

Abstract
Objective:
To identify pathways that influence endometrial cancer (EC) cell sensitivity to cisplatin and to characterize the BCL2 antagonist of cell death (BAD) pathway as a therapeutic target to increase cisplatin sensitivity.
Methods:
Eight EC cell lines (Ishikawa, MFE296, RL 95-2, AN3CA, KLE, MFE280, MFE319, HEC-1-A) were subjected to Affymetrix Human U133A GeneChip expression analysis of approximately 22,000 probe sets. In parallel, endometrial cell line sensitivity to cisplatin was quantified by MTS assay, and IC50 values were calculated. Pearson’s correlation test was used to identify genes associated with response to cisplatin. Genes associated with cisplatin responsiveness were subjected to pathway analysis. The BAD pathway was identified and subjected to targeted modulation, and the effect on cisplatin sensitivity was evaluated.
Results:
Pearson’s correlation analysis identified 1443 genes associated with cisplatin resistance (P<0.05), which included representation of the BAD-apoptosis pathway. Small interfering RNA (siRNA) knockdown of BAD pathway protein phosphatase PP2C expression was associated with increased phosphorylated BAD (serine-155) levels and a parallel increase in cisplatin resistance in Ishikawa (P=0.004) and HEC-1-A (P=0.02) cell lines. In contrast, siRNA knockdown of protein kinase A expression increased cisplatin sensitivity in the Ishikawa (P=0.02) cell line.
Conclusion:
The BAD pathway influences EC cell sensitivity to cisplatin, likely via modulation of the phosphorylation status of the BAD protein. The BAD pathway represents an appealing therapeutic target to increase EC cell sensitivity to cisplatin.
Keywords: cisplatin, endometrial cancer, microarray gene analysis, BAD pathway, targeted therapy
INTRODUCTION
In 2010, an estimated 43,470 new cases of endometrial cancer (EC) were diagnosed and 7,950 women died from the disease, such that EC represents the most common gynecologic malignancy [1]. Although the majority of patients with EC present with symptomatic, early-stage, and curable disease, a subset of patients present with advanced-stage disease or develop recurrent disease that is associated with much less favorable survival. Reported response rates to systemic hormonal and cytotoxic therapy for advanced or recurrent EC vary dramatically, such that overall survival is generally extremely poor. Both single-agent and combination chemotherapy regimens have antitumor activity but are not curative. Paclitaxel in combination with cisplatin and doxorubicin increased response rates and median survival, but is associated with increased toxicity [2]. Standard initial treatment for advanced or recurrent EC includes combinations of cisplatin, doxorubicin, and paclitaxel or carboplatin plus paclitaxel. Results of the recently completed Gynecologic Oncology Group protocol 209 (comparing doxorubicin, cisplatin, and paclitaxel to carboplatin plus paclitaxel in patients with stage III and IV or recurrent EC) are not yet available, but prior phase 2 studies seem to indicate similar activity with overall response rates of 40–62% for both regimens [3, 4]. Additionally, evidence is building suggesting that platinum sensitivity has relevance in the setting of recurrent EC as a means to predict prognosis and to potentially inform therapeutic decision-making.
In general, efficacy of systemic therapy for recurrent or advanced-stage EC is suboptimal. Moreover, no reliable clinical tools exist to enable specific therapies to be directed at the individual patients most likely to benefit. As a result of subsequent empiric treatment decision-making, many patients receive inactive, yet potentially toxic therapies, without substantial benefit. Such a dilemma underscores the importance of efforts to develop personalized-medicine approaches to gynecologic cancer, approaches that mandate an improved understanding of the biology that underlies chemo-response.
We and others have previously reported the genomic profiles associated with chemotherapy response to ovarian and other human cancers, as well as the status of oncogenic signaling pathways [5, 6]. In the present study, we sought to utilize global gene expression analysis to identify pathways that not only contribute to EC cell resistance to cisplatin but that may also represent therapeutic targets to increase sensitivity.
MATERIALS AND METHODS
Cell lines.
EC cell lines RL 95-2, AN3CA, KLE, and HEC-1-A were obtained from American Type Culture Collection (Manassas, VA); Ishikawa cells were purchased from Sigma Aldrich (St. Louis, MO); MEF 319, MEF 280, and MEF 296 cells were acquired from Deutsche Sammlung von Mikroorganismen und Zellkulturen, DSMZ (Braunschweig, Germany). MFE 280 and MFE 296 cell lines were maintained in 40% RPMI 1640, 40% MEM supplemented with 20% fetal bovine serum, and x1 insulin-transferrin-sodium selenite (ITS). The MFE 319 cell line was maintained in 40% RPMI 1640 and 40% MEM supplemented with 20% fetal bovine serum. The Ishikawa cell line was maintained in 95% MEM, 5% fetal bovine serum, and 1% nonessential amino acids. KLE cells were maintained in 90% DMEM/F12 and 10% fetal bovine serum. RL 95-2 cells were maintained in 90% DMEM/F12, 10% fetal bovine serum, and 0.00 5 mg/mL insulin. AN3CA cells were maintained in 90% MEM, 10% fetal bovine serum, 1% sodium pyruvate, and 1% nonessential amino acids. HEC-1-A cells were maintained in 90% McCoy’s 5A and 10% fetal bovine serum. All tissue culture reagents were obtained from Sigma Aldrich.
