

Abstract
Antibodies, particularly of the immunoglobulin G (IgG) isotype, are a group of biomolecules that are extensively used as affinity reagents for many applications in research, disease diagnostics, and therapy. Most of these applications require antibodies to be modified with specific functional moieties, including fluorophores, drugs, and proteins. Thus, a variety of methodologies have been developed for the covalent labeling of antibodies. The most common methods stably attach functional molecules to lysine or cysteine residues, which unavoidably results in heterogeneous products that cannot be further purified. In an effort to prepare homogeneous antibody conjugates, bioorthogonal handles have been site-specifically introduced via enzymatic treatment, genetic code expansion, or genetically encoded tagging, followed by functionalization using bioorthogonal conjugation reactions. The resulting homogeneous products have proven superior to their heterogeneous counterparts for both in vitro and in vivo usage. Nevertheless, additional chemical treatment or protein engineering of antibodies is required for incorporation of the bioorthogonal handles, processes that often affect antibody folding, stability, and/or production yield and cost. Accordingly, concurrent with advances in the fields of bioorthogonal chemistry and protein engineering, there is growing interest in site-specifically labeling native (nonengineered) antibodies without chemical or enzymatic treatments. In this review, we highlight recent strategies for producing site-specific native antibody conjugates and provide a comprehensive summary of the merits and disadvantages of these strategies.
Graphical Abstract
INTRODUCTION
Antibodies are a class of circulating Y-shaped proteins that are produced mainly by plasma cells and exploited by the immune system for targeting and neutralizing/eliminating foreign substances. Because of their superb ability to bind to specific molecules or parts of molecules, antibodies are recognized as a unique class of biological tools that are invaluable for both basic biochemical research and biomedical therapy. Antibodies are widely used in basic science laboratories for a variety of detection technologies, including Western blotting, immunocytochemistry, flow cytometry, microscopy, and others. Aside from their use in detection, monoclonal antibodies have been used for therapeutic applications since 1985, when muromonab-CD3 was first approved by the Food and Drug Administration (FDA) for the treatment of acute rejection of organ transplants.1 To further expand the utility of these tools, a variety of functional molecules, including fluorophores, enzymes, proteins, and other moieties, have been conjugated to antibodies. The science of antibody modification has advanced in parallel with the development of antibody–drug conjugates (ADCs) for targeted therapy of cancer.2
First-Generation Antibody Conjugation.
The concept of combining the specificity of antibodies with the toxicity of drugs to create a targeted pharmaceutical with increased therapeutic index can be traced to 1913, when Paul Ehrlich proposed to develop a “magic bullet” for selective targeting of tumors.3 Despite the simplicity and elegance of this concept, the first ADC was not available for clinical use until 2000, with FDA approval of Gemtuzumab ozogamicin (Mylotarg, Pfizer/Wyeth) for the treatment of acute myeloid leukemia.4 In 2011, Brentuximab vedotin (Adcetris, Seattle Genetics) was approved for the treatment of anaplastic large cell lymphoma/Hodgkin’s lymphoma.5 Two years later, Trastuzumab emtansine (Kadcyla, Genentech/Roche) was approved for the treatment of advanced human epidermal growth factor receptor 2 (HER2)-positive breast cancer.6 To date, more than 10 ADCs have been approved for various cancer treatments, including Inotuzumab ozogamicin; Polatuzumab vedotin; Enfortumab vedotin; Trastuzumab deruxtecan; Sacituzumab govitecan; Belantamab mafodotin; Moxetumomab pasudotox; and Loncastuximab tesirine.7,8 In these cases, covalent conjugation of a functional drug to the antibody was accomplished via the use of first-generation antibody conjugation technology, which makes use of either reduced cysteine residues (4 pairs of interchain disulfides of IgG1) or surface-exposed lysine residues (roughly 80 potential coupling sites) present in the antibody. These first-generation antibody conjugation methods utilize N-hydroxysuccinimide (NHS) or maleimide-mediated cross-linking for stable attachment of a functional molecule to the primary amine of lysine or the thiol group of cysteine, respectively. Because of the large number of potential attachment sites, these methodologies invariably yield heterogeneous products with variable drug-to-antibody ratios (DAR). These heterogeneous reagents have subsequently been shown to have suboptimal therapeutic indices compared to homogeneous site-specific ADCs.9,10
Second-Generation Antibody Conjugation.
The first systematic study demonstrating the benefits of site-specific ACDs was initiated by Junutula and co-workers at Genentech.10 These site-specific ACDs, called THIOMAB antibody–drug conjugates (TDCs), were produced by the introduction of a “hot” cysteine residue, followed by a global reduction of “hot” cysteine and interchain disulfides and subsequent oxidation in the presence of CuSO4 to regenerate interchain disulfides. Finally, drugs were conjugated to the reduced “hot” cysteine using maleimide chemistry. Compared to the average 3:1 DAR of the ADCs prepared using the first-generation antibody conjugation technology, the site-specific THIOMAB-derived anti-MUC16-monomethyl auristatin E (MMAE) was characterized by a reduced DAR of 2:1, yet exhibited improved in vivo efficacy in a mouse xenograft model of ovarian cancer. Importantly, the improved therapeutic index of the TDCs was accompanied by higher dose tolerance and increased serum stability in rats and cynomolgus monkeys.
Due to the growing ADC market and the observed advantages of site-specific ADCs, increased efforts have been directed toward the creation of site-specific ADCs generated using the second-generation antibody conjugation technologies. In general, the second-generation antibody conjugation methods require an initial site-specific introduction of a unique reactive moiety into the antibody, followed by the use of the bioorthogonal reaction counterpart for selective coupling of a functional molecule to this unique moiety. Genetic code expansion, protein tagging, and enzymatic treatments have all been used for site-specific introduction of a unique reactive moiety into proteins.11 Advances in strategies for site-specific incorporation of noncanonical amino acids (ncAAs) into proteins in both prokaryotic and eukaryotic cells provide an elegant approach toward the preparation of site-specific antibody conjugates.12–21 An ncAA with a bioorthogonal keto moiety, p-acetylphenylalanine (pAcF), was first chosen to explore this methodology. Using an aminoacyl-tRNA synthesis (aaRS)/tRNA pair that is orthogonal to the endogenous translation machinery of the host,22,23 pAcF was incorporated into trastuzumab at a site represented by an amber codon.24 The resulting bis-pAcF-containing trastuzumab, each on one heavy chain, was then coupled to the alkoxy-amine-derivatized drug monomethyl auristatin F, achieving a DAR of 2. Although this site-specific ADC exhibited antitumor efficacy comparable to that of a heterogeneous ADC prepared using the first-generation antibody conjugation technology, the ADC prepared by genetic code expansion exhibited an improved safety profile. Using a similar strategy, trastuzumab with an azido moiety was also recently prepared and conjugated to alkyne-modified drug using azide–alkyne cycloaddition, or click chemistry.25,26 This technology provides the additional advantage of enabling the preparation of first-in-class well-defined multiaction antibodies site-specifically conjugated with both a drug and a fluorophore.26 This combination provides a direct and general method for probing the biodistribution of therapeutically active ADCs.
In spite of the power of this genetic code expansion technology, the relatively low expression level of ncAA-containing antibody compared to wild-type antibody limits the utility of this methodology for studies in which large quantities of product are needed.12 To avoid the potential decrease in the yield of engineered antibodies, the desired bioorthogonal moiety contained in an antibody could be generated in situ via enzymatic treatment.27 The enzymatic strategy SMARTag is based on aldehyde tag technology in which a short peptide tag, CXPXR, is introduced at the desired protein position. The cysteine residue within the peptide sequence, but not other cysteines, can be oxidized to a formylglycine residue in the presence of the formylglycine generating enzyme (FGE).28 Bertozzi, Rabuka, and co-workers at Redwood Bioscience (now part of Catalent Pharma Solutions) found that antibodies containing a formylglycine residue can be used for site-specific conjugation of drugs via the use of bioorthogonal reactions.29 Oxime ligation and alkoxyamine- and hydrazine-Pictet-Spengler (HIPS) ligation30,31 are two examples of this type of coupling reaction. Using this technology, the Redwood Bioscience team performed a structure–activity relationship study on ADCs, revealing that the in vivo stability and efficacy of ADCs are conjugation-site-dependent.32 This strategy was recently used to prepare glycocalyx editing antibodies to reactivate immune response toward cancer cells both in vitro and in vivo.33,34
By taking advantage of enzyme-mediated covalent bond formation between proteins and small molecules, other investigators have used enzymes such as transglutaminase and sortase to prepare site-specific ADCs. Transglutaminase catalyzes amide bond formation between a glutamine residue and the primary amine of the molecule.35 Schibli’s group first used bacterial transglutaminase to carry out the site-specific antibody modification.36 Surprisingly, despite the abundance of glutamine residues in the antibody molecule, transglutaminase modified only Gln295 following peptide-N-glycosidase F (PNGase F)-mediated deglycosylation of nearby residue N297 or following the creation of an N297S mutation. Rather than utilizing this deglycosylation approach, Strop and others showed that the glutamine tag LLQGA could be site-specifically introduced into antibodies.37 The tagged antibodies could be subsequently coupled to amine-functionalized molecules with a DAR of 1.8–1.9. In a similar fashion, sortase can also be employed to site-specifically modify a residue within a desired protein sequence.38 Sortase is a bacterial transpeptidase that catalyzes the covalent attachment of polyglycines to the LPXTG protein tag. Based on this mechanism, the sortase recognition motif LPETG was site-specifically introduced into different antibodies and antibody fragments, followed by sortase-mediated linkage to polyglycine probes.39–41 Under optimal conditions, 90% conjugation was achieved using this strategy.
Avoiding the use of enzymes, the Pentelute group recently discovered that the four-residue π-clamp peptide FCPF provides a unique chemical environment for the selective reaction of the motif’s cysteine residue with perfluoroaromatic reagents.42 A site-specific ADC was prepared by first installing an FCPF tag into the C-terminus of the antibody heavy chain, followed by a reaction of the modified antibody with a perfluoroaryl-labeled drug under reducing conditions. The in vivo efficacy of the resulting site-specific antibody conjugates remains to be explored.
Next-Generation Antibody Conjugation.
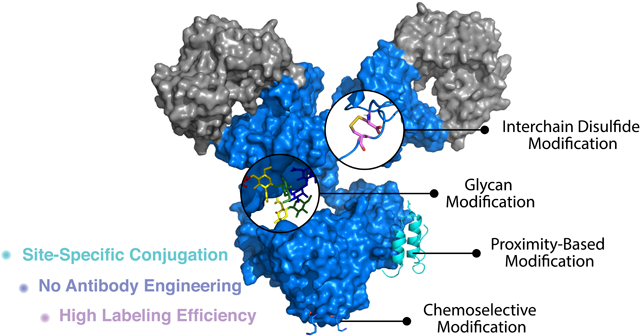
In general, the second-generation antibody conjugation technologies achieve their site-specific modifications by introducing a bioorthogonal moiety into the antibody. While homogeneous antibody conjugates can be prepared using these methods, the introduction of the bioorthogonal moieties requires additional chemical treatment or antibody engineering procedures that are often technically challenging and, in some cases, could possibly affect antibody folding and stability. Furthermore, these methods cannot be applied to native (nonengineered) antibodies, which account for more than 99% of commercial antibodies. Thus, there is growing interest in performing well-defined, site-specific antibody modifications that maintain antibody integrity, while achieving optimal therapeutic efficacy and production efficiency. Central to precision labeling of native antibodies is the ability to site-selectively modify a single, desired canonical amino acid residue without labeling identical canonical amino acids that appear in several other positions within the antibody. Recently, this site-selectivity has been achieved via the use of unique targeting moieties or by directing targeting reactivities through the use of proximity effects. Here, we review recently reported strategies that enable site-specific modification of native antibodies. These methods have been summarized into four categories, including interchain disulfide modification, glycan modification, chemo-selective modification, and proximity-based modification (Figure 1).
Figure 1.
