the electrocardiographic J wave and various clinical manifestations of Brugada and Early Repolarization syndromes (9).
Figure 3.

Prevalence and arrhythmic risk associated with the appearance of ECG J waves and clinical manifestations of Brugada and Early Repolarization syndromes. The yellow highlighted region estimates the prevalence of the J wave syndromes. Reproduced from (111), with permission.
Aizawa et al. reported that the response of J wave amplitude to atrial pacing is different between idiopathic ventricular fibrillation (IVF) and non-IVF patients. The authors suggested the presence of different mechanisms for the genesis of J waves: early repolarization in IVF patients and conduction delay in non-IVF patients.(20) Although the presence of sodium or calcium channel blockers might be expected to yield such results, the authors indicate that none of the participants in the study were administered any medication.
Similarities and difference between BrS and ERS
BrS and ERS have several clinical similarities, suggesting a closely related pathophysiology (21, 22, 23 ). Males predominate is observed in both syndromes (24, 25). Patients can remain totally asymptomatic until presenting with cardiac arrest. The highest incidence of VF or SCD occurs in the third decade of life in both syndromes (26). In general, the appearance of accentuated J waves and ST segment elevation is in association with bradycardia or pauses (27, 28); accordingly, VF often occurs during sleep or at a low level of physical activities (10, 29).
ERS and BrS also share similarities to pharmacological therapy. Electrical storms can be suppressed with β-adrenergic agonists (30-33). Chronic oral administration of quinidine (34, 35), bepridil (32), denopamine, (30, 36) and cilostazol (30, 37, 38) are reported to prevent VT/VF in both syndromes, secondary to inhibition of Ito and/or augmentation of ICa (2, 39, 40).
Differences between the two syndromes include: 1) the region of the heart most affected (RVOT in BrS vs. inferior LV in ERS); 2) the presence of subtle structural abnormalities in BrS, which as yet have not been reported in ERS (41); 3) the incidence of late potentials in signal-averaged ECGs (SAECG): 60% in BrS / 7% in ERS (10); 4) greater elevation of JO, JP or JT (ST segment elevation) in response to sodium channel blockers in BrS vs. ERS; 5) higher prevalence of atrial fibrillation in BrS vs. ERS (42) and differences in response to hypothermia and fever. Patients with ERS are at greater risk of VF during hypothermia(43-47) as well as in the event of an acute myocardial infarction.(48) BrS patients are known to be at greater risk of VF in febrile states(49, 50) as well as when accompanied by an ER pattern in the infero-lateral leads.(51) Available data suggest that mild therapeutic hypothermia to a temperature of 34°C can be used safely in cases of Brugada syndrome.(52, 53)
Genetics
ERS and BrS have been associated with variants in 10 and 23 genes, respectively (Table 2). The gene most often associated with BrS is SCN5A, accounting for 11-28% of cases depending largely on geographic location. Over 300 BrS-related variants in SCN5A, the gene encoding the cardiac Na channel, have been described (54, 55). Loss-of-function mutations in SCN5A contribute to the development of both BrS and ERS, as well as to various conduction diseases, Lenegre disease and Sick Sinus Syndrome.
Table 2:
Gene defects associated with Early Repolarization (ERS) and Brugada (BrS) syndromes.
| Genetic Defects Associated with ERS | ||||
|---|---|---|---|---|
| Locus | Gene/Protein | Ion Channel | ||
| ERS1 | 12p11.23 | KCNJ8, Kir6.1 | ↑IK-ATP | |
| ERS2 | 12p13.3 | CACNA1C, Cav1.2 | ↓ ICa | |
| ERS3 | 10p12.33 | CACNB2b, Cavß2b | ↓ ICa | |
| ERS4 | 7q21.11 | CACNA2D1, Cava2d1 | ↓ ICa | |
| ERS5 | 12p12.1 | ABCC9, SUR2A | ↑ IK-ATP | |
| ERS6 | 3p21 | SCN5A, Nav1.5 | ↓ INa | |
| ERS7 | 3p22.2 | SCN10A, Nav1.8 | ↓ INa | |
| ERS8 | 7q35 | KCNH2, hERG | ↑ IKr | |
| ERS9 | 19q13.1 | SCN1Bβ, Navβ1 | ↑ Ito | |
| ERS10 | 1p13.2 | KCND3, Kv4.3 | ↑ Ito | |
| Genetic Defects Associated with BrS | ||||
| Locus | Gene/Protein | Ion Channel | ||
| BrS1 | 3p21 | SCN5A, Nav1.5 | ↓ INa | |
| BrS2 | 3p24 | GPD1L | ↓ INa | |
| BrS3 | 12p13.3 | CACNA1C, Cav1.2 | ↓ ICa | |
| BrS4 | 10p12.33 | CACNB2b, Cavß2b | ↓ ICa | |
| BrS5 | 19q13.1 | SCN1B, Navß1 | ↓ INa | |
| BrS6 | 11q13-14 | KCNE3, MiRP2 | ↑ Ito | |
| BrS7 | 11q23.3 | SCN3B, Navß3 | ↓ INa | |
| BrS8 | 12p11.23 | KCNJ8, Kir6.1 | ↑ IK-ATP | |
| BrS9 | 7q21.11 | CACNA2D1, Cav a2d1 | ↓ ICa | |
| BrS10 | 1p13.2 | KCND3, Kv4.3 | ↑ Ito | |
| BrS11 | 17p13.1 | RANGRF, MOG1 | ↓ INa | |
| BrS12 | 3p21.2-p14.3 | SLMAP | ↓ INa | |
| BrS13 | 12p12.1 | ABCC9, SUR2A | ↑ IK-ATP | |
| BrS14 | 11q23 | SCN2B, Navß2 | ↓ INa | |
| BrS15 | 12p11 | PKP2, Plakophillin-2 | ↓ INa | |
| BrS16 | 3q28 | FGF12, FHAF1 | ↓ INa | |
| BrS17 | 3p22.2 | SCN10A, Nav1.8 | ↓ INa | |
| BrS18 | 6q | HEY2 (transcriptional factor) | ↑ INa | |
| BrS19 | 1p36.3 | KCNAB2, Kvβ2 | ↑ Ito | |
| BrS20 | 7q31.1 | TMEM168, trans-membrane | ↓ INa | |
| BrS21 | 3q29 | SLG1/SAP97 | ↑ Ito | |
| BrS22 | 17q12 | TCAP- Z-disk cytoskeletal protein | ↓ INa | |
| BrS23 | 1p13.3 | GSTM3 - Glutathione S-transferase transferase | ↓ INa | |
Listed in Chronological order of discovery. Modified from (133), with permission.
Juang and co-workers, using genome-wide copy number variation analysis, recently identified a 5.5 Kb deletion in the GSTM3 gene in 23% of SCN5A negative probands (vs. 1% in controls). The deletion results in a loss of function LOF of INa. This finding awaits confirmation but represents a potentially significant cause of BrS.(56) Recent studies have also uncovered variants in telethonin (TCAP), a Z-disk protein that maintains cytoskeletal integrity and participates in various signaling pathways in cardiomyocytes, associate with cases of BrS, ARVC and other J wave syndrome phenotypes. These variants were found to cause reduced expression of INa (57).
New susceptibility genes proposed and awaiting confirmation include the Transient Receptor Potential Melastatin Protein 4 gene (TRPM4)(58) and the KCND2 gene. Variants in KCNH2, KCNE5 and SEMA3A although not causative, are thought to be capable of modulating the substrate for the development of BrS (59-63). Loss-of-function mutations in HCN4 have been associated with BrS but may be modulatory by acting to unmask BrS by reducing heart rate (64).
Clatot et al. recently reported the interesting discovery that NaV and KV4.3 channels can modulate each other’s function via trafficking and gating mechanisms. This finding has important implications for improved understanding of both cardiac and neuronal syndromes. SCN5A variants associated with BrS were found to not only reduce INa, but to increase Ito. Moreover, BrS and spinocerebellar ataxia (SCA19/22) KCND3 variants associated with a gain of function of Ito, were found to significantly reduce INa, whereas the SCA19/22 KCND3 variants associated with a loss of function (LOF) of Ito significantly increased INa. Auxiliary subunits Navβ1, MiRP3 and KChIP2 also modulated INa/Ito balance. Co-immunoprecipitation and Duolink studies suggested that the two channels interact within the intracellular compartments and biotinylation showed that LOF SCN5A variants can increase Kv4.3 cell-surface expression (65).
Recent reappraisals of reported BrS-susceptibility genes whether by application of strict ClinGen evidence-based gene curation framework (66) or by means of testing for the increased burden of rare genetic variants in BrS patients compared to controls (67) have disputed causality of variants in all of the genes thus far associated with BrS, with the exception of SCN5A. The sole gene unequivocally implicated in causing BrS is SCN5A. Moreover, the often sporadic presentation of the disorder and the low disease penetrance in families with rare SCN5A variants, as well as the observation of phenotype-positive genotype-negative individuals in such families, suggests that BrS may be a disease with more complex genetics and that inheritance is likely not Mendelian. Collectively, these data suggest that the vulnerability to VF and SCD in BrS patients may not be due to a single mutation but rather to inheritance of multiple BrS-susceptibility variants (oligogenic) acting in concert through one or more mechanistic pathways.(68) Also contributing to the multifactorial phenotypic expression of the syndrome are the modulatory effects of hormonal factors including testosterone (69, 70) and thyroxine (71) as well as other environmental factors and structural remodeling involving development of fibrosis (41, 72).
Ionic and cellular mechanisms underlying the JWS
Experimental evidence indicates that the electrocardiographic J wave is the expression of a transmural voltage gradient of the action potential (AP) notch due to a prominent transient outward current (Ito) in ventricular epicardium but not endocardium (22, 73). The J wave and the underlying transmural gradient of the AP notch are much more prominent in the right vs. left ventricle, particularly in the region of the RVOT, because of the more prominent Ito-mediated AP notch in right ventricular epicardium (74). This distinction explains why BrS is a right ventricular disease. An end of QRS notch, resembling a J wave, has also been proposed to be due to intramural conduction delays. The distinction can be made on the basis of their response to heart rate, with the latter showing accentuation at faster rates (75, 76).