MTS assay.
Cells were seeded in 96-well plates at a density of 100 μM of 5–10 × 103 cells per mL and incubated overnight at 37°C. Cells were incubated with the indicated concentrations of cisplatin for 72 hours at 37°C. After addition of 20 μL of CellTiter 96R AQueous One solution (Promega) to each well, plates were incubated for 2.5 hours at 37°C. Absorbance was measured at 490 nm with a 96-well plate reader. All experimental wells and controls were set up in triplicate. The concentration at which proliferation was decreased by 50% was used to define the IC50. Cisplatin sensitivity/resistance was thus quantified, and genomic data were analyzed to identify biologic pathways associated with cisplatin sensitivity/resistance.
RNA extraction.
RNA was isolated from endometrial cell lines using Qiagen RNeasy Mini kit (Qiagen, Valencia, CA). Approximately 1 × 107 cells were disrupted using lysis buffer, and DNA was sheared by passing the lysate through 21-gauge needle 10 times. Total RNA was extracted in accordance with the Qiagen RNeasy Mini kit protocol. Quality of the RNA was measured using an Agilent 2100 Bioanalyzer (Agilent, Santa Clara, CA). Approximately 54,000 human mRNA genes were analyzed using the Affymetrix Human133A plus 2.0 GeneChip (Affymetrix, Santa Clara, CA). Messenger RNA and the target probes were prepared, hybridized, washed, and scanned according to the manufacturer’s instructions. The signal intensity measurements computed in the Affymetrix Microarray Analysis Suite (version 5.0) served as a relative indicator of the level of expression.
Statistical analysis of gene expression.
Affymetrix U133A plus 2.0 GeneChip expression data from the eight EC cell lines were subjected to background correction and normalization using the MAS5 algorithm implemented in Affymetrix Expression Console. Pearson’s correlation test was utilized to evaluate gene expression data and cell line cisplatin IC50 data. The genes with P<0.05 were considered to be significantly differentially expressed and used for pathway analysis.
Pathway analysis.
In an effort to provide a relevant biologic context to the genes identified to be associated with in vitro cisplatin responsiveness, an analysis of biologic pathway relationships was performed using commercially available software (MetaCore from GeneGo Inc). This literature-curated application correlates gene expression array data to relevant biological pathways, such that one can identify the networks, molecular mechanisms, and biological processes most relevant to developed data. A cutoff of P<0.05 was used to determine the statistical significance of the association between cisplatin sensitivity and resistance in an effort to identify potential activated molecular oncogenic pathway targets.
Western blot analysis.
Cells were harvested in media using a Cell Lifter, washed with cold PBS, and resuspended in cold HEPES lysis buffer (30 mM HEPES, pH 7.5, 10 mM NaCl, 5 mM MgCl2, 1 mM EGTA, 1% Triton X-100, and 10% glycerol), containing protease and phosphatase inhibitors (Sigma) on ice for 30 minutes. Lysates were clarified by centrifugation at 10,000 × g for 10 minutes at 0°C, and the supernatant was retained. Lysates were evaluated for protein concentration using the BCA method (Pierce, Rockford, IL). Proteins (50 μg) were separated on 10% SDS-PAGE gels and transferred to nitrocellulose membranes. Membranes were blocked in Tris-buffered saline containing 0.1% Tween 20 (TBST)-5% nonfat milk and incubated with primary antibody in TBST-5% nonfat milk overnight at 4°C. Membranes were washed three times for 10 minutes with TBST and incubated with the appropriate secondary antibody in TBST-5% non-fat milk for 120 minutes at room temperature. Antibody binding was visualized by chemiluminescence on autoradiography film.
siRNA electroporation.