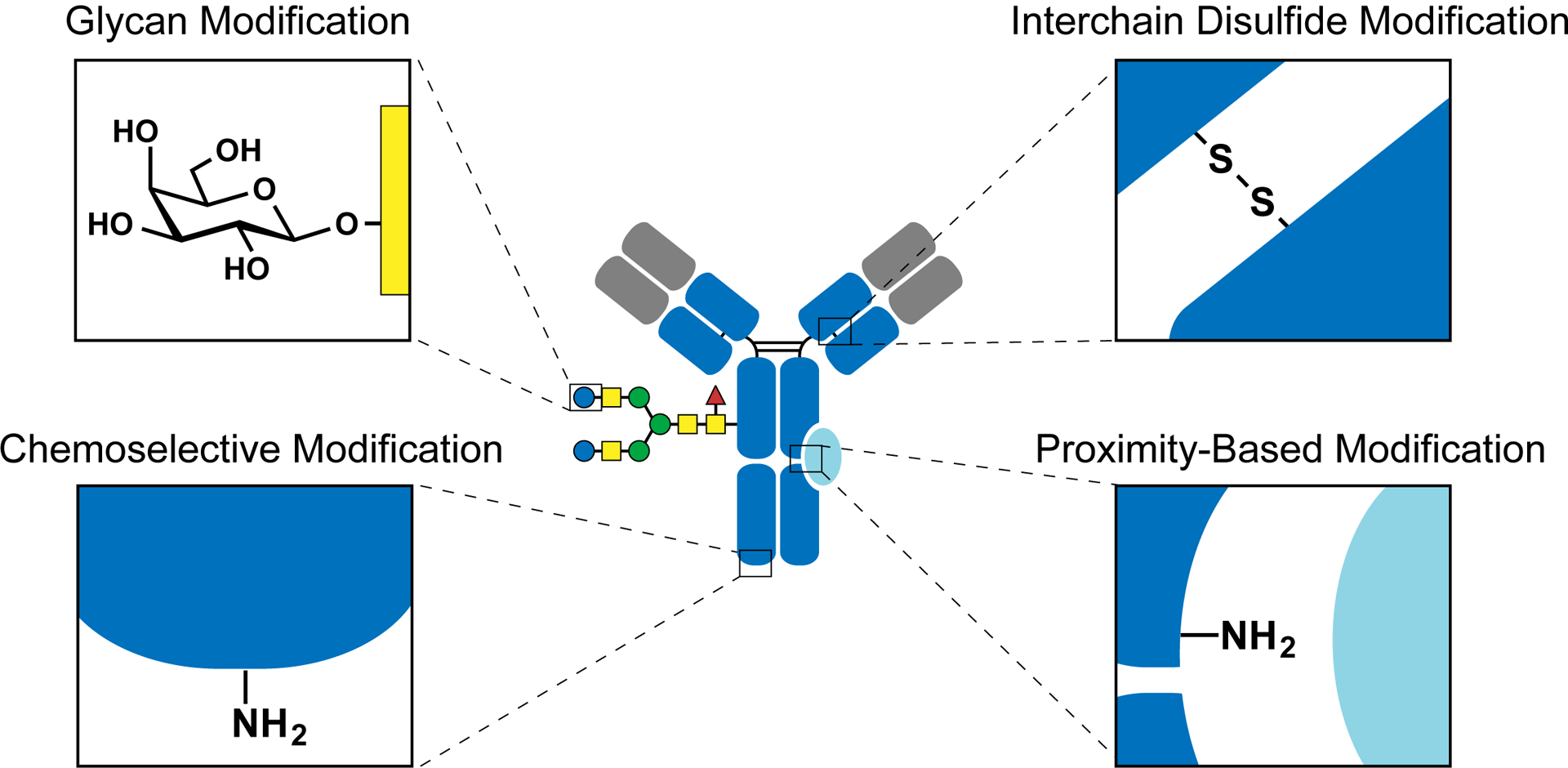
Scheme highlighting methods to site-specifically label native antibodies.
INTERCHAIN DISULFIDE MODIFICATION
Cysteine-based conjugation of a payload to an antibody relies on the reaction between a reduced sulfhydryl group in the antibody and a thiol-specific electrophile in the payload. For making ADCs, these methods have advantages over other methods based on nucleophilic lysine residues because the low abundance of cysteine residues confers a high level of site-specificity for the conjugation. The four interchain disulfide bonds of an IgG molecule can be reduced to yield eight free thiol groups, leading to the generation of a homogeneous ADC with a maximum DAR of 8. However, it is reported that higher drug loading (DAR = 6 or 8) can result in decreased therapeutic efficacy owing to higher plasma clearance rate9 and aggregation.43 Introduced in 1990 by the Smith and Lawton research group,44,45 disulfide rebridging offers a promising strategy for functionalizing antibody cystines in a site-specific fashion with less structural disturbance of the native state. The key steps for disulfide rebridging can be summarized as follows: (1) Reduction of disulfides to yield two activated cysteines without disulfide scrambling that results in the reconfiguration of interchain disulfide bonds in antibodies; (2) An initial reaction between one of the cysteines and the rebridging reagent; and (3) a second reaction between the remaining cysteine and the rebridging reagent–cysteine complex, thus retaining the native linkages of the antibody. In the following section, we will discuss current methods of disulfide rebridging, including reagents, potential problems, and limitations.
Disulfide Reactivation.
Reduction of the disulfide bond to generate a pair of free cysteines is generally the first step for disulfide rebridging. There are a total of 16 disulfide bonds in IgG, of which the 4 interchain ones are more solvent accessible than the 12 intrachain ones. The most commonly used cystine reducing agents including tris(2-carboxyethyl)phosphine (TCEP) hydrochloride, dithiothreitol (DTT), and 2-mercaptoethanol. The thiol-based reductants DTT and 2-mercaptoethanol require a neutral to basic working pH (>7) due to the intrinsic pKa value of the sulfhydryl group. After reduction, the unreacted DTT and 2-mercaptoethanol must be removed prior to the rebridging process; otherwise, the reductant will compete with the rebridging agent. As an alternative, the phosphine-based reagent TCEP has a wider effective reducing pH range (1.5 < pH < 8.5), and TCEP can be used in some cases along with the rebridging agent without competition due to its weaker nucleophilic nature toward common rebridging electrophiles (Michael acceptors, addition–elimination sub-stitutions) compared to thiol-based reductants. Generally, under nondenaturing conditions, only the four solvent-accessible interchain disulfides will be reduced, leaving the intrachain disulfides intact.46,47
Mono/Bis-Sulfone-Type Reagents.
In 1990, the Smith and Lawton research group reported the use of bis-sulfone for specific modification of antibodies (a1, Figure 2i).44,45 Bis-sulfone requires activation, by eliminating a sulfinate to yield the active monosulfone species that reacts with one of the free thiols generated by disulfide reduction. A second Michael acceptor is generated by elimination of the other sulfinate, and the remaining thiol then undergoes the second Michael addition to yield the bisthioether with a three-carbon linkage. In 2014, the first full-size ADC produced by disulfide rebridging was reported by Badescu et al.47 Utilizing this bis-sulfone strategy, Badescu successfully linked the cytotoxic payload MMAE to the disulfides of the native humanized antibody Trastuzumab, achieving a 78% yield of a species with the desired DAR of 4 (Figure 2i). The disulfide-rebridged ADC exhibited minimal degradation over 96 h in the presence of serum condition, while the traditional maleimide construct suffered significant decomposition. Remarkably, the rebridged ADC product can survive in 10 mM DTT for 30 min.47 However, another work shows that the glutathione (GSH) present inside cells at millimolar concentration was found to cleave bisthioether conjugates of the peptide hormone somatostatin.48 Wang et al. later reported that the revised reagent allyl sulfone, a monosulfone intermediate generated during bis-sulfone dialkylation, has similar functions but better conjugation efficiency than bis-sulfone (due to greater water solubility), and in addition avoids in situ activation (a2, Figure 2i).49 However, allyl sulfone-based disulfide rebridging methods have only been carried out in model proteins, and not yet in full-size IgGs.
Figure 2.
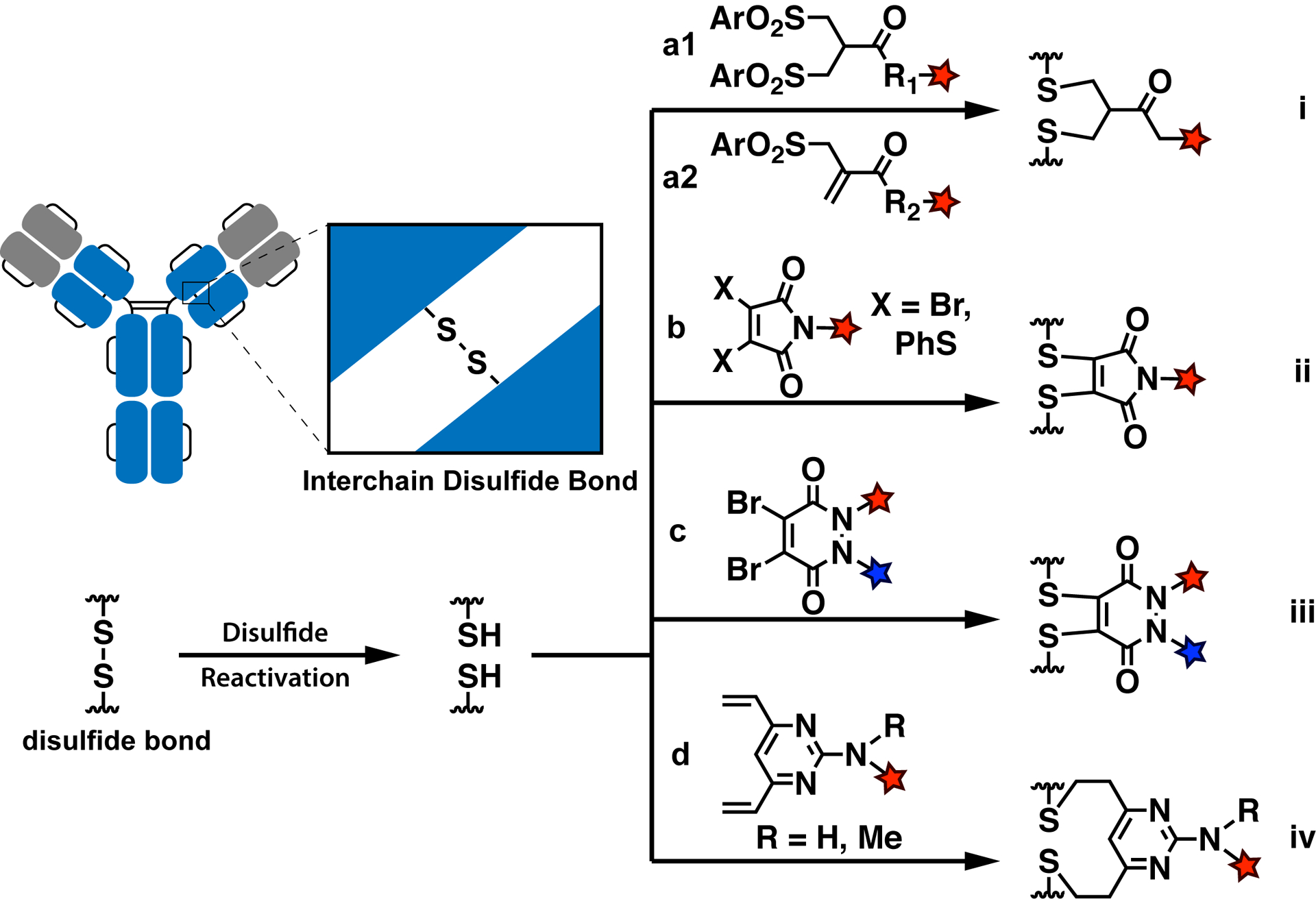
Site-specific antibody conjugation based on disulfide rebridging. Reducing agents are used to activate the solvent accessible interchain cystines, followed by rebridging with (i) mono/bis-sulfone-type reagents, (ii) 3,4-disubstituted maleimides, (iii) dibromopyridazinediones, and (iv) divinylpyrimidines.
