

Abstract
Traumatic brain injury (TBI) is a risk factor for dementia, with studies describing a mixed neurodegenerative pathology in late survivors. However, the mechanisms driving this post-TBI neurodegeneration remain elusive. Increasingly, blood brain barrier (BBB) disruption is recognized in a range of neurological disorders, including dementias; although little is known of the consequences of TBI on the BBB. From the Glasgow TBI Archive autopsy cases of single, moderate or severe TBI (n=70) were selected to include a range of survivals from acute (10h to 13days) to long-term (1 to 47years) survival, together with age-matched, uninjured controls (n=21). Multiple brain regions were examined for fibrinogen (FBG) and immunoglobulin G (IgG) immunohistochemistry. Following TBI, 40% of patients dying in the acute phase and 47% of those surviving a year or more from injury showed multi-focal, abnormal, perivascular and parenchymal FBG and IgG immunostaining localized to grey matter, with preferential distribution towards the crests of gyri and deep neocortical layers. In contrast, where present, controls showed only limited, localized immunostaining. These preliminary data demonstrate evidence of widespread BBB disruption in a proportion of TBI patients emerging in the acute phase and, intriguingly, persisting in a high proportion of late survivors.
Keywords: Traumatic brain injury, chronic traumatic encephalopathy, blood brain barrier, fibrinogen
Introduction
Traumatic brain injury (TBI) represents a major health concern, with 3.5 million cases in the United States each year (1). In addition to the immediate health impacts of the injury, TBI is acknowledged as the strongest environmental risk factor for the development of neurodegenerative disease, typically reported as Alzheimer’s disease (AD)(2–13) in type or, more recently, recognized as chronic traumatic encephalopathy (CTE)(14–16). Corresponding to this, autopsy studies in material from patients exposed to either single moderate or severe or repetitive mild brain injury reveal a complex of neurodegenerative pathologies including pathologies in tau, amyloid-β and TDP-43, neuronal loss, neuroinflammation and white matter degradation (13,15–20). However, the mechanisms driving these late, post-TBI neurodegenerative pathologies remain elusive.
Disruption in blood brain barrier (BBB) integrity is increasingly recognized in a variety of disorders, including ischemia (21), multiple sclerosis (22) and neurodegeneration (23, 24). In particular, imaging (25) and animal modelling (23, 26) studies in AD suggest BBB dysfunction is an early event in the course of disease and may contribute to its pathogenesis, perhaps as a result of impaired amyloid-β clearance or associated neuroinflammation (23,27–31). Furthermore, neuropathology studies report complex vascular changes associated with BBB dysfunction in AD (32–34), with evidence of a correlation between histological evidence of BBB leakage and Alzheimer-type pathologies in ‘normal’ ageing (35).
Following TBI, acute phase BBB disruption has been demonstrated in several animal models (36–48) though with conflicting data on the time-course of this disruption some studies suggesting BBB disruption is short lived, resolving within hours of injury (36–47), while others suggest a more dynamic, biphasic course following injury, with early phase BBB disruption at 3–6 hours post injury followed by later, further BBB at 1–3 (38, 48). In support of this ‘late’ BBB dysregulation after TBI is the observation of focal immunoglobulin G (IgG) deposition around callosal blood vessels ipsilateral to controlled cortical impact in mice at 3 months survival from injury (49).
Consistent with observations in animal models, evidence of acute BBB permeability following severe TBI in humans is provided by reports of elevations in CSF to serum albumin quotient (50–53) and in serum S100β levels (50–53) after injury. Of note, such evidence of acute BBB disruption following TBI might predict a population of patients with poor long-term outcome (52). Furthermore, neuroimaging evidence of BBB disruption has been reported in patients following TBI, even after clinically mild or moderate injury; in some cases persisting for years at the site of focal injury (contusions) and with greater frequency in patients with post-traumatic epilepsy (54). Intriguingly, imaging evidence of BBB disruption has been observed in American football participants, independent of clinical evidence of TBI, possibly as a result of exposure to ‘sub-concussive’ head impacts(55).
Thus there is evidence from animal models and limited clinical studies of BBB disruption following TBI. However, the extent and time course of this TBI-associated BBB disruption in humans remains poorly understood. In particular, evidence of BBB disruption at late time points post-injury has not been explored. Here, we utilize material from the Glasgow TBI Archive to characterize the extent, distribution and temporal dynamics of BBB disruption following closed head injury, with respect to age-matched, uninjured controls.
Materials and Methods
Case Selection and Brain Tissue Preparation
Ethical approval for this study was granted by the West of Scotland Research Ethics Service. From the Glasgow TBI Archive, cases with a history of single moderate or severe TBI aged 60 years or under at time of death were selected, representing a range of survival times from injury to include: acute cases with survival times of 10 hours to <14 days (n=27); intermediate cases with survival times of 14 days to <1 year (n=11); and long-term cases with survival times from 1 year to 47 years (n=32). Detailed reports from the original diagnostic autopsywere available for all cases , where necessary supplemented by forensic and clinical records, and confirmed a clinical history of moderate or severe TBI as defined by Glasgow Coma Scale at presentation. Age matched, uninjured controls aged 60 years or under at time of death and with no known history of neurological disease or history of TBI (n=21) were selected for comparison. Across the cohorts, post-mortem delays were comparable (p>0.05 in all comparisons across cohorts; Student t-test). Demographics, clinical data and detail on neuropathology findings at the original autopsy for each cohort are presented in Table 1.
Table 1.
Demographic and clinical data for all groups.
| TBI: Acute Survival (n=27) | TBI: Intermediate Survival (n=11) | TBI: Long-Term Survival (n=32) | Controls (n=21) |
||
|---|---|---|---|---|---|
| Mean age (Range) | 44.4 years (9–60) |
32 years (17–56) |
46.3 years (19–60) |
34.3 years (14–60) |
|
| Males | 17 (63%) |
11 (100%) |
31 (97%) |
12 (57.1%) |
|
| Mean PM Delay (Range) | 56.1 hours (3–240) |
74.7 hours (26–192) |
65.5 hours (9–184.5) |
71.6 hours (12–144) |
|
| Mean Survival Interval (Range) | 69.3 hours (6–216) |
72.8 days (14–279) |
7.8 years (1–47) |
Not applicable | |
| Cause of TBI | Fall | 15 (56%) |
2 (18%) |
15 (47%) |
Not applicable (No history TBI) |
| RTA | 7 (26%) |
5 (42%) |
5 (16%) |
||
| Assault | 4 (15%) |
3 (25%) |
8 (25%) |
||
| Unknown | 1 (4%) |
1 (8%) |
4 (12%) |
||
| Cause of Death | Head injury | 25(93%) | 4 (33%) | 0 | 0 |
| Bronchopneumonia | 2(7%) | 4(33%) | 7(22%) | 1(4.8%) | |
| ARDS | 0 | 2(17%) | 1(3%) | 0 | |
| Pulmonary thromboembolism | 0 | 1(8%) | 0 | 0 | |
| Heart disease | 0 | 0 | 6 (19%) | 4(16.67%) | |
| Alcohol related | 0 | 0 | 2(6%) | 0 | |
| Pyelonephritis | 0 | 0 | 1(3%) | 0 | |
| Multi-organ failure | 0 | 0 | 1(3%) | 0 | |
| GIT hemorrhage | 0 | 0 | 1(3%) | 0 | |
| Polytrauma | 0 | 0 | 1(3%) | 0 | |
| Drug overdose | 0 | 0 | 0 | 4(16.67%) | |
| SUDEP | 7(22%) | 8(38.1%) | |||
| Pulmonary edema | 0 | 0 | 1(3%) | 0 | |
| Septicemia | 0 | 0 | 0 | 2(8.3%) | |
| Inhalation of gastric contents | 0 | 0 | 0 | 2(8.3%) | |
| Unknown | 0 | 0 | 4(12%) | 0 | |
| TBI Pathologies | Skull fracture | 20(74%) | 6(54%) | 15(47%) | 0 |
| DAI | 11(40%) | 10(91%) | 6(19%) | 0 | |
| Brain Swelling | 13(48%) | 2(18%) | 0 | 0 | |
| SDH | 20(74%) | 4(36%) | 13(41%) | 0 | |
| EDH | 0 | 0 | 3(9%) | 0 | |
| SAH | 15(56%) | 0 | 1(3%) | 0 | |
Key: TBI = traumatic brain injury; SUDEP = sudden unexpected death in epilepsy; GIT = gastrointestinal tract; ARDS = acute respiratory distress syndrome; RTA = road traffic accident; DAI = Diffuse Axonal Injury; SDH = Subdural hemorrhage; EDH = Extradural hemorrhage; SAH = Subarachnoid hemorrhage
At the time of the original diagnostic autopsy, whole brains were immersion fixed in 10% formal saline for a minimum of 3 weeks then dissected, sampled following a standardized block selection protocol and processed to paraffin using standard techniques. Paraffin tissue blocks from a coronal slice of the cerebral hemispheres at the mid-thalamic level were identified for analysis to include: the thalamus; corpus callosum with adjacent cingulate and superior frontal gyri; hippocampus at the level of the lateral geniculate nucleus extending through the entorhinal cortex, collateral sulcus and fusiform gyrus; and the insular cortex. From these tissue blocks 8μm sections were prepared for Hematoxylin and eosin staining and immunohistochemistry.