The cisplatin-sensitive cell line, Ishikawa, and the cisplatin-resistant cell line, HEC-1-A, were subjected to small interfering RNA (siRNA) electroporetic transfection using the Nucleofector transfection kit according to manufacturer’s recommendations (Amaxa, Gaithersburg, MD). Cells (4 × 106) were suspended in 0.1 mL of electroporation buffer V each containing 1 μM siRNA and pulsed using program X-001. Pulsed cells were re-suspended in 0.5 mL of complete medium without antibiotics and incubated at 37°C for 15 minutes before experimentation. The Silencer negative control # 2 siRNA (catalog no. 4613, Ambion), a nonsense siRNA duplex, was used as a control. The effect on cisplatin-induced cell proliferation was evaluated by MTS assay. Two different siRNAs for each target were used in this study: protein kinase A (PKA; #6406, #6574 Cell Signaling) and PP2C (PPM1A #s10919, #s10920, Applied Biosystems).
RESULTS
Gene and pathway expression correlates with EC cell cisplatin sensitivity.
Cisplatin sensitivity/resistance was quantified by MTS, and the EC cell lines were ranked from most sensitive to most resistant by cisplatin IC50 as follows: Ishikawa, MFE 296, RL 95-2, AN3CA, KLE, MFE280, HEC-1-A, and MFE319 (Table 1). Pearson’s correlation of Affymetrix HGU133A expression data and cisplatin IC50 values identified 1443 genes (P<0.05) associated with in vitro cisplatin sensitivity/resistance. In an effort to place these differentially expressed genes into a relevant biologic contest, we used MetaCore from GeneGo Inc. MetaCore analysis identified a series of biologic pathways that correlated with cisplatin-resistant EC cells (P<0.002, Table 2). This analysis identified the BCL2 antagonist of cell death (BAD) protein and several members of the BAD apoptosis and survival pathway to be associated with EC cell line sensitivity to cisplatin (P=0.001) (Figure 1).
Table 1.
IC50 of 8 EC cell lines.
| Endometrial cell line | IC50 (μM), ±SE |
|---|---|
| Ishikawa | 22.9 ± 3.62 |
| MFE296 | 25.2 ± 6.46 |
| RL95-2 | 28.9 ± 4.31 |
| AN3CA | 32.1 ± 6.70 |
| KLE | 127.4 ± 38.87 |
| MFE280 | 321.2 ± 36.39 |
| HEC-1-A | 340.3 ± 24.83 |
| MFE319 | 383.7 ± 36.79 |
The 8 EC cell lines were seeded in 96-well plates at a density of 5–10 × 103 cells per mL and incubated overnight at 37°C. Cells were subsequently treated with cisplatin for 72 hours, and cell viability was analyzed using MTS assay.
Table 2.
Selection of the most represented biological categories and signaling pathways.
| Category | Pathway | P Value | Objects |
|---|---|---|---|
| Apoptosis and survival | HTR1A signaling | 2.7e-4 | 15/38 |
| IAP-proteins | 2.6e-2 | 9/29 | |
| BAD phosphorylation | 4.1e-2 | 10/36 | |
| Development | Leptin signaling via PI3K-dependent pathway | 6.0e-5 | 15/34 |
| A2A receptor signaling | 7.4e-3 | 12/37 | |
| IGF-a receptor signaling | 1.6e-3 | 15/44 | |
| GM-CSF signaling | 9.1e-3 | 14/47 | |
| Flt3 signaling | 1.8e-2 | 12/41 | |
| Cytoskeleton remodeling | Role of PKA in cytoskeleton reorganization | 4.8e-3 | 11/31 |
| Cytoskeleton remodeling, TGF,WNT and cytoskeletal remodeling | 1.2e-3 | 29/107 | |
| Integrin outside-in signaling | 7.7e-5 | 18/46 | |
| Cell adhesion | Integrin-mediated cell adhesion and migration | 2.0e-4 | 17/45 |
| Signal transduction | IP3 signaling | 5.7e-7 | 19/38 |
| cAMP signaling | 1.0e-2 | 11/34 | |
| Erk interactions | 1.5e-2 | 10/31 | |
| Regulation of lipid metabolism | Regulation of acetyl-CoA carboxylase 1 activity in keratinocytes, | 8.0e-5 | 6/7 |
| Regulation of acetyl-CoA carboxylase 1 activity in lipogenic tissues | 8.0e-5 | 6/7 | |
| Transcription | ChREBP regulation pathway | 8.5e-3 | 6/13 |
| Immune response | CD28 | 1.0e-4 | 17/43 |
| NFAT in immune response | 1.8e-5 | 18/42 | |
| BCR pathway | 9.5e-4 | 15/42 | |
| T cell receptor signaling | 2.1e-3 | 15/45 | |
| ICOS pathway in T-helper cell | 7.2e-4 | 14/37 | |
| NF-AT signaling and leukocyte interactions | 3.2e-3 | 13/38 | |
| CCR5 signaling in macrophages and T lymphocytes | 1.3e-2 | 13/44 |
Genes that showed differentially expressed between cisplatin-sensitive and –resistant EC cells were analyzed for biological pathway representation using GeneGo MetaCore software (P <0.002). IPS, inositol triphosphate.