The J wave is thus a reflection of early repolarization of the epicardial action potential in both right and left ventricles. It can manifest as a J point elevation (Fig 4A), a distinct J wave (Fig. 4B), a slurring of the terminal portion of the QRS (Fig. 4C), a distinct J wave with an ST segment elevation (Fig. 4D) or as a gigantic J wave appearing as an ST segment elevation (due to the influence of the Ito agonist NS5806; Fig. 4E). It is under these last set of conditions that we see the development of phase 2 reentry and polymorphic VT (Fig. 4F).
Figure 4.
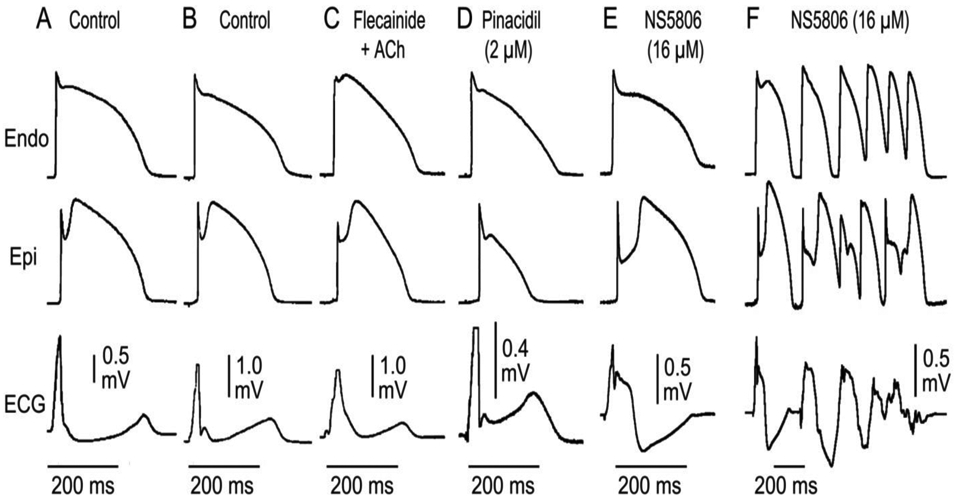
Electrocardiographic manifestations of early repolarization recapitulated in coronary perfused canine ventricular wedge preparations. Each panel shows action potential recordings from epicardium (Epi) and endocardium (Endo) and a transmural ECG. J waves are a reflection of early repolarization of ventricular epicardium and can manifest as a J point elevation (A), a distinct J wave (B), a slurring of the terminal part of the QRS recorded following exposure to flecainide + acetylcholine (C ), a distinct J wave with an ST segment recorded following exposure to pinacidil (D), a gigantic J wave appearing as an ST segment elevation recorded following exposure to the Ito agonist NS5806 (E). It is under these conditions that we observe the development of polymorphic VT (F). Modified from (134) with permission.
The cellular mechanisms underlying the J-wave syndromes have long been a matter of debate. (17, 77) Two principle hypotheses have been advanced in the case of BrS: 1) The repolarization hypothesis maintains that an outward shift in the balance of currents in the right ventricular epicardium leads to repolarization abnormalities, resulting in the development of phase 2 reentry, which generates closely-coupled premature beats capable of precipitating VT/VF (Figure 5); 2) the depolarization hypothesis maintains that delayed conduction in the RVOT plays a primary role in the development of the electrocardiographic and arrhythmic manifestations of the syndrome and that the J wave or ST segment elevation is due to a difference in activation time of RVOT vs. the remainder of RV. Figure 6 shows a schematic depicting the repolarization and depolarization hypothesis proposed to underlie the mechanism responsible for the Brugada syndrome phenotype.
Figure 5.
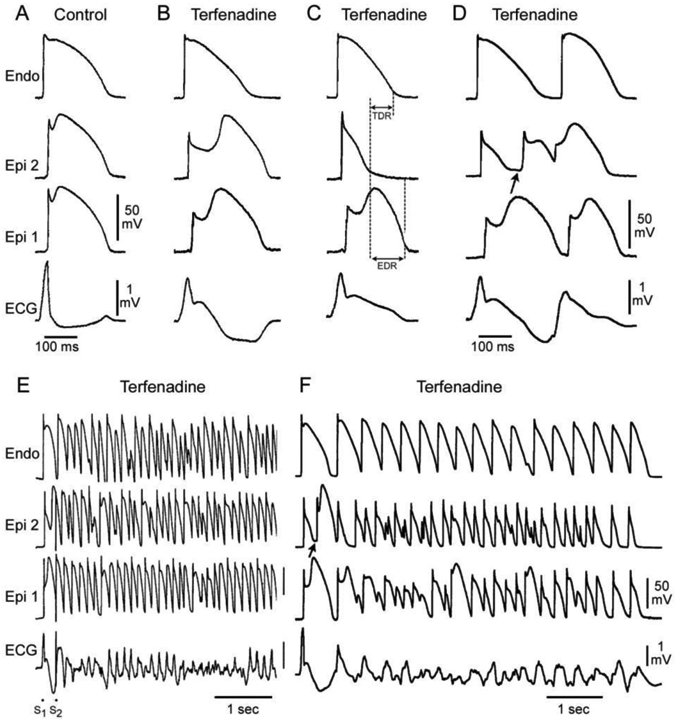
Cellular basis for the ECG and arrhythmic manifestation of BrS. Each panel shows transmembrane action potentials recorded from one endocardial (top) and two epicardial sites together with a transmural ECG recorded from a canine coronary-perfused right ventricular wedge preparation. A: Control (Basic cycle length (BCL) 400 msec). B: Combined sodium and calcium channel block with terfenadine (5 μM) accentuates the epicardial AP notch creating a transmural voltage gradient that manifests as an ST segment elevation or exaggerated J wave in the ECG. C: Continued exposure to terfenadine results in all-or-none repolarization at the end of phase 1 at some epicardial sites but not others, creating a local epicardial dispersion of repolarization (EDR) as well as a transmural dispersion of repolarization (TDR). D: Phase 2 reentry occurs when the epicardial AP dome propagates from a site where it is maintained to regions where it has been lost giving rise to a closely coupled extrasystole. E: Extrastimulus (S1-S2 = 250 msec) applied to epicardium triggers a polymorphic VT. F: Phase 2 reentrant extrasystole triggers a brief episode of polymorphic VT. Modified from (135), with permission.
Figure 6.

Schematic depicting the repolarization and depolarization hypotheses proposed to underlie the mechanism responsible for the Brugada syndrome phenotype.
Contributing to the outward shift in the balance of current responsible for the J wave is the presence of small-conductance calcium (Ca2+)-activated potassium (SK) channel current (ISK) in ventricular myocardium. Landaw et al.(78) recently suggested that colocalization of SK channels with L-type Ca2+ channels may preferentially sense Ca2+ in the subsarcolemmal or junctional space resulting in a “spiky” ISK, which can functionally play a role similar to that of Ito in promoting J-wave syndrome and ventricular arrhythmias.
Support for the repolarization hypothesis also derives from the observation that in BrS patients acceleration of heart rate leads to a reduction of the ST segment elevation, due to reduced availability of Ito and smaller RV epicardial AP notches at the faster rate. In contrast, in the depolarization hypothesis, acceleration of rate is expected to have the opposite effect (i.e., exacerbation of the ST segment elevation at fast rates) since acceleration is usually associated with slowing of conduction (77, 79).
In 2011, Nademanee et al. (80) reported a seminal study showing that radiofrequency (RF) ablation of epicardial sites displaying fractionated bipolar electrograms (EGs) and late potentials (LP) in the RVOT of patients with BrS suppresses the electrocardiographic and arrhythmic manifestations of BrS. It is noteworthy that much of the RVOT is generally ablated. These authors hypothesized that LP and fractionated electrograms are due to conduction delays within the RVOT (80).
Szél and co-workers(81) provided a direct test of this hypothesis and showed, using experimental models of BrS, that the electrophysiologic and arrhythmic manifestations BrS are due to repolarization defects rather than depolarization or conduction defects in that the late potentials are due to accentuation of the action potential notch and delayed appearance of the action potential plateau or to concealed phase 2 reentry (Figures 7 and 8).
Figure 7.

Cellular basis for the appearance of fractionated bipolar electrograms (EG). Accentuation of the action potential notch and delay in the appearance of the action potential plateau (2 nd upstroke) gives rise to fractionated epicardial electrogram in the setting of Brugada syndrome (BrS). Left panel: Shown are right precordial lead recordings, unipolar and bipolar EGs recorded form the right ventricular outflow tract of a BrS patient (from Nademanee et al. (11), with permission). Right panel: ECG, action potentials from endocardium (Endo) and two epicardial (Epi) sites, and a bipolar epicardial EG (Bipolar EG) all simultaneously recorded from a coronary-perfused right ventricular wedge preparation treated with NS5806 (5 μM) and verapamil (2 μM) to induce the Brugada phenotype. Basic cycle length=1000 ms. Reproduced from reference,(81) with permission.
Figure 8.