Hematoxylin and eosin staining
Hematoxylin and eosin (H&E) staining was performed on sections from all tissue blocks. Slides were deparaffinized in xylene and rehydrated to water followed by immersion for 10 minutes in hematoxylin (Mayer’s, Leica Microsystems, Wetzlar, Germany). After rinsing and immersion in Scott’s tap water substitute (Leica Microsystems, Wetzlar, Germany), slides were differentiated in 1% acid alcohol and rinsed. The sections were then immersed in 25% aqueous eosin Y solution (TCS Biosciences, Buckingham, UK) for 5 minutes, rinsed, then dehyrated, cleared and coverslipped.
Immunohistochemistry
Following deparaffinization and rehydration, sections were immersed in 3% aqueous H2O2 (15 minutes) to quench endogenous peroxidase activity. Antigen retrieval was performed via microwave pressure cooker for 8 minutes in preheated 0.1M Tris EDTA buffer. Subsequent blocking was achieved by applying 50μL of normal horse serum (Vector Labs, Burlingame, CA, USA) per 5 mL of Optimax buffer (BioGenex, San Ramon, CA, USA) for 30 minutes. Incubation with the primary antibody was then performed for 20 h at 4°C. Polyclonal rabbit anti-human antibodies for fibrinogen (FBG) and immunoglobulin G (IgG) (Dako, Carpinteria, CA, USA) were utilized at dilutions of 1:17,500 and 1:10,000 respectively. A biotinylated secondary antibody was then applied for 30 minutes followed by avidin biotin complex as per the manufacturer’s instructions (Vectastain Universal Elite kit, Vector Labs, Burlingame, CA, USA). Finally, visualization was achieved using the DAB peroxidase substrate kit (VectorLabs, Burlingame, CA, USA) followed by counterstaining with haematoxylin. Known positive tissue sections were run in parallel with test sections in all antibody runs in addition to sections with primary antibody omitted as standard controls for antibody specificity. All sections were viewed using a Leica DMRB light microscope (Leica Microsystems, Wetzlar, Germany). In addition, sections were scanned at 20x using a Hamamastu Nanozoomer 2.0-HT slide scanner, with the images viewed via the SlidePath Digital Image Hub application (Leica Microsystems, Wetzlar, Germany).
Analysis of Immunohistochemical Findings
All observations were conducted blind to demographic and clinical data by two independent observers (JH, AY). Anatomically distinct regions were defined for assessment to include the neocortical grey matter of the cingulate gyrus; cingulate sulcus; superior frontal gyrus; parahippocampal gyrus; collateral sulcus; fusiform gyrus and insular cortex. A subanalysis of neocortical grey matter was performed by dividing the cortex into superficial (layers I-II), mid (layers III-IV) and deep layers (V- VI). In addition, the white matter of the midline and lateral extent of the corpus callosum and internal capsule; hippocampal sectors CA1–4 and the thalamus (sub-divided into medial, intermediate and lateral regions) were assessed. Each anatomical region was reviewed and a semi-quantitative assessment of the frequency and intensity of immunoreactivity determined as absent (0), sparse (1), moderate (2) or extensive (3). Representative examples of immunohistochemical findings and the corresponding semi-quantitative score are shown in Figure 1.
Fig. 1. Representative examples of the patterns of FBG immunoreactivity encountered.
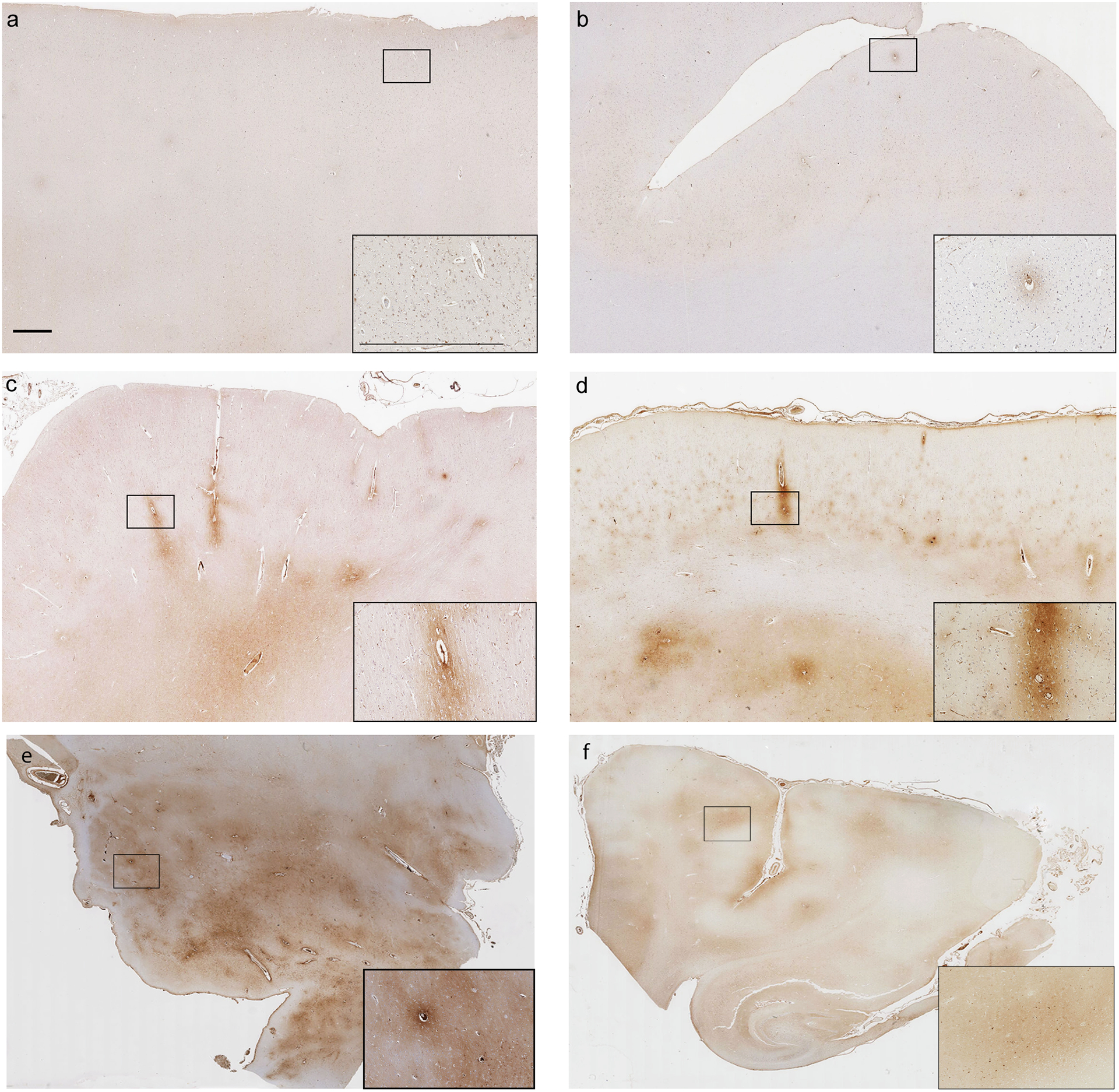
(a) Section from the superior frontal gyrus of a 47 year old male TBI patient who died 1 year following a fall. No abnormal FBG immunoreactivity is present (score of 0). (b) Limited, faint perivascular FBG immunoreactivity (score of 1) from the superior frontal gyrus of a 60 year old male TBI patient who died 16 years after a road traffic accident. (c) More widespread, moderate perivascular FBG immunoreactivity (score of 2) in the superior frontal gyrus of a 60 year old male TBI patient who survived 10 hours after a fall. (d) Extensive perivascular and adjacent parenchymal FBG immunoreactivity (score of 3) in the superior frontal gyrus of a 50 year old male TBI patient who survived 1 year after an assault. (e) Extensive perivascular FBG immunoreactivity in thalamic region of a 60 year old male TBI patient who survived 8 days after a fall. (f) Limited perivascular FBG immunoreactivity in parahippocampal region of a 56 year old female TBI patient who survived 24 hours after a road traffic accident. Scale bars = 1mm and apply to all corresponding images.