Figure 1.
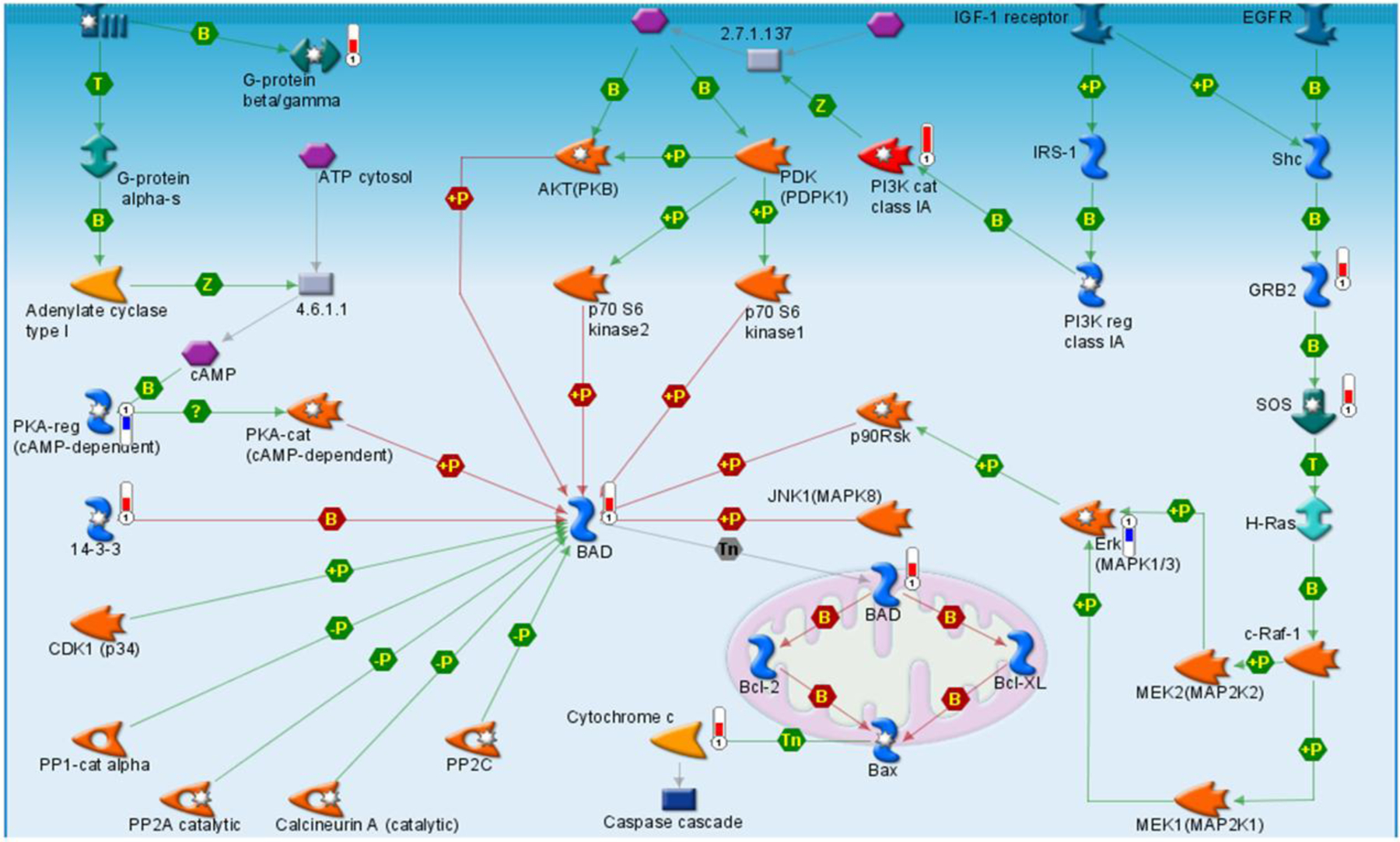
The BAD apoptosis and survival pathway is associated with cisplatin resistance. GeneGo Metacore Pathway analysis indicated that the BAD apoptosis and survival pathway was significantly associated with cisplatin resistance (P=0.0013). Expression levels of BAD pathway genes that increased (red) or decreased (blue) with cisplatin resistance are indicated. Depending on phosphorylation status, BAD is both a positive and negative regulator of apoptosis (BioCarta).
Modulation of BAD protein phosphorylation.