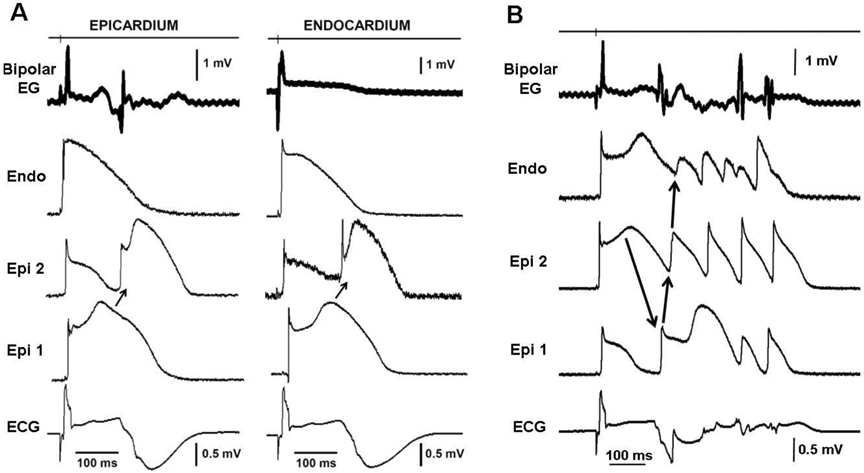
A: Concealed phase 2 reentry gives rise to late potentials and fractionated bipolar electrogram (Bipolar EG) activity recorded from epicardium but not endocardium (Endo) in an experimental model of Brugada syndrome. Each panel shows (from top to bottom) a bipolar epicardial (Epi) EG, action potentials recorded from Endo and two Epi sites and an ECG all simultaneously recorded from a coronary-perfused right ventricular (RV) wedge preparation exposed to NS5806 (5 μM) and verapamil (2 μM) to induce the Brugada phenotype. Heterogeneous loss of the dome at epicardium caused local re-excitation via a concealed phase 2 re-entry mechanism, leading to the development of late potentials and fractionated bipolar epicardial EGs. Basic cycle length = 1000 ms. B: Phase 2 Reentry-induced ventricular fibrillation. The phase 2 reentrant beat produced a closely coupled extrasystole that precipitated an episode of polymorphic tachycardia. Reproduced from (81), with permission.
Figure 7 shows the similarity between the low voltage fractionated electrical bipolar electrogram activity recorded by Nademanee and co-workers from the RVOT epicardium of patients with BrS with low voltage fractionated electrical activity recorded by Szél et al. from RV epicardium of a coronary-perfused wedge models of BrS. The study by Szél et al. showed that such fractionated electrical activity in these cases is not due to conduction delay within the RV but rather to temporal heterogeneities in the appearance of the epicardial action potential dome giving rise to undulations in the bipolar electrogram appearing as fractionated epicardial electrical activity (81).
Nagase and coworkers (82) studied 12 BrS patients who had experienced VF episodes and 17 control subjects. Using a multipolar catheter introduced into the left lateral coronary vein they recorded unipolar and bipolar electrograms from the LV epicardium. ER patterns were observed in the inferolateral ECG leads in three BrS patients and six control subjects. Prominent J waves were recorded from the LV epicardium using the unipolar leads and these coincided with late potentials recorded using the bipolar leads in the three BrS patients. These features were accentuated following administration of pilsicainide (n = 2) but diminished with atrial pacing (n = 3) and isoproterenol administration (n = 1). These authors concluded that these manifestations reflect depolarization abnormalities. depolarization feature. The experimental studies reported by Szel et al. and pictured in Figs. 7 and 8 would argue that these exact features are observed as a result of repolarization abnormalities and not depolarization abnormalities. Similar conclusions were arrived at by Konz and coworkers in studies involving experimental models of early repolarization syndrome.(83).
It remained to be explained why ablation of regions of the RVOT exhibiting fractionated electrogram activity and late potentials are effective in suppressing the ECG and arrhythmic manifestations of BrS. Patocskai et al.(84) presented evidence that ablation was effective because it eliminated the cells in the surface of the RVOT responsible for the repolarizations defects giving rise to a BrS phenotype (Figure 9).
Figure 9:

Radiofrequency ablation of epicardium (Epi) suppresses the electrocardiographic and arrhythmic manifestations of Brugada syndrome in coronary-perfused canine RV wedge model by eliminating the cells with the prominent action potential notch. The BrS phenotype is generated using a combination of the Ito blocker NS806 (8 μM) and the calcium channel blocker verapamil (1 μM). Column 1: Control. 2 Recorded 40 min after the addition of NS5806; 3-4: Recorded 20 and 40 min after the addition of verapamil. The addition of NS5806 and verapamil to the coronary perfusate induced pronounced J waves, phase 2 reentry and polymorphic VT . Bipolar electrogram recorded from RV epicardium shows late potentials and fractionated electrogram activity. 5: Recorded following 45 reintroduction of provocative agents following ablation of RV epicardium. AP recordings were obtained from midmyocardial (Mid) and subepicardial layers due to inactivation of the epicardium. Ablation of the outermost layer of the RV epicardium totally suppressed all ECG and arrhythmic manifestations of BrS. Calibrations of Bipolar EG and ECG an 1 and 2 mV, respectively. (Modified from reference,(92) with permission).
Additional evidence for the repolarization hypothesis derives from the observation by Kurita and coworkers that monophasic action potentials recorded from the epicardial and endocardial surfaces of the RVOT of a patient with BrS are nearly identical to transmembrane action potentials recorded from the epicardial and endocardial surfaces of the wedge model of BrS (85, 86). In both cases the action potential displays a prominent accentuation of the notch in epicardium, but not endocardium without any major transmural conduction delays (Figure 10). It is noteworthy that these electrophysiologic distinctions were not observed in an isolated heart explanted from a BrS patient after transplantation of a new heart. The epicardium of the explanted heart was very depressed, perhaps as a result of the 129 shocks delivered by the ICD in an attempt to control the cascade of electrical storms (87).
Figure 10.
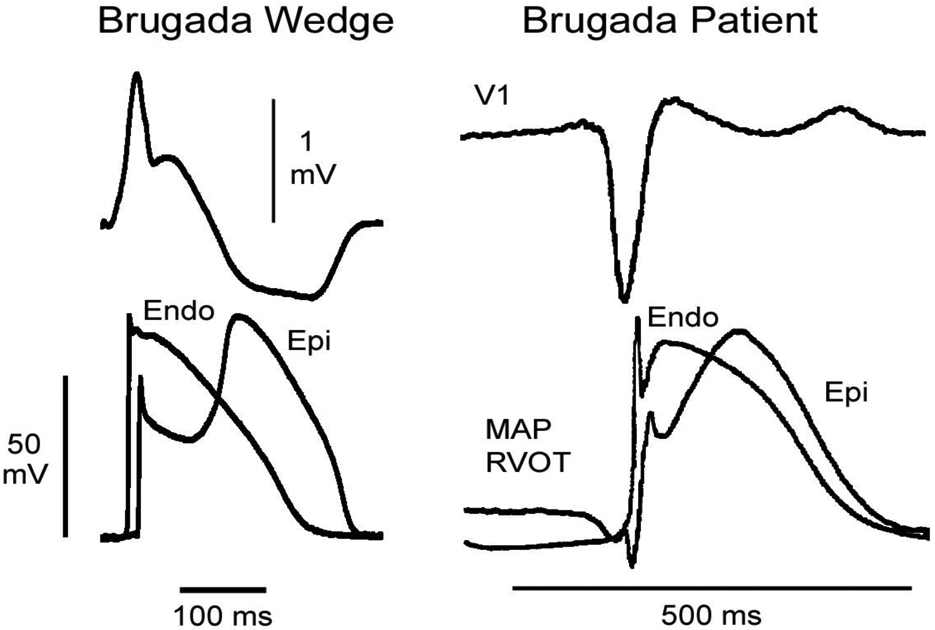
Comparison of transmembrane and monophasic action potentials and ECGs recorded from a wedge model of BrS and a patient diagnosed with BrS. Left panel shows a pseudo-ECG and transmembrane action potentials recorded from the epicardium and endocardium of a right ventricular wedge preparation in which the Brugada phenotype was elicited using 5 uM terfenadine to block INa and ICa, thus mimicking the genetic variants associated with BrS. Right panel shows lead V1 and monophasic action potentials recorded from epicardium and endocardium of the RVOT of a patient with BrS. Note that the apparent notch in the endocardial MAP is an “intrinsic potential” and not a true notch. Modified from (85, 86), with permission
In recent studies, our group has endeavored to create whole-heart models of Brugada syndrome and used these to provide a further test of the repolarization vs. depolarization hypotheses (Figure 11) (88, 89). Adult canine hearts were perfused in Langendorff mode and immersed in a volume-conducting chamber with preset electrodes to record a 12-lead ECG. Epi unipolar electrograms (EGs) were recorded from the RVOT, RV inferior wall, LV antero-basal region, and LV apex. Transmembrane APs were recorded from the RVOT using floating microelectrodes. The Ito agonist NS5806 (5-10 μM, NS), ajmaline (10 μM) or hypothermia (30-32°C) were used to elicit J waves. The Ito agonist and sodium channel blocker served to mimic the genetic defects associated with BrS. In the example illustrated in Figure 11, NS increased the Epi AP notch in the RVOT (compare panels A and B) leading to a prominent J wave in V1. Activation of the RVOT (Vmin; circled) of the EGs clearly did not contribute to inscription of the J wave. Fig. 11C, recorded 4 min later, shows loss of the AP dome at one site but not the other and the development of a closely-coupled extrasystole arising from the RVOT, consistent with a phase 2 reentry mechanism which, 1 min later, precipitated VT/VF (Fig. 11D) (89, 90). Worth mentioning, inhibition of Ito with 4-AP (0.5-1 mM) or with acacetin (5 μM) was found to greatly reduce or eliminate the manifestation of the J wave and to prevent arrhythmogenesis. Moreover, activation delay in the RVOT (relative to RV inferior wall) induced by ajmaline and/or hypothermia did not contribute to inscription of the J wave (90). In fact, RVOT activation (EGs Vmin) and the phase 0 of the RVOT APs are always in line with the onset of the J wave whether RVOT conduction is delayed or not.
Figure 11.