Statistical analysis
All data were analyzed using Minitab (v.16; Minitab Inc.). The Chi-square Test was used to assess differences in data between and within cohorts.
Results
Fibrinogen Immunoreactivity After TBI
Controls
In uninjured controls, where present, FBG immunoreactivity was observed in a predominantly perivascular distribution, highlighting small vessels throughout the neuropil. In addition, occasional, sparse immunoreactive neurons and glia were observed. In the overwhelming majority of controls (17 of 21), this FBG immunoreactivity was limited (score 0 or 1) (Figure 2a) and localized (Figure 3). In the remaining 4 controls, localized foci of moderate (score 2) levels of FBG immunoreactivity were observed in single tissue blocks; in two cases localized to the thalamus, one to the fusiform gyrus and the last to the superior frontal gyrus (Figure 4). Causes of death in these controls with localized, moderate FBG immunoreactivity were mixed; one sudden death in epilepsy, one sudden death of cardiac origin, one sepsis with multi-organ failure and one pneumonia.
Fig. 2. Representative images of FBG and IgG immunoreactivity following TBI and in controls.
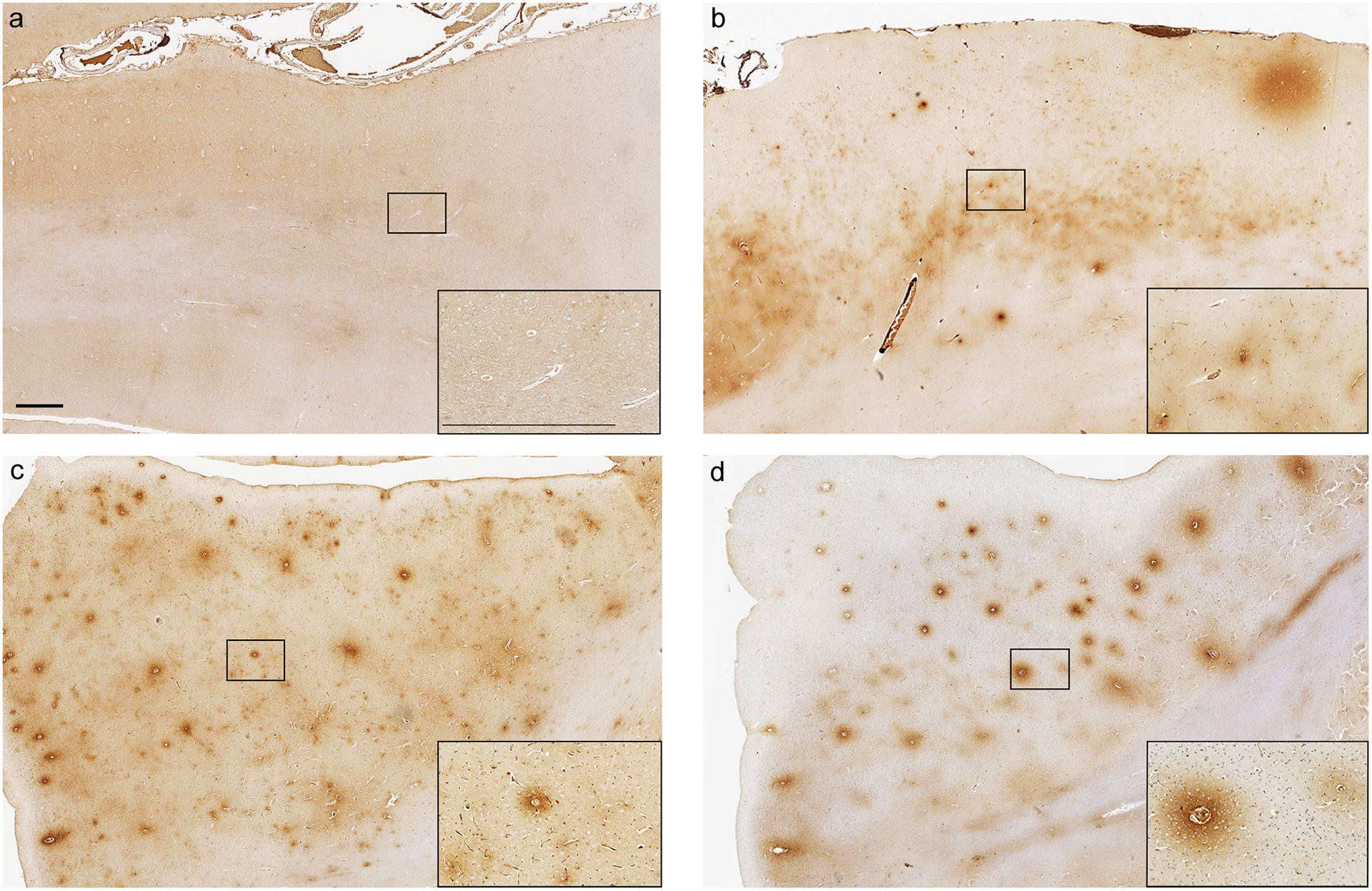
Absence of FBG immunoreactivity in the superior frontal gyrus of a 46 year old male with no history of TBI. (b) Extensive FBG immunoreactivity in the superior frontal gyrus, with preferential distribution of staining to the mid and deep cortical layers, in a 20 year old male TBI patient who survived 2 days following an assault. Extensive FBG (c) and IgG (d) immunoreactivity in the adjacent sections from the superior frontal gyrus of a 60 year old male TBI patient who survived 18 years following a fall. Scale bars = 1mm and apply to all corresponding images.
Fig. 3. Extent of abnormal FBG immunoreactivity following TBI at varying survivals versus controls.
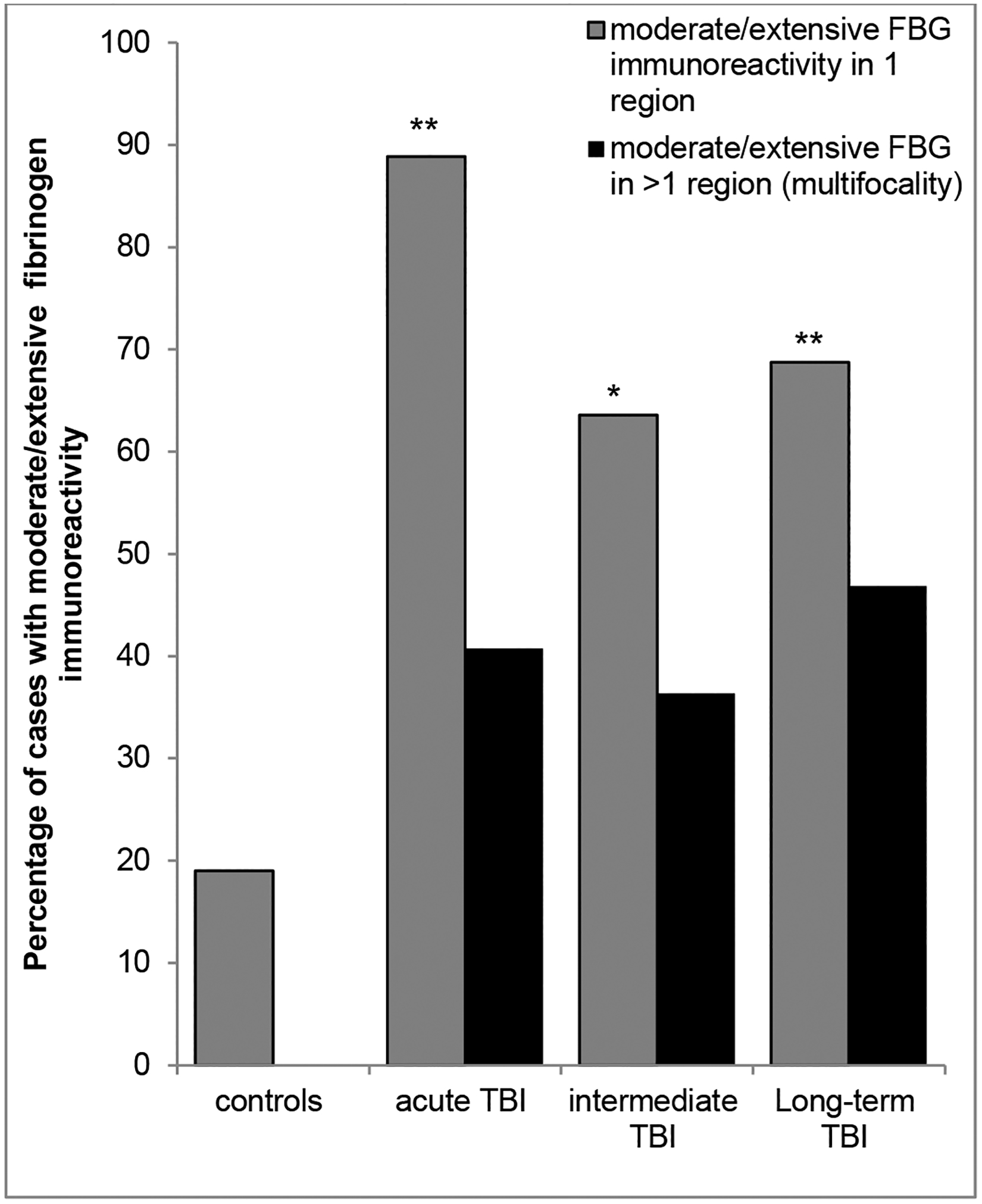
Whilst moderate or extensive FBG immunoreactivity was an uncommon observation in controls, occurring in just 4 of 21 cases (19%), it was a frequent observation following TBI at all survival time points assessed, being present in 88%, 62% and 69% of acute, intermediate and long-term survival cases respectively. Furthermore, in controls abnormal FBG immunostaining was restricted to single anatomical regions, in contrast to the often multifocal pathology in material following TBI. (*p<0.005; **p<0.001; Chi-square TBI cohort v control fibrinogen positivity).
