

Abstract
The opportunistic pathogen Pseudomonas aeruginosa chronically infects the lungs of patients with cystic fibrosis (CF). During infection the bacteria evolve and adapt to the lung environment. Here we use genomic, transcriptomic and phenotypic approaches to compare multiple isolates of P. aeruginosa collected more than 20 years apart during a chronic infection in a CF patient. Complete genome sequencing of the isolates, using short- and long-read technologies, showed that a genetic bottleneck occurred during infection and was followed by diversification of the bacteria. A 125 kb deletion, an 0.9 Mb inversion and hundreds of smaller mutations occurred during evolution of the bacteria in the lung, with an average rate of 17 mutations per year. Many of the mutated genes are associated with infection or antibiotic resistance. RNA sequencing was used to compare the transcriptomes of an earlier and a later isolate. Substantial reprogramming of the transcriptional network had occurred, affecting multiple genes that contribute to continuing infection. Changes included greatly reduced expression of flagellar machinery and increased expression of genes for nutrient acquisition and biofilm formation, as well as altered expression of a large number of genes of unknown function. Phenotypic studies showed that most later isolates had increased cell adherence and antibiotic resistance, reduced motility, and reduced production of pyoverdine (an iron-scavenging siderophore), consistent with genomic and transcriptomic data. The approach of integrating genomic, transcriptomic and phenotypic analyses reveals, and helps to explain, the plethora of changes that P. aeruginosa undergoes to enable it to adapt to the environment of the CF lung during a chronic infection.
Keywords: adaptive gene expression, antibiotic resistance, genetic bottleneck, genome evolution, genome deletion, mutator strain
Impact Statement
Pseudomonas aeruginosa is a bacterium that causes serious and often fatal infections in already ill people, such as those with the genetic disease cystic fibrosis (CF). P. aeruginosa infects the airways of people with CF, impairing lung function, reducing quality of life and shortening life expectancy. Infections can last for years or even decades, during which time the bacteria adapt and evolve to maximize their survival and growth in the lungs. To better understand this process we studied samples of P. aeruginosa from a CF patient who was infected with these bacteria for more than 20 years. We found that during the infection the genomes of the bacteria underwent many changes in genes affecting multiple properties, such as antibiotic resistance, metabolism, mobility and adherence. We compared expression of all of the genes in isolates obtained 20 years apart and found that the genome changes caused altered expression of very many genes. Lastly, we showed that genome and gene expression changes resulted in biological changes that helped the bacteria survive during the infection. This research advances understanding of how P. aeruginosa changes to successfully colonize the lung during infection, and may help develop new treatment strategies.
Data Summary
All raw fastq files from the 15 whole genome sequences and six RNA sequencing samples are available under BioProject accession number PRJNA588274. Genome accession numbers are listed in Table S3. The authors confirm that all supporting data and protocols have been provided within the article and through the supplementary data files.
Introduction
Pseudomonas aeruginosa is a Gram-negative bacterium present in a range of environments. It is also an opportunistic human pathogen, commonly infecting immune-compromised patients [1, 2]. P. aeruginosa is a major cause of increased morbidity in individuals with cystic fibrosis (CF), where it colonizes the lungs [2]. Patients are typically infected by P. aeruginosa from the environment, which can act as a reservoir for infection [3]. Infections in CF patients commonly become chronic, with the infecting bacteria adapting to survive antibiotic treatment and the environment of the host lung [4, 5].
In P. aeruginosa , antibiotic resistance is multifactorial with a range of genetic events responsible for the development of resistance [6, 7]. Mutations altering target proteins reduce the binding affinities of antibiotics. For example, mutations in gyrA or gyrB that encode subunits of DNA gyrase lead to fluoroquinolone resistance [8], and mutations in ftsI that encodes penicillin-binding protein 3 contribute to meropenem resistance [9, 10]. Decreased uptake of antibiotics can occur due to changes in membrane permeability. For example, mutations in oprD that encodes a porin contribute to carbapenem resistance [11] and changes to lipopolysaccharide (LPS) increase tolerance to polymyxins and aminoglycosides [12, 13]. Increased antibiotic efflux can arise due to increased expression of efflux systems, arising from mutations affecting regulatory proteins such as MexZ [6], and increased production of AmpC β-lactamase can result from mutations in genes such as mpl [6, 14]. Although antibiotic resistance in P. aeruginosa in CF predominantly occurs through mutations in chromosomally encoded genes, horizontal gene transfer can also introduce genes that confer a resistant phenotype [15, 16].
As well as resisting antibiotic treatment, during chronic infection in CF patients P. aeruginosa adapts to other challenges of the lung environment, including oxidative stress, the immune system, host withholding of micronutrients such as iron and zinc, and competition from other microbiota [17, 18]. The bacteria can undergo multiple phenotypic changes including changes to metabolism, loss of motility, increased production of the extracellular polysaccharide alginate, reduced virulence factor and siderophore production, emergence of auxotrophs, and occurrence of small colony variants and hypermutator strains [4, 5, 19–25]. The advent of high-throughput DNA sequencing technologies has allowed analysis of the genetic changes that underlie phenotypic changes during P. aeruginosa infections in CF. The first such study identified multiple mutations associated with antibiotic resistance and immune evasion over the course of 8 years of infection [26]. Subsequent studies have also followed the progression of genomic changes undergone by P. aeruginosa during chronic infection in CF [19, 21, 22, 27–31] and non-CF patients [32], including gene loss and acquisition [33], and have identified many other genes that undergo mutation during the course of infection. Some studies have also investigated associated transcriptomic and phenotypic changes [22, 34, 35]. However, our understanding of the relationship between genome-wide genetic changes, consequent changes to the transcriptome and the resulting phenotypic changes that facilitate adaptation to the lung environment is limited. The aim of this study, therefore, was to investigate in detail the evolution of P. aeruginosa during infection over the course of 20 years in the lungs of a patient with CF, comparing multiple isolates and merging phenotypic, transcriptional and genotypic changes to understand how P. aeruginosa evolved during the course of the infection.
Methods
Sample isolation
Multiple sputum samples were collected from a single patient located in Dunedin, New Zealand, in 1991 when the patient was aged 26 years, and from the same patient in 2011–2012. The patient was male with CF genotype Δ508/Δ508. Single P. aeruginosa colonies were obtained from sputum inoculated onto cetrimide agar [36]. A summary of lung function is provided in Table S1 (available in the online version of this article) and a summary of antibiotics given to the patient between first and last sampling is given in Table S2.
Minimum inhibitory concentration
Antibiotic resistance of all strains to multiple antibiotics were determined on Difco Mueller-Hinton (MH) agar [37]. Colonies were selected and grown in Difco MH broth. Overnight cultures were serially diluted and plated using an inoculum of approximately 104 c.f.u. per spot onto MH agar containing doubling concentrations of antibiotics. The minimum inhibitory concentration (MIC) was recorded as the lowest concentration of antibiotic that inhibited bacterial growth (mg l−1). Susceptibilities for meropenem (Penembact; Venus Remedies), tobramycin (Mylan New Zealand) and ciprofloxacin (Cipflox; Mylan New Zealand) were tested in duplicate.
Swimming motility
Motility of the bacteria was assessed using a previously described method [38]. In short, M8 medium containing 0.3 % agar was inoculated by stabbing into the agar with a pipette tip that had been dipped in culture (with an OD600nm of 1.0) and incubated at 37 °C for 24 h. Plates were then imaged and the area of motility was determined using ImageJ [39].
Quantification of pyoverdine production
Bacteria were grown in King’s B broth [40] in 96-well clear-bottomed, black walled tissue culture plates (Corning). The microtitre plates were incubated in a BMG FLUOstar Omega microplate reader at 37 °C with shaking at 200 r.p.m. for 24 h. Optical density (OD600nm) was recorded every 30 min, to monitor the growth of the isolates. Simultaneously, pyoverdine was measured spectrophotometrically using a fluorescence assay, with excitation at 410 nm and emission at 460 nm. The pyoverdine production values used were the fluorescence after 24 h normalized to the OD600 of the final culture.
Biofilm formation assay
Measurement of biofilm formation via crystal violet staining was performed as described previously [41, 42]. In short, isolates were grown in M63 medium supplemented with 0.4 % arginine to promote biofilm formation [42]. Cultures were grown for 24 h in U-bottomed 96-well plates (Corning), at 37 °C without shaking, and OD600nm was measured. Wells were washed out with water to remove planktonic cells and biofilms were stained with crystal violet. Acetic acid was used to dissolve the stain into an aqueous solution for measurement. Measurements were made at 550 nm and normalized to the OD600nm prior to staining.
Growth rate assay
Growth of bacterial cultures in LB or synthetic CF nedium (SCFM) [43] in 96-well plates, incubated at 37 °C with shaking, was measured as described previously [9]. The OD600 was measured every 30 min for 18 h for growth in LB, and for 24 h in SCFM.
DNA extraction and quality check
An isolated colony was inoculated in brain heart infusion (BHI) broth and grown at 35 °C without agitation. Cells were harvested by centrifugation for 10 min at 5000 g to have approximately 2×109 cells per pellet. Supernatant was discarded and pellets were placed at −80 °C until DNA extraction. DNA extractions were performed using DNeasy Blood and Tissue kit (Qiagen) following the manufacturer’s protocol, with the following additional steps. First, to avoid DNA shearing, all vortexing steps were replaced by tube inversions. After the bacterial lysis step, RNA was digested by adding 10 µl of RNase cocktail (Invitrogen) to the lysate and incubating at room temperature for 5 min. A second washing with 500 µl of AW2 buffer was performed instead of one wash. Finally, DNA was eluted twice with 50 µl of 10 mM Tris-HCl pH 8. Extracted DNA was quantified by the Qubit dsDNA BR method (Invitrogen) and purity (based on 260/280 and 260/230 absorbance ratios) was measured with a Nanodrop 2000 spectrophotometer (Thermo Fisher).
Illumina MiSeq genome analysis
Short paired-end reads (2×300 bp) were obtained for each sequenced strain by Illumina MiSeq sequencing as previously described [44, 45]. Paired-end sequencing reads were checked using FastQC version 0.11.5. Trimmomatic version 0.36 [46] was used to filter the raw Illumina short reads and remove adapter sequences. Post-trimming, all reads used subsequently were at least 20 nt long, with a minimum average phred score of >20, and paired. Draft genome assemblies were created using SPAdes version 3.12.0 [47] (Table S3).