To determine whether BAD protein phosphorylation status is a determinant of EC cell cisplatin-induced apoptosis, we evaluated cisplatin-induced cell growth arrest after siRNA depletion of the BAD protein kinase PKA and the BAD protein phosphatase PP2C. Depletion of the BAD pathway phosphatase (PP2C) in an EC cisplatin-sensitive (Ishikawa) and -resistant (HEC-1-A) cell line increased expression of phosphorylated BAD (serine-155) and induced a parallel increase in resistance to cisplatin-induced cell growth arrest when compared to cells transfected with non-targeting siRNA (Figure 2A and 2B). In contrast, siRNA depletion of the BAD pathway kinase (PKA) decreased expression of phosphorylated BAD protein and increased cell line sensitivity to cisplatin-induced cell growth arrest (Figure 3A and 3B).
Figure 2.
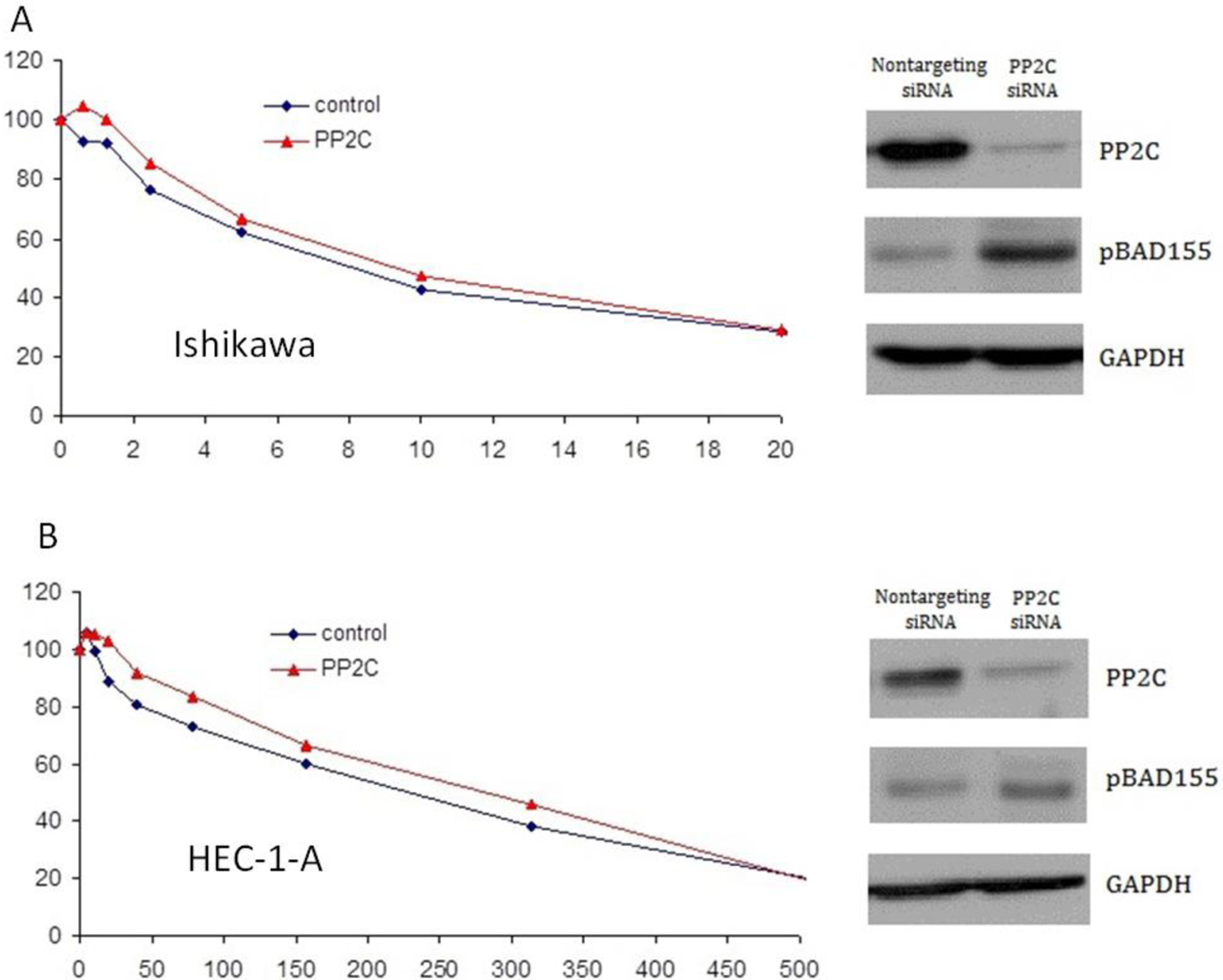
Increased expression of phosphorylated BAD (pBAD) decreases cisplatin sensitivity. Depletion of PP2C by siRNA in the endometrial cell lines Ishikawa (A) and HEC-1-A (B) resulted in increased expression of pBAD-serine-155 and decreased sensitivity to cisplatin-induced cell growth arrest.