Fig. 4. Regional distribution of FBG immunoreactivity following TBI versus controls.
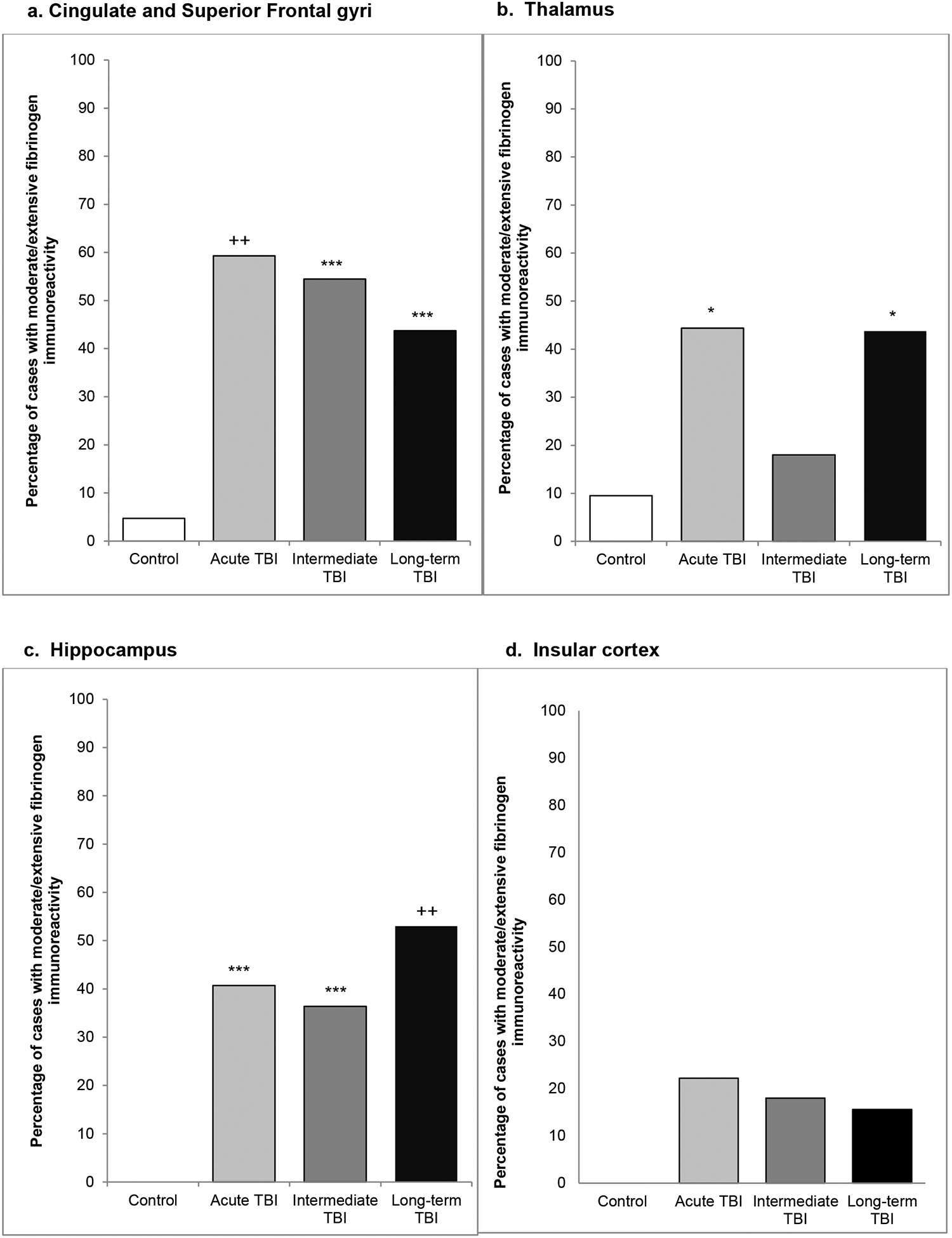
At all survival intervals and in each region analyzed there was evidence of BBB disruption following TBI, evidenced by moderate/ extensive FBG immunoreactivity, in a higher proportion of TBI survivors than matched, non-injured controls in material from the (a) cingulate/superior frontal gyri, (b) thalamus (c) hippocampus and (d) insular cortex. (*p<0.05; **p<0.01; ***p<0.005; +p<0.0005; ++p<0.0001; Chi-square TBI cohort v control)
Acute TBI survival
In material from patients dying acutely following TBI (10 hours to <14 days survival), in addition to perivascular FBG immunoreactivity, regions of more confluent, diffuse immunostaining were also present (Figure 2b). Indeed, in contrast to the typically limited abnormal FBG immunoreactivity observed in controls, in this acute TBI survival cohort 88% (p<0.001; ChiSq) of cases showed at least one anatomical region with moderate (score 2) or extensive (score 3) FBG immunoreactivity. Further, in contrast to the localized pathology in controls, following TBI increased FBG immunoreactivity often appeared multifocal, involving 2 or more regions in 11 out of 27 (40%) acute survival cases (Figure 3). Of the regions examined, moderate or extensive FBG immunoreactivity was detected most frequently in material from the cingulate and superior frontal gyri (16 cases), followed by thalamus (12 cases), medial temporal lobe (11 cases) and insular cortex (6 cases) (Figure 4). Within the neocortical material (cingulate, superior frontal, fusiform and parahippocampal) there was evidence of preferential anatomical distribution, with moderate or extensive FBG immunoreactivity more evident in the crests of gyri than in the depths of the adjacent sulci (Figure 5a) in both cingulate and medial temporal lobe cortical regions. Further, there was localization of abnormal FBG immunoreactivity towards mid (layers 3 and 4) and deep (layers 5 and 6) cortical layers rather than superficial (layers 1 and 2) cortical layers (Figure 2b; Figure 5b) in all cortical regions. Notably, only one acute survival case showed focal, moderate FBG immunoreactivity in the hippocampal formation (sector CA2), with no acute survival case showing more than localized, limited FBG immunoreactivity in the corpus callosum, similar to controls. No localization to a particular thalamic sub-region was evident, neither was the observed abnormal FBG staining localized to focal pathologies of TBI.
Fig. 5. Neocortical distribution of FBG following TBI.

(a) Across all survival time points there was a clear preferential distribution of abnormal FBG immunoreactivity to the crests of gyri when compared to the depths of sulci, as illustrated here for the superior frontal gyrus versus the adjacent cingulate sulcus (*p<0.01; **p<0.001; Chi-square sulcus versus gyrus). (b) Further, within the neocortical grey there was preferential distribution of abnormal staining to the mid (layers 3 and 4) and deep (layers 5 and 6) cortical layers when compared to superficial layers (layers 1 and 2). (+p<0.01; ++p<0.005; Chi-square deep versus superficial cortical layers).