Whole-heart model of Brugada syndrome. Shown are traces recorded from an adult canine heart perfused in Langendorff mode. The heart was paced from the septum (the atria was removed). Epi unipolar electrograms (EGs) were recorded from the RVOT, RV inferior wall, LV antero-basal region, and LV apex. APs were recorded from the RVOT. The Ito agonist NS5806 (7.5 μM, NS) was used to elicit J waves. NS increased the Epi AP notch in the RVOT (compare panels A and B) leading to a prominent J wave in V1. Activation of the RVOT (Vmin; circled) of the EGs did not contribute to inscription of the J wave. Panel C, recorded 4 min later, shows loss of the AP dome at one site but not the other and the development of a closely-coupled premature beats arising from the RVOT, consistent with a phase 2 reentry mechanism which, 1 min later, precipitated VT/VF (panel D), thus recapitulating the ECG and arrhythmic manifestations observed clinically in patients with BrS. Reproduced from (90), with permission.
Collectively, these findings provide compelling evidence against the depolarization hypothesis showing that the electrocardiographic and arrhythmic manifestations of BrS can be due exclusively to dispersion of repolarization secondary to accentuated repolarization during the early phases of the RV epicardial action potential.
Park and coworkers conducted the first in vivo test in a large mammal of the hypothesis that slowed conduction contributes to the BrS phenotype (91). These authors genetically engineered Yucatan minipigs to heterozygously express a nonsense mutation in SCN5A (E558X) originally identified in a child with Brugada syndrome. Atrial myocytes isolated from the SCN5AE558X/+ pigs showed a loss of function of INa accounting for the slowed conduction responsible for prolongation of the P wave, QRS complex and PR interval. Despite major conduction impairment throughout the ventricular myocardium, a BrS phenotype was never observed, not even after the administration of flecainide. These observations are expected owing to the lack of Ito in the pig, which is a prerequisite for the development of the repolarization abnormalities associated with BrS. These observations provide additional strong evidence for the repolarization hypothesis and against the depolarization hypothesis.
The strong similarity between BrS and ERS with respect to clinical manifestations and response to treatment lend further support for the repolarization hypothesis. Using an experimental model of ERS, Koncz et al. (83) provided evidence in support of the hypothesis that, similar to the mechanism operative in BrS, an accentuation of transmural gradients in the LV wall is responsible for the repolarization abnormalities underlying ERS, giving rise to J point elevation, distinct J waves, or slurring of the terminal part of the QRS. The repolarization defect was accentuated by cholinergic agonists and reduced by quinidine, isoproterenol, cilostazol and milrinone, accounting for the ability of these agents to reverse the repolarization abnormalities in patients with ERS (83, 92, 93). Greater intrinsic levels of Ito in the inferior LV were shown to underlie the greater vulnerability of the inferior LV wall to VT/VF (83). Using electrocardiographic imaging (ECGI), Rudy and co-workers provided additional evidence in support of the repolarization abnormalities by identifying abnormally short activation-recovery intervals (ARI) in the inferior and lateral regions of LV and a marked dispersion of repolarization (94). Recent ECGI mapping studies performed in an ERS patient during VF demonstrated VF rotors anchored in the inferior-lateral left ventricular wall (95).
Because the mechanisms underlying BrS and ERS are closely related, it is not surprising that the response to pharmacologic agents is similar as well. Quinidine, Phosphodiesterase III inhibitors (cilostazol and milrinone) and isoproterenol all suppress arrhythmogenesis associated with both ERS and BrS (5, 32, 36, 81, 96-99). All of these agents have been shown to correct the repolarization defects responsible for development of phase 2 reentry and VT/VF in experimental wedge models of BrS and ERS (83, 97, 100).
Structural Involvement
Debate continues as to the extent to which structural abnormalities contribute to the manifestation of BrS. Concealed structural abnormalities, such as histologic myocardial fibrosis of the RVOT, which may not become evident using conventional imaging techniques have been proposed to account for or contribute to delayed conduction and ventricular arrhythmias in BrS.(101, 102) Electron beam computed tomography (CT) and MRI studies conducted in BrS patients show subtle abnormalities, including wall motion abnormalities and reduced contractile function of the right ventricle (RV) and to a lesser extent of the left ventricle (LV), and dilatation of the RV outflow tract (RVOT). (103-106) In the only study to discriminate between patients with and without SCN5A variants, no difference was observed in RVOT dimensions or RV ejection fraction between patients with and without SCN5A variants. Slightly greater depression of LV dimensions and ejection fraction was observed in patients with SCN5A variants. (107) Cardiac dilatation and reduced contractility in all of these studies were attributed to structural changes (fibrosis, fatty degeneration). However, no signs of structural abnormalities could be detected. Antzelevitch and co-workers have proposed an alternative explanation for such findings.(96, 108, 109) Loss of the action potential dome, which has been shown in experimental models to create the arrhythmogenic substrate in Brugada syndrome, can lead to contractile changes, thus providing an explanation for the wall motion abnormalities observed. The all-or-none repolarization at the end of phase 1 of the epicardial action potential responsible for loss of the dome causes calcium channels to inactivate soon after they activate, leading to depletion of intracellular calcium, and loss of contractile function. This is expected to lead to wall motion abnormalities, particularly in the RVOT, dilatation of the RVOT region and reduced ejection fraction observed in patients with Brugada syndrome. This hibernation-like state, may also lead to mild structural changes, including intracellular lipid accumulation, vacuolization and connexin 43 redistribution, which could contribute to the arrhythmogenic substrate of the BrS. (109, 110). These observations suggest that some of the changes observed by recent studies (41, 101, 102) may be the result of rather than the cause of the BrS phenotype.
Approaches to therapy of JWS
Implantable Cardioverter Defibrillator
Approaches to therapy of BrS and ERS have gradually evolved in recent years as reported in several consensus reports (6, 111-113).
The most effective therapy for the prevention of SCD in high risk BrS and ERS patients is an implantable cardioverter defibrillator (ICD) (6, 7, 112-115). However, ICDs are associated with complications, especially in young active individuals (116, 117) and, over time, inappropriate shocks and lead failure are not uncommon (118).
Radiofrequency Ablation (RFA) Therapy
As noted above, Nademanee et al. (80) demonstrated that RFA of epicardial sites displaying late potentials (LP) and fractionated bipolar electrograms (EGs) in the RVOT of BrS patients can reduce arrhythmia risk and the ECG phenotype and that, over a period of weeks or months, the ablation makes VT/VF non-inducible in most patients. Additional evidence in support of the effectiveness of epicardial RVOT ablation was provided by Sacher et al., Shah et al. and Brugada and coworkers (119-121). With regards to ERS, little clinical data are available concerning the effectiveness of epicardial RFA (11).
A recent report from Scheinman’s group reported successfully ablating abnormal epicardial electrograms in the inferolateral left ventricle of patients with ERS (122). Voskoboinik et al. concluded that mapping and ablation of abnormal epicardial electrograms in the inferolateral left ventricle may be a potential future treatment strategy for patients diagnosed with ERS, just as mapping and ablation of the RVOT are of benefit in patients with BrS (122).
Pharmacologic approach to therapy
In that ICDs can lead to complications including dislodgement and inappropriate shocks and is unaffordable in many regions of the world, a pharmacologic approach to therapy is desirable. Because the presence of a prominent Ito is thought to be important for the development of both BrS and ERS, inhibition of this current, even partially, is thought to be effective irrespective of the underlying ionic or genetic basis. Unfortunately, ion-channel specific and cardio-selective Ito blockers are not currently available. The best drug currently available in the clinic capable of blocking Ito is quinidine. Quinidine was first recommended as therapy for BrS by our group in 1999 based on experimental evidence obtained from the coronary-perfused RV wedge model of BrS (23, 81, 123, 124). Clinical evidence for the effectiveness of quinidine has been reported in numerous studies and case reports. Hermida et al. reported 76% efficacy in prevention of VF induced by programmed electrical stimulation (PES) (34). Belhassen, Viskin and coworkers, who pioneered the use of quinidine in VF(125), recently reported a 90% efficacy in prevention of VF induction, despite the use of very aggressive PES protocols (125).
Agents capable of augmenting the L-type calcium channel current, such as β-adrenergic agents including isoproterenol or orciprenaline, are useful as well (5, 23, 32, 36, 99, 126). Increasing ICa prevents arrhythmogenesis associated with JWS by opposing the increased outward current forces, thus restoring the epicardial AP dome in both BrS (127) and ERS (92).
Another encouraging pharmacologic approach for BrS is cilostazol, a phosphodiesterase (PDE) III inhibitor (32, 36, 128) which normalizes the ST segment by augmenting ICa as well as by reducing Ito secondary to an increase in cAMP and heart rate (129). Of note, failure of cilostazol in the treatment of BrS has been described in a single case report (130).
Acacetin, a natural flavone, has been shown to inhibit Ito in human atrial myocytes in a frequency-dependent manner (131). The beneficial effect of this agent in experimental models of BrS and ERS has recently been demonstrated by our group (89, 90, 132). Figure 12 illustrates the effect of acacetin in two different wedge models of BrS designed to mimic the INa and ICa loss of function genetic defects associated with BrS. (90) The effectiveness of acacetin has also been demonstrated in the whole-heart model of BrS, but is yet to be studied clinically. (90)
Figure 12.