3,4-Disubstituted Maleimides.
The group of Baker and Caddick have utilized next-generation maleimides (NGMs), including 3,4-dihalomaleimides and dithiomaleimides, to rebridge and functionalize disulfides via a double addition–elimination mechanism (b, Figure 2ii).50,51 Compared to the mono/bis-sulfone-type reagents, which require weak basic conditions for activation, NGMs have a wider working pH range from 6.2 to 8.50 In addition, these NGMs exhibit fast reaction kinetics, an advantage for decreasing the chance of disulfide scrambling and antibody aggregation during the rebridging reaction. In 2014, Schumacher et al. demonstrated the use of dithiophenolmaleimide-modified doxorubicin to rebridge Trastuzumab disulfides after the reduction with TCEP or benzeneselenol (Figure 2ii).52 A DAR = 3.4 and 89% overall yield (76% full-size correctly folded antibody, 24% scrambled product) was achieved. In addition, control of the DAR over a range from 0 to 4 was achieved by fine-tuning the stoichiometry of reagents and the reaction time. In terms of stability, the rebridged antibody was prone to lose its cargo by transferring the dithiomaleimides to serum proteins via the process of cysteine addition–elimination. This stability issue was solved by hydrolysis of the maleimides under basic conditions (pH 8.4, 72 h),53,54 to yield maleamic acids, which are inert to thiols.54
Dibromopyridazinediones.
Following the success of NGMs, Maruani et al. utilized a similar rebridging mechanism in 2015 to design dibromopyridazinedione-type heterocycles (Figure 2iii).55 These reagents were used to carry out one-pot reactions that, compared to NGMs, resulted in near-complete retention of antibody structure with minimum disulfide scrambling.55 The two nitrogens on the pyridazinedione provide two modification opportunities (Figure 2iii). However, the incompatibility between dibromopyridazine-dione and the TCEP reducing agent needed to be compensated for by using a large excess of reagents. To address this problem, Lee et al. replaced the bromide leaving group with a TCEP-linked thiophenol to achieve one-step, single reagent disulfide rebridging.56 This successfully retained the advantage of one-pot in situ methods without the need for use of excess rebridging agents.56 To restrict the DAR to lower values for pharmacokinetic reasons, Lee et al.57 connected two dibromopyridazinedione with an appropriate-length linker to conjugate two disulfide pairs with a single bioorthogonal functionality, resulting in antibody products with a DAR of 2. Incubation of the resulting conjugates at either low or high pH does not cause decomposition of the dithiopyridazinedione products.55 The rebridged dithiopyridazinedione product is hydrolytically inert but sensitive to thiols at high concentrations.55 The rebridged product (0.4 μM) can survive in serum-mimicking conditions with 0.2 μM glutathione for 7 days,55 but can be quantitatively converted back to the reduced disulfide state using a 100 equiv excess of 2-mercaptoethanol.58
Divinylpyrimidines.
In 2019, Walsh et al.59 reported a new type of bis-Michael acceptor, divinylpyrimidine, for coupling functionalities to antibodies via reaction with reduced disulfides. This methodology resulted in good yields of rebridging product, as verified by LC-MS (Figure 2iv). It is worth noting that serious disulfide scrambling was observed. Compared to other types of rebridging agents, divinylpyrimidine possesses a relatively long disulfide bridge (7 carbons versus 2 or 3 carbons between 2 thiols), which potentiates disulfide scrambling. In spite of this, good reaction kinetics and high yield make the divinylpyrimidine reagent a good substitute for dibromopyridazinediones or NGMs when disulfides are rebridged in less complex systems such as fragment antigen-binding (Fab) or monocystine proteins.
Summary.
Despite extensive studies on mono/bis-sulfone, NGMs, and divinylpyrimidines that have facilitated the site-specific functionalization of interchain disulfides in native antibodies, certain pitfalls and limitations still exist. First, disulfide scrambling is unavoidable, affecting product efficacy, yield, and scalability. Several approaches have been proposed or applied to minimize disulfide scrambling, including precise control of reaction conditions, increasing rebridging kinetics,51 reduction and rebridging in situ,52,55 and use of an all-in-one TCEP-linked thiophenol derivative.56 However, these strategies need to be optimized case by case. Second, these rebridging reagents cannot distinguish between reduced disulfides and free thiols under reducing condition, limiting the use of disulfide rebridging to a subset of antibodies lacking free cysteines. Wilson et al.60 reported a trivalent arsenous (As(III)) acid derivative as a potential solution to generating orthogonality for disulfide thiols, since the monothiol arsenous acid complex is entropically unfavored. However, this usage was not demonstrated in antibody applications, and questions regarding in vivo toxicity at therapeutically relevant antibody concentrations still need to be addressed. Third, rebridging reagents exhibit poor water solubility and usually require varying ratios of cosolvent to carry out the reaction. This increases the chance of denaturing the antibody. In the case of bis-sulfone reagents, this problem can be alleviated by using the water-soluble monosulfone intermediate. Extra PEG linkers can be introduced for other constructs.
GLYCAN MODIFICATION
Immunoglobulin G antibodies (IgGs) are modified by two structurally heterogeneous N-glycosyl groups at the conserved Asn297 residue, located in the constant CH2 domain of the fragment crystallizable (Fc) region (Figure 3A and B). Because this oligosaccharide modification lies far away from the variable region of IgG, it serves as an attractive site for chemical or biological modifications of antibodies that do not affect antigen-binding properties. In general, oligosaccharide-based antibody conjugation is achieved by introducing a chemically reactive moiety into the N-glycosyl moiety, followed by conjugation to a substrate bearing another chemically reactive group.
Figure 3.
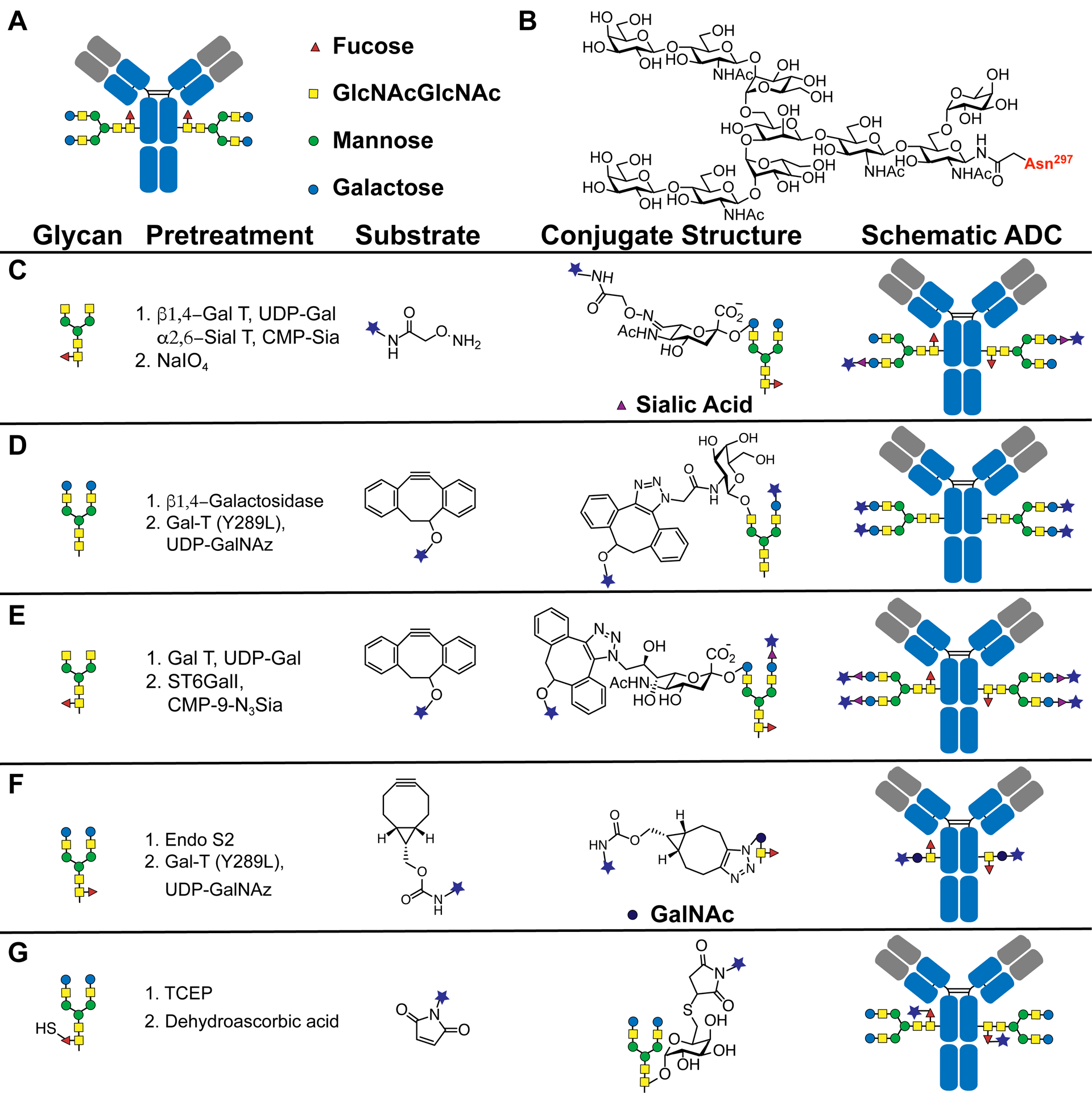
Preparation strategies for oligosaccharide-based antibody conjugation. (A) Schematic structure of an immunoglobulin G antibody with N-glycosylation at Asn297. (B) Chemical structure of N-glycosyl group. (C) Glycosylation remodeling through Gal T and Sial T followed by periodate oxidation and oxime-based chemical conjugation. (D) 1,4-Galactosidase-mediated removal of terminal galactose, followed by Gal-T mutant-mediated GalNAz attachment and catalyst-free azide/alkyne cycloaddition click chemistry. (E) Gal T-mediated terminal galactose attachment, then ST6GalI-mediated 9-azido-sialic acid attachment and generation of ADCs via click chemistry. (F) Endo S2-mediated removal of terminal glycans, followed by Gal-T mutant-mediated GalNAz attachment and generation of ADCs via copper-free click chemistry. (G) Metabolic incorporation of the 6-thiofucose peracetate, followed by generation of ADCs via maleimide chemistry.
Oxidation-Based Glycan Remodeling.
Oxidative cleavage of the cis-diol group at the distal terminus of an oligosaccharide generates an aldehyde group that is ready to react with an aminooxy-, hydrazine-, or hydrazide-functionalized payload to form a site-specific antibody conjugate. O’Shannessy and colleagues first used the periodate oxidation method to prepare antibodies with aldehyde groups that were then reacted with hydrazide-labeled biotin.61 Similar approaches were used to conjugate a variety of molecules, including radiolabeled organometallic complexes,62 toxins,63 and proteins/antibodies. Although these methods do achieve site-specific conjugation of native antibodies, the heterogeneity of antibody glycosylation greatly limits the applicability of this approach. Furthermore, high concentrations of periodate (10–30 mM) are known to result in undesirable oxidation of sensitive methionine residues, potentially compromising the serum half-lives, integrity, and efficacy of antibodies.64
To avoid this problem, Zhou et al. introduced a periodate-sensitive sialic acid moiety into the antibody N-glycan (Figure 3C).65 To achieve the well-defined modification, galactose and sialic acid residues were enzymatically incorporated into the native antibody oligosaccharide using β1,4-galactosyltransferase (Gal T) and α2,6-sialyltransferase (Sial T), respectively. MALDI-TOF analysis indicated that over 94% of the N-glycosyl residues were converted to the monosialylated biantennary form by treatment with Gal T and Sial T. These sialic acid residues were oxidized using 1 mM periodate to yield reactive aldehyde groups, which were reacted with aminooxy-functionalized cytotoxins to generate site-selective antibody conjugates with an average DAR of 1.6. Although these ADCs have significant antitumor efficacy in vitro and in vivo, low concentrations of periodate still compromise the neonatal Fc receptor (FcRn) binding of trastuzumab by about 25%.