Oxford Nanopore sequencing
One microgram of high-molecular-weight genomic DNA from each P. aeruginosa isolate was transferred into a 96-well microtitre PCR plate and volumes were adjusted to 48 µl using nuclease-free water. DNA repair and end-prep and native barcode ligation were performed as in Oxford Nanopore’s instructions for native barcoding genomic DNA version NBE_9065_v 109_revG_23May2018 using an LSK-109 sequencing kit, native barcode expansions and reagents (New England Biolabs) specified in the protocol. Barcoded and purified DNA was quantified with Qubit and 50 ng of each were pooled together to obtain ca. 900 ng of DNA in the pool. Volume was adjusted to 65 µl with nuclease-free water. Adapter ligation and clean-up were performed following the manufacturer’s instructions. Pooled libraries were purified with SparQ PureMag beads (QuantaBio) and eluted by suspending beads in 14 µl of 10 mM Tris-HCl (pH 8) on the MixMate (24 °C, 10 min, 1000 r.p.m.). One microlitre of eluate was kept for Qubit quantification. A total of 379 ng of pooled libraries was loaded on a MinION FLO-MIN106 flow cell (v. R9.4). Raw signal acquisition was performed with Oxford Nanopore’s proprietary software MinKNOW version 8.3.1 (Oxford Nanopore Technologies), with the basecalling option turned off, run length set at 72 h and time between mux scans set at 3 h. The raw sequencing data (in fast5 format) were basecalled and demultiplexed post-sequencing using Guppy v.4.2.2-gpu [48].
Hybrid genome assembly
Long nanopore reads of over 1000 bp in length and mean Phred score >9 were selected with NanoFilt v2.3.0 [49] and adapter sequences were trimmed with poreChop v0.2.4 (https://github.com/rrwick/Porechop). The Unicycler assembly pipeline v0.4.8 [50] was used to perform a hybrid genome assembly in which Illumina short reads were assembled into contigs and then bridged with MinION long reads to form complete and circular replicons. Assembly statistics (number of contigs, total bases, longest contigs, length N50, etc.) were calculated and compiled using assembly-stats v1.0.1 (https://github.com/sanger-pathogens/assembly-stats) and in-house Unix shell scripts.
Core genome analysis
To generate a core genome (defined as DNA that is present in all isolates), complete genome assemblies were compared using parsnp version 1.2 from the Harvest suite 1.1.2 [51]. E-S2239-15 was excluded from core genome generation as it is a genetic outlier. The core genome covered on average 96 % of the remaining assemblies. Genome comparisons used for the generation of Figs 2 and S1 were visualized using gingr version 1.2 from the Harvest suite, and an SNP cladogram from the differences between isolates was visualized using FigTree version 1.4.4 [52]. Assemblies were compared using BRIG version 0.95 [53]. Gene prediction was performed on genome assemblies using Prokka version 1.13 [54] using bacterial annotation and PAO1 homologue gene IDs as PAO1 is the most closely related reference strain. Multi-locus sequence typing (MLST) for each isolate was determined using pubMLST [55].
Novel region discovery
To identify new regions of DNA obtained from the transition of earlier to later isolates, Panseq version 3.1.0 [56] was used. Novel regions with a minimum length of 500 bp were identified by comparing early isolate E-S2239-16 with all other isolates. To identify any resistance-associated genes that may have been acquired by horizontal gene transfer, ResFinder version 2.3 [57] was used.
Variant analysis
Indels, duplications and small variants were analysed as previously described [9], using Breseq version 0.30.0 [58] with E-S2239-16 as a reference for all analyses. All Breseq outputs (variants called between the isolate of interest and E-S2239-16) were extracted and compared to each other using GDtools-GDcompare as part of the Breseq package.
RNA sequencing (RNAseq) analysis
Three biological replicate cultures of representative isolates E-S2239-16 and L-001–1C were grown in SCFM to exponential growth phase (OD600nm of 0.4–0.6) and RNA was extracted using the GeneJET RNA Purification kit, according to the manufacturer’s protocol. RNAseq ScriptSeq libraries were prepared from rRNA-depleted RNA samples using a ScriptSeq Complete Bacterial Kit and sequenced as 125 bp paired-end reads (average of 15 million reads per sample) using an Illumina HiSeq 2000 by New Zealand Genomics.
Reads were trimmed using Trimmomatic as for whole-genome sequencing. Trimmed reads were mapped onto the annotated E-S2239-16 genome using Kalisto version 0.44.0 [59], using 1000 bootstraps (Table S4). Differential gene expression was assessed using Sleuth version 0.30.0 [60] in R version 3.6.0 [61] (Fig. S2). A log2 transformation function was done in Sleuth to output log2 fold change between E-S2239-16 and L-001–1C, and a false discovery rate (FDR)-adjusted P value of ≤0.01 was considered to indicate significantly differentially expressed.
Results
A single P. aeruginosa lineage extends through a 20-year infection
Four earlier (1991 – sample prefix E) and 11 later (2012–2013 – sample prefix L) isolates of P. aeruginosa were obtained from an individual with CF (Table 1, Table S1). The patient suffered a decline in lung function, as shown by reduced forced expiratory volume (FEV1) and indicating advancing lung pathology, between collection dates of the earlier and later samples (Table S1). The patient was treated with a wide range of antibiotics (Tables S1 and S2) and the later isolates had on average higher antibiotic resistance than the earlier isolates (Table 1). The complete genome sequences of the isolates were determined, and details of genome assemblies are given in Table S3. A cladogram was reconstructed based on the DNA shared between isolates (Fig. 1a; Fig. S1). This showed that one isolate, E-S2239-15, is a genetic outlier (MLST type 110) and the remaining isolates are clonal (MLST type 244). The three other earlier isolates are closely related, with a median of 12 non-synonymous differences and 12 synonymous or intergenic differences (Tables S5 and S6). The later isolates are genetically very similar to three of the earlier isolates (Fig. 1b; Fig. S1; Tables S5 and S6) although they had higher genetic diversity (median of 190 non-synonymous differences for all pairwise comparisons between the later isolates) than the three clonal earlier isolates. There was no evidence of later isolates descended from E-S2239-15, nor was there any evidence of isolates that had been independently acquired after the earlier infecting strains. These findings indicate the same infecting lineage was present in the patient for over 20 years.
Table 1.
Antibiotic minimum inhibitory concentrations (mg l–1) of isolates in this study
|
Isolate |
Collection date (MM/YY) |
Ciprofloxacin |
Meropenem |
Tobramycin |
Ceftazidime |
|---|---|---|---|---|---|
|
E-S2239-16 |
12/91 |
0.5 |
0.25 |
0.25 |
1 |
|
E-MSB2494 |
12/91 |
1* |
0.25 |
0.25 |
1 |
|
E-MSB3405 |
12/91 |
1 |
0.125 |
0.25 |
0.5 |
|
E-S2239-15 |
12/91 |
1 |
1 |
2 |
1 |
|
L-001–1A |
08/12 |
2 |
16 |
8 |
4 |
|
L-001-1B |
08/12 |
4 |
16 |
2 |
16 |
|
L-001–1C |
08/12 |
2 |
8 |
8 |
4 |
|
L-001–2A |
10/12 |
2 |
8 |
8 |
8 |
|
L-001-2B |
10/12 |
2 |
8 |
8 |
4 |
|
L-001–3A |
11/12 |
2 |
8 |
4 |
4 |
|
L-001-3B |
11/12 |
2 |
8 |
2 |
4 |
|
L-001–4 |
01/13 |
2 |
8 |
2 |
4 |
|
L-001–5A |
04/13 |
1 |
16 |
4 |
4 |
|
L-001-5B |
04/13 |
2 |
8 |
2 |
16 |
|
L-001–6 |
05/13 |
2 |
16 |
2 |
8 |
*Antibiotic resistance is shown in bold type. Resistance breakpoints (eucast.org) are: ciprofloxacin, >0.5; meropenem, >8; tobramycin, >2, ceftazidime, >8.
Fig. 1.
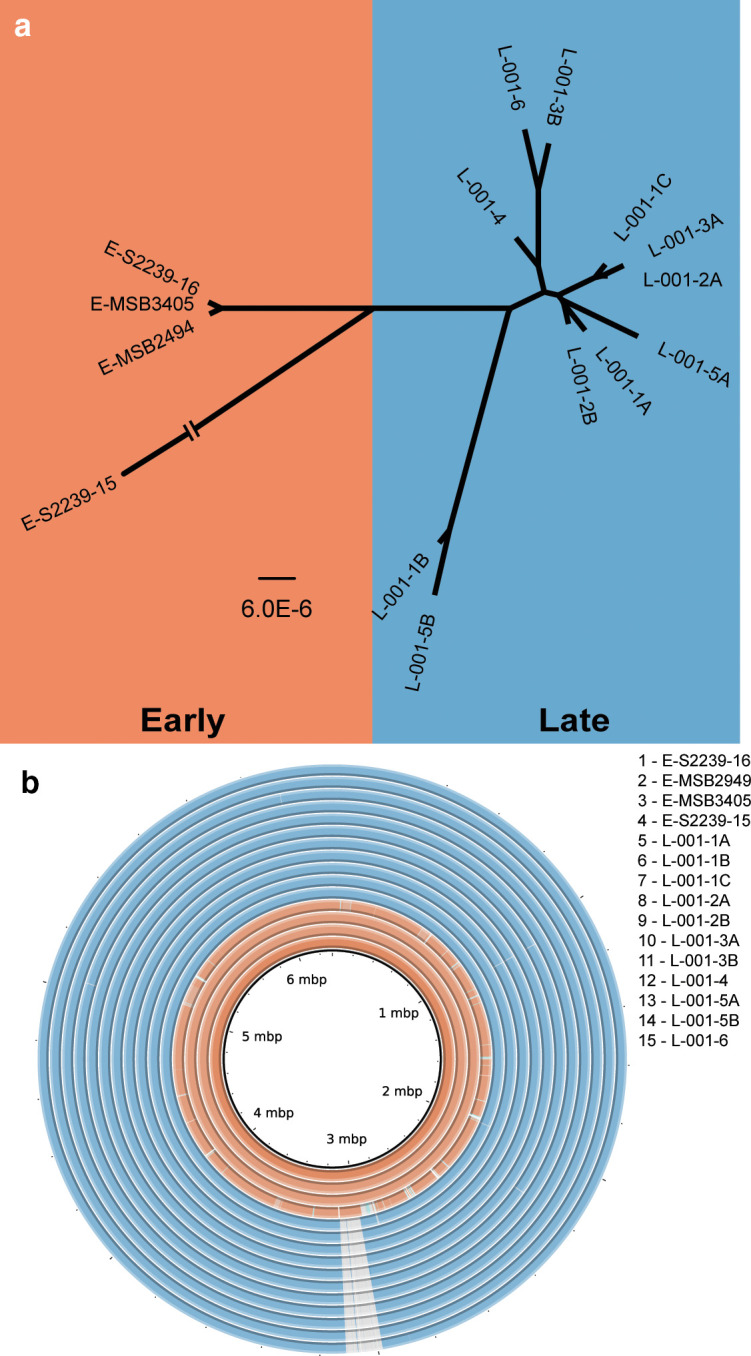
Whole genome analysis of earlier and later isolates. (a) Dendogram representing relationships between isolates, with branch lengths representing the number of nucleotide differences (bar, 6.0×10−6 nt differences per site). The strain E-S2239-15 branch is not to scale as it is distinct from all other isolates (Fig. 1). (b) Genome comparison of earlier (orange) and later (blue) isolates. Genomes were aligned using BRIG with the innermost ring being reference strain E-S2239-16. Grey hatching indicates absence of DNA that is present in the reference strain. Regions in darker shades are 100 % identical, with a lighter shade being 99 % identical.