Effect of acacetin to suppress the electrocardiographic and arrhythmic manifestations in two different models of Brugada syndrome. Each panel shows simultaneously recorded transmembrane action potentials from one endocardial (Endo) or subendocardial (Subendo) and two epicardial (Epi1, Epi2) sites together with a pseudo-ECG and bipolar surface electrograms (Bip EG) in arterially-perfused RV wedge preparations. BCL = 1000 msec. (A) Brugada phenotype induced by a combination of NS5806 and ajmaline. The provocative agents accentuated phase 1, leading to heterogeneous loss of the AP dome, thus giving rise to concealed P2R and a prominent type 1 ST segment elevation (col3). The delayed 2nd upstroke of the epicardial APs and concealed P2R give rise to late potentials in the bipolar electrograms recorded from epicardium but not endocardium (col2-4, Bip EG Epi and Bip EG Endo; red arrows). When P2R was able to propagate from its protected focus, it initiated VT/VF episodes (col5). Acacetin (10 μM) restored normal AP morphology secondary to diminution of the AP notch, thus abolishing all late potential activity (O), VT/VF, P2R and the Brugada- ECG-pattern (col6). (B) Brugada phenotype induced by the addition of NS5806 and verapamil to the coronary perfusate. The provocative agents accentuated phase 1, leading to heterogeneous loss of the AP dome in epicardium and the development of phase 2 reentry (col2-3, black arrows) and VT/VF (col3). Acacetin (10 μM) restored the epicardial AP dome, reduced the AP notch, and abolished all arrhythmic activity (col4-5). The effect of acacetin was reversed upon washout (col6). Reproduced from (90), with permission.
Funding:
Supported by NIH grants HL47678, HL138103, HL152201, W.W. Smith Charitable Trust and the Wistar and Martha Morris Fund.
Footnotes
Disclosures: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Brugada P, Brugada J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome: a multicenter report. JAmCollCardiol. 1992;20(6):1391–6. [DOI] [PubMed] [Google Scholar]
- 2.Nam GB, Kim YH, Antzelevitch C. Augmentation of J waves and electrical storms in patients with early repolarization. NEnglJ Med. 2008;358(19):2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Haissaguerre M, Derval N, Sacher F, Jesel L, Deisenhofer I, De Roy L, et al. Sudden cardiac arrest associated with early repolarization. NEnglJ Med. 2008;358(19):2016–23. [DOI] [PubMed] [Google Scholar]
- 4.Kanter RJ, Pfeiffer R, Hu D, Barajas-Martinez H, Carboni MP, Antzelevitch C. Brugada-like syndrome in infancy presenting with rapid ventricular tachycardia and intraventricular conduction delay. Circulation. 2012;125(1):14–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Antzelevitch C. The Brugada syndrome: ionic basis and arrhythmia mechanisms. JCardiovasc Electrophysiol. 2001;12(2):268–72. [DOI] [PubMed] [Google Scholar]
- 6.Priori SG, Blomstrom-Lundqvist C, Mazzanti A, Blom N, Borggrefe M, Camm J, et al. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC)Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur Heart J. 2015. [Google Scholar]
- 7.Antzelevitch C, Yan GX, Ackerman MJ, Borggrefe M, Corrado D, Guo J, et al. J-Wave syndromes expert consensus conference report: Emerging concepts and gaps in knowledge: Endorsed by the Asia Pacific Heart Rhythm Society (APHRS), the European Heart Rhythm Association (EHRA), the Heart Rhythm Society (HRS), and the Latin American Society of Cardiac Pacing and Electrophysiology (Sociedad Latinoamericana de Estimulacifin Cardiaca y Electro fi siologia [SOLAECE]). Europace : European pacing, arrhythmias, and cardiac electrophysiology : journal of the working groups on cardiac pacing, arrhythmias, and cardiac cellular electrophysiology of the European Society of Cardiology. 2016. [Google Scholar]
- 8.Macfarlane P; Antzelevitch C; Haissaguerre M; Huikuri H P M R R, Sacher F; Tikkanen J; Wellens H; Yan G-X Consensus Paper- Early Repolarization Pattern. J Amer Coll Cardiol. 2015;66(4):470–7. [DOI] [PubMed] [Google Scholar]
- 9.Antzelevitch C Y G, Ackerman MJ, Borggrefe M, Corrado D, Guo J, Gussak I, Hasdemir C, Horie M, Huikuri H, Ma C, Morita H, Nam GB, Sacher F, Shimizu W, Viskin S, Wilde AA. J-Wave Syndromes expert consensus conference report: Emerging concepts and gaps in knowledge. Journal of arrhythmia. 2016;32(5):315–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kawata H, Noda T, Yamada Y, Okamura H, Satomi K, Aiba T, et al. Effect of sodium-channel blockade on early repolarization in inferior/lateral leads in patients with idiopathic ventricular fibrillation and Brugada syndrome. Heart Rhythm. 2012;9(1):77–83. [DOI] [PubMed] [Google Scholar]
- 11.Nakagawa K, Nagase S, Morita H, Ito H. Left ventricular epicardial electrogram recordings in idiopathic ventricular fibrillation with inferior and lateral early repolarization. Heart Rhythm. 2014;11(2):314–7. [DOI] [PubMed] [Google Scholar]
- 12.McIntyre WF, Perez-Riera AR, Femenia F, Baranchuk A. Coexisting early repolarization pattern and Brugada syndrome: recognition of potentially overlapping entities. J Electrocardiol. 2012;45(3):195–8. [DOI] [PubMed] [Google Scholar]
- 13.Nam GB, Ko KH, Kim J, Park KM, Rhee KS, Choi KJ, et al. Mode of onset of ventricular fibrillation in patients with early repolarization pattern vs. Brugada syndrome. Eur Heart J. 2010;31(3):330–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kawada S, Morita H, Antzelevitch C, Morimoto Y, Nakagawa K, Watanabe A, et al. Shanghai Score System for Diagnosis of Brugada Syndrome: Validation of the Score System and System and Reclassification of the Patients. JACC Clin Electrophysiol. 2018;4(6):724–30. [DOI] [PubMed] [Google Scholar]
- 15.Tikkanen JT, Junttila MJ, Anttonen O, Aro AL, Luttinen S, Kerola T, et al. Early repolarization: electrocardiographic phenotypes associated with favorable long-term outcome. Circulation. 2011;123(23):2666–73. [DOI] [PubMed] [Google Scholar]
- 16.Take Y, Morita H. Fragmented QRS: What Is The Meaning? Indian Pacing Electrophysiol J. 2012;12(5):213–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morita H, Zipes DP, Wu J. Brugada syndrome: insights of ST elevation, arrhythmogenicity, and risk stratification from experimental observations. Heart Rhythm. 2009;6(11 Suppl):S34–S43. [DOI] [PubMed] [Google Scholar]
- 18.Morita H, Watanabe A, Kawada S, Miyamoto M, Morimoto Y, Nakagawa K, et al. Identification of electrocardiographic risk markers for the initial and recurrent episodes of ventricular fibrillation in patients with Brugada syndrome. Journal of cardiovascular electrophysiology. 2018;29(1):107–14. [DOI] [PubMed] [Google Scholar]
- 19.Morita H, Kusano KF, Miura D, Nagase S, Nakamura K, Morita ST, et al. Fragmented QRS as a marker of conduction abnormality and a predictor of prognosis of Brugada syndrome. Circulation. 2008;118(17):1697–704. [DOI] [PubMed] [Google Scholar]
- 20.Aizawa Y, Takatsuki S, Nishiyama T, Kimura T, Kohsaka S, Kaneko Y, et al. Tachycardia-Induced J-Wave Changes in Patients With and Without Idiopathic Ventricular Fibrillation. Circ Arrhythm Electrophysiol. 2017;10(7). [DOI] [PubMed] [Google Scholar]
- 21.Nam G-B. Idiopathic Ventricular Fibrillation, Early Repolarization and Other J Wave-Related Ventricular Fibrillation Syndromes. Circulation Journal. 2012;76(12):2723–31. [DOI] [PubMed] [Google Scholar]
- 22.Yan GX, Antzelevitch C. Cellular basis for the electrocardiographic J wave. Circulation. 1996;93(2):372–9. [DOI] [PubMed] [Google Scholar]
- 23.Yan GX, Antzelevitch C. Cellular basis for the Brugada syndrome and other mechanisms of arrhythmogenesis associated with ST-segment elevation. Circulation. 1999;100(15):1660–6. [DOI] [PubMed] [Google Scholar]
- 24.Benito B, Sarkozy A, Mont L, Henkens S, Berruezo A, Tamborero D, et al. Gender differences in clinical manifestations of Brugada syndrome. J Am Coll Cardiol. 2008;52(19):1567–73. [DOI] [PubMed] [Google Scholar]
- 25.Kamakura T, Kawata H, Nakajima I, Yamada Y, Miyamoto K, Okamura H, et al. Significance of non-type 1 anterior early repolarization in patients with inferolateral early repolarization syndrome. J Am Coll Cardiol. 2013;62(17):1610–8. [DOI] [PubMed] [Google Scholar]
- 26.Matsumoto AM. Fundamental aspects of hypogonadism in the aging male. Reviews in urology. 2003;5 Suppl 1:S3–s10. [PMC free article] [PubMed] [Google Scholar]
- 27.Kalla H, Yan GX, Marinchak R. Ventricular fibrillation in a patient with prominent J (Osborn) waves and ST segment elevation in the inferior electrocardiographic leads: a Brugada syndrome variant? J Cardiovasc Electrophysiol. 2000;11(1):95–8. [DOI] [PubMed] [Google Scholar]