Nonoxidation-Based Glycan Remodeling.
To avoid periodate treatment altogether, several groups have enzymatically modified oligosaccharides by introducing sugar residues with bioorthogonal groups. Boeggeman et al. demonstrated that galactose containing a ketone handle at the C2 position can be incorporated into the antibody oligosaccharide using the β1,4-galactosyltransferase mutant GalT (Y289L).66 Selective conjugation can then occur between the ketone handle and an aminooxy-containing derivative (biotin or fluorescent dye). However, the conversion yield and efficiency of this process require further evaluation. Zeglis et al. have also applied enzyme-mediated glycoengineering and strain-promoted azide–alkyne cycloaddition reaction to site-specifically radiolabel the modified oligosaccharide of a prostate-specific membrane antigen (PSMA) targeting antibody, J591 (Figure 3D).67 Specifically, they used β1,4-galactosidase to remove the terminal galactose and then used GalT (Y289L) to introduce azide-modified galactose as the terminal residue of the glycosyl group. A desferrioxamine (DFO)-containing chelator was then attached to the antibody using a strain-promoted azide–alkyne cycloaddition reaction. Finally, 89Zr-labeled ADCs can be generated by mixing chelator-modified antibodies with 89Zr. The 89Zr-labeled antibody generated via this method was selectively taken up by tumors and exhibited excellent tumor-to-background contrast in mouse models bearing PSMA-expressing prostate tumors.
As an alternative strategy, Li and co-workers used the Gal T to remodel antibody glycosyl groups, converting existing glycoforms to G2 glycoforms (Figure 3E).68 A sialic acid derivative bearing an azide at the C9 position was then incorporated into the N-glycan using the sialyltransferase ST6GalI. Subsequently, dibenzylcyclooctanol (DIBO)-modified biotin, fluorophores, or cytotoxic drugs were reacted with azide-containing antibodies to form antibody conjugates with DARs of 3.5–4.5. This glycosylation remodeling strategy provides an attractive alternative to the preparation of homologous ADCs with unaltered FcγRIIIa binding. To reach lower DAR for pharmacokinetic reasons, van Delft and co-workers reported a new chemoenzymatic conjugation strategy for generating ADCs with a DAR of 2 (Figure 3F).69 After deglycosylation and introducing GalNAz by endoglycosidase Endo S2 and galactosyl-transferase GalT (Y289L), respectively, the modified antibody can be ligated to the payload via copper-free click chemistry, resulting in highly stable and homogeneous ADCs with DARs around 2.
Metabolic Engineering of Antibody Oligosaccharides.
In addition to engineering antibody oligosaccharide modification using enzymatic and chemical strategies in vitro, ADCs can also be obtained by using metabolic engineering of oligosaccharide residues to introduce sugar analogues containing bioorthogonal reactive groups.70,71 Okeley and co-workers first reported efficient metabolic incorporation of the fucose analogue 6-thiofucose at the N-glycan terminus using fucosyltransferase VIII (Figure 3G).72 Specifically, 6-thiofucose peracetate, identified among approximately 200 synthetic fucose analogues,73 could replace fucose in the antibody oligosaccharide with 60–70% efficiency. Subsequently, the 6-thiofucose moiety was conjugated to MMAE via maleimide chemistry to produce an ADC with an average DAR of 1.3. The ADC obtained using this strategy maintains improved plasma stability and in vitro antitumor activity compared to ADCs prepared by interchain disulfide/maleimide conjugation chemistry.
Summary.
Although significant advances have been made in obtaining IgGs with glycoforms bearing bioorthogonal groups, most enzyme-mediated glycosylation engineering techniques are still in the lab-scale exploration period. Additionally, some unnatural components always need to be introduced into antibody glycans to produce ADCs. It has been reported that some glycans with subtle modifications are immunogenic in humans.74,75 Therefore, the in vivo stability and immunogenicity of glycan-engineered antibody conjugates remain to be fully evaluated.
CHEMOSELECTIVE MODIFICATION
In concept, performing chemical reactions that directly modify a specific amino acid residue based on chemoselectivity is the most straightforward means of modifying native antibodies. However, small differences in reactivity of different nucleophilic amino acids make it challenging to achieve the desired specificity of chemical modifications. In addition, due to the residue heterogeneity that exists between structurally similar antibodies, chemo- and regioselective modification methods usually work only in specific cases, limiting the generality of this approach. A widely used concept involves targeting the lysine with the lowest pKa via precise control of reaction pH. For example, N-terminal amines exhibit weaker basicity than other lysines and can be targeted using an average pH of 7.7 ± 0.5.76 In this section, we summarize the recent development of site-specific antibody modifications based on chemoselectivity.
N-Terminal Transamination.
In 2007, Scheck et al.77 reported the use of pyridoxal-5′-phosphate (PLP, vitamin B6) to convert N-terminal amines of immunoglobulin light chains to ketone moieties via a transamination reaction. The ketone can then be easily functionalized via oxime ligation. The less basic N-terminal amine (imine formation) and the more acidic N-terminal α-proton (tautomerization) underlie the chemoselectivity of the N-terminal amines compared to other side chains of lysine residues. A moderate conversion (47%) is achieved at an elevated temperature (50 °C). Based on the same transamination mechanism, Witus et al. utilized Rapoport’s salt (RS) to selectively convert the N-terminal amine of the immunoglobulin heavy chain to a ketone moiety under much milder conditions (37 °C), with an improved yield (67%, Figure 4A).78 Interestingly, these transamination reagents exhibit some residue preferences. PLP prefers light chain aspartate over heavy chain glutamine in murine IgGs, and RS prefers heavy chain glutamate over light chain aspartate in human IgGs. Further experiments have revealed that the optimal reaction partners for PLP and RS are the tripeptides, H-Ala-Lys-Thr and H-Glu-Glu-Ser, respectively. Since these N-terminal sequences are not present in native antibodies, genetic engineering is required for optimal reactivity.
Figure 4.
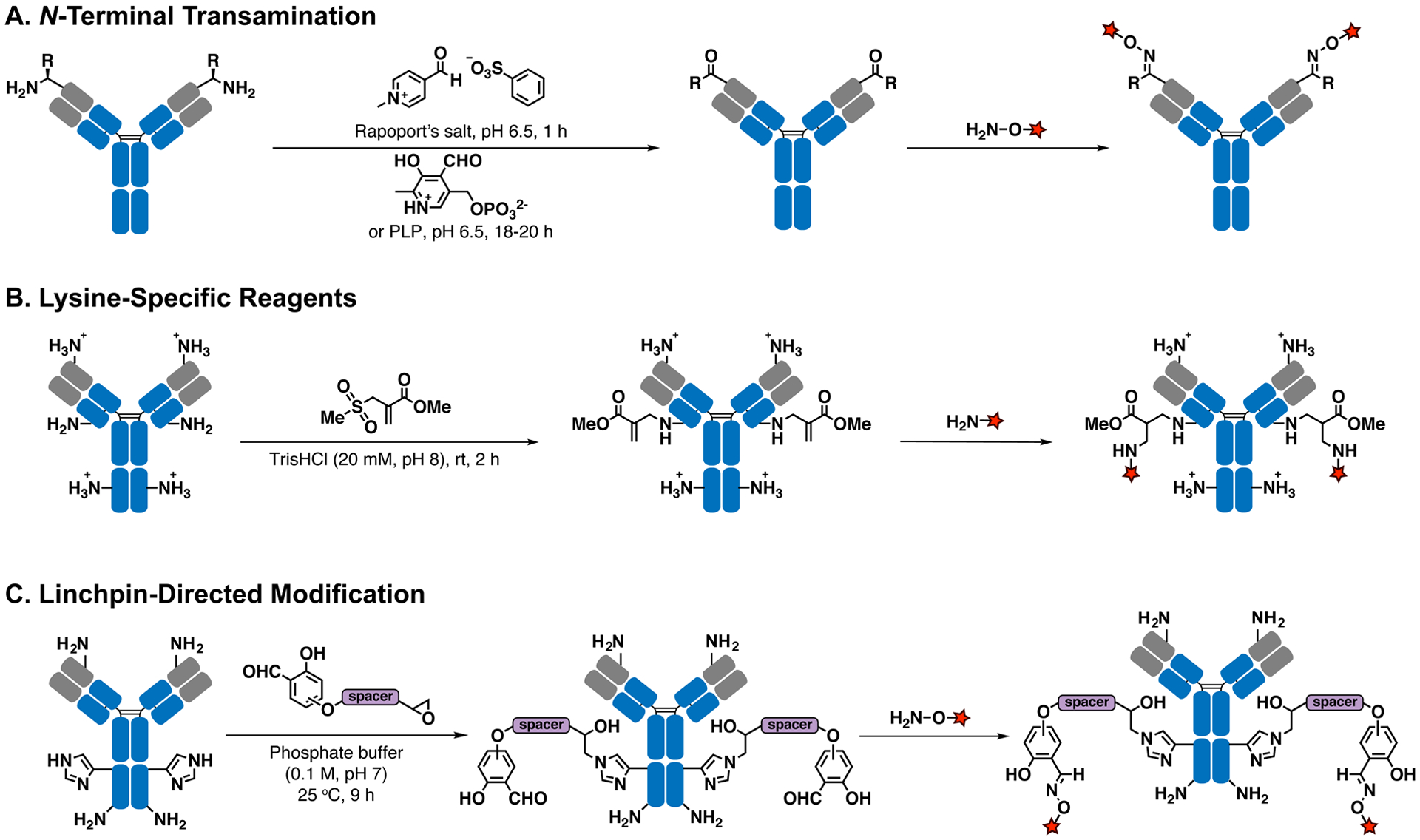
Chemoselective modification of native antibodies. (A) Chemical transamination at N-terminal amines using Rapoport’s salt or PLP. (B) pH-based regioselective lysine 1,4-addition reaction. (C) Lysine-guided linchpin-directed modification.
Lysine-Specific Reagents.
Several attempts based on control of reaction kinetics,79 protein site-specificity,80–82 or targeting hyper-reactive lysines83,84 have been used for selective modification of certain antibody lysine residues. However, since mechanisms underlying lysine selectivity are not well understood, these methods failed to achieve high yields. Thus, it is difficult to apply them for modification of antibodies or other proteins. To address this issue, Matos et al.85 have reported the use of sulfonyl acrylate-based reagents, combined with pH control, to achieve selective and high-yield (>95%) modification of a single lysine with the lowest pKa (Figure 4B). The sulfonyl oxygen stabilizes the positive charge generated upon amine addition to the double bond via a chairlike transition state. The less polar cysteine S–H group diminishes hydrogen bond interaction, thus explaining the chemoselectivity for lysine over cysteine. The regioselectivity is based on pKa control, as the lysine with the lowest pKa is the most likely to be reactive under neutral to slightly basic conditions. Lysine residues modified with acrylates can further conjugate to amine-containing compounds by aza-Michael addition. The generality of this modification strategy has been successfully demonstrated with five model proteins, including a humanized full-length trastuzumab antibody. The binding affinity and specificity of trastuzumab for human HER2 were unaffected after site-specific modification with the anticancer drug Crizotinib, as verified by flow cytometry. It is important to note that the success of modifying a specific lysine is determined by the intrinsic properties of the antibody. These differ on a case-by-case basis, and derivatization could fail if, for example, the modified lysine is crucial to antibody function.
Linchpin-Directed Modification.
Another reactivity-based method, known as linchpin-directed modification (LDM), was developed by Adusumalli et al.86 LDM utilizes a fast and reversible amine-reactive group to guide a slow and irreversible reaction between an epoxide group and a protein histidine residue (Figure 4C). While p-hydroxybenzaldehyde establishes a fast on-and-off equilibrium with lysine residues, the linked epoxide moiety will only react with a histidine that lies within a certain distance of the lysine residue. After irreversible linkage between histidine and epoxide, the free aldehyde can undergo oxime ligation for coupling other molecules of interest. Two ADCs, trastuzumab-doxorubicin and trastuzumab-emtansine, have been made using this technology. The binding properties and antitumor cell activity of both ADCs have been established using in vitro experiments. To expand the substrate range of LDM, Adusumalli et al.87 reported another version of an LDM reagent for modifying lysine instead of histidine. Switching the moderate leaving group from epoxide to 2,6-dibromo-4-(morpholine-4-carbonyl)phenol ester provided more options for optimization of substrates. Despite the variability encountered from case to case, LDM yields a modification efficiency of 40% for most antibodies.