Figure 3.

Decreased expression of phosphorylated BAD increases cisplatin sensitivity. Depletion of the BAD pathway kinase, PKA, by siRNA in Ishikawa (A) and HEC-1-A (B) cells resulted in increased sensitivity to cisplatin-induced cell growth arrest.
DISCUSSION
Advanced or recurrent EC has a poor prognosis, and conventional empiric treatment strategies with single-agent and combination cytotoxic therapies have produced little improvement in survival despite the potential for significant toxicity. Throughout treatment for recurrent EC, prolongation of survival and the successful maintenance of quality of life remain important goals. Improving our ability to manage the disease by optimizing the use of existing drugs and/or developing new agents is essential in this endeavor. To this end, individualizing treatments by identifying patients who will and will not respond to specific agents will potentially increase response rates and limit the incidence and severity of toxicities that compromise not only quality of life but also the ability to tolerate further therapies. We have defined a series of pathways, including BAD phosphorylation, 5-hydroxytryptamine receptor 1A (HTR1A) signaling, inhibitor of apoptosis proteins (IAP), leptin signaling via PI3K-dependent pathway, inositol triphosphate signaling, and integrin-mediated cell adhesion and migration, that may influence EC sensitivity to cisplatin-induced apoptosis.
Leptin signaling, integrin-mediated cell adhesion, and IAP pathways have previously been implicated in the proliferation of EC cells [7–9], and the α(v)β(6)-integrin and the IAP protein survivin have been shown to be overexpressed in EC patient tissue [7, 8]. Although HTR1A signaling has not been evaluated in EC, it has been found to be overexpressed in high-grade prostate tumors and may have a role in cellular proliferation [10].
BAD pathway genes have previously been implicated in human cancer development and resistance to therapy [11, 12], and the phosphorylation status of the BAD protein is known to influence cell apoptosis and survival [13, 14]. BAD phosphorylation has been reported to be associated with bladder cancer sensitivity to paclitaxel [15]; breast cancer sensitivity to epirubicin, paclitaxel, and tamoxifen [16–18]; and hepatocellular carcinoma sensitivity to sorafenib [12]. Furthermore, BAD expression has been associated with disease stage and disease free- and overall survival from lymphoma and colon cancer [19, 20].
The BCL2 family includes pro-apoptotic (e.g., BAD, BCL-Xs, Bak, Bax) and anti-apoptotic (e.g., BAG-1, Bcl-xL, Bcl-2, A1, MCL-1) members [21–23], each of which contain up to 4 BCL2-homology domains [21]. BAD has been shown to hetero-dimerize with Bcl-2 and Bcl-xL but not with Bax, Bcl-xs, A1, or Mcl-1. BAD dimerization with Bcl-xL results in Bcl-xL release of Bax, which increases mitochondrial membrane permeability that leads to apoptosis [14]. The ability of BAD to bind with other BCL2 family members is modulated by phosphorylation, which can occur at three principal sites, including serine-112, -136, and -155. Phosphorylated BAD cannot heterodimerize with Bcl-2 or Bcl-xL, such that Bcl-xL is free to bind Bax, which inhibits apoptosis [14]. BAD serine-136 phosphorylation is induced by protein kinase B (PKB/Akt) [24], whereas BAD serine-112 phosphorylation is induced by mitogen-activated protein kinase-activated protein kinase-1 (MAPKAP-K1, also called RSK) and PKA, and BAD serine-155 by PKA, which also inhibits Bcl-xL binding [25, 26]. Phosphatases PP1, PP2A, and PPM1 (PP2C/PPM1A) and calcineurin have pro-apoptotic effects due to their ability to de-phosphorylate BAD [27, 28].
In the present study, we sought to identify genes and gene pathways related to EC cell line cisplatin chemosensitivity. Furthermore, we evaluated the role of the BAD pathway protein kinase and phosphatase, PKA and PP2C, in determination of cisplatin chemosensitivity in EC cell lines. We demonstrated that selective manipulation of BAD phosphorylation status by manipulation of a kinase and phosphatase known to be key regulators in the phosphorylation of BAD provides further evidence that BAD phosphorylation is an important determinant of cisplatin sensitivity. Using an siRNA to PP2C, a phosphatase that functions to dephosphorylate BAD, we were not only able to increase phosphorylated BAD levels but also increased cisplatin resistance in EC cell lines. Conversely, using an siRNA to PKA kinases that function to phosphorylate BAD, we were able to demonstrate increased cisplatin sensitivity.
Our findings not only provide insights into the molecular basis of EC sensitivity to cytotoxic therapy but also support our previous findings that implicated the BAD pathway as an important determinant of human cancer chemosensitivity and also as a viable therapeutic target.