- 28.Aizawa Y, Sato A, Watanabe H, Chinushi M, Furushima H, Horie M, et al. Dynamicity of the J-wave in idiopathic ventricular fibrillation with a special reference to pause-dependent augmentation of the J-wave. J Am Coll Cardiol. 2012;59(22):1948–53. [DOI] [PubMed] [Google Scholar]
- 29.Nademanee K. Sudden unexplained death syndrome in southeast Asia. American Journal of Cardiology. 1997;79(6A):10–1. [DOI] [PubMed] [Google Scholar]
- 30.Shimizu W, Aiba T, Kurita T, Kamakura S. Paradoxic abbreviation of repolarization in epicardium of the right ventricular outflow tract during augmentation of Brugada-type ST segment elevation. J Cardiovasc Electrophysiol. 2001;12(12):1418–21. [DOI] [PubMed] [Google Scholar]
- 31.Suzuki H, Torigoe K, Numata O, Yazaki S. Infant case with a malignant form of Brugada syndrome. Journal of cardiovascular electrophysiology. 2000;11:1277–80. [DOI] [PubMed] [Google Scholar]
- 32.Ohgo T, Okamura H, Noda T, Satomi K, Suyama K, Kurita T, et al. Acute and chronic management in patients with Brugada syndrome associated with electrical storm of ventricular fibrillation. Heart Rhythm. 2007;4(6):695–700. [DOI] [PubMed] [Google Scholar]
- 33.Watanabe A, Fukushima Kusano K, Morita H, Miura D, Sumida W, Hiramatsu S, et al. Low-dose isoproterenol for repetitive ventricular arrhythmia in patients with Brugada syndrome. Eur Heart J. 2006;27(13):1579–83. [DOI] [PubMed] [Google Scholar]
- 34.Hermida JS, Denjoy I, Clerc J, Extramiana F, Jarry G, Milliez P, et al. Hydroquinidine therapy in Brugada syndrome. J Am Coll Cardiol. 2004;43(10):1853–60. [DOI] [PubMed] [Google Scholar]
- 35.Belhassen B, Viskin S. Pharmacologic approach to therapy of Brugada syndrome: quinidine as an alternative to ICD therapy? In: Antzelevitch C, Brugada P, Brugada J, Brugada R, editors. The Brugada Syndrome: From Bench to Bedside. Oxford: Blackwell Futura; 2004. p. 202–11. [Google Scholar]
- 36.Tsuchiya T, Ashikaga K, Honda T, Arita M. Prevention of ventricular fibrillation by cilostazol, an oral phosphodiesterase inhibitor, in a patient with Brugada syndrome. JCardiovascElectrophysiol. 2002;13(7):698–701. [DOI] [PubMed] [Google Scholar]
- 37.Hasegawa K, Ashihara T, Kimura H, Jo H, Itoh H, Yamamoto T, et al. Long-term pharmacological therapy of Brugada syndrome: is J-wave attenuation a marker of drug efficacy? InternMed. 2014;53(14):1523–6. [DOI] [PubMed] [Google Scholar]
- 38.Shinohara T, Ebata Y, Ayabe R, Fukui A, Okada N, Yufu K, et al. Combination therapy of cilostazol and bepridil suppresses recurrent ventricular fibrillation related to J-wave syndromes. Heart Rhythm. 2014;11(8):1441–5. [DOI] [PubMed] [Google Scholar]
- 39.Haissaguerre M, Chatel S, Sacher F, Weerasooriya R, Probst V, Loussouarn G, et al. Ventricular fibrillation with prominent early repolarization associated with a rare variant of KCNJ8/KATP channel. Journal of cardiovascular electrophysiology. 2009;20(1):93–8. [DOI] [PubMed] [Google Scholar]
- 40.Iguchi K, Noda T, Kamakura S, Shimizu W. Beneficial effects of cilostazol in a patient with recurrent ventricular fibrillation associated with early repolarization syndrome. Heart Rhythm. 2013;10(4):604–6. [DOI] [PubMed] [Google Scholar]
- 41.Nademanee K, Raju H, de Noronha SV, Papadakis M, Robinson L, Rothery S, et al. Fibrosis, Connexin-43, and Conduction Abnormalities in the Brugada Syndrome. J Am Coll Cardiol. 2015;66(18):1976–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Junttila MJ, Tikkanen JT, Kentta T, Anttonen O, Aro AL, Porthan K, et al. Early repolarization as a predictor of arrhythmic and nonarrhythmic cardiac events in middle-aged subjects. Heart Rhythm. 2014;11(10):1701–6. [DOI] [PubMed] [Google Scholar]
- 43.Noda T, Shimizu W, Tanaka K, Chayama K. Prominent J wave and ST segment elevation: serial electrocardiographic changes in accidental hypothermia. J CardiovascElectrophysiol. 2003;14(2):223. [DOI] [PubMed] [Google Scholar]
- 44.Bastiaenen R, Hedley PL, Christiansen M, Behr ER. Therapeutic hypothermia and ventricular fibrillation storm in early repolarization syndrome. Heart Rhythm. 2010;7(6):832–4. [DOI] [PubMed] [Google Scholar]
- 45.Federman NJ, Mechulan A, Klein GJ, Krahn AD. Ventricular fibrillation induced by spontaneous hypothermia in a patient with early repolarization syndrome. Journal of cardiovascular electrophysiology. 2013;24:586–8. [DOI] [PubMed] [Google Scholar]
- 46.Kowalczyk E, Kasprzak JD, Lipiec P. Giant J-wave and Brugada-like pattern in a patient with severe hypothermia. Acta Cardiol. 2014;69(1):66–7. [DOI] [PubMed] [Google Scholar]
- 47.RuDusky BM. The electrocardiogram in hypothermia-the J wave and the Brugada syndrome. Am J Cardiol. 2004;93(5):671–2. [DOI] [PubMed] [Google Scholar]
- 48.Patel RB, Ng J, Reddy V, Chokshi M, Parikh K, Subacius H, et al. Early repolarization associated with ventricular arrhythmias in patients with chronic coronary artery disease. Circ ArrhythmElectrophysiol. 2010;3(5):489–95. [DOI] [PubMed] [Google Scholar]
- 49.Amin AS, Meregalli PG, Bardai A, Wilde AA, Tan HL. Fever increases the risk for cardiac arrest in the Brugada syndrome. AnnInternMed. 2008;149(3):216–8. [DOI] [PubMed] [Google Scholar]
- 50.Rattanawong P, Vutthikraivit W, Charoensri A, Jongraksak T, Prombandankul A, Kanjanahattakij N, et al. Fever-Induced Brugada Syndrome Is More Common Than Previously Suspected: A Cross-Sectional Study from an Endemic Area. Ann Noninvasive Electrocardiol. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kawata H, Morita H, Yamada Y, Noda T, Satomi K, Aiba T, et al. Prognostic significance of early repolarization in inferolateral leads in Brugada patients with documented ventricular fibrillation: a novel risk factor for Brugada syndrome with ventricular fibrillation. Heart Rhythm. 2013;10(8):1161–8. [DOI] [PubMed] [Google Scholar]
- 52.Tan HL, Meregalli PG. Lethal ECG changes hidden by therapeutic hypothermia. Lancet. 2007;369(9555):78. [DOI] [PubMed] [Google Scholar]
- 53.Kurisu S, Inoue I, Kawagoe T, Ishihara M, Shimatani Y, Nakama Y, et al. Therapeutic hypothermia after out-of-hospital cardiac arrest due to Brugada syndrome. Resuscitation. 2008;79(2):332–5. [DOI] [PubMed] [Google Scholar]
- 54.Kapplinger J, Tester D, Alders M, Benito B, Berthet M, Brugada J, et al. An International Compendium of Mutations in the SCN5A-Encoded Cardiac Sodium Channel in Patients Referred for Brugada Syndrome Genetic Testing. Heart Rhythm. 2010;7:33–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ackerman MJ, Priori SG, Willems S, Berul C, Brugada R, Calkins H, et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Heart Rhythm. 2011;8(8):1308–39. [DOI] [PubMed] [Google Scholar]
- 56.Juang JJ, Binda A, Lee SJ, Hwang JJ, Chen WJ, Liu YB, et al. GSTM3 variant is a novel genetic modifier in Brugada syndrome, a disease with risk of sudden cardiac death. EBioMedicine. 2020;57:102843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Turker I, Makiyama T, Ueyama T, Shimizu A, Yamakawa M, Chen PS, et al. Telethonin variants found in Brugada syndrome, J-wave pattern ECG, and ARVC reduce peak Na(v) 1.5 currents in HEK-293 cells. Pacing Clin Electrophysiol. 2020. [DOI] [PubMed] [Google Scholar]
- 58.Liu H, Chatel S, Simard C, Syam N, Salle L, Probst V, et al. Molecular genetics and functional anomalies in a series of 248 Brugada cases with 11 mutations in the TRPM4 channel. PloS one. 2013;8(1):e54131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Verkerk AO, Wilders R, Schulze-Bahr E, Beekman L, Bhuiyan ZA, Bertrand J, et al. Role of sequence variations in the human ether-a-go-go-related gene (HERG, KCNH2) in the Brugada syndrome. Cardiovascular research. 2005;68(3):441–53. [DOI] [PubMed] [Google Scholar]
- 60.Wilders R, Verkerk AO. Role of the R1135H KCNH2 mutation in Brugada syndrome. IntJ Cardiol. 2010;144(1):149–51. [DOI] [PubMed] [Google Scholar]
- 61.Ohno S, Zankov DP, Ding WG, Itoh H, Makiyama T, Doi T, et al. KCNE5 (KCNE1L) variants are novel modulators of Brugada syndrome and idiopathic ventricular fibrillation. Circ ArrhythmElectrophysiol. 2011;4(3):352–61. [DOI] [PubMed] [Google Scholar]
- 62.Boczek NJ, Ye D, Johnson EK, Wang W, Crotti L, Tester DJ, et al. Characterization of SEMA3A-encoded semaphorin as a naturally occurring Kv4.3 protein inhibitor and its contribution to Brugada syndrome. Circ Res. 2014;115(4):460–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Perrin MJ, Adler A, Green S, Al-Zoughool F, Doroshenko P, Orr N, et al. Evaluation of genes encoding for the transient outward current (Ito) identifies the KCND2 gene as a cause of J-wave syndrome associated with sudden cardiac death. Circ Cardiovasc Genet. 2014;7(6):782–9. [DOI] [PubMed] [Google Scholar]
- 64.Ueda K, Nakamura K, Hayashi T, Inagaki N, Takahashi M, Arimura T, et al. Functional characterization of a trafficking-defective HCN4 mutation, D553N, associated with cardiac arrhythmia. J Biol Chem. 2004;279(26):27194–8. [DOI] [PubMed] [Google Scholar]