Acquisition and loss of DNA in the later isolates was analysed. All later isolates have lost a 125 kb region of the genome (Fig. 1b). This region includes 130 predicted genes, corresponding to PA2229–PA2359 in P. aeruginosa PAO1. A list of the deleted genes is included in Table S7. The deleted region overlaps with deletions that have occurred on multiple occasions during chronic P. aeruginosa infections in patients with CF or non-CF bronchiectasis [32, 62]. Many of the deleted genes encode regulatory proteins or enzymes that are involved in catabolic processes and nutrient acquisition, reflecting the myriad metabolic changes that occur in P. aeruginosa during adaptation to the nutrient conditions of the CF lung environment [63]. Genes required for synthesis of psl polysaccharide, one of three extracellular polysaccharides that can be made by P. aeruginosa and that influences biofilm formation in vitro [64–66], are also deleted. The loss of psl genes, in multiple isolates, suggests significant differences between biofilm architecture in vitro, and during long-term infection in the lung. The amb genes required for synthesis of a toxin, aminomethoxybutenoic acid (AMB), by P. aeruginosa [67] have also been lost, consistent with reduced virulence that is often associated with isolates from long-term infections [23].
All later isolates have a sequence of approximately 44 kb that shares 98 % identity to Pseudomonas phage LKA5. The absence of this phage from earlier isolates indicates that it was acquired during infection. We did not identify any acquired genes associated with antibiotic resistance or with known roles in pathogenicity.
Four later isolates also had an inversion of a large (0.9 Mb) portion of the genome relative to the earlier isolates (Fig. S3). Recombination endpoints were in rRNA (rrn) operons, which provide repeat sequences that can serve as inversion endpoints in P. aeruginosa [68]. Genomic inversions have been reported previously in isolates of P. aeruginosa from long-term infections in individuals with CF [69], although their impact of the bacteria is not fully understood.
Genetic differences between isolates reveal mutations that enhance infectivity
Identification of all genetic differences between the 15 clonal isolates was performed using earlier isolate E-S2239-16 as a reference. A total of 892 different variants were identified across all 15 isolates with an average of 298 variants per isolate, and an average of 321 mutations per late isolate (Table S5). Of these 892 variants, 562 were non-synonymous differences that affected a total of 515 genes. The remaining variants were either synonymous or changes in intergenic regions of the genome. In total, 552 non-synonymous variants were only present in later isolates and are very likely to be mutations that arose during infection. The later isolates showed a mutation rate of approximately 16 mutations per year relative to the earlier isolates, although this value may be an underestimate due to the probable population bottleneck between the earlier and later isolates.
Later isolates in our study contain a different variant than ancestral isolates (M185K) in the mutY gene. The MutY protein reduces the frequency of GC→TA transversions that can arise following oxidation of guanine bases in DNA, and mutY mutants of P. aeruginosa have up to a 7.5-fold higher mutation rate than the wild-type [70]. A high proportion of the single nucleotide mutations (79.2%) were GC→TA transversions (Table S5; Table S8) so that it is likely that the MutY M185K variant has reduced or no function.
A large number of genes can undergo mutation to increase antibiotic tolerance as P. aeruginosa evolves during infection in CF [19, 22, 28, 71]. Comparison of earlier and later isolates showed that many of these genes underwent mutation during infection (Table 2; Table S5). For example, resistance to meropenem is associated with mutations in the porin-encoding gene oprD [72], and all later isolates had a nonsense mutation in this gene. Most of the later isolates also had at least one mutation in the ftsI gene that encodes a meropenem target protein, PBP3 [73, 74], and mutations in this gene also contribute to meropenem resistance [10]. These findings are consistent with the higher MIC values of all later isolates for meropenem (Table 1). All later isolates also have a mutation in fusA1 that encodes elongation factor G, and nine later isolates have a mutation in fusA2. Consistent with the higher MICs of later isolates for tobramycin, mutations in fusA1 confer resistance to aminoglycosides [28, 75] and fusA2 mutations have also been suggested to be involved in aminoglycoside resistance [76]. Later isolates also have higher MICs for ciprofloxacin, and these isolates contained a DNA gyrase mutation (gyrB E469D) and mutations in gyrB are associated with fluoroquinolone resistance [8]. Curiously, none of the later isolates had mutations in gyrA associated with fluoroquinolone resistance, whereas two of the earlier isolates contained a gyrA variant (T83A) that increases tolerance to fluoroquinolones [8, 10, 77–79]. Lastly, mutations in mpl contribute to ceftazidime resistance [80] and all later isolates have a mutation in this gene and higher MICs for ceftazidime. Overall, the mutations that were present in the later isolates were consistent with higher resistance of the bacteria to antibiotics.
Table 2.
Non-synonymous changes in genes associated with adaptation to the lung environment, relative to earlier isolate E-S2239-16*
|
Gene |
Description |
E-MSB2494 |
E-MSB3405 |
L-001-1A |
L-001-1B |
L-001-1C |
L-001-2A |
L-001-2B |
L-001-3A |
L-001-3B |
L-001-4 |
L-001-5A |
L-001-5B |
L-001-6 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Antibiotic resistance |
|
|
||||||||||||
|
ftsI |
penicillin-binding protein 3 |
|
|
G63C |
G63C R504C |
G63C R504C |
G63C |
G63C R504C |
G63C |
G63C |
G63C |
G63C |
||
|
fusA1 |
elongation factor G |
|
|
G611V |
G611V |
G611V |
G611V |
G611V |
G611V |
G611V |
G611V |
G611V |
G611V |
G611V |
|
fusA2 |
elongation factor G |
|
|
G252V |
G252V |
G252V |
G252V |
G252V |
G252V |
G252V |
G252V |
G252V |
||
|
gyrA |
DNA gyrase subunit A |
A83T |
|
A83T |
A83T |
A83T |
A83T |
A83T |
A83T |
A83T |
A83T |
A83T |
A83T |
A83T |
|
gyrB |
DNA gyrase subunit B |
|
|
E468D |
E468D G89V |
E468D |
E468D |
E468D |
E468D |
E468D |
E468D |
E468D |
E468D G89V |
E468D |
|
mexB |
RND multidrug efflux transporter MexB |
|
|
643L |
643L |
643L |
643L |
643L |
643L |
643L |
643L |
643L |
643L |
643L |
|
mexZ |
transcriptional regulator MexZ |
|
|
Δ1bp |
Δ1bp |
Δ1 bp |
Δ1 bp |
Δ1 bp |
Δ1 bp |
Δ1 bp |
Δ1 bp |
Δ1 bp |
Δ1 bp |
Δ1 bp |
|
mpl |
UDP-N-acetylmuramate: l-alanyl-gamma-d-glutamyl-meso-diaminopimelate ligase |
|
|
E126 |
A20D |
E126 |
E126 |
E126 |
E126 |
E126 |
E126 |
E126 |
A20D |
E126 |
|
oprD |
outer membrane porin OprD |
|
|
E140 |
E182 |
E140 E182 Δ13 bp |
E140 E182 Δ13 bp |
E140 |
E140 E182 Δ13 bp |
E140 |
E140 |
E140 |
E182 |
E140 |
|
Physiology |
|
|
||||||||||||
|
algG |
alginate-c5-mannuronan-epimerase AlgG |
|
|
F398L A499T |
F398L A499T |
F398L A499T |
F398L A499T |
F398L A499T |
F398L A499T |
F398L A499T |
F398L A499T |
F398L A499T |
F398L A499T |
F398L A499T |
|
algU |
sigma factor AlgU |
Q48 |
Δ1bp |
Q30K |
A193S |
Q30K |
||||||||
|
ccoN1 |
Cytochrome c oxidase cbb3-type CcoN subunit |
|
|
S78 |
S78 |
S78 |
S78 |
S78 |
S78 |
D48G |
S78 |
S78 |
S78 |
D48G |
|
fleQ |
transcriptional regulator FleQ |
|
|
G382W |
G382W |
G382W |
G382W |
G382W |
G382W |
G382W |
G382W |
G382W |
G382W |
G382W |
|
fliA |
sigma factor FliA |
|
|
D49A |
D49A |
D49A |
||||||||
|
lasR |
transcriptional regulator LasR |
Q45 |
|
W195L |
G31 |
W195L |
W195L |
W195L |
W195L |
W195L |
W195L |
W195L |
G31 |
W195L |
|
mucA |
anti-sigma factor MucA |
|
|
118Q |
118Q |
118Q |
118Q |
118Q |
118Q R168C |
118Q |
118Q |
118Q |
118Q |
118Q |
|
mucB |
negative regulator for alginate biosynthesis MucB |
|
|
ins 1 bp |
ins 1 bp |
ins 1 bp |
ins 1 bp |
ins 1 bp |
ins 1 bp |
ins 1 bp |
ins 1 bp |
ins 1 bp |
ins 1 bp |
ins 1 bp |
|
mvaT |
transcriptional regulator MvaT |
|
|
W107R |
W107R |
W107R |
W107R |
W107R |
W107R |
W107R |
W107R |
W107R |
W107R |
W107R |
|
pvdA |
l-ornithine N5-oxygenase |
Q327 |
|
E195 |
||||||||||
|
pvdD |
pyoverdine synthetase D |
|
C1559Y |
A306S |
Δ12 bp |
C1559Y |
A306S |
|||||||
|
pvdH |
l-2,4-diaminobutyrate:2-ketoglutarate 4-aminotransferase |
|
|
ins 4 bp |
||||||||||
|
pvdN |
aminotransferase PvdN |
|
|
V274G |
Δ1 bp |
Δ1 bp |
||||||||
|
pvdS |
sigma factor PvdS |
|
|
E85 |
E85 |
|||||||||
|
wbpL |
glycosyltransferase WbpL |
|
ins 1 bp |
ins 1 bp |
ins 1 bp |
ins 1 bp |
ins 1 bp |
ins 1 bp |
ins 1 bp |
ins 1 bp |
ins 1 bp |
ins 1 bp |
ins 1 bp |
ins 1 bp |
*Variants in key genes are listed here. A complete listing of gene variants is given in Table S5.