RESEARCH HIGHLIGHTS.
Endometrial cancer cell sensitivity to cisplatin is associated with expression of the BCL2 antagonist of cell death (BAD) pathway, likely via phosphorylation of the BAD protein
Modulation of BAD phosphorylation, by targeted inhibition of BAD kinases and phosphatases influences endometrial cancer cell sensitivity to cisplatin-induced apoptosis
The BAD pathway represents an appealing therapeutic target to increase endometrial cancer cell sensitivity to cisplatin
Funding:
Ocala Royal Dames for Cancer Research, Inc., Phi Beta Psi Sorority, and The Sandra Ernst Fellowship
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICT OF INTEREST STATEMENT
J. M. Lancaster is a member of the speaker’s bureau for Amgen and OrthoBiotech.
REFERENCES
- [1].Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin 60:277–300. [DOI] [PubMed] [Google Scholar]
- [2].Fleming GF, Brunetto VL, Cella D, Look KY, Reid GC, Munkarah AR, Kline R, Burger RA, Goodman A, Burks RT. Phase III trial of doxorubicin plus cisplatin with or without paclitaxel plus filgrastim in advanced endometrial carcinoma: a Gynecologic Oncology Group Study. J Clin Oncol 2004;22:2159–66. [DOI] [PubMed] [Google Scholar]
- [3].Pectasides D, Xiros N, Papaxoinis G, Pectasides E, Sykiotis C, Koumarianou A, Psyrri A, Gaglia A, Kassanos D, Gouveris P, Panayiotidis J, Fountzilas G, Economopoulos T. Carboplatin and paclitaxel in advanced or metastatic endometrial cancer. Gynecol Oncol 2008;109:250–4. [DOI] [PubMed] [Google Scholar]
- [4].Scudder SA, Liu PY, Wilczynski SP, Smith HO, Jiang C, Hallum AV 3rd, Smith GB, Hannigan EV, Markman M, Alberts DS. Paclitaxel and carboplatin with amifostine in advanced, recurrent, or refractory endometrial adenocarcinoma: a phase II study of the Southwest Oncology Group. Gynecol Oncol 2005;96:610–5. [DOI] [PubMed] [Google Scholar]
- [5].Dressman HK, Berchuck A, Chan G, Zhai J, Bild A, Sayer R, Cragun J, Clarke J, Whitaker RS, Li L, Gray J, Marks J, Ginsburg GS, Potti A, West M, Nevins JR, Lancaster JM. An integrated genomic-based approach to individualized treatment of patients with advanced-stage ovarian cancer. J Clin Oncol 2007;25:517–25. [DOI] [PubMed] [Google Scholar]
- [6].Potti A, Dressman HK, Bild A, Riedel RF, Chan G, Sayer R, Cragun J, Cottrill H, Kelley MJ, Petersen R, Harpole D, Marks J, Berchuck A, Ginsburg GS, Febbo P, Lancaster J, Nevins JR. Genomic signatures to guide the use of chemotherapeutics. Nat Med 2006;12:1294–300. [DOI] [PubMed] [Google Scholar]
- [7].Ai Z, Yin L, Zhou X, Zhu Y, Zhu D, Yu Y, Feng Y. Inhibition of survivin reduces cell proliferation and induces apoptosis in human endometrial cancer. Cancer 2006;107:746–56. [DOI] [PubMed] [Google Scholar]
- [8].Hecht JL, Dolinski BM, Gardner HA, Violette SM, Weinreb PH. Overexpression of the alphavbeta6 integrin in endometrial cancer. Appl Immunohistochem Mol Morphol 2008;16:543–7. [DOI] [PubMed] [Google Scholar]
- [9].Liu Y, Lv L, Xiao W, Gong C, Yin J, Wang D, Sheng H. Leptin activates STAT3 and ERK1/2 pathways and induces endometrial cancer cell proliferation. J Huazhong Univ Sci Technolog Med Sci 2011;31:365–70. [DOI] [PubMed] [Google Scholar]
- [10].Dizeyi N, Bjartell A, Nilsson E, Hansson J, Gadaleanu V, Cross N, Abrahamsson PA. Expression of serotonin receptors and role of serotonin in human prostate cancer tissue and cell lines. Prostate 2004;59: 328–36. [DOI] [PubMed] [Google Scholar]
- [11].Lee JW, Soung YH, Kim SY, Nam SW, Kim CJ, Cho YG, Lee JH, Kim HS, Park WS, Kim SH, Lee JY, Yoo NJ, Lee SH. Inactivating mutations of proapoptotic Bad gene in human colon cancers. Carcinogenesis 2004;25: 1371–6. [DOI] [PubMed] [Google Scholar]