- 65.Clatot J, Neyroud N, Cox R, Souil C, Huang J, Guicheney P, et al. Inter-Regulation of K(v)4.3 and Voltage-Gated Sodium Channels Underlies Predisposition to Cardiac and Neuronal Channelopathies. Int J Mol Sci. 2020;21(14). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hosseini SM, Kim R, Udupa S, Costain G, Jobling R, Liston E, et al. Reappraisal of Reported Genes for Sudden Arrhythmic Death: An Evidence-Based Evaluation of Gene Validity for Brugada Syndrome. Circulation. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Le Scouarnec S, Karakachoff M, Gourraud JB, Lindenbaum P, Bonnaud S, Portero V, et al. Testing the burden of rare variation in arrhythmia-susceptibility genes provides new insights into molecular diagnosis for Brugada syndrome. Human molecular genetics. 2015. [DOI] [PubMed] [Google Scholar]
- 68.Bezzina CR, Barc J, Mizusawa Y, Remme CA, Gourraud JB, Simonet F, et al. Common variants at SCN5A-SCN10A and HEY2 are associated with Brugada syndrome, a rare disease with high risk of sudden cardiac death. Nat Genet. 2013;45(9):1044–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Matsuo K, Akahoshi M, Seto S, Yano K. Disappearance of the Brugada-type electrocardiogram after surgical castration: a role for testosterone and an explanation for the male preponderance? Pacing ClinElectrophysiol. 2003;In press(7 Pt 1):1151–3. [DOI] [PubMed] [Google Scholar]
- 70.Antzelevitch C. Molecular genetics of arrhythmias and cardiovascular conditions associated with arrhythmias. Pacing ClinElectrophysiol. 2003;26(11):2194–208. [DOI] [PubMed] [Google Scholar]
- 71.Korte AK, Derde L, van Wijk J, Tjan DH. Sudden cardiac arrest as a presentation of Brugada syndrome unmasked by thyroid storm. BMJ Case Rep. 2015;2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schwartz PJ, Ackerman MJ, Antzelevitch C, Bezzina CR, Borggrefe M, Cuneo BF, et al. Inherited cardiac arrhythmias. Nat Rev Dis Primers. 2020;6(1):58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Litovsky SH, Antzelevitch C. Transient outward current prominent in canine ventricular epicardium but not endocardium. CircRes. 1988;62(1):116–26. [DOI] [PubMed] [Google Scholar]
- 74.Di Diego JM, Sun ZQ, Antzelevitch C. Ito and action potential notch are smaller in left vs. right canine ventricular epicardium. AmJPhysiol. 1996;271:H548–H61. [DOI] [PubMed] [Google Scholar]
- 75.Antzelevitch C, Yan GX. J-wave syndromes: Brugada and early repolarization syndromes. Heart Rhythm. 2015;12(8):1852–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Huikuri HV, Juhani Junttila M. Clinical aspects of inherited J-wave syndromes. Trends in cardiovascular medicine. 2015;25(1):24–30. [DOI] [PubMed] [Google Scholar]
- 77.Wilde AA, Postema PG, Di Diego JM, Viskin S, Morita H, Fish JM, et al. The pathophysiological mechanism underlying Brugada syndrome: depolarization versus repolarization. J Mol Cell Cardiol. 2010;49(4):543–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Landaw J, Zhang Z, Song Z, Liu MB, Olcese R, Chen PS, et al. Small-conductance Ca(2+)-activated K(+) channels promote J-wave syndrome and phase-2 reentry. Heart Rhythm. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Antzelevitch C Y G. J wave syndrome: Brugada and Early Repolarization Syndromes. Heart Rhythm. 2015;12(8):1852–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nademanee K, Veerakul G, Chandanamattha P, Chaothawee L, Ariyachaipanich A, Jirasirirojanakorn K, et al. Prevention of ventricular fibrillation episodes in Brugada syndrome by catheter ablation over the anterior right ventricular outflow tract epicardium. Circulation. 2011;123(12):1270–9. [DOI] [PubMed] [Google Scholar]
- 81.Szel T, Antzelevitch C. Abnormal repolarization as the basis for late potentials and fractionated electrograms recorded from epicardium in experimental models of brugada syndrome. J Am Coll Cardiol. 2014;63(19):2037–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nagase S, Tanaka M, Morita H, Nakagawa K, Wada T, Murakami M, et al. Local Left Ventricular Epicardial J Waves and Late Potentials in Brugada Syndrome Patients with Inferolateral Early Repolarization Pattern. Frontiers in physiology. 2017;8:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Koncz I, Gurabi Z, Patocskai B, Panama BK, Szel T, Hu D, et al. Mechanisms underlying the development of the electrocardiographic and arrhythmic manifestations of early repolarization syndrome. J Mol Cell Cardiol. 2014;68C:20–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Patocskai B, Yoon N, Antzelevitch C. Mechanisms underlying epicardial radiofrequency ablation to suppress arrhythmogenesis in experimental models of Brugada syndrome. JACC: Clinical Electrophysiology. 2017;3(4):353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Antzelevitch C, Brugada P, Brugada J, Brugada R, Shimizu W, Gussak I, et al. Brugada syndrome: a decade of progress. CircRes. 2002;91(12):1114–9. [DOI] [PubMed] [Google Scholar]
- 86.Kurita T, Shimizu W, Inagaki M, Suyama K, Taguchi A, Satomi K, et al. The electrophysiologic mechanism of ST-segment elevation in Brugada syndrome. JAmCollCardiol. 2002;40(2):330–4. [DOI] [PubMed] [Google Scholar]
- 87.Coronel R, Casini S, Koopmann TT, Wilms-Schopman FJ, Verkerk AO, de Groot JR, et al. Right ventricular fibrosis and conduction delay in a patient with clinical signs of Brugada syndrome: a combined electrophysiological, genetic, histopathologic, and computational study. Circulation. 2005;112(18):2769–77. [DOI] [PubMed] [Google Scholar]
- 88.Di Diego JM, Argenziano M, Chen K, Tabler M, Antzelevitch C. In a Whole-Heart model of the Brugada Syndrome, Delayed Conduction in the RVOT “does not” Contribute to Inscription of the Electrocardiographic J wave / ST segment elevation. Heart Rhythm. 2018;15:S242. [Google Scholar]
- 89.Di Diego JM, Argenziano M, Tabler M, Chen AE, Messerschmidt V, Jerud E, et al. A Definitive Test Of The Repolarization Versus Depolarization Hypothesis In A Whole-heart Model Of Brugada Syndrome. Circulation. 2019;140:A9991. [Google Scholar]
- 90.Di Diego JM, Patocskai B, Barajas-Martinez H, Borbáth V, Ackerman MJ, Burashnikov A, et al. Acacetin suppresses the electrocardiographic and arrhythmic manifestations of the J wave syndromes. PloS one. 2020;15(11):e0242747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Park DS, Cerrone M, Morley G, Vasquez C, Fowler S, Liu N, et al. Genetically engineered SCN5A mutant pig hearts exhibit conduction defects and arrhythmias. J Clin Invest. 2015;125(1):403–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gurabi Z, Koncz I, Patocskai B, Nesterenko VV, Antzelevitch C. Cellular mechanism underlying hypothermia-induced ventricular tachycardia/ventricular fibrillation in the setting of early repolarization and the protective effect of quinidine, cilostazol, and milrinone. Circ Arrhythm Electrophysiol. 2014;7(1):134–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Patocskai B, Barajas-Martinez H, Hu D, Gurabi Z, Koncz I, Antzelevitch C. Cellular and ionic mechanisms underlying the effects of cilostazol, milrinone, and isoproterenol to suppress arrhythmogenesis in an experimental model of early repolarization syndrome. Heart Rhythm. 2016;13(6):1326–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ghosh S, Cooper DH, Vijayakumar R, Zhang J, Pollak S, Haissaguerre M, et al. Early repolarization associated with sudden death: insights from noninvasive electrocardiographic imaging. Heart Rhythm. 2010;7(4):534–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mahida S, Derval N, Sacher F, Berte B, Yamashita S, Hooks DA, et al. History and clinical significance of early repolarization syndrome. Heart Rhythm. 2015;12(1):242–9. [DOI] [PubMed] [Google Scholar]
- 96.Antzelevitch C, Brugada P, Brugada J, Brugada R, Nademanee K, Towbin JA. Clinical approaches to tachyarrhythmias. The Brugada syndrome. Armonk, NY: Futura Publishing Company, Inc.; 1999. 1999. [Google Scholar]
- 97.Yan GX, Antzelevitch C. Cellular basis for the Brugada syndrome and other mechanisms of arrhythmogenesis associated with ST segment elevation. Circulation. 1999;100(15):1660–6. [DOI] [PubMed] [Google Scholar]
- 98.Kyriazis K, Bahlmann E, van der SH, Kuck KH. Electrical storm in Brugada syndrome successfully treated with orciprenaline; effect of low-dose quinidine on the electrocardiogram. Europace. 2009;11(5):665–6. [DOI] [PubMed] [Google Scholar]
- 99.Schweizer PA, Becker R, Katus HA, Thomas D. Successful acute and long-term management of electrical storm in Brugada syndrome using orciprenaline and quinine/quinidine. Clin Res Cardiol. 2010;99(7):467–70. [DOI] [PubMed] [Google Scholar]