Intermediate TBI survival
Similar to material from patients dying acutely following TBI, regions of moderate or extensive FBG immunoreactivity were present in a greater number of intermediate (>2 weeks to 1 year post-TBI) TBI survival patients than in controls, with 7 of 11 (64%; p=0.004 ChiSq) demonstrating at least one anatomical sub-region with moderate or high FBG immunoreactivity; showing multifocality in 4 of these cases (Figure 3). Again, material from the region of the cingulate and superior frontal gyri most frequently showed increased staining (6 cases), followed by medial temporal lobe (4 cases), thalamus (2 cases) and insular cortex (2 cases) (Figure 4). As in acute survival material, there was a predilection for gyral crests over sulcal depths in the cingulate gyrus and medial temporal lobe and, though the small number of cases precludes formal statistical assessment, a trend towards preferential staining in mid to deep rather than superficial neocortical layers in all cortical regions (Figures 5a and 5b). There was no notable FBG immunoreactivity in the white matter of the corpus callosum, with only 2 cases showing more than mild staining in the hippocampus.
Long-term TBI survival
Once again, a higher proportion of long-term survival cases showed evidence of increased FBG immunoreactivity when compared to matched, non-injured controls (Figure 2c), with at least one anatomical region showing moderate or extensive FBG immunoreactivity in 22 of 32 cases (69%) (p<0.001; ChiSq). Again, multifocality was common, being present in 15 of the long term survival cases (47%) (Figure 3); this increased staining most frequent in material from the medial temporal lobe (17 cases), followed by cingulate and superior frontal gyri and thalamus (14 cases each) and insular cortex (5 cases) (Figure 4). As in acute and intermediate cases, a preferential distribution to the crests of gyri over depths of sulci was evident (Figure 5a). Further, within the crests of gyri, FBG immunoreactivity was greater in the mid and deeper layers than the superficial layers of the cortex. Thus, in the superior frontal gyrus 56% showed moderate or high FBG immunostaining in the deeper layers compared to just 12% in the superficial layers (p=0.001; ChiSq) (Figure 5b) this pattern was reflected in all cortical regions assessed. In the medial temporal lobe 40% showed high FBG immunostaining in the deep layers compared to 18% in the superficial layers (p=0.1379; ChiSq). In contrast to earlier survival time points and controls, 23% (p=0.023; ChiSq) of cases in this long-term survival cohort showed hippocampal FBG immunoreactivity greater than mild in at least one hippocampal sector, though with no clear preferential distribution to any particular sector. In keeping with this widespread increased FBG immunoreactivity in these long-term survival cases, 58% showed increased thalamic staining, versus just 10% of controls (p=0.001; ChiSq). However, once again, no increase in FBG immunoreactivity was detected in material from the corpus callosum.
Immunoglobulin G Immunoreactivity After TBI
Controls
Observations in the IgG stained material corroborated the observations in material stained for FBG (Figure 2d). Thus, at all survival points and in controls the extent and distribution of IgG immunoreactivity was comparable to FBG immunoreactivity with regards to greater perivascular and confluent, diffuse staining IgG immunoreactivity in a proportion of TBI cases at all survivals when compared to controls. Specifically, IgG immunoreactivity in the control cohort was limited with only 4 of 21 (19%) cases displaying moderate IgG immunoreactivity (score of 2); in two cases this was localised to the superior frontal gyrus, one case localised to the thalamus and one to the fusiform gyrus. Again, no multifocality was observed and causes of death included sudden death in epilepsy (n=2), drug overdose (n=1), and pneumonia (n=1).
Acute TBI survival
The acute TBI survival cohort demonstrated moderate or high IgG immunoreactivity in at least one anatomical sub-region in 63% of cases (p<0.005 ChiSq), with multifocality present in 10 out of 27 (37%). Of the regions examined, moderate or extensive IgG immunoreactivity was detected most frequently in material from the medial temporal lobe (10 cases), followed by the cingulate/superior frontal gyri and the thalamus (8 cases) then the insular cortex (2 cases). As with FBG immunoreactivity there was evidence of preferential anatomical distribution, with moderate or extensive IgG immunoreactivity more evident in the crests of gyri than in the depths of the adjacent sulci and in the mid to cortical deep layers when compared to the superficial cortical layers.
Intermediate TBI survival
Following intermediate survival from TBI, regions of moderate or widespread IgG immunoreactivity were present in a greater number than in controls with 6 of 11 (54%; p=0.05 ChiSq) demonstrating at least one anatomical sub-region with moderate/high IgG immunoreactivity, with multifocality present in 45%.
Long-term TBI survival
The long-term survival TBI cohort demonstrated 71% (23 out of 32 cases) of moderate/high IgG immunoreactivity in at least one anatomical sub-region (p=0.0002; ChiSq). Multifocality again was common with 14 of the 32 long-term TBI cases (43%) showing immunoreactivity in 2 or more sub-regions. Increased IgG staining was most frequent in the medial temporal lobe (13 cases each), followed by cingulate/ superior frontal gyri and thalamus (12 cases) and insular cortex (3 cases). The pattern and distribution of IgG immunoreactivity echoed the results in fibrinogen prepared material, with preferential distribution to the crests of gyri over the depths of sulci in both cingulate gyri and hippocampal gyri, and greater IgG immunoreactivity observed in the mid and deeper layers versus the superficial neocortical layers.
Association of BBB disruption with TBI-associated pathologies
Reviewing H&E stained sections from the multiple tissue blocks examined for immunohistochemical evidence of BBB disruption confirmed anticipated BBB disruption in association with acute, focal hemorrhagic pathologies, in addition to more widespread and diffuse BBB disruption independent of focal pathology. Specifically, in material from acute TBI survivors 21 TBI-associated focal hemorrhagic or contusional lesions were observed across 13 of the 27 cases, with virtually all (12 of 20 lesions) displaying some degree of extravascular fibrinogen immunoreactivity in the surrounding parenchyma. However, in the majority of these cases (9 of 13; 69%) BBB disruption was not confined to the region of focal pathology, with evidence of widespread, diffuse BBB also present in the remaining tissue blocks. Notably, in the 14 acute survival TBI cases without focal hemorrhage or contusion, 12 (86%) also displayed high fibrinogen immunoreactivity in at least one region examined.
In material from patients surviving beyond the acute phase of injury evidence of healed hemorrhages or contusions, with histological features consistent with lesions dating from the time of the original TBI, were present in 2 of 11 intermediate survival (comprising 6 individual lesions) and 8 of 32 long-term survival cases (comprising 12 distinct lesions). While not as extensive as observed in relation to focal hemorrhages or contusions in acute survivors, some evidence of BBB disruption was observed in the immediate locality of all 6 lesions in material from intermediate survivors. In contrast, evidence of focal BBB disruption in association with healed hemorrhages or contusions was less frequently observed in material from long-term survivors, with just 6 of 12 lesions associated with abnormal FBG or IgG immunoreactivity. Of note, both of the intermediate survival cases and all 8 of the long-term survival cases with BBB disruption in association with focal pathologies also displayed more widespread, diffuse BBB disruption remote from focal pathology. Further, 6 of 9 intermediate and 15 of 24 long-term survival cases showing no evidence of focal hemorrhagic or contusional pathology in the material examined displayed evidence of high FBG immunoreactivity in at least one region.
In addition to these focal hemorrhagic and contusional pathologies, histological evidence of diffuse hypoxic/ ischemic injury, microgliosis and/or astroglisosis in varying stages of evolution was identified in a proportion of TBI cases across all survivals. No correlation between these TBI-related pathologies and evidence of BBB disruption was observed.
Discussion
Here we demonstrate histological evidence of widespread, diffuse blood-brain barrier (BBB) disruption in survivors of a single moderate or severe TBI when compared to non-injured controls. Notably, evidence of increased and widespread extravascular immunoreactivity for fibrinogen (FBG) and immunoglobulin G (IgG) was observed not only in material from patients dying in the acute phase post-injury, but also in a high proportion of those surviving many years after injury. As such, these data raise the possibility that a single moderate or severe TBI is responsible for immediate and long-lasting alterations in BBB function following injury, which might contribute to post-TBI neurodegeneration.
Under normal conditions, the plasma proteins fibrinogen (340kDa) and IgG (150kDa) do not cross the BBB. However, we observed widespread, multifocal, perivascular and parenchymal FBG and IgG deposition in approximately half of patients dying within the first 2 weeks following a single moderate or severe TBI; indicating that these proteins had crossed the BBB in the acute phase following injury. Similar acute BBB leakage has been identified in various animal models of TBI, as revealed by the presence of either extravasated serum proteins or intravascularly injected labels in the brain parenchyma (36–44, 46, 56, 57) following injury. In these models often extensive, acute BBB leakage can be observed in the absence of hemorrhage, consistent with our observations in this human, autopsy-acquired tissue.