Summary.
In general, direct antibody labeling based on chemoselectivity is the most convenient strategy for antibody modification. Only a single reagent is needed to introduce the bioorthogonal handle in an initial step, which is followed by the second step of attaching a cargo to the antibody. The specific site of modification is determined by the intrinsic properties and 3D structure of the antibody. This variability from immunoglobulin to immunoglobulin could potentially lead to lower yields or impaired antibody function in some cases.
PROXIMITY-BASED MODIFICATION
An alternative, emerging strategy for achieving site-specific labeling of native antibody molecules takes advantage of proximity effects that direct a cross-linking reactant or catalyst to the desired amino acid residue. Several groups have employed ligands able to bind to a specific location on antibodies to deliver a photo-cross-linker, metallocatalyst, or other reactive groups capable of forming a site-selective covalent bond through proximity-induced reactivity.
Proximity-Directed Photo-Cross-Linking.
Because of the versatility of photo-cross-linking in the context of different chemical environments, photo-cross-linking was used to site-specifically label native antibodies. Benzoylphenylalanine (Bpa), a UV-inducible cross-linking amino acid, has been widely used for mapping protein interactions. Upon UV irradiation (<365 nm), Bpa within the protein of interest generates radicals that insert nonspecifically into nearby C–H bonds to form a covalent bond between the protein and its binding partner.88 Due to its chemical stability and potent UV-inducible cross-linking ability, Bpa is useful not only for revealing valuable information regarding molecular interactions but also for designing covalent protein-based agonists, antagonists, and inhibitors. In what was perhaps the first attempt to site-specifically label native antibodies using this approach, Jung and co-workers used cysteine engineering and maleimide chemistry to construct an antibody-binding peptide with a benzophenone derivative (Figure 5A).89 Specifically, amino acid residues in the Fc-binding domain of the peptide were first mutated to cysteines, followed by the reaction with maleimide-functionalized benzophenone. The resulting affinity peptide was able to direct the benzophenone-containing cross-linker to the antibody Fc domain. After UV irradiation, the benzophenone cross-linker formed a covalent bond with the antibody Fc domain to yield the site-specific conjugate. Under optimal conditions (UV irradiation at 365 nm for 1 h), roughly 50% of the antibody population was covalently labeled with one or two affinity peptides. The utility of this technology was demonstrated by covalent immobilization of antibodies directly onto various surfaces, including glass, gold chips, and magnetic particles. Several subsequent studies used different types of antibody-binding peptides to prepare site-specifically labeled native antibodies. These affinity peptides included derivatives of protein A (binding to the antibody Fc domain),90–92 derivatives of protein G (binding to the antibody Fab domain),93 and antibody-binding peptide FcIII94 evolved via phage display.95 Park et al. first demonstrated a biomedical application for this technology utilizing FcIII by conjugating an engineered Pseudomonas exotoxin A (PE24) to the Fc domain of trastuzumab, the human epidermal growth factor receptor 2 (HER2)-specific antibody.95 The resulting conjugate exhibited significantly higher cytotoxicity than unmodified trastuzumab toward HER2-overexpressing cells. In another example, Bpa-containing FcIII peptide with a free thiol residue was first coupled to trastuzumab to introduce a unique thiol group. This was followed by conjugation to maleimide-functionalized MMAE, with a DAR of 1.9.96 While photo-cross-linking efficiency has been significantly improved since the initial use of this strategy, the low degree of chemical selectivity of the photo-cross-linking reaction tends to generate heterogeneous products. In addition, 30–60 min of exposure to UV irradiation is known to cause protein damage that may impair antibody efficacy.97
Figure 5.
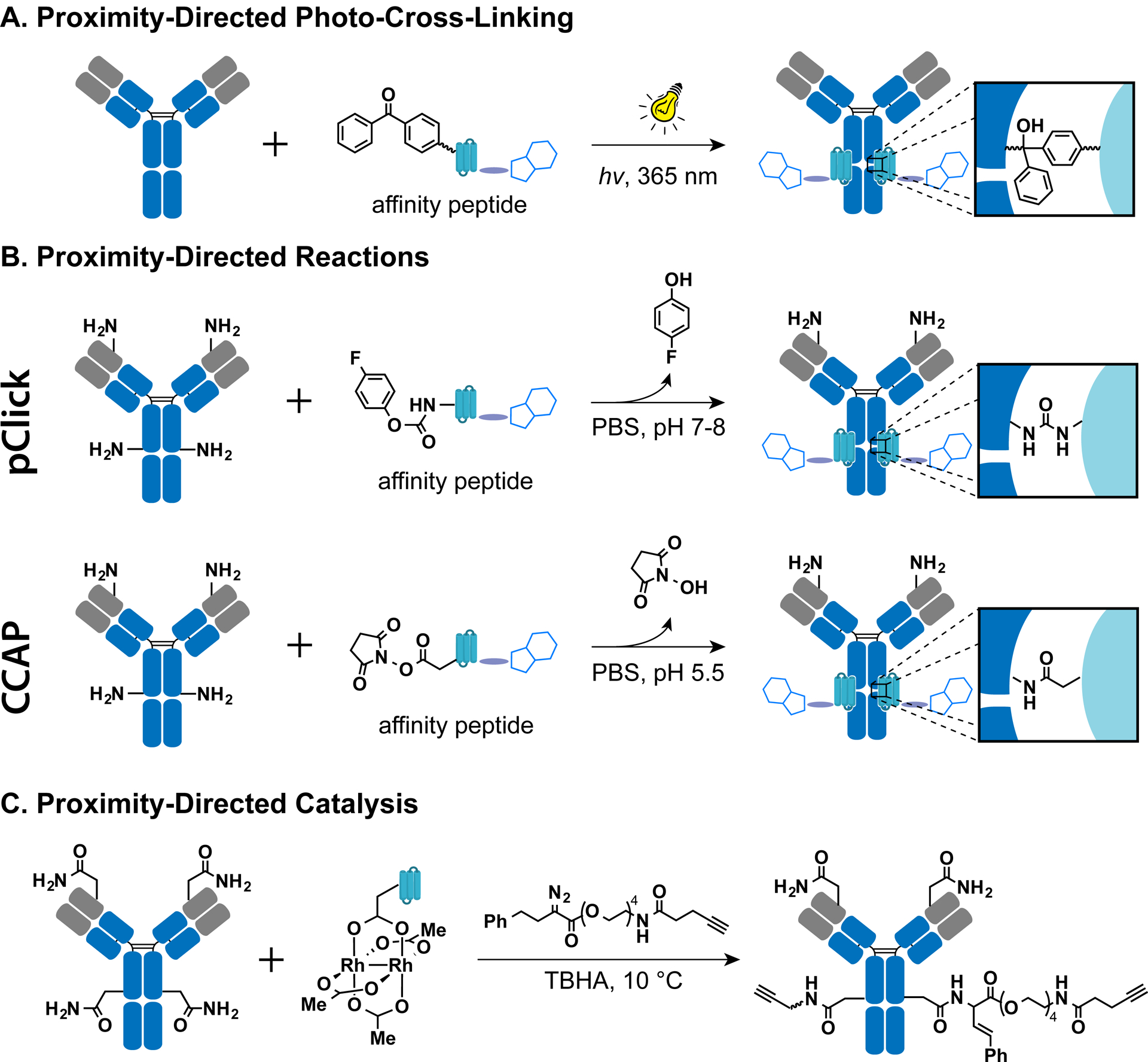
Antibody conjugates prepared by targeting canonical amino acids using (A) proximity-directed photo-cross-linking, (B) proximity-directed reactions, and (C) proximity-directed catalysis.
Proximity-Directed Reactions.
While most peptide-based antibody conjugation strategies rely on selective delivery of a UV-sensitive photo-cross-linker to a specific site in an antibody, the need for UV light can be avoided via delivery of a proximity-based reactive electrophile capable of forming a covalent bond with nucleophilic residues, for example, a specific lysine. This strategy provides the basis for proximity-induced antibody conjugation technology (pClick), which was first developed in our lab in 2018 as a general method for site-specific modification of native antibodies (Figure 5B).98 Specifically, a lysine-proximity probe, 4-fluorophenyl carba-mate lysine (FPheK), capable of covalent linkage to a proximal antibody lysine residue, was introduced at the Glu25 position of an affinity peptide (FB peptide) that has a well-characterized binding site within the antibody Fc region. Upon binding to the antibody, the FPheK-modified FB peptide covalently attaches to the antibody via a spontaneous cross-linking of FPheK to a specific lysine residue. The utility of this approach is demonstrated by site-specific conjugation of fluorescein to Trastuzumab, followed by in vitro characterization of the antibody conjugate activity. Most importantly, a 91–99% conjugation efficiency was achieved with several IgG species and subclasses, without the generation of any detectable nonspecific conjugation products. In another study, the FB peptide could be further shortened to 33 residues without compromising its binding affinity and conjugation efficiency.99 This chemically synthesized FB peptide was applied to the synthesis of ADCs and bispecific small molecule–antibody conjugates with both excellent in vitro cytotoxic activity and antitumor activity in mouse xenograft models.99 Furthermore, this strategy was recently employed to prepare the first bone-targeting antibody, which is able to enhance the biodistribution to the bone and dramatically increase the therapeutic effect to HER2+ breast cancer bone metastasis in murine models.100 In 2019, Ito’s group reported a similar lysine modification approach (CCAP) using N-hydroxysuccinimide ester as a proximity-based reactive probe (Figure 5B).101 By precise control of the pH and reactant concentrations, Fc-III peptide modified with an NHS-ester linker could conjugate to antibodies with a high efficiency. Interestingly, the resulting site-specific conjugates exhibit reduced binding affinity to FcRn, but enhanced affinity to FcγRIIIa. Ito and collaborators at Ajinomoto Co., Inc. further advanced this technology by preparing site-specific ADCs in which free thiols were introduced into native antibodies, which they termed AJI-CAP.102 These thiol-containing native antibodies were initially produced by using CCAP for conjugation of Fc-III peptide to a disulfide linker. Reduction of introduced disulfide bonds was subsequently carried out using TCEP. However, the antibody interchain disulfides were also reduced. An extra oxidation step with dehydroascorbic acid (DHAA) to regenerate the interchain disulfide bonds is required, with the increasing risk of disulfide scrambling. The resulting thiol-modified native antibodies were then conjugated to maleimide-functionalized drugs. Importantly, this strategy for preparing ADCs is “traceless”. In other words, no affinity peptide remains attached to the final products, since the disulfide-linked Fc-III peptide is removed in the TCEP reduction step. Besides utilizing the affinity moieties binding to antibody constant regions, the ligands interact with the variable region of antibodies have been used for site-specific labeling at the antibody variable region.103–105 Using the proximity-induced reactivity, covalent immune recruiters that can enhance immune recognition of different types of cancers were prepared recently.106
Central to the success of proximity-directed modification is the development of proximity-based probes that exhibit superb conjugation efficiency in the absence of nonspecific reactivities. Advances in proximity-based chemistry offer great opportunities for labeling native antibodies with payloads possessing a variety of chemical, physical, or biological characteristics under mild conditions. This technology does not require antibody engineering or additional UV/chemical/enzymatic treatments, and therefore is applicable to most commercial antibodies.
Proximity-Directed Catalysis.