- 100.Minoura Y, Di Diego JM, Barajas-Martinez H, Zygmunt AC, Hu D, Sicouri S, et al. Ionic and cellular mechanisms underlying the development of acquired Brugada syndrome in patients treated with antidepressants. Journal of cardiovascular electrophysiology. 2012;23(4):423–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nademanee K, Haissaguerre M, Hocini M, Nogami A, Cheniti G, Duchateau J, et al. Mapping and Ablation of Ventricular Fibrillation Associated With Early Repolarization Syndrome. Circulation. 2019;140(18):1477–90. [DOI] [PubMed] [Google Scholar]
- 102.Behr ER, Ben-Haim Y, Ackerman MJ, Krahn AD, Wilde AAM. Brugada syndrome and reduced right ventricular outflow tract conduction reserve: a final common pathway? Eur Heart J. 2021. [DOI] [PubMed] [Google Scholar]
- 103.Papavassiliu T, Wolpert C, Fluchter S, Schimpf R, Neff W, Haase KK, et al. Magnetic resonance imaging findings in patients with Brugada syndrome. J CardiovascElectrophysiol. 2004;15(10):1133–8. [DOI] [PubMed] [Google Scholar]
- 104.Papavassiliu T, Veltmann C, Doesch C, Haghi D, Germans T, Schoenberg SO, et al. Spontaneous type 1 electrocardiographic pattern is associated with cardiovascular magnetic resonance imaging changes in Brugada syndrome. Heart Rhythm. 2010;7(12):1790–6. [DOI] [PubMed] [Google Scholar]
- 105.Takagi M, Aihara N, Kuribayashi S, Taguchi A, Kurita T, Suyama K, et al. Abnormal response to sodium channel blockers in patients with Brugada syndrome: augmented localised wall motion abnormalities in the right ventricular outflow tract region detected by electron beam computed tomography. Heart (British Cardiac Society). 2003;89(2):169–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Catalano O, Antonaci S, Moro G, Mussida M, Frascaroli M, Baldi M, et al. Magnetic resonance investigations in Brugada syndrome reveal unexpectedly high rate of structural abnormalities. EurHeart J. 2009;30(18):2241–8. [DOI] [PubMed] [Google Scholar]
- 107.van Hoorn F, Campian ME, Spijkerboer A, Blom MT, Planken RN, van Rossum AC, et al. SCN5A mutations in Brugada syndrome are associated with increased cardiac dimensions and reduced contractility. PloS one. 2012;7(8):e42037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Antzelevitch C. Brugada syndrome: historical perspectives and observations. EurHeart J. 2002;23(8):676–8. [DOI] [PubMed] [Google Scholar]
- 109.Antzelevitch C. Brugada syndrome. Pacing ClinElectrophysiol. 2006;29(10):1130–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Slezak J, Tribulova N, Okruhlicova L, Dhingra R, Bajaj A, Freed D, et al. Hibernating myocardium: pathophysiology, diagnosis, and treatment. Can J Physiol Pharmacol. 2009;87(4):252–65. [DOI] [PubMed] [Google Scholar]
- 111.Antzelevitch C, Yan GX, Ackerman MJ, Borggrefe M, Corrado D, Guo J, et al. J-Wave syndromes expert consensus conference report: Emerging concepts and gaps in knowledge. Heart Rhythm. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Priori SG, Wilde AA, Horie M, Cho Y, Behr ER, Berul C, et al. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm. 2013;10(12):1932–63. [DOI] [PubMed] [Google Scholar]
- 113.Al-Khatib SM, Stevenson WG, Ackerman MJ, Bryant WJ, Callans DJ, Curtis AB, et al. 2017 AHA/ACC/HRS guideline for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Rhythm Society. Heart Rhythm. 2018;15(10):e73–e189. [DOI] [PubMed] [Google Scholar]
- 114.Brugada P, Brugada R, Brugada J. Sudden death in high-risk family members: Brugada syndrome. AmJCardiol. 2000;86(9 Suppl 1):K40–K3. [DOI] [PubMed] [Google Scholar]
- 115.Nademanee K, Veerakul G, Mower M, Likittanasombat K, Krittayapong R, Bhuripanyo K, et al. Defibrillator Versus beta-Blockers for Unexplained Death in Thailand (DEBUT): a randomized clinical trial. Circulation. 2003;107(17):2221–6. [DOI] [PubMed] [Google Scholar]
- 116.Sacher F, Probst V, Maury P, Babuty D, Mansourati J, Komatsu Y, et al. Outcome after implantation of a cardioverter-defibrillator in patients with Brugada syndrome: a multicenter study-part 2. Circulation. 2013;128(16):1739–47. [DOI] [PubMed] [Google Scholar]
- 117.Conte G, Sieira J, Ciconte G, de Asmundis C, Chierchia GB, Baltogiannis G, et al. Implantable cardioverter-defibrillator therapy in brugada syndrome: a 20-year single-center experience. Journal of the American College of Cardiology. 2015;65(9):879–88. [DOI] [PubMed] [Google Scholar]
- 118.Sacher F, Probst V, Bessouet M, Wright M, Maluski A, Abbey S, et al. Remote implantable cardioverter defibrillator monitoring in a Brugada syndrome population. Europace : European pacing, arrhythmias, and cardiac electrophysiology : journal of the working groups on cardiac pacing, arrhythmias, and cardiac cellular electrophysiology of the European Society of Cardiology. 2009;11(4):489–94. [DOI] [PubMed] [Google Scholar]
- 119.Shah AJ, Hocini M, Lamaison D, Sacher F, Derval N, Haissaguerre M. Regional substrate ablation abolishes Brugada syndrome. Journal of cardiovascular electrophysiology. 2011;22(11):1290–1. [DOI] [PubMed] [Google Scholar]
- 120.Sacher F, Jesel L, Jais P, Haissaguerre M. Insight into the mechanism of Brugada syndrome: epicardial substrate and modification during ajmaline testing. Heart Rhythm. 2014;11(4):732–4. [DOI] [PubMed] [Google Scholar]
- 121.Brugada J, Pappone C, Berruezo A, Vicedomini G, Manguso F, Ciconte G, et al. Brugada Syndrome Phenotype Elimination by Epicardial Substrate Ablation. Circ Arrhythm Electrophysiol. 2015. [DOI] [PubMed] [Google Scholar]
- 122.Voskoboinik A, Hsia H, Moss J, Vedantham V, Tanel RE, Patel A, et al. The many faces of early repolarization syndrome: A single-center case series. Heart Rhythm. 2020;17(2):273–81. [DOI] [PubMed] [Google Scholar]
- 123.Antzelevitch C Ion channels and ventricular arrhythmias: cellular and ionic mechanisms underlying the Brugada syndrome. Current Opinion in Cardiology. 1999;14(3):274–9. [DOI] [PubMed] [Google Scholar]
- 124.Minoura Y, Panama BK, Nesterenko VV, Betzenhauser M, Barajas-Martinez H, Hu D, et al. Effect of Wenxin Keli and quinidine to suppress arrhythmogenesis in an experimental model of Brugada syndrome. Heart Rhythm. 2013;10(7):1054–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Belhassen B, Rahkovich M, Michowitz Y, Glick A, Viskin S. Management of Brugada Syndrome: A 33-Year Experience Using Electrophysiologically-Guided Therapy with Class 1A Antiarrhythmic Drugs. Circ Arrhythm Electrophysiol. 2015. [DOI] [PubMed] [Google Scholar]
- 126.Kyriazis K, Bahlmann E, van der Schalk H, Kuck KH. Electrical storm in Brugada syndrome successfully treated with orciprenaline; effect of low-dose quinidine on the electrocardiogram. Europace : European pacing, arrhythmias, and cardiac electrophysiology : journal of the working groups on cardiac pacing, arrhythmias, and cardiac cellular electrophysiology of the European Society of Cardiology. 2009;11(5):665–6. [DOI] [PubMed] [Google Scholar]
- 127.Szel T, Koncz I, Antzelevitch C. Cellular mechanisms underlying the effects of milrinone and cilostazol to suppress arrhythmogenesis associated with Brugada syndrome. Heart rhythm : the official journal of the Heart Rhythm Society. 2013;10(11):1720–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Agac MT, Erkan H, Korkmaz L. Conversion of Brugada type I to type III and successful control of recurrent ventricular arrhythmia with cilostazol. Arch Cardiovasc Dis. 2014;107(8-9):476–8. [DOI] [PubMed] [Google Scholar]
- 129.Kanlop N, Chattipakorn S, Chattipakorn N. Effects of cilostazol in the heart. J Cardiovasc Med (Hagerstown). 2011;12(2):88–95. [DOI] [PubMed] [Google Scholar]
- 130.Abud A, Bagattin D, Goyeneche R, Becker C. Failure of cilostazol in the prevention of ventricular fibrillation in a patient with Brugada syndrome. J CardiovascElectrophysiol. 2006;17(2):210–2. [DOI] [PubMed] [Google Scholar]
- 131.Li GR, Wang HB, Qin GW, Jin MW, Tang Q, Sun HY, et al. Acacetin, a natural flavone, selectively inhibits human atrial repolarization potassium currents and prevents atrial fibrillation in dogs. Circulation. 2008;117(19):2449–57. [DOI] [PubMed] [Google Scholar]
- 132.Di Diego JM, Argenziano M, Tabler M, Chen K, Victoria Messerschmidt, Elliot Jerud, Antzelevitch C. A Whole-heart Model Of Brugada Syndrome. Heart Rhythm. 2020;17. [Google Scholar]
- 133.Antzelevitch C, Patocskai B. Brugada Syndrome: Clinical, Genetic, Molecular, Cellular, and Ionic Aspects. Curr Probl Cardiol. 2016;41(1):7–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Antzelevitch C, Yan GX. Ionic and cellular basis for arrhythmogenesis. In: Yan GX, Kowey PR, editors. Management of Cardiac Arrhythmias. 2nd. ed. New York, NY: Springer; 2011. p. 41–64. [Google Scholar]
- 135.Fish JM, Antzelevitch C. Role of sodium and calcium channel block in unmasking the Brugada syndrome. Heart Rhythm. 2004;1(2):210–7. [DOI] [PMC free article] [PubMed] [Google Scholar]