- [12].Galmiche A, Ezzoukhry Z, Francois C, Louandre C, Sabbagh C, Nguyen-Khac E, Descamps V, Trouillet N, Godin C, Regimbeau JM, Joly JP, Barbare JC, Duverlie G, Maziere JC, Chatelain D. BAD, a proapoptotic member of the BCL2 family, is a potential therapeutic target in hepatocellular carcinoma. Mol Cancer Res 2010;8:1116–25. [DOI] [PubMed] [Google Scholar]
- [13].Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 1997;91:231–41. [DOI] [PubMed] [Google Scholar]
- [14].Yang E, Zha J, Jockel J, Boise LH, Thompson CB, Korsmeyer SJ. Bad, a heterodimeric partner for Bcl-XL and Bcl-2, displaces Bax and promotes cell death. Cell 1995;80:285–91. [DOI] [PubMed] [Google Scholar]
- [15].Szanto A, Bognar Z, Szigeti A, Szabo A, Farkas L, Gallyas F Jr. Critical role of bad phosphorylation by Akt in cytostatic resistance of human bladder cancer cells. Anticancer Res 2009;29:159–64. [PubMed] [Google Scholar]
- [16].Yu B, Sun X, Shen HY, Gao F, Fan YM, Sun ZJ. Expression of the apoptosis-related genes BCL-2 and BAD in human breast carcinoma and their associated relationship with chemosensitivity. J Exp Clin Cancer Res 2010;29:107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Craik AC, Veldhoen RA, Czernick M, Buckland TW, Kyselytzia K, Ghosh S, Lai R, Damaraju S, Underhill DA, Mackey JR, Goping IS. The BH3-only protein Bad confers breast cancer taxane sensitivity through a nonapoptotic mechanism. Oncogene 2010;29:5381–91. [DOI] [PubMed] [Google Scholar]
- [18].Cannings E, Kirkegaard T, Tovey SM, Dunne B, Cooke TG, Bartlett JM. Bad expression predicts outcome in patients treated with tamoxifen. Breast Cancer Res Treat 2007;102:173–9. [DOI] [PubMed] [Google Scholar]
- [19].Troutaud D, Petit B, Bellanger C, Marin B, Gourin-Chaury MP, Petit D, Olivrie A, Feuillard J, Jauberteau MO, Bordessoule D. Prognostic significance of BAD and AIF apoptotic pathways in diffuse large B-cell lymphoma. Clin Lymphoma Myeloma Leuk 2010;10:118–24. [DOI] [PubMed] [Google Scholar]
- [20].Sinicrope FA, Rego RL, Foster NR, Thibodeau SN, Alberts SR, Windschitl HE, Sargent DJ. Proapoptotic Bad and Bid protein expression predict survival in stages II and III colon cancers. Clin Cancer Res 2008;14:4128–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell 2004;116:205–19. [DOI] [PubMed] [Google Scholar]
- [22].Dejean LM, Martinez-Caballero S, Guo L, Hughes C, Teijido O, Ducret T, Ichas F, Korsmeyer SJ, Antonsson B, Jonas EA, Kinnally KW. Oligomeric Bax is a component of the putative cytochrome c release channel MAC, mitochondrial apoptosis-induced channel. Mol Biol Cell 2005;16:2424–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Desagher S, Osen-Sand A, Nichols A, Eskes R, Montessuit S, Lauper S, Maundrell K, Antonsson B, Martinou JC. Bid-induced conformational change of Bax is responsible for mitochondrial cytochrome c release during apoptosis. J Cell Biol 1999;144:891–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].del Peso L, Gonzalez-Garcia M, Page C, Herrera R, Nunez G. Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science 1997;278:687–9. [DOI] [PubMed] [Google Scholar]
- [25].Lizcano JM, Morrice N, Cohen P. Regulation of BAD by cAMP-dependent protein kinase is mediated via phosphorylation of a novel site, Ser155. Biochem J 2000;349:547–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Tan Y, Demeter MR, Ruan H, Comb MJ. BAD Ser-155 phosphorylation regulates BAD/Bcl-XL interaction and cell survival. J Biol Chem 2000;275:25865–9. [DOI] [PubMed] [Google Scholar]
- [27].Klumpp S, Selke D, Krieglstein J. Protein phosphatase type 2C dephosphorylates BAD. Neurochem Int 2003;42:555–60. [DOI] [PubMed] [Google Scholar]
- [28].Yang L, Omori K, Suzukawa J, Inagaki C. Calcineurin-mediated BAD Ser155 dephosphorylation in ammonia-induced apoptosis of cultured rat hippocampal neurons. Neurosci Lett 2004;357:73–5. [DOI] [PubMed] [Google Scholar]