As an alternative to site-specific modification of native antibodies using proximity-directed reactants, Ball and co-workers used the affinity-peptide approach to deliver a metallocatalyst capable of carrying out the reaction between carboxamide and diazo groups (Figure 5C).107 An Fc-binding dirhodium metal-lopeptide was prepared by introducing three Glu residues into the Z domain of protein A, followed by metalation with a heteroleptic dirhodium complex. Upon binding to the antibody, this metallocatalyst was able to catalyze the derivatization of a nearby Asn312 residue with an alkyne-bearing diazo reagent. The resulting alkyne-functionalized native antibodies were then conjugated either to an azide-modified fluorophore or to drugs by means of a copper-catalyzed azide–alkyne cycloaddition (CuAAC) reaction. In another study, an enzyme-based catalysis was developed, which utilized sortase A mutant–protein G B1 domain fusion to guide the transpeptidation on specific lysines on IgGs.108 In the case of Cetuximab, as demonstrated in this study, the modification only happened on heavy chain K5, K123, K135, K292, and K441, and light chain K207, as verified by LC-MS analysis. Compared to the use of proximity-directed reactants, the advantage of proximity-directed catalysis is that the catalyst is not covalently attached to the final products and is therefore “traceless”. Furthermore, the proximity-directed catalytic approach allows lower molar ratios of ligand, potentially limiting off-target binding. These studies highlight both the feasibility and the benefits of proximity-directed catalysis in site-specific native antibody labeling, providing a promising new expansion of this field of study.
Summary.
Proximity-based modification methods allowing for the selective reaction between a proximity-based probe and a specific natural residue (e.g., lysine, cysteine, serine) of proteins are emerging as an important class of antibody conjugation methods. These strategies enable the preparation of site-specific antibody conjugates without the need for additional antibody engineering or additional treatments. Current efforts of the proximity-based modification focus on the evolution of new proximity-induced chemistry as well as smaller antibody binders with minimal disruption of antibody structure and function.
SUMMARY AND DISCUSSION
Due to the high therapeutic indices and outstanding biochemical properties of site-specific antibody conjugates, many current efforts have focused on strategies for site-specific labeling of antibodies. As a result, a variety of methods have been developed for specific covalent attachment of functional molecules to antibodies. Most of these methods require antibody engineering for site-specific installation of a unique reactive moiety, followed by selective modification of this moiety using bioorthogonal chemistry. While these technologies offer superb control of the conjugation site, drawbacks of the approaches include the technical challenge and lack of generality of strategies for antibody engineering. Thus, it is extremely attractive to consider methods for site-specific modification of native antibodies that do not include antibody engineering. Central to precision labeling of native antibodies is the ability to site-selectively modify a single desired amino acid residue with high efficiency and without off-target effects. Thanks to advances in bioorthogonal chemistry, enzyme engineering, and proximity chemistry, a variety of solutions have emerged for targeting unique interchain disulfides, sugars, and N-terminal residues, as well as for directing the reactivity of functional probes through the use of proximity effects. Despite the many choices for specific modification of the native antibodies, these strategies still pose some limitations in certain applications: Disulfide rebridging might suffer from disulfide scrambling and batch to batch variability; different glycol forms of IgGs and high price of saccharide analogues can significantly limit both the efficiency and the scale of glycan-based antibody labeling methods; fine modulation of chemical reactivity is required for both chemoselective and proximity-based antibody modifications. As the field progresses, the continued development and refinement of protein engineering and protein chemistry in conjugation with new design concepts will enable the production of well-defined native antibody conjugates that possess superior biochemical and therapeutic properties without the need for complex chemical or enzymatic treatments.
ACKNOWLEDGMENTS
This work was supported by the Cancer Prevention Research Institute of Texas (CPRIT, RR170014 to H.X.), NIH (R35-GM133706, R01-AI165079, and R21-CA255894 to H.X.), U.S. Department of Defense (W81XWH-21-1-0789 to H.X.), the Robert A. Welch Foundation (C-1970 to H.X. and C-1680 to Z.T.B.), National Science Foundation (CHE-1904865 to Z.T.B.), the Hamill Innovation Award (Hamill Foundation), the John S. Dunn Foundation Collaborative Research Award (Gulf Coast Consortia). H. X. is a Cancer Prevention & Research Institute of Texas (CPRIT) Scholar in Cancer Research.
Footnotes
The authors declare no competing financial interest.
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.bioconjchem.1c00342
Contributor Information
Kuan-Lin Wu, Department of Chemistry, Rice University, Houston, Texas 77005, United States.
Chenfei Yu, Department of Chemistry, Rice University, Houston, Texas 77005, United States.
Catherine Lee, Department of Chemistry, Rice University, Houston, Texas 77005, United States.
Chao Zuo, Department of Chemistry, Rice University, Houston, Texas 77005, United States.
Zachary T. Ball, Department of Chemistry, Rice University, Houston, Texas 77005, United States;.
Han Xiao, Department of Chemistry, Department of Biosciences, and Department of Bioengineering, Rice University, Houston, Texas 77005, United States;.
REFERENCES
- (1).Lu R-M; Hwang Y-C; Liu I-J; Lee C-C; Tsai H-Z; Li H-J; Wu H-C Development of Therapeutic Antibodies for the Treatment of Diseases. J. Biomed. Sci 2020, 27 (1), 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Khongorzul P; Ling CJ; Khan FU; Ihsan AU; Zhang J Antibody–Drug Conjugates: A Comprehensive Review. Mol. Cancer Res 2020, 18 (1), 3–19. [DOI] [PubMed] [Google Scholar]
- (3).Strebhardt K; Ullrich A Paul Ehrlich’s Magic Bullet Concept: 100 Years of Progress. Nat. Rev. Cancer 2008, 8 (6), 473–480. [DOI] [PubMed] [Google Scholar]
- (4).Bross PF; Beitz J; Chen G; Chen XH; Duffy E; Kieffer L; Roy S; Sridhara R; Rahman A; Williams G; Pazdur R Approval Summary: Gemtuzumab Ozogamicin in Relapsed Acute Myeloid Leukemia. Clin. Cancer Res 2001, 7 (6), 1490–1496. [PubMed] [Google Scholar]
- (5).de Claro RA; McGinn K; Kwitkowski V; Bullock J; Khandelwal A; Habtemariam B; Ouyang Y; Saber H; Lee K; Koti K; et al. U.S. Food and Drug Administration Approval Summary: Brentuximab Vedotin for the Treatment of Relapsed Hodgkin Lymphoma or Relapsed Systemic Anaplastic Large-Cell Lymphoma. Clin. Cancer Res 2012, 18 (21), 5845–5849. [DOI] [PubMed] [Google Scholar]
- (6).Amiri-Kordestani L; Blumenthal GM; Xu QC; Zhang L; Tang SW; Ha L; Weinberg WC; Chi B; Candau-Chacon R; Hughes P; et al. FDA Approval: Ado-Trastuzumab Emtansine for the Treatment of Patients with HER2-Positive Metastatic Breast Cancer. Clin. Cancer Res 2014, 20 (17), 4436–4441. [DOI] [PubMed] [Google Scholar]
- (7).Joubert N; Beck A; Dumontet C; Denevault-Sabourin C Antibody–Drug Conjugates: The Last Decade. Pharmaceuticals 2020, 13 (9), 245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Lin AY; Dinner SN Moxetumomab Pasudotox for Hairy Cell Leukemia: Preclinical Development to FDA Approval. Blood Advances 2019, 3 (19), 2905–2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Hamblett KJ; Senter PD; Chace DF; Sun MMC; Lenox J; Cerveny CG; Kissler KM; Bernhardt SX; Kopcha AK; Zabinski RF; et al. Effects of Drug Loading on the Antitumor Activity of a Monoclonal Antibody Drug Conjugate. Clin. Cancer Res 2004, 10 (20), 7063–7070. [DOI] [PubMed] [Google Scholar]
- (10).Junutula JR; Raab H; Clark S; Bhakta S; Leipold DD; Weir S; Chen Y; Simpson M; Tsai SP; Dennis MS; et al. Site-Specific Conjugation of a Cytotoxic Drug to an Antibody Improves the Therapeutic Index. Nat. Biotechnol 2008, 26 (8), 925–932. [DOI] [PubMed] [Google Scholar]
- (11).Agarwal P; Bertozzi CR Site-Specific Antibody–Drug Conjugates: The Nexus of Bioorthogonal Chemistry, Protein Engineering, and Drug Development. Bioconjugate Chem 2015, 26 (2), 176–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Huang Y; Liu T Therapeutic Applications of Genetic Code Expansion. Synthetic and Systems Biotechnology 2018, 3 (3), 150–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Liu CC; Schultz PG Adding New Chemistries to the Genetic Code. Annu. Rev. Biochem 2010, 79 (1), 413–444. [DOI] [PubMed] [Google Scholar]
- (14).Wang L Genetically Encoding New Bioreactivity. New Biotechnol. 2017, 38, 16–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Chin JW Expanding and Reprogramming the Genetic Code of Cells and Animals. Annu. Rev. Biochem 2014, 83 (1), 379–408. [DOI] [PubMed] [Google Scholar]
- (16).Xiao H; Schultz PG At the Interface of Chemical and Biological Synthesis: An Expanded Genetic Code. Cold Spring Harbor Perspect. Biol 2016, 8 (9), a023945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Chen Y; Loredo A; Gordon A; Tang J; Yu C; Ordonez J; Xiao H A Noncanonical Amino Acid-Based Relay System for Site-Specific Protein Labeling. Chem. Commun 2018, 54 (52), 7187–7190. [DOI] [PubMed] [Google Scholar]
- (18).Oller-Salvia B; Kym G; Chin JW Rapid and Efficient Generation of Stable Antibody–Drug Conjugates via an Encoded Cyclopropene and an Inverse-Electron-Demand Diels–Alder Reaction. Angew. Chem., Int. Ed 2018, 57 (11), 2831–2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Chen Y; Tang J; Wang L; Tian Z; Cardenas A; Fang X; Chatterjee A; Xiao H Creation of Bacterial Cells with 5-Hydroxytryptophan as a 21st Amino Acid Building Block. Chem 2020, 6 (10), 2717–2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Italia JS; Addy PS; Erickson SB; Peeler JC; Weerapana E; Chatterjee A Mutually Orthogonal Nonsense-Suppression Systems and Conjugation Chemistries for Precise Protein Labeling at up to Three Distinct Sites. J. Am. Chem. Soc 2019, 141 (15), 6204–6212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Addy PS; Erickson SB; Italia JS; Chatterjee A A Chemoselective Rapid Azo-Coupling Reaction (CRACR) for Unclickable Bioconjugation. J. Am. Chem. Soc 2017, 139 (34), 11670–11673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Wang L; Zhang Z; Brock A; Schultz PG Addition of the Keto Functional Group to the Genetic Code of Escherichia Coli. Proc. Natl. Acad. Sci. U. S. A 2003, 100 (1), 56–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Liu W; Brock A; Chen S; Chen S; Schultz PG Genetic Incorporation of Unnatural Amino Acids into Proteins in Mammalian Cells. Nat. Methods 2007, 4 (3), 239–244. [DOI] [PubMed] [Google Scholar]