Mutations in the later isolates (Table S5; Table 2) revealed other genes with a role in adaptation of P. aeruginosa to the lung environment. Isolates of P. aeruginosa from chronic infections can lack O-antigen polysaccharide [81] and all later isolates have a frameshift mutation in wbpL, probably preventing O-antigen synthesis. All later isolates had mutations in the global regulators lasR and mvaT that influence a range of phenotypes, including biofilm formation and production of virulence factors. The later isolates also had mutations in the muc and alg genes that are associated with alginate production, in fleQ and fliA associated with flagella production, and in genes associated with iron acquisition such as pvdD and other pvd genes that are involved in the production of a siderophore, pyoverdine.
Although many mutations were in genes with known functions, over half of the mutations were in genes of unknown function, or for which a possible function has been assigned on the basis of sequence similarity but not experimentally investigated. It is likely that mutations in some of these uncharacterized genes contribute to the phenotypes described above, and others may contribute to different phenotypic changes associated with adaptation to the lung environment or to different micro-niches within the lung.
Differential gene expression between an earlier and a later isolate
RNAseq of an earlier and a later isolate was carried out on bacteria grown in a synthetic CF sputum medium [43] to identify pathways and genes that had altered expression following 20 years of infection. Of 5630 genes expressed in both isolates, 34 had significantly lower expression in the later isolate and 170 had significantly higher expression (Table S7; Fig. S2). Genes of known function showing large differences in expression are summarized in Table 3. Most of the genes that have reduced expression are involved in flagellar synthesis, consistent with the presence of a mutation in the transcriptional regulator fleQ. The gene with the most highly reduced expression that is present in both isolates, sfnG, encodes an enzyme involved in oxygenation of dimethylsulfone [82], but the biological significance of its reduced expression in the context of infection in CF is not clear.
Table 3.
Differentially expressed genes between earlier isolate E-S2239-16 and later isolate L-001-1C*
|
Gene |
Log2 fold-change |
FDR adjusted P-value |
Gene description |
|---|---|---|---|
|
Reduced expression in later isolate | |||
|
Motility |
|
||
|
flgG |
−3.79 |
1.44×10−03 |
flagellar basal-body rod protein FlgG |
|
flgF |
−3.71 |
3.82×10−03 |
flagellar basal-body rod protein FlgF |
|
flgH |
−3.43 |
8.79×10−04 |
flagellar L-ring protein precursor FlgH |
|
flgI |
−3.34 |
2.46×10−05 |
flagellar P-ring protein precursor FlgI |
|
flgK |
−3.16 |
3.82×10−04 |
flagellar hook-associated protein 1 FlgK |
|
fleR |
−2.97 |
1.54×10−04 |
two-component response regulator |
|
flgJ |
−2.91 |
9.16×10−05 |
flagellar protein FlgJ |
|
fliF |
−2.68 |
4.18×10−07 |
flagella M-ring outer membrane protein precursor |
|
Physiology |
|
||
|
sfnG |
−4.93 |
3.01×10−20 |
FMNH2-dependent monooxygenase, SfnG |
|
mexI |
−2.85 |
1.05×10−04 |
RND efflux transporter |
|
Increased expression in later isolate | |||
|
Nutrient acquisition and utilization |
|
||
|
aceA |
4.14 |
9.76×10−08 |
isocitrate lyase AceA |
|
fptA |
2.95 |
3.85×10−03 |
Fe(III)-pyochelin outer membrane receptor |
|
pchA |
2.61 |
5.51×10−03 |
salicylate biosynthesis isochorismate synthase |
|
pchB |
2.45 |
3.67×10−06 |
salicylate biosynthesis protein PchB |
|
pchC |
2.77 |
1.98×10−06 |
pyochelin biosynthetic protein PchC |
|
pchD |
3.01 |
9.99×10−04 |
pyochelin biosynthesis protein PchD |
|
pchG |
2.27 |
2.10×10−03 |
pyochelin biosynthetic protein PchG |
|
rpmE2 |
4.69 |
9.94×10−08 |
Zinc-independent paralog of ribosomal L31 protein |
|
dksA2 |
4.23 |
3.17×10−12 |
transcriptional regulator |
|
zrmD |
3.10 |
1.13×10−17 |
secretion of zinc metallophore |
|
zrmC |
2.93 |
1.50×10−03 |
biosynthesis of zinc metallophore |
|
zrmB |
4.26 |
1.59×10−11 |
biosynthesis of zinc metallophore |
|
zrmA |
4.40 |
4.56×10−11 |
outer membrane receptor for zinc metallophore |
|
Cell surface and adhesion |
|
||
|
algA |
3.34 |
7.36×10−07 |
phosphomannose isomerase / guanosine 5'-diphospho-d-mannose pyrophosphorylase |
|
algD |
2.43 |
1.705×10−03 |
GDP-mannose 6-dehydrogenase AlgD |
|
cdrA |
2.28 |
2.53×10−03 |
cyclic diguanylate-regulated TPS partner A |
|
lecB |
3.02 |
2.86×10−05 |
fucose-binding lectin PA-IIL |
|
lptF |
3.80 |
1.21×10−06 |
lipotoxin F, LptF |
|
mucB |
2.80 |
3.16×10−07 |
negative regulator for alginate biosynthesis MucB |
|
Pathogenicity |
|
||
|
aprA |
3.24 |
2.44×10−04 |
alkaline metalloproteinase |
|
hcp1 |
2.67 |
9.39×10−15 |
Hcp1 |
|
tagQ1 |
2.71 |
1.02×10−09 |
TagQ1 |
|
tssB1 |
2.25 |
1.71×10−06 |
TssB1 |
|
tssC1 |
2.44 |
1.56×10−07 |
TssC1 |
|
Antibiotic resistance |
|
||
|
ampC |
2.97 |
2.42×10−09 |
β-lactamase |
|
mexZ |
2.73 |
5.11×10−09 |
negative regulator of mexXY efflux pump genes MexZ |
|
Stress response |
|
||
|
ibpA |
3.50 |
3.63×10−13 |
heat-shock protein IbpA |
|
katE |
2.98 |
1.44×10−07 |
catalase HPII |
|
lexA |
3.62 |
2.48×10−06 |
repressor protein LexA |
|
sulA |
3.38 |
4.52×10−04 |
SulA |
|
Lifestyle regulation |
|
||
|
pqsA |
2.48 |
1.02×10−05 |
PqsA |
|
pqsB |
2.32 |
1.34×10−03 |
PqsB |
|
pqsC |
2.18 |
3.40×10−04 |
PqsC |
|
pqsD |
2.44 |
7.65×10−06 |
3-oxoacyl-[acyl-carrier-protein] synthase III |
|
pqsE |
2.29 |
8.34×10−05 |
quinolone signal response protein |
|
sbrI |
1.51 |
2.51×10−03 |
SbrI |
|
sbrR |
1.33 |
1.43×10−03 |
SbrR |
*RNAseq was carried out on earlier isolate E-S2239-16 and later isolate L-001-1C. A complete listing of all genes is given in Table S7.
Genes that had significantly increased expression in the later isolate reveal a wide range of functions associated with host–pathogen interactions, including genes involved in nutrient acquisition. Amongst those with the highest increase in expression are multiple genes involved in zinc scavenging (zrmA-D, also known as cntI-O) and the zinc starvation response (dksA2 and rpmE2), consistent with high levels of expression of these genes by P. aeruginosa during chronic lung infection in CF [83]. The pch and fptA genes required for synthesis and uptake of an iron-scavenging siderophore, pyochelin, also had higher expression in the later isolate. The aceA gene that encodes isocitrate lyase also had higher expression in the later isolate, consistent with the key role of this gene in utilization of acetate and fatty acids as carbon sources [84].
As well as genes involved in nutrient acquisition and utilization, multiple genes involved in biofilm formation and adhesion to host cells had higher expression in the later isolate. These include genes associated with synthesis of cell-surface molecules, including lecB (lectin), cdrA (an adhesin) and lptF (an outer membrane protein associated with host cell attachment). Some though not all genes involved in alginate production also had higher expression in the later isolate.
Several genes associated with stress responses (lexA, sulA, katE, pfpI and ibpA) had higher expression in the later isolate, probably reflecting at least in part ongoing exposure of the bacteria to the host immune system. Perhaps as part of this response, or perhaps reflecting interactions with other bacteria, some Type VI secretion system genes (tssB1, tssC1, hcp1 and tagQ1) as well as the protease-encoding aprA gene had higher expression in the later strain. The complex nature of bacterial adaptation to the environment of the host lung is also shown by the higher expression of genes required for synthesis of the Pseudomonas quinolone signal (PQS). PQS is a quorum sensing molecule that contributes to regulation of a range of different functions, but is also cytotoxic, alters the host immune response and contributes directly to iron acquisition [85]. The SbrIR sigma/antisigma factor system that controls swarming activity also had higher expression in the later isolate.
Altered gene expression can also contribute to resistance of P. aeruginosa to antibiotics. Isolates of P. aeruginosa from chronically infected patients often have increased expression of the ampC gene, which is associated with resistance to cephalosporins such as ceftazidime. This is true of the later isolate, which had increased expression of ampC. A major factor in the resistance of P. aeruginosa to antibiotics is increased expression of efflux pump genes [6]. None of the efflux genes had significantly higher expression in the later isolate. The mexXY genes had increased expression, consistent with the presence of a mutation in the regulator gene mexZ, but this did not reach statistical significance (Table S7).
Transcriptomic differences between the earlier and later isolate demonstrate the wide range of changes that P. aeruginosa undergoes while adapting to the host lung environment. It is important to note that the majority of the 170 genes that had significantly higher expression in the later isolate, including some of those with the largest differences in expression, do not have well-defined functions. Intriguingly, two of the genes showing highest increase in expression are a non-coding RNA of unknown function (P8) and a tRNA gene (PA4581.1), but the biological significance of this is not known.
Phenotypic changes between earlier and later isolates
The patient was treated with a wide range of antibiotics during infection (Tables S1 and S2). Genome analysis (Table 2; Table S5) showed that later isolates had acquired mutations likely to increase the ability of the bacteria to tolerate carbapenem, fluoroquinolone, cephalosporin and aminoglycoside antibiotics. Resistance to meropenem (a carbapenem), ciprofloxacin (a fluoroquinolone), ceftazidime (a cephalosporin) and tobramycin (an aminoglycoside) was therefore tested for all isolates. Of the 11 later isolates, four were clinically resistant to all antibiotics tested (Table 1). Only two later isolates were not resistant to more than one class tested. Earlier isolates were all sensitive to tobramycin and meropenem and were inhibited by lower concentrations of ciprofloxacin than later isolates.
P. aeruginosa commonly adapts to the host lung environment through altered biofilm formation, reflected in altered adherence properties and associated with reduced motility [4, 26]. None of the later isolates showed swimming motility whereas three of the four ancestral isolates were motile (Fig. 2a). Nine of the 11 later isolates also showed increased adherence in an in vitro assay, whereas all earlier isolates showed no detectable adherence (Fig. 2b).
Fig. 2.