As regards possible structural correlates of this acute BBB disruption, ultrastructural studies in animal models of TBI have identified a variety of alterations in vascular endothelia in the acute phase following injury, including the formation of vacuoles, craters and microvilli (47, 57–59) some of which were later identified in human TBI (60, 61). These very rapid changes in vascular structure and function have been attributed to a range of mechanisms such as direct perturbation of the vessels by mechanical forces, including the immediate disruption of vascular endothelial cells. Additional, secondary insults following injury might also be detrimental to normal endothelial integrity and function, including acute rises in arterial pressure or intravascular thrombus formation (45, 56, 57). Finally, active physiological changes have also been described in both animals and humans following TBI, including increased transendothelial vesicular transport with otherwise intact tight junctions (47, 59, 61, 62) as well as alterations to other components of the BBB, such as early astrocyte disruption and swelling (45, 58).
Studies suggest that these acute BBB disruptions may confer worse long-term outcome following TBI clinically (52), although the relative role of this pathology is unknown. Indeed, acute BBB opening after TBI may simply be one of many common pathological features of a more severely injured brain, which collectively contribute to poor outcome. Nonetheless, specific deleterious mechanisms of BBB disruption following TBI may include influx of fluid together with chemical and protein mediators promoting vasogenic edema, disruption in the normal pathways to clear toxic metabolites, and a failure to deliver normal metabolites vital for function. In turn, the temporal course and relative contribution of these various consequences of BBB disruption may differ with the mode of injury.
Although data from animal models are reported as showing evidence of a biphasic BBB opening (38, 48), in the present study we identified no clear evidence of defined phases of BBB disruption across our various survival cohorts, in particular in the acute phase. Nevertheless, the possibility of distinct phases of BBB disruption following injury cannot be entirely excluded, with the diverse and heterogeneous nature of post-mortem TBI cases with varying intercurrent illnesses, survival times and causes of injury conceivably masking such an observation. Nonetheless, studies specifically directed at understanding the underlying mechanisms of this BBB dysfunction over time might reveal important potential therapeutic targets with defined windows of opportunity.
While a degree of acute BBB disruption was anticipated, the number of cases, often with extensive BBB disruption following long-term survival from TBI was, nonetheless, surprising. Specifically, extensive, multifocal, extravascular FBG and IgG deposition was observed in approximately half of those surviving a year or more from injury in our cohort. Notably, clearance of large soluble proteins from the brain parenchyma occurs via convective bulk flow of interstitial fluid over the order of hours to days, or more rapidly via reverse transcytosis, such as occurs with IgG (63). Thus, the observation of soluble plasma proteins in the brain parenchyma of patients exposed to TBI more than a year prior to death is consistent with on-going BBB disruption, rather than evidence of protein deposited at the time of injury and not cleared in the intervening period.
The pattern and distribution of chronic BBB disruption post-TBI was more frequently localized to the grey matter of the neocortical ribbon and deep grey nuclei, with a preferential distribution in the former to mid and deeper cortical layers over superficial layers and the crests of gyri over depths of sulci. This pattern indicates preferential vulnerability in specific locations within a more global and diffuse process, rather than a heterogeneous multifocal pathology. Undoubtedly this observation presents challenging and interesting data given the contrast with current descriptions of aspects of the neuropathology of post-TBI neurodegeneration and CTE where localization of tau pathologies to the depths of sulci is reported. However, at present, the pathologies of CTE are provisional, therefore should be taken as such. More research into these conflicting observations should be carried out in the future.
Notably our data demonstrate no association with evidence of widespread and diffuse BBB disruption and the presence of other TBI-associated pathologies. Specifically, BBB disruption was not localized to areas of focal TBI pathologies, such as contusions or hemorrhages. Further, there was no correlation between diffuse and widespread BBB disruption and diffuse ischemic pathology. Nevertheless, the number of cases and necessary heterogeneity in pathologies in this cohort limit the ability to determine whether specific diffuse primary or secondary pathologies in the acute phase can account for later patterns of BBB disruption. However, this will be worthy of exploration using larger post-mortem cohorts and, possibly, animal models.
In recent years, the chronic clinical and neuropathological sequelae of TBI have been increasingly reported, with particular attention paid to the association of TBI survival with increased risk of progressive neurodegenerative disorders, specifically CTE (14–16). Neuropathological examination of autopsy acquired material from patients with survival greater than a year from a single moderate or severe TBI reveals an increased frequency of neurodegeneration-associated pathologies when compared to material from matched controls, including amyloid-β plaques, neurofibrillary tangles, inflammation and white matter degradation (13, 17, 18, 20). Notably, the role of the BBB in neurodegeneration is of increasing interest, with evidence indicating that changes to the brain’s microvasculature may actively contribute to development of AD pathology (for detailed review see (23)). Various micro-vascular changes have been reported in AD including changes to vessel structure and density (33), BBB breakdown and leakage (32, 35), and secretion of potentially neurotoxic factors from the vascular endothelium (64). While the extravasation of serum proteins such as fibrinogen has also been shown to correlate with the presence of Alzheimer-type pathology(35), the cause or effect relationship between alterations in the vasculature and associated neurodegenerative pathologies is not well defined. However, increasing clinical and experimental data suggest vascular changes can occur early and may contribute to states of hypoperfusion promoting neuronal injury (23, 33, 65, 66). Studies to evaluate potential associations between BBB breakdown and other pathologies associated with neurodegenerative disease will be an important future consideration.
Perhaps the most widely studied aspect of BBB with regards to AD, is its role in the clearance and sequestration of amyloid beta to the peripheral circulation (28, 67–69). Interestingly, individuals with high serum fibrinogen have been shown at greater risk of AD (70, 71). Fibrinogen was also shown to accelerate inflammation and neurovascular damage in a mouse mode of AD (72). While the number of cases described in the present study precludes analysis of multiple co-variants, the interplay and temporal relationship between BBB disruption and subsequent progressive neurodegenerative pathologies will be an important direction for future investigation.
Conclusion
Here we present the first preliminary data indicating that after just a single moderate or severe TBI there is neuropathological evidence of widespread, diffuse, multifocal BBB disruption in just under one half of patients, even after many years of survival from injury. Given that vascular dysfunction may be an important contributor to neurodegenerative disorders, acute and chronic BBB alterations post-TBI will be important to examine in this context.
Support
This study was supported by; the National Institutes of Health (NIH) Grants NS038104, NS056202; the United States Department of Defense Grant No. PT110785; and The Sackler Institute Endowment Fund, Department of Neurology, Queen Elizabeth University Hospital, Glasgow
References
- 1.Coronado VG, McGuire LC, Sarmiento K, Bell J, Lionbarger MR, Jones CD, Geller AI, Khoury N, Xu L. Trends in traumatic brain injury in the U.S. and the public health response: 1995–2009. J Safety Res 2012:43;299–307. [DOI] [PubMed] [Google Scholar]