- (24).Axup JY; Bajjuri KM; Ritland M; Hutchins BM; Kim CH; Kazane SA; Halder R; Forsyth JS; Santidrian AF; Stafin K; et al. Synthesis of Site-Specific Antibody-Drug Conjugates Using Unnatural Amino Acids. Proc. Natl. Acad. Sci. U. S. A 2012, 109 (40), 16101–16106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Xiao H; Chatterjee A; Choi S; Bajjuri KM; Sinha SC; Schultz PG Genetic Incorporation of Multiple Unnatural Amino Acids into Proteins in Mammalian Cells. Angew. Chem., Int. Ed 2013, 52 (52), 14080–14083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Wissler HL; Ehlerding EB; Lyu Z; Zhao Y; Zhang S; Eshraghi A; Buuh ZY; McGuth JC; Guan Y; Engle JW; et al. Site-Specific Immuno-PET Tracer to Image PD-L1. Mol. Pharmaceutics 2019, 16 (5), 2028–2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Agarwal P; Bertozzi CR Site-Specific Antibody–Drug Conjugates: The Nexus of Bioorthogonal Chemistry, Protein Engineering, and Drug Development. Bioconjugate Chem 2015, 26 (2), 176–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Carrico IS; Carlson BL; Bertozzi CR Introducing Genetically Encoded Aldehydes into Proteins. Nat. Chem. Biol 2007, 3 (6), 321–322. [DOI] [PubMed] [Google Scholar]
- (29).Hudak JE; Barfield RM; de Hart GW; Grob P; Nogales E; Bertozzi CR; Rabuka D Synthesis of Heterobifunctional Protein Fusions Using Copper-Free Click Chemistry and the Aldehyde Tag. Angew. Chem., Int. Ed 2012, 51 (17), 4161–4165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Agarwal P; van der Weijden J; Sletten EM; Rabuka D; Bertozzi CR A Pictet-Spengler Ligation for Protein Chemical Modification. Proc. Natl. Acad. Sci. U. S. A 2013, 110 (1), 46–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Agarwal P; Kudirka R; Albers AE; Barfield RM; de Hart GW; Drake PM; Jones LC; Rabuka D Hydrazino-Pictet-Spengler Ligation as a Biocompatible Method for the Generation of Stable Protein Conjugates. Bioconjugate Chem 2013, 24 (6), 846–851. [DOI] [PubMed] [Google Scholar]
- (32).Drake PM; Albers AE; Baker J; Banas S; Barfield RM; Bhat AS; de Hart GW; Garofalo AW; Holder P; Jones LC; et al. Aldehyde Tag Coupled with HIPS Chemistry Enables the Production of ADCs Conjugated Site-Specifically to Different Antibody Regions with Distinct in Vivo Efficacy and PK Outcomes. Bioconjugate Chem 2014, 25 (7), 1331–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Xiao H; Woods EC; Vukojicic P; Bertozzi CR Precision Glycocalyx Editing as a Strategy for Cancer Immunotherapy. Proc. Natl. Acad. Sci. U. S. A 2016, 113 (37), 10304–10309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Gray MA; Stanczak MA; Mantuano NR; Xiao H; Pijnenborg JFA; Malaker SA; Miller CL; Weidenbacher PA; Tanzo JT; Ahn G; et al. Targeted Glycan Degradation Potentiates the Anticancer Immune Response in Vivo. Nat. Chem. Biol 2020, 16 (12), 1376–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Lin C-W; Ting AY Transglutaminase-Catalyzed Site-Specific Conjugation of Small-Molecule Probes to Proteins in Vitro and on the Surface of Living Cells. J. Am. Chem. Soc 2006, 128 (14), 4542–4543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Jeger S; Zimmermann K; Blanc A; Grünberg J; Honer M; Hunziker P; Struthers H; Schibli R Site-Specific and Stoichiometric Modification of Antibodies by Bacterial Transglutaminase. Angew. Chem., Int. Ed 2010, 49 (51), 9995–9997. [DOI] [PubMed] [Google Scholar]
- (37).Strop P; Liu S-H; Dorywalska M; Delaria K; Dushin RG; Tran T-T; Ho W-H; Farias S; Casas MG; Abdiche Y; et al. Location Matters: Site of Conjugation Modulates Stability and Pharmacokinetics of Antibody Drug Conjugates. Chem. Biol 2013, 20 (2), 161–167. [DOI] [PubMed] [Google Scholar]
- (38).Mao H; Hart SA; Schink A; Pollok BA Sortase-Mediated Protein Ligation: A New Method for Protein Engineering. J. Am. Chem. Soc 2004, 126 (9), 2670–2671. [DOI] [PubMed] [Google Scholar]
- (39).Paterson BM; Alt K; Jeffery CM; Price RI; Jagdale S; Rigby S; Williams CC; Peter K; Hagemeyer CE; Donnelly PS Enzyme-Mediated Site-Specific Bioconjugation of Metal Complexes to Proteins: Sortase-Mediated Coupling of Copper-64 to a Single-Chain Antibody. Angew. Chem., Int. Ed 2014, 53 (24), 6115–6119. [DOI] [PubMed] [Google Scholar]
- (40).Möhlmann S; Mahlert C; Greven S; Scholz P; Harrenga A In Vitro Sortagging of an Antibody Fab Fragment: Overcoming Unproductive Reactions of Sortase with Water and Lysine Side Chains. ChemBioChem 2011, 12 (11), 1774–1780. [DOI] [PubMed] [Google Scholar]
- (41).Swee LK; Guimaraes CP; Sehrawat S; Spooner E; Barrasa MI; Ploegh HL Sortase-Mediated Modification of ADEC205 Affords Optimization of Antigen Presentation and Immunization against a Set of Viral Epitopes. Proc. Natl. Acad. Sci. U. S. A 2013, 110 (4), 1428–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Zhang C; Welborn M; Zhu T; Yang NJ; Santos MS; Van Voorhis T; Pentelute BL π -Clamp-Mediated Cysteine Conjugation. Nat. Chem 2016, 8 (2), 120–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Beckley NS; Lazzareschi KP; Chih H-W; Sharma VK; Flores HL Investigation into Temperature-Induced Aggregation of an Antibody Drug Conjugate. Bioconjugate Chem 2013, 24 (10), 1674–1683. [DOI] [PubMed] [Google Scholar]
- (44).Liberatore FA; Comeau RD; McKearin JM; Pearson DA; Belonga BQ; Brocchini SJ; Kath J; Phillips T; Oswell K; Lawton RG Site-Directed Chemical Modification and Crosslinking of a Monoclonal Antibody Using Equilibrium Transfer Alkylating Crosslink Reagents. Bioconjugate Chem 1990, 1 (1), 36–50. [DOI] [PubMed] [Google Scholar]
- (45).Del Rosario RB; Wahl RL; Brocchini SJ; Lawton RG; Smith RH Sulfhydryl Site-Specific Crosslinking and Labeling of Monoclonal Antibodies by a Fluorescent Equilibrium Transfer Alkylation Crosslink Reagent. Bioconjugate Chem 1990, 1 (1), 51–59. [DOI] [PubMed] [Google Scholar]
- (46).Burns JA; Butler JC; Moran J; Whitesides GM Selective Reduction of Disulfides by Tris(2-Carboxyethyl)Phosphine. J. Org. Chem 1991, 56 (8), 2648–2650. [Google Scholar]
- (47).Badescu G; Bryant P; Bird M; Henseleit K; Swierkosz J; Parekh V; Tommasi R; Pawlisz E; Jurlewicz K; Farys M; et al. Bridging Disulfides for Stable and Defined Antibody Drug Conjugates. Bioconjugate Chem 2014, 25 (6), 1124–1136. [DOI] [PubMed] [Google Scholar]
- (48).Wang T; Ng DYW; Wu Y; Thomas J; TamTran T; Weil T Bis-Sulfide Bioconjugates for Glutathione Triggered Tumor Responsive Drug Release. Chem. Commun 2014, 50 (9), 1116–1118. [DOI] [PubMed] [Google Scholar]
- (49).Wang T; Riegger A; Lamla M; Wiese S; Oeckl P; Otto M; Wu Y; Fischer S; Barth H; Kuan SL; et al. Water-Soluble Allyl Sulfones for Dual Site-Specific Labelling of Proteins and Cyclic Peptides. Chem. Sci 2016, 7 (5), 3234–3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Smith MEB; Schumacher FF; Ryan CP; Tedaldi LM; Papaioannou D; Waksman G; Caddick S; Baker JR Protein Modification, Bioconjugation, and Disulfide Bridging Using Bromo-maleimides. J. Am. Chem. Soc 2010, 132 (6), 1960–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Schumacher FF; Nobles M; Ryan CP; Smith MEB; Tinker A; Caddick S; Baker JR In Situ Maleimide Bridging of Disulfides and a New Approach to Protein PEGylation. Bioconjugate Chem 2011, 22 (2), 132–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Schumacher FF; Nunes JPM; Maruani A; Chudasama V; Smith MEB; Chester KA; Baker JR; Caddick S Next Generation Maleimides Enable the Controlled Assembly of Antibody–Drug Conjugates via Native Disulfide Bond Bridging. Org. Biomol. Chem 2014, 12 (37), 7261–7269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Ryan CP; Smith MEB; Schumacher FF; Grohmann D; Papaioannou D; Waksman G; Werner F; Baker JR; Caddick S Tunable Reagents for Multi-Functional Bioconjugation: Reversible or Permanent Chemical Modification of Proteins and Peptides by Control of Maleimide Hydrolysis. Chem. Commun 2011, 47 (19), 5452–5454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Nunes JPM; Morais M; Vassileva V; Robinson E; Rajkumar VS; Smith MEB; Pedley RB; Caddick S; Baker JR; Chudasama V Functional Native Disulfide Bridging Enables Delivery of a Potent, Stable and Targeted Antibody–Drug Conjugate (ADC). Chem. Commun 2015, 51 (53), 10624–10627. [DOI] [PubMed] [Google Scholar]
- (55).Maruani A; Smith MEB; Miranda E; Chester KA; Chudasama V; Caddick S A Plug-and-Play Approach to Antibody-Based Therapeutics via a Chemoselective Dual Click Strategy. Nat. Commun 2015, 6 (1), 6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Lee MTW; Maruani A; Baker JR; Caddick S; Chudasama V Next-Generation Disulfide Stapling: Reduction and Functional Re-Bridging All in One. Chem. Sci 2016, 7 (1), 799–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Lee MTW; Maruani A; Richards DA; Baker JR; Caddick S; Chudasama V Enabling the Controlled Assembly of Antibody Conjugates with a Loading of Two Modules without Antibody Engineering. Chem. Sci 2017, 8 (3), 2056–2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Chudasama V; Smith MEB; Schumacher FF; Papaioannou D; Waksman G; Baker JR; Caddick S Bromopyridazinedione-Mediated Protein and Peptide Bioconjugation. Chem. Commun 2011, 47 (31), 8781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Walsh SJ; Omarjee S; Galloway WRJD; Kwan TT-L; Sore HF; Parker JS; Hyvönen M; Carroll JS; Spring DR A General Approach for the Site-Selective Modification of Native Proteins, Enabling the Generation of Stable and Functional Antibody–Drug Conjugates. Chem. Sci 2019, 10 (3), 694–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Wilson P; Anastasaki A; Owen MR; Kempe K; Haddleton DM; Mann SK; Johnston APR; Quinn JF; Whittaker MR; Hogg PJ; et al. Organic Arsenicals As Efficient and Highly Specific Linkers for Protein/Peptide–Polymer Conjugation. J. Am. Chem. Soc 2015, 137 (12), 4215–4222. [DOI] [PubMed] [Google Scholar]
- (61).O’Shannessy DJ; Dobersen MJ; Quarles RH A Novel Procedure for Labeling Immunoglobulins by Conjugation to Oligosaccharide Moieties. Immunol. Lett 1984, 8 (5), 273–277. [DOI] [PubMed] [Google Scholar]