Phenotypic analysis of isolates. Earlier samples are shown in orange, and later in blue. Three biological replicates were carried out for each experiment, with each point representing a biological replicate and median values shown as crossbars. A one-way ANOVA with post-hoc Dunnett’s test was carried out between early and late isolates, and Bonferroni-corrected P-values are stated. (a) Swimming motility of isolates on M8 media containing 0.3 % agar. ***P=4.12×10−13. (b) Cell adherence in a microtitre plate assay. Crystal violet staining of standing cultures was normalized to culture OD600. *P=0.034. (c) Growth of isolates in synthetic cystic fibrosis media (SCFM), summarized as area under the curve (AUC). *P=0.045. (d) Growth of isolates in LB broth. ns, No significant change. (e) Pyoverdine production normalized to culture OD600 following growth in King’s B broth. ***P=8.96×10−7.
Following long-term infection, P. aeruginosa can have altered growth in laboratory culture due to development of auxotrophy or of small colony variants [86, 87]. Isolates were tested for growth in nutrient-defined SCFM medium that is representative of the chemical composition of CF sputum [43]. Three of the later isolates showed reduced growth in SCFM compared to earlier isolates (Fig. 2c). There was no clear pattern of difference in growth between the earlier and later isolates in nutrient-rich medium, although one later isolate grew more slowly than any other (Fig. 2d).
During the early stages of infection in CF patients P. aeruginosa acquires iron by secreting a siderophore, pyoverdine, but during chronic infections the bacterium becomes less dependent on pyoverdine, transitioning to utilizing haem and other iron sources [88–91]. Earlier and later isolates were tested for pyoverdine production in an iron-limited growth environment. Two earlier isolates showed high levels of pyoverdine production. All later isolates, and one earlier isolate, showed impaired production of pyoverdine, consistent with reduced dependence on pyoverdine-mediated iron acquisition as infection progresses (Fig. 2e).
Discussion
The availability of isolates of P. aeruginosa from the same patient over a 20 year timeframe has allowed us to integrate genomic, transcriptomic and phenotypic approaches to shed light on the changes that occur in these bacteria during prolonged infection. All of the later isolates in our study shared a high amount of genetic similarity with three of the earlier isolates, making it extremely likely that the later isolates are descended from a common ancestor closely related to the earlier group and indicating that a single lineage was able to maintain an infection over more than 20 years (Fig. 1a; Fig. S1). However, a genetically distinct co-infecting earlier isolate was not represented in the later isolates and was probably lost from the infection. These findings are consistent with studies from geographically separate centres [19–21], showing that CF patients acquire a colonizing strain of P. aeruginosa that evolves throughout the course of infection. Many of the pathways that underwent mutational change in the patient in this study have also undergone evolutionary change during chronic P. aeruginosa infections in other CF patients [20, 21, 26, 30].
Genome sequencing data indicate that a population bottleneck occurred during infection. All later isolates contain an identical 125 kb deletion not present in earlier isolates (Fig. 1b) and many genes in these isolates had identical alleles that are different from any of the ancestral isolates (Table 2, Table S5). This bottleneck could have occurred due to a major but temporary reduction in the number of infecting bacteria, or because a mutation arose that conferred a significant competitive advantage. Genetic bottlenecks, which significantly reduce genetic diversity within a population, have not been reported in other longitudinal studies of P. aeruginosa genome evolution during chronic infection [20–22, 30] and it remains to be determined how often they occur. Nonetheless the later isolates have a significant degree of genetic diversity (Table S5, Table S6), consistent with diversification of P. aeruginosa after the bottleneck event and with generation of different morphotypes and adaptation to micro-niches during chronic infection within the CF lung [23, 29, 30, 92]. In comparison, the genetically related earlier isolates have relatively little genetic diversity, consistent with a shorter amount of time for adaptation to the lung environment. Deletions have recently been identified as occurring at a significant frequency in clinical isolates of P. aeruginosa , with up to 15 % of isolates containing large deletions [9, 32, 33, 93]. Large deletions in the P. aeruginosa genome can contribute to phage resistance [94] and to resistance to carbapenems and other β-lactam-based antibiotics [14, 95]. The 125 kb deletion identified here overlaps with the deleted regions identified in earlier studies but whether this deletion confers resistance to phage or antibiotics has not yet been determined. The 0.9 Mb inversion of part of the genome in some of the later isolates also represents a major genome rearrangement. The occurrence of genome inversions during chronic infection has been little studied, although there is one previous report [69]. How often such inversions occur, and how they affect the phenotype of infecting bacteria, remains to be determined.
The mean mutation rate for P. aeruginosa during infection in CF has been estimated to be between 2.6 and 10 mutations per year [19, 21] but this rate is higher in strains with defects in DNA repair (so-called ‘hypermutators’) [96]. Later isolates contain a different variant than earlier isolates of the MutY protein that normally reduces the incidence of GC→TA transversions [70]. The relatively high mutation rate of P. aeruginosa in this patient is probably due to reduced activity of MutY, with the consequent increase in mutation rate facilitating an increased rate of genetic diversification and adaptation of the bacteria during the course of the infection. One of the genes with the highest increases in expression in the later isolate is the anti-mutator gene pfpI that is upregulated in response to mutY mutations [97], consistent with the MutY variant present in later isolates being disfunctional.
Transcriptomic profiling of an earlier and a later isolate showed that substantial changes in gene expression occurred during the course of infection. This contrasts with the high similarity in transcriptomic profiles of different isolates of P. aeruginosa collected at the same time from a CF lung [98]. Many of the genes with higher expression in the later isolate have been noted in studies comparing P. aeruginosa in laboratory culture with those in late-stage infections within CF lungs [99, 100]. Although many of the genes with altered expression levels in the later isolate have known functions, a large proportion of these genes have undefined or unconfirmed functions consistent with other studies and demonstrating the complex nature of CF lung-adapted P. aeruginosa [98–100]. Most of the other genes with significantly reduced expression have no known function, emphasizing our incomplete understanding of how P. aeruginosa adapts to the lung environment.
During infection P. aeruginosa became resistant to all the antibiotic classes routinely used in treatment of the patient (Table 1) and both genome and transcriptome analysis were used to understand the basis of resistance. Resistance arose through mutations, with no evidence of acquisition of antibiotic resistance genes. All later isolates have non-synonymous mutations in resistance-related genes including gyrB, fusA1, mpl and oprD that are associated with resistance to fluoroquinolones, aminoglycosides, cephalosporins and carbapenems, respectively. The later isolate also had significantly increased expression of ampC, a change that is associated with increased resistance to cephalosporins. It is likely that increased expression is due to mutations in mpl that are present in all later isolates, as mutations in this gene increase expression of ampC and contribute to ceftazidime resistance [14, 101]. Antibiotic resistance in P. aeruginosa is also commonly associated with increased expression of efflux pump genes [6], but we did not observe widespread differences in efflux gene expression between an earlier and a later isolate. One exception was expression of the mexXY efflux-encoding operon that had increased expression, albeit not significant, in the later isolate consistent with the presence of a frameshift mutation in the mexZ repressor-encoding gene. Expression of mexXY genes is highly varied in P. aeruginosa isolated from patients with CF due in part to mutations in mexZ, which are associated with aminoglycoside resistance [102–104]. Curiously, early isolates contain a premature stop codon in mexB that encodes a component of the MexAB-OprM efflux pump, which is involved in resistance of P. aeruginosa to a range of antibiotics [105, 106]. This stop codon has undergone mutation to a leucine codon in later isolates, potentially reflecting selection for a functional efflux pump. Our findings demonstrate the multifactorial nature of antibiotic resistance development during chronic infection in the CF lung, with a complex interplay between mutations and changes to gene expression.
All the later isolates in our study had lost flagellar-mediated motility (Fig. 2a), a phenotype commonly associated with biofilm formation and avoidance of the host immune system. Transcriptomic analysis demonstrated a significant reduction of expression of flagellar genes in a later isolate (Table 3; Table S7) consistent with a lack of motility. All of the later isolates have a mutation in fleQ that encodes a sigma factor required for expression of flagellar synthesis genes [107], providing a molecular explanation for loss of motility. Most of the later isolates showed enhanced adherence in a microtitre plate assay, a common property of P. aeruginosa isolated from chronic infections [108]. The genetic basis for this complex phenotype is not clear, although it is noteworthy that a number of genes that may influence biofilm formation, including global regulators (PQS, Las and Sbr) and cell surface adhesins (LecB and CdrA), had higher expression in the later isolate. Later isolates also had mutations in genes required for synthesis of lipopolysaccharide, Pel polysaccharide and alginate, all of which might influence attachment and biofilm formation.
P. aeruginosa from chronic infections often produce copious amounts of extracellular alginate due to mutations in regulatory genes such as mucA and mucB that control the alginate synthesis pathway [109], but the only isolate to have a mucoid phenotype was the early isolate E-S2239-16. Indeed, a premature stop codon in mucA in that strain has been replaced by a glutamine-coding codon that would restore production of full-length protein with potential to repress alginate gene expression in later isolates. All later isolates also have mutations in the alginate regulatory gene mucB and in some cases the regulatory gene algU, as well as mutations in genes encoding alginate-encoding enzymes required for alginate synthesis. Expression of some alginate synthesis genes was upregulated in the later isolate although this did not result in a mucoid phenotype. The relationship between alginate genotype, gene expression and phenotype is clearly complex in the isolates studied here.
Host colonization is also influenced by quorum sensing, which controls production of a range of virulence factors. Later isolates had mutations in the quorum-sensing regulator lasR that is often mutated in chronically infected CF patients [4, 110–112], and expression of the LasR-regulated apr gene that encodes alkaline protease was higher in the later isolate. Expression of the pqs genes that encode the PQS quorum sensing molecule, which regulates a wide range of infection-associated phenotypes [112], was also higher in the later isolate. Later isolates also contained a mutation in mvaT that is associated with quorum sensing and virulence factor production, as well as with biofilm formation [113, 114]. Clearly, quorum sensing pathways evolve during chronic infection, although the complex interplay between quorum sensing pathways mean that further work will be needed to dissect out the effects of these changes on phenotype.
Adaptation to the host environment was also reflected in metabolic changes. The later isolate had much higher expression of isocitrate lyase, a key enzyme in the glyoxylate pathway, reflecting the use of lipids as a carbon source during infection [84]. During infections, P. aeruginosa counters zinc starvation by upregulating expression of zinc acquisition genes [83], and these genes were amongst those with the greatest increase in expression in the later isolate. P. aeruginosa has multiple mechanisms to acquire iron. Genes for synthesis of an iron-scavenging siderophore, pyochelin, also had higher expression in the later isolate, indicating that pyochelin has an important role in iron acquisition during the later stages of infection. Conversely, during infection in the CF lung, P. aeruginosa transitions away from the use of the siderophore pyoverdine for iron acquisition, instead using other sources of iron [88–91]. Two of the earlier isolates but none of the later isolates have a high production of pyoverdine (Fig. 2e). The later isolates have mutations in different pyoverdine genes (Table S5), indicating that loss of pyoverdine production has occurred independently on several occasions although transcription of pvd genes was similar in the earlier and later isolate (Table S7).