- 2.Molgaard CA, Stanford EP, Morton DJ, Ryden LA, Schubert KR, Golbeck AL. Epidemiology of head trauma and neurocognitive impairment in a multi-ethnic population. Neuroepidemiology 1990:9;233–42. [DOI] [PubMed] [Google Scholar]
- 3.Mortimer JA, French LR, Hutton JT, Schuman LM. Head injury as a risk factor for Alzheimer’s disease. Neurology 1985:35;264–7. [DOI] [PubMed] [Google Scholar]
- 4.Mortimer JA, van Duijn CM, Chandra V, Fratiglioni L, Graves AB, Heyman A, Jorm AF, Kokmen E, Kondo K, Rocca WA, Head trauma as a risk factor for Alzheimer’s disease: a collaborative re-analysis of case-control studies. EURODEM Risk Factors Research Group. Int J Epidemiol 1991:20 Suppl 2;S28–35. [DOI] [PubMed] [Google Scholar]
- 5.Graves AB, White E, Koepsell TD, Reifler BV, van Belle G, Larson EB, Raskind M. The association between head trauma and Alzheimer’s disease. Am J Epidemiol1990:131;491–501.1990:131;491–501. [DOI] [PubMed] [Google Scholar]
- 6.O’Meara ES, Kukull WA, Sheppard L, Bowen JD, McCormick WC, Teri L, Pfanschmidt M, Thompson JD, Schellenberg GD, Larson EB. Head injury and risk of Alzheimer’s disease by apolipoprotein E genotype. Am J Epidemiol 1997:146;373–84. [DOI] [PubMed] [Google Scholar]
- 7.Salib E, Hillier V. Head injury and the risk of Alzheimer’s disease: a case control study. Int J Geriatr Psych 1997:12;363–8. [DOI] [PubMed] [Google Scholar]
- 8.Guo Z, Cupples LA, Kurz A, Auerbach SH, Volicer L, Chui H, Green RC, Sadovnick AD, Duara R, DeCarli C, Johnson K, Go RC, Growdon JH, Haines JL, Kukull WA, Farrer LA. Head injury and the risk of AD in the MIRAGE study. Neurology 2000:54;1316–23. [DOI] [PubMed] [Google Scholar]
- 9.Schofield PW, Tang M, Marder K, Bell K, Dooneief G, Chun M, Sano M, Stern Y, Mayeux R. Alzheimer’s disease after remote head injury: an incidence study. J Neurol Neurosurg Psychiatry 1997:62;119–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Plassman BL, Havlik RJ, Steffens DC, Helms MJ, Newman TN, Drosdick D, Phillips C, Gau BA, Welsh-Bohmer KA, Burke JR, Guralnik JM, Breitner JC. Documented head injury in early adulthood and risk of Alzheimer’s disease and other dementias. Neurology 2000:55;1158–66. [DOI] [PubMed] [Google Scholar]
- 11.Fleminger S, Oliver DL, Lovestone S, Rabe-Hesketh S, Giora A. Head injury as a risk factor for Alzheimer’s disease: the evidence 10 years on; a partial replication. J J Neurol Neurosurg Psychiatry 2003:74;857–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lye TC, Shores EA. Traumatic brain injury as a risk factor for Alzheimer’s disease: a review. Neuropsychol Rev 2000:10;115–29. [DOI] [PubMed] [Google Scholar]
- 13.Johnson VE, Stewart W, Smith DH. Traumatic brain injury and amyloid-beta pathology: a link to Alzheimer’s disease? Nat Rev Neurosci 2010:11;361–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Corsellis JA, Bruton CJ, Freeman-Browne D. The aftermath of boxing. Psychol Med 1973:3;270–303. [DOI] [PubMed] [Google Scholar]
- 15.Smith DH, Johnson VE, Stewart W. Chronic neuropathologies of single and repetitive TBI: substrates of dementia? Nat Rev Neurol 2013:9;211–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McKee AC, Stern RA, Nowinski CJ, Stein TD, Alvarez VE, Daneshvar DH, Lee HS, Wojtowicz SM, Hall G, Baugh CM, Riley DO, Kubilus CA, Cormier KA, Jacobs MA, Martin BR, Abraham CR, Ikezu T, Reichard RR, Wolozin BL, Budson AE, Goldstein LE, Kowall NW, Cantu RC. The spectrum of disease in chronic traumatic encephalopathy. Brain 2013:136;43–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johnson VE, Stewart W, Smith DH. Widespread tau and amyloid-beta pathology many years after a single traumatic brain injury in humans. Brain Pathol 2012:22;142–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Johnson VE, Stewart JE, Begbie FD, Trojanowski JQ, Smith DH, Stewart W. Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain 2013:136;28–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ramlackhansingh AF, Brooks DJ, Greenwood RJ, Bose SK, Turkheimer FE, Kinnunen KM, Gentleman S, Heckemann RA, Gunanayagam K, Gelosa G, Sharp DJ. Inflammation after trauma: microglial activation and traumatic brain injury. Ann Neurol 2011:70;374–83. [DOI] [PubMed] [Google Scholar]
- 20.Johnson VE, Stewart W, Smith DH. Axonal pathology in traumatic brain injury. Exp Neurol 2013:246;35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sandoval KE, Witt KA. Blood-brain barrier tight junction permeability and ischemic stroke. Neurobiol Dis 2008:32;200–19. [DOI] [PubMed] [Google Scholar]
- 22.Kirk J, Plumb J, Mirakhur M, McQuaid S. Tight junctional abnormality in multiple sclerosis white matter affects all calibres of vessel and is associated with blood-brain barrier leakage and active demyelination. J Pathol 2003:201;319–27. [DOI] [PubMed] [Google Scholar]
- 23.Zlokovic BV. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat Rev Neurosci 2011:12;723–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bell RD, Zlokovic BV. Neurovascular mechanisms and blood-brain barrier disorder in Alzheimer’s disease. Acta Neuropathol 2009:118;103–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Starr JM, Farrall AJ, Armitage P, McGurn B, Wardlaw J. Blood-brain barrier permeability in Alzheimer’s disease: a case-control MRI study. Psychiatry Res 2009:171;232–41. [DOI] [PubMed] [Google Scholar]
- 26.Ujiie M, Dickstein DL, Carlow DA, Jefferies WA. Blood-brain barrier permeability precedes senile plaque formation in an Alzheimer disease model. Microcirculation 2003:10;463–70. [DOI] [PubMed] [Google Scholar]
- 27.Grammas P Neurovascular dysfunction, inflammation and endothelial activation: implications for the pathogenesis of Alzheimer’s disease. J Neuroinflammation 2011:8;26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shibata M, Yamada S, Kumar SR, Calero M, Bading J, Frangione B, Holtzman DM, Miller CA, Strickland DK, Ghiso J, Zlokovic BV. Clearance of Alzheimer’s amyloid-beta(1–40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J Clin Invest 2000:106;1489–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zlokovic BV, Deane R, Sagare AP, Bell RD, Winkler EA. Low-density lipoprotein receptor-related protein-1: a serial clearance homeostatic mechanism controlling Alzheimer’s amyloid beta-peptide elimination from the brain. J Neurochem 2010:115;1077–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Deane R, Wu ZH, Sagare A, Davis J, Yan SD, Hamm K, Xu F, Parisi M, LaRue B, Hu HW, Spijkers P, Guo H, Song XM, Lenting PJ, Van Nostrand WE, Zlokovic BV. LRP/amyloid beta-peptide interaction mediates differential brain efflux of A beta isoforms. Neuron 2004:43;333–44. [DOI] [PubMed] [Google Scholar]
- 31.Jaeger LB, Dohgu S, Sultana R, Lynch JL, Owen JB, Erickson MA, Shah GN, Price TO, Fleegal-Demotta MA, Butterfield DA, Banks WA. Lipopolysaccharide alters the blood-brain barrier transport of amyloid beta protein: a mechanism for inflammation in the progression of Alzheimer’s disease. Brain Behav Immun 2009:23;507–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zipser BD, Johanson CE, Gonzalez L, Berzin TM, Tavares R, Hulette CM, Vitek MP, Hovanesian V, Stopa EG. Microvascular injury and blood-brain barrier leakage in Alzheimer’s disease. Neurobiol Aging 2007:28;977–86. [DOI] [PubMed] [Google Scholar]
- 33.Brown WR, Thore CR. Review: cerebral microvascular pathology in ageing and neurodegeneration. Neuropathol Appl Neurobiol 2011:37;56–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Buee L, Hof PR, Bouras C, Delacourte A, Perl DP, Morrison JH, Fillit HM. Pathological alterations of the cerebral microvasculature in Alzheimer’s disease and related dementing disorders. Acta Neuropathol 1994:87;469–80. [DOI] [PubMed] [Google Scholar]
- 35.Viggars AP, Wharton SB, Simpson JE, Matthews FE, Brayne C, Savva GM, Garwood C, Drew D, Shaw PJ, Ince PG. Alterations in the blood brain barrier in ageing cerebral cortex in relationship to Alzheimer-type pathology: a study in the MRC-CFAS population neuropathology cohort. Neurosci Lett 2011:505;25–30. [DOI] [PubMed] [Google Scholar]
- 36.Barzo P, Marmarou A, Fatouros P, Corwin F, Dunbar J. Magnetic resonance imaging-monitored acute blood-brain barrier changes in experimental traumatic brain injury. J Neurosurg 1996:85;1113–21. [DOI] [PubMed] [Google Scholar]