- (62).Rodwell JD; Alvarez VL; Lee C; Lopes AD; Goers JW; King HD; Powsner HJ; McKearn TJ Site-Specific Covalent Modification of Monoclonal Antibodies: In Vitro and in Vivo Evaluations. Proc. Natl. Acad. Sci. U. S. A 1986, 83 (8), 2632–2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Vallabhajosula S; Nikolopoulou A; Babich JW; Osborne JR; Tagawa ST; Lipai I; Solnes L; Maresca KP; Armor T; Joyal JL; et al. 99mTc-Labeled Small-Molecule Inhibitors of Prostate-Specific Membrane Antigen: Pharmacokinetics and Biodis-tribution Studies in Healthy Subjects and Patients with Metastatic Prostate Cancer. J. Nucl. Med 2014, 55 (11), 1791–1798. [DOI] [PubMed] [Google Scholar]
- (64).Yamasaki RB; Osuga DT; Feeney RE Periodate Oxidation of Methionine in Proteins. Anal. Biochem 1982, 126 (1), 183–189. [DOI] [PubMed] [Google Scholar]
- (65).Zhou Q; Stefano JE; Manning C; Kyazike J; Chen B; Gianolio DA; Park A; Busch M; Bird J; Zheng X; et al. Site-Specific Antibody–Drug Conjugation through Glycoengineering. Bioconjugate Chem 2014, 25 (3), 510–520. [DOI] [PubMed] [Google Scholar]
- (66).Boeggeman E; Ramakrishnan B; Kilgore C; Khidekel N; Hsieh-Wilson LC; Simpson JT; Qasba PK Direct Identification of Nonreducing GlcNAc Residues on N-Glycans of Glycoproteins Using a Novel Chemoenzymatic Method. Bioconjugate Chem 2007, 18 (3), 806–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Zeglis BM; Davis CB; Aggeler R; Kang HC; Chen A; Agnew BJ; Lewis JS Enzyme-Mediated Methodology for the Site-Specific Radiolabeling of Antibodies Based on Catalyst-Free Click Chemistry. Bioconjugate Chem 2013, 24 (6), 1057–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Li X; Fang T; Boons G-J Preparation of Well-Defined Antibody–Drug Conjugates through Glycan Remodeling and Strain-Promoted Azide–Alkyne Cycloadditions. Angew. Chem., Int. Ed 2014, 53 (28), 7179–7182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Delft FLV; Geel RV; Wijdeven MA Modified Antibody, Antibody-Conjugate and Process for the Preparation Thereof. International Patent WO2014065661A1, May 1, 2014. [Google Scholar]
- (70).Campbell CT; Sampathkumar S-G; Yarema KJ Metabolic Oligosaccharide Engineering: Perspectives, Applications, and Future Directions. Mol. BioSyst 2007, 3 (3), 187–194. [DOI] [PubMed] [Google Scholar]
- (71).Agard NJ; Bertozzi CR Chemical Approaches To Perturb, Profile, and Perceive Glycans. Acc. Chem. Res 2009, 42 (6), 788–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Okeley NM; Toki BE; Zhang X; Jeffrey SC; Burke PJ; Alley SC; Senter PD Metabolic Engineering of Monoclonal Antibody Carbohydrates for Antibody–Drug Conjugation. Bioconjugate Chem 2013, 24 (10), 1650–1655. [DOI] [PubMed] [Google Scholar]
- (73).Alley SC; Jeffrey SC; Sussman D; Benjamin DR; Toki B; Burke PJ Methods and Compositions for Making Antibodies and Antibody Derivatives with Reduced Core Fucosylation. US Patent US8163551B2, April 24, 2012. [Google Scholar]
- (74).Du J; Meledeo MA; Wang Z; Khanna HS; Paruchuri VDP; Yarema KJ Metabolic Glycoengineering: Sialic Acid and Beyond. Glycobiology 2009, 19 (12), 1382–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Ghaderi D; Taylor RE; Padler-Karavani V; Diaz S; Varki A Implications of the Presence of N -Glycolylneuraminic Acid in Recombinant Therapeutic Glycoproteins. Nat. Biotechnol 2010, 28 (8), 863–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Grimsley GR; Scholtz JM; Pace CN A Summary of the Measured PK Values of the Ionizable Groups in Folded Proteins. Protein Sci 2008, 18 (1), 247–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Scheck RA; Francis MB Regioselective Labeling of Antibodies through N-Terminal Transamination. ACS Chem. Biol 2007, 2 (4), 247–251. [DOI] [PubMed] [Google Scholar]
- (78).Witus LS; Netirojjanakul C; Palla KS; Muehl EM; Weng C-H; Iavarone AT; Francis MB Site-Specific Protein Transamination Using N -Methylpyridinium-4-Carboxaldehyde. J. Am. Chem. Soc 2013, 135 (45), 17223–17229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Chen X; Muthoosamy K; Pfisterer A; Neumann B; Weil T Site-Selective Lysine Modification of Native Proteins and Peptides via Kinetically Controlled Labeling. Bioconjugate Chem 2012, 23 (3), 500–508. [DOI] [PubMed] [Google Scholar]
- (80).Choi S; Connelly S; Reixach N; Wilson IA; Kelly JW Chemoselective Small Molecules That Covalently Modify One Lysine in a Non-Enzyme Protein in Plasma. Nat. Chem. Biol 2010, 6 (2), 133–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Asano S; Patterson JT; Gaj T; Barbas CF Site-Selective Labeling of a Lysine Residue in Human Serum Albumin. Angew. Chem., Int. Ed 2014, 53 (44), 11783–11786. [DOI] [PubMed] [Google Scholar]
- (82).Patterson JT; Wilson HD; Asano S; Nilchan N; Fuller RP; Roush WR; Rader C; Barbas CF Human Serum Albumin Domain I Fusion Protein for Antibody Conjugation. Bioconjugate Chem 2016, 27 (10), 2271–2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Hacker SM; Backus KM; Lazear MR; Forli S; Correia BE; Cravatt BF Global Profiling of Lysine Reactivity and Ligandability in the Human Proteome. Nat. Chem 2017, 9 (12), 1181–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Nanna AR; Li X; Walseng E; Pedzisa L; Goydel RS; Hymel D; Jr TRB; Roush WR; Rader C Harnessing a Catalytic Lysine Residue for the One-Step Preparation of Homogeneous Antibody-Drug Conjugates. Nat. Commun 2017, 8 (1), 1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Matos MJ; Oliveira BL; Martínez-Sáez N; Guerreiro A; Cal PMSD; Bertoldo J; Maneiro M; Perkins E; Howard J; Deery MJ; et al. Chemo- and Regioselective Lysine Modification on Native Proteins. J. Am. Chem. Soc 2018, 140 (11), 4004–4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Adusumalli SR; Rawale DG; Singh U; Tripathi P; Paul R; Kalra N; Mishra RK; Shukla S; Rai V Single-Site Labeling of Native Proteins Enabled by a Chemoselective and Site-Selective Chemical Technology. J. Am. Chem. Soc 2018, 140 (44), 15114–15123. [DOI] [PubMed] [Google Scholar]
- (87).Adusumalli SR; Rawale DG; Thakur K; Purushottam L; Reddy NC; Kalra N; Shukla S; Rai V Chemoselective and Site-Selective Lysine-Directed Lysine Modification Enables Single-Site Labeling of Native Proteins. Angew. Chem., Int. Ed 2020, 59 (26), 10332–10336. [DOI] [PubMed] [Google Scholar]
- (88).Dorman G; Prestwich GD Benzophenone Photophores in Biochemistry. Biochemistry 1994, 33 (19), 5661–5673. [DOI] [PubMed] [Google Scholar]
- (89).Jung Y; Lee JM; Kim J; Yoon J; Cho H; Chung BH Photoactivable Antibody Binding Protein: Site-Selective and Covalent Coupling of Antibody. Anal. Chem 2009, 81 (3), 936–942. [DOI] [PubMed] [Google Scholar]
- (90).Konrad A; Eriksson Karlström A; Hober S Covalent Immunoglobulin Labeling through a Photoactivable Synthetic Z Domain. Bioconjugate Chem 2011, 22 (12), 2395–2403. [DOI] [PubMed] [Google Scholar]
- (91).Yu F; Järver P; Nygren P-Å Tailor-Making a Protein A-Derived Domain for Efficient Site-Specific Photocoupling to Fc of Mouse IgG1. PLoS One 2013, 8 (2), No. e56597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Perols A; Karlström AE Site-Specific Photoconjugation of Antibodies Using Chemically Synthesized IgG-Binding Domains. Bioconjugate Chem 2014, 25 (3), 481–488. [DOI] [PubMed] [Google Scholar]
- (93).Kanje S; von Witting E; Chiang SCC; Bryceson YT; Hober S Site-Specific Photolabeling of the IgG Fab Fragment Using a Small Protein G Derived Domain. Bioconjugate Chem 2016, 27 (9), 2095–2102. [DOI] [PubMed] [Google Scholar]
- (94).DeLano WL Convergent Solutions to Binding at a Protein-Protein Interface. Science 2000, 287 (5456), 1279–1283. [DOI] [PubMed] [Google Scholar]
- (95).Park J; Lee Y; Ko BJ; Yoo TH Peptide-Directed Photo-Cross-Linking for Site-Specific Conjugation of IgG. Bioconjugate Chem 2018, 29 (10), 3240–3244. [DOI] [PubMed] [Google Scholar]
- (96).Vance N; Zacharias N; Ultsch M; Li G; Fourie A; Liu P; LaFrance-Vanasse J; Ernst JA; Sandoval W; Kozak KR; et al. Development, Optimization, and Structural Characterization of an Efficient Peptide-Based Photoaffinity Cross-Linking Reaction for Generation of Homogeneous Conjugates from Wild-Type Antibodies. Bioconjugate Chem 2019, 30 (1), 148–160. [DOI] [PubMed] [Google Scholar]
- (97).Qi P; Volkin DB; Zhao H; Nedved ML; Hughes R; Bass R; Yi SC; Panek ME; Wang D; DalMonte P; et al. Characterization of the Photodegradation of a Human IgG1Mono-clonal Antibody Formulated as a High-Concentration Liquid Dosage Form. J. Pharm. Sci 2009, 98 (9), 3117–3130. [DOI] [PubMed] [Google Scholar]
- (98).Yu C; Tang J; Loredo A; Chen Y; Jung SY; Jain A; Gordon A; Xiao H Proximity-Induced Site-Specific Antibody Conjugation. Bioconjugate Chem 2018, 29 (11), 3522–3526. [DOI] [PubMed] [Google Scholar]
- (99).Cao YJ; Yu C; Wu K-L; Wang X; Liu D; Tian Z; Zhao L; Qi X; Loredo A; Chung A; Xiao H Synthesis of Precision Antibody Conjugates Using Proximity-Induced Chemistry. Theranostics 2021, DOI: 10.7150/thno.62444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (100).Tian Z; Wu L; Yu C; Chen Y; Xu Z; Bado I; Loredo A; Wang L; Wang H; Wu K-L; et al. Harnessing the Power of Antibodies to Fight Bone Metastasis. Science Advances 2021, 7 (26), eabf2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (101).Kishimoto S; Nakashimada Y; Yokota R; Hatanaka T; Adachi M; Ito Y Site-Specific Chemical Conjugation of Antibodies by Using Affinity Peptide for the Development of Therapeutic Antibody Format. Bioconjugate Chem 2019, 30 (3), 698–702. [DOI] [PubMed] [Google Scholar]
- (102).Yamada K; Shikida N; Shimbo K; Ito Y; Khedri Z; Matsuda Y; Mendelsohn BA AJICAP: Affinity Peptide Mediated Regiodivergent Functionalization of Native Antibodies. Angew. Chem., Int. Ed 2019, 58 (17), 5592–5597. [DOI] [PubMed] [Google Scholar]
- (103).Metzger H; Wofsy L; Singer SJ Affinity Labeling of the Active Sites of Antibodies to the 2,4-Dinitrophenyl Hapten*. Biochemistry 1963, 2 (5), 979–988. [DOI] [PubMed] [Google Scholar]
- (104).Chmura AJ; Orton MS; Meares CF Antibodies with Infinite Affinity. Proc. Natl. Acad. Sci. U. S. A 2001, 98 (15), 8480–8484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (105).Meares CF The Chemistry of Irreversible Capture. Adv. Drug Delivery Rev 2008, 60 (12), 1383–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (106).Lake B; Serniuck N; Kapcan E; Wang A; Rullo AF Covalent Immune Recruiters: Tools to Gain Chemical Control Over Immune Recognition. ACS Chem. Biol 2020, 15 (4), 1089–1095. [DOI] [PubMed] [Google Scholar]
- (107).Ohata J; Ball ZT A Hexa-Rhodium Metallopeptide Catalyst for Site-Specific Functionalization of Natural Antibodies. J. Am. Chem. Soc 2017, 139 (36), 12617–12622. [DOI] [PubMed] [Google Scholar]
- (108).Yu W; Gillespie KP; Chhay B; Svensson A-S; Nygren P-Å; Blair IA; Yu F; Tsourkas A Efficient Labeling of Native Human IgG by Proximity-Based Sortase-Mediated Isopeptide Ligation. Bioconjugate Chem 2021, 32, 1058. [DOI] [PMC free article] [PubMed] [Google Scholar]