In conclusion, this study reveals the relationship between genomic, transcriptomic and phenotypic changes undergone by P. aeruginosa during long-term infection in the lungs of a person with CF. Adaptations included a trend towards increased antibiotic resistance, altered nutrient acquisition and altered biofilm production, and were accelerated through an increased mutation rate. While many of the genomic and transcriptomic alterations affect known phenotypes, many affect genes with no known function, showing that an understanding of how P. aeruginosa evolves during infection in CF is still far from complete. Comparable studies of infections in other patients, combined with experimental and bioinformatic approaches, can be expected to shed further light on how P. aeruginosa adapts and maintains infections in the CF lung.
Supplementary Data
Funding information
This research was supported by grants from the New Zealand Health Research Council (17/372) and the Otago Medical Research Foundation (AG 330). S.J.T.W. was supported by a Postgraduate Scholarship from the University of Otago. Work in RCL's laboratory is funded by the Canadian Cystic Fibrosis foundation and by the Canadian Institute for Health Research. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
The authors wish to express their heartfelt thanks to Richard Sutton, who died of complications of lung transplantation on 27 October 2014, and always gave very freely of his time for medical education and scientific research.
Author contributions
S.W. carried out investigation, data curation, formal analysis, methodology, validation, co-wrote the original draft and reviewed and edited the manuscript. J.G. carried out data curation, formal analysis, investigation and visualization. L.M. carried out investigation. M.P. carried out investigation. B.B. provided resources and reviewed the manuscript. R.L. obtained funding, carried out formal analysis, project administration and supervision, obtained funding, co-wrote the original draft and reviewed and edited the manuscript. I.L. conceived the study, carried out formal analysis, project administration and supervision, obtained funding, co-wrote the original draft and reviewed and edited the manuscript.
Conflicts of interest
The authors declare that there are no conflicts of interest.
Ethical statement
Sputum samples were collected with the approval of the New Zealand Health and Disability Ethics Committees (NTY/10/12/106) and with written consent from the patient.
Footnotes
Abbreviations: CF, cystic fibrosis; LPS, lipopolysaccharide; MH, Mueller-Hinton; MIC, minimum inhibitory concentration; RNAseq, RNA sequencing; SCFM, Synthetic Cystic Fibrosis Medium.
All supporting data, code and protocols have been provided within the article or through supplementary data files. Eight supplementary tables and three supplementary figures are available with the online version of this article.
References
- 1.Mulani MS, Kamble EE, Kumkar SN, Tawre MS, Pardesi KR. Emerging strategies to combat ESKAPE pathogens in the era of antimicrobial resistance: a review. Front Microbiol. 2019;10:539. doi: 10.3389/fmicb.2019.00539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Talwalkar JS, Murray TS. The approach to Pseudomonas aeruginosa in cystic fibrosis. Clin Chest Med. 2016;37:69–81. doi: 10.1016/j.ccm.2015.10.004. [DOI] [PubMed] [Google Scholar]
- 3.Ramsay KA, Wardell SJT, Patrick WM, Brockway B, Reid DW, et al. Genomic and phenotypic comparison of environmental and patient-derived isolates of Pseudomonas aeruginosa suggest that antimicrobial resistance is rare within the environment. J Med Microbiol. 2019;68:1591–1595. doi: 10.1099/jmm.0.001085. [DOI] [PubMed] [Google Scholar]
- 4.Winstanley C, O’Brien S, Brockhurst MA. Pseudomonas aeruginosa evolutionary adaptation and diversification in cystic fibrosis chronic lung infections. Trends Microbiol. 2016;24:327–337. doi: 10.1016/j.tim.2016.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Camus L, Vandenesch F, Moreau K. From genotype to phenotype: adaptations of Pseudomonas aeruginosa to the cystic fibrosis environment. Microb Genom. 2021;7:000513. doi: 10.1099/mgen.0.000513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lister PD, Wolter DJ, Hanson ND. Antibacterial-resistant Pseudomonas aeruginosa: clinical impact and complex regulation of chromosomally encoded resistance mechanisms. Clin Microbiol Rev. 2009;22:582–610. doi: 10.1128/CMR.00040-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pang Z, Raudonis R, Glick BR, Lin TJ, Cheng Z. Antibiotic resistance in Pseudomonas aeruginosa: mechanisms and alternative therapeutic strategies. Biotechnol Adv. 2019;37:177–192. doi: 10.1016/j.biotechadv.2018.11.013. [DOI] [PubMed] [Google Scholar]
- 8.Rehman A, Patrick WM, Lamont IL. Mechanisms of ciprofloxacin resistance in Pseudomonas aeruginosa: new approaches to an old problem. J Med Microbiol. 2019;68:1–10. doi: 10.1099/jmm.0.000873. [DOI] [PubMed] [Google Scholar]
- 9.Wardell SJT, Rehman A, Martin LW, Winstanley C, Patrick WM, et al. A large-scale whole-genome comparison shows that experimental evolution in response to antibiotics predicts changes in naturally evolved clinical Pseudomonas aeruginosa . Antimicrob Agents Chemother. 2019;63:01619. doi: 10.1128/AAC.01619-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lopez-Causape C, Cabot G, Del Barrio-Tofino E, Oliver A. The versatile mutational resistome of Pseudomonas aeruginosa . Front Microbiol. 2018;9:685. doi: 10.3389/fmicb.2018.00685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kohler T, Michea-Hamzehpour M, Epp SF, Pechere JC. Carbapenem activities against Pseudomonas aeruginosa: respective contributions of OprD and efflux systems. Antimicrob Agents Chemother. 1999;43:424–427. doi: 10.1128/AAC.43.2.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huszczynski SM, Lam JS, Khursigara CM. The role of Pseudomonas aeruginosa lipopolysaccharide in bacterial pathogenesis and physiology. Pathogens. 2019;9:E6. doi: 10.3390/pathogens9010006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krahn T, Gilmour C, Tilak J, Fraud S, Kerr N. Determinants of intrinsic aminoglycoside resistance in Pseudomonas aeruginosa . Antimicrob Agents Chemother. 2012;56:5591–5602. doi: 10.1128/AAC.01446-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cabot G, Florit-Mendoza L, Sanchez-Diener I, Zamorano L, Oliver A. Deciphering beta-lactamase-independent beta-lactam resistance evolution trajectories in Pseudomonas aeruginosa . J Antimicrob Chemother. 2018;73:3322–3331. doi: 10.1093/jac/dky364. [DOI] [PubMed] [Google Scholar]
- 15.Poole K. Pseudomonas aeruginosa: resistance to the max. Front Microbiol. 2011;2:65. doi: 10.3389/fmicb.2011.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cazares A, Moore MP, Hall JPJ, Wright LL, Grimes M. A megaplasmid family driving dissemination of multidrug resistance in Pseudomonas. Nat Commun. 2020;11:1370. doi: 10.1038/s41467-020-15081-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hector A, Griese M, Hartl D. Oxidative stress in cystic fibrosis lung disease: an early event, but worth targeting? Eur Respir J. 2014;44:17–19. doi: 10.1183/09031936.00038114. [DOI] [PubMed] [Google Scholar]
- 18.Lopes SP, Azevedo NF, Pereira MO. Microbiome in cystic fibrosis: Shaping polymicrobial interactions for advances in antibiotic therapy. Crit Rev Microbiol. 2015;41:353–365. doi: 10.3109/1040841X.2013.847898. [DOI] [PubMed] [Google Scholar]
- 19.Marvig RL, Johansen HK, Molin S, Jelsbak L. Genome analysis of a transmissible lineage of Pseudomonas aeruginosa reveals pathoadaptive mutations and distinct evolutionary paths of hypermutators. PLoS Genet. 2013;9:e1003741. doi: 10.1371/journal.pgen.1003741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bianconi I, D’Arcangelo S, Esposito A, Benedet M, Piffer E. Persistence and microevolution of Pseudomonas aeruginosa in the cystic fibrosis lung: a single-patient longitudinal genomic study. Front Microbiol. 2018;9:3242. doi: 10.3389/fmicb.2018.03242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Klockgether J, Cramer N, Fischer S, Wiehlmann L, Tummler B. Long-term microevolution of Pseudomonas aeruginosa differs between mildly and severely affected cystic fibrosis lungs. Am J Respir Cell Mol Biol. 2018;59:246–256. doi: 10.1165/rcmb.2017-0356OC. [DOI] [PubMed] [Google Scholar]
- 22.Marvig RL, Dolce D, Sommer LM, Petersen B, Ciofu O. Within-host microevolution of Pseudomonas aeruginosa in Italian cystic fibrosis patients. BMC Microbiol. 2015;15:218. doi: 10.1186/s12866-015-0563-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cullen L, McClean S. Bacterial adaptation during chronic respiratory infections. Pathogens. 2015;4:66–89. doi: 10.3390/pathogens4010066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Faure E, Kwong K, Nguyen D. Pseudomonas aeruginosa in chronic lung infections: how to adapt within the host? Front Immunol. 2018;9:2416. doi: 10.3389/fimmu.2018.02416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rossi E, La Rosa R, Bartell JA, Marvig RL, Haagensen JAJ. Pseudomonas aeruginosa adaptation and evolution in patients with cystic fibrosis. Nat Rev Microbiol. 2021;19:331–342. doi: 10.1038/s41579-020-00477-5. [DOI] [PubMed] [Google Scholar]
- 26.Smith EE, Buckley DG, Wu Z, Saenphimmachak C, Hoffman LR. Genetic adaptation by Pseudomonas aeruginosa to the airways of cystic fibrosis patients. Proc Natl Acad Sci U S A. 2006;103:8487–8492. doi: 10.1073/pnas.0602138103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lopez-Causape C, Rojo-Molinero E, Mulet X, Cabot G, Moya B. Clonal dissemination, emergence of mutator lineages and antibiotic resistance evolution in Pseudomonas aeruginosa cystic fibrosis chronic lung infection. PLoS One. 2013;8:e71001. doi: 10.1371/journal.pone.0071001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lopez-Causape C, Sommer LM, Cabot G, Rubio R, Ocampo-Sosa AA. Evolution of the Pseudomonas aeruginosa mutational resistome in an international Cystic Fibrosis clone. Sci Rep. 2017;7:5555. doi: 10.1038/s41598-017-05621-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hoboth C, Hoffmann R, Eichner A, Henke C, Schmoldt S. Dynamics of adaptive microevolution of hypermutable Pseudomonas aeruginosa during chronic pulmonary infection in patients with cystic fibrosis. J Infect Dis. 2009;200:118–130. doi: 10.1086/599360. [DOI] [PubMed] [Google Scholar]