- 37.Habgood MD, Bye N, Dziegielewska KM, Ek CJ, Lane MA, Potter A, Morganti-Kossmann C, Saunders NR. Changes in blood-brain barrier permeability to large and small molecules following traumatic brain injury in mice. Eur J Neurosci 2007:25;231–8. [DOI] [PubMed] [Google Scholar]
- 38.Baldwin SA, Fugaccia I, Brown DR, Brown LV, Scheff SW. Blood-brain barrier breach following cortical contusion in the rat. J Neurosurg 1996:85;476–81. [DOI] [PubMed] [Google Scholar]
- 39.Shapira Y, Setton D, Artru AA, Shohami E. Blood-brain barrier permeability, cerebral edema, and neurologic function after closed head injury in rats. Anesth Analg 1993:77;141–8. [DOI] [PubMed] [Google Scholar]
- 40.Rinder L, Olsson Y. Studies on vascular permeability changes in experimental brain concussion. I. Distribution of circulating fluorescent indicators in brain and cervical cord after sudden mechanical loading of the brain. Acta Neuropathologica 1968:11;183–200. [DOI] [PubMed] [Google Scholar]
- 41.Shreiber DI, Smith DH, Meaney DF. Immediate in vivo response of the cortex and the blood-brain barrier following dynamic cortical deformation in the rat. Neurosci Lett 1999:259;5–8. [DOI] [PubMed] [Google Scholar]
- 42.Smith DH, Soares HD, Pierce JS, Perlman KG, Saatman KE, Meaney DF, Dixon CE, McIntosh TK. A model of parasagittal controlled cortical impact in the mouse: cognitive and histopathologic effects. J Neurotrauma 1995:12;169–78. [DOI] [PubMed] [Google Scholar]
- 43.Cortez SC, McIntosh TK, Noble LJ. Experimental fluid percussion brain injury: vascular disruption and neuronal and glial alterations. Brain Res 1989:482;271–82. [DOI] [PubMed] [Google Scholar]
- 44.Ommaya AK, Rockoff SD, Baldwin M. Experimental Concussion; a First Report. J J Neurosurg 1964:21;249–65. [DOI] [PubMed] [Google Scholar]
- 45.Hekmatpanah J, Hekmatpanah CR. Microvascular alterations following cerebral contusion in rats. Light, scanning, and electron microscope study. J Neurosurg 1985:62;888–97. [DOI] [PubMed] [Google Scholar]
- 46.Hicks RR, Smith DH, Lowenstein DH, Saint Marie R, McIntosh TK. Mild experimental brain injury in the rat induces cognitive deficits associated with regional neuronal loss in the hippocampus. J Neurotrauma 1993:10;405–14. [DOI] [PubMed] [Google Scholar]
- 47.Povlishock JT, Becker DP, Sullivan HG, Miller JD. Vascular-Permeability Alterations to Horseradish-Peroxidase in Experimental Brain Injury. Brain Res 1978:153;223–39. [DOI] [PubMed] [Google Scholar]
- 48.Baskaya MK, Rao AM, Dogan A, Donaldson D, Dempsey RJ. The biphasic opening of the blood-brain barrier in the cortex and hippocampus after traumatic brain injury in rats. Neurosci Lett 1997:226;33–6. [DOI] [PubMed] [Google Scholar]
- 49.Glushakova OY, Johnson D, Hayes RL. Delayed increases in microvascular pathology after experimental traumatic brain injury are associated with prolonged inflammation, blood-brain barrier disruption, and progressive white matter damage. J Neurotrauma 2014:31;1180–93. [DOI] [PubMed] [Google Scholar]
- 50.Saw MM, Chamberlain J, Barr M, Morgan MP, Burnett JR, Ho KM. Differential disruption of blood-brain barrier in severe traumatic brain injury. Neurocrit Care 2014:20;209–16. [DOI] [PubMed] [Google Scholar]
- 51.Stahel PF, Morganti-Kossmann MC, Perez D, Redaelli C, Gloor B, Trentz O, Kossmann T. Intrathecal levels of complement-derived soluble membrane attack complex (sC5b-9) correlate with blood-brain barrier dysfunction in patients with traumatic brain injury. J Neurotrauma 2001:18;773–81. [DOI] [PubMed] [Google Scholar]
- 52.Ho KM, Honeybul S, Yip CB, Silbert BI. Prognostic significance of blood-brain barrier disruption in patients with severe nonpenetrating traumatic brain injury requiring decompressive craniectomy. J Neurosurg 2014:121;674–9. [DOI] [PubMed] [Google Scholar]
- 53.Blyth BJ, Farhavar A, Gee C, Hawthorn B, He H, Nayak A, Stocklein V, Bazarian JJ. Validation of serum markers for blood-brain barrier disruption in traumatic brain injury. J Neurotrauma 2009:26;1497–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tomkins O, Feintuch A, Benifla M, Cohen A, Friedman A, Shelef I. Blood-brain barrier breakdown following traumatic brain injury: a possible role in posttraumatic epilepsy. Cardiovasc Psychiatry Neurol 2011:2011;765923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Weissberg I, Veksler R, Kamintsky L, Saar-Ashkenazy R, Milikovsky DZ, Shelef I, Friedman A. Imaging Blood-Brain Barrier Dysfunction in Football Players. Jama Neurol 2014:71;1453–5. [DOI] [PubMed] [Google Scholar]
- 56.Povlishock JT, Kontos HA, Wei EP, Rosenblum WI, Becker DP. Changes in the cerebral vasculature after hypertension and trauma: a combined scanning and transmission electron microscopic analysis. Adv Exp Med Biol 1980:131;227–41. [DOI] [PubMed] [Google Scholar]
- 57.Wei EP, Dietrich WD, Povlishock JT, Navari RM, Kontos HA. Functional, morphological, and metabolic abnormalities of the cerebral microcirculation after concussive brain injury in cats. Circulation research 1980:46;37–47. [DOI] [PubMed] [Google Scholar]
- 58.Maxwell WL, Irvine A, Adams JH, Graham DI, Gennarelli TA. Response of cerebral microvasculature to brain injury. J Pathol 1988:155;327–35. [DOI] [PubMed] [Google Scholar]
- 59.Vaz R, Sarmento A, Borges N, Cruz C, Azevedo T. Experimental traumatic cerebral contusion: morphological study of brain microvessels and characterization of the oedema. Acta neurochirurgica 1998:140;76–81. [DOI] [PubMed] [Google Scholar]
- 60.Rodriguez-Baeza A, Reina-de la Torre F, Poca A, Marti M, Garnacho A. Morphological features in human cortical brain microvessels after head injury: a three-dimensional and immunocytochemical study. Anat Rec A Discov Mol Cell Evol Biol 2003:273;583–93. [DOI] [PubMed] [Google Scholar]
- 61.Vaz R, Sarmento A, Borges N, Cruz C, Azevedo I. Ultrastructural study of brain microvessels in patients with traumatic cerebral contusions. Acta Neurochir 1997:139;215–20. [DOI] [PubMed] [Google Scholar]
- 62.Maxwell WL, Whitfield PC, Suzen B, Graham DI, Adams JH, Watt C, Gennarelli TA. The cerebrovascular response to experimental lateral head acceleration. Acta Neuropath 1992:84;289–96. [DOI] [PubMed] [Google Scholar]
- 63.Zhang Y, Pardridge WM. Mediated efflux of IgG molecules from brain to blood across the blood-brain barrier. J Neuroimmunol 2001:114;168–72. [DOI] [PubMed] [Google Scholar]
- 64.Grammas P, Moore P, Weigel PH. Microvessels from Alzheimer’s disease brains kill neurons in vitro. Am J Pathol 1999:154;337–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hirao K, Ohnishi T, Hirata Y, Yamashita F, Mori T, Moriguchi Y, Matsuda H, Nemoto K, Imabayashi E, Yamada M, Iwamoto T, Arima K, Asada T. The prediction of rapid conversion to Alzheimer’s disease in mild cognitive impairment using regional cerebral blood flow SPECT. Neuroimage 2005:28;1014–21. [DOI] [PubMed] [Google Scholar]
- 66.Johnson NA, Jahng GH, Weiner MW, Miller BL, Chui HC, Jagust WJ, Gorno-Tempini ML, Schuff N. Pattern of cerebral hypoperfusion in Alzheimer disease and mild cognitive impairment measured with arterial spin-labeling MR imaging: initial experience. Radiology 2005:234;851–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, Yarasheski KE, Bateman RJ. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science 2010:330;1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sutcliffe JG, Hedlund PB, Thomas EA, Bloom FE, Hilbush BS. Peripheral reduction of beta-amyloid is sufficient to reduce brain beta-amyloid: implications for Alzheimer’s disease. J Neurosci Res 2011:89;808–14. [DOI] [PubMed] [Google Scholar]
- 69.Eisele YS, Obermuller U, Heilbronner G, Baumann F, Kaeser SA, Wolburg H, Walker LC, Staufenbiel M, Heikenwalder M, Jucker M. Peripherally applied Abeta-containing inoculates induce cerebral beta-amyloidosis. Science 2010:330;980–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.van Oijen M, Witteman JC, Hofman A, Koudstaal PJ, Breteler MM. Fibrinogen is associated with an increased risk of Alzheimer disease and vascular dementia. Stroke 2005:36;2637–41. [DOI] [PubMed] [Google Scholar]
- 71.Xu G, Zhang H, Zhang S, Fan X, Liu X. Plasma fibrinogen is associated with cognitive decline and risk for dementia in patients with mild cognitive impairment. IntJ Clin Pract 2008:62;1070–5. [DOI] [PubMed] [Google Scholar]
- 72.Paul J, Strickland S, Melchor JP. Fibrin deposition accelerates neurovascular damage and neuroinflammation in mouse models of Alzheimer’s disease. J Exp Med 2007:204;1999–2008. [DOI] [PMC free article] [PubMed] [Google Scholar]