- 30.Marvig RL, Sommer LM, Molin S, Johansen HK. Convergent evolution and adaptation of Pseudomonas aeruginosa within patients with cystic fibrosis. Nat Genet. 2015;47:57–64. doi: 10.1038/ng.3148. [DOI] [PubMed] [Google Scholar]
- 31.Bragonzi A, Paroni M, Nonis A, Cramer N, Montanari S. Pseudomonas aeruginosa microevolution during cystic fibrosis lung infection establishes clones with adapted virulence. Am J Respir Crit Care Med. 2009;180:138–145. doi: 10.1164/rccm.200812-1943OC. [DOI] [PubMed] [Google Scholar]
- 32.Hilliam Y, Moore MP, Lamont IL, Bilton D, Haworth CS. Pseudomonas aeruginosa adaptation and diversification in the non-cystic fibrosis bronchiectasis lung. Eur Respir J. 2017;49:1602108. doi: 10.1183/13993003.02108-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gabrielaite M, Johansen HK, Molin S, Nielsen FC, Marvig RL. Gene loss and acquisition in lineages of Pseudomonas aeruginosa evolving in cystic fibrosis patient airways. mBio. 2020;11:e02359-20. doi: 10.1128/mBio.02359-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lucchetti-Miganeh C, Redelberger D, Chambonnier G, Rechenmann F, Elsen S. Pseudomonas aeruginosa genome evolution in patients and under the hospital environment. Pathogens. 2014;3:309–340. doi: 10.3390/pathogens3020309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bartell JA, Sommer LM, Haagensen JAJ, Loch A, Espinosa R. Evolutionary highways to persistent bacterial infection. Nat Commun. 2019;10:629. doi: 10.1038/s41467-019-08504-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brown VI, Lowbury EJ. Use of an improved cetrimide agar medium and other culture methods for Pseudomonas aeruginosa . J Clin Pathol. 1965;18:752–756. doi: 10.1136/jcp.18.6.752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wiegand I, Hilpert K, Hancock RE. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat Protoc. 2008;3:163–175. doi: 10.1038/nprot.2007.521. [DOI] [PubMed] [Google Scholar]
- 38.Ha DG, Kuchma SL, O’Toole GA. Plate-based assay for swimming motility in Pseudomonas aeruginosa . Methods Mol Biol. 2014;1149:59–65. doi: 10.1007/978-1-4939-0473-0_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.King EO, Ward MK, Raney DE. Two simple media for the demonstration of pyocyanin and fluorescin. J Lab Clin Med. 1954;44:301–307. [PubMed] [Google Scholar]
- 41.O’Toole GA. Microtiter dish biofilm formation assay. J Vis Exp. 2011;30:2437. doi: 10.3791/2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Coffey BM, Anderson GG. Biofilm formation in the 96-well microtiter plate. Methods Mol Biol. 2014;1149:631–641. doi: 10.1007/978-1-4939-0473-0_48. [DOI] [PubMed] [Google Scholar]
- 43.Palmer KL, Aye LM, Whiteley M. Nutritional cues control Pseudomonas aeruginosa multicellular behavior in cystic fibrosis sputum. J Bacteriol. 2007;189:8079–8087. doi: 10.1128/JB.01138-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Freschi L, Vincent AT, Jeukens J, Emond-Rheault JG, Kukavica-Ibrulj I. The Pseudomonas aeruginosa pan-genome provides new insights on its population structure, horizontal gene transfer, and pathogenicity. Genome Biol Evol. 2019;11:109–120. doi: 10.1093/gbe/evy259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Freschi L, Jeukens J, Kukavica-Ibrulj I, Boyle B, Dupont MJ. Clinical utilization of genomics data produced by the international Pseudomonas aeruginosa consortium. Front Microbiol. 2015;6:1036. doi: 10.3389/fmicb.2015.01036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wick RR, Judd LM, Holt KE. Performance of neural network basecalling tools for Oxford Nanopore sequencing. Genome Biol. 2019;20:129. doi: 10.1186/s13059-019-1727-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.De Coster W, D’Hert S, Schultz DT, Cruts M, Van Broeckhoven C. NanoPack: visualizing and processing long-read sequencing data. Bioinformatics. 2018;34:2666–2669. doi: 10.1093/bioinformatics/bty149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wick RR, Judd LM, Gorrie CL, Holt KE. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput Biol. 2017;13:e1005595. doi: 10.1371/journal.pcbi.1005595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Treangen TJ, Ondov BD, Koren S, Phillippy AM. The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol, Research Support, US Gov’t, Non-PHS. 2014;15:524. doi: 10.1186/s13059-014-0524-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rambaut A, Drummond A. FigTree version 1.4. 0. 2012. [Google Scholar]
- 53.Alikhan NF, Petty NK, Ben Zakour NL, Beatson SA. BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics. 2011;12:402. doi: 10.1186/1471-2164-12-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30:2068–2069. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 55.Jolley KA, Bray JE, Maiden MCJ. Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications. Wellcome Open Res. 2018;3:124. doi: 10.12688/wellcomeopenres.14826.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Laing C, Buchanan C, Taboada EN, Zhang Y, Kropinski A. Pan-genome sequence analysis using Panseq: an online tool for the rapid analysis of core and accessory genomic regions. BMC Bioinformatics. 2010;11:461. doi: 10.1186/1471-2105-11-461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zankari E, Hasman H, Cosentino S, Vestergaard M, Rasmussen S. Identification of acquired antimicrobial resistance genes. J Antimicrob Chemother. 2012;67:2640–2644. doi: 10.1093/jac/dks261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Deatherage DE, Barrick JE. Identification of mutations in laboratory-evolved microbes from next-generation sequencing data using breseq. Methods Mol Biol. 2014;1151:165–188. doi: 10.1007/978-1-4939-0554-6_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bray NL, Pimentel H, Melsted P, Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol. 2016;34:525–527. doi: 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- 60.Pimentel H, Bray NL, Puente S, Melsted P, Pachter L. Differential analysis of RNA-seq incorporating quantification uncertainty. Nat Methods. 2017;14:687–690. doi: 10.1038/nmeth.4324. [DOI] [PubMed] [Google Scholar]
- 61.R Development Core Team Vienna, Austria: R Foundation for Statistical Computing; 2017. [Google Scholar]
- 62.Moore MP, Lamont IL, Williams D, Paterson S, Kukavica-Ibrulj I. Transmission, adaptation and geographical spread of the Pseudomonas aeruginosa Liverpool epidemic strain. Microb Genom. 2021;7 doi: 10.1099/mgen.0.000511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.La Rosa R, Johansen HK, Molin S. Adapting to the airways: metabolic requirements of Pseudomonas aeruginosa during the infection of cystic fibrosis patients. Metabolites. 2019;9:10. doi: 10.3390/metabo9100234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Friedman L, Kolter R. Two genetic loci produce distinct carbohydrate-rich structural components of the Pseudomonas aeruginosa biofilm matrix. J Bacteriol. 2004;186:4457–4465. doi: 10.1128/JB.186.14.4457-4465.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jackson KD, Starkey M, Kremer S, Parsek MR, Wozniak DJ. Identification of psl, a locus encoding a potential exopolysaccharide that is essential for Pseudomonas aeruginosa PAO1 biofilm formation. J Bacteriol. 2004;186:4466–4475. doi: 10.1128/JB.186.14.4466-4475.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Matsukawa M, Greenberg EP. Putative exopolysaccharide synthesis genes influence Pseudomonas aeruginosa biofilm development. J Bacteriol. 2004;186:4449–4456. doi: 10.1128/JB.186.14.4449-4456.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lee X, Fox A, Sufrin J, Henry H, Majcherczyk P. Identification of the biosynthetic gene cluster for the Pseudomonas aeruginosa antimetabolite L-2-amino-4-methoxy-trans-3-butenoic acid. J Bacteriol. 2010;192:4251–4255. doi: 10.1128/JB.00492-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Klockgether J, Munder A, Neugebauer J, Davenport CF, Stanke F. Genome diversity of Pseudomonas aeruginosa PAO1 laboratory strains. J Bacteriol. 2010;192:1113–1121. doi: 10.1128/JB.01515-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Romling U, Schmidt KD, Tummler B. Large chromosomal inversions occur in Pseudomonas aeruginosa clone C strains isolated from cystic fibrosis patients. FEMS Microbiol Lett. 1997;150:149–156. doi: 10.1111/j.1574-6968.1997.tb10363.x. [DOI] [PubMed] [Google Scholar]
- 70.Mandsberg LF, Ciofu O, Kirkby N, Christiansen LE, Poulsen HE. Antibiotic resistance in Pseudomonas aeruginosa strains with increased mutation frequency due to inactivation of the DNA oxidative repair system. Antimicrob Agents Chemother. 2009;53:2483–2491. doi: 10.1128/AAC.00428-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Greipel L, Fischer S, Klockgether J, Dorda M, Mielke S. Molecular epidemiology of mutations in antimicrobial resistance loci of Pseudomonas aeruginosa isolates from cystic fibrosis airways. Antimicrob Agents Chemother. 2016;60:6726–6734. doi: 10.1128/AAC.00724-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Livermore DM. Of Pseudomonas, porins, pumps and carbapenems. J Antimicrob Chemother. 2001;47:247–250. doi: 10.1093/jac/47.3.247. [DOI] [PubMed] [Google Scholar]
- 73.Yang Y, Bhachech N, Bush K. Biochemical comparison of imipenem, meropenem and biapenem: permeability, binding to penicillin-binding proteins, and stability to hydrolysis by beta-lactamases. J Antimicrob Chemother. 1995;35:75–84. doi: 10.1093/jac/35.1.75. [DOI] [PubMed] [Google Scholar]
- 74.Davies TA, Shang W, Bush K, Flamm RK. Affinity of doripenem and comparators to penicillin-binding proteins in Escherichia coli and Pseudomonas aeruginosa . Antimicrob Agents Chemother. 2008;52:1510–1512. doi: 10.1128/AAC.01529-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bolard A, Plesiat P, Jeannot K. Mutations in Gene fusA1 as a Novel Mechanism of Aminoglycoside Resistance in Clinical Strains of Pseudomonas aeruginosa. Antimicrob Agents Chemother. 2018;62:01817. doi: 10.1128/AAC.01835-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Colque CA, Albarracin Orio AG, Feliziani S, Marvig RL, Tobares AR, et al. Hypermutator Pseudomonas aeruginosa Exploits Multiple Genetic Pathways To Develop Multidrug Resistance during Long-Term Infections in the Airways of Cystic Fibrosis Patients. Antimicrob Agents Chemother. 2020;64:02119. doi: 10.1128/AAC.02142-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kos VN, Deraspe M, McLaughlin RE, Whiteaker JD, Roy PH. The resistome of Pseudomonas aeruginosa in relationship to phenotypic susceptibility. Antimicrob Agents Chemother. 2015;59:427–436. doi: 10.1128/AAC.03954-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Qin X, Zhou C, Zerr DM, Adler A, Addetia A. Heterogeneous antimicrobial susceptibility characteristics in Pseudomonas aeruginosa isolates from cystic fibrosis patients. mSphere. 2018;3:e00615-17. doi: 10.1128/mSphere.00615-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rehman A, Jeukens J, Levesque RC, Lamont IL. Gene-gene interactions dictate ciprofloxacin resistance in pseudomonas aeruginosa and facilitate prediction of resistance phenotype from genome sequence data. Antimicrob Agents Chemother. 2021:02696–20. doi: 10.1128/AAC.02696-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Moya B, Dotsch A, Juan C, Blazquez J, Zamorano L. β-lactam resistance response triggered by inactivation of a nonessential penicillin-binding protein. PLoS Pathog. 2009;5:e1000353. doi: 10.1371/journal.ppat.1000353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.King JD, Kocincova D, Westman EL, Lam JS. Review: Lipopolysaccharide biosynthesis in Pseudomonas aeruginosa . Innate Immun. 2009;15:261–312. doi: 10.1177/1753425909106436. [DOI] [PubMed] [Google Scholar]
- 82.Wicht DK. The reduced flavin-dependent monooxygenase SfnG converts dimethylsulfone to methanesulfinate. Arch Biochem Biophys. 2016;604:159–166. doi: 10.1016/j.abb.2016.07.001. [DOI] [PubMed] [Google Scholar]
- 83.Mastropasqua MC, Lamont I, Martin LW, Reid DW, D’Orazio M. Efficient zinc uptake is critical for the ability of Pseudomonas aeruginosa to express virulence traits and colonize the human lung. J Trace Elem Med Biol. 2018;48:74–80. doi: 10.1016/j.jtemb.2018.03.009. [DOI] [PubMed] [Google Scholar]
- 84.Hagins JM, Scoffield JA, Suh SJ, Silo-Suh L. Influence of RpoN on isocitrate lyase activity in Pseudomonas aeruginosa . Microbiology (Reading) 2010;156:1201–1210. doi: 10.1099/mic.0.033381-0. [DOI] [PubMed] [Google Scholar]
- 85.Lin J, Cheng J, Wang Y, Shen X. The Pseudomonas Quinolone Signal (PQS): Not just for quorum sensing anymore. Front Cell Infect Microbiol. 2018;8:230. doi: 10.3389/fcimb.2018.00230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Barth AL, Pitt TL. The high amino-acid content of sputum from cystic fibrosis patients promotes growth of auxotrophic Pseudomonas aeruginosa . J Med Microbiol. 1996;45:110–119. doi: 10.1099/00222615-45-2-110. [DOI] [PubMed] [Google Scholar]
- 87.Haussler S, Tummler B, Weissbrodt H, Rohde M, Steinmetz I. Small-colony variants of Pseudomonas aeruginosa in cystic fibrosis. Clin Infect Dis. 1999;29:621–625. doi: 10.1086/598644. [DOI] [PubMed] [Google Scholar]
- 88.Nguyen AT, O’Neill MJ, Watts AM, Robson CL, Lamont IL. Adaptation of iron homeostasis pathways by a Pseudomonas aeruginosa pyoverdine mutant in the cystic fibrosis lung. J Bacteriol. 2014;196:2265–2276. doi: 10.1128/JB.01491-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Konings AF, Martin LW, Sharples KJ, Roddam LF, Latham R. Pseudomonas aeruginosa uses multiple pathways to acquire iron during chronic infection in cystic fibrosis lungs. Infect Immun. 2013;81:2697–2704. doi: 10.1128/IAI.00418-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Marvig RL, Damkiær S, Khademi SMH, Markussen TM, Molin S, et al. Within-host evolution of Pseudomonas aeruginosa reveals adaptation toward iron acquisition from hemoglobin. MBio. 2014;5:e00966–14.:e00966-14. doi: 10.1128/mBio.00966-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Martin LW, Reid DW, Sharples KJ, Lamont IL. Pseudomonas siderophores in the sputum of patients with cystic fibrosis. Biometals. 2011;24:1059–1067. doi: 10.1007/s10534-011-9464-z. [DOI] [PubMed] [Google Scholar]
- 92.Sousa AM, Pereira MO. Pseudomonas aeruginosa diversification during infection development in cystic fibrosis lungs-A review. Pathogens. 2014;3:680–703. doi: 10.3390/pathogens3030680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rau MH, Marvig RL, Ehrlich GD, Molin S, Jelsbak L. Deletion and acquisition of genomic content during early stage adaptation of Pseudomonas aeruginosa to a human host environment. Environ Microbiol. 2012;14:2200–2211. doi: 10.1111/j.1462-2920.2012.02795.x. [DOI] [PubMed] [Google Scholar]
- 94.Shen M, Zhang H, Shen W, Zou Z, Lu S. Pseudomonas aeruginosa MutL promotes large chromosomal deletions through non-homologous end joining to prevent bacteriophage predation. Nucleic Acids Res. 2018;46:4505–4514. doi: 10.1093/nar/gky160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cabot G, Zamorano L, Moya B, Juan C, Navas A. Evolution of Pseudomonas aeruginosa antimicrobial resistance and fitness under low and high mutation rates. Antimicrob Agents Chemother. 2016;60:1767–1778. doi: 10.1128/AAC.02676-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Oliver A. Mutators in cystic fibrosis chronic lung infection: Prevalence, mechanisms, and consequences for antimicrobial therapy. Int J Med Microbiol. 2010;300:563–572. doi: 10.1016/j.ijmm.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 97.Mandsberg LF, Macia MD, Bergmann KR, Christiansen LE, Alhede M. Development of antibiotic resistance and up-regulation of the antimutator gene pfpI in mutator Pseudomonas aeruginosa due to inactivation of two DNA oxidative repair genes (mutY, mutM) FEMS Microbiol Lett. 2011;324:28–37. doi: 10.1111/j.1574-6968.2011.02383.x. [DOI] [PubMed] [Google Scholar]
- 98.Kordes A, Preusse M, Willger SD, Braubach P, Jonigk D. Genetically diverse Pseudomonas aeruginosa populations display similar transcriptomic profiles in a cystic fibrosis explanted lung. Nat Commun. 2019:3397. doi: 10.1038/s41467-019-11414-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Cornforth DM, Dees JL, Ibberson CB, Huse HK, Mathiesen IH, et al. Pseudomonas aeruginosa transcriptome during human infection. Proc Natl Acad Sci U S A. 2018;115:E5125–E5134. doi: 10.1073/pnas.1717525115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Rossi E, Falcone M, Molin S, Johansen HK. High-resolution in situ transcriptomics of Pseudomonas aeruginosa unveils genotype independent patho-phenotypes in cystic fibrosis lungs. Nat Commun. 2018;9:3459. doi: 10.1038/s41467-018-05944-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tsutsumi Y, Tomita H, Tanimoto K. Identification of novel genes responsible for overexpression of ampC in Pseudomonas aeruginosa PAO1. Antimicrob Agents Chemother. 2013;57:5987–5993. doi: 10.1128/AAC.01291-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Masuda N, Sakagawa E, Ohya S, Gotoh N, Tsujimoto H. Substrate specificities of MexAB-OprM, MexCD-OprJ, and MexXY-oprM efflux pumps in Pseudomonas aeruginosa . Antimicrob Agents Chemother. 2000;44:3322–3327. doi: 10.1128/AAC.44.12.3322-3327.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Li XZ, Plesiat P, Nikaido H. The challenge of efflux-mediated antibiotic resistance in Gram-negative bacteria. Clin Microbiol Rev. 2015;28:337–418. doi: 10.1128/CMR.00117-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Martin LW, Robson CL, Watts AM, Gray AR, Wainwright CE. Expression of Pseudomonas aeruginosa antibiotic resistance genes varies greatly during infections in cystic fibrosis patients. Antimicrob Agents Chemother. 2018;62:e01718–01789. doi: 10.1128/AAC.01789-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sennhauser G, Bukowska MA, Briand C, Grutter MG. Crystal structure of the multidrug exporter MexB from Pseudomonas aeruginosa . J Mol Biol. 2009;389:134–145. doi: 10.1016/j.jmb.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 106.Li XZ, Nikaido H, Poole K. Role of mexA-mexB-oprM in antibiotic efflux in Pseudomonas aeruginosa . Antimicrob Agents Chemother. 1995;39:1948–1953. doi: 10.1128/AAC.39.9.1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jyot J, Dasgupta N, Ramphal R. FleQ, the major flagellar gene regulator in Pseudomonas aeruginosa, binds to enhancer sites located either upstream or atypically downstream of the RpoN binding site. J Bacteriol. 2002;184:5251–5260. doi: 10.1128/JB.184.19.5251-5260.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Deligianni E, Pattison S, Berrar D, Ternan NG, Haylock RW. Pseudomonas aeruginosa cystic fibrosis isolates of similar RAPD genotype exhibit diversity in biofilm forming ability in vitro . BMC Microbiol. 2010;10:38. doi: 10.1186/1471-2180-10-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Jones AK, Fulcher NB, Balzer GJ, Urbanowski ML, Pritchett CL. Activation of the Pseudomonas aeruginosa AlgU regulon through mucA mutation inhibits cyclic AMP/Vfr signaling. J Bacteriol. 2010;192:5709–5717. doi: 10.1128/JB.00526-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Feltner JB, Wolter DJ, Pope CE, Groleau MC, Smalley NE. LasR variant cystic fibrosis isolates reveal an adaptable quorum-sensing hierarchy in Pseudomonas aeruginosa . mBio. 2016;7:e01513-16. doi: 10.1128/mBio.01513-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kostylev M, Kim DY, Smalley NE, Salukhe I, Greenberg EP. Evolution of the Pseudomonas aeruginosa quorum-sensing hierarchy. Proc Natl Acad Sci U S A. 2019;116:7027–7032. doi: 10.1073/pnas.1819796116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Azimi S, Klementiev AD, Whiteley M, Diggle SP. Bacterial quorum sensing during infection. Annu Rev Microbiol. 2020;74:201–219. doi: 10.1146/annurev-micro-032020-093845. [DOI] [PubMed] [Google Scholar]
- 113.Diggle SP, Winzer K, Lazdunski A, Williams P, Camara M. Advancing the quorum in Pseudomonas aeruginosa: MvaT and the regulation of N-acylhomoserine lactone production and virulence gene expression. J Bacteriol. 2002;184:2576–2586. doi: 10.1128/JB.184.10.2576-2586.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Vallet-Gely I, Sharp JS, Dove SL. Local and global regulators linking anaerobiosis to cupA fimbrial gene expression in Pseudomonas aeruginosa . J Bacteriol. 2007;189:8667–8676. doi: 10.1128/JB.01344-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.